
Use of silylmethoxy groups as inducers of efficient room temperature phosphorescence from precious-metal-free organic luminophores†
Masaki
Shimizu
 *,
Takumi
Kinoshita
,
Ryosuke
Shigitani
,
Yusuke
Miyake
and
Kunihiko
Tajima
*,
Takumi
Kinoshita
,
Ryosuke
Shigitani
,
Yusuke
Miyake
and
Kunihiko
Tajima
Faculty of Molecular Chemistry and Engineering, Kyoto Institute of Technology, 1 Hashikami-cho, Matsugasaki, Sakyo-ku, Kyoto 606-8585, Japan. E-mail: mshimizu@kit.ac.jp
First published on 14th December 2017
Abstract
We designed and characterized 1,4-diaroyl-2,5-bis(silylmethoxy)benzenes as precious-metal-free organic luminophores that efficiently phosphoresce at room temperature. The benzene derivatives in crystals emit green phosphorescence with quantum yields up to 0.45 under ambient conditions. The luminescence quantum yield increases with increasing number of intermolecular interactions in the crystal, such as hydrogen bonding and CH–π interactions. The luminescence lifetimes are inversely proportional to temperature over the −80 to 80 °C range, demonstrating the potential of the benzene derivatives as materials for temperature sensing. Poly(methyl methacrylate) films doped with these luminophores also exhibit intense green phosphorescence at room temperature under vacuum, while they emit very faint blue fluorescence under ambient conditions. Electron spin resonance spectroscopy of a UV-excited diphenylmethylsilyl-derivative in toluene at 77 K reveals a triplet diradical species, whose electronic distribution is similar to that of naphthalene, indicating that the triplet diradical is distributed over almost ten atoms.
Introduction
Phosphorescence is a radiative transition from the lowest triplet excited state (T1) to the ground state (S0). Due to its spin-forbidden nature, the lifetime of phosphorescence is much longer than that of fluorescence. Accordingly, room temperature phosphorescence (RTP) serves as a versatile tool for time-resolved bio-imaging and bio-sensing, because the effects of the short-lived auto-fluorescence background and fluorescent non-target species are easily excluded.1 The T1 energy is readily transferred to triplet molecular oxygen in the ground state, which results in the quenching of phosphorescence and the generation of singlet molecular oxygen. Hence, luminophores exhibiting RTP can be applied to molecular oxygen sensing2 and photodynamic therapy.3 Therefore, the design and development of organic luminophores that exhibit highly efficient RTP are crucial for advances in areas such as analytical technologies and medicine.To achieve efficient RTP, first, intersystem crossing (ISC) from the singlet excited state (S1) to T1 needs to be faster than internal conversion and fluorescence, both of which are spin-allowed transitions from S1 to S0. Since ISC is promoted by large spin–orbit coupling, the incorporation of heavy atoms such as iodine or bromine, precious metals such as iridium or platinum, or arylcarbonyl groups into luminophores effectively accelerates ISC and efficiently generates T1. While precious metals provide the additional advantage of accelerating the rate of phosphorescence, they are limited and expensive resources, and their complexes are toxic. Therefore, increasing attention is being paid to the development of precious-metal-free organic phosphors.4 However, as the rate of radiative decay (phosphorescence) of a metal-free phosphor is very slow, its RTP is easily quenched through non-radiative decay processes such as molecular vibrations that occur at room temperature, and collisions with the surrounding molecules. Accordingly, the construction of rigid environments that suppress possible intramolecular phosphor motion is the second requirement for achieving efficient RTP. Early approaches that satisfied this requirement involved the adsorption of phosphors onto solid substrates such as filter paper or silica gel, and the inclusion of phosphors into cyclodextrin or surfactants, although most of these studies did not disclose the luminescence efficiency of the resulting RTP.5 Recent approaches include the crystallization of phosphors6 and the co-crystallization of phosphors with low molecular weight molecules that fix the phosphor conformation through intermolecular interactions such as halogen- or hydrogen-bonding;7 these approaches have led to the development of several highly efficient RTP materials. The dispersion or cross-linking of phosphors in a polymer matrix is the third approach for facilitating efficient RTP.8 Considering that polymer films exhibit good processabilities that are not available to crystals and have found many bio- and chemical sensing applications,9 the development of phosphorescent polymers is also an intriguing subject of investigation from the viewpoint of practical applications. On the other hand, most phosphors that exhibit efficient RTP in crystals are non- or poorly phosphorescent when dispersed in polymer films. Therefore, the creation of a polymer film that exhibits efficient RTP with a metal-free phosphor as the dopant is an important frontier in the field of materials chemistry.
We recently demonstrated that crystals of 1,4-dibenzoyl-2,5-disiloxybenzenes 1 exhibit RTP with excellent quantum yields of 0.46–0.64 and persistent lifetimes of 76.0–98.3 ms (Scheme 1).10 Considering that T1 states with longer lifetimes, in theory, increase the chance of non-radiative de-excitation, the simultaneous realization of excellent luminescence efficiencies (Φ > 0.4) and long lifetimes (τ > 10 ms) during RTP is unusual. In other words, the efficient RTP of 1 indicates that the T1 state of 1 is very stable against radiationless relaxation. X-ray diffraction analysis of single crystals of 1 disclosed that each molecule in the crystal is locked through several intermolecular interactions, such as hydrogen bonding and CH–π interactions, leading to the inhibition of thermal vibrations. Based on electron spin resonance studies and time-dependent density functional theory (TD-DFT) calculations, we proposed that the T1 of 1 is generated as a triplet diradical species T1 (1), in which one radical develops over the oxygen of the silyl ether moiety, and is electronically stabilized by σ–n conjugation with the Si–C σ-bond attached to the ether oxygen. We then realized that reversing the arrangement of the Si and C atoms attached to the ether oxygen atom (see T1 (2) in Scheme 1) should also provide similar stabilization to the oxyradical in T1 by σ–n conjugation.11 Hence, we envisaged that 1,4-diaroyl-2,5-bis(silylmethoxy)benzenes 2 would serve as new RTP materials; indeed, this proved to be the case. Herein, we report the preparation, structures, and photophysical properties of 2, and demonstrate that crystals of 2 in air, and 2 doped in poly(methyl methacrylate) (PMMA) films under vacuum, exhibit green RTP with high quantum yields and persistent lifetimes.
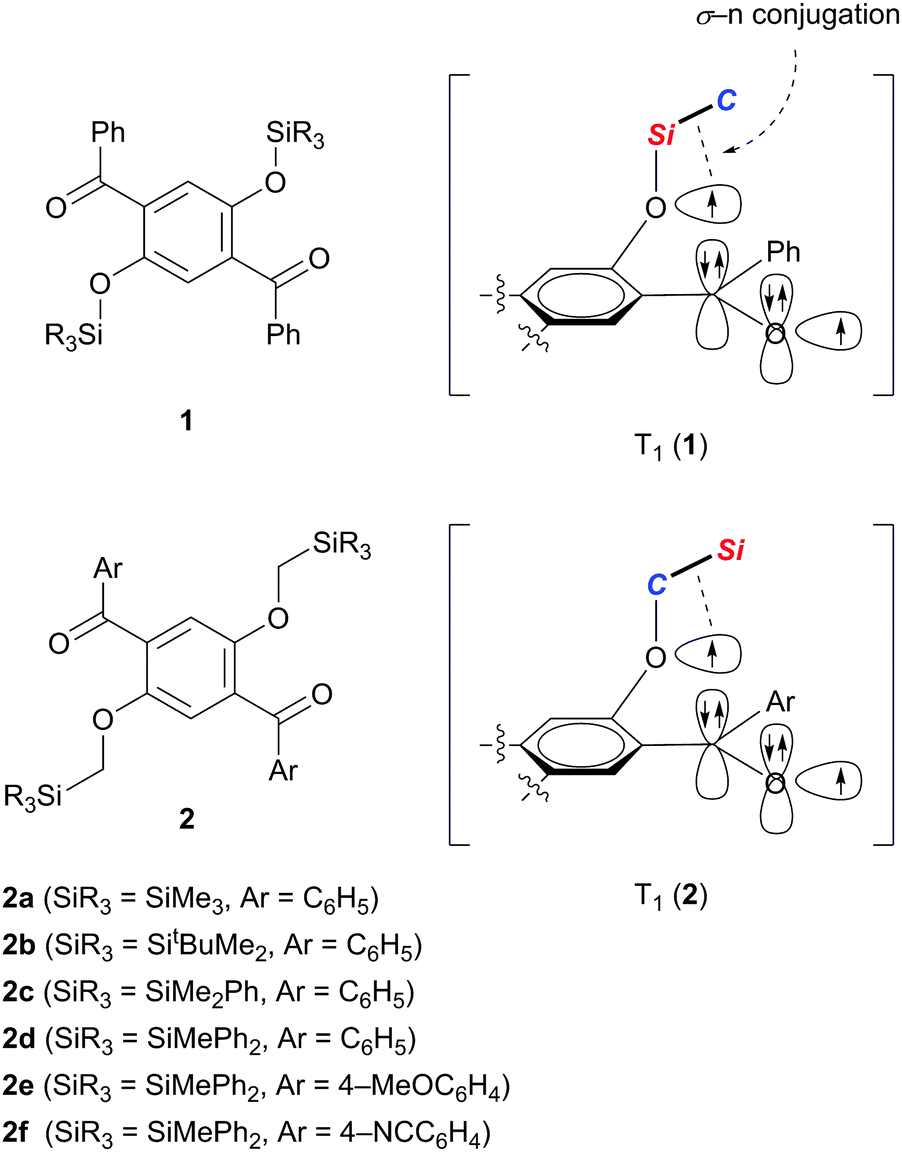 | ||
| Scheme 1 Molecular structures and orbital descriptions of the T1 states of 1 and 2. | ||
Results and discussion
Preparation and structures
The designed luminophores 2 were prepared from 1,4-diaroyl-2,5-dihydroxybenzenes 3 and R3SiCH2Cl in the presence of K2CO3 in DMF upon heating at 90–100 °C (Scheme 2). The moderate yields of 2d, 2e, and 2f are ascribed to the formation of the corresponding 1,4-diaroyl-2,5-dimethoxybenzenes, which are presumably formed through the expected silylmethylation followed by desilylation.12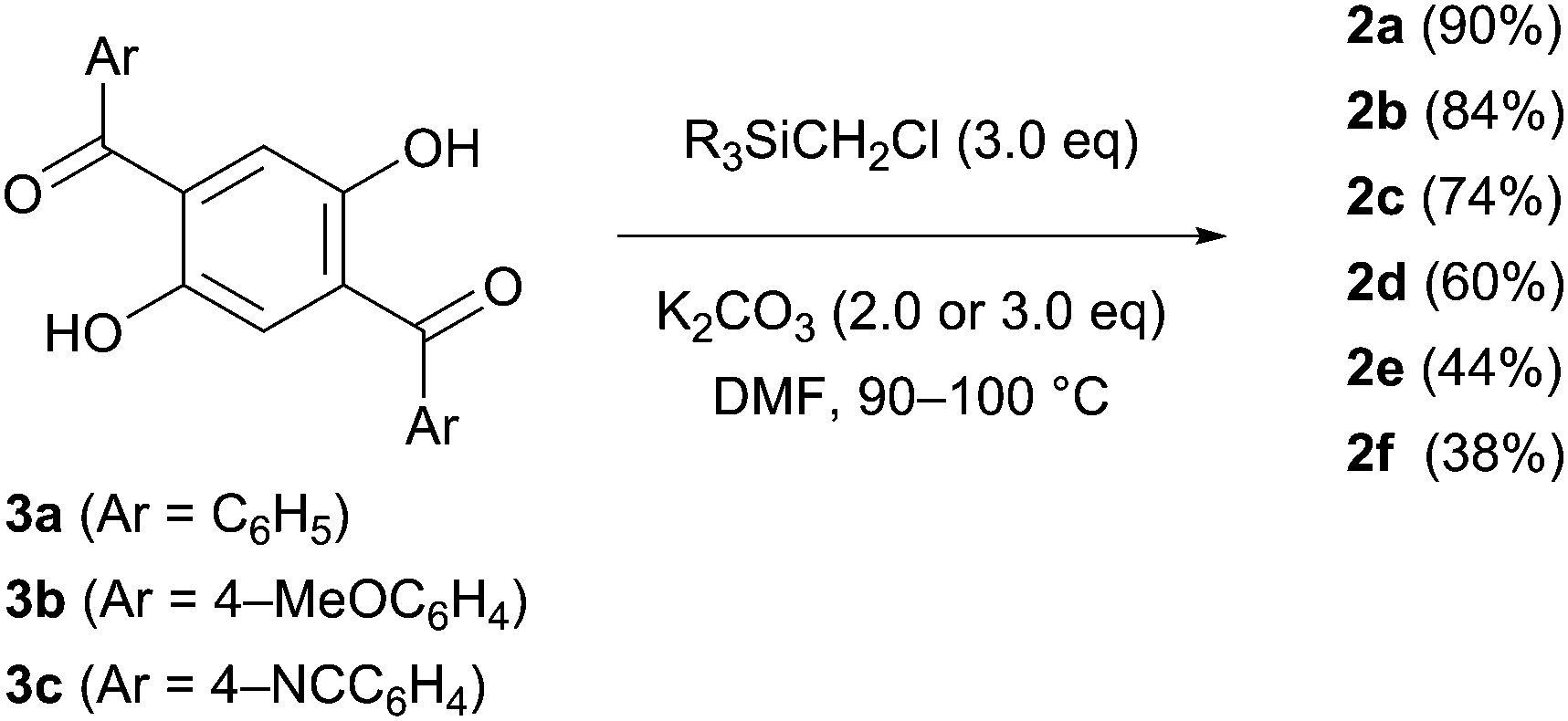 | ||
| Scheme 2 Preparation of 2. | ||
Single crystals of 2a, 2b, and 2d, suitable for X-ray diffraction analysis, were obtained by recrystallization from a mixed solution of hexane and CH2Cl2;132a, 2b, and 2d adopt similar conformations in which the two Si–C–O linkages and the central benzene ring are arranged in an almost coplanar fashion, and the two aroyl groups are twisted in vertically opposite directions with respect to the central benzene ring. The molecular structure of 2d, as a representative example, is shown in Fig. 1; the structures of 2a and 2b are available in the ESI.†
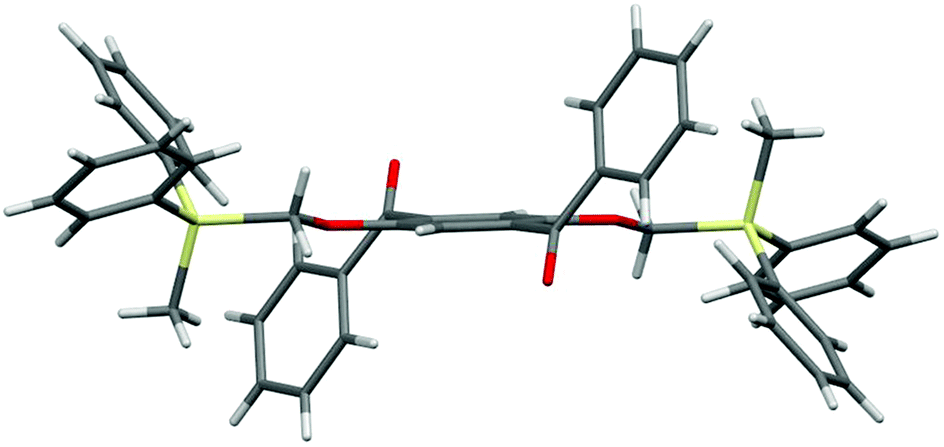 | ||
| Fig. 1 Molecular structure of 2d. | ||
Crystal packing diagrams of 2a, 2b, and 2d are shown in Fig. 2. Each molecule of 2a interacts with the surrounding molecules through eight hydrogen bonds (Fig. 2a), suggesting that the molecular motions/vibrations of 2a in the crystal are relatively restricted. A unit cell of the crystal of 2b contains two molecules, each of which forms only two hydrogen bonds (Fig. 2b), indicating that multiple molecular motion/vibration opportunities exist that result in the non-radiative decay of T1. Four hydrogen bonds and eight CH–π interactions are observed per molecule in the crystal of 2d (Fig. 2c); consequently, the conformation of 2d in the crystal is firmly locked. Thus, the number of intermolecular interactions which is an important factor in enhancing the conformational rigidity of the phosphor in the crystal varies according to the triorganosilyl group.
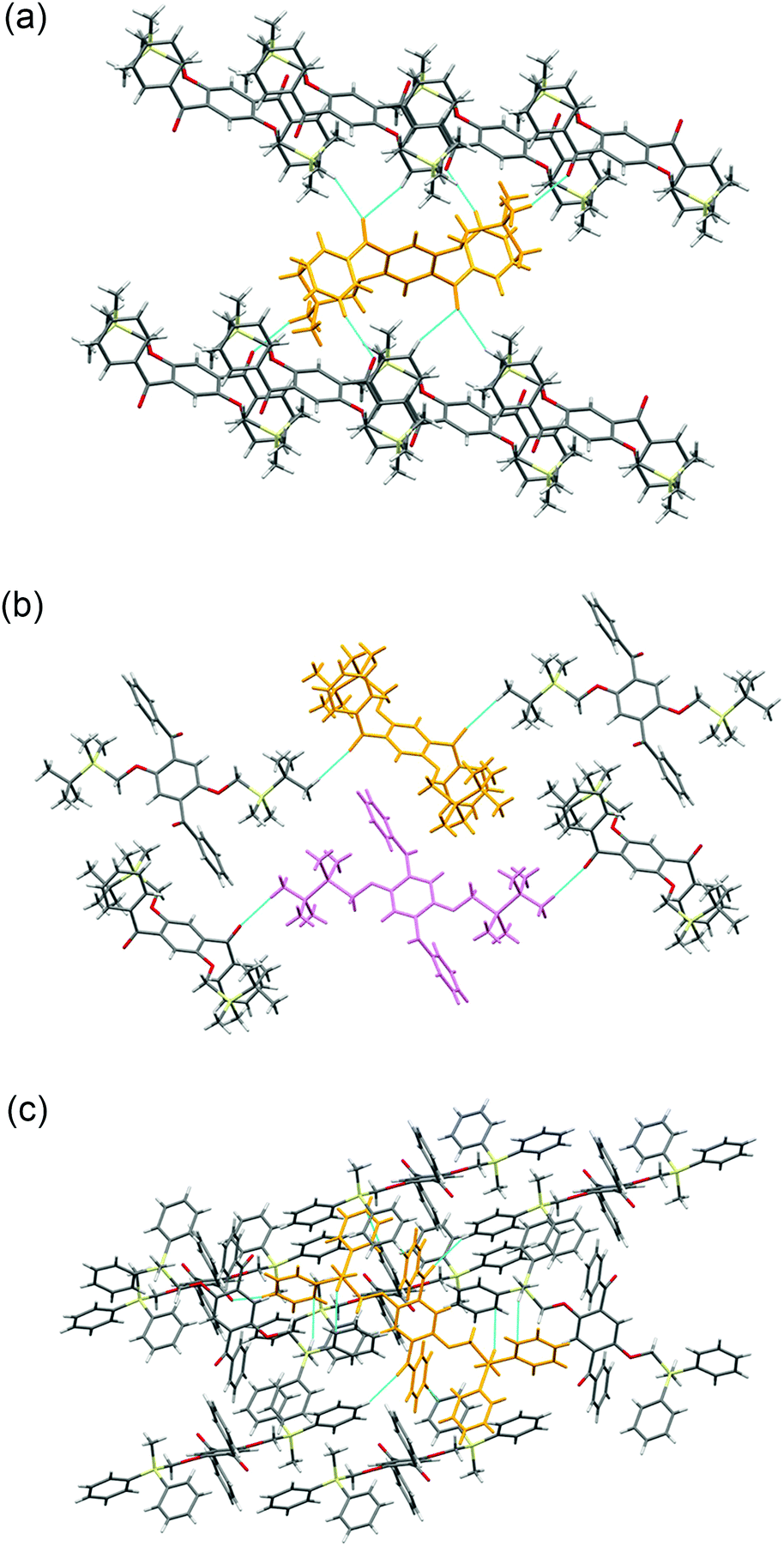 | ||
| Fig. 2 Crystal packing diagrams of (a) 2a, (b) 2b, and (c) 2d. The dotted blue lines indicate intermolecular interactions. | ||
Photophysical properties
Absorption spectra of 2 acquired in 2-methyltetrahydrofuran (2-MeTHF) at 298 K are displayed in Fig. 3. The spectra of dibenzoyl derivatives 2a–2d (Ar = C6H5) are almost identical, with absorption maxima at 345–348 nm. Among these compounds, the 4-methoxy- and 4-cyanophenylcarbonyl derivatives 2e and 2f exhibit blue- and red-shifted spectra, respectively, suggesting that the weak absorption bands observed for 2 involve intramolecular charge-transfer (ICT) transitions from ether oxygen atoms to the aroyl groups. Compared to the absorption spectra of 1 that exhibit absorption maxima at 330–337 nm,14 the spectra of 2 are red-shifted, implying that the electron-donating ability of a silylmethyl-substituted oxygen is slightly greater than that of a silyl ether oxygen for ICT.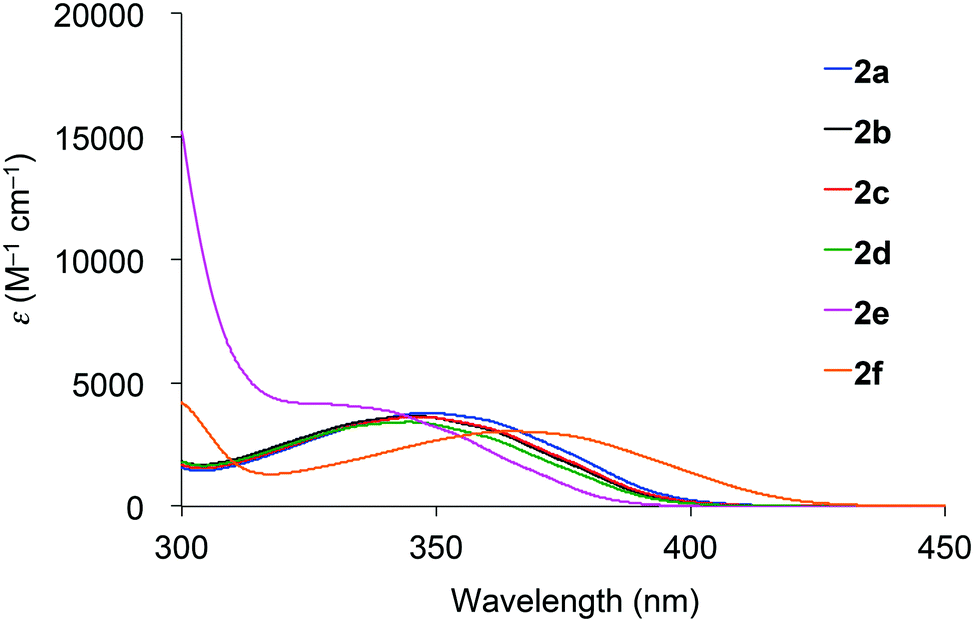 | ||
| Fig. 3 Absorption spectra of 2 in 2-MeTHF at 298 K. | ||
Crystals of 2a and 2c–2e are green luminescent under ambient conditions with good-to-high quantum yields (Table 1). On the other hand, no RTP was observed with crystalline 2b or 2f.15 It is remarkable that a quantum yield in excess of 0.4 and a lifetime longer than 10 ms are simultaneously exhibited by 2d. The excellent quantum yield (0.45) of 2d is similar to that (0.46) of 1 (SiR3 = SiPh3, Ar = C6H5) and much higher than that (0.14) of 1,4-dibenzoyl-2,5-dibromobenzenes.16 Accordingly, the Ph2MeSiCH2O group serves as a superior inducer of RTP in 1,4-diaroylbenzene-based π-conjugated systems. Fig. 4 displays a luminescence image of 2d in the crystal and the luminescence spectra of 2a and 2c–2e, which exhibit small bands at 426–440 nm and strong bands at 513–522 nm. Short and long luminescence lifetimes,17 which are of the order of nano- and milliseconds, respectively, are observed for each luminescent crystal. Similar photoluminescence features, involving two emission bands in the blue and green regions, with nano- and millisecond-order lifetimes, are observed in the luminescence spectra of 2 in 2-MeTHF at 77 K (Table 1 and Fig. S3, ESI†). Therefore, we conclude that the green emissions from crystals of 2 are due to phosphorescence, while the small bands at shorter wavelengths are due to prompt fluorescence. The quantum yields of crystalline 2a, 2b, and 2d, the crystal structures of which are described above, were determined to be 0.15, 0.02, and 0.45, respectively (Table 1). The magnitude of the quantum yield is related to the number of intermolecular interactions observed in the crystal-packed structures. This relationship clearly demonstrates that intermolecular interactions in the crystal are factors that suppress non-radiative decay processes, i.e., facilitation of efficient RTP.
| 2 | Crystals at 298 K | In 2-MeTHF at 77 K | In PMMA films at 298 K | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Under vacuum | In air | |||||||||||||
| λ em (nm) | Φ | τ F (ns) | τ P (ms) | λ em (nm) | Φ | τ F (ns) | τ P (ms) | λ em (nm) | Φ | τ F (ns) | τ P (ms) | λ em (nm) | Φ | |
| a Excited at 330 nm for 2a–2d, 320 nm for 2e, and 350 nm for 2f. Absolute quantum yields (Φ) were determined using a calibrated integrating sphere system. The luminescence lifetimes (τ) were determined as intensity-averaged lifetimes when the emission decay was multi-exponential. b NE: no emission. | ||||||||||||||
| 2a | 426, 513 | 0.15 | 0.1 | 12 | 429, 521 | 0.78 | 2.2 | 175 | 541 | 0.17 | 0.8 | 92 | 450 | 0.02 |
| 2b | 437, 517 | 0.02 | — | — | 423, 521 | 0.79 | 1.6 | 167 | 537 | 0.17 | 0.9 | 94 | 457 | 0.02 |
| 2c | 433, 517 | 0.24 | 0.2 | 32 | 426, 517 | 0.79 | 1.5 | 167 | 540 | 0.18 | 0.8 | 99 | 463 | 0.02 |
| 2d | 435, 516 | 0.45 | 0.1 | 58 | 430, 521 | 0.78 | 1.8 | 163 | 530 | 0.17 | 0.7 | 88 | 466 | 0.02 |
| 2e | 442, 522 | 0.22 | 0.4 | 26 | 420, 505 | 0.92 | 0.4 | 102 | 523 | 0.24 | 0.3 | 71 | 433 | 0.01 |
| 2f | NEb | — | — | — | 467, 533 | 0.64 | 6.3 | 141 | 549 | 0.13 | 0.9 | 69 | 468 | 0.02 |
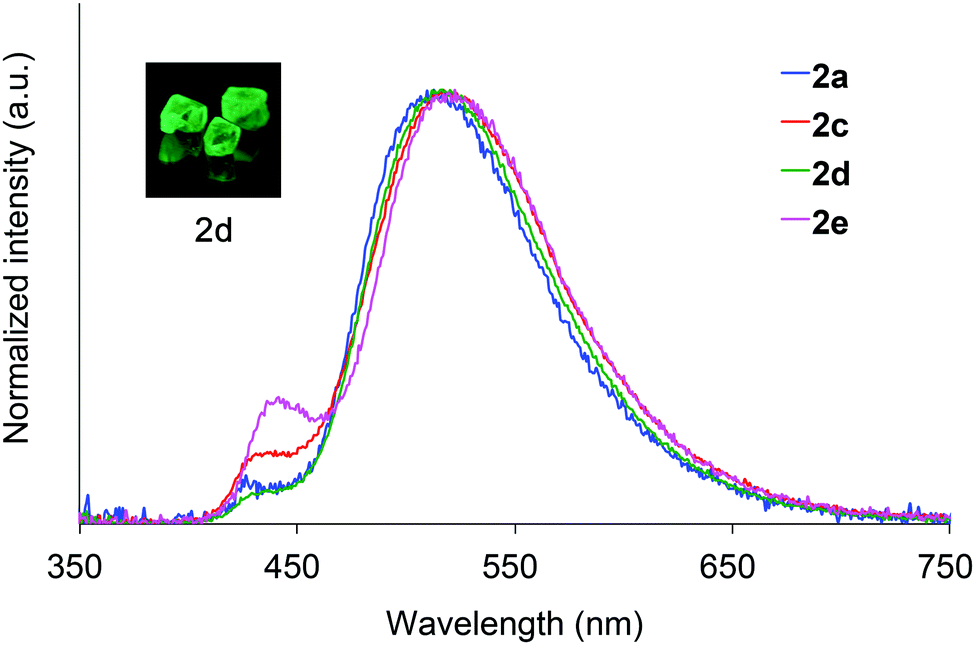 | ||
| Fig. 4 Photoluminescence spectra of crystalline 2a and 2c–2e, and a luminescence image of 2d. | ||
With crystals of 2d exhibiting RTP with the highest quantum yield in this study, we investigated the relationship between the phosphorescence lifetime and temperature over the −80 to 80 °C range. As shown in Fig. 5, the lifetime is essentially inversely proportional to temperature, with a correlation coefficient (R2) of 0.99186. Hence, compounds 2 are potentially excellent materials for phosphorescence-based temperature sensing.18
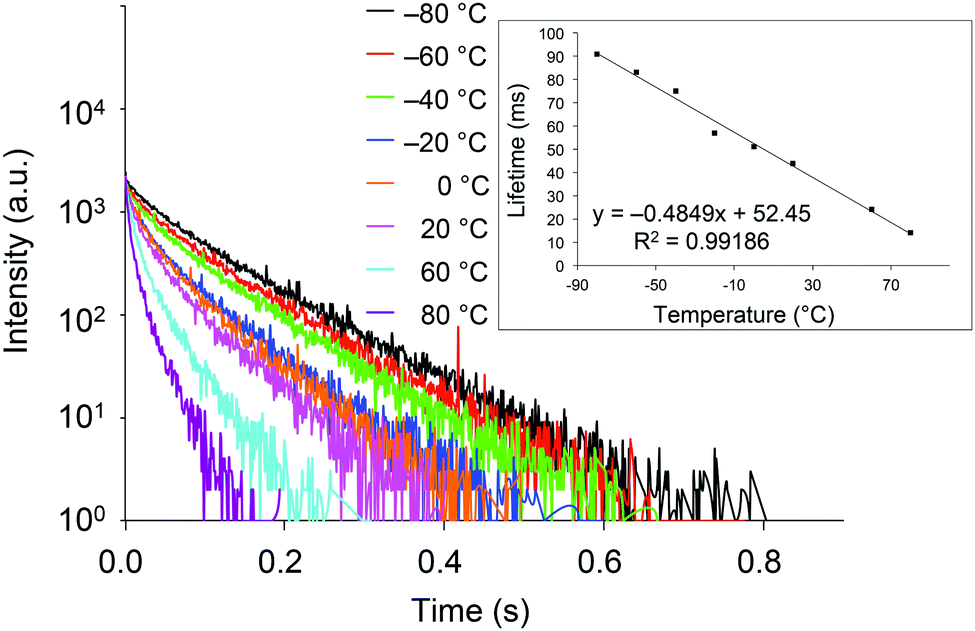 | ||
| Fig. 5 Transient luminescence decay curves of crystalline 2d at various temperatures. Inset: Temperature dependence of the phosphorescence lifetime. | ||
We next investigated the photoluminescence of 2 dispersed in PMMA films at 298 K. PMMA films doped with 1.0 wt% of 2 were prepared in a cuvette equipped with a J. Young high-vacuum valve. Under vacuum, all 2, including 2b and 2f whose crystals showed no emissions, exhibited efficient green RTP with long lifetimes (Table 1 and Fig. 6), along with weak prompt fluorescence in the blue region. The luminescence quantum yields of the doped films ranged from 0.13 to 0.24, which are as high as those of other polymer films doped with precious-metal-free phosphors.8 The brilliantly emissive behavior of 2 in polymer films contrasts sharply with that of 1, since 1 dispersed in polymer films was almost non-emissive under vacuum.19 When the green-emissive films of 2 under vacuum were exposed to air (oxygen), the green color immediately disappeared and a faint blue emission (λem = 433–468 nm and Φ = 0.01–0.02) was observed instead.20 The spectra of 2 (except for 2d) in PMMA films under vacuum and in air are shown in Fig. S4–S8 (ESI†). Evacuation of the blue-emissive cuvette restored the intense green emission. The spectra and luminescence images of a 2d-doped PMMA film are shown in Fig. 7 as typical examples. Hence, the RTP of 2 in PMMA films are very sensitive to molecular oxygen as anticipated from the intrinsic nature of their T1 states. The spectral differences in the absence and presence of molecular oxygen are significant. Accordingly, 2-doped polymer films may serve as useful media for oxygen sensing.2
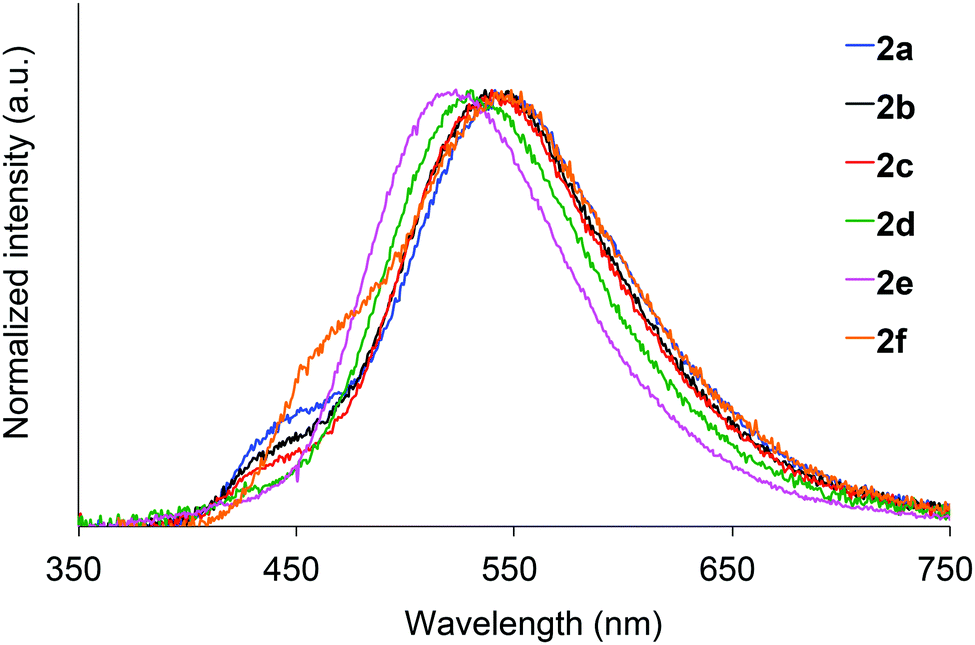 | ||
| Fig. 6 Photoluminescence spectra of 2 dispersed in PMMA films under vacuum at 298 K. | ||
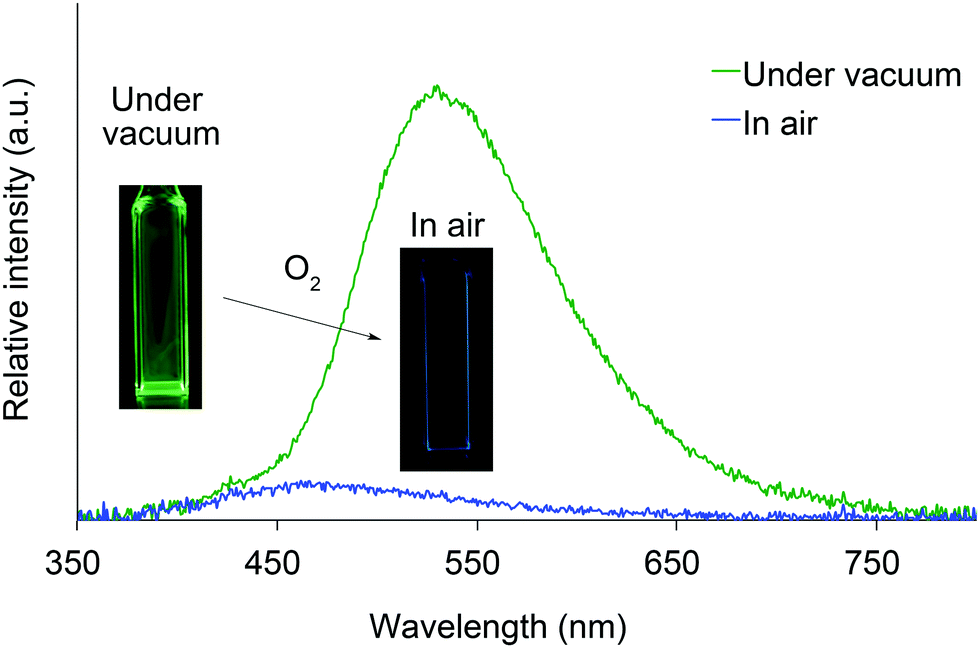 | ||
| Fig. 7 Photoluminescence spectra and luminescence images of 2d dispersed in a PMMA film under vacuum and in air at 298 K. | ||
The electron spin resonance (ESR) spectrum of 2d irradiated by UV was measured in toluene at 77 K to confirm the generation of a triplet diradical species (Fig. 8). There are six signals under magnetic fields of approximately 230, 260, 290, 350, 385, and 415 mT, which are derived from the ΔmS = ±1 transitions of the electron spins in the triplet state. The zero splitting parameters D and E were determined to be 0.088 and 0.010 cm−1, respectively. The intense signal near 166 mT is derived from the ΔmS = ±2 transition, and its g value was determined to be 3.95. As the D and E parameters are comparable to those of the triplet state of 1 (SiR3 = SitBuPh2 and Ar = C6H5) and naphthalene in biphenyl,21 the electron distributions of the diradicals in the triplet state of 2 are very similar to those of 1 and naphthalene, indicating that the distribution is spread over almost ten atoms. These results indicate that the silylmethoxy groups in 2 are essential components of the triplet diradical species in the same manner as the siloxy groups are in 1.
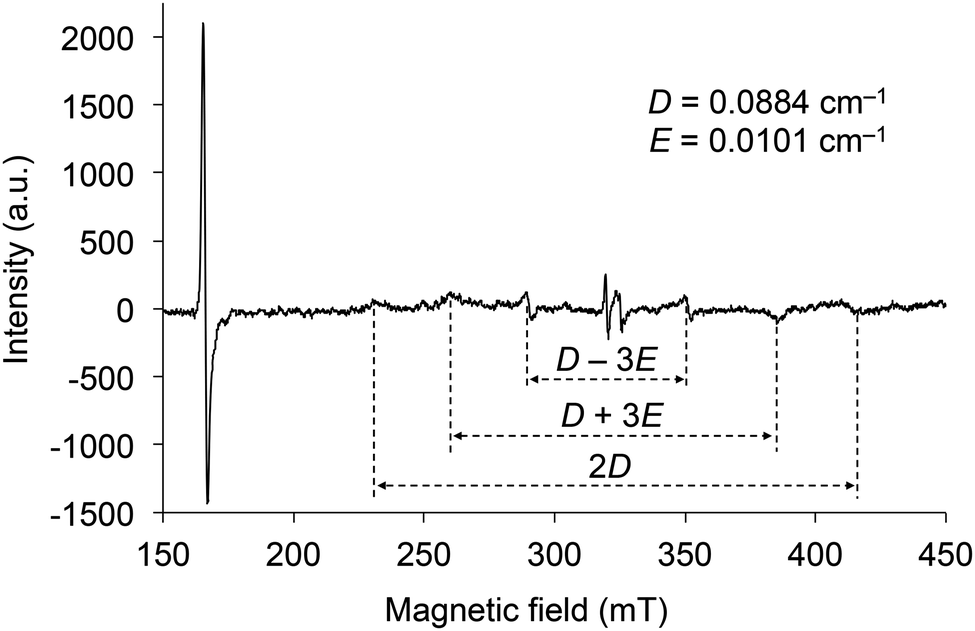 | ||
| Fig. 8 X-band ESR spectrum of 2d irradiated by UV in toluene at 77 K. | ||
Experimental
General
Melting and decomposition points were determined with a TG/DTA6200 Seiko Instrument. 1H NMR spectra were recorded with a Varian Mercury 400 (400 MHz) spectrometer. The chemical shifts of 1H NMR signals are expressed in parts per million relative to the internal tetramethylsilane standard (δ = 0 ppm) or chloroform (δ = 7.26 ppm). Splitting patterns are indicated as follows: s, singlet; d, doublet; t, triplet; and m, multiplet. 13C NMR spectra were recorded with a Varian Mercury 400 (100 MHz) spectrometer with tetramethylsilane (δ = 0 ppm) or chloroform-d (δ = 77.0 ppm) as an internal standard. Chemical shifts are given in parts per million relative to the internal standards. IR attenuated total reflectance (IRATR) spectra were recorded using a Shimadzu FTIR-8400 spectrometer equipped with a Specac® Quest ATR diamond accessory. Fast atom bombardment high-resolution mass spectrometry (FAB-HRMS) analyses were performed with a JEOL JMS-700 spectrometer. TLC analyses were performed using Merck Kieselgel 60 F254 silica plates. Silica gel column chromatography was carried out using a Merck Kieselgel 60 (230–400 meshes). All reactions were carried out under argon. Compounds 3a–3c were prepared according to a reported procedure.10Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to obtain 2a (0.22 g, 0.44 mmol, 90% yield) as a colorless solid.
:1) to obtain 2a (0.22 g, 0.44 mmol, 90% yield) as a colorless solid.
2a: M.p.: 180 °C. Td: 259 °C. TLC: Rf 0.27 (hexane/EtOAc 20:1). 1H NMR (CDCl3, 400 MHz): δ −0.26 (s, 18H), 3.46 (s, 4H), 7.14 (s, 2H), 7.45 (t, J = 7.6 Hz, 4H), 7.56 (t, J = 7.6 Hz, 2H), 7.83 (d, J = 7.6 Hz, 4H). 13C NMR (CDCl3, 100 MHz): δ −3.6, 62.1, 112.5, 128.4, 129.6, 131.4, 133.1, 138.0, 152.9, 196.7. IR: ν = 3059, 2951, 2899, 2880, 2826, 1659, 1599, 1582, 1485, 1451, 1398, 1240, 1190, 1015, 951, 845, 818, 756, 723, 692, 679 cm−1. HRMS (FAB): [M + H]+ calcd for C28H35O4Si2, 491.2074; found, 491.2066.
2b: M.p.: 224 °C. Td: 279 °C. TLC: Rf 0.46 (hexane/EtOAc 10:1). 1H NMR (CDCl3, 400 MHz): δ −0.35 (s, 12H), 0.71 (s, 18H), 3.53 (s, 4H), 7.13 (s, 2H), 7.44 (t, J = 7.6 Hz, 4H), 7.56 (tt, J = 7.6, 1.2 Hz, 2H), 7.84 (dd, J = 7.6, 1.2 Hz, 4H). 13C NMR (CDCl3, 100 MHz): δ −7.9, 16.1, 26.4, 59.9, 112.2, 128.4, 129.8, 131.4, 133.2, 137.6, 152.8, 196.7. IR: ν = 3051, 2949, 2926, 2882, 2855, 1665, 1593, 1580, 1487, 1468, 1449, 1400, 1239, 1196, 1017, 951, 835, 820, 799, 772, 725, 691, 683 cm−1. HRMS (FAB): [M + H]+ calcd for C34H47O4Si2, 575.3013; found, 575.3014.
2c: M.p.: 150 °C. Td: 332 °C. TLC: Rf 0.29 (hexane/EtOAc 10:1). 1H NMR (CDCl3, 400 MHz): δ −0.01 (s, 12H), 3.65 (s, 4H), 7.11 (s, 2H), 7.22–7.34 (m, 10H), 7.43 (t, J = 7.6 Hz, 4H), 7.56 (tt, J = 7.6, 1.2 Hz, 2H), 7.83 (dd, J = 7.6, 1.2 Hz, 4H). 13C NMR (CDCl3, 100 MHz): δ −5.2, 61.6, 112.5, 127.8, 128.4, 129.4, 129.7, 131.5, 133.2, 133.7, 136.2, 137.7, 152.7, 196.5. IR: ν = 3067, 2961, 2907, 2882, 2826, 1663, 1597, 1582, 1485, 1449, 1427, 1402, 1240, 1200, 1115, 1018, 951, 839, 818, 768, 723, 691, 681 cm−1. HRMS (FAB): [M]+ calcd for C38H38O4Si2, 614.2309; found, 614.2307.
2d: M.p.: 215 °C. Td: 358 °C. TLC: Rf 0.39 (toluene). 1H NMR (CDCl3, 400 MHz): δ 0.21 (s, 6H), 3.95 (s, 4H), 7.14 (s, 2H), 7.22–7.36 (m, 24H), 7.50 (t, J = 7.6 Hz, 2H), 7.77 (d, J = 7.6 Hz, 4H); 13C NMR (CDCl3, 100 MHz): δ −6.3, 60.9, 112.5, 127.9, 128.4, 129.7, 129.7, 131.6, 133.3, 134.2, 134.6, 137.5, 152.6, 196.3. IR: ν = 3067, 3050, 2999, 2953, 2895, 2820, 1657, 1595, 1582, 1483, 1451, 1427, 1398, 1234, 1190, 1111, 1020, 955, 812, 770, 723, 689, 681, 667 cm−1. HRMS (FAB): [M]+ calcd for C48H42O4Si2, 738.2622; found, 738.2629.
2e: M.p.: 215 °C. Td: 367 °C. TLC: Rf 0.45 (hexane/EtOAc 5:2). 1H NMR (CDCl3, 400 MHz): δ 0.29 (s, 6H), 3.84 (s, 6H), 3.96 (s, 4H), 6.81 (d, J = 8.4 Hz, 4H), 7.11 (s, 2H), 7.24 (t, J = 7.6 Hz, 8H), 7.29–7.35 (m, 12H), 7.76 (d, J = 8.4 Hz, 4H). 13C NMR (CDCl3, 100 MHz): δ −6.2, 55.4, 60.8, 112.3, 113.6, 127.8, 129.6, 130.5, 131.7, 132.2, 134.3, 134.6, 152.3, 163.7, 194.9. IR: ν = 3069, 3051, 3011, 2965, 2928, 2907, 2884, 2838, 1645, 1595, 1572, 1510, 1487, 1425, 1397, 1250, 1192, 1171, 1111, 1017, 955, 845, 833, 812, 779, 737, 698 cm−1. HRMS (FAB): [M + H]+ calcd for C50H47O6Si2, 799.2911; found, 799.2917.
2f: M.p.: 217 °C. Td: 366 °C. TLC: Rf 0.38 (hexane/EtOAc 4:1). 1H NMR (CDCl3, 400 MHz): δ 0.23 (s, 6H), 3.97 (s, 4H), 7.22 (s, 2H), 7.25–7.32 (m, 16H), 7.39–7.45 (m, 8H), 7.70 (d, J = 8.0 Hz, 4H). 13C NMR (CDCl3, 100 MHz): δ −6.2, 60.6, 113.0, 116.2, 118.1, 128.1, 129.5, 130.0, 131.0, 132.0, 133.6, 134.4, 140.4, 153.0, 194.6. IR: ν = 3071, 3021, 3005, 2959, 2903, 2882, 2824, 2230, 1663, 1485, 1427, 1406, 1279, 1234, 1200, 1173, 1109, 1011, 953, 858, 823, 785, 729, 714, 692 cm−1. HRMS (FAB): [M]+ calcd for C50H40N2O4Si2, 788.2527; found, 788.2523.
X-ray diffraction analysis of single crystals
Data were measured on a Bruker SMART APEX diffractometer (MoKα radiation, λ = 0.71073 Å) at 300 K. The structures were solved by direct methods using SHELXTL programs and refined with full-matrix least-squares on F2.Measurement of the absorption and luminescence spectra
2-Methyltetrahydrofuran (2-MeTHF), purchased from TCI Chemicals Co. Ltd, was distilled and degassed with argon before use. The absorption spectra were measured with a Shimadzu UV-2550 spectrometer. The luminescence spectra and absolute quantum yields were recorded using a calibrated integrating sphere with a Hamamatsu Photonics C9920-02 Absolute PL Quantum Yield Measurement System, and the luminescence lifetimes were measured with a Hamamatsu Photonics Quantaurus-Tau instrument. The temperature for lifetime measurements was controlled with a cryostat system, CoolSpeK UV USP-203 (UNISOKU Co., Ltd).Preparation of doped PMMA films
PMMA (99 mg, purchased from Wako Chemicals, no. 138-02735), CH2Cl2 (1 mL), and then the sample (1 mg) were added to a glass tube. The resulting solution was injected into a cuvette equipped with a J. Young high-vacuum valve. The cuvette was placed horizontally and slowly evacuated while the solution was gently swung up and down the sides of the cuvette. After thorough evaporation of the solvent, the cuvette was maintained under vacuum overnight to yield a thin film of doped PMMA. The photoluminescence properties of the films under vacuum were measured directly using the evacuated cuvette.Electron spin resonance (ESR) measurements
Compound 2d was dissolved in distilled toluene and sealed in a silica tube of 5 mm diameter under vacuum. Electron spin resonance spectra of 2d at 77 K were measured during UV light irradiation of the sample. X-band ESR spectra were recorded using a JEOL TE100 spectrometer. Static magnetic fields were calibrated with standard samples. A spot light mercury lamp (LC8, Hamamatsu) equipped into the ESR cavity was used as the photoexcitation light source (280–400 nm).Conclusions
We demonstrated that 1,4-diaroyl-2,5-bis(silylmethoxy)benzenes are new precious-metal-free phosphors that exhibit green RTP with good-to-high quantum yields and persistent lifetimes in their crystalline states. The RTP-inducing effect of the Ph2MeSiCH2O group is similar to that of Ph3SiO, and much better than that of Br, a typical heavy atom that promotes phosphorescence. The luminescence lifetimes of the crystals are inversely proportional to temperature with an almost perfect correlation coefficient. These luminescence characteristics make these phosphors attractive candidates for use in lifetime-based temperature sensing applications. Furthermore, these phosphors exhibit efficient RTP under vacuum even when they are dispersed in PMMA films. Considering that 1,4-dibenzoyl-2,5-disiloxybenzenes in polymer films did not exhibit any RTP, the realization of RTP in doped-polymer films is a remarkable feature of these silylmethoxy-based phosphors. Further modifications of the developed phosphors and their applications to time-resolved imaging and sensing are the subject of further research.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research on Innovative Areas “π-System Figuration: Control of Electron and Structural Dynamism for Innovative Functions” from MEXT, Japan, JSPS KAKENHI Grant Number 15H03795, the Nagase Science and Technology Foundation, and the Ogasawara Foundation for the Promotion of Science and Engineering.Notes and references
- (a) Q. Zhao, C. Huang and F. Li, Chem. Soc. Rev., 2011, 40, 2508–2524 RSC; (b) G. Marriott, R. M. Clegg, D. J. Arndt-Jovin and T. M. Jovin, Biophys. J., 1991, 60, 1374–1387 CrossRef CAS PubMed.
- (a) E. Roussakis, Z. Li, A. J. Nichols and C. L. Evans, Angew. Chem., Int. Ed., 2015, 54, 8340–8362 CrossRef CAS PubMed; (b) X.-d. Wang and O. S. Wolfbeis, Chem. Soc. Rev., 2014, 43, 3666–3761 RSC; (c) M. Quaranta, S. M. Borisov and I. Klimant, Bioanal. Rev., 2012, 4, 115–157 CrossRef PubMed; (d) G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747–751 CrossRef CAS PubMed; (e) P. Lehner, C. Staudinger, S. M. Borisov and I. Klimant, Nat. Commun., 2014, 5, 4460 CAS; (f) C. A. DeRosa, S. A. Seaman, A. S. Mathew, C. M. Gorick, Z. Fan, J. N. Demas, S. M. Peirce and C. L. Fraser, ACS Sensors, 2016, 1, 1366–1373 Search PubMed.
- (a) J. P. Celli, B. Q. Spring, I. Rizvi, C. L. Evans, K. S. Samkoe, S. Verma, B. W. Pogue and T. Hasan, Chem. Rev., 2010, 110, 2795–2838 CrossRef CAS PubMed; (b) J. M. Dabrowski and L. G. Arnaut, Photochem. Photobiol. Sci., 2015, 14, 1765–1780 RSC; (c) W. Fan, P. Huang and X. Chen, Chem. Soc. Rev., 2016, 45, 6488–6519 RSC.
- Reviews on metal-free organic phosphors: (a) S. Mukherjee and P. Thilagar, Chem. Commun., 2015, 51, 10988–11003 RSC; (b) Y. Liu, G. Zhan, Z.-W. Liu, Z.-Q. Bian and C.-H. Huang, Chin. Chem. Lett., 2016, 27, 1231–1240 CrossRef CAS; (c) S. Wang, W. Z. Yuan and Y. Zhang, in Aggregation-Induced Emission: Materials and Applications Volume 2, American Chemical Society, 2016, vol. 1227, ch. 1, pp. 1–26 Search PubMed; (d) M. Baroncini, G. Bergamini and P. Ceroni, Chem. Commun., 2017, 53, 2081–2093 RSC . See also; (e) S. Hirata, Adv. Opt. Mater., 2017, 5, 1700116 CrossRef; (f) Q. Li and Z. Li, Adv. Sci., 2017, 4, 1600484 CrossRef PubMed.
- (a) E. M. Schulman and C. Walling, Science, 1972, 178, 53–54 CAS; (b) E. M. Schulman and C. Walling, J. Phys. Chem., 1973, 77, 902–905 CrossRef CAS; (c) R. A. Paynter, S. L. Wellons and J. D. Winefordner, Anal. Chem., 1974, 46, 736–738 CrossRef CAS; (d) S. L. Wellons, R. A. Paynter and J. D. Winefordner, Spectrochim. Acta, Part A, 1974, 30, 2133–2140 CrossRef; (e) C. D. Ford and R. J. Hurtubise, Anal. Chem., 1980, 52, 656–662 CrossRef CAS; (f) S. M. Ramasamy and R. J. Hurtubise, Anal. Chem., 1982, 54, 2477–2481 CrossRef CAS; (g) L. J. C. Love, M. Skrilec and J. G. Habarta, Anal. Chem., 1980, 52, 754–759 CrossRef CAS; (h) S. Scypinski and L. J. Cline Love, Anal. Chem., 1984, 56, 322–327 CrossRef CAS; (i) S. Scypinski and L. J. Cline Love, Anal. Chem., 1984, 56, 331–336 CrossRef CAS; (j) A. Muñoz de la Peña, M. C. Mahedero and A. Bautista Sánchez, Analusis, 2000, 28, 670–678 CrossRef; (k) J. Joseph and A. A. Anappara, Phys. Chem. Chem. Phys., 2017, 19, 15137–15144 RSC.
- (a) C.-R. Wang, Y.-Y. Gong, W.-Z. Yuan and Y.-M. Zhang, Chin. Chem. Lett., 2016, 27, 1184–1192 CrossRef CAS; (b) W. Z. Yuan, X. Y. Shen, H. Zhao, J. W. Y. Lam, L. Tang, P. Lu, C. Wang, Y. Liu, Z. Wang, Q. Zheng, J. Z. Sun, Y. Ma and B. Z. Tang, J. Phys. Chem. C, 2010, 114, 6090–6099 CrossRef CAS; (c) G.-P. Yong, Y.-M. Zhang, W.-L. She and Y.-Z. Li, J. Mater. Chem., 2011, 21, 18520–18522 RSC; (d) Y. Gong, Y. Tan, H. Li, Y. Zhang, W. Yuan, Y. Zhang, J. Sun and B. Z. Tang, Sci. China: Chem., 2013, 56, 1183–1186 CrossRef CAS; (e) S. K. Maity, S. Bera, A. Paikar, A. Pramanik and D. Haldar, Chem. Commun., 2013, 49, 9051–9053 RSC; (f) Y. Gong, L. Zhao, Q. Peng, D. Fan, W. Z. Yuan, Y. Zhang and B. Z. Tang, Chem. Sci., 2015, 6, 4438–4444 RSC; (g) J. Li, Y. Jiang, J. Cheng, Y. Zhang, H. Su, J. W. Y. Lam, H. H. Y. Sung, K. S. Wong, H. S. Kwok and B. Z. Tang, Phys. Chem. Chem. Phys., 2015, 17, 1134–1141 RSC; (h) S. Kuno, H. Akeno, H. Ohtani and H. Yuasa, Phys. Chem. Chem. Phys., 2015, 17, 15989–15995 RSC; (i) P. Xue, J. Sun, P. Chen, P. Wang, B. Yao, P. Gong, Z. Zhang and R. Lu, Chem. Commun., 2015, 51, 10381–10384 RSC; (j) G.-P. Yong, C. Shen, C. Zhang and Y.-M. Zhao, J. Mater. Chem. C, 2015, 3, 9048–9052 RSC; (k) Y. Gong, G. Chen, Q. Peng, W. Z. Yuan, Y. Xie, S. Li, Y. Zhang and B. Z. Tang, Adv. Mater., 2015, 27, 6195–6201 CrossRef CAS PubMed; (l) A. Sakai, E. Ohta, Y. Matsui, S. Tsuzuki and H. Ikeda, ChemPhysChem, 2016, 17, 4033–4036 CrossRef CAS PubMed; (m) W. Zhao, Z. He, Jacky W. Y. Lam, Q. Peng, H. Ma, Z. Shuai, G. Bai, J. Hao and Ben Z. Tang, Chemistry, 2016, 1, 592–602 CrossRef CAS; (n) H. Shi, Z. An, P. Z. Li, J. Yin, G. Xing, T. He, H. Chen, J. Wang, H. Sun, W. Huang and Y. Zhao, Cryst. Growth Des., 2016, 16, 808–813 CrossRef CAS; (o) Z. He, W. Zhao, J. W. Y. Lam, Q. Peng, H. Ma, G. Liang, Z. Shuai and B. Z. Tang, Nat. Commun., 2017, 8, 416 CrossRef PubMed; (p) N.-N. Zhang, C. Sun, X.-M. Jiang, X.-S. Xing, Y. Yan, L.-Z. Cai, M.-S. Wang and G.-C. Guo, Chem. Commun., 2017, 53, 9269–9272 RSC; (q) E. Lucenti, A. Forni, C. Botta, L. Carlucci, C. Giannini, D. Marinotto, A. Previtali, S. Righetto and E. Cariati, J. Phys. Chem. Lett., 2017, 8, 1894–1898 CrossRef CAS PubMed; (r) S. Kuno, T. Kanamori, Z. Yijing, H. Ohtani and H. Yuasa, ChemPhotoChem, 2017, 1, 102–106 CrossRef CAS; (s) J. Yang, Z. Ren, Z. Xie, Y. Liu, C. Wang, Y. Xie, Q. Peng, B. Xu, W. Tian, F. Zhang, Z. Chi, Q. Li and Z. Li, Angew. Chem., Int. Ed., 2017, 56, 880–884 CrossRef CAS PubMed; (t) Y. Xie, Y. Ge, Q. Peng, C. Li, Q. Li and Z. Li, Adv. Mater., 2017, 29, 1606829 CrossRef PubMed; (u) B. Xu, H. Wu, J. Chen, Z. Yang, Z. Yang, Y.-C. Wu, Y. Zhang, C. Jin, P.-Y. Lu, Z. Chi, S. Liu, J. Xu and M. Aldred, Chem. Sci., 2017, 8, 1909–1914 RSC; (v) H. Wu, C. Hang, X. Li, L. Yin, M. Zhu, J. Zhang, Y. Zhou, H. Agren, Q. Zhang and L. Zhu, Chem. Commun., 2017, 53, 2661–2664 RSC; (w) Y. Shoji, Y. Ikabata, Q. Wang, D. Nemoto, A. Sakamoto, N. Tanaka, J. Seino, H. Nakai and T. Fukushima, J. Am. Chem. Soc., 2017, 139, 2728–2733 CrossRef CAS PubMed.
- (a) O. Bolton, K. Lee, H. J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205–210 CrossRef CAS PubMed; (b) H. Y. Gao, X. R. Zhao, H. Wang, X. Pang and W. J. Jin, Cryst. Growth Des., 2012, 12, 4377–4387 CrossRef CAS; (c) X. Pang, H. Wang, X. R. Zhao and W. J. Jin, CrystEngComm, 2013, 15, 2722–2730 RSC; (d) H. Wang, R. X. Hu, X. Pang, H. Y. Gao and W. J. Jin, CrystEngComm, 2014, 16, 7942–7948 RSC; (e) O. Bolton, D. Lee, J. Jung and J. Kim, Chem. Mater., 2014, 26, 6644–6649 CrossRef CAS; (f) S. d’Agostino, F. Grepioni, D. Braga and B. Ventura, Cryst. Growth Des., 2015, 15, 2039–2045 CrossRef; (g) D. Lee, J. Jung, D. Bilby, M. S. Kwon, J. Yun and J. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 2993–2997 CrossRef CAS PubMed; (h) J. Wei, B. Liang, R. Duan, Z. Cheng, C. Li, T. Zhou, Y. Yi and Y. Wang, Angew. Chem., Int. Ed., 2016, 55, 15589–15593 CrossRef CAS PubMed; (i) K. Murakami, Y. Ooyama, S. Watase, K. Matsukawa, S. Omagari, T. Nakanishi, Y. Hasegawa, K. Inumaru and J. Ohshita, Chem. Lett., 2016, 45, 502–504 CrossRef CAS.
- (a) D. Lee, O. Bolton, B. C. Kim, J. H. Youk, S. Takayama and J. Kim, J. Am. Chem. Soc., 2013, 135, 6325–6329 CrossRef CAS PubMed; (b) S. Hirata, K. Totani, J. Zhang, T. Yamashita, H. Kaji, S. R. Marder, T. Watanabe and C. Adachi, Adv. Funct. Mater., 2013, 23, 3386–3397 CrossRef CAS; (c) M. S. Kwon, D. Lee, S. Seo, J. Jung and J. Kim, Angew. Chem., Int. Ed., 2014, 53, 11177–11181 CrossRef CAS PubMed; (d) M. S. Kwon, Y. Yu, C. Coburn, A. W. Phillips, K. Chung, A. Shanker, J. Jung, G. Kim, K. Pipe, S. R. Forrest, J. H. Youk, J. Gierschner and J. Kim, Nat. Commun., 2015, 6, 8947 CrossRef PubMed; (e) M. S. Kwon, J. H. Jordahl, A. W. Phillips, K. Chung, S. Lee, J. Gierschner, J. Lahann and J. Kim, Chem. Sci., 2016, 7, 2359–2363 RSC; (f) H. Chen, X. Yao, X. Ma and H. Tian, Adv. Opt. Mater., 2016, 4, 1397–1401 CrossRef CAS; (g) X. Chen, C. Xu, T. Wang, C. Zhou, J. Du, Z. Wang, H. Xu, T. Xie, G. Bi, J. Jiang, X. Zhang, J. N. Demas, C. O. Trindle, Y. Luo and G. Zhang, Angew. Chem., Int. Ed., 2016, 55, 9872–9876 CrossRef CAS PubMed; (h) T. Zhang, H. Chen, X. Ma and H. Tian, Ind. Eng. Chem. Res., 2017, 56, 3123–3128 CrossRef CAS; (i) K. Yoshida, Y. Kuwahara, K. Miyamoto, S. Nakashima, H. Jintoku, M. Takafuji and H. Ihara, Chem. Commun., 2017, 53, 5044–5047 RSC; (j) R. Huang, J. Avo, T. Northey, E. Chaning-Pearce, P. L. dos Santos, J. S. Ward, P. Data, M. K. Etherington, M. A. Fox, T. J. Penfold, M. N. Berberan-Santos, J. C. Lima, M. R. Bryce and F. B. Dias, J. Mater. Chem. C, 2017, 5, 6269–6280 RSC.
- W. Guan, W. Zhou, J. Lu and C. Lu, Chem. Soc. Rev., 2015, 44, 6981–7009 RSC.
- M. Shimizu, R. Shigitani, M. Nakatani, K. Kuwabara, Y. Miyake, K. Tajima, H. Sakai and T. Hasobe, J. Phys. Chem. C, 2016, 120, 11631–11639 CAS.
- (a) M. Kira, H. Nakazawa and H. Sakurai, Chem. Lett., 1986, 497–500 CrossRef CAS; (b) J. Yoshida, T. Maekawa, T. Murata, S. Matsunaga and S. Isoe, J. Am. Chem. Soc., 1990, 112, 1962–1970 CrossRef CAS.
- At first, we attempted the silylmethylation of 3 using NaH as a base and dimethylsulfoxide as solvent, according to a reported procedure; J. J. Eisch, J. E. Galle, A. Piotrowski and M. R. Tsai, J. Org. Chem., 1982, 47, 5051–5056 CrossRef CAS . However, the products were only 1,4-diaroyl-2,5-dimethoxybenzenes. Changing the solvent to dimethylformamide allowed us to obtain the desired products 2.
- CCDC 1570763, 1570805, and 1570764 contain the supplementary crystallographic data for 2a, 2b, and 2d, respectively†.
- For example, the absorption maxima of 1a (SiR3 = SitBuMe2, Ar = C6H5), 1b (SiR3 = SitBuPh2, Ar = C6H5), and 1c (SiR3 = SiiPr3, Ar = C6H5) are 330, 331, and 337 nm, respectively.
- Under vacuum at room temperature, crystals of 2b exhibited weak fluorescence at 436 nm and strong phosphorescence at 524 nm with a total quantum yield of 0.11. On the other hand, crystals of 2f did not luminesce at all even under vacuum at room temperature.
- M. Shimizu, A. Kimura and H. Sakaguchi, Eur. J. Org. Chem., 2016, 467–473 CrossRef CAS.
- The luminescence lifetimes (τ) were determined as intensity-averaged lifetimes when the luminescence decay is multi-exponential. B. Valeur, Molecular Fluorescence–Principles and Applications, Wiley-VCH, Weinheim, 2002, pp. 171–172 Search PubMed.
- (a) E. J. McLaurin, L. R. Bradshaw and D. R. Gamelin, Chem. Mater., 2013, 25, 1283–1292 CrossRef CAS; (b) S. Uchiyama, A. Prasanna de Silva and K. Iwai, J. Chem. Educ., 2006, 83, 720–727 CrossRef CAS.
- The observation that 1 in polymer films were almost non-luminescent is unpublished.
- The spectra of 2 in PMMA films under vacuum and in air are shown in the ESI†.
- D values were 0.099 (1) and 0.09921 (naphthalene in biphenyl), and E values were 0.016 (1) and 0.01548 (naphthalene in biphenyl). R. W. Brandon, R. E. Gerkin and C. A. Hutchison, J. Chem. Phys., 1962, 37, 447–448 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1570763, 1570764 and 1570805. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qm00524e |
| This journal is © the Partner Organisations 2018 |