
An efficient method for retro-Claisen-type C–C bond cleavage of diketones with tropylium catalyst†
M. A.
Hussein
a,
V. T.
Huynh
b,
R.
Hommelsheim
 c,
R. M.
Koenigs
*c and
T. V.
Nguyen
*a
c,
R. M.
Koenigs
*c and
T. V.
Nguyen
*a
aSchool of Chemistry, University of New South Wales, Sydney, Australia. E-mail: t.v.nguyen@unsw.edu.au
bSchool of Chemistry, University of Sydney, Australia
cInstitute of Organic Chemistry, RWTH Aachen University, Germany. E-mail: rene.koenigs@rwth-aachen.de
First published on 19th October 2018
Abstract
The retro-Claisen reaction is frequently used in organic synthesis to access ester derivatives from 1,3-dicarbonyl precursors. The C–C bond cleavage in this reaction is usually promoted by a number of transition-metal Lewis acid catalysts or organic Brønsted acids/bases. Herein we report a new convenient and efficient method utilizing the tropylium ion as a mild and environmentally friendly organocatalyst to mediate retro-Claisen-type reactions. Using this method, a range of synthetically valuable substances can be accessed via solvolysis of 1,3-dicarbonyl compounds.
In recent years, the retro-Claisen reaction (Scheme 1) has often been used as an alternative approach to access ester products from dicarbonyl compounds.1 Esters from retro-Claisen reactions normally have different structural features to their analogues produced by other traditional esterification methods, hence offering a unique synthetic value for this type of chemical transformation.2 The C–C bond cleavage3 in this reaction has been promoted by a wide range of transition metal Lewis acids4 such as In(III),2,5 Fe(III),6 Pd(0)7 and Ag(I) catalysts.8 More recently, there have been a number of interesting movements to use non-transition metal Brønsted bases9 or organocatalytic systems10 to trigger the retro-Claisen reaction to reduce environmental footprints. These new developments have significantly advanced the field, however there are still limitations in the efficiency, regioselectivity and substrate scope of the retro-Claisen reaction. Herein we report a new method utilizing a tropylium salt to mediate this chemical transformation. The method can be used for the synthesis of alkenyl thioethers and it can be conveniently implemented in flow chemistry for the large-scale solvolysis of 1,3-dicarbonyl compounds (Scheme 1).
 | ||
| Scheme 1 retro-Claisen reactions. | ||
The tropylium ion11 possesses an interesting combination of stability and reactivity due to its unique non-benzenoid aromatic cation structure.12 Based on our recent works with tropylium ion-promoted chemistry13 and reactions of carbonyl compounds,14 we believed that the tropylium ion could act as a Lewis acid catalyst to activate 1,3-dicarbonyl substrates for retro-Claisen reactions. The tropylium ion could also enhance the Brønsted acidity of protic reagents to promote the reaction via a Lewis acid assisted Brønsted acid catalytic pathway,13g which has been rarely studied in the past.15
Thus, we set out to investigate the catalytic activity of tropylium tetrafluoroborate (1) in the retro-Claisen type solvolysis of 2-acetylcyclopentanone (2a) as a model substrate. Our initial reactions on the hydrolytic transformation of 2a using 10 mol% catalyst 1 met with very promising outcomes (Table 1, entries 1–4). Similar to a range of other catalytic retro-Claisen reactions, elevated temperatures were required to effectively promote the C–C bond cleavage. A quick optimization study on catalyst loadings and solvents showed that 10 mol% of catalyst 1 delivered the best efficiency (Table 1, entries 4–13). The optimal reaction conditions were reflected in entry 4 where we were able to carry out the ring-opening hydrolysis of diketone 2a within 16 hours and obtain product 4a in 99% yield after purification. Increasing the amount of water actually disfavoured the formation of this product (entry 9, Table 1).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Mol% cat. | Solvent | T (°C) | Timeb | Yieldc (%) |
| a Conditions: diketone 2a (1 mmol), water (3a, 2 mmol) and cat. 1 (0.1 mmol) in the indicated solvent (0.6 mL) under N2 atmosphere. b Reaction time (hour) until no further or total consumption of substrate 2a. c Yield of the isolated product. d 0.5 mL water was used. e 10 mol% of a Brønsted acid catalyst was used instead of tropylium catalyst 1.16 | |||||
| 1 | 10 | No solvent | rt | 48 | 18 |
| 2 | 10 | No solvent | 60 | 24 | 52 |
| 3 | 10 | No solvent | 80 | 24 | 96 |
| 4 | 10 | No solvent | 100 | 16 | 99 |
| 5 | 5 | No solvent | 100 | 16 | 62 |
| 6 | 2.5 | No solvent | 100 | 16 | 56 |
| 7 | 1 | No solvent | 100 | 16 | 38 |
| 8 | No cat. | No solvent | 100 | 16 | Traces |
| 9d | 10 | Water as solvent | 100 | 16 | 80 |
| 10 | 10 | MeCN | Reflux | 16 | 45 |
| 11 | 10 | Toluene | Reflux | 16 | 46 |
| 12 | 10 | DCE | Reflux | 16 | 70 |
| 13 | 10 | TFE | Reflux | 12 | 99 |
| 14 | 10 | TFE | rt | 24 | 98 |
| 15 | 10 | Water as solvent | rt | 48 | 21 |
| 16 | 10 | MeCN | rt | 48 | 62 |
| 17 | 10 | Toluene | rt | 48 | 37 |
| 18 | 10 | DCE | rt | 48 | 64 |
| 19e | 10% HBF4 | No solvent | 100 | 24 | 67 |
| 20e | 10% TfOH | No solvent | 100 | 24 | 78 |
| 21e | 10% HBF4 | TFE | rt | 48 | 59 |
| 22e | 10% TfOH | TFE | rt | 48 | 76 |
We subsequently examined the possibility of performing this chemical transformation at ambient temperature, which is more desirable for sustainable synthetic protocols. Based on our prior experience with tropylium-promoted chemistry, we identified the highly ionizing solvent trifluoroethanol (TFE) as an effective medium for this reaction. Indeed, TFE solvent could not only mediate the reaction smoothly at high temperature (Table 1, entry 13) but also at room temperature, albeit taking longer reaction time (entry 14). Other organic solvents gave unsatisfactory reaction outcomes at room temperature even with extended reaction times (entries 15–18). In brief, we established two practical protocols to facilitate the retro-Claisen type hydrolysis of 2-acetylcyclopentantone (2a), the solvent-free method requires elevated temperature (Table 1, entry 4, procedure A) while the use of TFE can mediate the reaction at room temperature with similar reaction efficiency (Table 1, entry 14, procedure B).
We subsequently used these newly developed procedures to perform a range of hydrolysis, alcoholytic and aminolytic reactions on cyclic diketones (Scheme 2). 2-Acetylcyclopentanone (2a), 1,3-cyclohexanedione (2b, also see page S18 in the ESI†)17 and 2-methyl-1,3-cyclohexanedione (2c) were smoothly ring-opened with water, alcohols and amines to afford the corresponding products in good to excellent yields (2a → 4a–o, 2b → 4p–r and 2c → 4s–w, respectively). Secondary alcohols (4g, 4i) and secondary amines (4m, 4n, 4w) generally gave lower product yields than their primary analogues.17 Treatment of substrate 2a with the sterically challenging tert-butyl alcohol under reaction conditions in procedure A led to the formation of hydrolysis product 4a, presumably due to water being formed from the tropylium ion-catalyzed dehydration reaction of the tertiary alcohol.
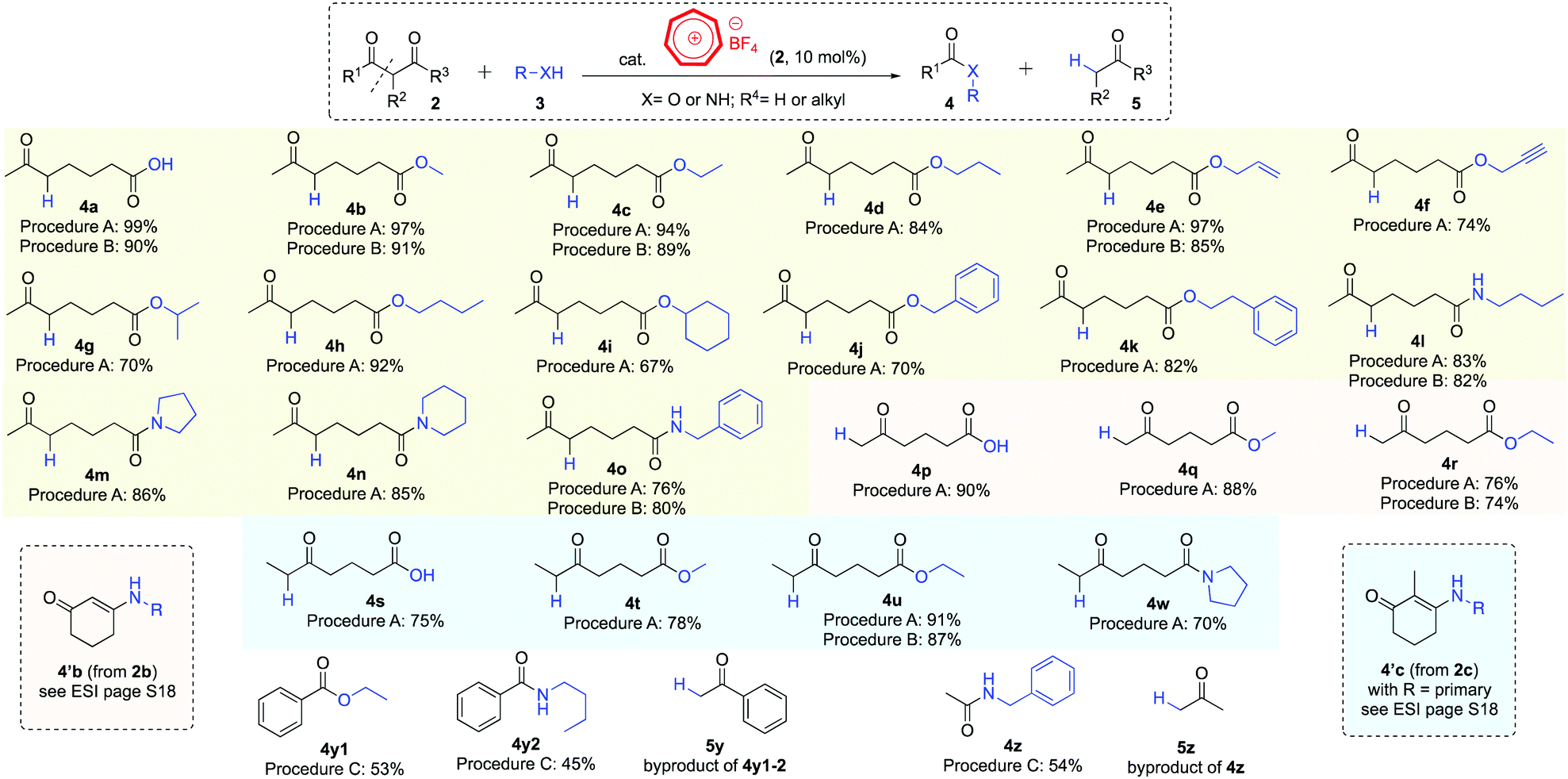 | ||
| Scheme 2 Tropylium-promoted retro-Claisen reactions. | ||
Tropylium ion can also promote the retro-Claisen solvolysis of acyclic substrates such as dibenzoylmethane (2d) and acetylacetone (2e). However, the reactions proved to be sluggish using procedures A or B so we adapted the conditions specified in entry 13 (Table 1, procedure C), which worked to give the cleaved products (4y–z) with moderate efficiency.
Our newly developed tropylium-catalyzed retro-Claisen method was amenable to large-scale synthesis using continuous flow chemistry setup. Indeed, after some reaction parameter optimization,16 we were able to carry out the alcoholysis and aminolysis of selected diketone substrates on multi-gram scale (Scheme 3, left). The tropylium catalyst loading could be reduced to 5 mol%, which is another improvement from batch conditions. Six products 4c–4w were synthesized in high to excellent yields by flowing a mixture of the reagents and tropylium catalyst in TFE solutions into a 10 mL tubular reactor heated to 150 °C with retention time of 30 minutes. This simple and efficient flow protocol offers a alternative practical approach to the retro-Claisen solvolysis reactions of diketone substrates.
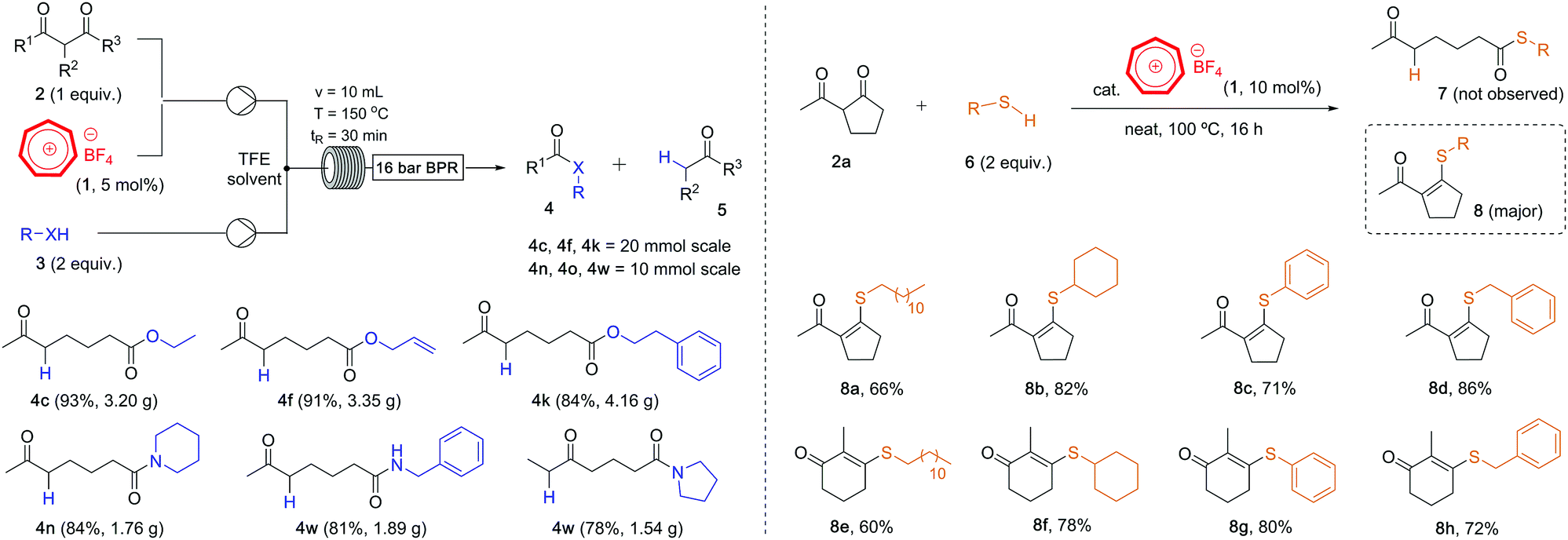 | ||
| Scheme 3 (left) retro-Claisen alcoholysis and aminolysis in continuous flow chemistry; (right) thio–enolization with mercaptan reagents. | ||
When we explored the possibility of using thiols as nucleophiles in this transformation, the outcomes were interesting. Instead of cleaving off the C–C bond to form the corresponding ketothioesters 7, we obtained thio–enol ether products 8 (Scheme 3, right), most likely via the nucleophilic addition of thiols to the carbonyl group followed by dehydration reaction. The position of the C–C double bond was confirmed by 2D-NMR.16 This reactivity is common for a range of diketone and thiol substrates,18 as we obtained their condensation products 8 in good to high yields. Although this reaction looked rather simple, this is, to the best of our knowledge, the first time thio–enol ethers are produced directly from diketones in a dehydrative fashion. These products belong to a broader family of alkenyl thioethers, which are interesting structural scaffolds19 and valuable synthetic precursors for C–C coupling reactions.20 Our protocol could serve as an alternative approach to access tetrasubstituted alkenes with thioether substituent instead of the addition of thiols to alkynes.21
To gain more information on how the tropylium ion activates the diketone substrates for these C–C cleavage reactions, we carried out a series of mechanistic studies on substrate 2a. 1H and 13C NMR spectra of mixtures of substrate 2a and tropylium salt 1 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 10:1 ratios) revealed clear evidence that tropylium ion coordinates to substrate 2a,16 presumably via the diketone complex form 9 or the enol complex form 9′ (Scheme 4, top). The progress of the tropylium-promoted reactions between 2a and water or methanol was also monitored by NMR spectroscopy; unfortunately the crude mixtures were too messy for any useful mechanistic insights to be deducted. The enolization of substrate 2a (to 2a′) and its subsequent reaction intermediates also contributed to the complication of spectroscopic signals. Indeed, when we treated substrate 2a with deuterated water and methanol, the level of deuteration on products 4aD/4bD indicated that uncontrolled enolization occurred during these reactions (Scheme 4, bottom).16 Changes in reaction temperatures and reaction times led to different deuterium contents in 4aD/4bD.
:1 or 10:1 ratios) revealed clear evidence that tropylium ion coordinates to substrate 2a,16 presumably via the diketone complex form 9 or the enol complex form 9′ (Scheme 4, top). The progress of the tropylium-promoted reactions between 2a and water or methanol was also monitored by NMR spectroscopy; unfortunately the crude mixtures were too messy for any useful mechanistic insights to be deducted. The enolization of substrate 2a (to 2a′) and its subsequent reaction intermediates also contributed to the complication of spectroscopic signals. Indeed, when we treated substrate 2a with deuterated water and methanol, the level of deuteration on products 4aD/4bD indicated that uncontrolled enolization occurred during these reactions (Scheme 4, bottom).16 Changes in reaction temperatures and reaction times led to different deuterium contents in 4aD/4bD.
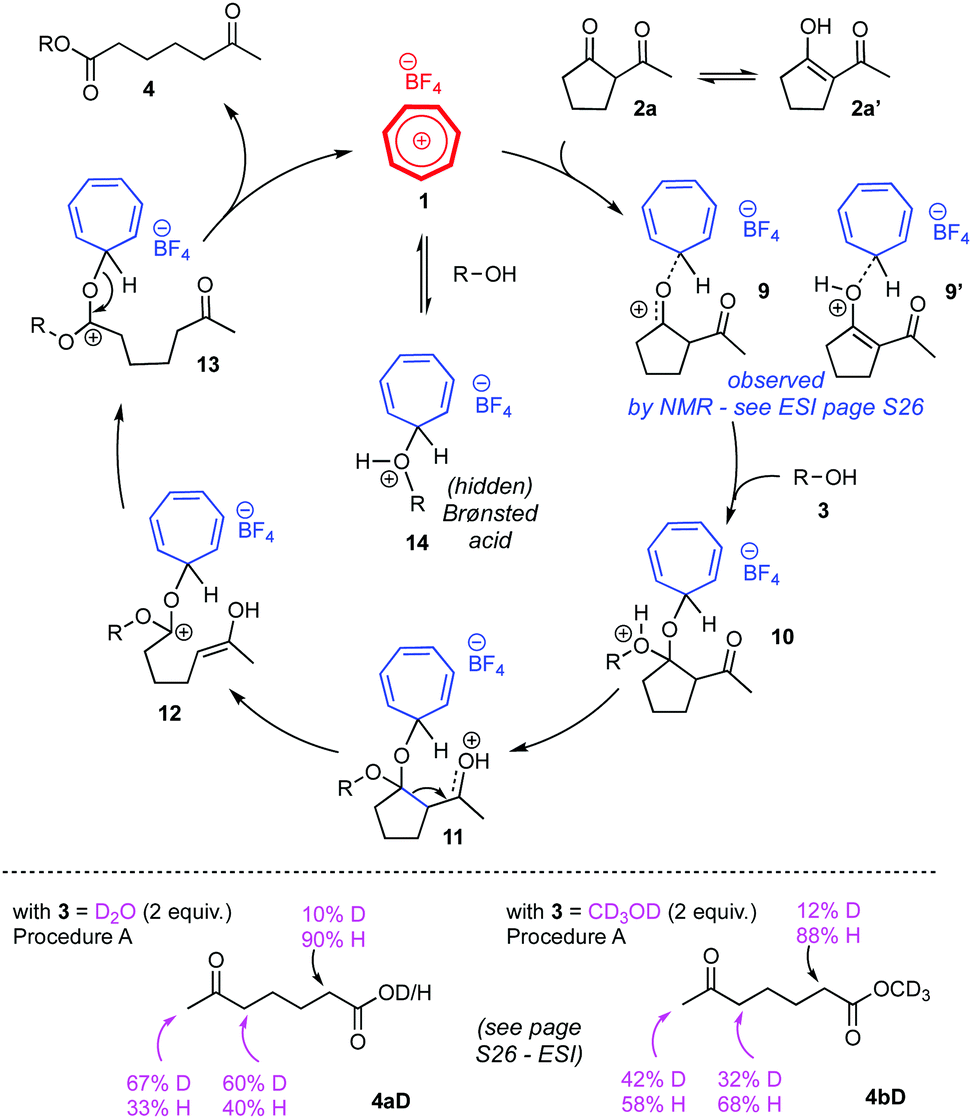 | ||
| Scheme 4 Proposed mechanism. | ||
Based on these studies and prior knowledge in this field, we propose that the retro-Claisen reaction occurs through the mechanistic pathways depicted in Scheme 4 (top). Again, enolization could happen for intermediates 10–13 during the course of reaction. We cannot rule out the possibility that tropylium ion can coordinate to the alcohol/water reagent itself to generate a strong Brønsted acid (14), which can in turn facilitate the retro-Claisen reaction. However, comparative reactions with strong Brønsted acid such as HBF4 or TfOH (see Table 1, entries 19–22) showed much lower efficiency than the tropylium-promoted protocol, even at longer reaction times. Therefore, we believe that the hidden Brønsted acid catalytic pathway, if indeed exists, is not the predominant process to mediate the retro-Claisen reaction.
In conclusion, we have developed a new convenient method for C–C bond cleavage of 1,3-diketone compounds using tropylium tetrafluoroborate as an organic Lewis acid promoter. A wide range of carboxylic acid, ester and amide products were efficiently obtained in batch or flow using this approach with water, alcohols and amines, respectively. Replacement of these solvolytic reagents with mercaptans led to the formation of a range of new alkenyl thioethers.
The authors thank the Australian Research Council (grant DE150100517) for financial support. MAH thanks the Iraqi HCED for sponsoring his PhD scholarship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Jukic, D. Sterk and Z. Casar, Curr. Org. Synth., 2012, 9, 488–512 CrossRef CAS.
- A. Kawata, K. Takata, Y. Kuninobu and K. Takai, Angew. Chem., Int. Ed., 2007, 46, 7793–7795 CrossRef CAS PubMed.
- H. Liu, M. Feng and X. Jiang, Chem. – Asian J., 2014, 9, 3360–3389 CrossRef CAS PubMed.
- F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613–8661 CrossRef CAS PubMed.
- Y. Kuninobu, A. Kawata, T. Noborio, S.-I. Yamamoto, T. Matsuki, K. Takata and K. Takai, Chem. – Asian J., 2010, 5, 941–945 CrossRef CAS PubMed.
- (a) C. B. Rao, D. C. Rao, D. C. Babu and Y. Venkateswarlu, Eur. J. Org. Chem., 2010, 2855–2859 CrossRef CAS; (b) S. Wang, Y. Yu, X. Chen, H. Zhu, P. Du, G. Liu, L. Lou, H. Li and W. Wang, Tetrahedron Lett., 2015, 56, 3093–3096 CrossRef CAS.
- A. J. Grenning and J. A. Tunge, J. Am. Chem. Soc., 2011, 133, 14785–14794 CrossRef CAS PubMed.
- R. Visbal, A. Laguna and M. C. Gimeno, Chem. – Eur. J., 2016, 22, 4189–4195 CrossRef CAS PubMed.
- (a) D. Yang, Y. Zhou, N. Xue and J. Qu, J. Org. Chem., 2013, 78, 4171–4176 CrossRef CAS PubMed; (b) F. Xie, F. Yan, M. Chen and M. Zhang, RSC Adv., 2014, 4, 29502–29508 RSC; (c) Y. Zhou, D. Yang, G. Luo, Y. Zhao, Y. Luo, N. Xue and J. Qu, Tetrahedron, 2014, 70, 4668–4674 CrossRef CAS; (d) G.-X. Cai, J. Wen, T.-T. Lai, D. Xie and C.-H. Zhou, Org. Biomol. Chem., 2016, 14, 2390–2394 RSC; (e) I. Deb and D. Seidel, Tetrahedron Lett., 2010, 51, 2945–2947 CrossRef CAS.
- Y. Zhu, L. Zhang and S. Luo, J. Am. Chem. Soc., 2016, 138, 3978–3981 CrossRef CAS PubMed.
- (a) G. Merling, Chem. Ber., 1891, 24, 3108–3126 CrossRef; (b) W. Von, E. Doering and L. H. Knox, J. Am. Chem. Soc., 1954, 76, 3203–3206 CrossRef; (c) W. Von, E. Doering and L. H. Knox, J. Am. Chem. Soc., 1957, 79, 352–356 CrossRef.
- (a) R. V. Williams, W. D. Edwards, P. Zhang, D. J. Berg and R. H. Mitchell, J. Am. Chem. Soc., 2012, 134, 16742–16752 CrossRef CAS PubMed; (b) D. J. M. Lyons, R. D. Crocker, M. Blümel and T. V. Nguyen, Angew. Chem., Int. Ed., 2017, 56, 1466–1484 CrossRef CAS PubMed.
- (a) T. V. Nguyen and A. Bekensir, Org. Lett., 2014, 16, 1720–1723 CrossRef CAS PubMed; (b) T. V. Nguyen and M. Hall, Tetrahedron Lett., 2014, 55, 6895–6898 CrossRef CAS; (c) T. V. Nguyen and D. J. M. Lyons, Chem. Commun., 2015, 51, 3131–3134 RSC; (d) D. J. M. Lyons, R. D. Crocker, D. Enders and T. V. Nguyen, Green Chem., 2017, 3993–3996 RSC; (e) D. Lyons, R. Crocker and V. Nguyen Thanh, Chem. – Eur. J., 2018, 24, 10959–10965 CrossRef CAS PubMed; (f) G. Oss, S. D. de Vos, K. N. H. Luc, J. B. Harper and T. V. Nguyen, J. Org. Chem., 2018, 83, 1000–1010 CrossRef CAS PubMed; (g) G. Oss, J. Ho and V. Nguyen Thanh, Eur. J. Org. Chem., 2018, 3974–3981 CrossRef CAS; (h) U. P. N. Tran, G. Oss, D. P. Pace, J. Ho and T. V. Nguyen, Chem. Sci., 2018, 9, 5145–5151 RSC.
- (a) D. Enders and T. V. Nguyen, Tetrahedron Lett., 2012, 53, 2091–2095 CrossRef CAS; (b) M. Blumel, R. D. Crocker, J. B. Harper, D. Enders and T. V. Nguyen, Chem. Commun., 2016, 52, 7958–7961 RSC; (c) M. Blümel, J.-M. Noy, D. Enders, M. H. Stenzel and T. V. Nguyen, Org. Lett., 2016, 18, 2208–2211 CrossRef PubMed.
- (a) G. Shen, H. Zhou, P. Du, S. Liu, K. Zou and Y. Uozumi, RSC Adv., 2015, 5, 85646–85651 RSC; (b) Q. Xing, P. Li, H. Lv, R. Lang, C. Xia and F. Li, Chem. Commun., 2014, 50, 12181–12184 RSC.
- See the ESI† for more details.
- Diketone 2b predominantly formed enamine adducts with amine substrates, see the ESI† page S18 for more details. Diketone 2c only formed enamine adducts with primary amines.
- Reactions between acyclic 1,3-diketone substrates (dibenzoylmethane, benzoylacetone and 1-benzoyl-3,3,3-trifluoroacetone) and mercaptans (cyclohexyl thiol and benzyl thiol) did not work, giving mainly starting materials back and a mixture of unidentifiable minor products.
- (a) G. Rosenkranz, S. Kaufmann and J. Romo, J. Am. Chem. Soc., 1949, 71, 3689–3694 CrossRef CAS; (b) A. F. Hirsch, Chem. Phys. Lipids, 1976, 17, 399–401 CrossRef CAS; (c) A. Kondoh, K. Takami, H. Yorimitsu and K. Oshima, J. Org. Chem., 2005, 70, 6468–6473 CrossRef CAS PubMed; (d) I. Fabre, T. Poisson, X. Pannecoucke, I. Gillaizeau, I. Ciofini and L. Grimaud, Catal. Sci. Technol., 2017, 7, 1921–1927 RSC.
- L. Jeanne-Julien, E. Astier, R. Lai-Kuen, G. Genta-Jouve and E. Roulland, Org. Lett., 2018, 20, 1430–1434 CrossRef CAS PubMed.
- P. Villuendas, S. Ruiz, P. Vidossich, A. Lledós and E. P. Urriolabeitia, Chem. – Eur. J., 2018, 24, 13124–13135 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, analytical data and NMR spectra are provided. See DOI: 10.1039/c8cc07329e |
| This journal is © The Royal Society of Chemistry 2018 |