
Near-field infrared nanospectroscopy and super-resolution fluorescence microscopy enable complementary nanoscale analyses of lymphocyte nuclei†
Godwin C.
Ajaezi
 ab,
Max
Eisele
c,
Fabio
Contu
de,
Sadhana
Lal
fg,
Aline
Rangel-Pozzo
d,
Sabine
Mai
dfg and
Kathleen M.
Gough
*ab
ab,
Max
Eisele
c,
Fabio
Contu
de,
Sadhana
Lal
fg,
Aline
Rangel-Pozzo
d,
Sabine
Mai
dfg and
Kathleen M.
Gough
*ab
aChemistry, University of Manitoba, Winnipeg, MB, Canada. E-mail: Kathleen.Gough@umanitoba.ca
bBiomedical Engineering Program, University of Manitoba, Winnipeg, MB, Canada
cNeaspec GmbH, Bunsenstrasse 5, 82512 Martinsried, Germany
dPhysiology and Pathophysiology, University of Manitoba, Winnipeg, MB, Canada
eDepartment of Biomedical Sciences, Unit of Biology and Genetics, University of Cagliari, Italy
fGenomic Centre for Cancer Research and Diagnosis, Winnipeg, MB, Canada
gCell Biology, Research Institute of Oncology and Hematology, CancerCare Manitoba, Winnipeg, MB, Canada
First published on 11th October 2018
Abstract
Recent super-resolution fluorescence microscopy (3D-Structured Illumination Microscopy, 3D-SIM) studies have revealed significantly altered nuclear organization between normal lymphocyte nuclei and those of classical Hodgkin's Lymphoma. Similar changes have been found in Multiple Myeloma (MM) nuclei, as well as in a premalignant condition, Monoclonal Gammopathy of Unknown Significance (MGUS). Using 3D-SIM, an increase in DNA-poor and DNA-free voids was evident in reconstructed 3D-SIM images of diseased nuclei at 40 nm pixel resolution (x,y: 40 nm, z: 80 nm). At best, far-field FTIR imaging yields spatially resolved images at ∼500 nm spatial resolution; however, near-field infrared imaging breaks the diffraction limit at a scale comparable to that of 3D-SIM, providing details on the order of 30 nm spatial resolution. We report here the first near-field IR imaging of lymphocyte nuclei, and far-field IR imaging results of whole lymphocytes and nuclei from normal human blood. Cells and nuclei were mounted on infrared-compatible substrates, including CaF2, undoped silicon wafers, and gold-coated silicon wafers. Thermal source far-field FTIR images were obtained with an Agilent-Cary 620 microscope, 15× objective, 0.62 NA and 64 × 64 array Focal Plane Array detector (University of Manitoba), or with a similar microscope equipped with both 15× and 25× (0.81 NA) objectives, 128 × 128 FPA and either thermal source or synchrotron source (single beam) infrared light at the Advanced Light Source (ALS), LBNL, Berkeley CA. Near-field IR spectra were acquired at the ALS, on the in-house SINS equipment, as well as with a Neaspec system, both illuminated with synchrotron light. Finally, some near-field IR spectra and images were acquired at Neaspec GmbH, Germany. Far-field IR spectra of normal cells and nuclei showed the characteristic bands of DNA and proteins. Near-field IR spectra of nuclei showed variations in bands assigned to protein and nucleic acids including single and double-stranded DNA. Near-field IR images of nuclei enabled visualization of protein and DNA distribution in spatially-resolved chromosome territories and nuclear pores.
Introduction
Conventional and super resolution fluorescence (SRF) imaging are important tools in the analysis of normal and diseased cells. With the advent of SRF imaging, the architecture of the nucleus, in particular that of nuclear DNA organization and of chromosome territories (CTs) in the interphase nucleus, were studied for the first time at a spatial resolution of 40 nm.1–5 An increase in DNA-free or DNA-poor spaces was identified in the nuclei of classical Hodgkin's Lymphoma, both in the mononucleated Hodgkin (H) cells and in the bi- and multi-nucleated Reed-Sternberg (RS) cells when compared to normal (NL) lymphocytes. Similar DNA-poor spaces have since been identified in multiple myeloma (MM) cells and their precursor cells: monoclonal gammopathy of undetermined significance (MGUS),3 as well as in the nuclei of buccal cells in Alzheimer's Disease.5 The biological significance of these spaces may relate to the state/stage and aggressiveness of the disease. For example, MGUS has fewer DNA-poor spaces than MM and both have more than control lymphocytes.3 In MM, the organization of CTs was examined comparatively using conventional 3D microscopy (with the Abbe limit of 200 nm) and 3D-SIM. Shared and distinct CTs were identified.4 The formation of DNA-poor spaces may be a general step in the development of many diseases, but the detailed changes may be disease-specific. The analysis of these DNA-poor and DNA-free regions is limited by the lack of tools for detecting their biochemical composition at a nm length scale.Infrared spectroscopy has been a mainstay for chemical analysis for many decades6 but the rich chemical information delivered by this technique has been restricted to diffraction-limited spectrochemical images, on the order of a micron, at best. Near-field infrared techniques have recently brought the spatial resolution capability of this technique into the same length scale as SRF.7–12
We present here the first scattering Scanning Nearfield Optical Microscopy (sSNOM) infrared study of NL nuclei as the first step in a larger study of normal and diseased nuclei. Sample preparation protocols have been devised in order to obtain bare nuclei that are thin enough for true nanoscale analysis.
Experimental
Materials
Peripheral blood was obtained from healthy donors who signed informed consent according to the institutional approval of University of Manitoba's Health Research Ethics Board Protocol (#HS14085(H2011:336)).The Hodgkin's derived cell line HDLM-2 was grown in RPMI-1640 medium, supplemented with 20% fetal bovine serum (FBS), 1% L-glutamine, and 1% penicillin–streptomycin (Invitrogen/Gibco, Burlington, Canada). Cells were incubated at 37 °C with 5% CO2 in a humidified atmosphere, washed with PBS and gently placed onto poly-L-lysine coated slides.
Immunohistochemistry for the upstream binding factor (UBF) in NL and HL-derived cell line HDLM-2 was performed using primary UBF antibody (ab) (rabbit polyclonal, H-300, sc-9131, Santa Cruz, Dallas, USA) and secondary Goat anti-rabbit Alexa 488 ab (Molecular Probes, Eugene, USA) with a dilution of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :60 and 1:500 respectively in 4% BSA/4 × SSC blocking solution.
:60 and 1:500 respectively in 4% BSA/4 × SSC blocking solution.
Cells underwent fixation in 3.7% formaldehyde/1 × PBS and permeabilization with 0.1% Triton X-100, followed by three 1× PBS washes, with shaking at room temperature (RT). Incubation with primary ab was performed for 45 minutes at RT and followed by three 1× PBS washes, with shaking at RT. Incubation with secondary ab was performed for 30 minutes at RT and followed by three 1× PBS washes with shaking at RT. Cells were counterstained with 50 μl of 1 μg mL−1 of DAPI (4′,6-diamidino-2-phenylindole) for 5 min at RT. A drop of Vectashield (Vector Labs, Burlingame, CA) was used to embed the slides to prevent photo-bleaching.
To obtain the bare lymphocyte nuclei, a washed cell pellet was treated with NP-40 in modified RSB buffer (10 mM Tris, pH 7.2, 10 mM NaCl, 3 mM MgCl2; 1:2500). The resuspended cells were homogenized using a tissue homogenizer (DWK Life Sciences Kimble, Kontes™ Dounce) and centrifuged (180g, 5 min). After resuspension in 1 mL NS, a 70 μl aliquot of stripped nuclei was cytofuged onto an IR substrate (200 g, 5 min).
Methods
Some cells measured at BL 5.4 were then examined with a Neaspec NanoFTIR illuminated with synchrotron radiation at BL 2.4, ALS. The second order amplitude and phase back scattered light were recorded by co-addition of 600 scans for both background and sample. Post-processing of data was carried out with neaPLOTTER software (Neaspec, Germany) to obtain absorption spectra.
Results and discussion
Our motivation for this work is to establish a robust methodology for near-field infrared imaging of nuclei, in order to compare normal and diseased cells. As has been shown through SRF, normal nuclei and those of diseased nuclei display distinctly different nuclear organization. This was established using quantitative imaging of telomeres, centromeres, chromosomes and their orientation.1–5,13–19 These data indicate that the nuclear organization is significantly altered in diseased cells and may serve as a structural biomarker for disease states and progression.20–22SRF of NL and HL nuclei
Superresolution fluorescence images show the nuclear organization of DNA of a normal interphase nucleus, and disturbed architecture in H and RS nuclei (Fig. 1). The distinct organization of the genome and the presence vs. absence of DNA-poor spaces in the form of increased interchromatin and circular DNA-poor spaces in the malignant cells are depicted in Fig. 1A (normal lymphocyte) and 1B–D (Hodgkin's lymphoma), respectively. The nucleolar phosphoprotein UBF is located in the nucleolus; however, DNA-poor spaces of H and RS cells do not contain UBF.1 Green-filled arrow heads indicate nucleoli containing UBF; black-filled arrowheads indicate DNA-poor spaces lacking UBF. As expected, the normal nucleus (Fig. 1A) is much smaller than the H and RS.1 The nucleoli are also much smaller; here, only one small nucleolus is visible. The large H and RS cells contain multiple large nucleoli and DNA-poor circular regions; only the more prominent examples are denoted (Fig. 1B–D). Videos of the concatenated z-stack images show normal nucleoli with UBF and DNA-poor regions throughout the full volume of NL, H and RS nuclei (ESI, video 1 NL, video 2 H, video 3 RS-binucleate and video 4 RS-tetranucleate†). A similar H cell stained with nucleolin instead of UBF provides an alternative visualization of these DNA-poor regions (ESI Fig. 1, video 5†).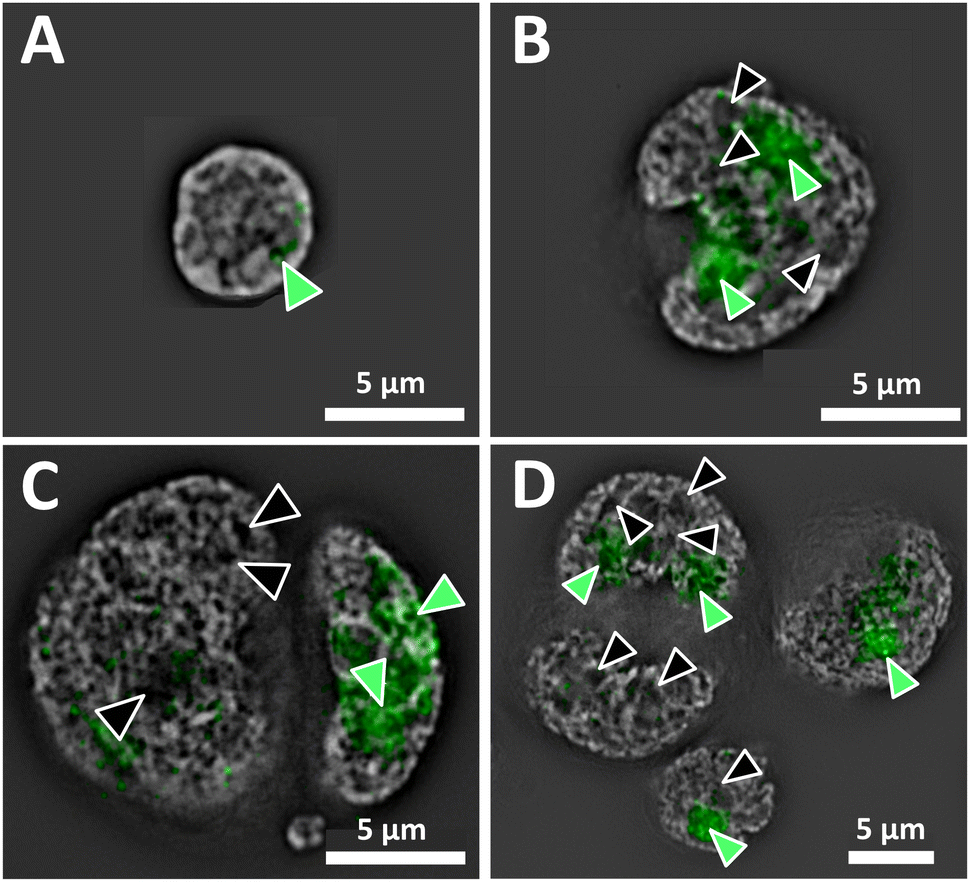 | ||
| Fig. 1 Single z-plane widefield SIM images of nuclei stained with DAPI (grayscale) and UBF (green) from (A) normal isolated peripheral-blood lymphocyte (NL), (B) Hodgkin (H) cell, (C) bi-nuclear and (D) tetra-nuclear Reed-Sternberg (RS) cells from the cHL derived cell line HDLM-2. Green-filled arrowheads indicate nucleoli containing UBF; black-filled arrowheads indicate DNA-poor spaces not containing UBF. | ||
AFM of whole cells and nuclei for near-field IR
Initial preparations of whole lymphocytes were found to be inadequate for NFIR spectroscopy. In early tests, 2 to 4 μL aliquots of resuspended lymphocytes were deposited on AuSi wafers and air-dried (Fig. 2A). AFM measurements showed that these cells beaded up on the surface and were at least 2 μm high; they proved to be too thick for good NFIR. Not only was the signal weak, but lensing of the synchrotron illumination created spectral artefacts (data not shown). Subsequently, suspensions of whole cells were cytofuged onto the substrate, yielding a maximum height of about 1 μm for deposited whole cells (Fig. 2B). Synchrotron near-field spectra (SINS) could be obtained from points around the edges, but the signal was still poor and dominated by cytoplasm (data not shown). Finally, stripped nuclei were prepared and cytofuged onto AuSi (Fig. 2C). Relative height profiles for each preparation are shown in Fig. 2D. The cytofuged stripped nuclei were ideal for NFIR measurements, particularly in thinner regions where height dropped to ∼30 nm.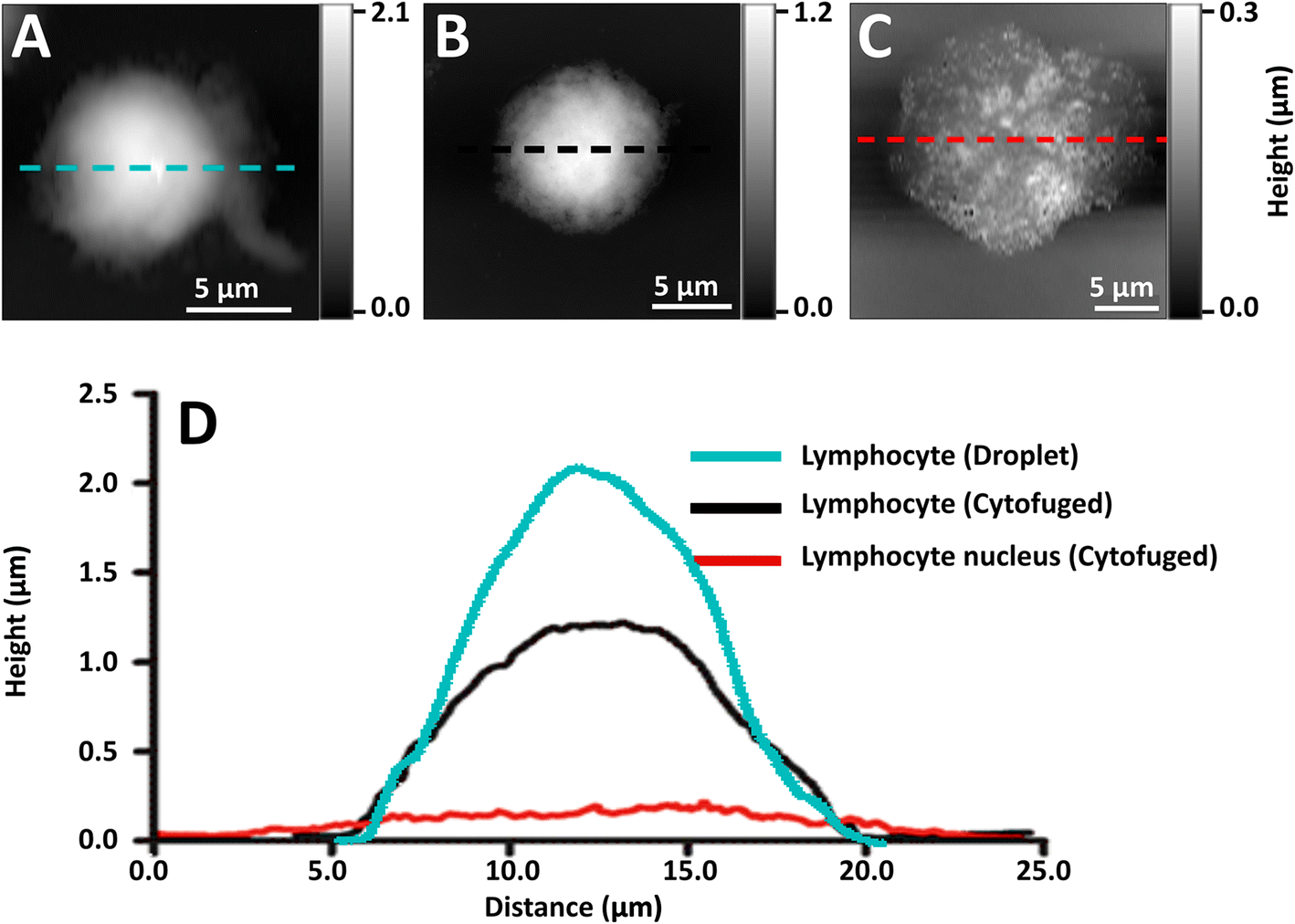 | ||
| Fig. 2 AFM topographic images of (A) whole NL from droplet preparation, (B) whole NL prepared by cyto-centrifugation of cell suspension, (C) stripped NL nucleus cytofuged onto AuSi IR substrate, and (D) corresponding AFM profiles of the whole lymphocytes and nucleus. | ||
Far field FTIR spectra
Far field FTIR (FFIR) spectrochemical FPA images of whole lymphocytes and nuclei on AuSi wafers were taken on cells and nuclei prepared for near-field IR. Correlated spectra for nuclei on AuSi wafers were not feasible (ESI, Fig. 2†) as the well-known Electric Field Standing Wave effect was significantly pronounced for these thin targets imaged in transflection mode off AuSi wafers.23,24 Nuclei prepared by cytofuging onto 1 mm thick CaF2 windows offered greater opportunity for comparison but the samples were too thin to give adequate signal except when multiple nuclei were overlaid (ESI, Fig. 3†), and they could not be imaged with NFIR. The ∼300 nm thick individual nuclei were identifiable by the broad OH–NH stretch band, but little detail could be discerned in the 1800–900 cm−1 region. However, if it is possible to define differences between normal and diseased nuclei with NFIR, we can envision the practical application of FFIR using a more concentrated sample preparation, with classification guided by NFIR analyses.NFIR of nuclei
Near-field IR spectra of nuclei were obtained in one of three ways: (1) illumination of AFM tip with synchrotron source light at BL 5.4 (SINS), (2) similar data collection but with Neaspec instrument at BL 2.4, both at the ALS; and (3) with mid-IR laser sources, Neaspec GmbH, Germany. Finally, single wavelength images were obtained with mid-IR laser sources, Neaspec GmbH, Germany.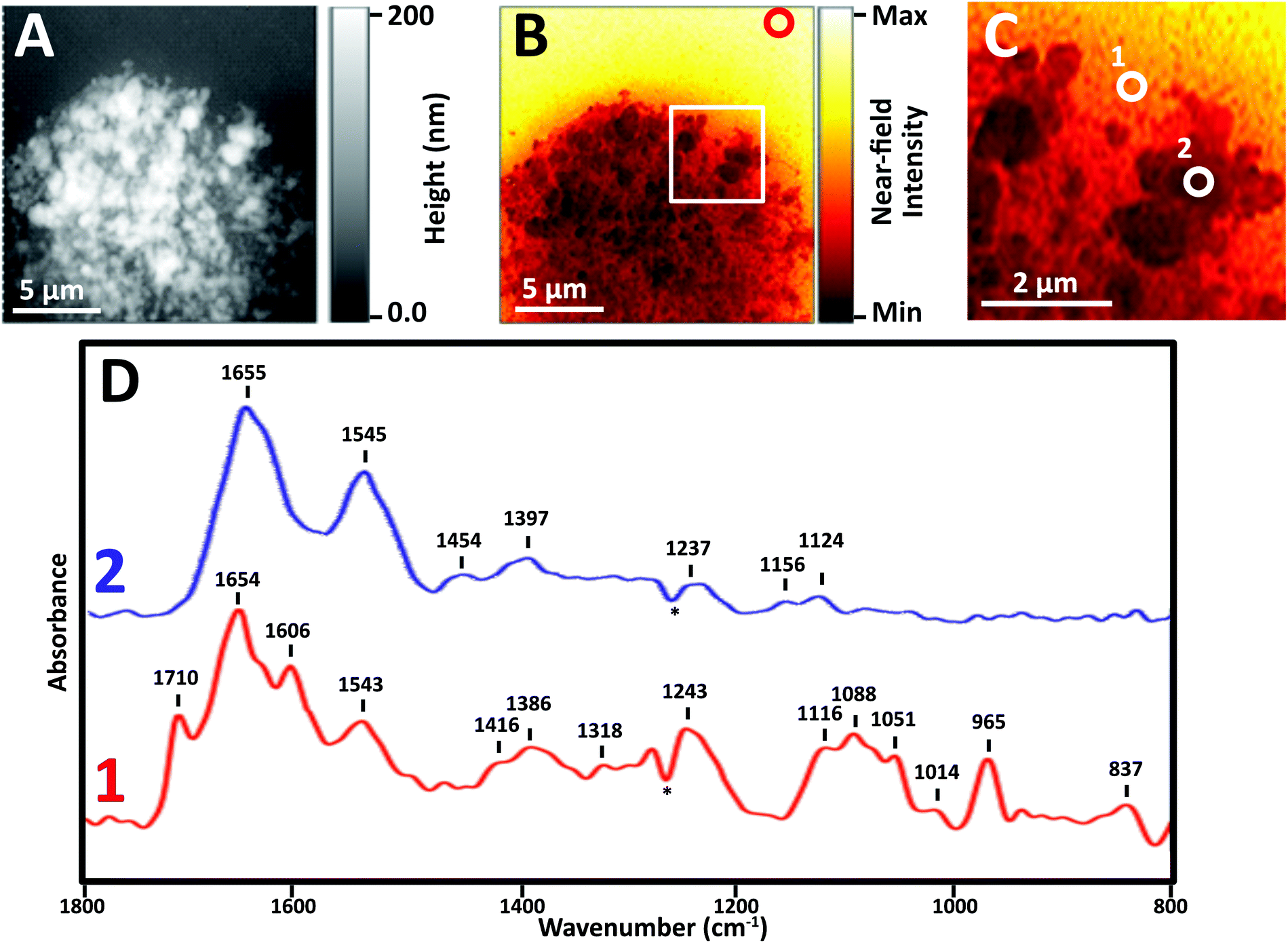 | ||
| Fig. 3 AFM topography and Near-field IR of stripped NL nucleus cytofuged onto AuSi wafer. (A) AFM topography shown in grey scale; (B) false-colour image showing total integrated broad band infrared absorption; red circle shows location for background spectrum; (C) region in B enlarged for clarity to show locations for two spectral acquisitions. (D) Corresponding near-field IR spectra recorded with mid-IR lasers, at positions 1 and 2. | ||
Images revealed physically distinct structures within the nucleus (Fig. 3). From the topographic scan, the stripped nucleus exhibited regions with maximum height of about 0.17 μm (Fig. 3A), corresponding to regions with the densest infrared absorbance (Fig. 3B). The thickness at the periphery of the nucleus was far less than 1 μm; the appearance and density of thinner regions at the edge suggested that they corresponded to nuclear pores. The thicker, denser regions have the appearance of chromosome territories (CTs), which would be rich in protein and DNA.
In general, thinner locations were found to be the best locations for the collection of strong, clear infrared spectra (Fig. 3C), as can be seen in full spectra recorded at two positions: the thinner region, position 1, AFM height 30 nm, and Position 2, AFM height 120 nm (Fig. 3D). There are numerous compilations of nucleic acid infrared spectra and conformational marker bands for nucleic acids vary with pH, salt concentrations and hydration; the sharp strong bands seen in this sSNOM NFIR spectrum represent dehydrated nucleic acids in saline. Observed bands and likely assignments proposed here are based on a recent review of extensive studies of DNA-forms, Table 1.25
| Peak (cm−1) | Assignmenta |
|---|---|
| a Band assignments are based on analyses summarized in ref. 25. | |
| 837 | A-DNA, B-DNA |
| 965 | ν(C-O) ribose, B-DNA |
| 1014 | DNA, RNA, ν(C–O) ribose |
| 1051 | DNA, RNA, ν(C–O–C) ribose |
| 1088 | DNA, νsPO2− |
| 1116–1120 | δ(CH2), ν(CO), ν(C–C) ribose RNA |
| 1156–1160 | δ(CH2), ν(CO), ν(C–C) ribose RNA, protein |
| 1237, 1243 | ν asP02− |
| 1265 | Negative band due to PDMS, ref. 26 |
| 1386, 1397 | CH2, CH3 deformation |
| 1416 | A-DNA, B-DNA, Z-DNA deoxyribose |
| 1454 | δ(CH2), δ(CH3) |
| 1545 | Amide II |
| 1606 | ν(NH2), Adenine, DNA |
| 1655 | Amide I α-helix |
| 1710 |
ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), B-DNA. A-DNA base pair vibrations O), B-DNA. A-DNA base pair vibrations |
The sample height at position 1 was only 30 nm; the signal strength was greater and the spectrum showed dramatically clear DNA signatures corresponding to single (1116 cm−1) and double (1710 cm−1) stranded DNA (Table 1). Both components can be expected in nuclear pores regions, supporting the probable assignment as nuclear pore.
At position 2, AFM height 120 nm, the spectrum was dominated by Amide I and II bands at 1655 and 1545 cm−1, along with weaker bands typical of CH deformations (lipids, protein side chains) and phosphates at 1465, 1400 and 1235 cm−1. None of the characteristic nucleic acid bands were evident. In spite of the dense DNA present, the interphase CT is rich in proteins, so the spectrum can be assumed to be from CT. We note that the near-field intensity response was relatively low at position 2, probably owing to the thickness of >100 nm (vide infra).
A dip at 1265 cm−1, marked with an asterisk (*) appears in most near-field IR spectra and is due to tip contamination with PDMS26 from the original packaging. That signal is enhanced when the contaminated bare tip is close to clean gold, i.e., when the background spectrum is being acquired; hence, the final appearance in the processed sample spectra is a negative peak.
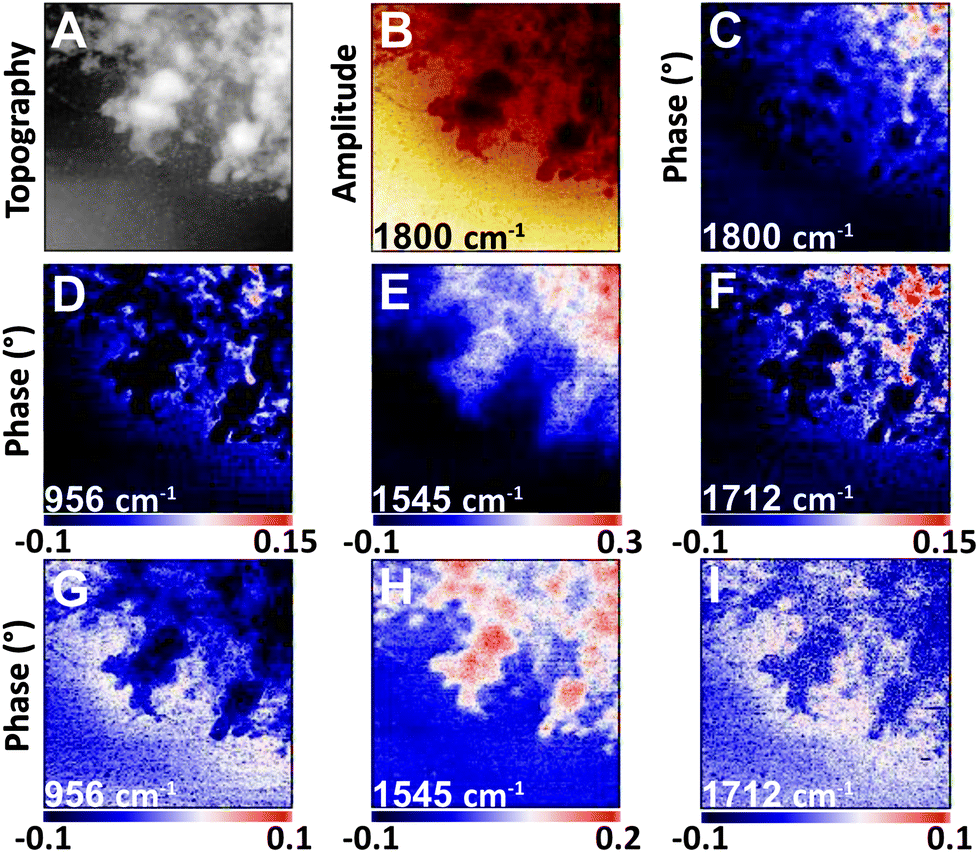 | ||
| Fig. 4 NFIR sSNOM single wavelength images of nucleus region from Fig. 3. Top row shows (A) AFM topography, (B) total integrated IR absorbance and (C) 2nd order phase shift under 1800 cm−1 illumination. Middle row (D–F) shows 2nd order phase shifts at given wavelengths; bottom row (G–I) shows same phase shifts corrected for thickness. | ||
The nuclear pores and dense chromatin regions are identifiable through the topography from the AFM (Fig. 4A) and the total integrated absorbance (Fig. 4B). The phase shift contains the majority of the near-field IR absorbance signal.7–12 A considerable phase shift is recorded in what should be a non-absorbing wavelength: 1800 cm−1, across the field of view (Fig. 4C). The magnitude of the phase shift diminishes as sample thickness decreases below 50 nm, probably owing to coupling between the gold-coated AFM tip and the gold coated substrate. The magnitude of the raw second order phase shift responses at 956 cm−1 (DNA, ribose), 1545 cm−1 (Amide II) and 1712 cm−1 (DNA) would be affected by this shift (Fig. 4D–F). This background distortion was subtracted (Fig. 4G–I) to allow proper visualization of the protein and DNA distributions. The thicker regions, ascribed to interphase chromatin region, are seen to be rich in protein, while the nuclear pores contain the greatest DNA signals.
000 Ohm cm resistance) were chosen over the more common doped silicon to avoid accidentally hitting a plasma frequency directly in the spectral range to be probed, as this would result in significant confounding absorption profiles. Spectra and single wavelength phase shift images acquired on the Neaspec instrument (Neaspec GmbH, Germany) are shown in Fig. 5.
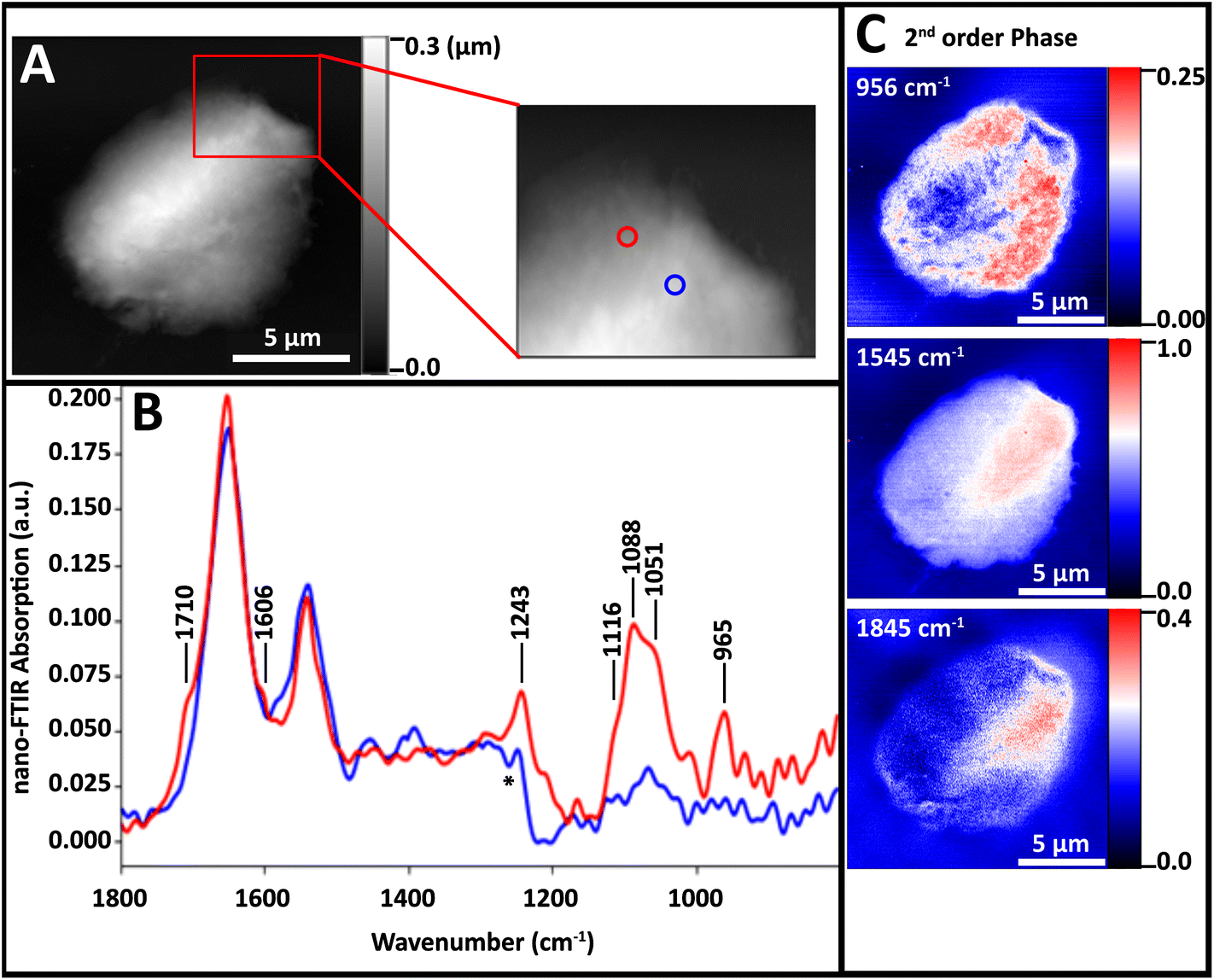 | ||
| Fig. 5 Spectra and single wavelength 2nd order phase shift images of a NL nucleus on undoped Si wafer. (A) AFM topography images and enlarged region of lymphocyte nucleus on undoped polished Si wafer. (B) Spectra acquired from points denoted by blue and red circles in A. (C) False colour images showing 2nd order phase shifts at 956, 1545 and 1845 cm−1. | ||
The nuclei did not lie as flat on this substrate as on AuSi; they remained slightly beaded up (Fig. 5A). Spectra from a thicker region (blue) and thinner region (red) were both dominated by protein Amide I and II bands (Fig. 5B). At the thinner location (blue spectrum), bands attributable to DNA are evident. The red spectrum in Fig. 5B includes bands at 1712, 1606, 1243, 1120–1050 and 960 cm−1 but these are much weaker than those in Fig. 3. That spectrum was acquired at a point where the 965 cm−1 band was close to maximum (Fig. 5C, top). The Amide II band seems to be strongest in the thick region (Fig. 5C, middle) but much of this is an artefact of sample thickness, since the increasing shift with increasing thickness increases persists even on the undoped Si wafer for thicknesses greater than 300 nm (Fig. 5C, bottom).
The DNA-poor circular regions are often located toward the periphery of the diseased nuclei.1 Our preparation method of cytoplasmic strip followed by cytofuge deposition delivers bare nuclei of generally appropriate thickness. We conclude that this preparation along with the AuSi substrate is to be preferred, as it allows us to exploit tip–substrate coupling and obtain maximum signal from regions of critical interest.
Variations in spectra obtained with NFIR
Many sSNOM near-field IR spectra were recorded from four whole lymphocytes and six stripped nuclei; for the latter, about 20 spots were sampled per nucleus, mainly at the target periphery. With optimization of the sample preparation methods, the quality of the spectra improved. Representative spectra of the 5 stripped nuclei for which data have been collected illustrate the frequently observed variations (Fig. 6).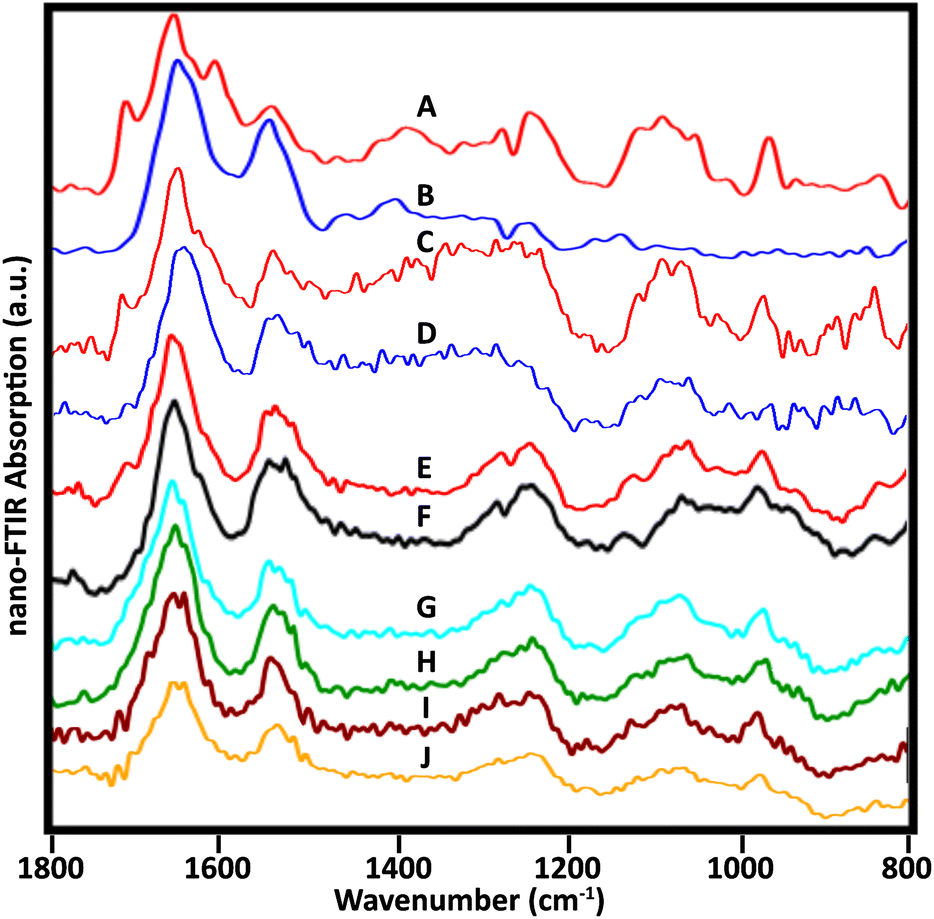 | ||
| Fig. 6 Near-field infrared spectra acquired from stripped nuclei of several NL. (A, B) Spectra as in Fig. 3, cell 1, recorded with mid-IR lasers, Neaspec, GmbH; (C, D) SINS spectra from similar locations in the same cell, BL 5.4, ALS. Spectra from three other cells recorded with the Neaspec instrument at BL 2.4, ALS: (E, F) cell 2; (G, H) cell 3; (I, J) cell 4. A dip at 1265 owing to PDMS is visible in A, B, E and F. | ||
Spectra from the same location in one nucleus were acquired at ALS, BL 5.4, with synchrotron illumination, and at Neaspec GmbH, with mid-IR lasers. The latter, shown in Fig. 3, with band assignments, are included here for comparison (Fig. 6A and B). The red spectra (Fig. 6A and C) are taken from the nuclear pore region; the blue spectra from chromatin-rich region (Fig. 6B and D). Importantly, both experiments yielded the same results, demonstrating an excellent correspondence between spectra obtained with the mid-IR lasers and with synchrotron illumination, though the latter are noisier. The DNA band at 1710 cm−1 is also detectable in a suspected nuclear pore region of another cell (Fig. 6E) but not at a denser location (Fig. 6F), just as for the first nucleus. Pairs of spectra from two other nuclei (Fig. 6G, H and I, J) were acquired in regions where thickness was 50–80 nm, using the Neaspec instrument at BL 2.4, ALS. While the 1710 cm−1 band was not resolved in any of these spectra, the broad band around 960 cm−1 is quite clear. A strong band in this region is associated with DNA, but could have contributions from non-DNA phosphate.25 These are the first such results and much more data must be collected to establish the variety of signatures in a statistically meaningful way. These first results show the enormous potential of the approach.
Conclusions
The spatial resolution of superresolution fluorescence is a close match with that achievable with sSNOM type near-field infrared spectroscopy. We have developed a satisfactory method for preparing normal whole lymphocytes and stripped nuclei for near-field IR imaging at 30 nm voxel resolution (x,y defined by AFM tip, z defined by thickness. The NFIR spectra of points within stripped nuclei showed characteristic absorbances from protein, single and double stranded DNA, as well as hydrocarbon, ribose and phosphate signatures. NFIR images of bare nuclei at select wavelengths showed differences in distribution ascribable to nuclear pores and chromosome territories of the interphase nucleus. Comparison of sSNOM NFIR spectroscopy of cells mounted onto gold-coated wafers and onto undoped silicon wafers revealed an advantage for the former, believed to be due to enhancement at thinner regions (<30 nm) due to tip–substrate dipole coupling. The combination of super resolution fluorescence imaging with near-field infrared spectroscopy and spectrochemical imaging allows unprecedented insight into the nanoscale architecture in normal lymphocyte nuclei. We anticipate that it will be similarly effective in diseased cells.Conflicts of interest
SM is a Director and Chair of the Clinical and Scientific Advisory Board of 3D Signatures Inc. There are no other potential conflicts to declare.Acknowledgements
The authors are grateful to Dr Hans Bechtel (ALS, LBNL, Berkeley) for assistance in the collection of near-field IR data and images; to Dr Christiaan H. Righolt (Vaccine and Drug Evaluation Centre, Department of Community Health Sciences, The University of Manitoba) for conducting the original SRF experiments; and to Mr Benoit Girouard (Department of Chemistry, The University of Manitoba) for assistance in creating the final figures. This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231. The authors acknowledge funding from the Natural Sciences and Engineering Research Council (NSERC) of Canada (KMG), CancerCare Manitoba (761036238) and the Canadian Institutes of Health Research (123379) (SM). GCA is grateful to the University of Manitoba for a UofM Graduate Fellowship and a Graduate Enhancement of Tri-council Stipend Award; FC is grateful to the Regione Autonoma della Sardegna for fellowship funding.References
- C. H. Righolt, A. Guffei, H. Knecht, I. T. Young, S. Stallinga, L. J. van Vliet and S. Mai, J. Cell. Biochem., 2014, 115, 1441–1448 CrossRef CAS PubMed.
- C. H. Righolt, H. Knecht and S. Mai, J. Cell Biochem., 2016, 117, 1633–1637 CrossRef CAS PubMed.
- C. Sathitruangsak, C. H. Righolt, L. Klewes, P. Tammur, T. Ilus, A. Tamm, M. Punab, A. Olujohungbe and S. Mai, J. Cell Biochem., 2015, 116, 704–710 CrossRef CAS PubMed.
- C. Sathitruangsak, C. H. Righolt, L. Klewes, D. Tung Chang, R. Kotb and S. Mai, Int. J. Cancer, 2017, 140, 400–410 CrossRef CAS PubMed.
- A. Garcia, D. Huang, A. Righolt, C. Righolt, M. C. Kalaw, S. Mathur, E. McAvoy, J. Anderson, A. Luedke, J. Itorralba and S. Mai, J. Cell. Physiol., 2017, 232, 2387–2395 CrossRef CAS PubMed.
- P. R. Griffiths and J. A. De Haseth, Fourier Transform Infrared Spectrometry, John Wiley & Sons, 2nd edn, 2007 Search PubMed.
- R. Hillenbrand, B. Knoll and F. Keilmann, J. Microsc., 2001, 202, 77–83 CrossRef CAS PubMed.
- F. Keilmann and R. Hillenbrand, Philos. Trans. R. Soc., A, 2004, 362, 787–806 CrossRef CAS PubMed.
- I. Amenabar, S. Poly, W. Nuansing, E. H. Hubrich, A. A. Govyadinov, F. Huth, R. Krutokhvostov, L. Zhang, M. Knez, J. Heberle, A. M. Bittner and R. Hillenbrand, Nat. Commun., 2013, 4, 1–9 Search PubMed.
- H. A. Bechtel, E. A. Muller, R. L. Olmon, M. C. Martin and M. B. Raschke, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7191–7196 CrossRef CAS PubMed.
- S. Mastel, A. A. Govyadinov, T. V. de Oliveira, I. Amenabar and R. Hillenbrand, Appl. Phys. Lett., 2015, 106, 023113 CrossRef.
- R. Wiens, C. R. Findlay, S. G. Baldwin, L. Kreplak, J. M. Lee, S. P. Veres and K. M. Gough, Faraday Discuss., 2016, 187, 555–573 RSC.
- S. F. Louis, B. J. Vermolen, Y. Garini, I. T. Young, A. Guffei, Z. Lichtensztejn, F. Kuttler, T. C. Y. Chuang, S. Moshir, V. Mougey, A. Y. C. Chuang, P. Donald Kerr, T. Fest, P. Boukamp and S. Mai, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 9613–9618 CrossRef CAS PubMed.
- B. J. Vermolen, Y. Garini, S. Mai, V. Mougey, T. Fest, T. C.-Y. Chuang, A. Y.-C. Chuang, L. Wark and I. T. Young, Cytometry, Part A, 2005, 67, 144–150 CrossRef CAS PubMed.
- R. Sarkar, A. Guffei, B. J. Vermolen, Y. Garini and S. Mai, Cytometry, Part A, 2007, 71, 386–392 CrossRef PubMed.
- C. H. Righolt, V. Raz, B. J. Vermolen, R. W. Dirks, H. J. Tanke and I. T. Young, Int. J. Mol. Imaging, 2011, 2011, 723283 Search PubMed.
- S. Mathur, A. Glogowska, E. McAvoy, C. Righolt, J. Rutherford, C. Willing, U. Banik, M. Ruthirakuhan, S. Mai and A. Garcia, J. Alzheimer's Dis., 2014, 39, 35–48 Search PubMed.
- A. Garcia, S. Mathur, M. C. Kalaw, E. McAvoy, J. Anderson, A. Luedke, J. Itorralba and S. Mai, J. Alzheimer's Dis., 2017, 58, 139–145 Search PubMed.
- A.-K. Schmälter, C. H. Righolt, A. Kuzyk and S. Mai, Transl. Oncol., 2015, 8, 417–423 CrossRef PubMed.
- H. Knecht, N. Kongruttanachok, B. Sawan, J. Brossard, S. Prévost, E. Turcotte, Z. Lichtensztejn, D. Lichtensztejn and S. Mai, Transl. Oncol., 2012, 5, 269–277 CrossRef PubMed.
- M. Gadji, A. J. Adebayo Awe, P. Rodrigues, R. Kumar, D. S. Houston, L. Klewes, T. N. Dièye, E. M. Rego, R. F. Passetto, F. M. de Oliveira and S. Mai, Clin. Cancer Res., 2012, 18, 3293–3304 CrossRef CAS PubMed.
- A. Kuzyk, J. Gartner and S. Mai, Transl. Oncol., 2016, 9, 348–356 CrossRef PubMed.
- P. Bassan, A. Sachdeva, A. Kohler, C. Hughes, A. Henderson, J. Boyle, J. H. Shanks, M. Brown, N. W. Clarke and P. Gardner, Analyst, 2012, 137, 1370–1377 RSC.
- T. G. Mayerhöfer and J. Popp, Spectrochim. Acta, Part A, 2018, 191, 283–289 CrossRef PubMed.
- B. R. Wood, Chem. Soc. Rev., 2016, 45, 1980–1998 RSC.
- D. Cai, A. Neyer, R. Kuckuk and H. M. Heise, J. Mol. Struct., 2010, 976, 274–281 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8an01341a |
| This journal is © The Royal Society of Chemistry 2018 |