
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Functional biodegradable polymers via ring-opening polymerization of monomers without protective groups
Greta
Becker
ab and
Frederik R.
Wurm
 *a
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: wurm@mpip-mainz.mpg.de
bGraduate School Materials Science in Mainz, Staudinger Weg 9, 55128 Mainz, Germany
First published on 17th September 2018
Abstract
Biodegradable polymers are of current interest and chemical functionality in such materials is often demanded in advanced biomedical applications. Functional groups often are not tolerated in the polymerization process of ring-opening polymerization (ROP) and therefore protective groups need to be applied. Advantageously, several orthogonally reactive functions are available, which do not demand protection during ROP. We give an insight into available, orthogonally reactive cyclic monomers and the corresponding functional synthetic and biodegradable polymers, obtained from ROP. Functionalities in the monomer are reviewed, which are tolerated by ROP without further protection and allow further post-modification of the corresponding chemically functional polymers after polymerization. Synthetic concepts to these monomers are summarized in detail, preferably using precursor molecules. Post-modification strategies for the reported functionalities are presented and selected applications highlighted.
 Greta Becker | Greta Becker studied biomedical chemistry at the University of Mainz, Germany, and at the Polymer Science and Engineering Department, the University of Massachusetts in Amherst, USA. She carried out her PhD research in the group of Dr Frederik R. Wurm at the Max Planck Institute for Polymer Research in Mainz, Germany and received her PhD degree in 2017. Great developed a series of degradable and polyfunctional polyphosphoesters for bioapplications. She was supported by a fellowship through funding of the Excellence Initiative (DFG/GSC 266) in the context of the graduate school of excellence “MAINZ” (Material Sciences in Mainz) and is currently working for Kuraray Europe GmbH. |
 Frederik R. Wurm | Frederik R. Wurm (Priv.-Doz. Dr habil.) is currently heading the research group “Functional Polymers” at the Max Planck Institute for Polymer Research (MPIP), Mainz (D). In his interdisciplinary research, Frederik designs polymeric materials with molecular-defined functions. Controlling the monomer sequence and chemical functionality allowed designing materials for degradable polymers, nanocarriers with controlled blood interactions, and phosphorus flame-retardants. He has published more than 150 research articles to date. He was awarded the Reimund Stadler Award in 2016, the Lecturer Award of the German Chemical Industry Fund in 2017, the European Young Chemist Award and the Georg Manecke Prize of the German Chemical Society in 2014. Frederik received his PhD in 2009 (JGU Mainz, D). After a two-year stay at EPFL (CH) as a Humboldt fellow, he joined the department “Physical Chemistry of Polymers” at MPIP and finished his habilitation in Macromolecular Chemistry in 2016. |
1. Introduction
Biodegradable polymers are of great current interest for biomedical applications, e.g. for drug and gene delivery systems, bioengineering scaffolds or as bioadhesives. They employ binding motifs within their backbone, inspired by natural biopolymers, e.g. polysaccharides, polyhydroxyalkanoates, polypeptides or (deoxy)ribonucleic acids (DNA and RNA). A broad range of synthetic biodegradable polymer classes has been developed so far, including polyesters, polyamides, polycarbonates, poly(phosphoester)s, polyphosphazenes, poly(ester amide)s, poly(ester-ether)s, poly(ester-anhydride)s, poly(ester urethane)s, poly(ester urea)s, polyacetals, polyorthoesters, polydioxanones and polyiminocarbonates, which can be obtained by step-growth polycondensation or -addition or chain-growth polymerization.1 Especially, when it comes to advanced applications, chemical functionality in such materials is demanded, e.g. to attach labels or other molecules along the backbone.With a plethora of modern catalysts, the chain growth approach has a higher control over polymer molar masses and dispersities. Different mechanisms are available, including cationic, anionic, enzymatic, coordinative and radical ring-opening polymerization (ROP). Copolymerization of different cyclic monomers with pendant alkyl or aryl groups gives access to a variety of polymeric materials with a broad range of different physical properties, e.g. varying hydrophilicity/hydrophobicity, crystallinity, solubility, mechanical strength, degradation behavior or thermal stability. Such degradable polymers are also important for the future of sustainable polymers and plastics.2 Properties and features, as well as their advantages and drawbacks of the different classes of synthetic biodegradable polymers, are beyond our scope and are extensively discussed in several reviews.2–6
While copolymerization of alkylated and arylated monomers adjusts the materials properties, fine-tuning of the polymers is often demanded for specific applications: additional attachment of bioactive molecules, redox- or pH-sensitive functionalities or cross-linkable groups might be required for their applications. On the one hand, (especially) ionic ROP might be sensitive to impurities and tolerates only certain chemical functionalities. The sensitivity to moisture and thereby the exclusion of water as reaction solvent is a drawback. On the other hand, also bioactive molecules (e.g. carbohydrates, peptides or proteins) can be sensitive or undergo side-reactions that they do not tolerate the polymerization process or conditions, e.g. organic solvents, high temperatures or required catalysts. Great effort has been made in the last decades, developing cyclic monomers with orthogonal chemical functions, which do not interfere the polymerization process. These monomers can be divided into two groups: (I) orthogonally reactive groups that do not interfere with the polymerization but can be post-modified afterward; (II) active groups, e.g. photo- or redox-active.
In this review, we summarize synthetic strategies to orthogonally reactive cyclic monomers reported in the literature that allows subsequent post-polymerization modification. We highlight the general concepts, preferably using precursor molecules, which can be used to prepare these monomers and thereby chemically functional biodegradable polymers by ROP (Table 1). A comparison on the synthetic ease of the different monomer classes will be given, that helps to choose the polymer class of choice for the desired application. We further display post-modification strategies with selected applications.
| Polymer class | General structure | Cyclic monomers |
|---|---|---|
| Polyesters |
|
Lactone |
| Macrolactone | ||
| Glycolide | ||
| Lactide | ||
| Hemilactide | ||
| O-Carboxyanhydride (OCA) | ||
| Polyamides |
|
Lactam |
| α-N-Carboxy anhydride (α-NCA) | ||
| γ-N-Carboxy anhydride (γ-NCA) | ||
| Poly(ester amide)s |
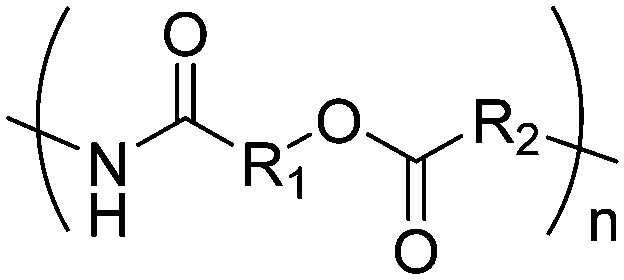
|
Esteramide |
| Polycarbonates |
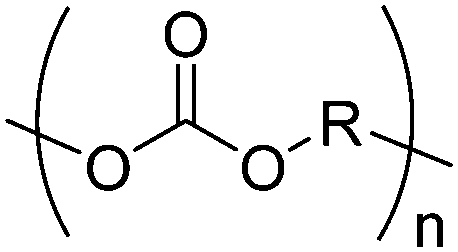
|
Trimethylene carbonate (TMC) |
| Polyphosphoesters |
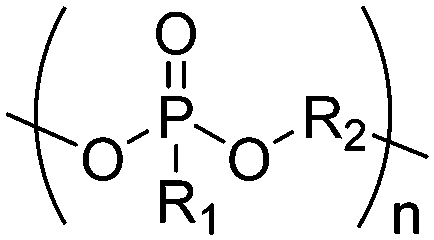
|
Phosphate |
| Phosphite | ||
| Phosphonate | ||
| Polyphosphazenes |
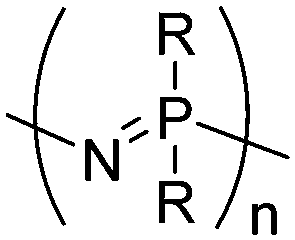
|
Hexachlorophosphazene |
The scope of the review is to be a handbook on the preparation of orthogonally reactive cyclic monomers to deliver a “toolbox” on how functional synthetic biodegradable polymers are prepared and post-modified. Tables after each section summarize the monomers discussed in the text, together with literature references and some comments.
All the herein discussed polymer classes are potentially degradable or biodegradable, due to certain linkages in the backbone. The degradation profile is one of the most important features of these polymers, depending on their area of application. Several of the examples given in this review are claimed to be degradable, due to labile ester or amide linkages in the backbone, although degradation behavior was not studied in detail. Degradation is possible by acidic, alkaline, enzymatic, microbial or oxidative cleavage of ester/amide bonds. The comparison of degradation rates and conditions is difficult, as the degradation profiles depend on various factors: the hydrophilicity or hydrophobicity, water-solubility, crystallinity, glass transition, and/or glass transition temperature, processing, size, geometry (in bulk, as foams, thin fibers, nanoparticles, micelles, in solution, etc.), porosity and water diffusion (Table 2). In addition, the degree of polymerization, sterics of any substituents, polymer architecture, and solubility of degradation products have a strong impact on the degradation rates as well. Another factor that makes the comparison even in one polymer class difficult are additional post-modifications, e.g. with hydrophobic, polar or charged groups that further alter the degradation profiles.
| Degradation | Influencing parameters of the… | ||
|---|---|---|---|
| Polymer | Sample | Procedure | |
| – Hydrolytic: | – Hydrophilicity/hydrophobicity | – Processing | – Choice of enzyme: |
| • Acidic | – Water-solubility | – Size/geometry: | • Origin |
| • Basic | – The degree of polymerization | • Bulk | • Activity |
| – Enzymatic | – The glass transition temperature | • Foam | • Selectivity |
| – Microbial | – Crystallinity | • Fibers | – Physiological/non-physiological conditions |
| – Oxidative | – Sterics of substituents | • Nanoparticles | – pH: |
| – Architecture (linear/branched/cross-linked) | • Micelles | • Acidic | |
| – The solubility of degradation products | • In solution | • Basic | |
| – Post-modifications | – Porosity | • Molarity | |
| – Water diffusion | – Buffer: | ||
| • Buffer-system | |||
| • Capacity | |||
| – Concentration | |||
| – Temperature | |||
| – In vitro/in vivo | |||
The protocols for polymer degradation are diverse and lack standardized conditions, which makes most degradation studies non-comparable with each other (detailed information can be found in a recent review).7 Most common degradation mechanism for the polymers discussed herein is hydrolysis of the polymer backbone. In most cases either acidic or basic hydrolysis are conducted under non-physiological conditions, i.e. at very low or high pH values that do not occur in natural environments. Furthermore, the chemical nature of the buffer solution, buffer-capacity, temperature, and concentration of polymer (if water-soluble) or shape of the specimen is different for most studies. For the enzymatic degradation, different enzymes can be applied, which may stem from different organisms and vary in their activity. Even batch-to-batch variations of the very same enzyme makes standardization of in vitro degradations difficult (overview of parameters shown in Table 1).
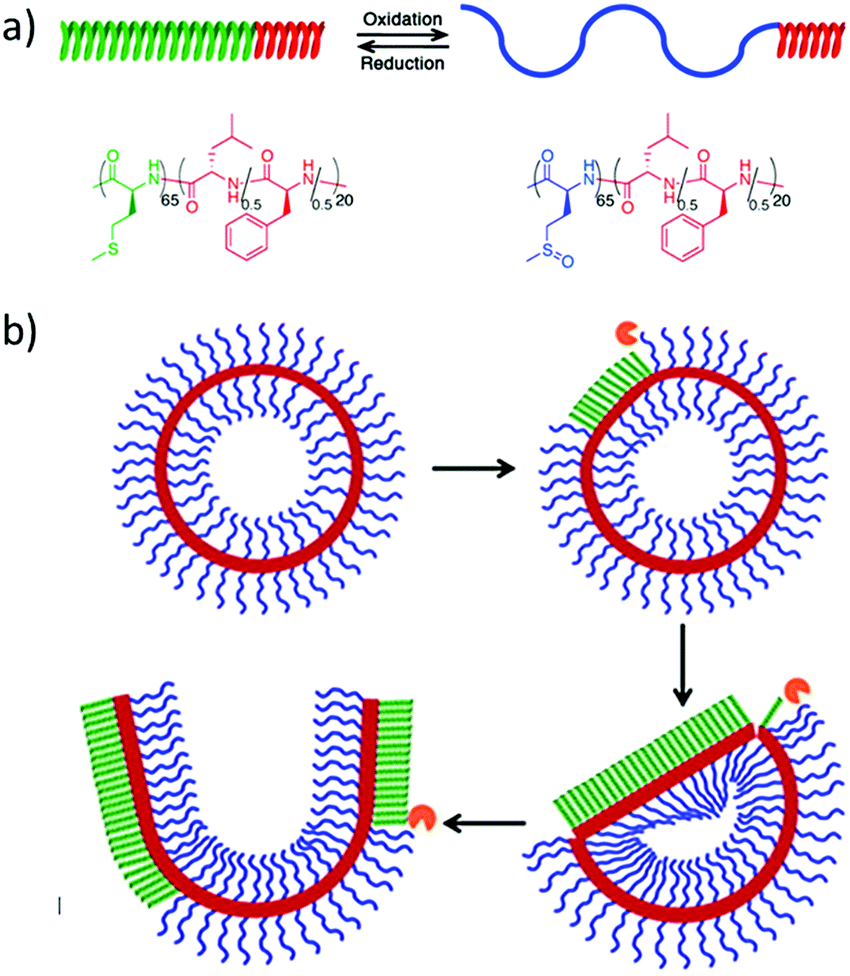
Trying to summarize some general aspects of degradation profile, herein we give some examples of non-functionalized polymers. Hydrolysis or enzymatic degradation are the typical degradation mechanism for such materials, with kinetics being very dependent on the environment and the chemical structure and/or crystallinity of the polymers. While polycaprolactone shows rather a slow degradation rate (within 2–3 years), due to its crystallinity, polylactide (depending on the chirality and composition) undergoes loss of mass within 6–16 months; polyglycolide (45–55% crystallinity) is known to lose mass within 6–24 months. Copolymers of poly(D,L-lactide-co-glycolide) are reported to degrade faster, depending on the composition ratio, within 5–6 months. Polyesters hydrolyze under acidic and basic conditions;8 in contrast, some polyphosphoesters can be very stable under acidic conditions but degrade in the presence of a base. For polyphosphates, a typical water-soluble example is poly(methyl ethylene phosphate); while being stable at low pH, degradation of triester to diester bonds occurs under alkaline conditions within 5 h (at pH 12.3) to 21 months (pH 7.3). (Note: these are degradation times for 50% cleavage of the ester bonds in the main chain of the polymer.)9 Polyphosphonates with the P–C bond in the side chain show similar degradation profiles under neutral and basic conditions. Complete degradation was observed after 1 hour at pH 12.10 Contrary, polyphosphoramidates undergo hydrolysis in basic and acidic media.11–15 While hydrolysis almost exclusively proceeds at the P–N bond under acidic and nearly neutral conditions, P–O, as well as P–N bond cleavage, occurs under basic conditions, still with a higher probability for P–N cleavage.14 94% cleavage of main-chain polyphosphoramidates to diesters has been shown at pH 3.0 within 12.5 days.11 The degradation profile of polyphosphazenes strongly depends on the substituents and ranges from hydrolytically stable (with hydrophobic, bulky alkoxy side groups) to hydrolytically unstable (with hydrophilic amino substituents). Degradation of the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N-backbone is commonly accelerated in acidic media, but they are rather inert under basic conditions.16,17 The biodegradation of synthetic aliphatic polyamides is known to be low due to high crystallinity. Enzymatic or microbial degradation has been shown.18Fig. 1 gives a rough overview of the systematic order of degradability of the discussed polymer classes, however, the data has to be taken as an estimation only, as many factors as mentioned above may influence absolute values. A recent review summarizes the degradation of polylactides.19 We refer the interested reader to separate reviews concentrating on the degradation of synthetic polymers.3,4,16,17,20–23
N-backbone is commonly accelerated in acidic media, but they are rather inert under basic conditions.16,17 The biodegradation of synthetic aliphatic polyamides is known to be low due to high crystallinity. Enzymatic or microbial degradation has been shown.18Fig. 1 gives a rough overview of the systematic order of degradability of the discussed polymer classes, however, the data has to be taken as an estimation only, as many factors as mentioned above may influence absolute values. A recent review summarizes the degradation of polylactides.19 We refer the interested reader to separate reviews concentrating on the degradation of synthetic polymers.3,4,16,17,20–23
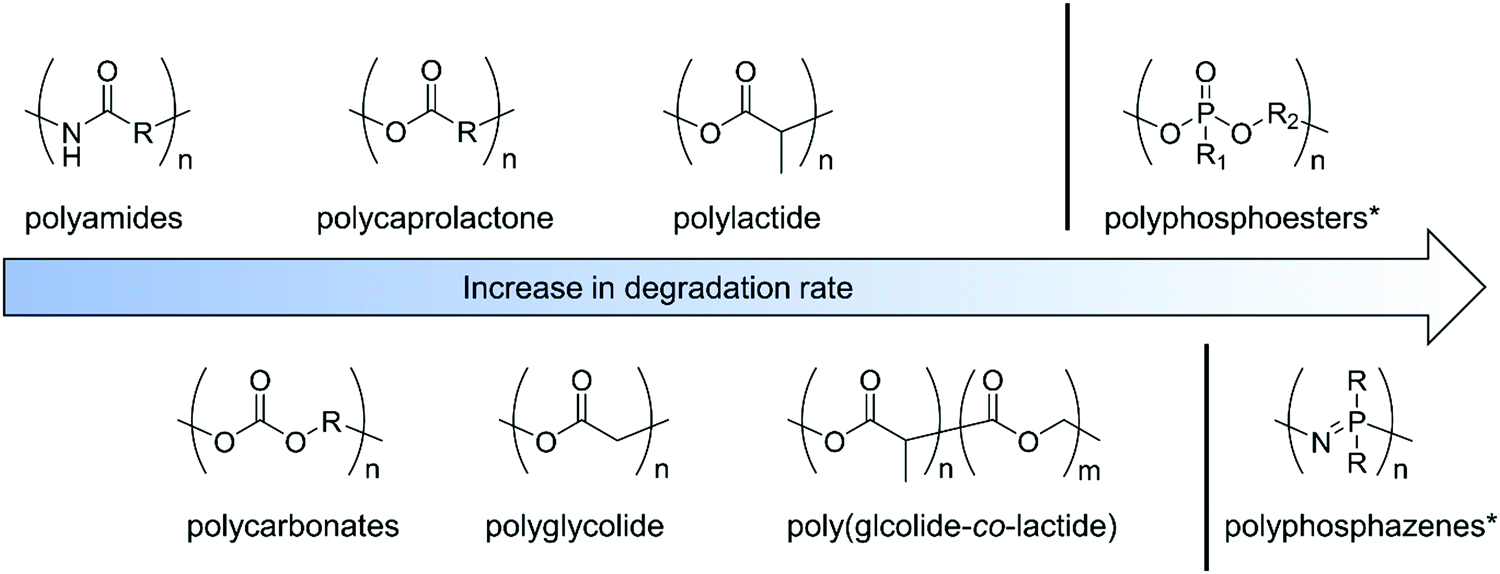 | ||
Fig. 1 Overview of the systematic order of degradability of the discussed polymer classes. *![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Note: degradation profiles of polyphosphazenes and polyphosphoesters depend strongly on the nature of the substituents. Note: degradation profiles of polyphosphazenes and polyphosphoesters depend strongly on the nature of the substituents. | ||
2. Overview of orthogonally reactive groups
ROP only tolerates some additional chemical functionalities in the monomers. Since alcohols, thiols, amines or carboxylic acids interfere with the propagation and serve as initiators or terminating agents, they need to be protected before polymerization. Commonly used protecting groups, e.g. benzyl, benzoyl, ethers, silyl ethers, acetals, urethanes, sulfonamides or esters are applied in cyclic monomers for ROP. Removal of protection groups is conducted after the polymerization under alkaline or acidic conditions, or by hydrogenation and often demands harsh conditions, which might also degrade the polymer backbone. We do not further consider these protected monomers in the review and concentrate exclusively on orthogonally reactive functionalities (Fig. 2). For protected monomers, we refer to other reviews, specializing in the respective polymer class.24–28 The same functional groups can also be installed into the initiator structure, in order to prepare end-functionalized polymers (telechelics).29,30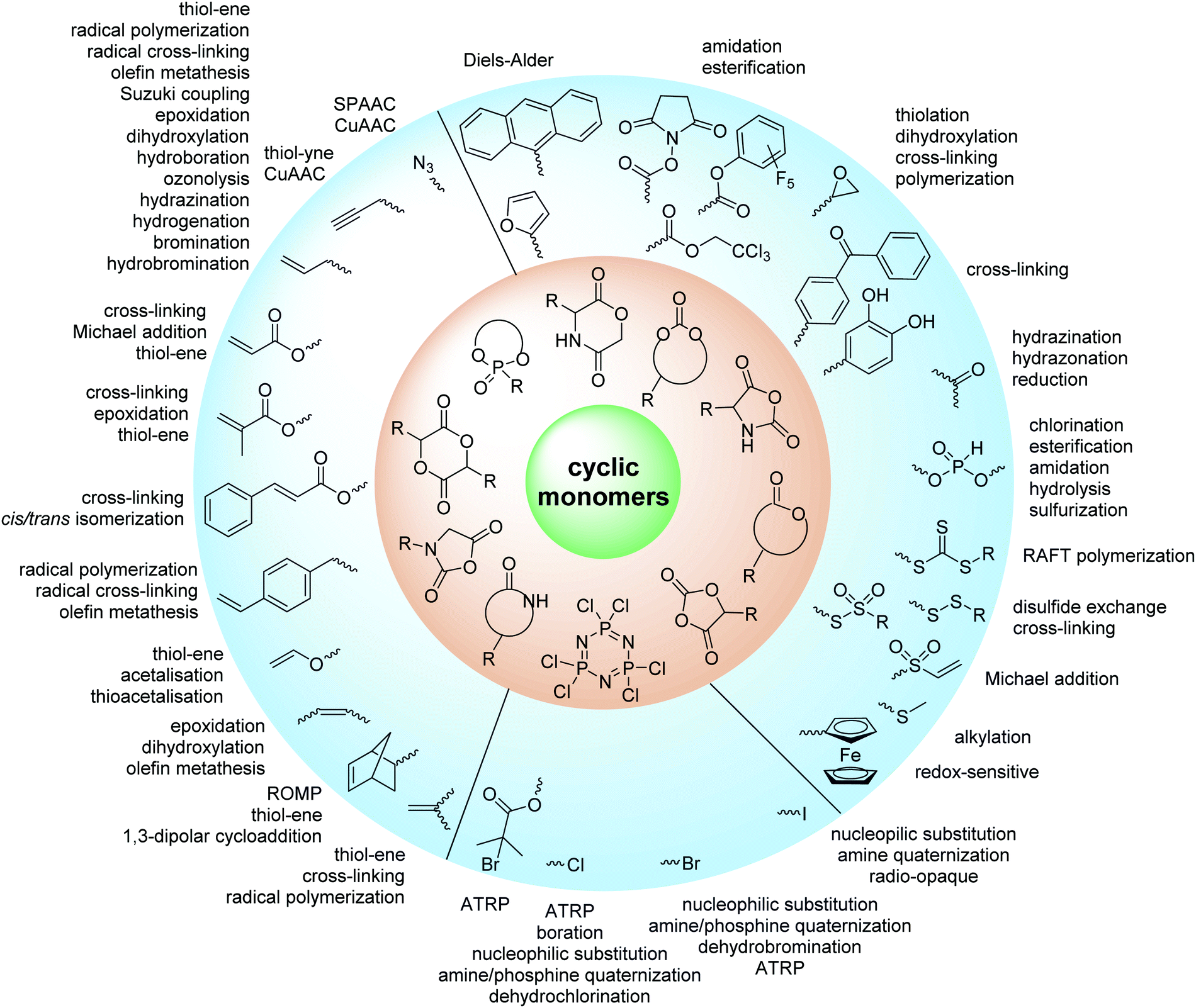 | ||
| Fig. 2 Overview on cyclic monomers discussed in the review with orthogonally reactive groups. | ||
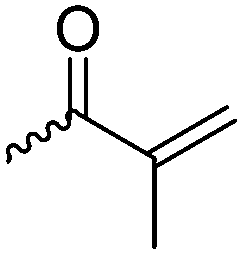
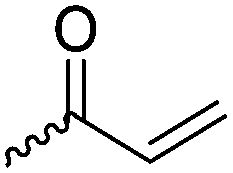
For the orthogonal functions, alkynes and alkenes are by far most frequently reported in the literature and are used in the monomers and initiator structures. Alkynes undergo the copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC, Huisgen 1,3-dipolar cycloaddition) with azides or can react in a thiol–yne reaction with thiols in often quantitative yield and mild conditions. Also, azides as the “counterpart” are reported as functionality in monomers. Besides CuAAC, they can be additionally modified with DBCO derivatives in a strain-promoted alkyne–azide cycloaddition (SPAAC),31 which turns the reaction into a copper-free functionalization and is especially interesting for biomedical applications. Alkenes are accessible for probably the most modification reactions: besides thiol–ene reaction and Michael addition, epoxidation (e.g. by mCPBA), dihydroxylation, hydroboration, ozonolysis, hydrazination, hydrogenation, bromination, hydrobromination, and others are applicable. Especially epoxidation opens a platform for diverse further reaction. Also, few monomers are reported that directly carry an epoxide, which under certain conditions does not interfere with ROP. Epoxides can cross-link the materials, react with thiols, be dihydroxylated or further polymerized. If alkene functions are vinylidenes, the cyclic monomers are bifunctional for radical polymerization or can serve as cross-linkers as well. They furthermore can be used for olefin cross-metathesis or Suzuki coupling. Acrylate, methacrylate and styrenic functions likewise can be radically polymerized, cross-link the materials, undergo thiol–ene reaction and Michael addition or are accessible for olefin cross-metathesis. Cinnamoyl groups serve as cross-linkers. Likewise, methylidene functions can be polymerized or cross-link materials and undergo thiol–ene reaction, which can be also achieved with norbornene groups, additionally suitable for 1,3-dipolar cycloadditions and ring-opening metathesis polymerization (ROMP). Completing the group of double bond-containing functionalities, internal double bonds are accessible for epoxidation, dihydroxylation, and cross-metathesis, while vinyl ethers are interesting reaction partners for thiol–ene reaction, acetal- and thioacetalisation.
Halogenated monomers are a second important category, especially with bromide or chloride substituents. Nucleophilic substitution e.g. with sodium azide and quaternization of tertiary amines or phosphines has been reported, as well as dehydrohalogenation or boration. Iodide substituted monomers play a minor part, but can also be used for nucleophilic substitution and quaternization of amines, or are used as a radio-opaque function, e.g. for contrast agents. Such halogenated polymers have also been used extensively as initiators for atom transfer radical polymerization (ATRP) to prepare graft or brush (co)polymers. Several bromo isobutyrate-containing monomers were developed for the same purpose, as well as trithiocarbonate monomers for reversible addition–fragmentation chain transfer polymerization (RAFT).
A third category includes more exotic, but at the same time very interesting and partly unexpected chemical functionality: besides the trithiocarbonate-containing monomers for RAFT polymerization, several further sulfur-containing monomers are introduced, bearing disulfide or S-sulfonyl groups. The functional groups do not interfere with ROP and can be considered as “protected thiols”. Functionalization is achieved with thiols by disulfide exchange reaction without any prior deprotection reaction: dynamic and redox-responsive cross-linking are accessible. Methyl-thioether functions can undergo reversible alkylation reaction, additionally implementing cationic charges. Vinyl sulfonyl moieties can react in Michael addition reactions. P–H bonds of cyclic H-phosphonate monomers are suitable for modification by esterification, amidation (after chlorination), hydrolysis and sulfurization. The P–H bond has not been reported yet in pendant chains. Ketones within the ring the cyclic monomer are accessible for reduction, hydrazination, and hydrazonation reactions. Benzophenone groups can be used as photo-cross-linkers by a C,H-insertion crosslinking reaction (CHic mechanism32,33) with CH groups. Cross-linking can also be achieved by catechol functions (1,2-dihydroxybenzene), either reversible by metal ion complexation or covalently by reaction with amine, thiols or other catechols after oxidation to quinone intermediates. In addition, active ester-containing monomers have been reported, such as trichloroethyl-, NHS- (N-hydroxysuccinimide) and pentafluorophenyl-ester groups, which undergo amidation and esterification reactions with alcohols or amines after polymerization.34,35 Finally, anthracene and furan derivatives are suitable for [4+2] cycloaddition Diels–Alder reactions. However, reports on this thermally reversible modification by additive/catalyst-free cycloaddition are rare, which might be a further potential for future applications.
In general, also orthogonal reactions have limitations. This starts with the degree of conversion, which is not always quantitative and leaves non-reacted groups behind. In addition, polymerization conditions of functional monomers need to be carefully selected. For instance, pentafluorophenyl esters can only be polymerized using acid catalysis, thiol derivatives in some cases are complicated to work with (for example due to formation of disulfides), catechol-functionalized compounds should be handled under oxygen-free atmosphere to avoid oxidation, and epoxides can also be ring-opened during the polymerization, if not carefully handled (e.g. at elevated temperatures). Moreover, the effectiveness of the post-functionalization methods can vary.36 Radical thiol–ene and thiol–yne reactions often need a large excess of the thiol to prevent unwanted crosslinking reactions. For click reactions, the removal of the copper catalyst needs to be taken into account, etc. Such issues need always to be considered and are sometimes not clearly mentioned in publications. We refer to some general reviews about post-polymerization modifications that can be considered as additional reading.25,36–38
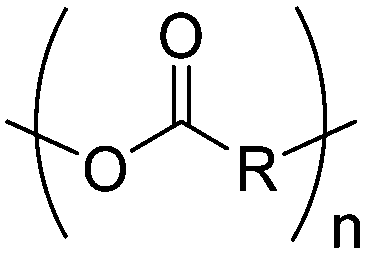
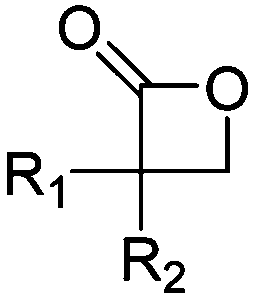
3. Polyesters
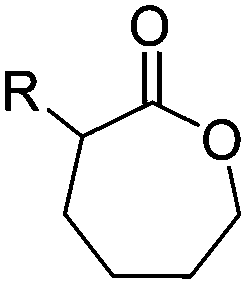
Aliphatic polyesters from ROP probably display the broadest group of fully synthetic biodegradable polymers. A variety of lactones with different ring sizes are available resulting in poly(ε-caprolactone)s, poly(δ-valerolactone)s, poly(γ-butyrolactone)s, poly(β-butyrolactone)s, poly(β-propiolactone)s, and poly(α-propiolactone)s. Macrolactone monomers are likewise polymerizable. Furthermore, cyclic diesters (lactides, glycolides and O-carboxy anhydrides (OCAs)) produce the commercialized poly(α-hydroxy acid)s (PAHAs) polylactides (PLAs) and polyglycolides (PGAs) (Table 3). Several reviews about functional aliphatic poly(ester)s have been published and might be considered for further reading.26,39,40| Monomer | R = | No. | Post-modification | Ref. |
|---|---|---|---|---|
| ε-Caprolactones | ||||
| α-Substituted | ||||
|
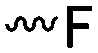
|
A1 | – No modification | 48 |
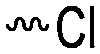
|
A2 | – Nucleophilic substitution with sodium azide and click chemistry | 46 and 50 | |
| – ATRP macroinitiator for “grafting from” of MMA | ||||
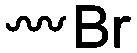
|
A3 | – Nucleophilic substitution with sodium azide and click chemistry | 49 and 51 | |
| – ATRP macroinitiator | ||||
|
A5 | – Radio-opaque properties | 53 | |
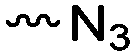
|
A4 | – Click chemistry | 52 | |
|
A7 | – Click reaction with cyclodextrin or Gd3+-complexes | 55, 58 and 59 | |
|
A8 | – Click reaction to core-cross-linked micelles | 56 | |
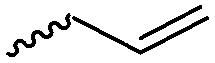
|
A6 | – Thiol–ene reaction with amines, dyes, sugars or zwitterions | 54 and 57 | |
|
A9 | – End-chain cross-linker for macrocyclic polyester | 60 | |
|
A10 | – No modification | 61 | |
| ε-Substituted | ||||
|
|
A7b | – Click reaction to couple cyclodextrin | 55 and 59 |
| β-Substituted | ||||
|
|
A11 | – No modification | 63 |
|
A12 | – Epoxidation and thiol–ene reaction to cross-link | 64 | |
| γ-Substituted | ||||
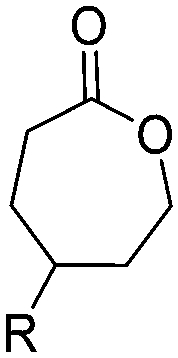
|
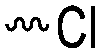
|
A13 | – Nucleophilic substitution with sodium azide and click chemistry with a cholesterol derivative for cell scaffolds and foams | 65 |
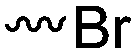
|
A14 | – Quaternization with pyridine | 66–68 | |
| – Elimination, epoxidation, and ring-opening to diols | ||||
| – Nucleophilic substitution with sodium azide and click chemistry | ||||
|
A15 | – Hydrazination or hydrogenation | 69–71, 151 and 152 | |
| – Reduction to alcohols and use as macroinitiator or coupling of maleic anhydride | ||||
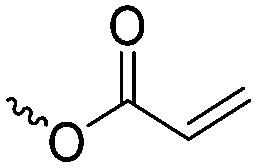
|
A16 | – Bifunctional polymerization | 72–75 | |
| – Electrografting onto metal surfaces | ||||
| – 2D- and 3D-microstructured resins | ||||
| – Michael-addition of thiols | ||||
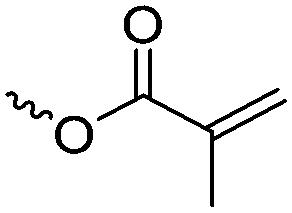
|
A17 | – Photo-cross-linking | 73 and 76 | |
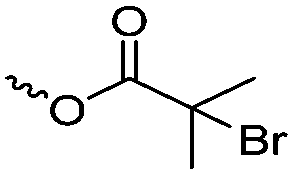
|
A18 | – ATRP initiator or macroinitiator | 77 | |
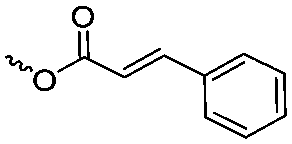
|
A19 | – Photo-cross-linking | 78 | |
| δ-Valerolactones | ||||
|
|
A20 | – Dihydroxylation with NMO/OsO4 | 80 and 85 |
|
A22 | – Click chemistry with PEG-, GRGDS-, phosphorylcholine or benzophenone-azides | 79, 82, 84 and 85 | |
|
A21 | – Dihydroxylation and PEG “grafting to” | 81 | |

|
A23 | – Radical copolymerization with methacrylates to form networks | 86 | |
|
A24 | – No polymerization | 87 | |
|
A25 | – No polymerization | 87 | |
|
A26 | – No polymerization | 87 | |
| γ-Butyrolactones | ||||
|

|
A27 | – Copolymerization with CL and cross-linking with methacrylate | 92 and 93 |
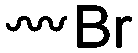
|
A28 | – Co- and terpolymerization with CL or TMCs and grafting from of methacrylates | 95 | |
| β-Propiolactones | ||||
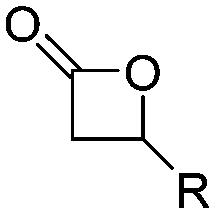
|
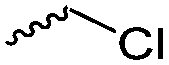
|
A29 | – Tacticity studies | 96 |
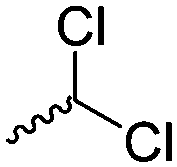
|
A30 | – Tacticity studies | 96 | |
|
A31 | – Tacticity studies | 96 and 98 | |
|
A32 | – Tacticity and property studies | 98 | |
|
A33 | – Tacticity and property studies | 99 | |
|
A34 | – Tacticity and property studies | 99 | |
|
A39 | – Selective polymerization studies | 103 | |
|
A40 | – Selective polymerization studies | 103 | |
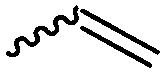
|
A41 | – Selective polymerization studies | 103 | |
|
A42 | – Hydroboration or olefin cross metathesis | 104–107 | |
|
A43 | – Epoxidation and sulfonation to polymers inducing bone formation; thiol–ene reaction to macroinitiator for “grafting from” | 108 and 110–112 | |
|
A44 | – Epoxidation | 108 | |
|
A45 | – No modification | 109 | |
| β-Butyrolactones | ||||
|
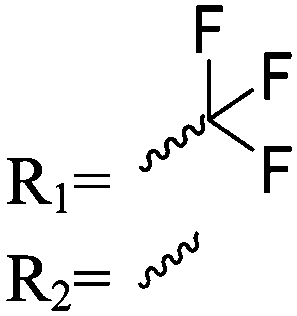
|
A35 | – Tacticity and property studies | 97 |
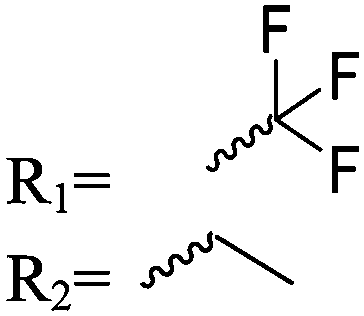
|
A36 | – Tacticity and property studies | 97 | |
|
|
A37 | – Quaternization with pyridine | 100 and 101 |
|
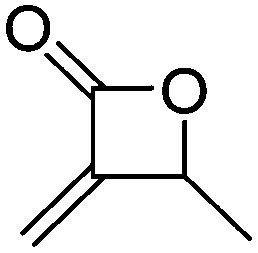
A38 | – Polymerization studies | 102 | |
|
A46 | – Radical cross-linking or thiol–ene reaction | 113 and 114 | |
| Macrolactones | ||||
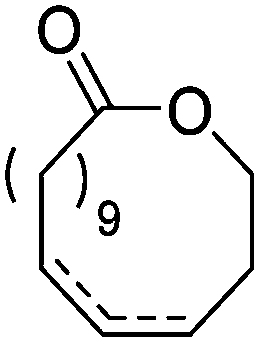
|
A47 | – Thiol–ene reaction for functionalization with pendant chains, cross-linking or functionalization with ATRP initiators for “grafting from” of tert-butyl acrylate | 119 and 122 | |
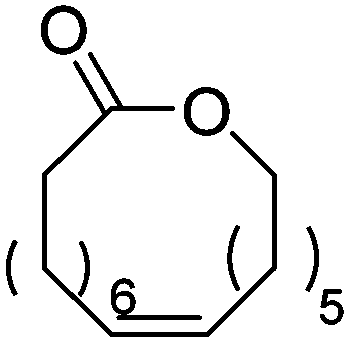
|
A48 | – Epoxidation | 125 | |
|
A49 | – No modification | 125 | |
|
A50 | – Radical cross-linking | 126 | |
| Mono-substituted glycolides | ||||
|
|
A51 | – Epoxidation, dihydroxylation | 127 |
|
A54 | – Click chemistry with PEG–azide | 128 | |
| Mono-substituted hemilactides | ||||
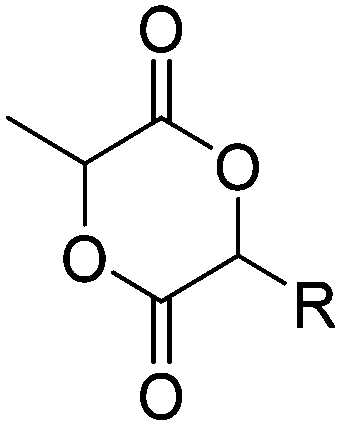
|
|
A52 | – Thiol–ene reaction with amines for gene delivery | 133 and 135 |
| – Photo-cross-linking of nanoparticles/capsules | ||||
|
A55 | – Click chemistry with PEG-/palitaxcel-azide | 129 | |
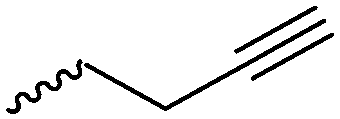
|
A58 | – Click chemistry with dansyl-azide | 138 | |
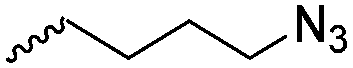
|
A59 | – Click chemistry with dansyl-alkyne | 138 | |
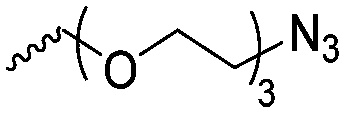
|
A60 | – Click chemistry with dansyl-alkyne | 138 | |
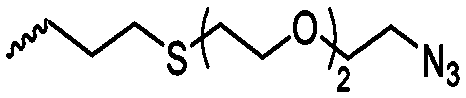
|
A61 | – Click chemistry with dye/cell internalizing peptide | 139 and 140 | |
| – Staudinger condensation with Tap-GRGDS | ||||
|
A53 | – Cross-linking | 134 | |
|
A62 | – ROMP | 130 and 131 | |
| – Click reaction with tetrazine derivatives | ||||
|
A64 | – No modification | 142 | |
|
A65 | – No modification | 142 | |
|
A63 | – No modification | 141 | |
| Di-substituted glycolides | ||||
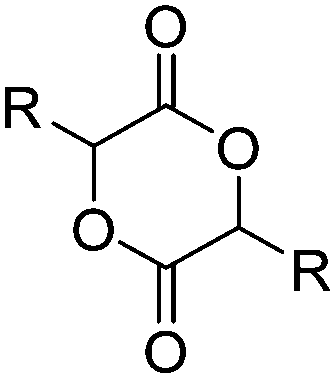
|
|
A56 | – Click chemistry with azide derivatives | 136 |
|
A57 | – Formation of double bonds | 137 | |
| – Thiol–ene reaction | ||||
| O-Carboxyanhydrides (OCAs) | ||||
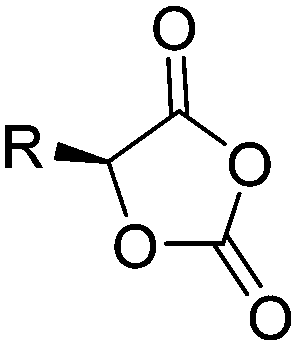
|
|
A66 | – Cross-linking with di-azides to redox- or light-responsive micelles | 145 and 147–149 |
| – Thiol–yne reaction to form polyelectrolytes for gene delivery | ||||
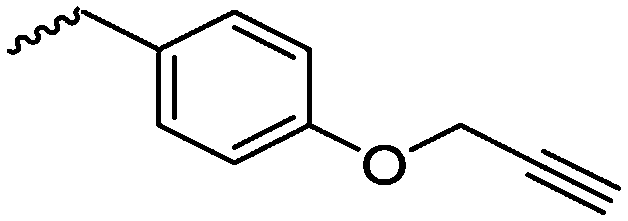
|
A67 | – No modification | 146 | |
| Morpholinones | ||||
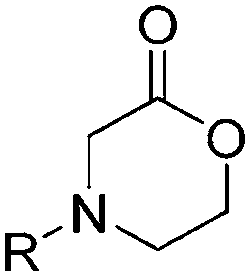
|
– R = N-acyl morpholin-2-ones polymerize readily, but the N-aryl or N-alkyl substituted morpholin-2-ones do not polymerize. | 144 | ||
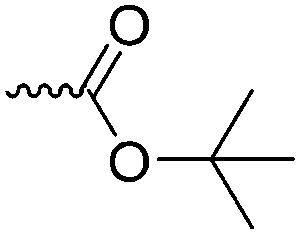
|
A68 | – After removal of BOC group water-soluble. | 144 | |
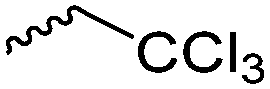
A less explored synthetic route to polyesters is the radical ring-opening polymerization (RROP) of cyclic ketene acetals (CKAs), which was summarized recently41 and is not further considered in this review. However, as radical polymerization is interesting also on the industrial scale, this strategy might be used also for the development of degradable functional polyesters. To the best of our knowledge, only methylidene-functionalized or chlorinated monomers are reported so far,41 resulting in halogenated polyesters or polymers with internal double bonds, but no postmodification was reported. In addition, less explored are poly(ester-ether)s and polythioesters. Five-, six- as well as seven-membered cyclic lactone ethers have been polymerized via ROP to poly(ester-ether)s. Reported substituents are mainly alkyl- or aryl chains or protected functions. To the best of our knowledge, orthogonally reactive monomers are not available so far but should be considered as a further development of lactone monomers. ε-Thiolactones and β-thiolactones can be polymerized by a base-catalyzed ring-opening polymerization to polythioesters. However, no functional reactive monomers have been reported to the best of our knowledge.42 The combination of polyaddition and ring-opening of different cyclic monomers offers a further strategy to novel functional materials with tunable properties: the reaction of lactone monomers with diamines has been reported.43 Polyesters can also be obtained by alternating ring-opening copolymerization (ROCOP) of epoxides and anhydrides. We exclude the technique of polyaddition of orthogonally reactive epoxides or anhydrides and point to several recent reviews.39,44,45
3.1 Lactones
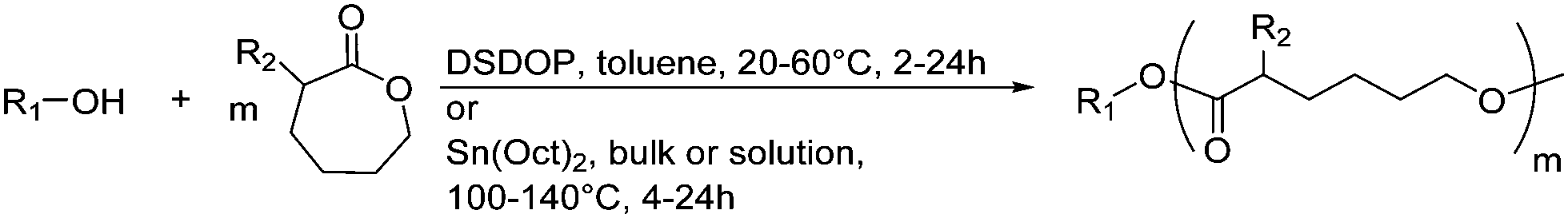 | ||
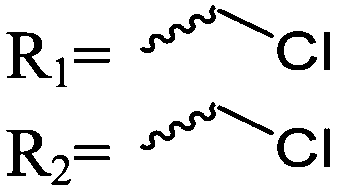
| Scheme 1 General polymerization scheme of caprolactones to polyesters (R1 and R2 represent non-specified alkyl or aryl substituents). | ||
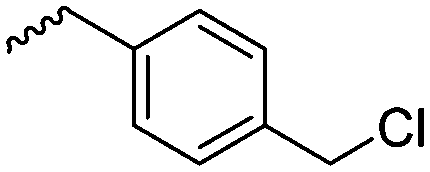
3.1.1.1 α-Substituted-ε-caprolactones. α-Halogenated caprolactones can be prepared by the Baeyer–Villiger oxidation of 2-halogenated-cyclohexanone with meta-chloroperoxybenzoic acid (mCPBA) (yield: 45–70%, Scheme 2A).46 Mohamod and coworkers48 polymerized α-fluoro caprolactone (A1) as homo- or copolymer with caprolactone. α-Chloro- (A2)46 and α-bromo-caprolactone (A3)49 was polymerized and substituted with azides, which allowed further postmodification with alkynes in a Huisgen 1,3-dipolar cycloaddition.49 These graft-polymers were used as macroinitiators for ATRP of methyl methacrylate (MMA)50 or hydroxyethyl methacrylate (HEMA)51 with the chlorides or bromides as initiating sites. Azide-functional poly(ε-CL) was also directly synthesized by an α-azido-ε-CL (A4),52 which was prepared by substitution of A2 or A3 with sodium azide.
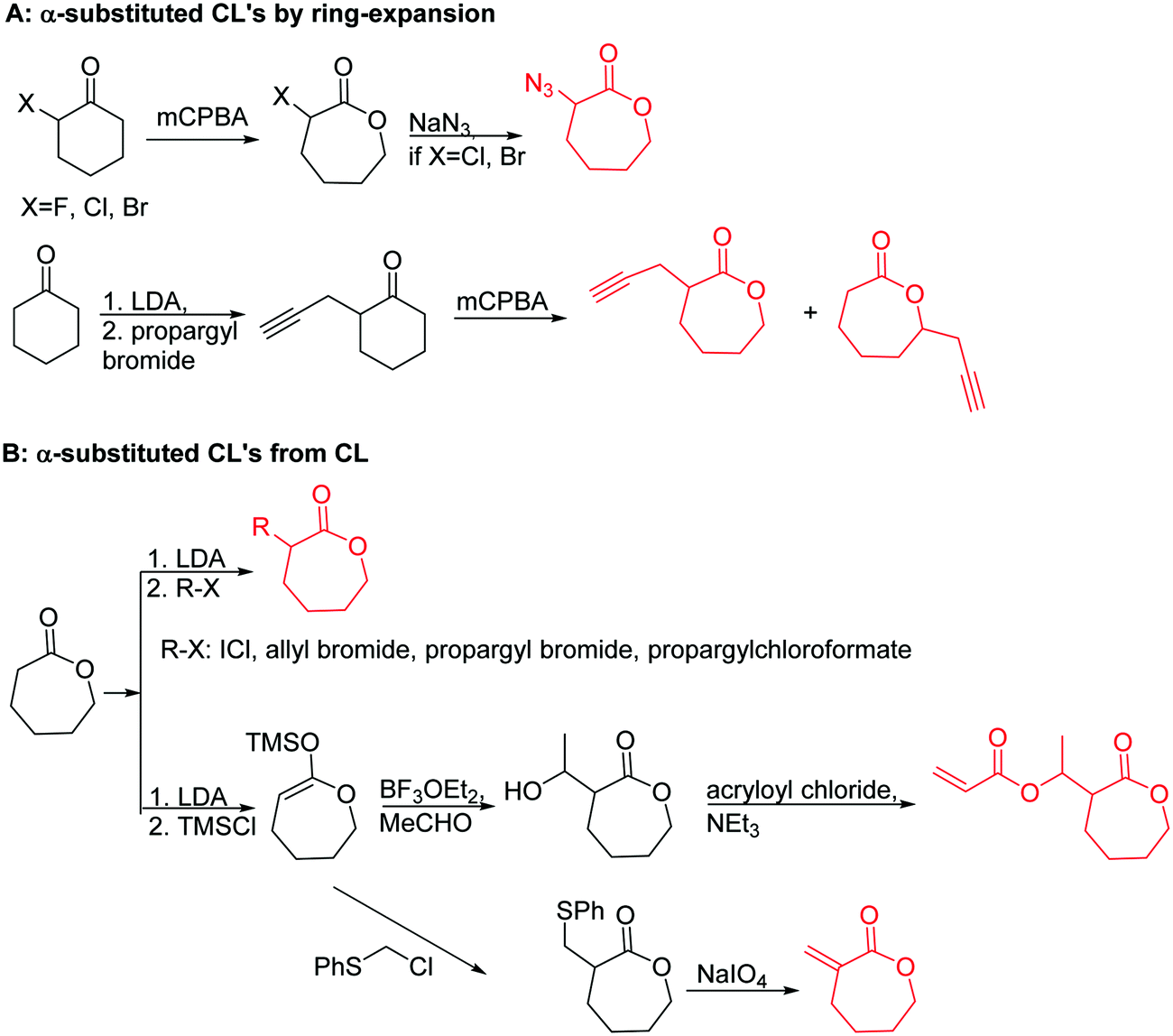 | ||
| Scheme 2 Synthetic strategies for the synthesis of α-substituted ε-caprolactones: (A) by ring-expansion via the Baeyer–Villiger oxidation of cyclohexanones; (B) by substitution of ε-caprolactone. | ||
A further general strategy is the functionalization of the α-position of ε-caprolactone by deprotonation with LDA (lithium diisopropylamide) and subsequent reaction with an electrophile (Scheme 2B). α-Iodo-caprolactone (A5) was obtained in this way by iodination with ICl (yield: 29%).53 The authors claimed the resulting copolymers to exhibit radio-opacity properties with potential application in temporary reconstructing material or drug delivery because of visualization via routine X-ray radioscopy.
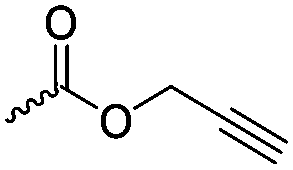
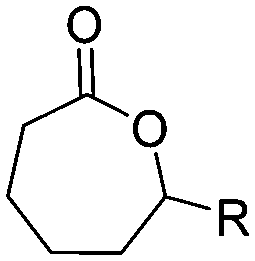
α-Alkene and -alkyne functionalized caprolactones are used for the purpose of thiol–ene reaction and click reaction, introducing charged functionalities or bulky molecules, such as dyes or sugars. Following the described strategy, deprotonated ε-caprolactone reacts with allyl bromide, propargyl bromide or propargyl chloroformate to yield α-allyl-ε-caprolactone (A6, yield: 65%),54 α-propargyl-ε-caprolactone (A7)55 and α-propargyl carboxylate-ε-caprolactone (A8).56 After copolymerization of A6 with ε-CL, Coudane and coworkers54 attached Boc-protected-amines as the pendant chains by the radical thiol–ene reaction. They proved deprotection of the amine without degradation of the backbone and subsequent reaction with fluorescein isothiocyanate (FITC). They claimed the water-soluble cationic polyesters as interesting materials for gene delivery. Maynard and coworkers57 recently reported trehalose- and carboxybetaine-substituted poly(CL) and used it as a polymeric excipient for the stabilization of the therapeutic protein G-CSF for storage at 4 °C and at heat stressor temperatures of 60 °C. Copolymers of A7 were functionalized with the clinically used diethylenetriaminepentaacetic acid (DTPA)/Gd3+ complex, resulting in MRI-visible polymers as a hydrophobic contrast agent.58 PEG-block copolymers of α-propargyl carboxylate-ε-caprolactone formed micelles, which were core-cross-linked by a difunctional azide-cross-linker.56 An alternative strategy to produce α-propargyl-ε-caprolactone (A7, yield: 14%) starts with deprotonation of cyclohexanone and subsequent substitution with propargyl bromide, followed by the Baeyer–Villiger oxidation to expand the ring to ε-caprolactone (Scheme 2A).59 However, a mixture of α- and ε-substituted caprolactones (A7 and A7b, isomeric mixture yield: 30/70) were obtained. Ritter and coworkers prepared polymers and attached cyclodextrins via click reaction to form supramolecular organogels.55
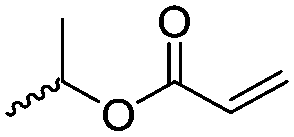
Lecomte and coworkers60 reported an acrylate-substituted CL (A9) using it as end chain comonomer to form macrocyclic polyesters. The macrocycles were formed by UV-crosslinking of the acrylates. A9 was synthesized in three steps (Scheme 2B): deprotonation of caprolactone and addition of trimethylsilyl chloride formed a trimethylsilylketene acetal, which further reacted in a Mukaiyama aldol reaction with acetaldehyde. Esterification of the formed hydroxylactone with acryloyl chloride yielded the final monomer α-(1-acryloyloxyethyl)-ε-caprolactone (A9). Ritter and coworkers61 reported the ROP of α-methylidene-ε-caprolactone (A10), while the monomer has been polymerized at the vinyl functionality before. Homopolymerization yielded only low molar masses, copolymerization with caprolactone clearly higher molecular weight polymers. They claimed the polymers to be radically cross-linkable, however, did not report on further details. The monomer was synthesized by O-silylation, followed by thioalkylation with α-chloro thioanisole and completed by oxidative sulfur (Scheme 2B) removal (overall yield: 39%).62
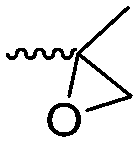
3.1.1.2 β-Substituted-ε-caprolactones. Hillmyer and coworkers reported two β-substituted CLs derived from natural carvone: 7-methyl-4-(2-methyl oxirane-2-yl)oxepan-2-one (A11)63 and dihydrocarvide (A12)64 (Scheme 3). A11 was synthesized in two steps: hydrogenation of carvone resulted dihydrocarvone; epoxidation and ring-expansion yielded the final monomer (second step 25% yield). The monomer was homo- and copolymerized with CL, however, both rings reacted under the reported polymerization conditions (in bulk or solution, 20–120 °C, diethylzinc (ZnEt2) or tin(II) 2-ethyl hexanoate (SnOct2) catalyst). To obtain polymers with epoxides as pendant groups, monomer A12 was obtained by ring-expansion of dihydrocarvone using Oxone® (triple salt 2·KHSO5·KHSO4·K2SO4).64 Poly(dihydrocarvide-co-carvomenthide) was subsequently epoxidized and crosslinked with a dithiol to prove the possibility of post-modification.
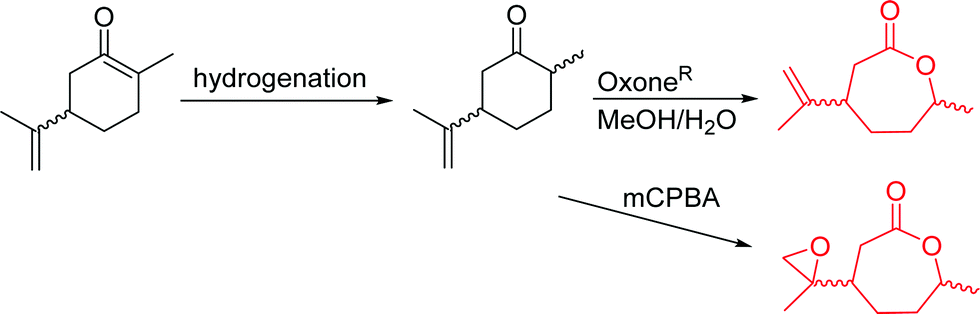 | ||
| Scheme 3 Synthetic route to β-substituted CLs from carvone. | ||
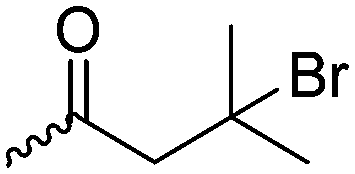
3.1.1.3 γ-Substituted-ε-caprolactones. γ-Halogenated caprolactones are synthesized starting from 4-halogenated-cyclohexanol (obtained by halogenation of 7-oxabicyclo[2.2.1]heptane); the alcohol was oxidized to a cyclic ketone to yield the monomer γ-chloro-ε-caprolactone (A13)65 or γ-bromo-ε-caprolactone (A14)66 after a Baeyer–Villiger oxidation (yield for the last step: 62%; Scheme 4A). Hegmann and coworkers65 used A13 in a copolymerization with caprolactone and lactide, substituted the chloride with an azide and attached cholesterol derivatives. The copolymers were used as cell scaffolds and foams. Jérôme and coworkers67 quaternized copolymers of caprolactone and A14 with pyridine, applied a debromination or epoxidation, and subsequent ring-opening to obtain hydrophilic poly(CL) substituted with diols in the backbone. P(A14) was substituted by an azide and amine-groups were then coupled by click chemistry to obtain pH-sensitive star-shaped polyesters.68 A γ-azide-ε-caprolactone has not been reported yet to the best of our knowledge. γ-Keto-ε-caprolactone (A15) was reported by the same group,69 obtained by ring extension of 1,4-cyclohexanedione (Scheme 4B). Qiao and coworkers70 functionalized the keto groups by hydrazine chemistry to introduce hydroxyl groups and coupled 4-nitrophenyl chloroformate. The activated ester was reacted with primary amines, e.g. of the cell adhesive peptide GRGDS. Lang and coworkers71 reduced the carbonyl group in copolymers to hydroxyl groups, using them as a macroinitiator for further grafting of lactide.
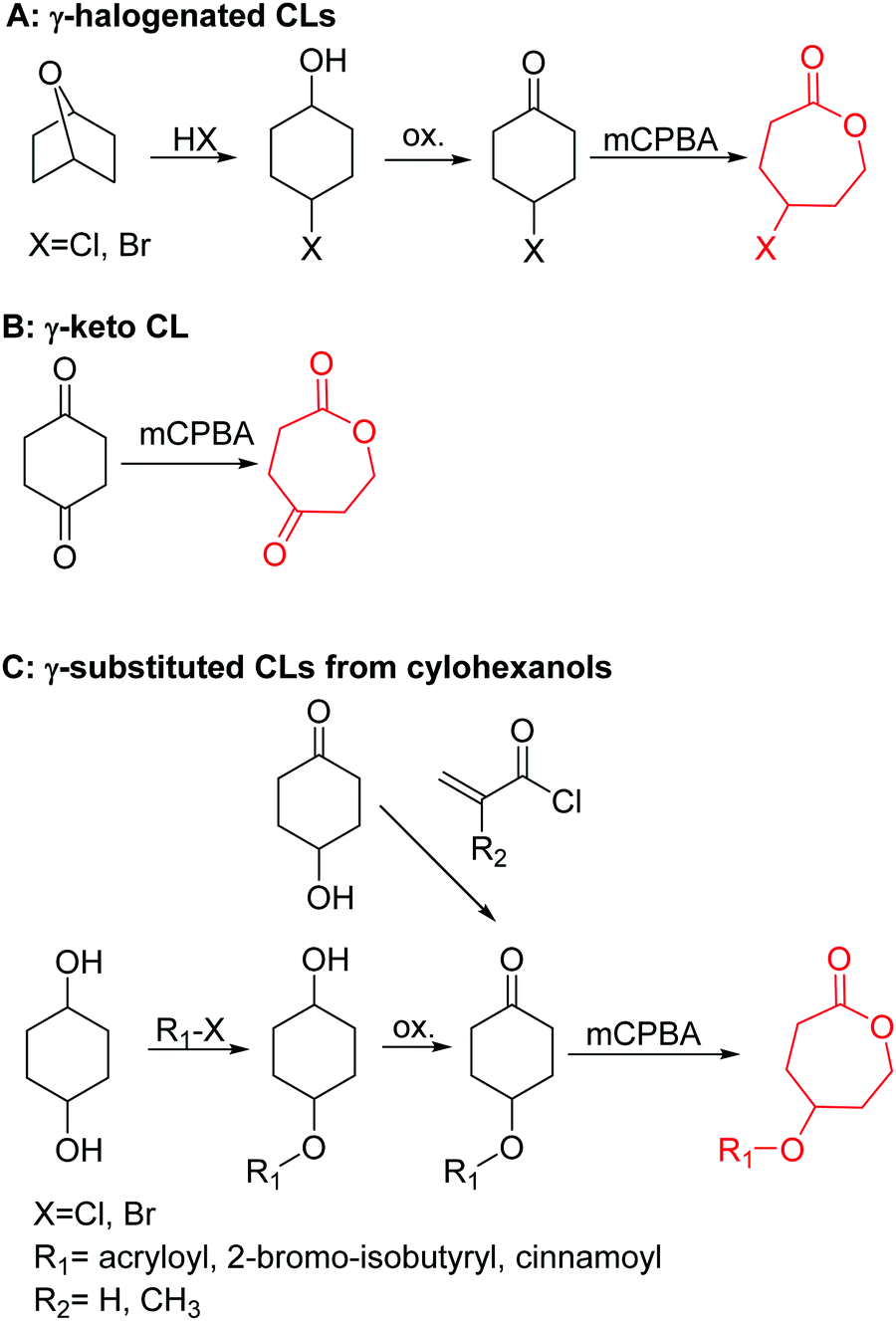 | ||
| Scheme 4 Synthetic strategies for the synthesis of γ-substituted ε-caprolactones: (A) for γ-halogenated CLs; (B) for γ-keto CL; (C) for γ-substituted CLs from cyclohexanols. | ||
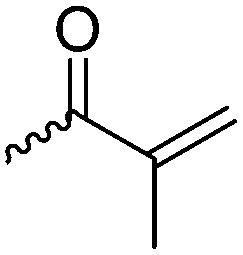
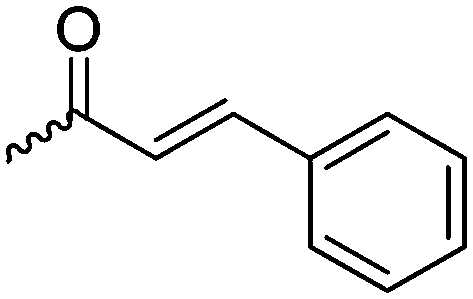
γ-Acryloyloxy ε-caprolactone (A16), a bifunctional monomer for ROP and radical polymerization,72 was prepared in two or three steps (Scheme 4C): 1,4-cyclohexanediol reacted with acryloyl chloride. The resulting monoalcohol was oxidized and the ring extended to yield the monomer (in an overall yield of 36%). A shorter alternative strategy started with the reaction of acryloyl chloride with 2-hydroxycyclohexan-1-one and subsequent ring extension (overall monomer yield 24%).73 Besides using the monomer for both ROP and ATRP, copolymers were grafted onto metal surfaces,74 used as 2D or 3D microstructured resins73 or were post-modified by Michael-addition of thiols.75 A similar monomer, γ-methacryloyloxy-ε-caprolactone (A17), was prepared by the same method, using methacryloyl chloride instead (overall monomer yield 29%),73 which were cross-linked upon UV-irradiation e.g. to form microparticles.76 γ-(2-Bromo-2-methyl propionate)-ε-caprolactone (A18) carries a classical ATRP initiating group and was presented by Hedrick and coworkers.77 A γ-cinnamate-modified caprolactone (A19) was recently reported by Budhlall and coworkers,78 which can undergo cis/trans isomerization and [2+2] cycloaddition upon UV-irradiation. Homo- or copolymers were used as thermoresponsive and semicrystalline networks after photochemical cross-linking.
Emrick and coworkers80 reported the first functionally substituted monomer, α-allyl-δ-valerolactone (A20), synthesized by the same strategy as α-substituted ε-caprolactones: lithiation of δ-valerolactone in α-position with LDA and subsequent reaction with allyl bromide yielded A20 in one step (yield: 71%, Scheme 5). A20 was copolymerized with ε-caprolactone or δ-valerolactone, as well as homopolymerization obtained polymers in good conversion and narrow molecular weight distributions. The alkenes were quantitatively dihydroxylated with NMO/OsO4 to obtain more hydrophilic poly(ester)s. The group also introduced a α-cyclopentene-δ-valerolactone (A21):81A20 was lithiated and allylated to yield α,α-diallyl-δ-valerolactone. Ring-closing metathesis using a Grubbs catalyst gave A21. The cyclopentene substituted lactone was not able to homopolymerize; copolymerization with ε-caprolactone was realized with the incorporation of ca. 20% of A21. The pendant group was converted to cis-1,2-diols by dihydroxylation with OsO4 and showed longer bench-life stability compared to the diol-containing poly(ester)s from pendant allyl groups probably due to the higher rigidity of the monomer units. PEG was grafted onto the copolymers. Finally, Emrick and coworkers82 also used α-propargyl-δ-valerolactone (A22)83 (synthesized by the same strategy as A21) and obtained homo- as well as copolymers with ε-caprolactone. They functionalized the polymers by click chemistry with a PEG–azide, an oligopeptide-azide (GRGDS-N3), a phosphorylcholine derivative84 or a benzophenone group, to produce photopatternable aliphatic polyester.79 Harth and coworkers85 used A20 and A22 to form multifunctional polyester nanoparticles.
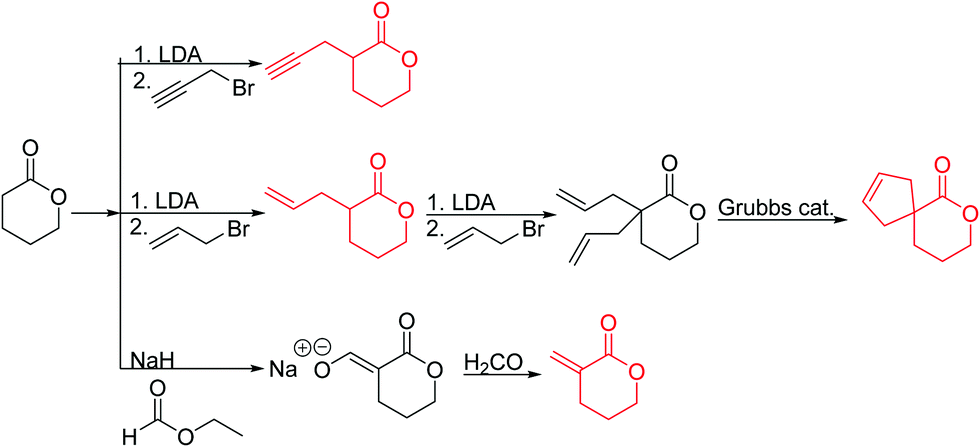 | ||
| Scheme 5 Synthetic strategy for the synthesis of substituted δ-valerolactones. | ||
α-Methylidene-δ-valerolactone (A23) has usually been polymerized as “vinyl monomer”. Ritter and coworkers86 reported the first polymerization by ring-opening. Formylation of δ-valerolactone, subsequent formyl transfer and elimination of a carboxylate anion yielded A23 (yield: 57%, Scheme 5). The monomer was copolymerized with δ-valerolactone, and networks obtained by free radical polymerization of the methylidene functionality with different methacrylates.
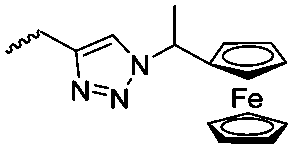
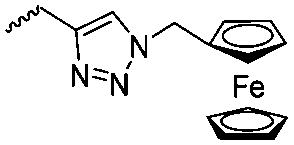
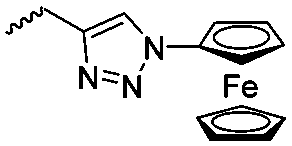
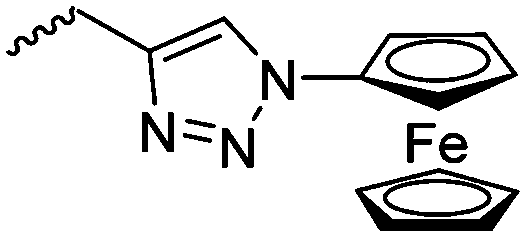
Diaconescu and coworkers reported a series of three different α-ferrocenyl-δ-valerolactones (A24–A26) and six ferrocenyl-substituted trimethylene carbonate (TMC) monomers (C5–C10), all obtained by click chemistry of azide-functionalized ferrocene to A22, 5-(propynyl)-1,3-dioxane-2-one and propargyl 5-methyl-2-oxo-1,3-dioxane-5-carboxylate (C11) (see also below).87 While all TMC monomers were polymerizable with DBU/TU as the catalyst (1,8-diazabicycloundec-7-ene and 1-(3,5-bis(trifluoromethyl)phenyl)-3-cyclohexyl thiourea), A24–A26 were not able to be polymerized neither as homo- nor copolymers.
000 atm) and can be copolymerized with other lactone monomers.90 Chen and coworkers91 recently successfully obtained poly(γ-butyrolactone) via ROP with a La[N(SiMe3)2]3/R–OH catalyst system at −40 °C in THF with a number molecular weight of 30 kg mol−1, 90% monomer conversion and control over linear or cyclic topology.
Since polymerization of γ-butyrolactone remains difficult, only a few functional monomers have been reported so far. α-Methylidene-γ-butyrolactone (A27) is widely used as vinyl-comonomer; Ritter and coworkers92 reported copolymerization with caprolactone in a ROP for the first time, Chen and coworkers93 recently reported homopolymerization. They used the methylidene function for crosslinking of the polymers with a methacrylate to transparent polyester networks. The monomer was synthesized in two steps by the same strategy as A23 (Scheme 6): formylation of γ-butyrolactone, subsequent formyl transfer and elimination of a carboxylate anion yielded A27.94 Albertsson and coworkers95 recently reported the copolymerization of α-bromo-γ-butyrolactone (A28), which is commercially available at Sigma Aldrich. Due to the high selectivity and reactivity of modern organocatalysts at ambient reaction temperatures, the authors were able to polymerize co- and terpolymers, with trimethylene carbonate (TMC), C47 or ε-caprolactone with an alcohol as the initiator and diphenyl phosphate (DPP) as the catalyst at 30 °C for 48 h. Grafting of methyl acrylate via Cu(0)-mediated CRP (controlled radical polymerization) on the copolymers was demonstrated.
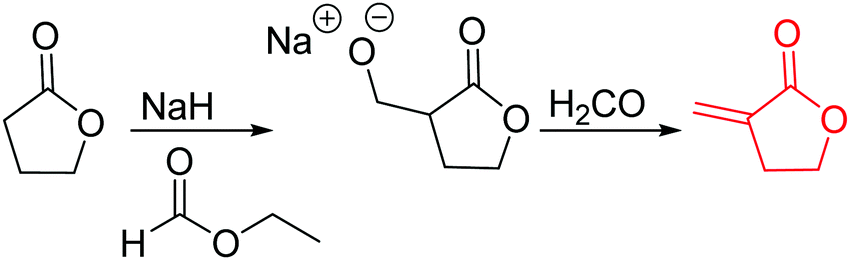 | ||
| Scheme 6 Synthetic strategy for the synthesis of α-methylidene-γ-butyrolactone. | ||
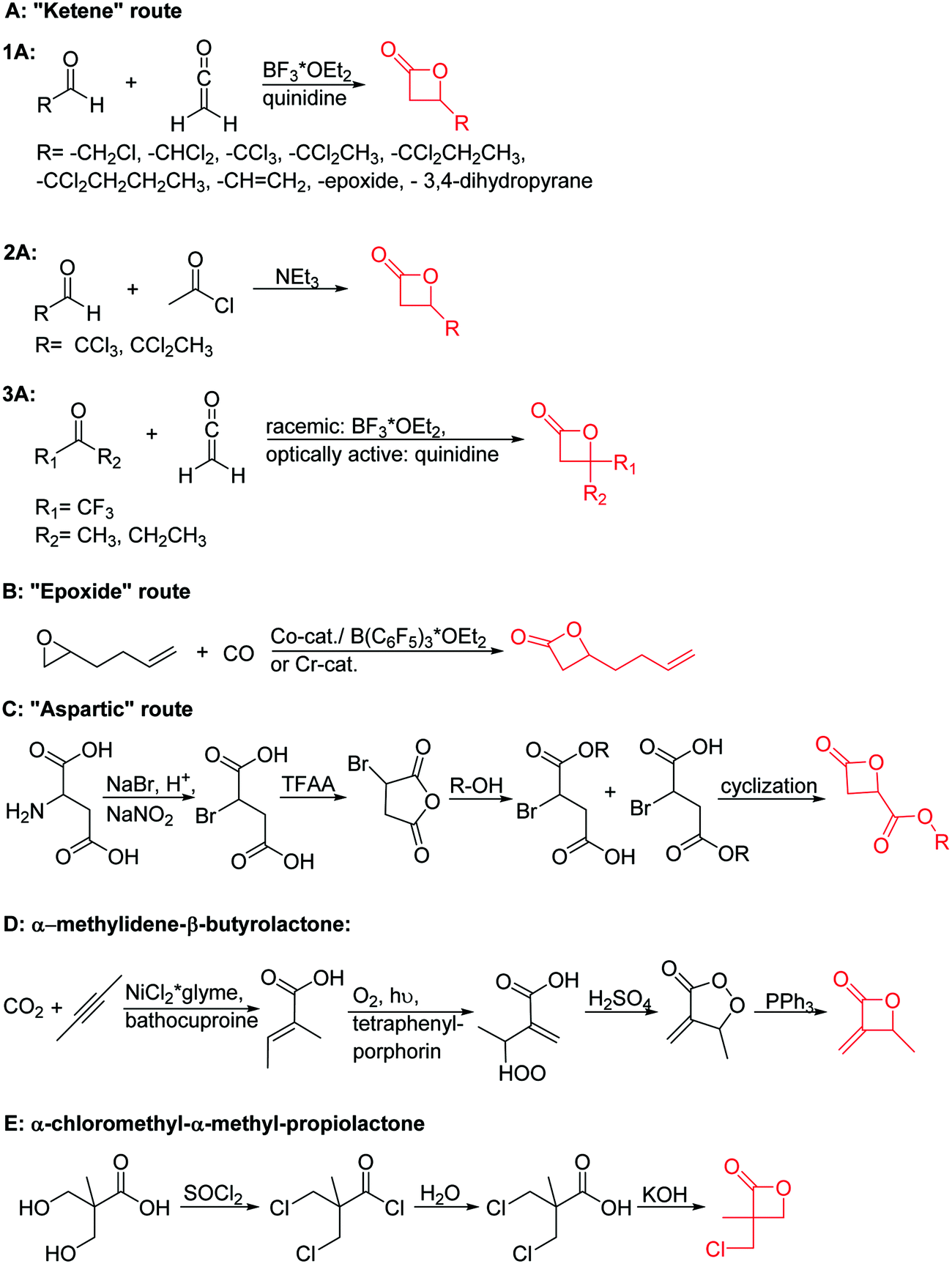 | ||
| Scheme 7 Synthetic strategies to substituted of β-propio- and β-butyrolactones: (A) “ketene” route, e.g. for halogenated lactones; (B) “epoxide” route yielding β-heptenolactone; (C) “aspartic” route; (D) synthesis of α-methylene-β-butyrolactone; (E) synthesis of an α-disubstituted propiolactone. | ||
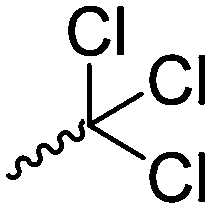
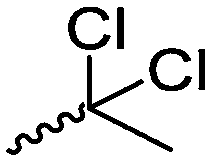
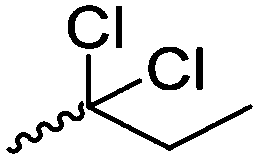
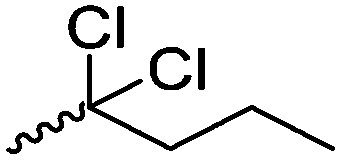
Mono-, di-, and tri-halogenated propiolactones and their polymerization are reported extensively in the literature. Modification after polymerization has not been reported so far. Tani and coworkers96 synthesized β-chloromethyl- (A29), β-dichloromethyl- (A30) β-trichloromethyl-β-propiolactone (A31) by [2+2] cycloaddition from ketene and the corresponding mono-, di- or trichlorinated acetaldehyde and intensively investigated in their polymerization behavior and tacticity of obtained polymers (Schemes 7, 1A). Prud’Homme and coworkers further introduced several chlorinated and fluorinated propiolactones (A32–A36),97–99 partially being β-disubstituted at the lactone ring (Scheme 7, 1A–3A). For racemic mixtures of the monomers, they used the corresponding halogenated aldehyde (or halogenated acetone or butanone for β-di substituted lactones), acetyl chloride and triethylamine, for optically active monomers they used the synthetic route using ketene and the chiral catalyst quinidine. Li and coworkers100,101 copolymerized α-chloromethyl-α-methyl-propiolactone (A37) with caprolactone (Scheme 7, E). Quaternization with pyridine resulted in polymers with increased hydrophilicity. Chlorination of 2,2′-bis(hydroxymethyl)propionic acid with thionyl chloride, hydrolysis of the formed acyl chloride and cyclization under basic conditions yielded the monomer. A α,α-bis-chloromethyl-propiolactone monomer (A38) has been reported by Kuriyama and coworkers102 and copolymerized with β-propiolactone. Post-modification has not been shown.
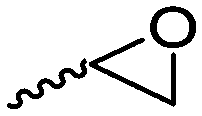
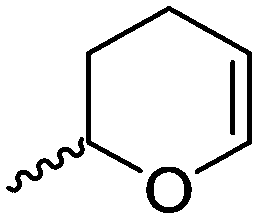
Cherdron and coworkers103 presented several β-substituted lactones, carrying pendant groups suitable for other polymerization techniques (epoxide (A39), 3,4-dihydropyrane (A40), vinyl (A41)). They showed selective lactone polymerization, but also did not use the further functionality for post-modification reactions. They followed the general synthetic strategy using ketene and a corresponding aldehyde (Scheme 7, 1A).
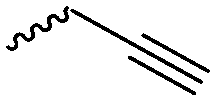
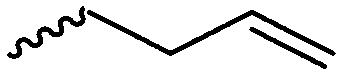
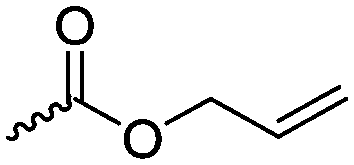
The polymerization of β-heptenolactone (A42) (or also called allyl-β-butyrolactone) is only rarely reported in the literature, which is probably due to an inconvenient synthetic route of the monomer or use of special zinc or yttrium catalysts for polymerization. But-3-en-1-yl-epoxide reacted with carbon monoxide in the presence of a Co-based catalyst at 6.2 MPa/80 °C104 or an active Cr-catalyst at 1 atm/22 °C105 to the monomer (Scheme 7, B). While Guillaume and coworkers106 functionalized poly(β-heptenolactone) by hydroboration, Shaver and coworkers107 recently post-modified the polymer by olefin cross-metathesis with 15 different alkene cross-partners producing a whole library of poly(ester)s with different functionalities.
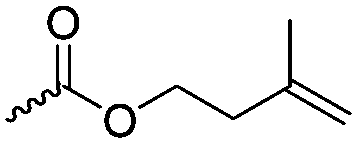
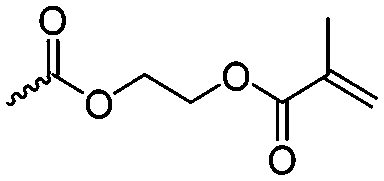
Guérin and coworkers108,109 developed three functional monomers for unsaturated poly(β-maleic acid) derivatives: allyl malolactonate (4-allyloxycarbonyl-2-oxetanone, A43), 3-methyl-3-butenyl malolactonate (4-[3-methyl-3-butenyloxycarbonyl]-2-oxetanone, A44) and 2-methylethenoyloxyethyl malolactonate (4-[2-methylethenoyloxyethyl-oxycarbonyl]-2-oxetanone, A45). While the ketene route gave only low yields, the “aspartic route” was applied (Scheme 7, C): aspartic acid was brominated and bromosuccinic acid anhydride formed. Esterification with an appropriate alcohol (allyl alcohol, 3-methyl-3-buten-1-ol or 2-hydroxyethyl methacrylate) opened the anhydride and formed a mixture of the corresponding mono-bromo succinic acid esters, and the major product was lactonizable. Epoxidation and subsequent sulfonation have been carried out. The copolymers were able to induce new bone formation and muscle regeneration in in vivo models.110,111 Thiol–ene reactions with mercaptoethanol converted the copolymers into macroinitiators to “graft from” polycaprolactone.112
Lu and coworkers113 recently reported a novel methylene functionalized monomer, α-methylidene-β-butyrolactone (A46), synthesized from carbon dioxide and 2-butyne in four steps (Scheme 7, D). After formation of tiglic acid, catalyzed by NiCl2*glyme and bathocuproine, an allylic peroxide was formed by photooxygenation. Dehydration formed a peroxylactone, which yielded A46 after deoxygenation. The vinylidene functional group was used for radical cross-linking or thiol–ene reaction.114
Heise and coworkers functionalized the olefins in polyglobalide via thiol–ene reaction with different thiols.120,122 In another study, dithiol-cross-linked polyglobalide films were further reacted with mercaptohexanol to attach ATRP initiators.119 Such films were further grafted with tert-butyl acrylate and proteins were conjugated to the deprotected grafts. Möller and coworkers125 polymerized A48 and oxidized the internal double bonds by Baeyer–Villiger oxidation using mCPBA to the epoxides. They showed, that a strategy vice versa is also possible: after epoxidation of A48, the monomer A49 (AmE) was polymerized with Novozyme 435, while the epoxides remained intact. Kobayashi and coworkers126 reported already in 2001 the enzymatic ROP of 2-methylene-4-oxa-12-dodecanolide (A50) by lipase and subsequent radical crosslinking of the polymers.
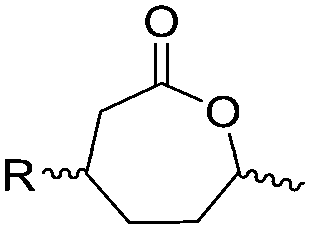
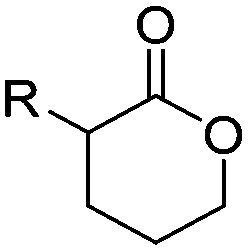
3.2 Cyclic diester monomers
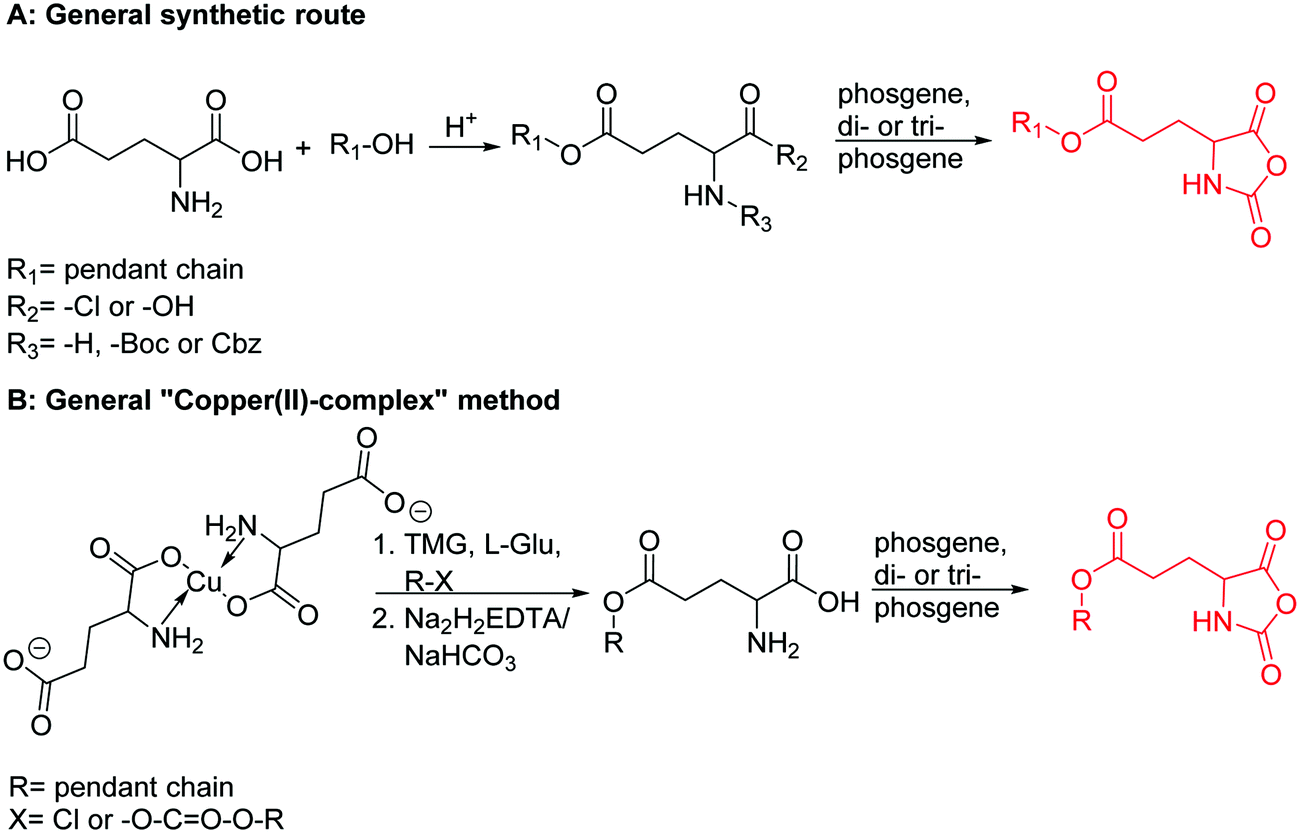
Poly(α-hydroxy acid)s (PAHAs) are obtained from cyclic diester monomers (Scheme 8). Poly(lactic acid) (PLA), poly(glycolic acid) (PGA), and their copolymers (PLGA) are accessible from renewable resources. They are typically prepared by ROP of the cyclic dimers of lactic and glycolic acid (lactides and glycolides). However, the lack of structural diversity of lactides and glycolides limits the preparation of functional poly(α-hydroxy acid)s. Synthesis of substituted 1,4- dioxane-2,5-diones can be complicated and reactivity in ROP is often poor. They are usually polymerized with Sn(Oct)2 at 110–130 °C for 2–24 h in bulk127 or toluene,123 with 4-dimethylamino pyridine (DMAP) at 35 °C for 18–48 h in DCM,128,129 or with TBD or DBU at room temperature for 2 min to 24 h in DCM,122,130,131 using primary alcohols as initiator.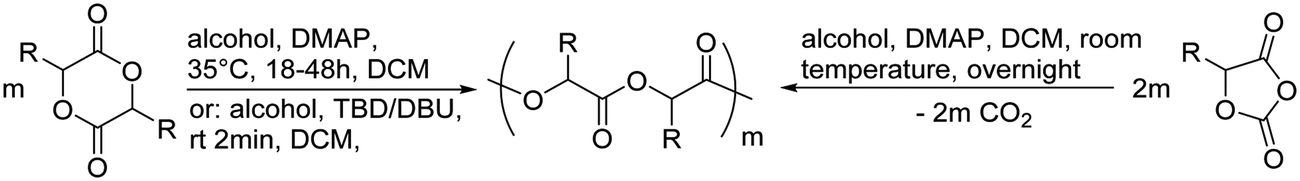 | ||
| Scheme 8 General scheme for the polymerization of lactides/glycolides and O-carboxy anhydrides (OCAs) to poly(α-hydroxy acid)s (PAHAs) (for definition of R-group, please see the main manuscript). | ||
O-Carboxy anhydrides (OCAs) are suitable alternatives for the preparation of functionalized PAHAs under mild conditions and were recently summarized in an excellent article.132
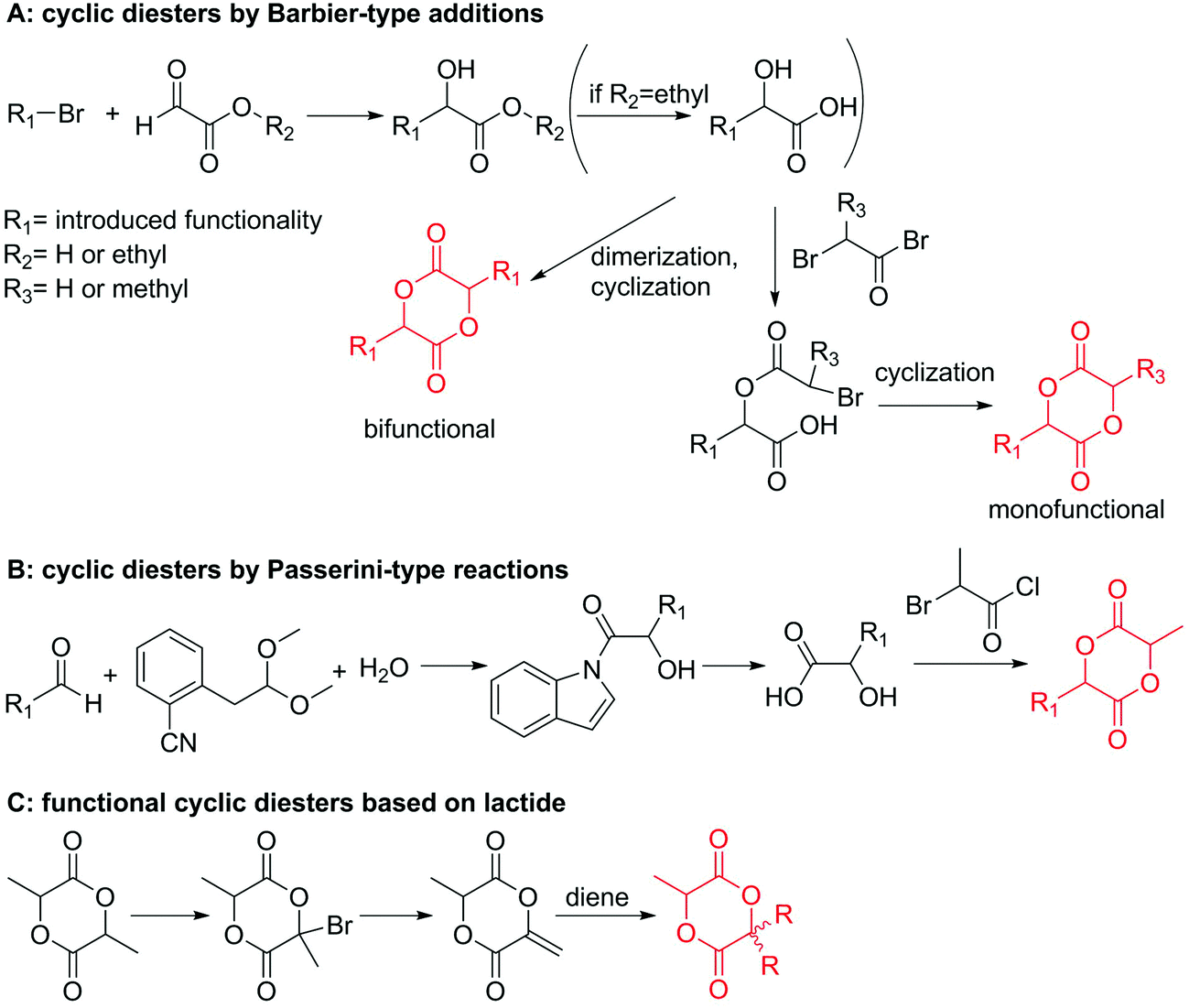 | ||
| Scheme 9 Synthetic strategies to functional lactides and glycolides: (A) by a Barbier-type addition; (B) by a Passerini-type reaction, (C) by functionalization of lactide. | ||
Hennink and coworkers127 polymerized an allyl functional glycolide (A51) and showed epoxidation with NMO/OsO4 and subsequent hydrolysis to diols. The allyl lactide analog A52 has been reported by Cheng and coworkers.133 They photochemically crosslinked PEG–PLA block copolymers via thiol–ene reaction with a dithiol-crosslinker to obtain nanoparticles. Pfeifer and coworkers135 used cationic modified PEG–PLA-block copolymers for gene delivery. The monomer was also further functionalized by olefin cross-metathesis with an epoxy alkene and further hydrogenated to the saturated epoxy lactide (A53).134 An orthogonal reactive iron-based catalyst was applied for the polymerization of A53, which selectively polymerizes the diester cycle if the catalyst is in the iron(II) form. The oxidized catalyst (iron(III)-species) instead selectively polymerizes the epoxide. The bifunctional epoxy diester was selectively polymerized to an epoxy-functional polyester (Fig. 3). After oxidation of the catalyst and removal of solvent, the epoxy-functions were polymerized to cross-link the polymers.
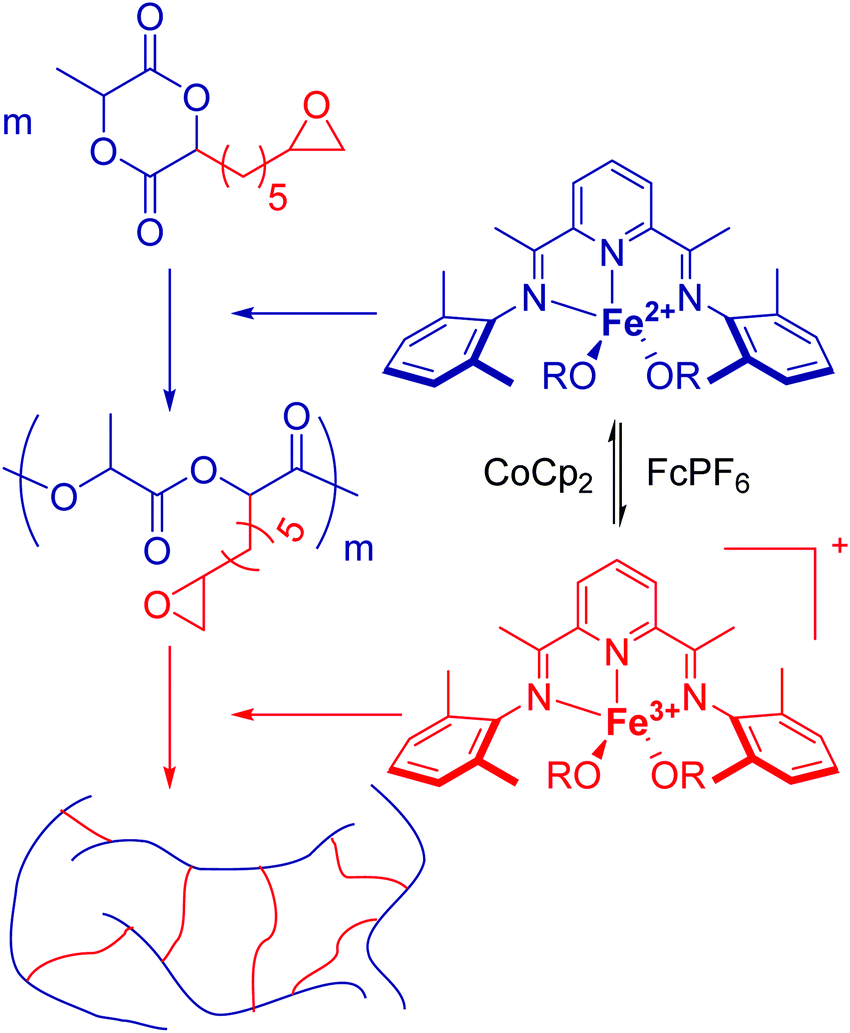 | ||
| Fig. 3 Selective polymerization of a bifunctional monomer by the redox-controlled iron catalyst. Reproduced from ref. 134 with permission from The Royal Society of Chemistry, Copyright 2016. | ||
Coudane and coworkers128 reported an alkyne functional glycolide (3-(2-propynyl)-1,4-dioxane-2,5-dione, A54), and modified PLGA-copolymers with PEG–azides. Cheng and coworkers129 reported the analogous alkyne lactide A55. They grafted PEG–paclitaxel–azide conjugates onto PLA-copolymers. A disubstituted alkynated glycolide (A56) has been used by Baker and coworkers136 for the polymerization of homopolymers and random or block copolymers, which were functionalized by click chemistry with PEG550–azide and azidododecane, to obtain thermoresponsive materials exhibiting lower critical solution temperatures (LCST) from room temperature to >60 °C. A facilitated synthesis of difunctional halogenide monomers was reported by Collard and coworkers to yielding 3,6-bis(chloromethyl)-1,4-dioxane-2,5-dione (A57):137 3-chloropropane-1,2-diol was oxidized to the glycolic acid derivative and subsequently dimerized and cyclized. Polymers were modified by dehydrochlorination to methylidene functions and further reacted with thiol derivatives by radical or nucleophilic thiol addition.
Yang and coworkers138 reported a further alkyne-functionalized lactide A58, synthesized by an alternative route: several commercially available aldehydes were reacted in a Passerini-type condensation to obtain the glycolic acid derivative (Scheme 9B). PLA-copolymers of A58 were modified with dansyl-azide as prove of concept. They additionally reported two azide-functionalized monomers (A59 and A60), synthesized by the same route. Copolymers were modified with dansyl alkyne. Overall yields for the synthetic strategy of A58–A60 were 6–16%. Weck and coworkers139 introduced an azido-tri(ethylene glycol) functional lactide (A61). Polymers were modified with a fluorescent dye (7-nitrobenzoxadiazole, NBD) and a cell internalization peptide gH625 by click chemistry, and proved cellular uptake. The group as well showed modification by Staudinger condensation with Tap-GRGDS.140
Modification of lactides without ring-opening is rare. Hillmyer and coworkers131 realized a bifunctional norbornene/lactide monomer (A62) suitable for ROP as well as ROMP by bromination and elimination of a lactide with an overall yield of 35% (Scheme 9C). The formed alkene reacted in a Diels–Alder reaction with cyclopentadiene and formed the bifunctional monomer. Dove and coworkers130 showed that the norbornenes in such copolymers were able to react with tetrazine derivatives. The norbornene-tetrazine reaction allowed post-modification under mild conditions at room temperature and without the addition of a catalyst or additives. The monomer was further functionalized with azide derivatives (A63), e.g. PEG-N3141 and polymerized. Two more lactides were realized by the same strategy using cyclohexa-1,3-diene (A64) and isoprene (A65) as diene for the Diels–Alder reaction, but the corresponding polymers were not further post-modified.142
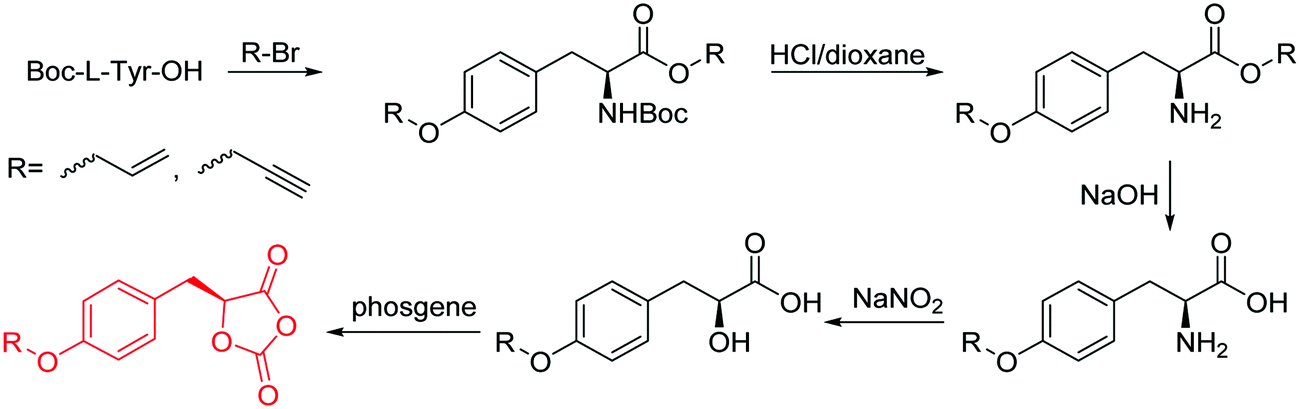 | ||
| Scheme 10 Synthetic route to O-carboxy anhydrides (OCAs) from amino acids. | ||
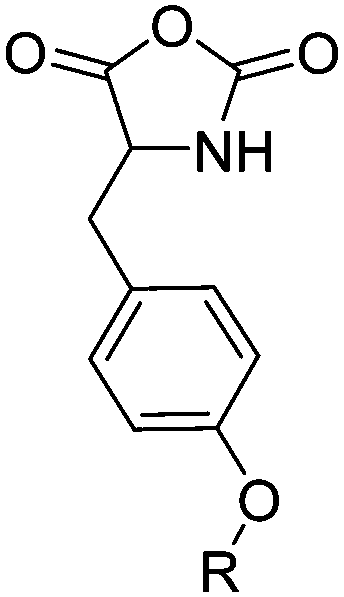
The class of monomer has mainly been explored in the last decade and two orthogonally reactive OCAs have been reported so far: L-Tyr-alkynyl- (A66)145 and L-Tyr-allyl-OCA (A67).146 Cheng and coworkers used boc-protected L-Tyr-OH and reacted it with propargyl bromide to introduce the alkyne (or allyl bromide for the analogs alkene, Scheme 10).145 After the release of the amine group and formation of the α-hydroxy acid by diazotation with sodium nitrite, carbonylation, and cyclization yielded the final monomer. PEG-block copoly(ester) of A66 were core-crosslinked with a di-azide-cross-linker to redox-147 or light-responsive148 poly(ester) micelles; homopolymers were post-modified by thiol–yne reaction with cysteamine to polyelectrolytes for gene delivery and cell-penetration.149L-Tyr-allyl-OCA (A67) has not been used for post-modification so far.
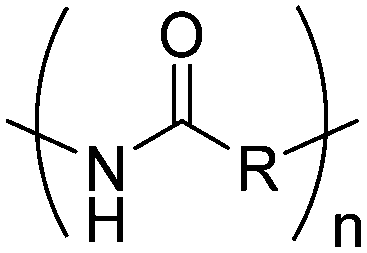
4. Polyamides
Polyamides from cyclic monomers can be obtained from cyclic lactams (polyamide-3 to polyamide-6), from α-N-carboxy anhydrides (NCAs) (polypeptides or polyamide-2) and N-substituted glycine N-carboxy anhydrides (NNCAs) (polypeptoids), or from cyclic diamides and ester amides (Table 4). Besides synthetic polypeptides, most of them exhibit only poor biodegradability, but under harsh alkaline conditions or in the presence of special microorganisms can be degraded.| Monomer | R = | No. | Post-modification | Ref. |
|---|---|---|---|---|
| Lactams | ||||
|
|
B1 | – No modification | 155 and 156 |
|
|
B2 | – Thermal or photochemical cross-linking | 153 |
| Glutamic acid-based NCAs | ||||
|
|
B3 | – Click chemistry with PEG-, carbohydrate-, amine- or cyclodextrin-azides; photochemical thiol–yne reaction to introduce carboxy groups | 165–169 |
|
B4 | – Click chemistry to introduce alkyl chains of different lengths | 170 | |
|
B5 | – Click reaction with amine/guanidines for gene delivery | 171 | |
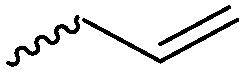
|
B6 | – Epoxidation and cross-linking; oxidation to carboxy functionalities; photochemical thiol–ene reaction to introduce carboxy groups | 172 and 173 | |
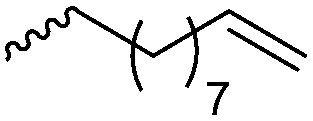
|
B7 | – Epoxidation and cross-linking | 172 | |
| – Oxidation to carboxy functionalities | ||||
|
B8 | – Thiol–ene reaction with cysteamine to elongate the distance between charged groups and backbone | 174 | |
|
B9 | – Radical cross-linking | 175–178 | |
| – Ozonolysis to alcohols and aldehydes and hydroamination | ||||
| – Oxidation to diols and carboxy groups | ||||
| – Olefin metathesis | ||||
| – Suzuki coupling | ||||
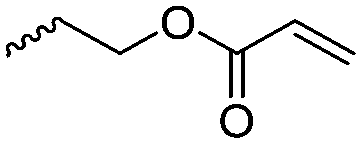
|
B10 | – Formation of films by photo-cross-linking | 179 | |
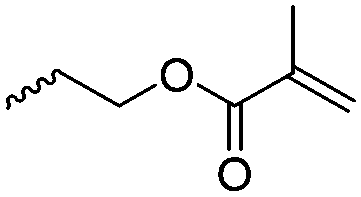
|
B11 | – Formation of films by photo-cross-linking | 179 | |
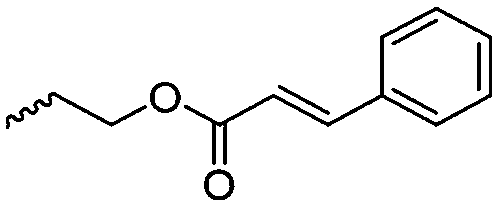
|
B12 | – Formation of films by photo-cross-linking | 179 | |
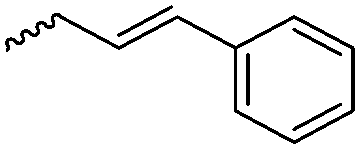
|
B19 | – Photo-cross-linking to stable micelles for drug delivery | 180 and 181 | |
|
B20 | – No modification | 182 | |

|
B21 | –ATRP macroinitiator | 183 and 184 | |
| – Quaternization with diamines to form nanogels for drug delivery | ||||
|
B22 | – Derivatization with NaN3 and click chemistry with carbohydrates, arginine or imidazolium | 185–189 | |
| – Quaternization of phosphine and pyridinium salts | ||||
|
B23 | – Derivatization with NaN3 and click chemistry with arginine | 186, 188 and 190 | |
| – Quaternization of phosphine and pyridinium salts | ||||
|
B24 | – Derivatization with NaN3 and click chemistry with arginine | 186 | |
|
B25 | – Nucleophilic substitution with 1-alkylimidazolium salts to LCST- and UCST-type polypeptides | 191 | |
|
B26 | – Amidation with amines for gene delivery | 192 and 193 | |
| Tyrosine-based NCAs | ||||
|
|
B13 | – Formation of films by photo-cross-linking | 179 |
|
B14 | – Formation of films by photo-cross-linking | 179 | |
|
B15 | – Formation of films by photo-cross-linking | 179 | |
| Lysine-based NCAs | ||||
|
|
B16 | – Formation of films by photo-cross-linking | 179 and 194 |
| – Thiol–ene reaction for cross-linking | ||||
|
B17 | – Formation of films by photo-cross-linking | 179 and 195 | |
| – Thiol–ene reaction | ||||
|
B18 | – Formation of films by photo-cross-linking | 179 | |
|
B28 | –ATRP macroinitiator | 196 | |
|
B29 | – Click chemistry | 197 | |
| Ornithine-based NCAs | ||||
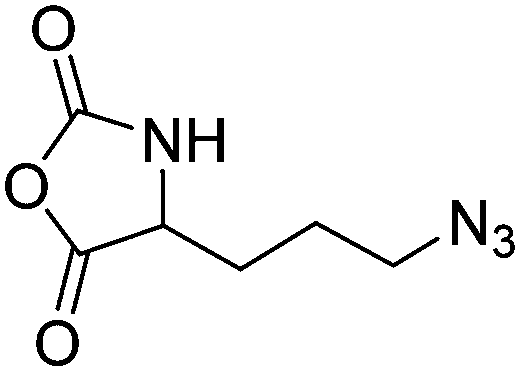
|
B30 | – Click chemistry | 197 | |
| Serine-based NCAs | ||||
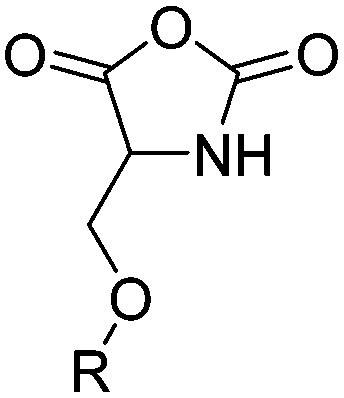
|
|
B31 | – Thiol–ene reaction with cysteamine to cell-penetrating peptides | 198 |
|
B32 | – Modification degrades the polymer | 199 | |
| Homoserine-based NCAs | ||||
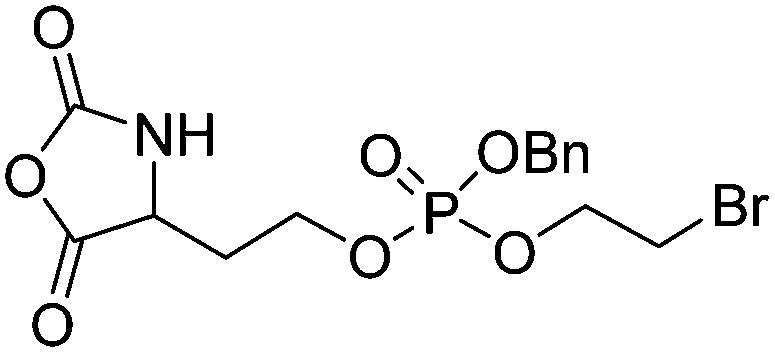
|
B33 | – Amination to form poly(L-phosphorylchloline homoserine) | 199 | |
| Cysteine-based NCAs | ||||
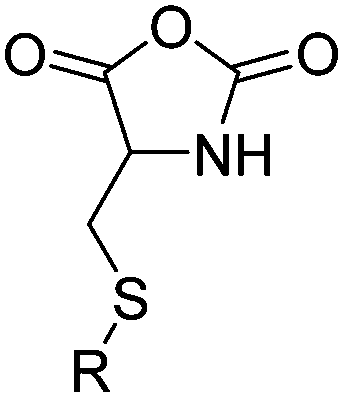
|
|
B34 | – Michael-type addition of polar-, charged- or carbohydrate-thiols forming glycopeptides, coatings, and hydrogels | 200 |
|
B35 | – Nucleophilic substitution with imidazolium salts | 201 | |
|
B36 | – Reaction with thiols to form asymmetric disulfides | 202 and 203 | |
|
B37 | – Reaction with thiols to form asymmetric disulfides | 202 | |
| Methionine-based NCAs | ||||
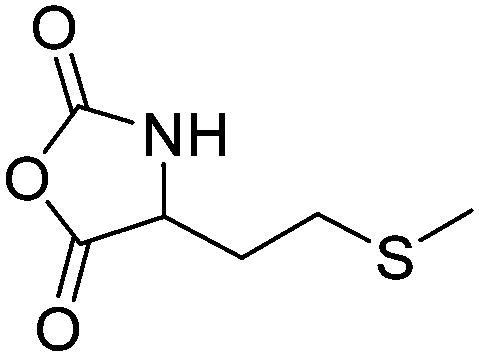
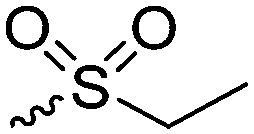
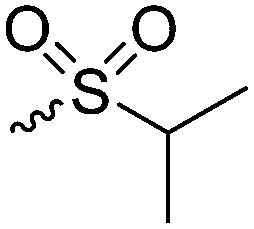
|
B38 | – Alkylation with bromide, iodide and triflate derivatives and triggered dealkylation with sulfur nucleophiles | 204–207 | |
| – Oxidation to sulfoxides causing a change of copolymer conformation | ||||
| – Reaction with epoxides to β-alkyl-β-hydroxyethyl sulfonium products | ||||
| DOPA-based NCAs | ||||
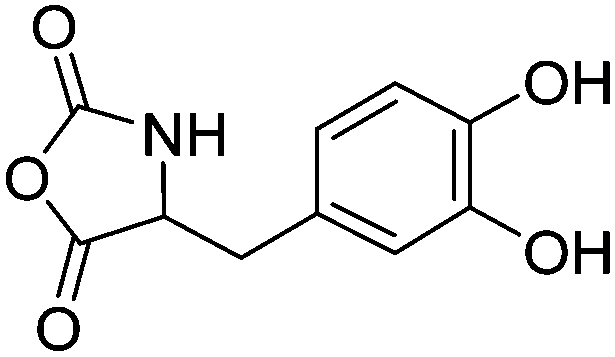
|
B39 | – Oxidative cross-linking and tissue adhesion | 194, 210 and 211 | |
| Unnatural amino acid-based NCAs | ||||
|
|
B40 | – Reduction or bromination | 212–214 |
| – Glycosylation by thiol–ene reaction | ||||

|
B41 | – No modification | 215 | |
|
B42 | – Glycosylation by click chemistry | 214 and 216–219 | |
| – Photochemical thiol–yne reaction | ||||
| γ-NCAs | ||||
|
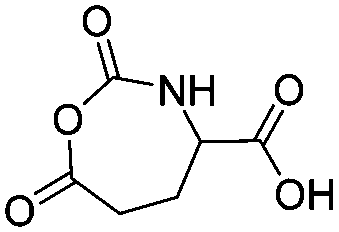
B27 | – No modification | 160 | |
| NNCAs | ||||
|
|
B43 | – Thiol–ene reaction with thioglycerol and –glucose | 224 and 225 |
|
B44 | – Click chemistry with PEG–azide | 226 and 227 | |
| – Thermal cross-linking | ||||
| Cyclic esteramides | ||||
|
B45 | – Thiol–ene reaction with charged or polar thiols | 223 | |
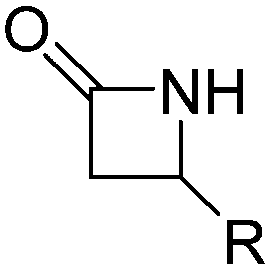
4.1 Lactams
β-Lactam (2-azetidinone), γ-butyrolactam (2-pyrrolidone), δ-valerolactam (2-piperidone) and ε-caprolactam (2-azepanone) are the main unsubstituted lactams used in the ROP. The number of orthogonally reactive lactam monomers is very limited so far. This might be attributed to the low solubility of the products in most common organic solvents, due to H-bonding or crystallization. ε-Caprolactam is typically polymerized in anionic ROP in bulk at temperatures of 140 °C above the melting point of the monomer (70 °C) within 15 min, with the polymer precipitating from the melt (Scheme 11).153N-Acyl or N-carbamoyl lactams as activators like hexamethylene-1,6-dicarbamoyl-caprolactam are commonly added to promote the polymerization. For more details, we refer to an excellent book chapter.154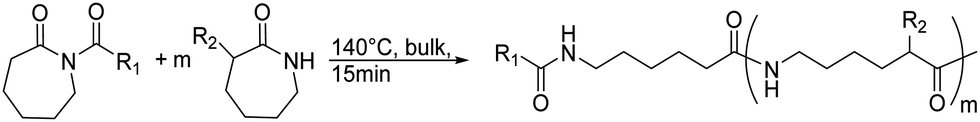 | ||
| Scheme 11 General polymerization protocol for caprolactams to poly(amides)s with an N-acyl lactam as an activator (R1 and R2 represent non-specified alkyl substituents). | ||
Vinyl lactam monomers are reported for all lactams, however, were only used for polymerization of the vinyl functionality. 4-Vinylazetidin-2-one (B1) is the only bifunctional monomer, whose anionic ROP was reported (in DMSO with potassium 2-pyrrolidone, at 25–30 °C, 2 h).155B1 was synthesized by reaction of 1,3-butadiene with chlorosulfonyl isocyanate and subsequent saponification (Scheme 12A).156 However, no post-modification has been reported so far.
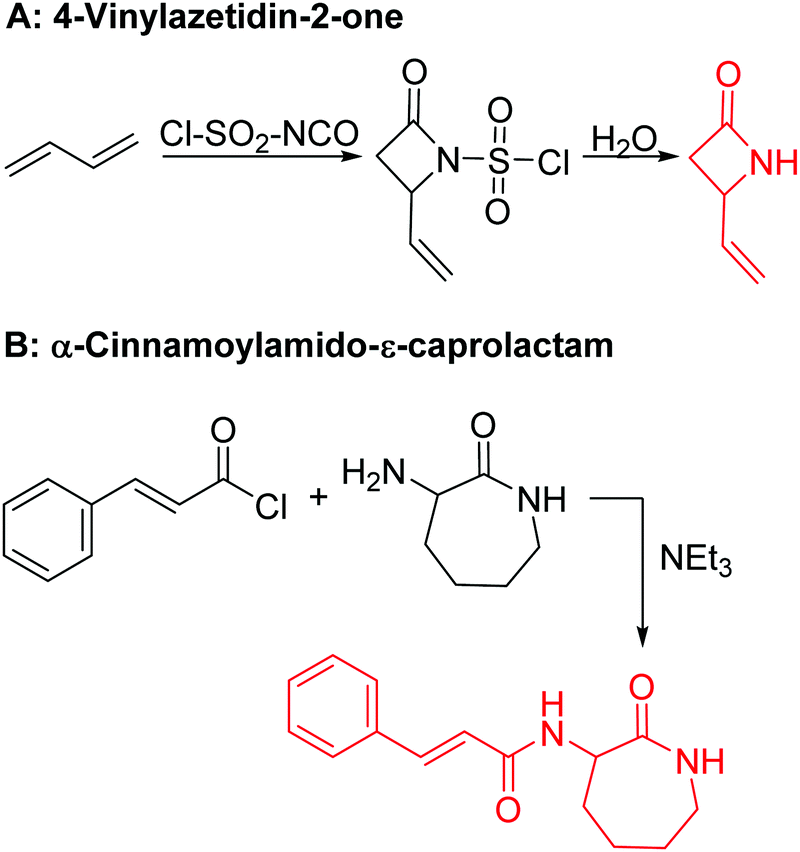 | ||
| Scheme 12 Synthetic route to functional lactams: (A) 4-vinylazetidin-2-one B1 and (B) α-cinnamoylamido-ε-caprolactam B2. | ||
While a few protected functional ε-caprolactam monomers (with an amine, carboxy, and carbonyl groups) are reported, Carlotti and coworkers153 synthesized a reactive monomer, bearing a cinnamoyl functionality. α-Cinnamoylamido-ε-caprolactam (B2) was synthesized in one step from cinnamoyl chloride and α-amino-ε-caprolactam (yield: 80%) (Scheme 12B). Copolymers with ε-caprolactam were cross-linked thermally (at 140 °C) or photochemically (at 364 nm) and again de-cross-linked photochemically (at 254 nm). α-Amino-ε-caprolactam might be an interesting precursor for future ε-caprolactams with other functional groups, e.g. to tailor the degradation rates. Polyamides can also be post-modified at the amide group by N-alkylation with formaldehyde,157 by epoxides or 2-bromoethylamine,158 isocyanates or acid chlorides.159
An interesting approach might be the synthesis of monomers from natural and renewable sources, e.g. macrolactones or carbohydrates. However, only a limited number of such examples is available up to date. Only a few functional OCA monomers for the synthesis of polyesters are reported, and expansion the monomer class is promising.
4.2 N-Carboxyanhydrides
Synthetic polypeptides are accessible by ROP of N-carboxy anhydrides (Leuchs’ anhydrides, NCAs), which are N-analogs to poly(α-hydroxy acid)s from O-carboxy anhydrides (OCAs). α-NCAs (5-membered rings) dominate the literature, while β-NCAs (6-membered rings) are only reported without additional functional groups to date. A single functional γ-NCA monomer (7-membered ring) has been reported in 1978,160 bearing a pendant carboxyl moiety (see below and Scheme 15).A variety of protected, functionalized, and orthogonally reactive α-NCA monomers are reported. They are excellently summarized in two recent reviews.161,162 Primary amines are the common initiators163 for the ROP of NCAs, but also alcohols can be applied (as the initiation of the OH-group is slower than that of an amino-group, either the alcohol needs to be activated or broad molecular weight distributions will be obtained)164 (Schemes 13 and 14).
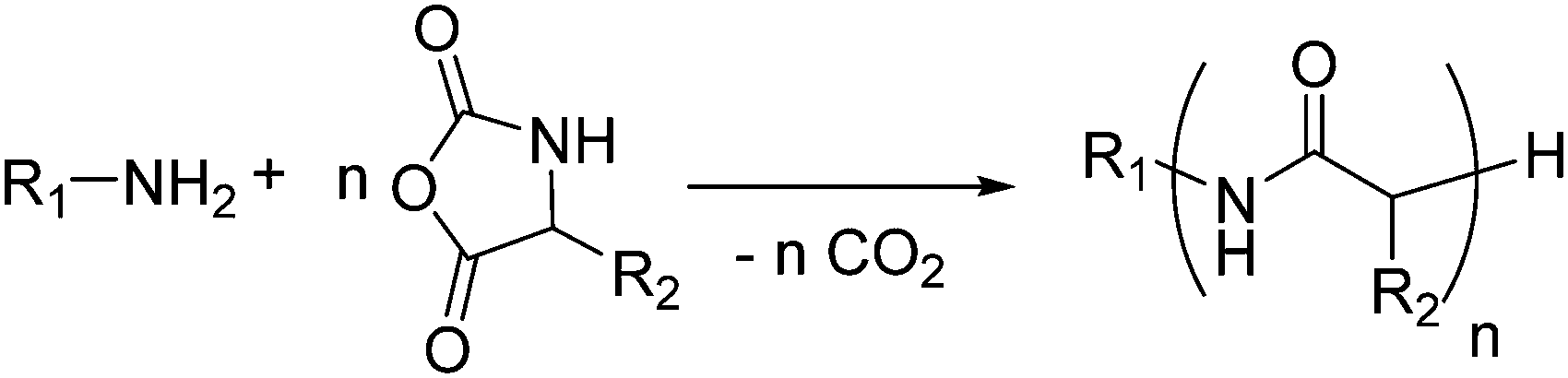 | ||
| Scheme 13 General protocol for the polymerization of NCAs to polypeptides with amines as the initiator (R1 and R2 represent non-specified substituents). | ||
 | ||
| Scheme 14 General synthetic routes to functional NCAs, depicted with glutamic acid. | ||
Several alkene-functionalized NCAs have been reported to date. Daly and coworkers172 prepared γ-allyl-L-glutamate (B6) and γ-(9-decenyl)-L-glutamate NCA (B7) and studied their homo- and copolymerization and epoxidation of the double bonds with mCPBA. Gelation was obtained by cross-linking of the epoxides with TFA, oxidation to carboxylic acids was shown with KMnO4/NaHCO3. Zhang and coworkers173 used B6 in copolymers for photochemical thiol–ene reaction and introduction of carboxy groups. γ-(4-Allyloxylbenzyl)-L-glutamate NCA (B8) has been reported by Cheng and coworkers,174 using the copper(II)-complex method and 4-allyloxylbenzyl chloride. Thiol–ene reaction in homopolymers of B8 with cysteamine to poly(γ-(4-aminoethylthiopropoxyl)benzyl-L-glutamate) exhibited a charge-backbone distance of 17 σ-bonds. Remarkably high helicity of 81% was observed for polypeptides with a DP of 10 at pH 2. Compared to the polymer with an elongated charge-backbone distance of 17 σ-bonds, poly(γ-(4-(1-hexanol-6-aminomethyl))benzyl-L-glutamate) with a DP of 10 and a charge-backbone distance of 11 σ-bonds showed mixed conformation of β-sheets and only 26% α-helices. γ-(4-Vinyl benzyl)-L-glutamate NCA was introduced by the group of Schouten.175 The olefins were used for radical cross-linking of terpolymer surface-grafted films,175 or transformation by ozonolysis into alcohols176 or aldehydes177 and subsequent hydroamination with primary amines, to yield polypeptides suitable for gene transfection.178 Oxidation to diols or carboxy functionalities by osmium tetroxide, olefin metathesis reaction with cis-1,4-dichlorobutene and Suzuki coupling were shown by Cheng and coworkers.176
Kamogawa and coworkers179 reported already in 1975 a whole library of photoreactive glutamic acid, tyrosine and lysine based NCAs with pendant acryloyl, methacryloyl and cinnamoyl groups (B10–B18). They showed polymerization of all NCAs and photochemical cross-linking of films, whereas the photosensitivity decreased for acryloyl ≈ methacryloyl > cinnamoyl, and polyglutamate > polylysine > polytyrosine. A further photo-cross-linkable monomer, also containing a cinnamoyl function, γ-cinnamoyl-L-glutamate NCA (B19), was already reported before by Iwakura and coworkers in 1974.180 Jing and coworkers181 photochemically crosslinked self-assembled polypeptide-block-PEG micelles of B19, loaded with paclitaxel as stable drug carriers. Iwakura and coworkers reported a further potentially photoactive benzophenone-containing NCA, γ-p-benzoyl benzyl-L-glutamate NCA and its homo- and copolymerization with γ-p-benzyl-L-glutamate NCA (B20).182 They studied the orientation of benzophenone groups in the side chains and conformation of polymers. The further photochemical reaction of the pendant groups has not been shown so far.
Several chlorinated monomers with varying length of alkyl spacers and suitable for nucleophilic substitution were reported: homo- and copolymers of a γ-(2-chloroethyl)-L-glutamate NCA (B21) were used as ATRP macroinitiator to graft oligoethylene methacrylate or to form nanogels for drug delivery.183 Cross-linking of the gels was achieved through quaternization reaction of 2,2′-dithiobis(N,N-dimethyl ethylamine) (dTbDEA) with the chloride functionalities.184 Polymers with the analogous γ-(3-chloropropyl)-L-glutamate (B22), γ-(6-chlorohexyl)-L-glutamate (B23), and γ-(8-chlorooctyl)-L-glutamate NCA (B24) were further derivatized to the respective azides and modified with carbohydrates,185 arginine,186 or imidazolium187 derivatives by click chemistry. These poly(arginine) mimics with hydrophobic side chains of different lengths exhibited helix-related cell-penetrating properties and high DNA and siRNA delivery efficiencies in various mammalian cells. Quaternization of the chloride substituted polypeptides with triethylphosphine yielded cell-penetrating peptides,188 quaternization with pyridinium salts UCST-type polypeptides,189 and with 1-methyl benzimidazole helical antimicrobial polypeptides.190 Tang and coworkers191 reported γ-(4-chloromethyl benzyl)-L-glutamate NCA (B25), synthesized by the copper(II) complex strategy. These chloride-substituted polypeptides were modified with 1-alkyl imidazolium (methyl or n-butyl) and various counter-anions (i.e. Cl−, F−, BF4−). The polypeptides exhibited LCST- or UCST-type behavior in organic solvents or in water. Poly(peptide)s of an active ester functionalized monomer, γ-trichloroethyl-L-glutamate NCA (B26)192 were post-modified by amidation with different amine derivatives and their properties examined as gene delivery vectors.193 Higashi and coworkers reported already in 1978 a carboxylated γ-NCA (B27),160 after the reaction of the glutamic acid with diphosgene (Scheme 15). They proved successful polymerization by viscosity measurements and characteristic amide signals in IR spectra, which are also observed for Nylon-4. Modification of the carboxyl function has not been reported.
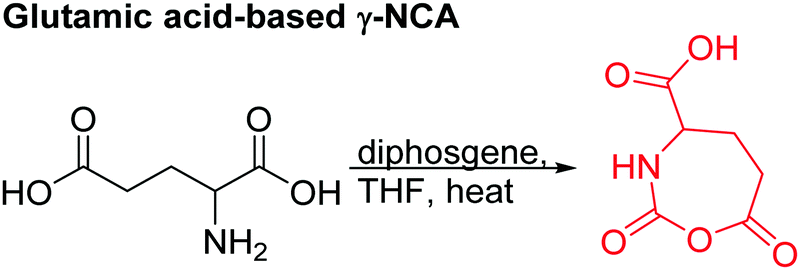 | ||
| Scheme 15 Synthetic strategy to a glutamic acid-based γ-NCA B27. | ||
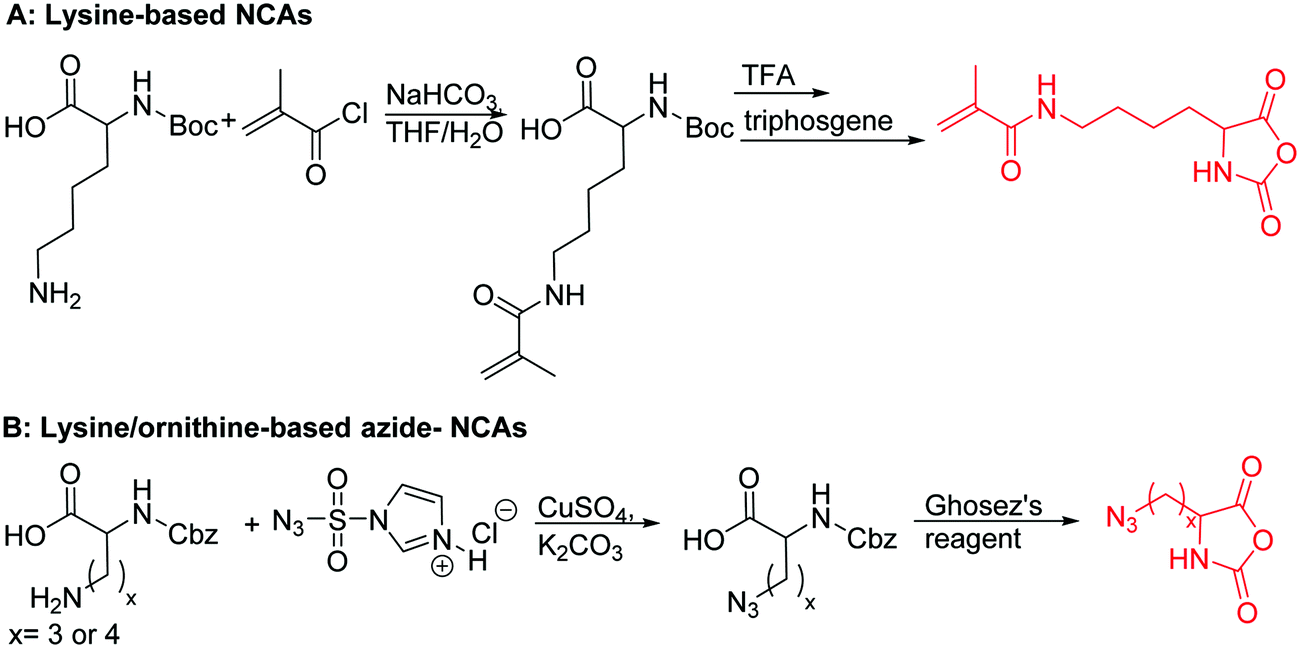 | ||
| Scheme 16 Synthetic routes to lysine/ornithine-based NCAs. | ||
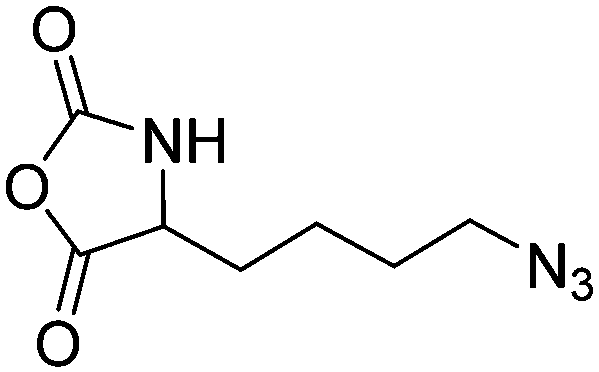
Polymers of ε-N-bromoisobutyryl-L-lysine NCA (B28) were used as ATRP macroinitiators by Li and coworkers,196 and two polypeptide bottlebrushes with polystyrene or poly(oligoethylene glycol methacrylate) prepared, exhibiting an α-helical conformation in appropriate solvents.
Deming and coworkers197 reported azide-containing monomers: azido-norleucine (B29) and azido-norvaline NCAs (B30). α-N-Carboxybenzyl lysine or ornithine was reacted with imidazole-1-sulfonyl-azide·HCl, CuSO4, and K2CO3 to form the azide derivative (Scheme 16B). The derivatives were converted to NCAs, using the Ghosez's reagent (1-chloro-N,N′-2-trimethyl-1-propenylamine). Various alkyne derivatives were attached to azide-substituted pol(peptide)s through click chemistry with >95% conversion.
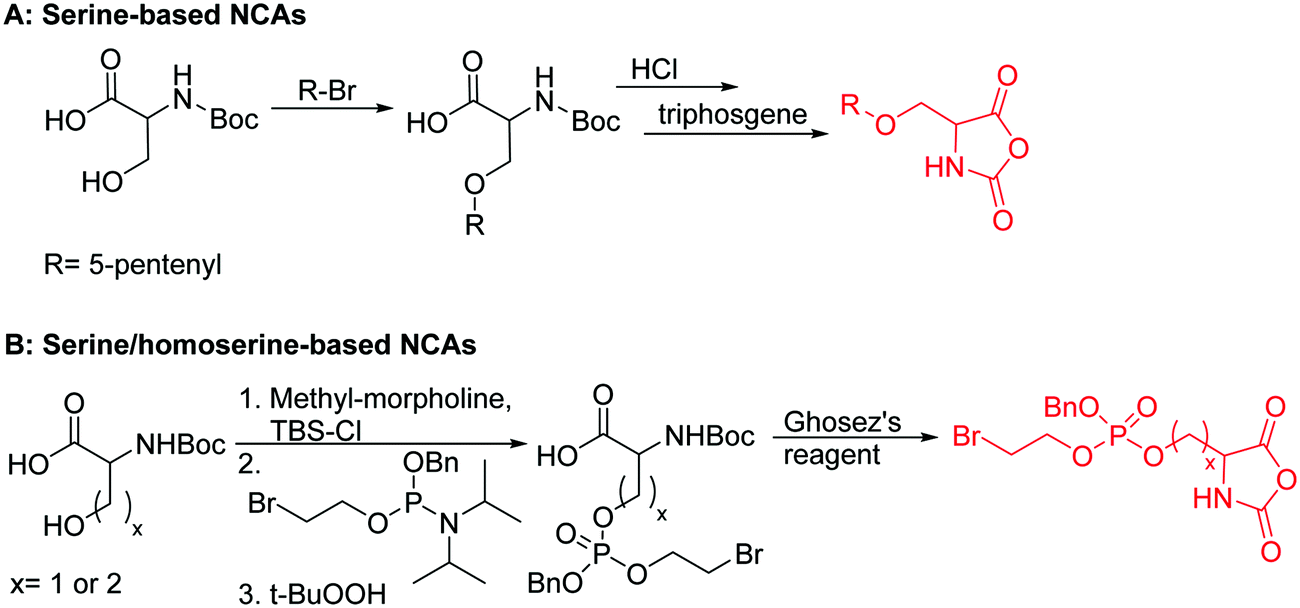 | ||
| Scheme 17 Synthetic routes to serine/homoserine-based NCAs. | ||
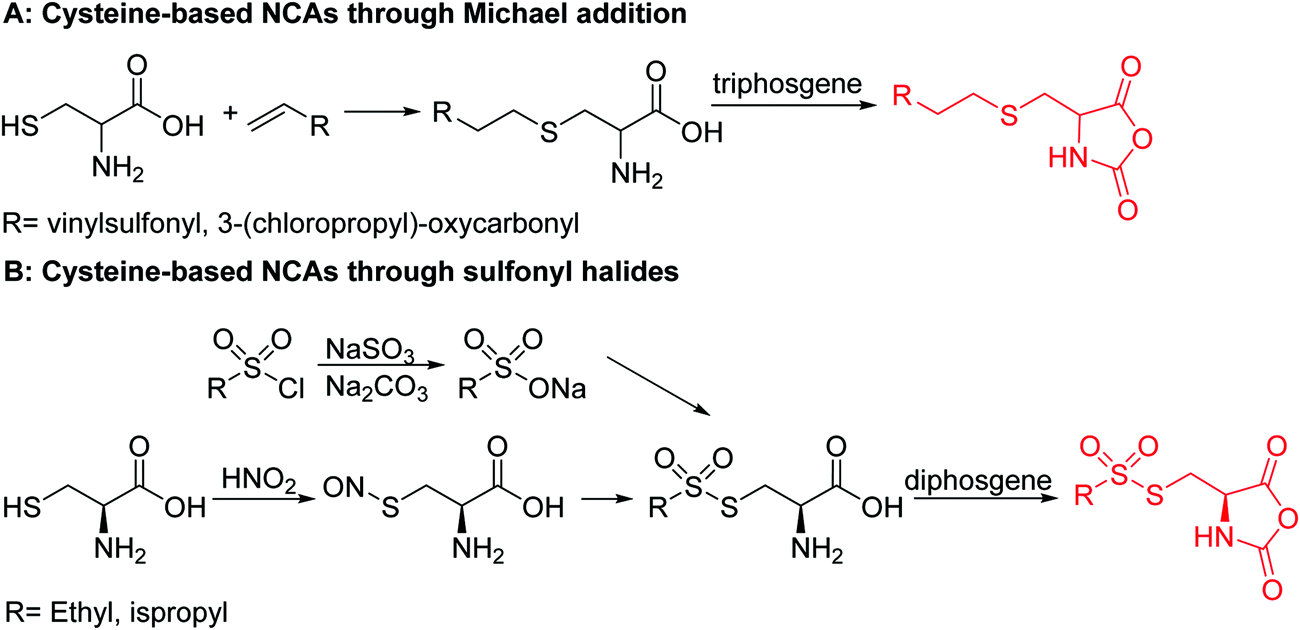 | ||
| Scheme 18 Synthetic protocols for cysteine-based NCAs. | ||
Barz and coworkers202 recently reported two interesting S-sulfonyl based NCA monomers: S-(ethylsulfonyl)-L-cysteine NCA (B36) and S-(isopropylsulfonyl)-L-cysteine NCA (B37) (Scheme 18B). Disulfides are reversible and therefore attractive for biomedical applications. They are stable under extracellular conditions, but cleavable inside of cells. Usually, disulfide formation has been achieved by oxidation of thiols (with long reaction times, often not 100% conversion and the lack of formation of asymmetric disulfides) or by the formation of reactive thiols (chlorinated or nitroso-thiols). A major drawback of activated thiols is the limited stability against aminolysis and hydrolysis. The group of Barz introduced two monomers with a protective and at the same time activating group, which was stable during ROP. Asymmetric disulfide formation was chemoselectively achieved by post-modification with appropriate thiols. An alkyl sulfonyl chloride was hydrolyzed to an alkyl sulfinic acid sodium salt and reacted with S-nitrosocysteine (generated in situ from L-cysteine) forming a thiosulfonate, which reacted with diphosgene to yield the final NCAs (B36 and B37) (Scheme 18B). Quantitative post-modification of polymers from B36 with benzylmercaptan was proven within 60 s, without degradation or side reactions. The approach opens a way to reversible conjugation of drugs as well as cross-linking to form nanostructures.203
 | ||
| Fig. 4 The enzyme-triggered release of cargos from methionine sulfoxide containing vesicles: (a) structure and redox properties of poly(L-methionine)-b-poly(L-leucine-stat-L-phenylalanine) peptides, (b) the possible effect of enzymatic reduction of vesicle surface of sulfoxide segments to methionine segments for a change of conformation and cause of vesicle ruptures. Adapted from ref. 207 with permission from the American Chemical Society, Copyright 2017. | ||
Alkene- or alkyne-substituted NCAs from unnatural amino acids compared to analog NCAs from natural amino acid display the distinct benefit, that ester or amide linkages are absent in the pendant chains. Pendant chains in polypeptides from naturally amino acid-based NCAs might be cleaved by hydrolysis or aminolysis at these linkages and thereby molecules introduced by post-modification detached. A challenging drawback of unnatural amino acid-based NCAs is the synthesis of the needed amino acids, which might be the reason for only a few reported monomers in literature.
4.3 Cyclic diamide monomers
To the best of our knowledge, no orthogonally functional cyclic diamides suitable for ROP have been reported so far. They are usually used for polycondensation reaction, where the ring remains in the backbone of the polymer. Elias and coworkers copolymerized the unfunctionalized monomer 2,5-dioxopiperazine (DOP) with caprolactam,220 Hildgen and coworkers with allyl glycidyl ether.2214.4 Cyclic ester amide monomers
Morpholine-2,5-diones, resulting in polydepsipeptides, have been functionalized with different alkyl chains or protected groups. However, they are less reactive as lactides.222 To the best of our knowledge, Klok and coworkers223 reported the only orthogonally reactive ester amide monomer, L-allylglycine-morpholine-2,5-dione (B45). Further functional morpholine-2,5-diones employ protecting groups. A recent review summarizes the properties of poly(ester amide)s.43 The synthetic strategy is analog to the synthesis of functional glycolides and lactides: a corresponding α-amino acid and 2-bromo acetyl bromide reacted and the final monomer was realized by intramolecular cyclization of the intermediate in 13% yield (Scheme 19). Klok and coworkers showed modification by thiol–ene reaction with different fluorinated, carboxylated, aminated or dihydroxylated thiols with conversions between 15–100%. | ||
| Scheme 19 Synthetic strategy to functional morpholino-2,5,-dione monomer (B45). | ||
5. Polycarbonates
Aliphatic polycarbonates find broad application in biomedical devices and drug delivery. They are typically prepared by phosgene condensation or ester exchange, by addition polymerization of epoxides with carbon dioxide in ring-opening copolymerization (ROCOP)228 or by ROP of 6-membered cyclic carbonates, i.e. trimethylene carbonate (TMC). Poly(trimethylene carbonate) (PTMC) and poly(dimethyl trimethylene carbonate) (PDTC) display poor hydrophilicity and slow degradation rates. A broad range of functional TMCs, which we present here, has been explored to alter these properties and increase hydrophilicity and degradation. Polymerization of the five-membered cyclic carbonates is thermodynamically unfavorable.229 Polymerization above 150 °C results in poly(ether-carbonate)s and decarboxylation. Five-membered vinylene carbonate230 and vinylethylene carbonate231 are radically polymerized at the vinyl function, while the cyclic carbonate remains unaffected. Polymerization of seven-membered rings is possible but less relevant. Reported functional monomers are limited to alkyl- or aryl-pendant chains, or protected functionalities; reactive functionalities have not been reported up to date. For more details, we also refer to a review of Rokicki.229 The copolymerization of epoxides with anhydrides or carbon dioxide to polycarbonates is an attractive strategy. Functional epoxides and their synthetic protocols are well-established,232 and the potential of the combination with carbon dioxide228 or anhydrides45 are exploited (Table 5).| Monomer | R = | No. | Post-modification | Ref. |
|---|---|---|---|---|
| 5-Monosubstituted TMCs | ||||
|
|
C1 | – Epoxidation with mCPBA, hydrazination and doxorubicin attachment for drug delivery | 236 and 238–241 |
| – Photochemical thiol–ene reaction with mercaptoethanol | ||||
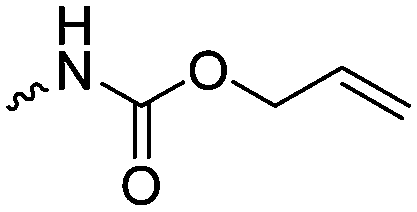
|
C2 | – No modification | 237 | |
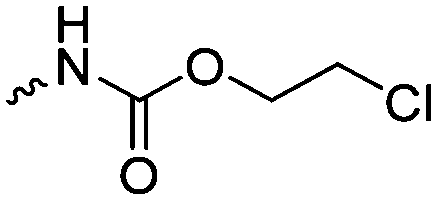
|
C3 | – No modification | 237 | |
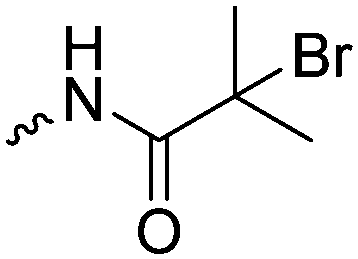
|
C4 | – Macroinitiator for ATRP polymerization of HEMA | 242 | |
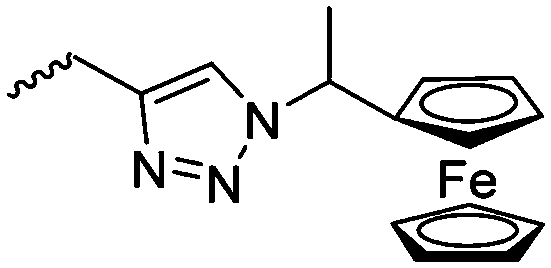
|
C5 | – Study of redox potential | 87 | |
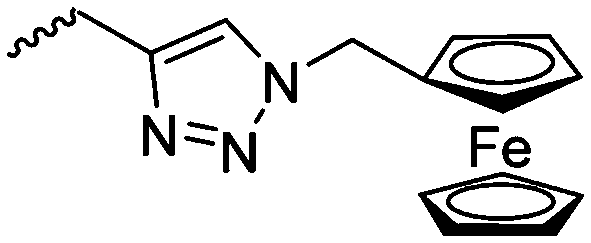
|
C6 | – Study of redox potential | 87 | |
|
C7 | – Study of redox potential | 87 | |
| Disubstituted TMCs | ||||
|
|
C11 | – Click chemistry with carbohydrates, immobilization of TSP50 proteins or hemoglobin as oxygen carrier or reversible light-responsive micelles with spiropyran modification | 243–248 |
| – Thiol–yne reaction | ||||
| – Functionalization with decaborane for BNCT | ||||
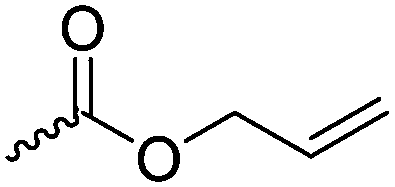
|
C13 | – Epoxidation and hydrolyzation to diols or reaction with alcohols, amines or thiols | 250–259 | |
| – Thiol–ene reaction to attach folic acid, nucleobases, for cross-linking and gelation | ||||
| – Ozonolysis and reduction to aldehydes and aldehyde-aminooxy click reaction | ||||
|
C14 | – Modification by Michael-addition with polar and charged groups | 260–262 | |
| – Photo-cross-linking to obtain micelles and fibers | ||||
|
C15 | – No modification | 260 | |
|
C16 | – No modification | 263 | |
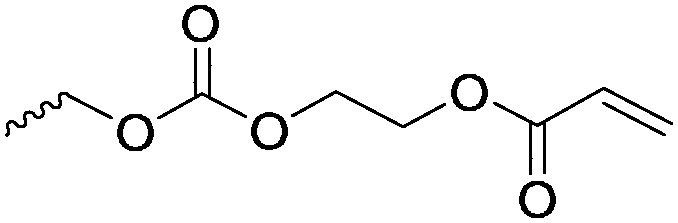
|
C17 | – No modification | 263 | |
|
C18 | – No modification | 263 | |
|
C19 | – Photo-cross-linking | 264 | |
|
C20 | – Michael-type reaction to attach polar or charged groups, GRGDC peptide or PEG-SH | 265 | |
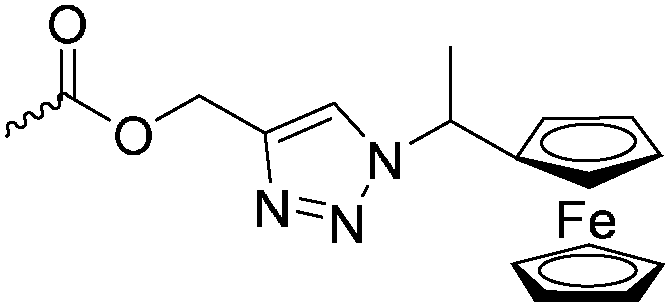
|
C8 | – Study of redox potential | 87 | |
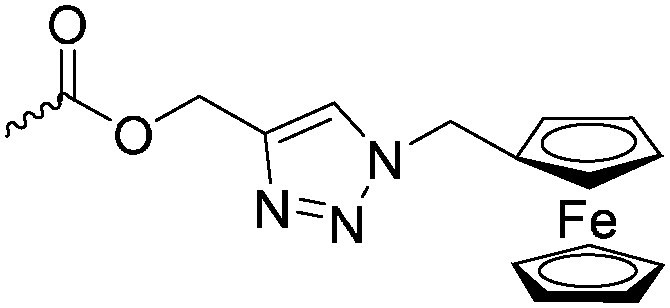
|
C9 | – Study of redox potential | 87 | |
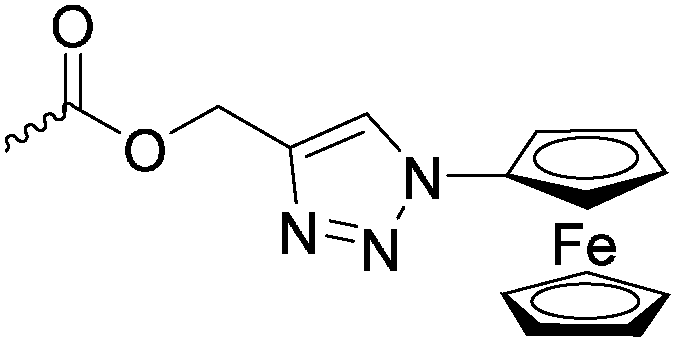
|
C10 | – Study of redox potential | 87 | |
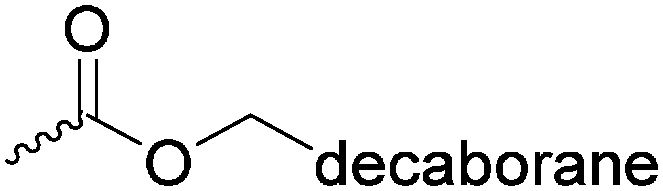
|
C12 | – PEG-copolymer nanoparticles as boron vector for BNCT | 249 | |

|
C21 | – No modification | 266 | |
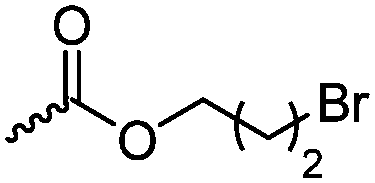
|
C28 | – Nucleophilic substitution with bis-tertiary amines, pyridines or imidazoles as gene delivery vector and for antimicrobial applications | 268, 269, 271, 272 and 274 | |
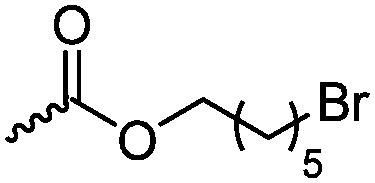
|
C31 | – Nucleophilic substitution with pyridines or imidazoles for antimicrobial applications | 272 | |
|
C22 | – No modification | 266 | |
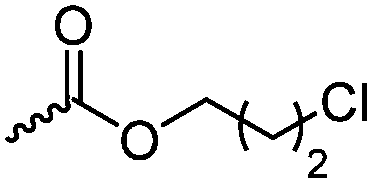
|
C27 | – Nucleophilic substitution with bis-tertiary amines, pyridines or imidazoles as gene delivery vector and for antimicrobial applications | 268, 270, 272 and 273 | |
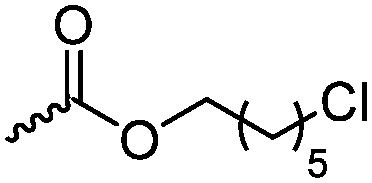
|
C29 | – Nucleophilic substitution with pyridines or imidazoles for antimicrobial applications | 272 and 273 | |
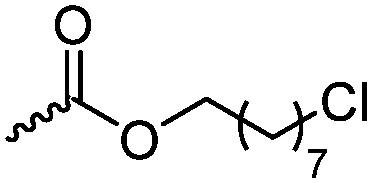
|
C30 | – Nucleophilic substitution with primary amines for antimicrobial applications | 273 | |
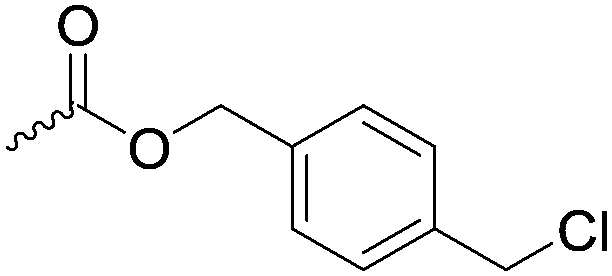
|
C32 | – Nucleophilic substitution with primary amines for antimicrobial applications | 274–277 | |
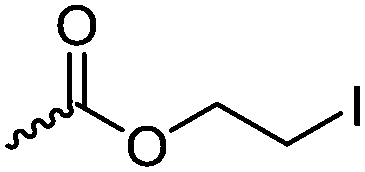
|
C26 | – Nucleophilic substitution with bis-tertiary amines as gene delivery vector | 268 | |
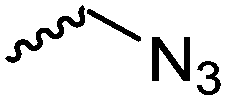
|
C25 | – Click reaction with alkyne derivatives | 267 | |
| – No polymerization is shown of azide monomer | ||||
|
C33 | – Functionalization with different amines | 277–281 | |
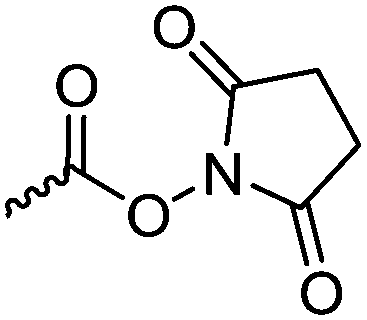
|
C34 | – Functionalization with amines to introduce charged groups or bioactive molecules | 282 | |
|
C35 | – Functionalization with epoxide derivatives to introduce galactose, tocopherol or carbazols | 283 | |
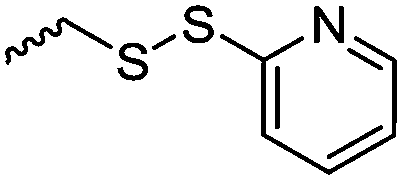
|
C36 | – Thiol–disulfide exchange reaction with thiols to reduction-sensitive self-assembled micelles or nanoparticles | 284 and 285 | |
|
C37 | – Thiol–disulfide exchange reaction for formation of dynamic, stimuli-responsive and self-healing hydrogels | 286 and 287 | |
|
C38 | – Thiol–disulfide exchange reaction for formation of core-cross-linked micelles or hydrogels | 287 | |
|
C39 | – RAFT macroinitiator | 288 | |
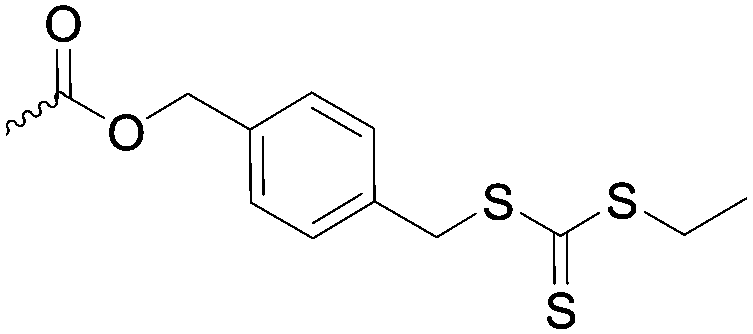
|
C40 | – RAFT macroinitiator | 289 | |
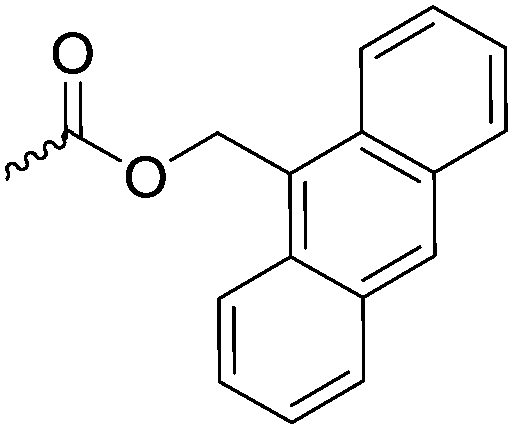
|
C41 | – Diels–Alder reaction with maleimide-polymer derivatives to grafted copolymers | 290 | |
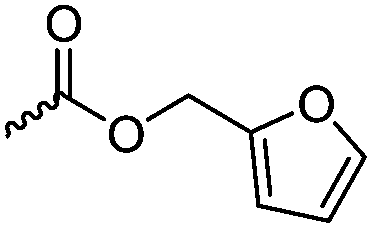
|
C42 | – Cross-linking with maleimide-comonomers by Diels–Alder reaction to materials for nanoimprinting | 291 | |
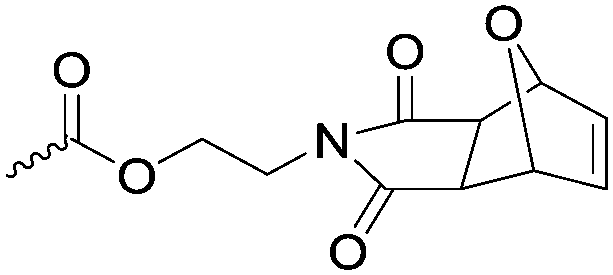
|
C43 | – Cross-linking with furfuryl-comonomers by Diels–Alder reaction to materials for nanoimprinting | 291 | |
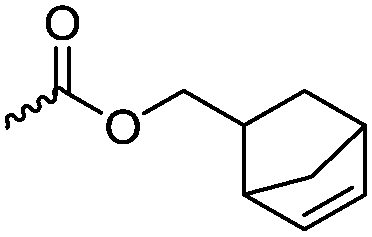
|
C44 | – Click reaction with azides | 292 | |
| – Thiol–ene reaction | ||||
| – Inverse electron demand Diels–Alder reaction with tetrazines | ||||
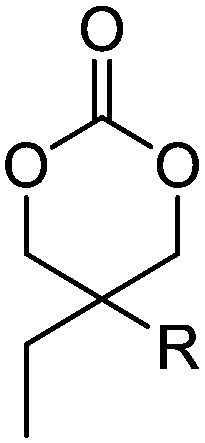
|
|
C45 | – No modification | 235 |
|
C46 | – No modification | 235 | |
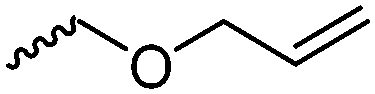
|
C47 | – Thermal or photochemical cross-linking | 293–295 | |
| – Thiol–ene reaction with PEG-SH for the formation of LCST-type polycarbonates | ||||
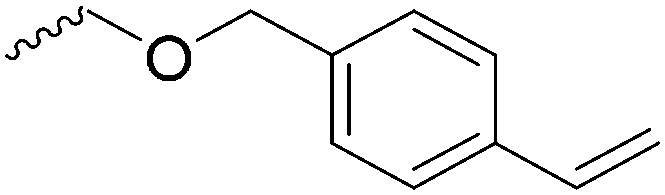
|
C49 | – Cross-linking with styrene | 297 | |
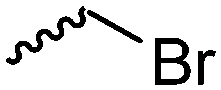
|
C23 | – No modification | 266 | |
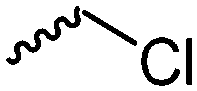
|
C24 | – No modification | 266 | |

|
C50 | – Click reaction with alkynes | 267 | |
| – No polymerization is shown of azide monomer | ||||
|
C51 | – No modification | 267 | |
|
C48 | – Functionalization with thiols or benzylamine | 296 | |
|
|
C52 | – Quaternization with tertiary amines for antimicrobial applications | 298–301 |
| – Azidation and click reaction to form amphiphilic graft copolymers, micelles or nanoparticles for drug delivery | ||||
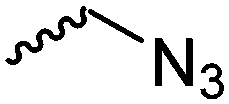
|
C53 | – Click reaction with alkynes core-cross-linked micelles or hydrogels for drug delivery or cell tissue engineering | 302–304 | |

|
C54 | – Self-cross-linking of micelles for drug delivery and intracellular de-cross-linking | 305 | |
|
C55 | – Epoxidation with p-CPBA | 306 and 307 | |
5.1 Trimethylene carbonate
Various mono- and disubstituted trimethylene carbonate (TMC) monomers have been reported in the literature. Dove and coworkers published an excellent review about TMCs in 2013,25 including general synthetic strategies, functional monomers, and their polymerization, as well as post-modification reactions and applications. Here, we restrict to general synthesis strategies for orthogonally-reactive monomers and selected some representative examples for further applications.While homopolymerization of the most functional monomers is possible, often copolymerization with alkyl-chain containing TMCs is conducted to adjust the functional group density. Furthermore, disubstituted monomers mostly contain a methyl or ethyl substituent, which has an impact on the thermal properties and solubility of the products. Alcohols are commonly used as initiators. A variety of synthetic protocols for the polymerization of TMCs are reported, including the polymerization with tin(II) octoate in bulk at ca. 110–120 °C for 4–48 h. Modern synthesis protocols rely on organocatalysis with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in bulk or solution at temperatures between 25 and 110 °C for 1–3 days, with a catalytic system of DBU or sparteine with a thiourea cocatalyst N-cyclohexyl-N′-(3,5-bis(trifluoromethyl)phenyl)thiourea (TU) at room temperature and in solution for 2–4 h or with the more reactive base 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) in bulk or solution at temperatures ranging from 0–25 °C over a period of 1 min to 2 h (Scheme 20). Commonly used solvents are dichloromethane, chloroform or toluene, depending on the solubility of TMCs and applied reaction temperatures.
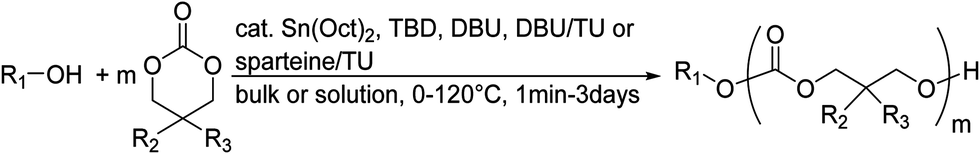 | ||
| Scheme 20 General protocol for the organocatalytic polymerization of TMCs to poly(carbonates)s with alcohols as initiators. | ||
A variety of synthetic strategies has been reported for the synthesis of substituted TMCs, including 2–5 steps with overall yields between 17–70%. A synthetic strategy relies on 2,2-bishydroxy(methyl)propionic acid (bis-MPA) as feedstock (Scheme 21), which was esterified to introduce the functional group, e.g. with alkylhalogenides (Scheme 21, route I). Following cyclization with ethyl chloroformate under basic conditions produced the TMCs. Hedrick and coworkers233 introduced an alternative route for a versatile precursor (5-methyl-5-carboxy-2-oxo-1,3-dioxane), yielding functional monomers in one further step (Scheme 21, route II). The carboxyl function of bis-MPA was protected with benzyl bromide, cyclization achieved, e.g. with triphosgene, and the carboxylic acid functionality deprotected again by hydrogenation (overall yield of precursor 59%). The precursor was either directly esterified again to introduce functionalities as pendant chains or transformed into a more reactive acid chloride and then esterified (overall yields: 15–41%). An alternative precursor, carrying a pentafluorophenyl ester protecting group,34,234 was synthesized on a gram to kilogram scale, easily handled and stored (yield: 75% Scheme 21, route III). It was reacted with suitable nucleophiles, as alcohols or amines in a transesterification reaction to functional TMCs. A further alternative is the use of imidazole intermediates using 1,1′-carbonyl diimidazole (CDI) as a key reagent (yield of intermediate: 67% Scheme 21, route IV). The intermediates were robust and bench stable.235 Alternatively, the hydroxyl functions of bis-MPA was first protected, e.g. with 2,2-dimethoxypropane (DMP) (Scheme 21, route V). Esterification of the carboxyl group introduced subsequently the pendant functionality. After deprotection of the hydroxyl groups, cyclization was achieved with ethyl chloroformate under basic conditions to yield the final monomers.
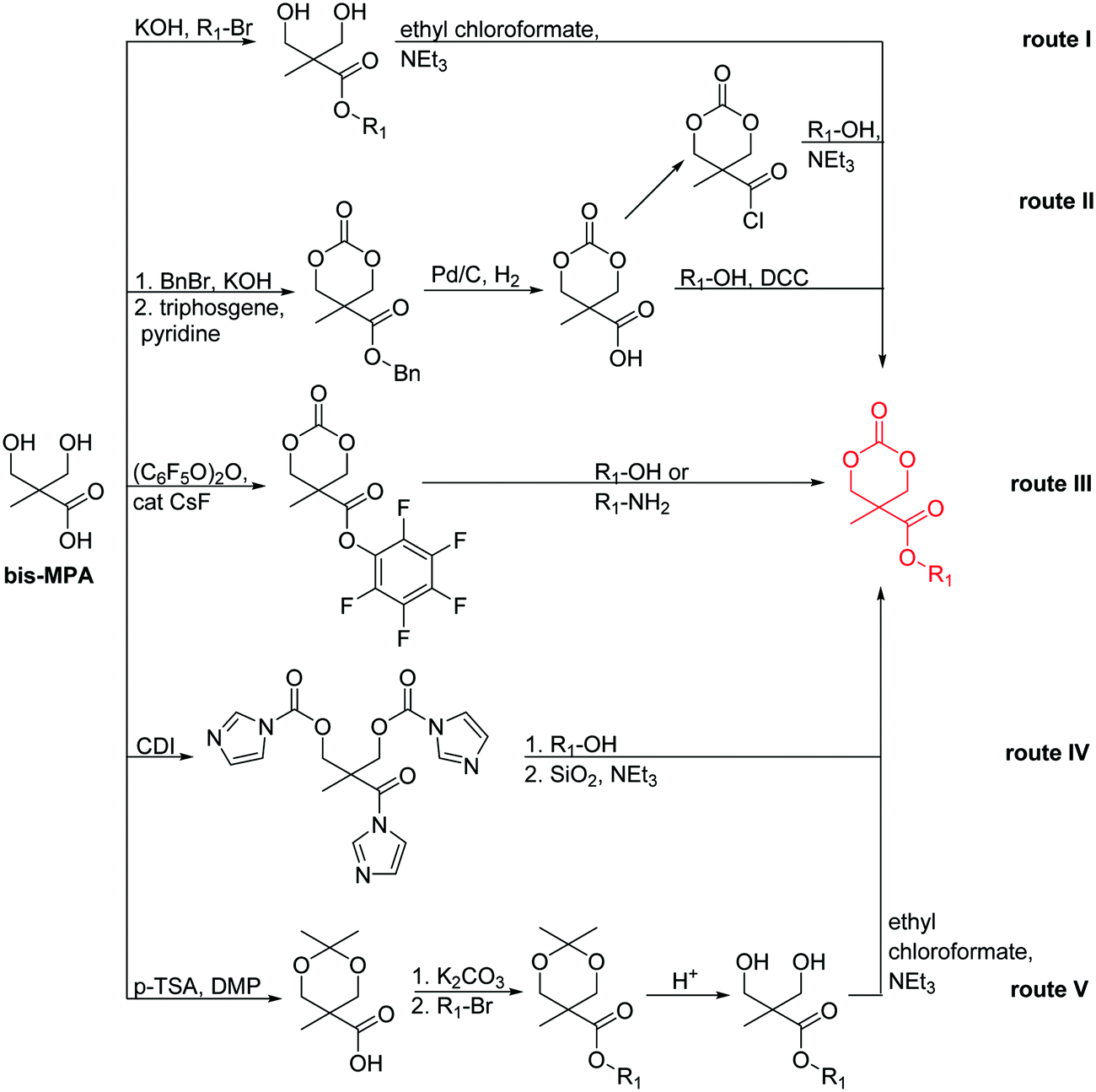 | ||
| Scheme 21 Synthetic strategies to trimethylene carbonates (TMCs) from 2,2-bishydroxy(methyl)propionic acid (bis-MPA). | ||
Another general route uses glycerol or trimethylolalkanes (such as trimethylolpropane) as feedstock (Scheme 22), which are protected as acetals with benzaldehyde (Scheme 22, route VI), acetone, or 2,2-dimethoxypropane (DMP) (Scheme 22, route VII). The pendant hydroxyl group was substituted with alkylhalogenides or acid chlorides, the acetal hydrolyzed, and the final monomer formed by reaction with triphosgene or ethyl chloroformate under basic conditions.236 Alternatively, the starting compound was either directly transformed to the cyclic carbonate and afterward esterified to introduce the pendant functionality (Scheme 22, route VIII) or first, an oxetane-ring was generated (Scheme 22, routes IX–XI). After halogenation of the remaining hydroxyl group and ring-opening of the oxetane, cyclization was achieved, e.g. with ethyl chloroformate or 1,1′-carbonyldiimidazole (CDI) (overall yield: 31–47%, Scheme 22, route IX). Alternatively, the alkyl halogenide was reacted with sodium hydrogen sulfide and the resulting thiol modified either by thiol–ene reaction or disulfide formation (Scheme 22, routes X (yield: 16%) and XI (yield: 16%)). For more details, we also refer to the review of Dove.25
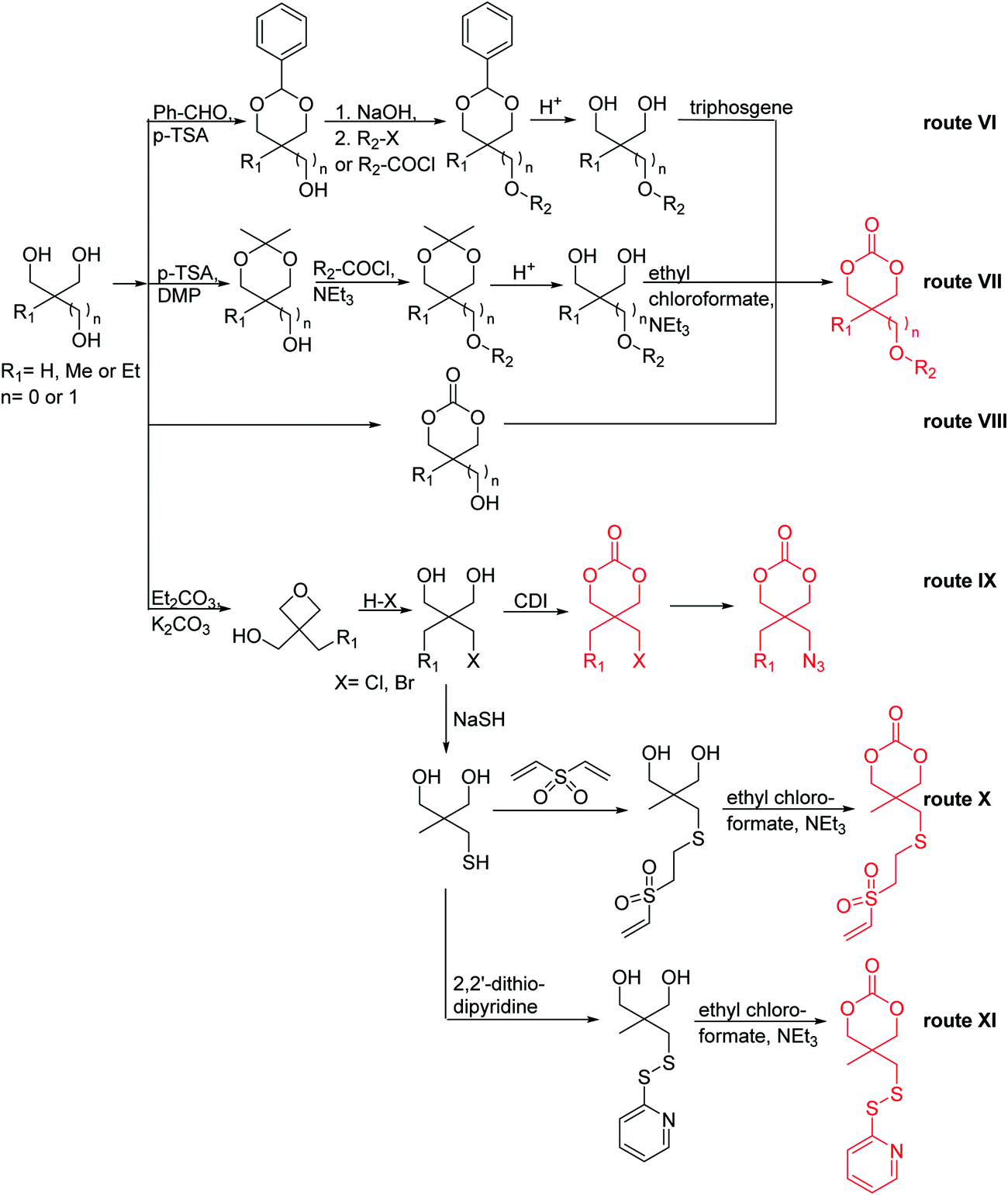 | ||
| Scheme 22 Synthetic strategies to trimethylene carbonates (TMCs) derived from trimethylolalkanes. | ||
Yang and coworkers237 reported a strategy synthesizing monosubstituted TMCs from 2-aminopropane-1,3-diols (serinol) (Scheme 23, route XII; yields: 32–69%). Substitution of the amine with chloroformates, acyl halides or N-carbonyloxy succinimide derivatives introduced the pendant chain, followed by cyclization with ethyl chloroformate.
 | ||
| Scheme 23 Synthetic strategy to trimethylene carbonates (TMCs) derived from serinol. | ||
Diaconescu and coworkers87 reported several ferrocene-containing TMCs (5-substituted (C5–C7) and 5-methyl-5′-substituted (C8–C10)), polymerized them and studied their redox potential for biological studies. Proceeding from 2-(prop-2-yn-1-yl)-propane-1,3-diol, cyclization was achieved with triphosgene. Ferrocene-azide derivatives were attached by click chemistry.
Zhong and coworkers reported (meth)acrylated MTCs C14 and C15. The acrylate was copolymerized with lactide or caprolactone and modified by Michael-addition with thiols of varying polar and charged groups,260 or folate-conjugated paclitaxel-loaded PEG–PC–PLA-triblock copolymer micelles photo-crosslinked for drug delivery.261 Amsden and coworkers262 electrospun lactide-copolymers of C14 and photo-crosslinked them yielding fibrous crimped scaffolds to culture cells. Hedrick and coworkers263 introduced additional (meth)acrylated and styrene functionalized monomers (C16–C18), synthesized from the pentafluorophenyl ester precursor (Scheme 22, route III). They showed polymerization of C16 and C18, but no further chemical modification. Jing and coworkers264 introduced a cinnamate-functionalized monomer (C19), which was photo-crosslinked after the ROP. A vinyl sulfone monomer (C20) was synthesized by Zhong and coworkers265 from 3-methyl-3-oxetanemethanol and divinyl sulfone (Scheme 22, route X). Copolymers and coatings were functionalized by selective Michael-type reaction with thiol-containing molecules to introduce polar and charged groups, GRGDC peptide, or thiolated poly(ethylene glycol) (PEG-SH).
Bowden and coworkers266 synthesized a series of 5-methyl or 5-ethyl-5′-halide-functional MTCs (halide = chloride or bromide, C21–C24) (Scheme 22, route IX), however, they did not perform post-functionalization. In another report, the bromide monomer C21 was transformed to an azide monomer (C25)267 and used as a precursor for modification by click reaction with alkyne derivatives and subsequent ROP. Hedrick and coworkers268 further reported 2-iodo-ethyl (C26), 3-chloro-propyl (C27) and 3-bromo-propyl (C28) MTC monomers, synthesized from the acid chloride MTC precursor (route II). Copolymers were functionalized with a bis-tertiary amine (TMEDA). While for the quaternization of the 3-chloropropyl substituted polymers from C27 90 °C was required, the 3-bromo-propyl (C28) and 2-iodo-ethyl (C26) substituted polymers were functionalized at room temperature in high conversion >90%. Chloride substituted polymers (C27) showed no gel formation because of the low reactivity of chlorine substituents. The iodide and bromide substituted polymers from C26 and C28 were more difficult to handle as undesired cross-linking occurred. Bromide-substituted copolymers from C28 with 50% 5-methyl-5-ethyloxycarboxyl-1,3-dioxan-2-one comonomer instead did not show cross-linking. The cationic polycarbonate was able to bind and complex DNA for generating nanoparticles and application as a gene delivery vector were studied.268 Coatings of surface grafted diblock copolymers of PEG and bromide substituted-polycarbonates from C28, quaternized with trimethylamine, exhibited antibacterial and antifouling properties and effectively killed Staphylococcus aureus (MSSA) and methicillin-resistant S. aureus (MRSA) on the coatings.269 The polymer coating prevented blood protein adhesion and no significant hemolysis was observed. Nanoparticles of cationic polycarbonates disrupted microbial walls/membranes selectively and efficiently and thus inhibited the growth of Gram-positive bacteria (MRSA and fungi).270 Galactose-functionalized cationic polycarbonates were applied for targeted gene delivery to hepatocytes.271 Yang and coworkers investigated in different pyridines and imidazoles as quaternizing agents272 for polycarbonates or the effect of hydrophobicity273 of the polymers from 3-chloropropyl (C27), -hexyl (C29) or -octyl (C30) and 3-bromo propyl (C28) or -hexyl (C31) MTCs, and their impact on antimicrobial properties.272 In other studies, cationic (co)polymers of a benzyl chloride-substituted MTC (5-methyl-5-(4-chloromethyl)benzyl carboxyl-1,3-dioxan-2-one, C32), again quaternized with different amines, were investigated with respect to their antimicrobial behavior,274 activity, and selectivity.275 Here, functionalization was faster compared to 3-bromo propyl substituted polymers from C28. Polymers were additionally functionalized by quaternization with phosphines, the chloride in polymers from C32 was substituted with NaN3 and subsequently clicked to various alkynes,276 or functionalized with boronic acid derivatives.277
Besides the use of a pentafluorophenyl ester MTC (C33) as a precursor for preparation of functionalized monomers (Scheme 22, route III),234C33 itself was polymerized to highly reactive PCs. Hedrick and coworkers proved post-modification with different amines278,279 to use them e.g. as stealth materials,280 with persistent radicals, or with boronic acid derivatives.277 They self-assembled them to nanoparticles or used them as MRI agents.281 Liu and coworkers282 reported a further active ester MTC monomer, functionalized with a NHS ester (5-methyl-5′-(succinimide-N-oxycarbonyl)-1,3-dioxan-2-one, C34), which was copolymerized with caprolactone and the copolymers modified by aminolysis with ethylene diamine to yield hydrophilic amido-amine pendant chains. They proposed facilitated attachment of bioactive molecules, targeting ligands, and covalent incorporation of prodrugs for the active ester copolymers.
A thioether-substituted monomer C35 was recently reported by Hedrick and coworkers,283 that was incorporated into homo-, di- and triblock copolymers, which were functionalized with epoxides, yielding sulfonium-functionalized PCs. Alkene functionalities, polar groups, galactose, tocopherol, and carbazols were introduced in the pendant chains. Zhong and coworkers reported a pyridyl disulfide-functionalized monomer (C36, Scheme 22, route XI), which was copolymerized with caprolactone. The polymers were functionalized in a thiol–disulfide exchange reaction with PEG-SH284 and self-assembled into reduction-sensitive micelles for active intracellular drug release or with thiolated lactobionic acid285 to reduction-sensitive shell-sheddable glyconanoparticles for efficient hepatoma-targeting delivery of doxorubicin. Two dithiolane-functionalized monomers C37 and C38 were recently synthesized by Waymouth and coworkers from 5-methyl-5′-carboxylic acid-trimethylene carbonate, oxalyl chloride, and 2-hydroxyethyl 4-carboxylate-4′-methyl-1,2-dithiolane286 (C37) or 2-hydroxyethyl-5-(1,2-dithiolan-3-yl)pentanoate (C38).287 Water-soluble triblock PEG–PC-copolymers were cross-linked with dithiols or with each other by reversible ring-opening of the pendant 1,2-dithiolanes and dynamic hydrogels obtained,286 or self-assembled into core-crosslinked (flower-bridged) micelles (Fig. 5).287 Depending on the pendant dithiolane chains, the hydrogels were dynamic, adaptable, and self-healing or rigid, resilient, and brittle. Cross-linked flower micelles dissociated upon the addition of acetone. Micelles cross-linked by a thiol and capped with maleimide persisted in acetone, and micelles cross-linked by a thiol, capped with maleimide, and then treated with dithiothreitol dissociated in acetone. Two trithiocarbonate functionalized monomers C39288 and C40289 were reported, serving as RAFT macroinitiator after ROP. N-Isopropylacrylamide (NiPAAm) (or methyl acrylate and tetrahydropyran acrylate)289 were grafted from the poly(carbonate)s.
 | ||
| Fig. 5 Self-assembly of dithiolane-functionalized TMC–PEG–TMC block copolymers into flower micelles and thiol-initiated cross-linking of the micelles. Adapted from ref. 287 with permission from the American Chemical Society, Copyright 2017. | ||
Tunca and coworkers290 reported an anthracene containing MTC monomer (C41), suitable for Diels–Alder reactions with dienophiles. Polymers were grafted with a furan-protected maleimide-terminated-poly(methyl methacrylate) or poly(ethylene glycol) or a mixture of both to yield well-defined polycarbonate graft or heterograft copolymers with an efficiency over 97%. Nelson and coworkers291 chose a similar approach, using a furfuryl-containing TMC monomer (C42). They provided copolymers with a second counterpart monomer: a Diels–Alder-protected maleimide-containing comonomer (C43). The maleimide was thermally deprotected and furan released at 130 °C. At 90 °C furfuryl functionalities in the copolymers reacted with the maleimide functions and covalently cross-linked the material. The polymer films were used for the thermally induced nanoimprinting process. Dove and coworkers292 introduced a monomer with a norbornene-functionality (C44). They presented different post-modification reactions on the homopolymers: (i) functionalization with azides via a 1,3-dipolar cycloaddition, (ii) inverse electron demand Diels–Alder reaction with tetrazines and (iii) radical thiol–ene coupling.
 | ||
| Scheme 24 The general protocol for the organocatalyzed ROP of cyclic phosphates to polyphosphoesters. | ||
Bowden and coworkers266 reported two 5-ethyl-5′-halide-functional TMCs (halogenide = chloride (C23) or bromide (C24)). C24 was transformed to an azide functionalized monomer267 (C50) allowing further click chemistry. The authors showed selected monomers to be polymerizable, e.g. a α-methyl vinyl triazole monomer (C51). However, the post-polymerization modification has not been shown so far.
Meng and coworkers305 introduced a dithiolane TMC (C54), synthesized from 2,2-bis(bromomethyl)-1,3-propanediol and NaSH and subsequent cyclization with ethyl chloroformate. PEG–PC copolymers self-assembled to micelles and crosslinked by disulfide exchange reaction, encapsulating doxorubicin (DOX). In vivo studies of the micelles in malignant B16 melanoma-bearing C57BL/6 mice with a dosage of 30 mg DOX eq. per kg effectively suppressed tumor growth, prolonged mice survival time and did not cause systemic toxicity. The drug was released within the tumor cells after reductive cleavage by glutathione.
Gross and coworkers306 reported 2,2-(2-pentene-1,5-diyl)trimethylene carbonate (C55), synthesized in one step from cyclohexene-4,4-dimethanol and ethyl chloroformate. They extensively studied the polymerization behavior and showed epoxidation of the pendant vinyl group with pCPBA (22–95% conversion, <2% hydrolysis of epoxides to diols). They suggested further hydrolysis to diols, reaction with alcohols or amines or initiation of ring-opening polymerization.307
6. Polyphosphoesters
Polyphosphoesters (PPEs) have gained interest in the last two decades for biomedical applications due to their biocompatibility and -degradability. Depending on the number of methylene spacers in the backbone, PPEs with a broad range of hydrophobic to hydrophilic properties have been reported with various degradation behavior. The binding motif around the phosphorus atom (P–O, P–H, P–C or P–N) divides PPEs into further subclasses of polyphosphates, -phosphites, -phosphonates, or -phosphoramidates. The pentavalency of the phosphorus atom allows the facile introduction of alkyl-, aryl- or functional side chains and thus further tuning of the polymer properties. PPEs can be obtained by ROP from 5- and 6-membered cyclic phosphates, H-phosphonates, phosphonates or phosphoramidates (Table 6). Orthogonally functional cyclic phosphoramidates has not been reported so far. We refer to two recent reviews for detailed information on PPEs.311,312| Monomer | R = | No. | Post-modification | Ref. |
|---|---|---|---|---|
| Phosphates | ||||
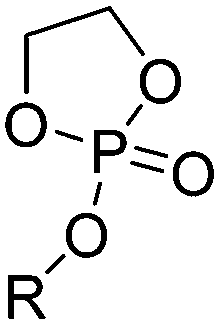
|
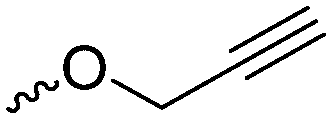
|
D1 | – Click chemistry with PEG–azide to self-assembled block copolymers | 315 |
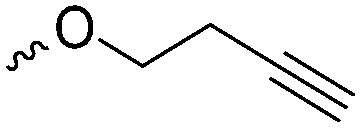
|
D2 | – Click chemistry or thiol–yne reaction to form charged micelles, silver- or paclitaxel-loaded nanoparticles, or block copolymers as a templating agent for calcium carbonate particles | 316–325 | |
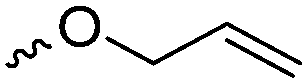
|
D3 | – Thiol–ene reaction to attach cysteamine for siRNA delivery or subsequent attachment of doxorubicin by hydrazine chemistry and dimethyl maleic anhydride for pH-responsivity | 326–328 | |

|
D4 | – Thiol–ene reaction to conjugate paclitaxel with an acid-labile linker for drug delivery | 314, 329 and 330 | |
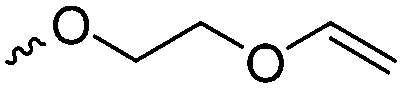
|
D5 | – Thiol–ene reaction, acetalization or thio-acetalization | 331 | |
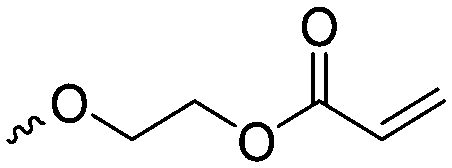
|
D6 | – Michael-type addition of charged thiols | 332 | |
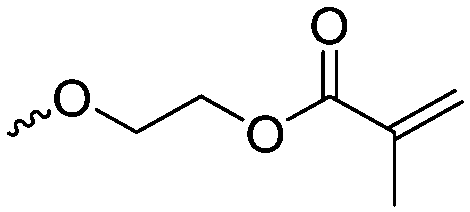
|
D7 | – Copolymers as cross-linker for copolymerization with 2-methacryloyloxyethyl phosphorylcholine (MPC) to hydrogels | 333–336 | |
| – Photo-cross-linking to hydrogels | ||||
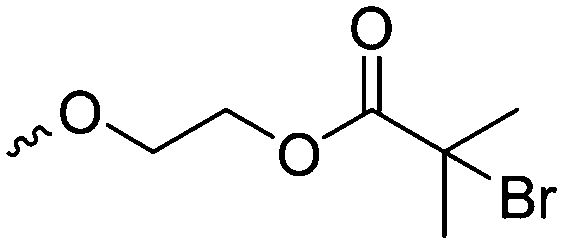
|
D8 | –ATRP-macroinitiator to graft poly(2-methacryloyloxyethyl phosphorylcholine) | 337 and 338 | |
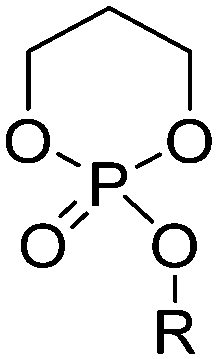
|
|
D9 | – No modification | 339 |
|
D10 | – No modification | 339 | |

|
D11 | – No modification | 339 | |
| H-Phosphonates | ||||
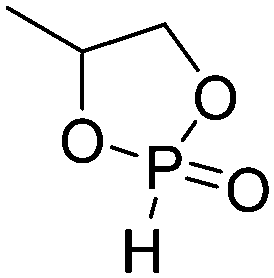
|
D12 | – Oxidation with N2O4 to poly(phosphoric acid)s | 340–347 | |
| – Reaction with amines to poly(phosphoramidate)s as a gene carrier | ||||
| – Chlorination and reaction with alcohols to polyphosphates as a gene carrier | ||||
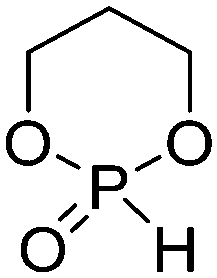
|
D13 | – Oxidation with N2O4 to poly(phosphoric acid)s | 348–353 | |
| – Chlorination and reaction with amines to polyphosphoramidates | ||||
| – Sulfurization to polyphosphorothioates | ||||
| Phosphonates | ||||
|
|
D14 | – Thiol–ene reaction with cysteine or 3-mercaptopropionic acid to form thermoresponsive terpolymers | 10 and 354 |
| Phostones | D15 | – Potential for functional R groups (to date only ethyl and butyl have been reported) | 357 | |
|
||||
| Phosphazenes | ||||
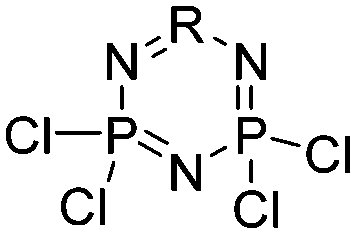
|
|
D16 | – Nucleophilic substitution with alcohols and amines | 16 and 358 |
|
D17 | – Nucleophilic substitution with alcohols and amines | 361 and 363 | |
|
D18 | – Nucleophilic substitution with alcohols and amines | 323 and 362 | |
|
D19 | – Nucleophilic substitution with alcohols and amines | 365 and 366 | |
|
D20 | – Nucleophilic substitution with alcohols and amines | 365 and 366 | |
6.1 Cyclic phosphates
PPEs can be prepared from 5-membered cyclic phosphates. Most monomers are prepared via 2-chloro-2-oxo-1,3,2-dioxaphospholane (COP) as the precursor. COP is either commercially available (but relatively expensive) or can be synthesized in two steps by the reaction of phosphorus trichloride with ethylene glycol, followed by oxidation (Scheme 26A). The oxidation procedure, however, was proven to be time-consuming and care has to be taken that the P–Cl-intermediate does not hydrolyze, e.g. if the oxygen gas is not dry. We recently optimized the reaction, using CoCl2 as catalyst and oxygen from the dried air instead of elemental oxygen, making the synthesis process more convenient and shorten the reaction time from days to hours.313 The final monomers are obtained by esterification of COP with an appropriate alcohol (yield for esterification step 30–70%). Their purification is sometimes challenging due to the high ring-strain: column chromatography over deactivated silica or distillation at reduced pressure are typical purification methods, but sometimes reduce the yields. If the starting COP is highly pure and the alcohol used in the final step can be removed in vacuo, the monomers are obtained pure enough without additional purification steps. Some orthogonally reactive monomers have been reported in literature up to date. Because of a growing interest in PPEs for biomedical applications, we expect the report of several further monomers in the common years.Modern polymerization techniques for PPEs rely on the use of organocatalysts such as TBD, DBU, or an additional use of a thiourea cocatalyst (TU), but also classical basic, acidic or metal-based catalysts (Sn(Oct)2) may be applied.314 Primary alcohols are typically used as the initiator. Depending on the reactivity of catalyst, polymerizations can be conducted in dichloromethane at 0 °C for a period of 1 min to several hours. The reaction is terminated after ca. 80% conversion of monomers to avoid transesterification side reactions during a later stage of polymerization (Scheme 24).
A propargyl-functionalized phosphate monomer (2-(prop-2-yn-1-yloxy)-2-oxo-1,3,2-dioxaphospholane, D1) was reported by Wang and coworkers,315 block copolymers with caprolactone were synthesized and PEG-N3 clicked on the alkyne. The polymers self-assembled to nanoparticles with high biocompatibility. Contrary, Wooley and coworkers316 reported the unsuccessful synthesis of D1 with isolated yield <20% and suspected the reaction via a SN2′ mechanism to be responsible for decomposition of the monomer (nucleophilic attack at the acetylene proton and subsequent loss of the pendant chain at the phosphate group). They pointed out the absence or loss of the terminal acetylene proton in the 1H-NMR spectra provided by Wang and coworkers and suggested loss or partial loss of the alkynyl functionality. Instead, Wooley et al. installed an additional methylene spacer and reported the butynyl monomer 2-(but-3-yn-1-yloxy)-2-oxo-1,3,2-dioxaphospholane (D2) with high yield and purity.316 The monomer was polymerized (homopolymers or copolymers with caprolactone or cyclic phosphates) and post-modification by click chemistry or thiol–yne reaction was applied to prepare non-ionic, cationic, anionic, and zwitterionic micelles,317,318 covalently labeled core–shell polymeric nanoparticles with fluorescent contrast agents for theranostic applications,319 PEG-b-PPE-based paclitaxel conjugates for ultra-high paclitaxel-loaded multifunctional nanoparticles,320,321 or silver-bearing degradable polymeric nanoparticles showing in vitro antimicrobial activity (Fig. 6).322,323 For this purpose, silver cations were chelated into the corona or incorporated into the core using AgOAc, or SCC22 or SCC10. The placements of the silver species within the nanoparticle frameworks are proposed locations that have not been confirmed experimentally. Frank and coworkers used D2 to form anionic hydrophilic PPE-copolymers as efficient templating agent for calcium carbonate particles.324,325
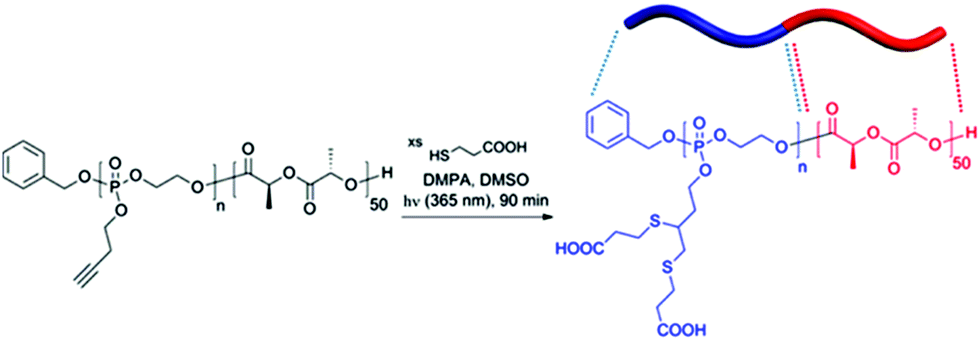 | ||
| Fig. 6 Synthetic route to silver-bearing degradable polymeric nanoparticles: post-polymerization modification of alkyne-functionalized PPE-b-PLLA via thiol–yne “click” reaction to prepare anionic amphiphilic diblock copolymer, reproduced from ref. 322 with permission from the American Chemical Society, Copyright 2017. | ||
Wang and coworkers326 reported a cyclic phosphate carrying an allyl pendant group (2-(prop-2-en-1-yloxy)-2-oxo-1,3,2-dioxaphospholane, D3), which was polymerized with a PEG-macroinitiator. The block copolymers self-assembled into nanoparticles and the allyl groups were post-modified by thiol–ene reaction with cysteamine and partially further reacted with dimethyl maleic anhydride to introduce pH-responsivity. Additionally, doxorubicine was conjugated to the polymers by hydrazine chemistry or the polymers were used for siRNA delivery.327 In contrast, Lecomte and coworkers328 used the pendant allyl chain of D3 as a protective group to produce polyphosphodiesters after selective deprotection with 1.5 eq. sodium benzene-thiolate. Quantitative deprotection without degradation was reported within 2 h in DMF/H2O (50:50, v/v) at room temperature. A butenyl monomer (2-butenoxy-2-oxo-1,3,2-dioxaphospholane, D4) was reported by Lecomte and coworkers314 and block copolymers with 2-isobutoxy-2-oxo-1,3,2-dioxaphospholane were prepared to demonstrate the control over copolymerizations of cyclic phosphates. Further post-modification has not been shown. Wooley and coworkers329 synthesized PEG-b-PPE of D4 and conjugated Paclitaxel with acid-labile linkages by thiol–ene reaction for drug delivery. Junkers and coworkers330 developed a flow microreactor to attach thiols upon UV-irradiation to butenyl-containing PPEs of D4. Wooley and coworkers331 also introduced a monomer with a reactive vinyl ether moiety (D5), which was either reacted in a thiol–ene reaction, thio-acetalization with thiols or acetalization with alcohols. While they reported quantitative conversion for thiol–ene reaction, acetalization, and thio- acetalization resulted in rather low conversions of 18% and 8%, respectively. Ni and coworkers332 reported a functional phosphate monomer carrying an acrylate group (D6, care has to be taken as the monomer undergoes quick radical polymerization during workup) and prepared block copolymers with caprolactone. The pendant acrylates were quantitatively functionalized by nucleophilic addition chemistry with thiols to introduce hydrophilic chains with hydroxyl, carboxyl, amine, and amino acid functionalities with low cytotoxicity. Stable micelles in aqueous solution, loaded with doxorubicin, opened the way to drug delivery carriers. While after 2 days at 37 °C, 30% release of cargo was observed in PBS-buffer (pH 7.4, 0.01 M), over 70% was reported in the presence of 0.2 mg mL−1 phosphodiesterase I. Iwasaki et al.333 used a methacryloyl phosphate monomer (D7) to obtain copolymers with 2-isopropyl-2-oxo-1,3,2-dioxaphospholane. They were used as cross-linker and copolymerized with 2-methacryloyloxyethyl phosphorylcholine (MPC) to prepare hydrogels. 100% degradation of the PPE-crosslinker polymer was observed within 6 days at pH 11, and 50% within 15 days at pH 7.4. 50% of the hydrogels degraded after 44 days at pH 7.4 (PBS-buffer).334 As highly porous hydrogels, they are suitable for 3D cell cultivation and increased proliferation of mouse osteoblastic cell (MC3T3-E1) was reported with increasing amount of PPEs in the hydrogels.335 The methacrylates as well were photo-cross-linked to obtain hydrogels.336
Iwasaki et al.337,338 copolymerized a cyclic bromoisobutylate phosphate (2-(2-oxo-1,3,2-dioxaphosphoroyloxyethyl-2-bromoisobutylate), D8) with 2-isopropyl-2-oxo-1,3,2-dioxaphospholane. The polymer was used as a macroinitiator for ATRP polymerization, to form amphiphilic PPEs with poly(2-methacryloyloxyethyl phosphorylcholine)-grafted chains.
6-Membered phosphate monomers are synthesized by a similar strategy, using 2-chloro-2-oxo-1,3,2-dioxaphosphorinane as the precursor, which is further substituted with an appropriate alcohol (Scheme 25B). Penczek and coworkers339 reported in 1977 three potentially orthogonally reactive monomers with 2,2,2-trichloroethyl (D9), 2,2,2-trifluoroethyl (D10) or 2-cyanoethyl (D11) functionalities as pendant chains (yields were not reported), but did not further modified the corresponding polymers, which were obtained by cationic polymerization in bulk. 6-Membered cyclic phosphates as monomers are less considered in the literature compared to 5-membered phosphates, due to a lower ring strain.
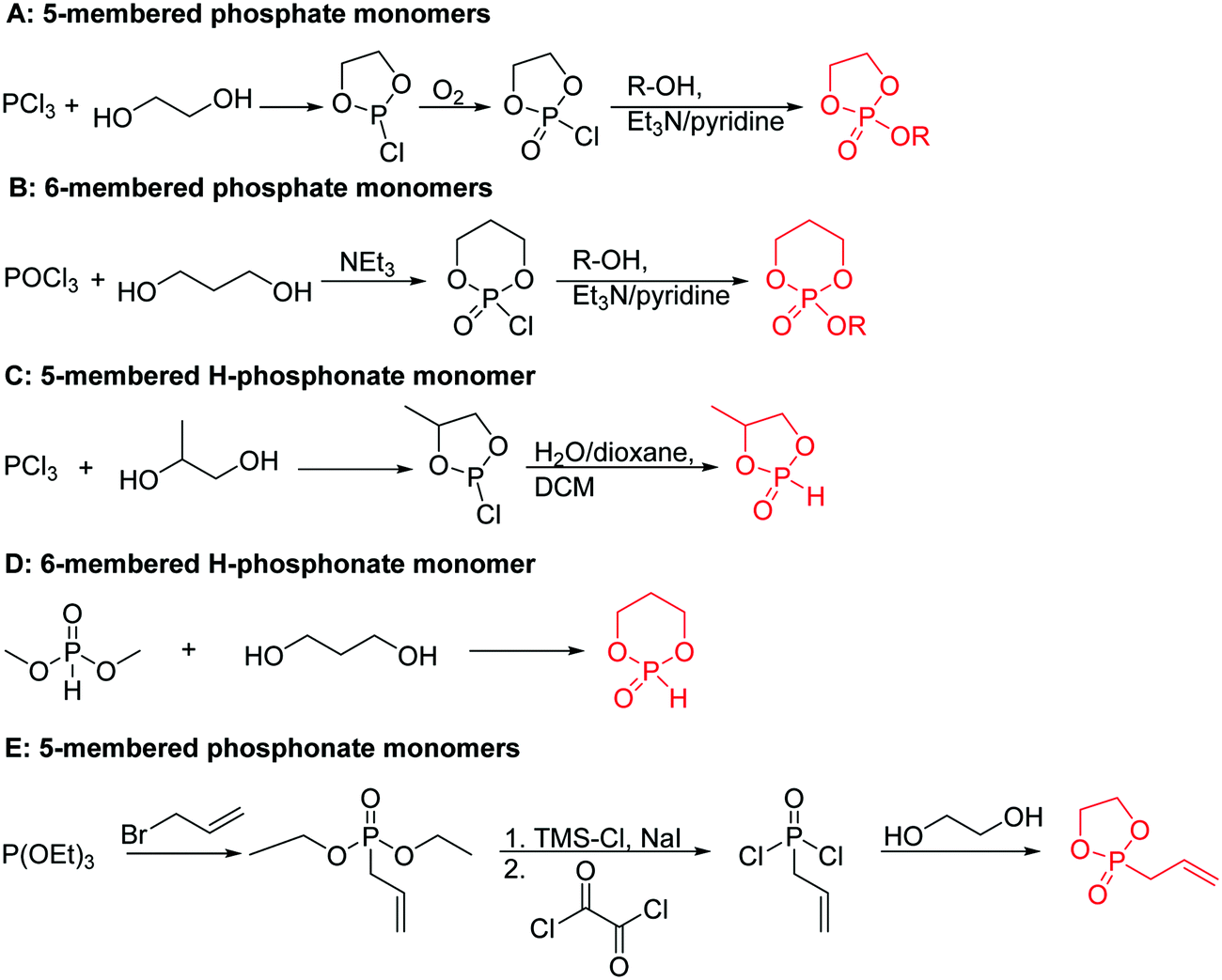 | ||
| Scheme 25 Synthetic strategies for cyclic phosphate, phosphonate, and H-phosphonate monomers. | ||
6.2 Cyclic H-phosphonates
A cyclic 5-membered H-phosphonate monomer, 4-methyl-2-oxo-2-hydro-1,3,2-dioxaphospholane (D12), was polymerized by Vogl and coworkers,340 starting from 1,2-propandiol and PCl3. 4-Methyl-2-chloro-1,3,2-dioxaphospholane was oxidized by a mixture of water/dioxane to obtain the phosphite (yield has not been reported) (Scheme 25C). The monomer was also synthesized from optically active propandiol. The polymer p(D12) was converted by various strategies to (i) poly(phosphoric acid)s by oxidation with dinitrogen tetroxide, (ii) poly(phosphoramidate)s341,342 by an Atherton–Todd reaction with primary or secondary amine (30–60% conversion), and (iii) polyphosphates343 by chlorination and subsequent substitution with an appropriate alcohol (quantitative conversion), e.g. to poly(2-aminoethyl propylene phosphate) (PPE-EA). Leong and Mao used the polyphosphates343,344 and polyphosphoarmidates342,345 (with charged groups346) in several reports as gene carrier and transfection agent, or to form plasmid-templated DNA-block copolymer nanoparticles.347A 6-membered H-phosphonate monomer, 2-hydro-2-oxo-1,3,2-dioxaphosphorinane (D13),348 was polymerized by Penczek and coworkers349 (Scheme 25D). It was prepared by transesterification from dimethyl phosphite and 1,3-propylene glycol (yield: 79%348). Penczek showed conversion to poly(phosphoric acid)s by oxidation with dinitrogen tetroxide, to polyphosphoramidates by chlorination with gaseous chlorine and subsequent reaction with primary amines (C- or N-substituted imidazoles, adenine or uracil),350–352 or to polyphosphorothioates353 by sufurization with sulfur in the presence of lutidine.
6.3 Cyclic phosphonates
A 5-membered cyclic allyl phosphonate monomer (2-allyl-2-oxo-1,3,2-dioxaphospholane, D14) was recently reported by Wurm and coworkers.10,354 For cyclic phosphonates the pendant group was introduced in the first step of a three-step reaction (Scheme 25E): after reaction of allyl bromide with triethyl phosphite, allyl phosphonic acid dichloride was formed by reaction with chlorotrimethylsilane and oxalyl chloride. Ring-closing reaction with ethylene glycol formed the final phosphonate monomer with an overall yield of 28% after distillation (as for cyclic phosphates, purification of the highly strained monomers can reduce the yield). Terpolymers were quantitatively modified with cysteine by a thiol–ene reaction and their thermoresponsive behavior analyzed.354 PEG-b-PPEs were modified with 3-mercaptopropionic acid. The block copolymers exhibited UCST behavior and were self-assembled into temperature-dependent dynamic aggregates.10 Polyphosphonates are prepared by using the organocatalyst DBU and primary alcohols as an initiator in dichloromethane at 0 °C for 16 hours with monomer conversions of 90–96%.354–356 Very recently also phostones have been polymerized to polyphosphonates with the P–C bond in the main-chain, which might be extended to functional derivatives.357 Their great benefit compared to the other cyclic phosphates or phosphonates is the relatively easy one-step synthesis, which might be expanded to allyl-functionalized phostones, for example (D15). It was proven that their degradation proceeds much slower than the corresponding side-chain polyphosphonates.7. Polyorganophosphazenes
Polyorganophosphazenes are a polymer class of inorganic–organic hybrid polymers and exhibit a high diversity of properties due to the high versatility of possible organic substituents (Table 6). The phosphorus-nitrogen backbone is substituted by two organic side groups on the phosphorus atoms. Polymerization techniques and application of polyorganophosphazenes were recently summarized in excellent reviews.16,358,359The most widely used method to prepare polyorganophosphazenes is the thermal ROP of hexachlorophosphazene (D16) with no control over the polymer molar mass and thus generally high molecular weights (>106 Da) and broad molecular weight distributions to the precursor polydichloro-phosphazene [NPCl2]. Living cationic polymerization of trichlorophosphoranimine (Cl3PNSiMe3) with two equivalents of phosphorus pentachloride by a chain growth mechanism in solution at room temperature results in higher control of polymer molar mass via the feed monomer to initiator ratio with lower dispersities. We refer the reader to a separate review.16
D15 is commercially available (at Sigma Aldrich) or can be synthesized from phosphorous pentachloride and ammonium chloride and subsequent sublimation from a mixture of oligomers (Scheme 26). The precursor rapidly hydrolyzes in the presence of water but can be completely post-modified under dry conditions with amines, alcohols, thiols or alkylation agents such as RMgX or RLi. Mixed, stepwise substitution with two or more organic substituents on the same macromolecule was possible and gives access to a library of polymers with varied properties.16 The complete modification is important to prevent uncontrolled cross-linking or degradation due to unreacted P–Cl groups. ROP was achieved with partially or completely substituted cyclophosphazene, but side reactions as ring expansion, decomposition or no reaction at all were observed. Generally, the probability for polymerization decreased with increasing number of organic substituents.360
 | ||
| Scheme 26 Synthetic route to hexachlorophosphazene and polyorganophosphazenes. | ||
Several further cyclic phosphazene monomers with a substitution of a phosphorus atom by carbon or sulfur were reported yielding cyclocarbophosphazene N3P2CCl5 (D17) from phosphorous pentachloride, ammonium chloride and cyanamide (10% yield)361 or cyclothiophosphazene N3P2SCl5 (D18) (yield 40–50%).362 Polychlorocarbophosphazenes363 and polychlorothiophosphazenes323 were obtained by thermal polymerization (120 °C, 4–6 h and 90 °C, 4 h, respectively), and were more reactive to nucleophilic substitution by aryloxides than classical polyphosphazene.361,362,364 Polycarbophosphazenes exhibited higher glass transition temperatures than polyphosphazene analog due to less torsional mobility in the backbone,361,364 polythiophosphazenes362 less hydrolytic stability. Polymerization (165–180 °C, 4 h) of cyclothionylphosphazenes N3P2SOCl5 (D19) and N3P2SOFCl4 (D20) yielded hydrolytically stable polythionylphosphazenes.365,366
8. Summary & outlook
The ring-opening polymerization gives access to well-defined polymers with narrow molecular weight distributions. However, as the reaction conditions for ROP typically do not tolerate nucleophiles and need inert conditions (in most cases), many functional groups in the monomers need to be protected. The deprotection step after the polymerization can be challenging, when also the polymer backbone can be degraded. In contrast, orthogonally reactive groups that do not interfere with the polymerization conditions are efficient alternatives to protective group chemistry. They can be post-modified by efficient reactions, which do not degrade the polymer backbone. Typical functional groups are alkenes, alkynes, and halogens, but several further functions have been summarized in this review.Most functional cyclic monomers are synthesized in several reaction steps: the orthogonal functional groups are mostly introduced in the first step, making the synthesis of most functional monomers lengthy with variable yields. The use of “precursor monomers” would be beneficial, introducing the desired functionality in the last reaction step, but such strategies are missing. It is however possible for the preparation of functional phosphate monomers, that rely on the commercially available precursor 2-chloro-2-oxo-1,3,2-dioxaphospholane. The design of similar precursor strategies for other material classes would give faster access to multifunctional polymers with tailored properties (such as solubility or degradation profile).
The presented general concepts of post-modifications can be used for all summarized polymer classes, which are:
– nucleophilic substitution of halides,
– alkyne–azide click chemistry,
– thiol–ene and –yne reactions
– Diels–Alder reactions
They often yield quantitative conversion under mild conditions (e.g. excluding acidic or alkaline conditions or heavy-metal catalysts).
A broad variety of orthogonally reactive functionalities for cyclic monomers for the ROP and post-modification opportunities has been reported so far, which give access to diverse chemically functional biodegradable polymers and promising applications. There is still plenty of scope for further developments.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. B. is recipient of a fellowship through funding of the Excellence Initiative (DFG/GSC 266) in the context of the graduate school of excellence “MAINZ” (Material Sciences in Mainz). Funding of the Deutsche Forschungsgemeinschaft (DFG) to F. R. W. (WU750/6-1) is gratefully acknowledged. Open Access funding provided by the Max Planck Society.References
- M. Okada, Prog. Polym. Sci., 2002, 27, 87–133 CrossRef
.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354 CrossRef PubMed
- B. D. Ulery, L. S. Nair and C. T. Laurencin, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 832–864 CrossRef PubMed
- L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef
- B. Guo and P. X. Ma, Sci. China: Chem., 2014, 57, 490–500 CrossRef
- A. Tardy, J. Nicolas, D. Gigmes, C. Lefay and Y. Guillaneuf, Chem. Rev., 2017, 117, 1319–1406 CrossRef PubMed
- T. Haider, C. Völker, J. Kramm, K. Landfester and F. R. Wurm, Angew. Chem., Int. Ed., 2018 DOI:10.1002/anie.201805766
-
M. Rahman, in Polyester, ed. H. E.-D. M. Saleh, InTech, Rijeka, 2012, ch. 05, DOI:10.5772/47765
- J. Baran and S. Penczek, Macromolecules, 1995, 28, 5167–5176 CrossRef
- T. Wolf, T. Rheinberger, J. Simon and F. R. Wurm, J. Am. Chem. Soc., 2017, 139, 11064–11072 CrossRef PubMed
- H. Wang, L. Su, R. Li, S. Zhang, J. Fan, F. Zhang, T. P. Nguyen and K. L. Wooley, ACS Macro Lett., 2017, 6, 219–223 CrossRef
- S. Zhang, H. Wang, Y. Shen, F. Zhang, K. Seetho, J. Zou, J.-S. A. Taylor, A. P. Dove and K. L. Wooley, Macromolecules, 2013, 46, 5141–5149 CrossRef PubMed
- A. Cankaya, M. Steinmann, Y. Bulbul, I. Lieberwirth and F. R. Wurm, Polym. Chem., 2016, 7, 5004–5010 RSC
- J. Baran, K. Kaluzynski, R. Szymanski and S. Penczek, Biomacromolecules, 2004, 5, 1841–1848 CrossRef PubMed
- M. Steinmann, M. Wagner and F. R. Wurm, Chem. – Eur. J., 2016, 22, 17329–17338 CrossRef PubMed
- S. Rothemund and I. Teasdale, Chem. Soc. Rev., 2016, 45, 5200–5215 RSC
- I. Teasdale and O. Brüggemann, Polymers, 2013, 5, 161 CrossRef PubMed
- J. Rydz, W. Sikorska, M. Kyulavska and D. Christova, Int. J. Mol. Sci., 2015, 16, 564 CrossRef PubMed
- M. A. Elsawy, K.-H. Kim, J.-W. Park and A. Deep, Renewable Sustainable Energy Rev., 2017, 79, 1346–1352 CrossRef
- K. Leja and G. Lewandowicz, Pol. J. Environ. Stud., 2010, 19, 255–266 Search PubMed
- M. A. Woodruff and D. W. Hutmacher, Prog. Polym. Sci., 2010, 35, 1217–1256 CrossRef
- A. Banerjee, K. Chatterjee and G. Madras, Mater. Sci. Technol., 2014, 30, 567–573 CrossRef
- V. M. Pathak and Navneet, Bioresour. Bioprocess., 2017, 4, 15 CrossRef
- H. Tian, Z. Tang, X. Zhuang, X. Chen and X. Jing, Prog. Polym. Sci., 2012, 37, 237–280 CrossRef
- S. Tempelaar, L. Mespouille, O. Coulembier, P. Dubois and A. P. Dove, Chem. Soc. Rev., 2013, 42, 1312–1336 RSC
- C. Jérôme and P. Lecomte, Adv. Drug Delivery Rev., 2008, 60, 1056–1076 CrossRef PubMed
- X. Lou, C. Detrembleur and R. Jérôme, Macromol. Rapid Commun., 2003, 24, 161–172 CrossRef
- C. K. Williams, Chem. Soc. Rev., 2007, 36, 1573–1580 RSC
- S. M. Guillaume, Eur. Polym. J., 2013, 49, 768–779 CrossRef
-
S. M. Guillaume, Handbook of Telechelic Polyesters, Polyethers and Polycarbonates, Pan Stanford Publishing, 2017 Search PubMed
- J. Dommerholt, F. P. J. T. Rutjes and F. L. van Delft, Top. Curr. Chem., 2016, 374, 16 CrossRef PubMed
- C. K. Pandiyarajan, O. Prucker and J. Rühe, Macromolecules, 2016, 49, 8254–8264 CrossRef
- O. Prucker, C. A. Naumann, J. Rühe, W. Knoll and C. W. Frank, J. Am. Chem. Soc., 1999, 121, 8766–8770 CrossRef
- J. M. W. Chan, H. Sardon, A. C. Engler, J. M. García and J. L. Hedrick, ACS Macro Lett., 2013, 2, 860–864 CrossRef
- P. Theato, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6677–6687 CrossRef
- M. A. Gauthier, M. I. Gibson and H.-A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef PubMed
- A. Das and P. Theato, Chem. Rev., 2016, 116, 1434–1495 CrossRef PubMed
-
F. Moldenhauer and P. Theato, in Multi-Component and Sequential Reactions in Polymer Synthesis, ed. P. Theato, 2015, vol. 269, pp. 133–162 Search PubMed
- R. Tong, Ind. Eng. Chem. Res., 2017, 56, 4207–4219 CrossRef
- H. Seyednejad, A. H. Ghassemi, C. F. van Nostrum, T. Vermonden and W. E. Hennink, J. Controlled Release, 2011, 152, 168–176 CrossRef PubMed
- S. Agarwal, Polym. Chem., 2010, 1, 953–964 RSC
- P. Espeel and F. E. Du Prez, Eur. Polym. J., 2015, 62, 247–272 CrossRef
- M. Winnacker and B. Rieger, Polym. Chem., 2016, 7, 7039–7046 RSC
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC
- J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef PubMed
- R. Riva, S. Schmeits, C. Jérôme, R. Jérôme and P. Lecomte, Macromolecules, 2007, 40, 796–803 CrossRef
-
A. Duda, in Polymer Science: A Comprehensive Reference, ed. M. Möller, Elsevier, Amsterdam, 2012, pp. 213–246, DOI:10.1016/B978-0-444-53349-4.00104-7
- T. F. Al-Azemi and A. A. Mohamod, Polymer, 2011, 52, 5431–5438 CrossRef
- N. Xu, R. Wang, F.-S. Du and Z.-C. Li, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3583–3594 CrossRef
- S. Lenoir, R. Riva, X. Lou, C. Detrembleur, R. Jérôme and P. Lecomte, Macromolecules, 2004, 37, 4055–4061 CrossRef
- B. Massoumi, Polym. Sci., Ser. B, 2015, 57, 85–92 CrossRef
- R. Riva, S. Schmeits, F. Stoffelbach, C. Jerome, R. Jerome and P. Lecomte, Chem. Commun., 2005, 5334–5336, 10.1039/B510282K
- S. E. Habnouni, V. Darcos and J. Coudane, Macromol. Rapid Commun., 2009, 30, 165–169 CrossRef PubMed
- V. Darcos, S. Antoniacomi, C. Paniagua and J. Coudane, Polym. Chem., 2012, 3, 362–368 RSC
- O. Jazkewitsch, A. Mondrzyk, R. Staffel and H. Ritter, Macromolecules, 2011, 44, 1365–1371 CrossRef
- S. M. Garg, X.-B. Xiong, C. Lu and A. Lavasanifar, Macromolecules, 2011, 44, 2058–2066 CrossRef
- E. M. Pelegri-O’Day, S. J. Paluck and H. D. Maynard, J. Am. Chem. Soc., 2017, 139, 1145–1154 CrossRef PubMed
- S. El Habnouni, B. Nottelet, V. Darcos, B. Porsio, L. Lemaire, F. Franconi, X. Garric and J. Coudane, Biomacromolecules, 2013, 14, 3626–3634 CrossRef PubMed
- O. Jazkewitsch and H. Ritter, Macromolecules, 2011, 44, 375–382 CrossRef
- H. Li, R. Jéröme and P. Lecomte, Polymer, 2006, 47, 8406–8413 CrossRef
- S. Sinnwell and H. Ritter, Macromolecules, 2006, 39, 2804–2807 CrossRef
- K. Takeda, I. Imaoka and E. Yoshii, Tetrahedron, 1994, 50, 10839–10848 CrossRef
- J. R. Lowe, W. B. Tolman and M. A. Hillmyer, Biomacromolecules, 2009, 10, 2003–2008 CrossRef PubMed
- J. R. Lowe, M. T. Martello, W. B. Tolman and M. A. Hillmyer, Polym. Chem., 2011, 2, 702–708 RSC
- Y. Gao, T. Mori, S. Manning, Y. Zhao, A. d. Nielsen, A. Neshat, A. Sharma, C. J. Mahnen, H. R. Everson, S. Crotty, R. J. Clements, C. Malcuit and E. Hegmann, ACS Macro Lett., 2016, 5, 4–9 CrossRef
- C. Detrembleur, M. Mazza, O. Halleux, P. Lecomte, D. Mecerreyes, J. L. Hedrick and R. Jérôme, Macromolecules, 2000, 33, 14–18 CrossRef
- C. Detrembleur, M. Mazza, X. Lou, O. Halleux, P. Lecomte, D. Mecerreyes, J. L. Hedrick and R. Jérôme, Macromolecules, 2000, 33, 7751–7760 CrossRef
- R. Riva, W. Lazzari, L. Billiet, F. Du Prez, C. Jérôme and P. Lecomte, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1552–1563 CrossRef
- J.-P. Latere, P. Lecomte, P. Dubois and R. Jérôme, Macromolecules, 2002, 35, 7857–7859 CrossRef
- E. L. Prime, Z. A. A. Hamid, J. J. Cooper-White and G. G. Qiao, Biomacromolecules, 2007, 8, 2416–2421 CrossRef PubMed
- W. Dai, J. Zhu, A. Shangguan and M. Lang, Eur. Polym. J., 2009, 45, 1659–1667 CrossRef
- D. Mecerreyes, J. Humes, R. D. Miller, J. L. Hedrick, C. Detrembleur, P. Lecomte, R. Jérôme and J. San Roman, Macromol. Rapid Commun., 2000, 21, 779–784 CrossRef
- C. Vaida, P. Mela, H. Keul and M. Möller, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6789–6800 CrossRef
- X. Lou, C. Jérôme, C. Detrembleur and R. Jérôme, Langmuir, 2002, 18, 2785–2788 CrossRef
- J. Rieger, K. Van Butsele, P. Lecomte, C. Detrembleur, R. Jerome and C. Jerome, Chem. Commun., 2005, 274–276, 10.1039/B411565A
- C. Vaida, P. Mela, K. Kunna, K. Sternberg, H. Keul and M. Möller, Macromol. Biosci., 2010, 10, 925–933 CrossRef PubMed
- D. Mecerreyes, B. Atthoff, K. A. Boduch, M. Trollsås and J. L. Hedrick, Macromolecules, 1999, 32, 5175–5182 CrossRef
- A. Garle, S. Kong, U. Ojha and B. M. Budhlall, ACS Appl. Mater. Interfaces, 2012, 4, 645–657 CrossRef PubMed
- D. Chen, C.-C. Chang, B. Cooper, A. Silvers, T. Emrick and R. C. Hayward, Biomacromolecules, 2015, 16, 3329–3335 CrossRef PubMed
- B. Parrish, J. K. Quansah and T. Emrick, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1983–1990 CrossRef
- B. Parrish and T. Emrick, Macromolecules, 2004, 37, 5863–5865 CrossRef
- B. Parrish, R. B. Breitenkamp and T. Emrick, J. Am. Chem. Soc., 2005, 127, 7404–7410 CrossRef PubMed
- J. L. Herrmann and R. H. Schlessinger, J. Chem. Soc., Chem. Commun., 1973, 711–712, 10.1039/C39730000711
- B. M. Cooper, D. Chan-Seng, D. Samanta, X. Zhang, S. Parelkar and T. Emrick, Chem. Commun., 2009, 815–817, 10.1039/B817600K
- A. E. v. d. Ende, E. J. Kravitz and E. Harth, J. Am. Chem. Soc., 2008, 130, 8706–8713 CrossRef PubMed
- O. Stöhr and H. Ritter, Macromol. Chem. Phys., 2014, 215, 426–430 CrossRef
- B. M. Upton, R. M. Gipson, S. Duhovic, B. R. Lydon, N. M. Matsumoto, H. D. Maynard and P. L. Diaconescu, Inorg. Chem. Front., 2014, 1, 271–277 RSC
- K. N. Houk, A. Jabbari, H. K. Hall and C. Alemán, J. Org. Chem., 2008, 73, 2674–2678 CrossRef PubMed
- G. A. R. Nobes, R. J. Kazlauskas and R. H. Marchessault, Macromolecules, 1996, 29, 4829–4833 CrossRef
- K. Tada, Y. Numata, T. Saegusa and J. Furukawa, Makromol. Chem., 1964, 77, 220–228 CrossRef
- M. Hong and E. Y. X. Chen, Nat. Chem., 2016, 8, 42–49 CrossRef PubMed
- J. Zhou, A. M. Schmidt and H. Ritter, Macromolecules, 2010, 43, 939–942 CrossRef
- X. Tang, M. Hong, L. Falivene, L. Caporaso, L. Cavallo and E. Y. X. Chen, J. Am. Chem. Soc., 2016, 138, 14326–14337 CrossRef PubMed
- A. W. Murray and R. G. Reid, Synthesis, 1985, 35–38 CrossRef
- J. Undin, P. Olsén, J. Godfrey, K. Odelius and A.-C. Albertsson, Polymer, 2016, 87, 17–25 CrossRef
- M. Iida, T. Araki, K. Teranishi and H. Tani, Macromolecules, 1977, 10, 275–284 CrossRef
- C. Lavallée, A. Leborgne, N. Spassky and R. E. Prud'homme, J. Polym. Sci., Part A: Polym. Chem., 1987, 25, 1315–1328 CrossRef
- C. Lavallee, G. Lemay, A. Leborgne, N. Spassky and R. E. Prud'homme, Macromolecules, 1984, 17, 2457–2462 CrossRef
- R. Voyer and R. E. Prud'Homme, J. Polym. Sci., Part A: Polym. Chem., 1986, 24, 2773–2787 CrossRef
- X.-Q. Liu, Z.-C. Li, F.-S. Du and F.-M. Li, Macromol. Rapid Commun., 1999, 20, 470–474 CrossRef
- X.-Q. Liu, M.-X. Wang, Z.-C. Li and F.-M. Li, Macromol. Chem. Phys., 1999, 200, 468–473 CrossRef
- Y. Yamashita, T. Tsuda, H. Ishida, A. Uchikawa and Y. Kuriyama, Makromol. Chem., 1968, 113, 139–146 CrossRef
- V. H. Ohse and H. Cherdron, Makromol. Chem., 1966, 95, 283–295 CrossRef
- J. T. Lee, P. J. Thomas and H. Alper, J. Org. Chem., 2001, 66, 5424–5426 CrossRef PubMed
- J. W. Kramer, E. B. Lobkovsky and G. W. Coates, Org. Lett., 2006, 8, 3709–3712 CrossRef PubMed
- C. Guillaume, N. Ajellal, J.-F. Carpentier and S. M. Guillaume, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 907–917 CrossRef
- F. Sinclair, L. Chen, B. W. Greenland and M. P. Shaver, Macromolecules, 2016, 49, 6826–6834 CrossRef
- M.-A. Leboucher-Durand, V. Langlois and P. Guérin, React. Funct. Polym., 1996, 31, 57–65 CrossRef
- R. Bizzarri, F. Chiellini, R. Solaro, E. Chiellini, S. Cammas-Marion and P. Guérin, Macromolecules, 2002, 35, 1215–1223 CrossRef
- V. Jeanbat-Mimaud, C. Barbaud, J.-P. Caruelle, D. Barritault, S. Cammas-Marion, V. Langlois and P. Guérin, C. R. Acad. Sci., Ser. IIc: Chim., 1999, 2, 393–401 CrossRef
- J.-P. Caruelle, D. Barritault, V. Jeanbat-Mimaud, S. Cammas-Marion, V. Langlois, P. Guerin and C. Barbaud, J. Biomater. Sci., Polym. Ed., 2000, 11, 979–991 CrossRef
- O. Coulembier, P. Degée, P. Gerbaux, P. Wantier, C. Barbaud, R. Flammang, P. Guérin and P. Dubois, Macromolecules, 2005, 38, 3141–3150 CrossRef
- Y.-C. Xu, H. Zhou, X.-Y. Sun, W.-M. Ren and X.-B. Lu, Macromolecules, 2016, 49, 5782–5787 CrossRef
- Y.-C. Xu, W.-M. Ren, H. Zhou, G.-G. Gu and X.-B. Lu, Macromolecules, 2017, 50, 3131–3142 CrossRef
- W. Saiyasombat, R. Molloy, T. M. Nicholson, A. F. Johnson, I. M. Ward and S. Poshyachinda, Polymer, 1998, 39, 5581–5585 CrossRef
-
A. Duda and A. Kowalski, in Handbook of Ring-Opening Polymerization, Wiley-VCH Verlag GmbH & Co. KGaA, 2009, ch. 1, pp. 1–51, DOI:10.1002/9783527628407
- S. Kobayashi, Proc. Jpn. Acad., Ser. B, 2010, 86, 338–365 CrossRef PubMed
- S. Kobayashi, Polym. Adv. Technol., 2015, 26, 677–686 CrossRef
- Z. Ates, F. Audouin, A. Harrington, B. O'Connor and A. Heise, Macromol. Biosci., 2014, 14, 1600–1608 CrossRef PubMed
- Z. Ates and A. Heise, Polym. Chem., 2014, 5, 2936–2941 RSC
- M. Claudino, I. van der Meulen, S. Trey, M. Jonsson, A. Heise and M. Johansson, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 16–24 CrossRef
- Z. Ates, P. D. Thornton and A. Heise, Polym. Chem., 2011, 2, 309–312 RSC
- I. van der Meulen, Y. Li, R. Deumens, E. A. J. Joosten, C. E. Koning and A. Heise, Biomacromolecules, 2011, 12, 837–843 CrossRef PubMed
- I. van der Meulen, M. de Geus, H. Antheunis, R. Deumens, E. A. J. Joosten, C. E. Koning and A. Heise, Biomacromolecules, 2008, 9, 3404–3410 CrossRef PubMed
- C. Vaida, H. Keul and M. Möller, Green Chem., 2011, 13, 889–899 RSC
- H. Uyama, S. Kobayashi, M. Morita, S. Habaue and Y. Okamoto, Macromolecules, 2001, 34, 6554–6556 CrossRef
- M. Leemhuis, N. Akeroyd, J. A. W. Kruijtzer, C. F. van Nostrum and W. E. Hennink, Eur. Polym. J., 2008, 44, 308–317 CrossRef
- F. Coumes, V. Darcos, D. Domurado, S. Li and J. Coudane, Polym. Chem., 2013, 4, 3705–3713 RSC
- Y. Yu, J. Zou, L. Yu, W. Ji, Y. Li, W.-C. Law and C. Cheng, Macromolecules, 2011, 44, 4793–4800 CrossRef
- I. A. Barker, D. J. Hall, C. F. Hansell, F. E. Du Prez, R. K. O'Reilly and A. P. Dove, Macromol. Rapid Commun., 2011, 32, 1362–1366 CrossRef PubMed
- F. Jing and M. A. Hillmyer, J. Am. Chem. Soc., 2008, 130, 13826–13827 CrossRef PubMed
- B. Martin Vaca and D. Bourissou, ACS Macro Lett., 2015, 4, 792–798 CrossRef
- J. Zou, C. C. Hew, E. Themistou, Y. Li, C.-K. Chen, P. Alexandridis and C. Cheng, Adv. Mater., 2011, 23, 4274–4277 CrossRef PubMed
- K. R. Delle Chiaie, L. M. Yablon, A. B. Biernesser, G. R. Michalowski, A. W. Sudyn and J. A. Byers, Polym. Chem., 2016, 7, 4675–4681 RSC
- C. H. Jones, C.-K. Chen, M. Chen, A. Ravikrishnan, H. Zhang, A. Gollakota, T. Chung, C. Cheng and B. A. Pfeifer, Mol. Pharmaceutics, 2015, 12, 846–856 CrossRef PubMed
- X. Jiang, E. B. Vogel, M. R. Smith and G. L. Baker, Macromolecules, 2008, 41, 1937–1944 CrossRef
- P. P. Kalelkar, G. R. Alas and D. M. Collard, Macromolecules, 2016, 49, 2609–2617 CrossRef
- M. Rubinshtein, C. R. James, J. L. Young, Y. J. Ma, Y. Kobayashi, N. C. Gianneschi and J. Yang, Org. Lett., 2010, 12, 3560–3563 CrossRef PubMed
- D. E. Borchmann, R. Tarallo, S. Avendano, A. Falanga, T. P. Carberry, S. Galdiero and M. Weck, Macromolecules, 2015, 48, 942–949 CrossRef
- D. E. Borchmann, N. ten Brummelhuis and M. Weck, Macromolecules, 2013, 46, 4426–4431 CrossRef PubMed
- J. A. Castillo, D. E. Borchmann, A. Y. Cheng, Y. Wang, C. Hu, A. J. García and M. Weck, Macromolecules, 2012, 45, 62–69 CrossRef PubMed
- G. L. Fiore, F. Jing, J. V. G. Young, C. J. Cramer and M. A. Hillmyer, Polym. Chem., 2010, 1, 870–877 RSC
- I. J. Smith and B. J. Tighe, J. Polym. Sci., Polym. Chem. Ed., 1976, 14, 949–960 CrossRef
- R. J. Pounder and A. P. Dove, Polym. Chem., 2010, 1, 260–271 RSC
- Z. Zhang, L. Yin, C. Tu, Z. Song, Y. Zhang, Y. Xu, R. Tong, Q. Zhou, J. Ren and J. Cheng, ACS Macro Lett., 2013, 2, 40–44 CrossRef PubMed
- R. Wang, J. Zhang, Q. Yin, Y. Xu, J. Cheng and R. Tong, Angew. Chem., Int. Ed., 2016, 55, 13010–13014 CrossRef PubMed
- H. Wang, L. Tang, C. Tu, Z. Song, Q. Yin, L. Yin, Z. Zhang and J. Cheng, Biomacromolecules, 2013, 14, 3706–3712 CrossRef PubMed
- Y. Li, Y. Niu, D. Hu, Y. Song, J. He, X. Liu, X. Xia, Y. Lu and W. Xu, Macromol. Chem. Phys., 2015, 216, 77–84 CrossRef
- Z. Zhang, L. Yin, Y. Xu, R. Tong, Y. Lu, J. Ren and J. Cheng, Biomacromolecules, 2012, 13, 3456–3462 CrossRef PubMed
- T. R. Blake and R. M. Waymouth, J. Am. Chem. Soc., 2014, 136, 9252–9255 CrossRef PubMed
- J. Li, Y. Zhang, J. Chen, C. Wang and M. Lang, J. Macromol. Sci., Part A: Pure Appl. Chem., 2009, 46, 1103–1113 CrossRef
- J. B. Gilroy, T. Gädt, G. R. Whittell, L. Chabanne, J. M. Mitchels, R. M. Richardson, M. A. Winnik and I. Manners, Nat. Chem., 2010, 2, 566 CrossRef PubMed
- D. Tunc, C. Le Coz, M. Alexandre, P. Desbois, P. Lecomte and S. Carlotti, Macromolecules, 2014, 47, 8247–8254 CrossRef
-
S. Russo and E. Casazza, in Polymer Science: A Comprehensive Reference, ed. M. Möller, Elsevier, Amsterdam, 2012, pp. 331–396, DOI:10.1016/B978-0-444-53349-4.00109-6
- V. E. Schmidt, Angew. Makromol. Chem., 1970, 14, 185–202 CrossRef
- R. Graf, Angew. Chem., 1968, 80, 179–189 CrossRef
- J. Lin, C. Winkelman, S. D. Worley, R. M. Broughton and J. F. Williams, J. Appl. Polym. Sci., 2001, 81, 943–947 CrossRef
- X. Jia, M. Herrera-Alonso and T. J. McCarthy, Polymer, 2006, 47, 4916–4924 CrossRef
- E. Perry and J. Savory, J. Appl.
Polym. Sci., 1967, 11, 2473–2483 CrossRef
- N. Honda, T. Kawai and F. Higashi, Makromol. Chem., 1978, 179, 1643–1646 CrossRef
- D. Huesmann, K. Klinker and M. Barz, Polym. Chem., 2017, 8, 957–971 RSC
- T. J. Deming, Chem. Rev., 2016, 116, 786–808 CrossRef PubMed
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and G. Sakellariou, Chem. Rev., 2009, 109, 5528–5578 CrossRef PubMed
- Š. Gradišar, E. Žagar and D. Pahovnik, ACS Macro Lett., 2017, 6, 637–640 CrossRef
- A. C. Engler, H.-i. Lee and P. T. Hammond, Angew. Chem., Int. Ed., 2009, 48, 9334–9338 CrossRef PubMed
- C. Xiao, C. Zhao, P. He, Z. Tang, X. Chen and X. Jing, Macromol. Rapid Commun., 2010, 31, 991–997 CrossRef PubMed
- A. C. Engler, A. Shukla, S. Puranam, H. G. Buss, N. Jreige and P. T. Hammond, Biomacromolecules, 2011, 12, 1666–1674 CrossRef PubMed
- Y.-C. Lin, P.-I. Wang and S.-W. Kuo, Soft Matter, 2012, 8, 9676–9684 RSC
- Y. Huang, Y. Zeng, J. Yang, Z. Zeng, F. Zhu and X. Chen, Chem. Commun., 2011, 47, 7509–7511 RSC
- W. Liu, M. Zhu, J. Xiao, Y. Ling and H. Tang, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3425–3435 CrossRef
- R. Zhang, N. Zheng, Z. Song, L. Yin and J. Cheng, Biomaterials, 2014, 35, 3443–3454 CrossRef PubMed
- D. S. Poché, S. J. Thibodeaux, V. C. Rucker, I. M. Warner and W. H. Daly, Macromolecules, 1997, 30, 8081–8084 CrossRef
- H. Tang and D. Zhang, Polym. Chem., 2011, 2, 1542–1551 RSC
- Y. Zhang, H. Lu, Y. Lin and J. Cheng, Macromolecules, 2011, 44, 6641–6644 CrossRef PubMed
- J. Luijten, D. Y. Groeneveld, G. W. Nijboer, E. J. Vorenkamp and A. J. Schouten, Langmuir, 2007, 23, 8163–8169 CrossRef PubMed
- H. Lu, Y. Bai, J. Wang, N. P. Gabrielson, F. Wang, Y. Lin and J. Cheng, Macromolecules, 2011, 44, 6237–6240 CrossRef PubMed
- H. Lu, J. Wang, Y. Bai, J. W. Lang, S. Liu, Y. Lin and J. Cheng, Nat. Commun., 2011, 2, 206 CrossRef PubMed
- N. P. Gabrielson, H. Lu, L. Yin, D. Li, F. Wang and J. Cheng, Angew. Chem., Int. Ed., 2012, 51, 1143–1147 CrossRef PubMed
- N. Masato and K. Hiroyoshi, Bull. Chem. Soc. Jpn., 1975, 48, 2588–2591 CrossRef
- Y. Kadoma, A. Ueno, K. Takeda, K. Uno and Y. Iwakura, J. Polym. Sci., Part B: Polym. Lett., 1974, 12, 715–718 Search PubMed
- L. Yan, L. Yang, H. He, X. Hu, Z. Xie, Y. Huang and X. Jing, Polym. Chem., 2012, 3, 1300–1307 RSC
- A. Ueno, F. Toda and Y. Iwakura, Chem. Lett., 1974, 97–100 CrossRef
- J. Ding, C. Xiao, Z. Tang, X. Zhuang and X. Chen, Macromol. Biosci., 2011, 11, 192–198 CrossRef PubMed
- J. Ding, W. Xu, Y. Zhang, D. Sun, C. Xiao, D. Liu, X. Zhu and X. Chen, J. Controlled Release, 2013, 172, 444–455 CrossRef PubMed
- H. Tang and D. Zhang, Biomacromolecules, 2010, 11, 1585–1592 CrossRef PubMed
- H. Tang, L. Yin, K. H. Kim and J. Cheng, Chem. Sci., 2013, 4, 3839–3844 RSC
- Q. Hu, Y. Deng, Q. Yuan, Y. Ling and H. Tang, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 149–153 CrossRef
- Z. Song, N. Zheng, X. Ba, L. Yin, R. Zhang, L. Ma and J. Cheng, Biomacromolecules, 2014, 15, 1491–1497 CrossRef PubMed
- Y. Wu, X. Wang, Y. Ling and H. Tang, RSC Adv., 2015, 5, 40772–40778 RSC
- M. Xiong, M. W. Lee, R. A. Mansbach, Z. Song, Y. Bao, R. M. Peek, C. Yao, L.-F. Chen, A. L. Ferguson, G. C. L. Wong and J. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13155–13160 CrossRef PubMed
- Y. Deng, Y. Xu, X. Wang, Q. Yuan, Y. Ling and H. Tang, Macromol. Rapid Commun., 2015, 36, 453–458 CrossRef PubMed
- K. Ishikawa and T. Endo, J. Am. Chem. Soc., 1988, 110, 2016–2017 CrossRef
- P. Dubruel, L. Dekie and E. Schacht, Biomacromolecules, 2003, 4, 1168–1176 CrossRef PubMed
- D. Lu, H. Wang, T. E. Li, Y. Li, F. Dou, S. Sun, H. Guo, S. Liao, Z. Yang, Q. Wei and Z. Lei, ACS Appl. Mater. Interfaces, 2017, 9, 16756–16766 CrossRef PubMed
- Q. Xu, C. He, C. Xiao, S. Yu and X. Chen, Polym. Chem., 2015, 6, 1758–1767 RSC
- Y. Liu, P. Chen and Z. Li, Macromol. Rapid Commun., 2012, 33, 287–295 CrossRef PubMed
- A. J. Rhodes and T. J. Deming, ACS Macro Lett., 2013, 2, 351–354 CrossRef
- H. Tang, L. Yin, H. Lu and J. Cheng, Biomacromolecules, 2012, 13, 2609–2615 CrossRef PubMed
- I. Yakovlev and T. J. Deming, J. Am. Chem. Soc., 2015, 137, 4078–4081 CrossRef PubMed
- J. Zhou, P. Chen, C. Deng, F. Meng, R. Cheng and Z. Zhong, Macromolecules, 2013, 46, 6723–6730 CrossRef
- J. Xiao, M. Li, W. Liu, Y. Li, Y. Ling and H. Tang, Eur. Polym. J., 2017, 88, 340–348 CrossRef
- O. Schäfer, D. Huesmann, C. Muhl and M. Barz, Chem. – Eur. J., 2016, 22, 18085–18091 CrossRef PubMed
- O. Schäfer, D. Huesmann and M. Barz, Macromolecules, 2016, 49, 8146–8153 CrossRef
- J. R. Kramer and T. J. Deming, Biomacromolecules, 2012, 13, 1719–1723 CrossRef PubMed
- E. G. Gharakhanian and T. J. Deming, Biomacromolecules, 2015, 16, 1802–1806 CrossRef PubMed
- J. R. Kramer and T. J. Deming, Chem. Commun., 2013, 49, 5144–5146 RSC
- A. R. Rodriguez, J. R. Kramer and T. J. Deming, Biomacromolecules, 2013, 14, 3610–3614 CrossRef PubMed
- M. Yu and T. J. Deming, Macromolecules, 1998, 31, 4739–4745 CrossRef PubMed
- W. D. Fuller, M. S. Verlander and M. Goodman, Biopolymers, 1978, 17, 2939–2943 CrossRef
- A. Sulistio, A. Blencowe, J. Wang, G. Bryant, X. Zhang and G. G. Qiao, Macromol. Biosci., 2012, 12, 1220–1231 CrossRef PubMed
- D. Lu, H. Wang, T. E. Li, Y. Li, X. Wang, P. Niu, H. Guo, S. Sun, X. Wang, X. Guan, H. Ma and Z. Lei, Chem. Mater., 2017, 29, 5493–5503 CrossRef
- K. Schlögl and H. Fabitschowitz, Monatsh. Chem. Verw. Teile Anderer Wiss., 1954, 85, 1060–1076 CrossRef
- J. Sun and H. Schlaad, Macromolecules, 2010, 43, 4445–4448 CrossRef
- K.-S. Krannig and H. Schlaad, J. Am. Chem. Soc., 2012, 134, 18542–18545 CrossRef PubMed
- R. M. Guinn, A. O. Margot, J. R. Taylor, M. Schumacher, D. S. Clark and H. W. Blanch, Biopolymers, 1995, 35, 503–512 CrossRef PubMed
- K. Schlögl and H. Pelousek, Monatsh. Chem. Verw. Teile Anderer Wiss., 1960, 91, 227–237 CrossRef
- J. Huang, G. Habraken, F. Audouin and A. Heise, Macromolecules, 2010, 43, 6050–6057 CrossRef
- J. Huang, C. Bonduelle, J. Thévenot, S. Lecommandoux and A. Heise, J. Am. Chem. Soc., 2012, 134, 119–122 CrossRef PubMed
- J. Jacobs, N. Gathergood, J. P. A. Heuts and A. Heise, Polym. Chem., 2015, 6, 4634–4640 RSC
- U. Haberthür and H.-G. Elias, Makromol. Chem., 1971, 144, 193–212 CrossRef
- V. Nadeau, G. Leclair, S. Sant, J.-M. Rabanel, R. Quesnel and P. Hildgen, Polymer, 2005, 46, 11263–11272 CrossRef
- A. C. Fonseca, M. H. Gil and P. N. Simões, Prog. Polym. Sci., 2014, 39, 1291–1311 CrossRef
- N. Franz and H.-A. Klok, Macromol. Chem. Phys., 2010, 211, 809–820 CrossRef
- J. W. Robinson and H. Schlaad, Chem. Commun., 2012, 48, 7835–7837 RSC
- L. Guo, S. H. Lahasky, K. Ghale and D. Zhang, J. Am. Chem. Soc., 2012, 134, 9163–9171 CrossRef PubMed
- J. W. Robinson, C. Secker, S. Weidner and H. Schlaad, Macromolecules, 2013, 46, 580–587 CrossRef
- S. H. Lahasky, W. K. Serem, L. Guo, J.
C. Garno and D. Zhang, Macromolecules, 2011, 44, 9063–9074 CrossRef
- M. Taherimehr and P. P. Pescarmona, J. Appl. Polym. Sci., 2014, 131, 41141 CrossRef
- G. Rokicki, Prog. Polym. Sci., 2000, 25, 259–342 CrossRef
- L. Ding, Y. Li, Y. Li, Y. Liang and J. Huang, Eur. Polym. J., 2001, 37, 2453–2459 CrossRef
-
D. C. Webster and A. L. Crain, Functional Polymers, 1998, ch. 21, vol. 704, pp. 303–320 Search PubMed
- J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Chem. Rev., 2016, 116, 2170–2243 CrossRef PubMed
- R. C. Pratt, F. Nederberg, R. M. Waymouth and J. L. Hedrick, Chem. Commun., 2008, 114–116, 10.1039/B713925J
- D. P. Sanders, K. Fukushima, D. J. Coady, A. Nelson, M. Fujiwara, M. Yasumoto and J. L. Hedrick, J. Am. Chem. Soc., 2010, 132, 14724–14726 CrossRef PubMed
- J. V. Olsson, D. Hult, Y. Cai, S. Garcia-Gallego and M. Malkoch, Polym. Chem., 2014, 5, 6651–6655 RSC
- G. Liu, F. He, Y. P. Wang, J. Feng and R. X. Zhuo, Chin. Chem. Lett., 2006, 17, 137–139 Search PubMed
- S. Venkataraman, N. Veronica, Z. X. Voo, J. L. Hedrick and Y. Y. Yang, Polym. Chem., 2013, 4, 2945–2948 RSC
- F. He, Y.-P. Wang, G. Liu, H.-L. Jia, J. Feng and R.-X. Zhuo, Polymer, 2008, 49, 1185–1190 CrossRef
- T. Jiang, Y.-M. Li, Y. Lv, Y.-J. Cheng, F. He and R.-X. Zhuo, Colloids Surf., B, 2013, 111, 542–548 CrossRef PubMed
- P. G. Parzuchowski, M. Jaroch, M. Tryznowski and G. Rokicki, Macromolecules, 2008, 41, 3859–3865 CrossRef
- T. Jiang, Y. Li, Y. Lv, Y. Cheng, F. He and R. Zhuo, J. Mater. Sci.: Mater. Med., 2014, 25, 131–139 CrossRef PubMed
- X. Zhang, H. Wang and Y. Dai, Mater. Lett., 2017, 198, 144–147 CrossRef
- C. Lu, Q. Shi, X. Chen, T. Lu, Z. Xie, X. Hu, J. Ma and X. Jing, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3204–3217 CrossRef
- Q. Shi, X. Chen, T. Lu and X. Jing, Biomaterials, 2008, 29, 1118–1126 CrossRef PubMed
- Q. Shi, Y. Huang, X. Chen, M. Wu, J. Sun and X. Jing, Biomaterials, 2009, 30, 5077–5085 CrossRef PubMed
- D. Hu, H. Peng, Y. Niu, Y. Li, Y. Xia, L. Li, J. He, X. Liu, X. Xia, Y. Lu and W. Xu, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 750–760 CrossRef
- S. Tempelaar, I. A. Barker, V. X. Truong, D. J. Hall, L. Mespouille, P. Dubois and A. P. Dove, Polym. Chem., 2013, 4, 174–183 RSC
- H. Xiong, D. Zhou, Y. Qi, Z. Zhang, Z. Xie, X. Chen, X. Jing, F. Meng and Y. Huang, Biomacromolecules, 2015, 16, 3980–3988 CrossRef PubMed
- H. Xiong, X. Wei, D. Zhou, Y. Qi, Z. Xie, X. Chen, X. Jing and Y. Huang, Bioconjugate Chem., 2016, 27, 2214–2223 CrossRef PubMed
- B. D. Mullen, C. N. Tang and R. F. Storey, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1978–1991 CrossRef
- C.-F. Wang, Y.-X. Lin, T. Jiang, F. He and R.-X. Zhuo, Biomaterials, 2009, 30, 4824–4832 CrossRef PubMed
- D. M. Stevens, S. Tempelaar, A. P. Dove and E. Harth, ACS Macro Lett., 2012, 1, 915–918 CrossRef PubMed
- X. Hu, X. Chen, S. Liu, Q. Shi and X. Jing, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1852–1861 CrossRef
- H. Kuang, S. Wu, Z. Xie, F. Meng, X. Jing and Y. Huang, Biomacromolecules, 2012, 13, 3004–3012 CrossRef PubMed
- H. Kuang, S. Wu, F. Meng, Z. Xie, X. Jing and Y. Huang, J. Mater. Chem., 2012, 22, 24832–24840 RSC
- V. X. Truong, I. A. Barker, M. Tan, L. Mespouille, P. Dubois and A. P. Dove, J. Mater. Chem. B, 2013, 1, 221–229 RSC
- I. A. Barker, M. P. Ablett, H. T. J. Gilbert, S. J. Leigh, J. A. Covington, J. A. Hoyland, S. M. Richardson and A. P. Dove, Biomater. Sci., 2014, 2, 472–475 RSC
- K. Olofsson, M. Malkoch and A. Hult, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2370–2378 CrossRef
- G. S. Heo, S. Cho and K. L. Wooley, Polym. Chem., 2014, 5, 3555–3558 RSC
- W. Chen, H. Yang, R. Wang, R. Cheng, F. Meng, W. Wei and Z. Zhong, Macromolecules, 2010, 43, 201–207 CrossRef
- J. Xiong, F. Meng, C. Wang, R. Cheng, Z. Liu and Z. Zhong, J. Mater. Chem., 2011, 21, 5786–5794 RSC
- F. Chen, J. W. S. Hayami and B. G. Amsden, Biomacromolecules, 2014, 15, 1593–1601 CrossRef PubMed
- D. P. Sanders, D. J. Coady, M. Yasumoto, M. Fujiwara, H. Sardon and J. L. Hedrick, Polym. Chem., 2014, 5, 327–329 RSC
- X. Hu, X. Chen, H. Cheng and X. Jing, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 161–169 CrossRef
- R. Wang, W. Chen, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, Macromolecules, 2011, 44, 6009–6016 CrossRef
- J. Mindemark and T. Bowden, Polymer, 2011, 52, 5716–5722 CrossRef
- J. Mindemark and T. Bowden, Polym.
Chem., 2012, 3, 1399–1401 RSC
- Z. Y. Ong, K. Fukushima, D. J. Coady, Y.-Y. Yang, P. L. R. Ee and J. L. Hedrick, J. Controlled Release, 2011, 152, 120–126 CrossRef PubMed
- X. Ding, C. Yang, T. P. Lim, L. Y. Hsu, A. C. Engler, J. L. Hedrick and Y.-Y. Yang, Biomaterials, 2012, 33, 6593–6603 CrossRef PubMed
- F. Nederberg, Y. Zhang, J. P. K. Tan, K. Xu, H. Wang, C. Yang, S. Gao, X. D. Guo, K. Fukushima, L. Li, J. L. Hedrick and Y.-Y. Yang, Nat. Chem., 2011, 3, 409–414 CrossRef PubMed
- Z. Y. Ong, C. Yang, S. J. Gao, X.-Y. Ke, J. L. Hedrick and Y. Y. Yang, Macromol. Rapid Commun., 2013, 34, 1714–1720 CrossRef PubMed
- V. W. L. Ng, J. P. K. Tan, J. Leong, Z. X. Voo, J. L. Hedrick and Y. Y. Yang, Macromolecules, 2014, 47, 1285–1291 CrossRef
- A. C. Engler, J. P. K. Tan, Z. Y. Ong, D. J. Coady, V. W. L. Ng, Y. Y. Yang and J. L. Hedrick, Biomacromolecules, 2013, 14, 4331–4339 CrossRef PubMed
- V. W. L. Ng, X. Ke, A. L. Z. Lee, J. L. Hedrick and Y. Y. Yang, Adv. Mater., 2013, 25, 6730–6736 CrossRef PubMed
- W. Chin, C. Yang, V. W. L. Ng, Y. Huang, J. Cheng, Y. W. Tong, D. J. Coady, W. Fan, J. L. Hedrick and Y. Y. Yang, Macromolecules, 2013, 46, 8797–8807 CrossRef
- R. J. Ono, S. Q. Liu, S. Venkataraman, W. Chin, Y. Y. Yang and J. L. Hedrick, Macromolecules, 2014, 47, 7725–7731 CrossRef
- N. H. Park, Z. X. Voo, Y. Y. Yang and J. L. Hedrick, ACS Macro Lett., 2017, 6, 252–256 CrossRef
- A. C. Engler, J. M. W. Chan, D. J. Coady, J. M. O’Brien, H. Sardon, A. Nelson, D. P. Sanders, Y. Y. Yang and J. L. Hedrick, Macromolecules, 2013, 46, 1283–1290 CrossRef
- J. M. W. Chan, X. Ke, H. Sardon, A. C. Engler, Y. Y. Yang and J. L. Hedrick, Chem. Sci., 2014, 3294–3300, 10.1039/C4SC00789A
- A. C. Engler, X. Ke, S. Gao, J. M. W. Chan, D. J. Coady, R. J. Ono, R. Lubbers, A. Nelson, Y. Y. Yang and J. L. Hedrick, Macromolecules, 2015, 48, 1673–1678 CrossRef
- J. M. W. Chan, R. J. Wojtecki, H. Sardon, A. L. Z. Lee, C. E. Smith, A. Shkumatov, S. Gao, H. Kong, Y. Y. Yang and J. L. Hedrick, ACS Macro Lett., 2017, 6, 176–180 CrossRef
- Y. Zhou, R.-X. Zhuo and Z.-L. Liu, Macromol. Rapid Commun., 2005, 26, 1309–1314 CrossRef
- N. H. Park, M. Fevre, Z. X. Voo, R. J. Ono, Y. Y. Yang and J. L. Hedrick, ACS Macro Lett., 2016, 5, 1247–1252 CrossRef
- W. Chen, Y. Zou, J. Jia, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, Macromolecules, 2013, 46, 699–707 CrossRef
- W. Chen, Y. Zou, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, Biomacromolecules, 2014, 15, 900–907 CrossRef PubMed
- G. A. Barcan, X. Zhang and R. M. Waymouth, J. Am. Chem. Soc., 2015, 137, 5650–5653 CrossRef PubMed
- X. Zhang and R. M. Waymouth, J. Am. Chem. Soc., 2017, 139, 3822–3833 CrossRef PubMed
- L. Mespouille, F. Nederberg, J. L. Hedrick and P. Dubois, Macromolecules, 2009, 42, 6319–6321 CrossRef
- R. J. Williams, R. K. O'Reilly and A. P. Dove, Polym. Chem., 2012, 3, 2156–2164 RSC
- A. Dag, M. Aydin, H. Durmaz, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4476–4483 CrossRef
- W. Thongsomboon, M. Sherwood, N. Arellano and A. Nelson, ACS Macro Lett., 2013, 2, 19–22 CrossRef
- R. J. Williams, I. A. Barker, R. K. O’Reilly and A. P. Dove, ACS Macro Lett., 2012, 1, 1285–1290 CrossRef
- S. Kühling, H. Keul and H. Höcker, Makromol. Chem., 1990, 191, 1611–1622 CrossRef
- J. Mindemark, L. Imholt, J. Montero and D. Brandell, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2128–2135 CrossRef
- A. W. Thomas, P. K. Kuroishi, M. M. Perez-Madrigal, A. K. Whittaker and A. P. Dove, Polym. Chem., 2017, 8, 5082–5090 RSC
- P. K. Kuroishi, M. J. Bennison and A. P. Dove, Polym. Chem., 2016, 7, 7108–7115 RSC
- T. Miyagawa, M. Shimizu, F. Sanda and T. Endo, Macromolecules, 2005, 38, 7944–7949 CrossRef
- Q. J. Zhang, W. P. Zhu and Z. Q. Shen, Chin. Chem. Lett., 2010, 21, 1255–1258 CrossRef
- W. Zhu, Y. Wang, S. Sun, Q. Zhang, X. Li and Z. Shen, J. Mater. Chem., 2012, 22, 11785–11791 RSC
- W. Zhu, Y. Wang, Q. Zhang and Z. Shen, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4886–4893 CrossRef
- W. Zhu, Y. Wang, X. Cai, G. Zha, Q. Luo, R. Sun, X. Li and Z. Shen, J. Mater. Chem. B, 2015, 3, 3024–3031 RSC
- X. Zhang, Z. Zhong and R. Zhuo, Macromolecules, 2011, 44, 1755–1759 CrossRef
- X. Zhang, H. Dong, S. Fu, Z. Zhong and R. Zhuo, Macromol. Rapid Commun., 2016, 37, 993–997 CrossRef PubMed
- J. Xu, T. M. Filion, F. Prifti and J. Song, Chem. – Asian J., 2011, 6, 2730–2737 CrossRef PubMed
- Y. Zou, Y. Fang, H. Meng, F. Meng, C. Deng, J. Zhang and Z. Zhong, J. Controlled Release, 2016, 244, 326–335 CrossRef PubMed
- X. Chen, S. P. McCarthy and R. A. Gross, Macromolecules, 1997, 30, 3470–3476 CrossRef
- X. Chen, S. P. McCarthy and R. A. Gross, Macromolecules, 1998, 31, 662–668 CrossRef
- S. Venkataraman, V. W. L. Ng, D. J. Coady, H. W. Horn, G. O. Jones, T. S. Fung, H. Sardon, R. M. Waymouth, J. L. Hedrick and Y. Y. Yang, J. Am. Chem. Soc., 2015, 137, 13851–13860 CrossRef PubMed
- A. Pascual, J. P. K. Tan, A. Yuen, J. M. W. Chan, D. J. Coady, D. Mecerreyes, J. L. Hedrick, Y. Y. Yang and H. Sardon, Biomacromolecules, 2015, 16, 1169–1178 CrossRef PubMed
- J. P. K. Tan, D. J. Coady, H. Sardon, A. Yuen, S. Gao, S. W. Lim, Z. C. Liang, E. W. Tan, S. Venkataraman, A. C. Engler, M. Fevre, R. Ono, Y. Y. Yang and J. L. Hedrick, Adv. Healthcare Mater., 2017, 6, 1601420 CrossRef PubMed
- T. Steinbach and F. R. Wurm, Angew. Chem., Int. Ed., 2015, 54, 6098–6108 CrossRef PubMed
- K. N. Bauer, H. T. Tee, M. M. Velencoso and F. R. Wurm, Prog. Polym. Sci., 2017, 73, 61–122 CrossRef
- G. Becker and F. R. Wurm, Tetrahedron, 2017, 73, 3536–3540 CrossRef
- B. Clément, B. Grignard, L. Koole, C. Jérôme and P. Lecomte, Macromolecules, 2012, 45, 4476–4486 CrossRef
- Y.-C. Wang, Y.-Y. Yuan, F. Wang and J. Wang, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 487–494 CrossRef
- S. Zhang, A. Li, J. Zou, L. Y. Lin and K. L. Wooley, ACS Macro Lett., 2012, 1, 328–333 CrossRef PubMed
- S. Zhang, J. Zou, F. Zhang, M. Elsabahy, S. E. Felder, J. Zhu, D. J. Pochan and K. L. Wooley, J. Am. Chem. Soc., 2012, 134, 18467–18474 CrossRef PubMed
- Y. H. Lim, G. S. Heo, S. Cho and K. L. Wooley, ACS Macro Lett., 2013, 2, 785–789 CrossRef PubMed
- T. P. Gustafson, Y. H. Lim, J. A. Flores, G. S. Heo, F. Zhang, S. Zhang, S. Samarajeewa, J. E. Raymond and K. L. Wooley, Langmuir, 2014, 30, 631–641 CrossRef PubMed
- F. Zhang, S. Zhang, S. F. Pollack, R. Li, A. M. Gonzalez, J. Fan, J. Zou, S. E. Leininger, A. Pavía-Sanders, R. Johnson, L. D. Nelson, J. E. Raymond, M. Elsabahy, D. M. P. Hughes, M. W. Lenox, T. P. Gustafson and K. L. Wooley, J. Am. Chem. Soc., 2015, 137, 2056–2066 CrossRef PubMed
- S. Zhang, J. Zou, M. Elsabahy, A. Karwa, A. Li, D. A. Moore, R. B. Dorshow and K. L. Wooley, Chem. Sci., 2013, 4, 2122–2126 RSC
- Y. H. Lim, K. M. Tiemann, G. S. Heo, P. O. Wagers, Y. H. Rezenom, S. Zhang, F. Zhang, W. J. Youngs, D. A. Hunstad and K. L. Wooley, ACS Nano, 2015, 9, 1995–2008 CrossRef PubMed
- T. A. Aweda, S. Zhang, C. Mupanomunda, J. Burkemper, G. S. Heo, N. Bandara, M. Lin, C. S. Cutler, C. L. Cannon, W. J. Youngs, K. L. Wooley and S. E. Lapi, J. Labelled Compd. Radiopharm., 2015, 58, 234–241 CrossRef PubMed
- E. Y. Zeynep, D. Antoine, C. Brice, B. Frank and J. Christine, J. Mater. Chem. B, 2015, 3, 7227–7236 RSC
- Z. Ergul Yilmaz, T. Cordonnier, A. Debuigne, B. Calvignac, C. Jerome and F. Boury, Int. J. Pharm., 2016, 513, 130–137 CrossRef PubMed
- J.-Z. Du, X.-J. Du, C.-Q. Mao and J. Wang, J. Am. Chem. Soc., 2011, 133, 17560–17563 CrossRef PubMed
- X.-Z. Yang, J.-Z. Du, S. Dou, C.-Q. Mao, H.-Y. Long and J. Wang, ACS Nano, 2012, 6, 771–781 CrossRef PubMed
- B. Clément, D. G. Molin, C. Jérôme and P. Lecomte, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2642–2648 CrossRef
- J. Zou, F. Zhang, S. Zhang, S. F. Pollack, M. Elsabahy, J. Fan and K. L. Wooley, Adv. Healthcare Mater., 2014, 3, 441–448 CrossRef PubMed
- E. Baeten, S. Vanslambrouck, C. Jérôme, P. Lecomte and T. Junkers, Eur. Polym. J., 2016, 80, 208–218 CrossRef
- Y. H. Lim, G. S. Heo, Y. H. Rezenom, S. Pollack, J. E. Raymond, M. Elsabahy and K. L. Wooley, Macromolecules, 2014, 47, 4634–4644 CrossRef PubMed
- H. Shao, M. Zhang, J. He and P. Ni, Polymer, 2012, 53, 2854–2863 CrossRef
- Y. Iwasaki, S. Komatsu, T. Narita, K. Akiyoshi and K. Ishihara, Macromol. Biosci., 2003, 3, 238–242 CrossRef
- Y. Iwasaki, C. Nakagawa, M. Ohtomi, K. Ishihara and K. Akiyoshi, Biomacromolecules, 2004, 5, 1110–1115 CrossRef PubMed
- C. Wachiralarpphaithoon, Y. Iwasaki and K. Akiyoshi, Biomaterials, 2007, 28, 984–993 CrossRef PubMed
- J.-Z. Du, T.-M. Sun, S.-Q. Weng, X.-S. Chen and J. Wang, Biomacromolecules, 2007, 8, 3375–3381 CrossRef PubMed
- Y. Iwasaki and K. Akiyoshi, Macromolecules, 2004, 37, 7637–7642 CrossRef
- Y. Iwasaki and K. Akiyoshi, Biomacromolecules, 2006, 7, 1433–1438 CrossRef PubMed
- G. Lapienis and S. Penczek, Macromolecules, 1977, 10, 1301–1306 CrossRef
- T. Biela, S. Penczek, S. Slomkowski and O. Vogl, Makromol. Chem., Rapid Commun., 1982, 3, 667–671 CrossRef
- J.-C. Brosse, D. Derouet, L. Fontaine and S. Chairatanathavorn, Makromol. Chem., 1989, 190, 2339–2345 CrossRef
- J. Wang, P.-C. Zhang, H.-F. Lu, N. Ma, S. Wang, H.-Q. Mao and K. W. Leong, J. Controlled Release, 2002, 83, 157–168 CrossRef PubMed
- J. Wang, H.-Q. Mao and K. W. Leong, J. Am. Chem. Soc., 2001, 123, 9480–9481 CrossRef PubMed
- S.-W. Huang, J. Wang, P.-C. Zhang, H.-Q. Mao, R.-X. Zhuo and K. W. Leong, Biomacromolecules, 2004, 5, 306–311 CrossRef PubMed
- J. Wang, P.-C. Zhang, H.-Q. Mao and K. W. Leong, Gene Ther., 2002, 9, 1254–1261 CrossRef PubMed
- P.-C. Zhang, J. Wang, K. W. Leong and H.-Q. Mao, Biomacromolecules, 2005, 6, 54–60 CrossRef PubMed
- X. Jiang, W. Qu, D. Pan, Y. Ren, J.-M. Williford, H. Cui, E. Luijten and H.-Q. Mao, Adv. Mater., 2013, 25, 227–232 CrossRef PubMed
- A. A. Oswald, Can. J. Chem., 1959, 37, 1498–1504 CrossRef
- K. Kaluzynski, J. Libisowski and S. Penczek, Macromolecules, 1976, 9, 365–367 CrossRef
- J. Pretula, K. Kaluzynski and S. Penczek, Macromolecules, 1986, 19, 1797–1799 CrossRef
- G. Łapienis, S. Penczek, G. P. Aleksiuk and V. A. Kropachev, J. Polym. Sci., Part A: Polym. Chem., 1987, 25, 1729–1736 CrossRef
- G. Łapienis and S. Penczek, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 1519–1526 CrossRef
- O. Kuznetsova, K. Kaluzynski, G. Lapienis, J. Pretula and S. Penczek, J. Bioact. Compat. Polym., 1999, 232–242 CrossRef
- T. Wolf, T. Rheinberger and F. R. Wurm, Eur. Polym. J., 2017, 95, 756–765, DOI:10.1016/j.eurpolymj.2017.05.048
- T. Wolf, T. Steinbach and F. R. Wurm, Macromolecules, 2015, 48, 3853–3863 CrossRef
- T. Steinbach, S. Ritz and F. R. Wurm, ACS Macro Lett., 2014, 3, 244–248 CrossRef
- K. N. Bauer, L. Liu, D. Andrienko, M. Wagner, E. K. Macdonald, M. P. Shaver and F. R. Wurm, Macromolecules, 2018, 51, 1272–1279 CrossRef
- I. Teasdale and O. Brüggemann, Polymers, 2013, 5, 161–187 CrossRef PubMed
- R. S. Ullah, L. Wang, H. Yu, N. M. Abbasi, M. Akram, Z. ul-Abdin, M. Saleem, M. Haroon and R. U. Khan, RSC Adv., 2017, 7, 23363–23391 RSC
-
M. Gleria and R. De Jaeger, in New Aspects in Phosphorus Chemistry, ed. J.-P. Majoral, Springer Berlin Heidelberg, Berlin, Heidelberg, 2005, pp. 165–251, DOI:10.1007/b100985
- H. R. Allcock, S. M. Coley, I. Manners, O. Nuyken and G. Renner, Macromolecules, 1991, 24, 2024–2028 CrossRef
- J. A. Dodge, I. Manners, H. R. Allcock, G. Renner and O. Nuyken, J. Am. Chem. Soc., 1990, 112, 1268–1269 CrossRef
- I. Manners, H. R. Allcock, G. Renner and O. Nuyken, J. Am. Chem. Soc., 1989, 111, 5478–5480 CrossRef
- H. R. Allcock, S. M. Coley and C. T. Morrissey, Macromolecules, 1994, 27, 2904–2911 CrossRef
- M. Liang and I. Manners, J. Am. Chem. Soc., 1991, 113, 4044–4045 CrossRef
- Y. Ni, A. Stammer, M. Liang, J. Massey, G. J. Vancso and I. Manners, Macromolecules, 1992, 25, 7119–7125 CrossRef
| This journal is © The Royal Society of Chemistry 2018 |