DOI:
10.1039/C7QM00289K
(Research Article)
Mater. Chem. Front., 2017,
1, 2559-2568
A new class of gold(III) complexes with saturated poly(benzyl ether) dendrons for solution-processable blue-green-emitting organic light-emitting devices
Received
28th June 2017
, Accepted 31st August 2017
First published on 19th September 2017
Abstract
A new class of gold(III) complexes with saturated poly(benzyl ether) dendrons has been synthesized, characterized, and applied as phosphorescent dopants in the fabrication of solution-processable organic light-emitting devices (OLEDs). The incorporation of the poly(benzyl ether) dendron into the cyclometalated gold(III) center can effectively perturb the packing of the molecules and minimizes the intermolecular interactions for suppressing excimeric emission. In addition, blue-green-emitting OLEDs can be realized, representing the first example of a blue-green-emitting device based on saturated dendrimer containing alkynylgold(III) complexes. Taking advantage of the diverse and well-developed synthetic routes for saturated dendrons, this work provides a simple means to develop a new class of gold(III) complexes emitting in the blue region.
Introduction
Organic light-emitting devices (OLEDs) have received enormous attention due to their potential applications in large-sized, flexible panel displays and solid-state lighting systems.1,2 Extensive studies on the development of phosphorescent metal complexes such as iridium(III)3–7 and platinum(II)8–12 systems, and their applications for the fabrication of highly efficient OLEDs have been reported.13–18 The incorporation of a heavy metal atom can facilitate strong spin orbit coupling, and harvest both singlet and triplet excited states. This yields a significant improvement in the internal quantum efficiency of up to unity.19,20 Nowadays, high performance iridium(III) and platinum(II) complexes with emission spanning from blue to red have been successfully developed by perturbing the nature of their excited states via systematic modification of cyclometalated or auxiliary ligands.16 On the other hand, the development of phosphorescent gold(III) emitters with an isoelectronic configuration and isostructural square-planar geometry as platinum(II) complexes is relatively less explored. It is believed that the presence of low-energy d–d ligand field excited states would quench the luminescence excited state through thermal equilibration or energy transfer. This limitation has been overcome through the incorporation of strong σ-donating ligands on the gold(III) center, which would render the metal center less electrophilic as well as result in the enhancement of the luminescence properties by increasing the energy of the d–d states.21 Since then, a number of green- to red-emitting gold(III) complexes with bidentate, tridentate or tetradentate ligands have been reported.22–31 However, blue-emitting OLEDs based on gold(III) complexes have scarcely been demonstrated with only one example in the literature reported by us very recently,32 possibly due to the presence of the dimeric or excimeric emission arising from the π–π stacking of the cyclometalated ligands in the solid-state thin films, that inevitably leads to a bathochromic shift of the emission energies. Such observations have been commonly observed in the d8 square-planar metal complexes.22,33–37
Recent demonstrations on the design and synthesis of phosphorescent dendrimers have opened up a new and interesting direction for the preparation of solution-processable phosphorescent OLEDs (PHOLEDs).38–42 For instance, incorporation of conjugated carbazole moieties has been shown to generate highly efficient phosphorescent gold(III) dendritic complexes.35 In such dendrimers, the tert-butyl surface groups can effectively enhance the solubility. Meanwhile, higher generation dendrimers can minimize the intermolecular interactions between the molecules that could eventually lead to emission quenching or triplet–triplet annihilation and reduce the bathochromic shift in emission.35,43 Respectable device performance with maximum current and power efficiencies of 24.0 cd A−1 (corresponding to an external quantum efficiency (EQE) of 7.8%) and 14.5 lm W−1 has been achieved.35 In order to fine-tune the emission color into the blue region, here we report a novel class of saturated poly(benzyl ether) dendritic alkynylgold(III) complexes 1–3 and their application studies in the fabrication of PHOLEDs. In particular, saturated dendrimers can be readily prepared by the copper(I)-catalysed click reaction with reasonable yield. Taking advantages of the diverse and well-developed synthetic routes of the saturated poly(benzyl ether) dendrimers, this work opens up a simple method for the design and synthesis of a new class of saturated poly(benzyl ether) dendritic gold(III) complexes. Unlike the previously reported carbazole-based gold(III) dendrimers which exhibit high electroluminescence (EL) performance in the green to yellow region, bluish-green-emitting solution-processable OLEDs have been realized for the first time. In particular, this work represents the first report on a new class of alkynylgold(III) complexes with saturated dendrimers.
Results and discussion
Synthesis and characterization
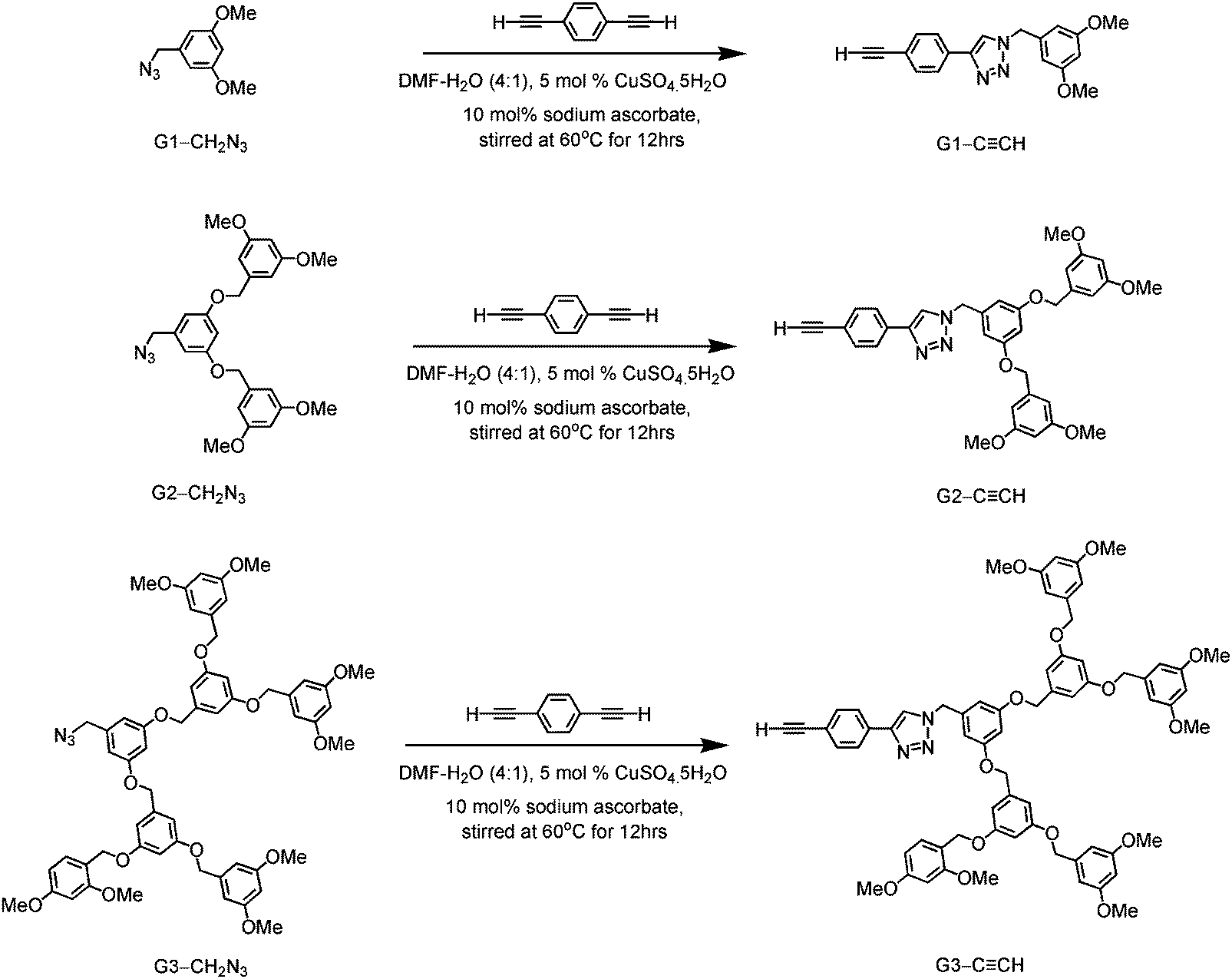
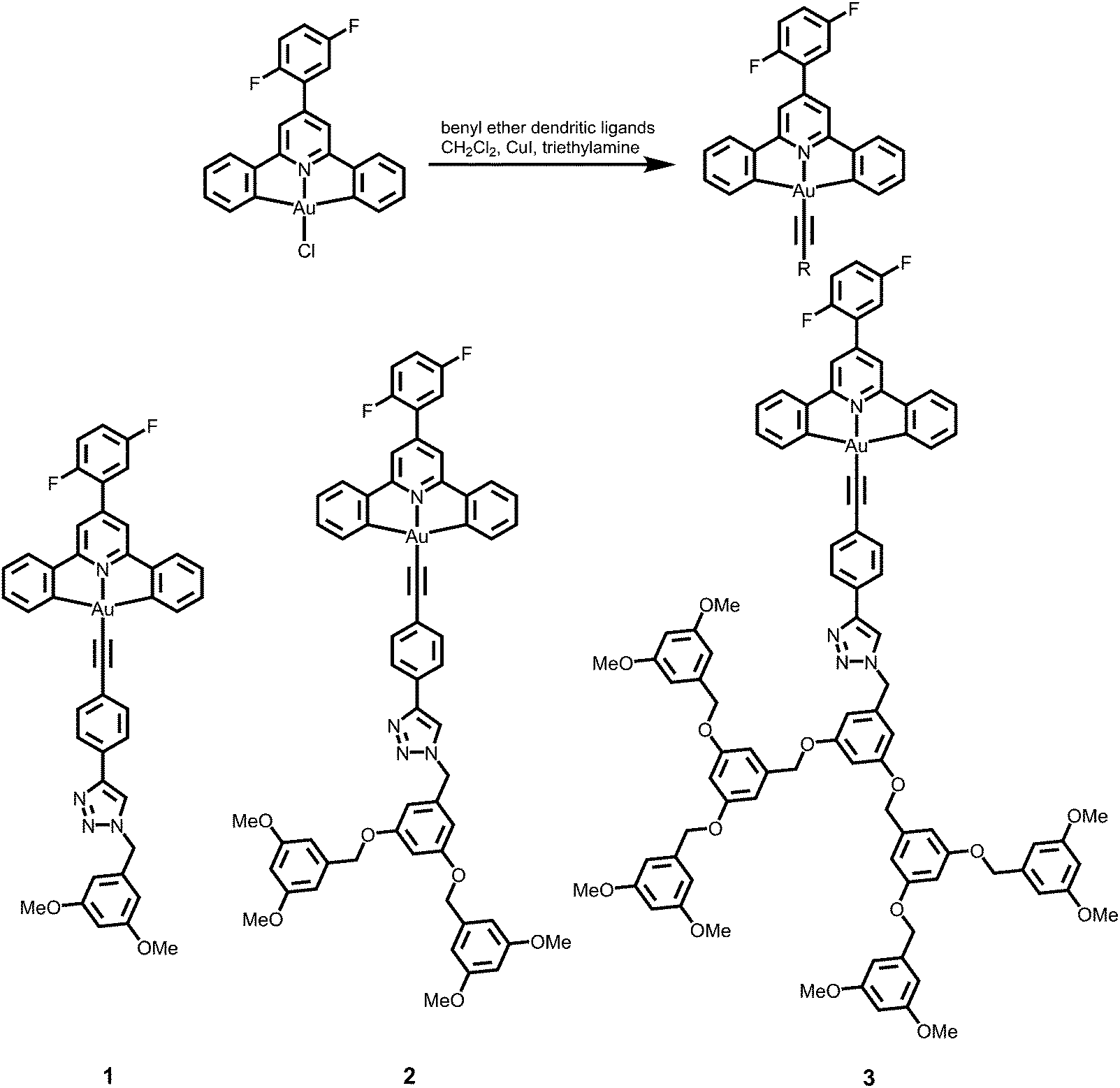
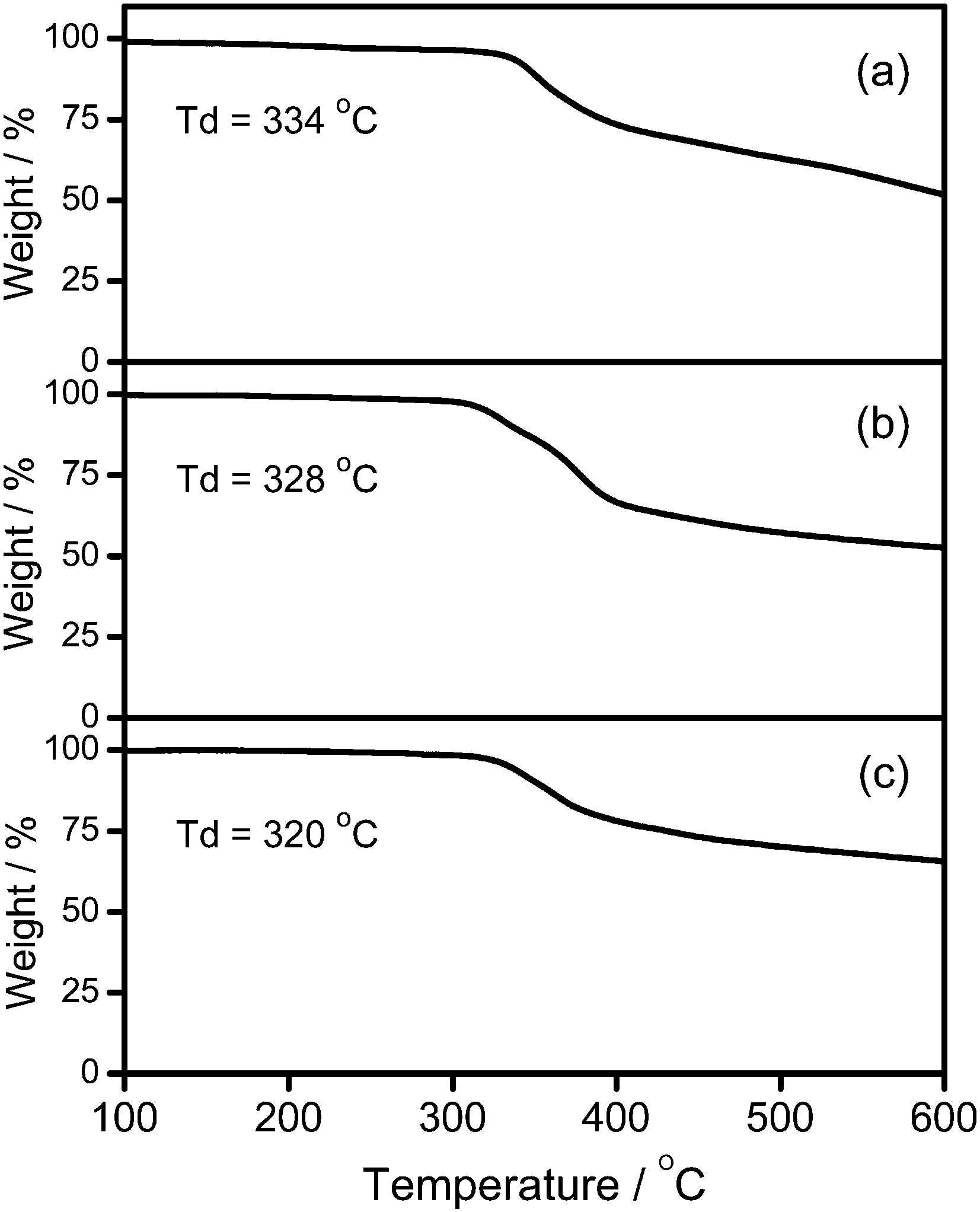
A new class of solution-processable phosphorescent dendritic alkynylgold(III) complexes 1–3 has been successfully synthesized by the reaction of [Au(2,5-F2C6H3–C^N^C)Cl] with poly(benzyl ether) dendritic ligands (G1-C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH, G2-CCH and G3-CCH) in the presence of a catalytic amount of copper(I) iodide in triethylamine and degassed dichloromethane (Schemes 1 and 2). The targeted complexes are then purified by column chromatography. Subsequent diffusion of diethyl ether vapor into concentrated dichloromethane solutions of the complexes gives 1–3 as pale yellow solids. The incorporation of the dendritic ligands into the gold(III) center has rendered the products soluble in common organic solvents such as chloroform and dichloromethane. The identities of complexes 1–3 have been confirmed by 1H NMR spectroscopy, FAB-mass spectrometry, IR spectroscopy, and satisfactory elemental analyses. Weak absorption bands at ca. 2146–2150 cm−1 appear in their IR spectra, which are in accordance with the characteristic of the ν(CC) stretching frequency of a terminal alkynyl-metal coordination mode. These new poly(benzyl ether) dendrimers have been isolated as thermally stable solids, as confirmed by thermogravimetric analysis (TGA) study. The TGA traces of all the complexes are shown in Fig. 1. All the complexes feature high thermal stability with Td > 320 °C under a nitrogen atmosphere.
CH, G2-CCH and G3-CCH) in the presence of a catalytic amount of copper(I) iodide in triethylamine and degassed dichloromethane (Schemes 1 and 2). The targeted complexes are then purified by column chromatography. Subsequent diffusion of diethyl ether vapor into concentrated dichloromethane solutions of the complexes gives 1–3 as pale yellow solids. The incorporation of the dendritic ligands into the gold(III) center has rendered the products soluble in common organic solvents such as chloroform and dichloromethane. The identities of complexes 1–3 have been confirmed by 1H NMR spectroscopy, FAB-mass spectrometry, IR spectroscopy, and satisfactory elemental analyses. Weak absorption bands at ca. 2146–2150 cm−1 appear in their IR spectra, which are in accordance with the characteristic of the ν(CC) stretching frequency of a terminal alkynyl-metal coordination mode. These new poly(benzyl ether) dendrimers have been isolated as thermally stable solids, as confirmed by thermogravimetric analysis (TGA) study. The TGA traces of all the complexes are shown in Fig. 1. All the complexes feature high thermal stability with Td > 320 °C under a nitrogen atmosphere.
 |
| | Scheme 1 Synthetic routes of (poly benzyl)ether-based dendritic ligands. | |
 |
| | Scheme 2 Synthetic route of poly(benzyl ether)-based dendritic alkynylgold(III) complexes. | |
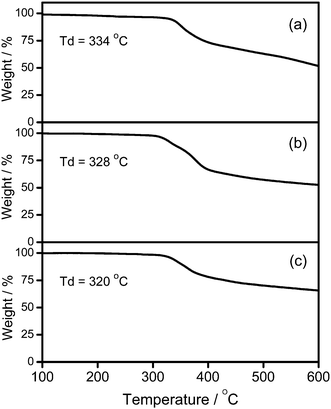 |
| | Fig. 1 Thermogravimetric curves of (a) 1, (b) 2, and (c) 3. | |
Photophysical properties
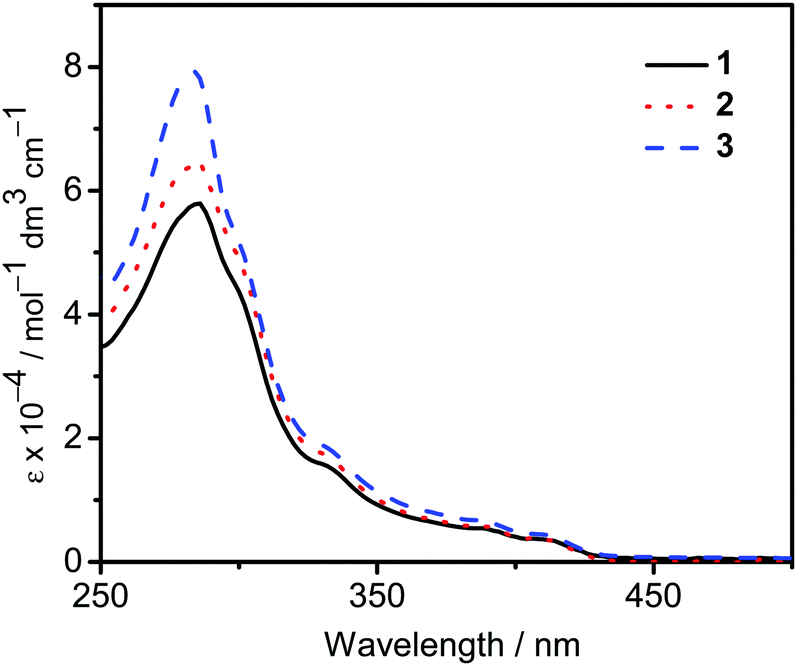
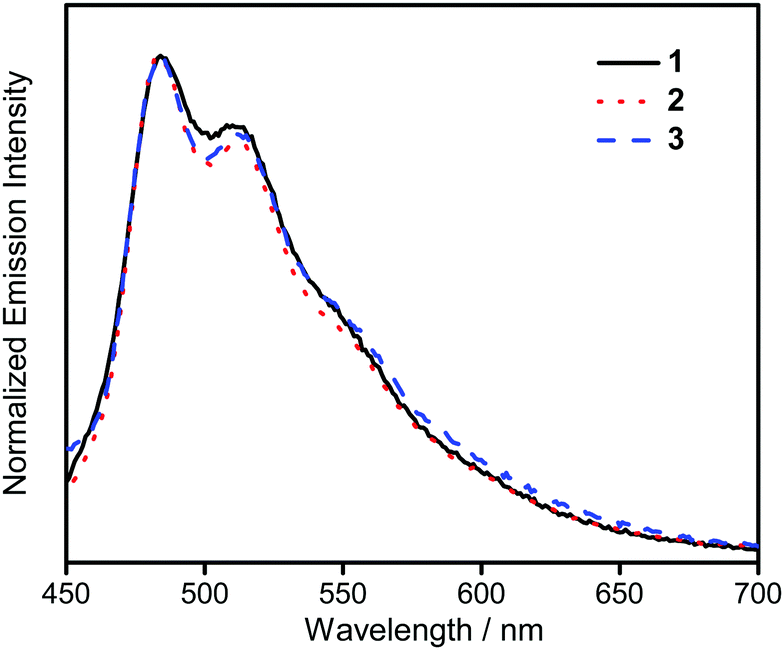
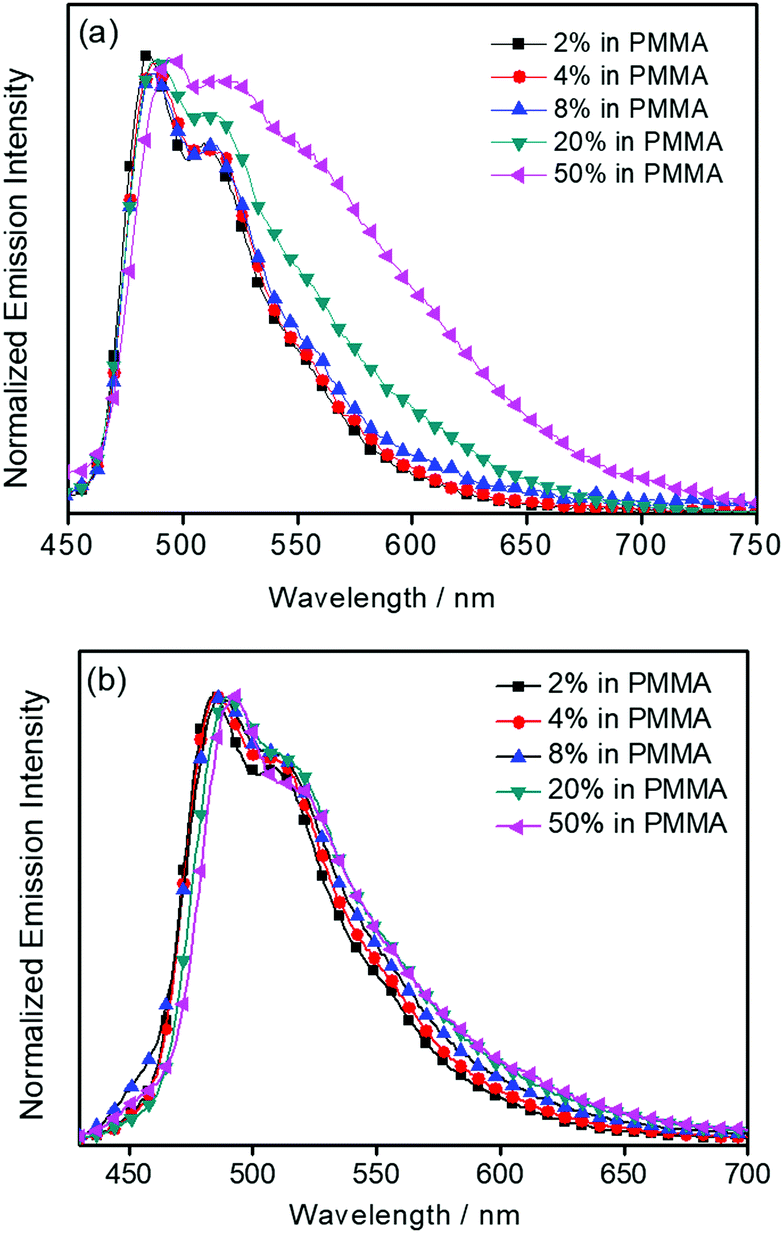
The UV-vis absorption spectra of 1–3 in dichloromethane at 298 K have been studied. In general, all the complexes show characteristic vibronic-structured absorption bands at 280–340 nm with a moderately intense vibronic-structured band at 390–410 nm with molar extinction coefficients (ε) on the order of 104 dm3 mol−1 cm−1. The electronic absorption data are summarized in Table 1 and the electronic absorption spectra are shown in Fig. 2. The high-energy absorption bands at wavelength (λ) ≤ 340 nm are mainly attributed to the spin-allowed intraligand (IL) [π → π*] transition of the benzyloxy units.44 The molar extinction coefficients at approximately 285 nm increase from 1 to 3, which is ascribed to an increase in the number of benzyloxy units. For instance, the ε at 285 nm for complexes 1, 2 and 3 are 5.77 × 104, 6.48 × 104 and 7.93 × 104 dm3 mol−1 cm−1, respectively. The low-energy vibronic-structured band shows vibrational progressional spacings of ca. 1300 cm−1, corresponding to the skeletal vibrational mode of the 2,5-F2C6H3–C^N^C ligand.33,35 Similar to our previously reported structurally related gold(III) complexes,24 the low-energy absorptions are assigned as IL [π → π*] transition of the 2,5-F2C6H3–C^N^C ligand, with IL charge transfer (ILCT) character from the aryl ring to the pyridine unit.24,33,34 Upon excitation at λ ≥ 350 nm in dichloromethane solution at 298 K, 1–3 feature a vibronic-structured emission band with a maximum at ca. 480 nm (Fig. 3). Interestingly, the introduction of an increasing content of poly(benzyl ether) dendrons onto the alkynyl moiety results in a minor perturbation of the emission maxima. The progressional spacings of ca. 1300 cm−1, which are characteristics of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN stretching frequencies of the tridentate 2,5-F2C6H3–C^N^C ligand, indicate the involvement of the tridentate ligand in the excited state origin.24 The luminescence origin of 1–3 has been assigned as a metal-perturbed 3IL [π → π*] state of the tridentate 2,5-F2C6H3–C^N^C ligand, with mixing of ILCT character from the phenyl ring to the pyridine unit.24 In addition, the solid-state thin film emission properties of complexes 2 and 3 have been studied. Fig. 4a and b show the normalized emission spectra of thin films of 2 and 3 doped into poly(methyl methacrylate) (PMMA) at different concentrations. Different from the case in dichloromethane, where the emission energies are almost unchanged, the solid-state emission spectra of the dendritic gold(III) complexes are dramatically changed upon the incorporation of the saturated poly(benzyl ether) dendrons. Particularly, a red emission tail is observed for 2 with increasing dopant concentrations from 8 wt% to 50 wt%. This observation could be due to the excimeric emission resulting from the π-stacking of the 2,5-F2C6H3–C^N^C moieties with higher order and a better packing of molecules at higher concentrations.33–37 Interestingly, the issue of the bathochromic problem can be greatly alleviated by the introduction of the second-generation of poly(benzyl ether) dendrons onto the alkynyl moiety in 3. There is no unwanted excimeric emission observed for 3, even if the dopant concentration increases from 8 wt% to 50 wt%. Notably, the emission band of 3 (i.e. 50 wt%) is almost the same as that in solution, suggesting that the π-stacking of the 2,5-F2C6H3–C^N^C moieties between the emissive cores has been greatly suppressed by the bulky poly(benzyl ether) dendrons. This encapsulation effect could also minimize the intermolecular interactions between the molecules that lead to emission quenching or triplet–triplet annihilation, as well as improve the color purity of the OLEDs.11,40 Unlike in the solid-state thin films, it should be mentioned that the luminescence quantum yield of 3 is lower than that of 2 in the solution state. The lower luminescence quantum yield of 3 in the solution state is possibly due to the incorporation of a higher generation saturated dendrimer onto the gold(III) metal center that would increase the floppiness of the molecules, and hence the rotational and vibrational degrees of freedom. These would inevitably decrease the luminescence quantum yield in the solution state.
C and CN stretching frequencies of the tridentate 2,5-F2C6H3–C^N^C ligand, indicate the involvement of the tridentate ligand in the excited state origin.24 The luminescence origin of 1–3 has been assigned as a metal-perturbed 3IL [π → π*] state of the tridentate 2,5-F2C6H3–C^N^C ligand, with mixing of ILCT character from the phenyl ring to the pyridine unit.24 In addition, the solid-state thin film emission properties of complexes 2 and 3 have been studied. Fig. 4a and b show the normalized emission spectra of thin films of 2 and 3 doped into poly(methyl methacrylate) (PMMA) at different concentrations. Different from the case in dichloromethane, where the emission energies are almost unchanged, the solid-state emission spectra of the dendritic gold(III) complexes are dramatically changed upon the incorporation of the saturated poly(benzyl ether) dendrons. Particularly, a red emission tail is observed for 2 with increasing dopant concentrations from 8 wt% to 50 wt%. This observation could be due to the excimeric emission resulting from the π-stacking of the 2,5-F2C6H3–C^N^C moieties with higher order and a better packing of molecules at higher concentrations.33–37 Interestingly, the issue of the bathochromic problem can be greatly alleviated by the introduction of the second-generation of poly(benzyl ether) dendrons onto the alkynyl moiety in 3. There is no unwanted excimeric emission observed for 3, even if the dopant concentration increases from 8 wt% to 50 wt%. Notably, the emission band of 3 (i.e. 50 wt%) is almost the same as that in solution, suggesting that the π-stacking of the 2,5-F2C6H3–C^N^C moieties between the emissive cores has been greatly suppressed by the bulky poly(benzyl ether) dendrons. This encapsulation effect could also minimize the intermolecular interactions between the molecules that lead to emission quenching or triplet–triplet annihilation, as well as improve the color purity of the OLEDs.11,40 Unlike in the solid-state thin films, it should be mentioned that the luminescence quantum yield of 3 is lower than that of 2 in the solution state. The lower luminescence quantum yield of 3 in the solution state is possibly due to the incorporation of a higher generation saturated dendrimer onto the gold(III) metal center that would increase the floppiness of the molecules, and hence the rotational and vibrational degrees of freedom. These would inevitably decrease the luminescence quantum yield in the solution state.
Table 1 Photophysical properties of complexes 1–3
| Complex |
Medium (T/K) |
Absorption λmax/nm (εmax/dm3 mol−1 cm−1) |
Emission λmax/nm (τ0/μs) |
Φ
sol
|
Φ
film
|
The relative luminescence quantum yield in solution was measured at room temperature using [Ru(bpy)3]Cl2 in aqueous solution as the reference (excitation wavelength = 436 nm, Φlum = 0.042).
Φ
film of the gold(III) compound doped into MCP excited at a wavelength of 320 nm.
Measured in EtOH–MeOH–CH2Cl2 (40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:1, v/v). :10:1, v/v).
|
|
1
|
CH2Cl2 (298) |
284 (57800), 302 (41500), 334 (15000), 390 (5340), 410 (3665) |
484, 515, 556 (<0.1) |
4 × 10−3 |
|
| Glass (77)c |
|
476, 510, 555 (90) |
|
|
|
|
|
2
|
CH2Cl2 (298) |
286 (64850), 302 (46660), 334 (16585), 390 (5706), 410 (3705) |
484, 515, 556 (<0.1) |
8 × 10−3 |
|
| Glass (77)c |
|
476, 510, 550 (99) |
|
|
| Thin film (298) |
|
|
| 5% in MCP |
490, 520, 550 |
0.15 |
| 10% in MCP |
490, 520, 554 |
0.16 |
| 15% in MCP |
490, 520, 558 |
0.13 |
| 20% in MCP |
494, 530, 560 |
0.04 |
| 2% in PMMA |
484, 510, 536 |
|
| 4% in PMMA |
486, 512, 536 |
|
| 8% in PMMA |
486, 514, 536 |
|
| 20% in PMMA |
490, 516, 550 |
|
| 50% in PMMA |
492, 524, 566 |
|
|
|
|
3
|
CH2Cl2 (298) |
284 (79250), 302 (49245) 334 (17850), 390 (6515), 410 (4465) |
484, 515, 556 (<0.1) |
4 × 10−3 |
|
| Glass (77)c |
|
492, 526, 574 (88) |
|
|
| Thin film (298) |
|
|
| 5% in MCP |
490, 522, 556 |
0.20 |
| 10% in MCP |
490, 522, 556 |
0.19 |
| 15% in MCP |
490, 522, 556 |
0.16 |
| 20% in MCP |
492, 524, 562 |
0.09 |
| 2% in PMMA |
484, 510, 554 |
|
| 4% in PMMA |
486, 512, 554 |
|
| 8% in PMMA |
486, 514, 554 |
|
| 20% in PMMA |
490, 514, 554 |
|
| 50% in PMMA |
490, 518, 554 |
|
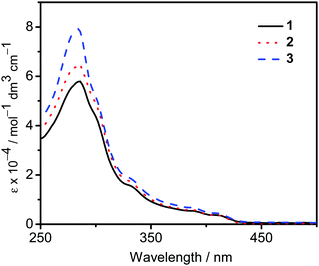 |
| | Fig. 2 UV-Vis absorption spectra of complexes 1–3 in dichloromethane at 298 K. | |
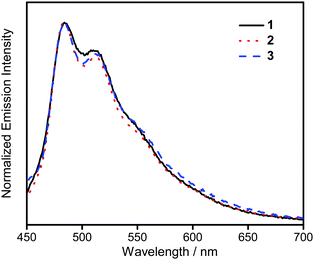 |
| | Fig. 3 Normalized PL spectra of complexes 1–3 in dichloromethane at 298 K. | |
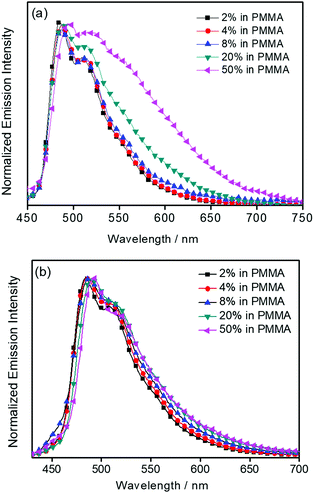 |
| | Fig. 4 Normalized PL spectra of thin films of (a) 2, and (b) 3 doped into PMMA at different concentrations (wt%) at 298 K. | |
Electrochemical properties
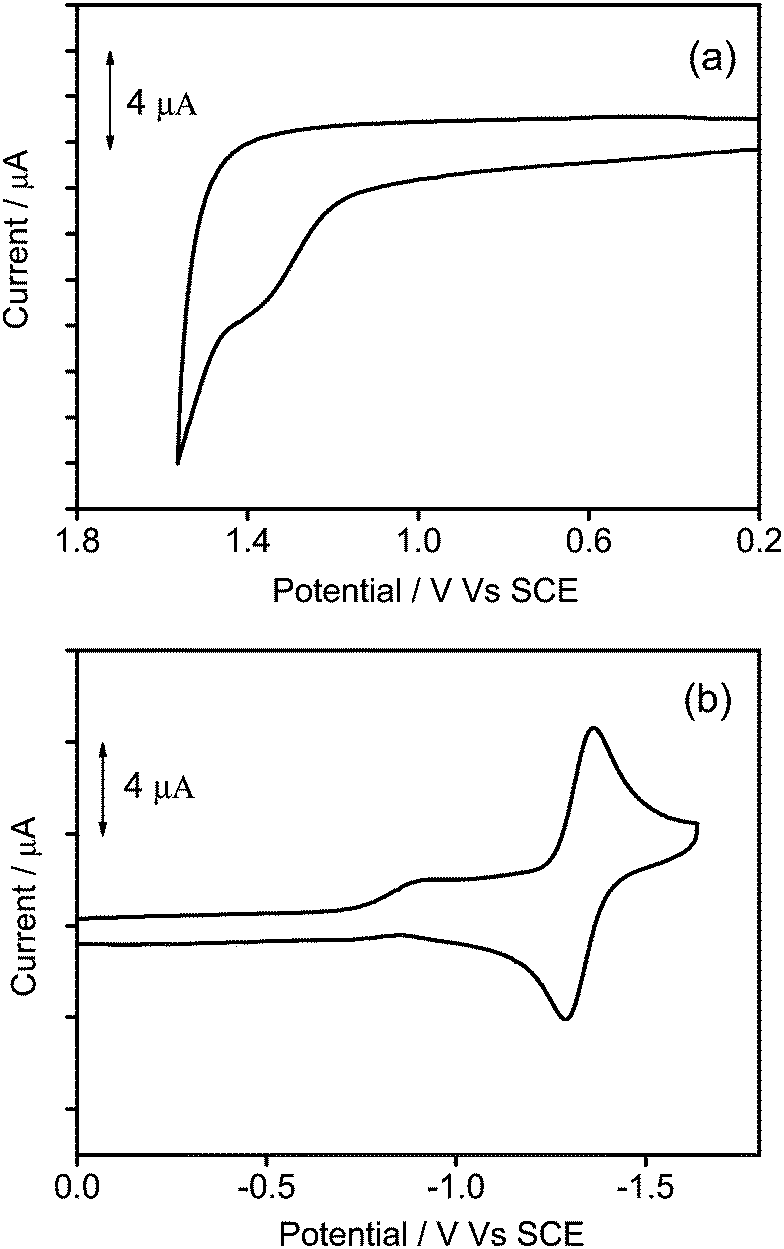
To gain further insights into the electrochemical properties, the cyclic voltammetry of 1–3 in dichloromethane (0.1 mol dm−3 nBu4NPF6) has been investigated and their highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels are estimated. All the complexes show a quasi-reversible reduction couple at −1.32 V vs. a standard calomel electrode (SCE), and an irreversible first oxidation wave at +1.38 to +1.54 V vs. SCE. The electrochemical data are summarized in Table 2, and the representative cyclic voltammograms are shown in Fig. 5. Similar to previous reports on related alkynylgold(III) complexes,24,33,34 the reduction is attributed to the ligand-centered reduction of the cyclometalated ligand. For the irreversible oxidative wave, the potentials for oxidation become more positive (+1.38 to +1.54 V vs. SCE) with increasing dendrimer generations, and similar potentials for the first oxidation have also been observed in other poly(benzyl ether) dendrimers or alkynylgold(III) complexes.24,40,44 In view of this, the first oxidation wave is assigned to the alkynyl ligand-centered oxidation.
Table 2 Electrochemical data for 1–3a
| Complex |
Oxidation [Epa/V vs. SCE]b |
Reduction E1/2/V vs. SCEc (ΔEpd/mV) |
E
HOMO
/eV |
E
LUMO
/eV |
|
In CH2Cl2 solution with 0.1 M nBu4NPF6 as a supporting electrolyte at a 298 K working electrode, glassy carbon; scan rate = 100 mV s−1.
E
pa refers to the anodic peak potential for the irreversible oxidation waves.
E
1/2 = (Epa + Epc)/2; Epa and Epc are the peak anodic and peak cathodic potentials, respectively.
ΔEp = (Epa − Epc).
E
HOMO and ELUMO levels were calculated from electrochemical potentials, i.e. EHOMO = −e(4.8 V + Epa); ELUMO = −e(4.8 V + Epc).
|
|
1
|
[+1.38] |
−1.32 (70) |
−6.16 |
−3.48 |
|
2
|
[+1.54] |
−1.32 (60) |
−6.35 |
−3.48 |
|
3
|
[+1.55] |
−1.32 (60) |
−6.34 |
−3.48 |
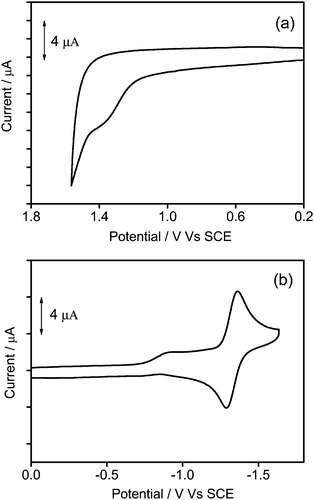 |
| | Fig. 5 Cyclic voltammograms for the (a) oxidation and (b) reduction scans of 2 in dichloromethane (0.1 M nBu4NPF6). | |
OLED fabrication and characterization
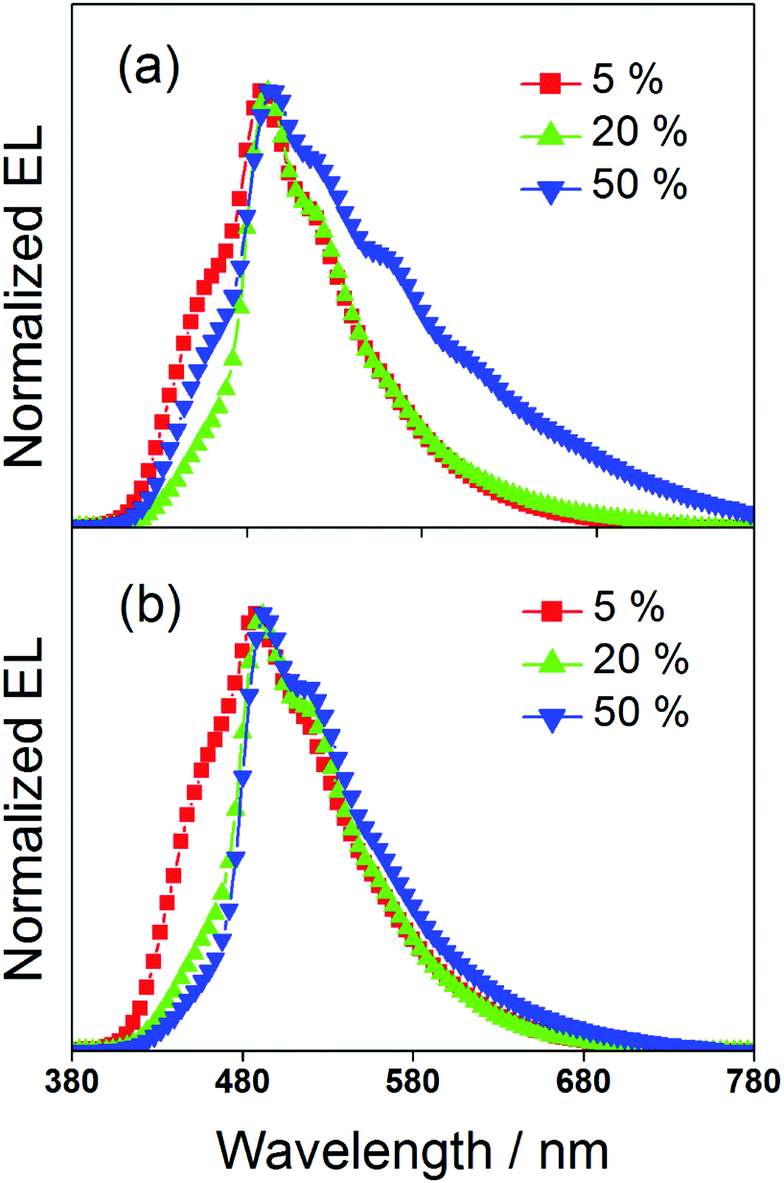
To study the EL performance of complexes 2 and 3, solution-processable OLEDs with the configuration of indium tin oxide (ITO)/poly(ethylene-dioxythiophene):poly(styrene sulfonic acid) (PEDOT:PSS)/x% dendrimer:1,3-bis(carbazol-9-yl)benzene (MCP)/bis(2-methyl-8-quinolinolate)-(4-phenyl-phenolato)aluminium (BAlq)/LiF/Al were constructed, in which PEDOT:PSS and BAlq were used as hole-transporting and electron-transporting layers, respectively. The emissive layer was prepared by spin-coating a solution of dendrimer:MCP blend at different concentrations in chloroform. Fig. 6 depicts the normalized EL spectra of the devices at a current density of 20 mA cm−2. Except for the observation of an EL band at ∼480 nm that was originated from the emission of the electron-transporting BAlq layer due to inefficient electron trapping by the gold(III) complexes, the EL spectra of all the devices were almost identical to their emission spectra in MCP thin films. This confirms the occurrence of efficient exothermic energy transfer from the MCP triplet state to the triplet state of the dendrimers. As elucidated by the photoluminescence studies, the triplet energies of the gold(III) dendrimers are ∼2.5 eV. The higher triplet energy of MCP (2.9 eV) can effectively confine both charge carriers and triplet excitons within the emissive layer for light emission. It should be noted that the emission energies of the present gold(III) complexes with saturated dendrons are found to be independent of the dopant concentration. In particular, the emission maxima for all the devices are almost constant over a wide range of dopant concentrations from 5 wt% to 50 wt%. More notably, no undesirable excimer emission is observed for devices made from 3, in excellent agreement with the emission spectra in solid-state thin films. This is not the case for the previously reported carbazole-based alkynylgold(III) complexes,37 in which significant bathochromic shifts of ∼983 cm−1 and 1203 cm−1 were observed for first and second generation dendrimeric complexes, respectively. Such concentration independence of the present gold(III) complexes is anticipated to be due to the encapsulation effect of the poly(benzyl ether) dendrons that can greatly avoid the π-stacking of the 2,5-F2C6H3–C^N^C moieties between the emissive cores. Taking advantage of this, blue-green-emitting devices with peak maxima at ca. 490 nm can be realized and this class of gold(III) dendrimers can potentially become candidates in the very limited library of blue-green-emitting gold(III) complexes.31 To the best of our knowledge, this is the first example of blue-green emitting devices based on saturated dendrimer-containing gold(III) complexes.
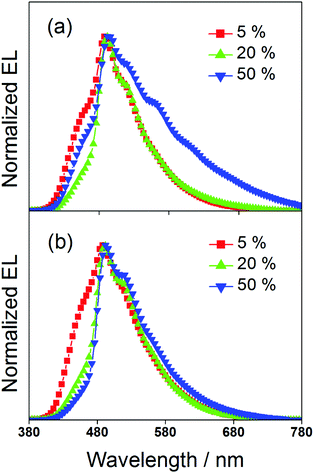 |
| | Fig. 6 Normalized EL spectra of devices made from 2 and 3. | |
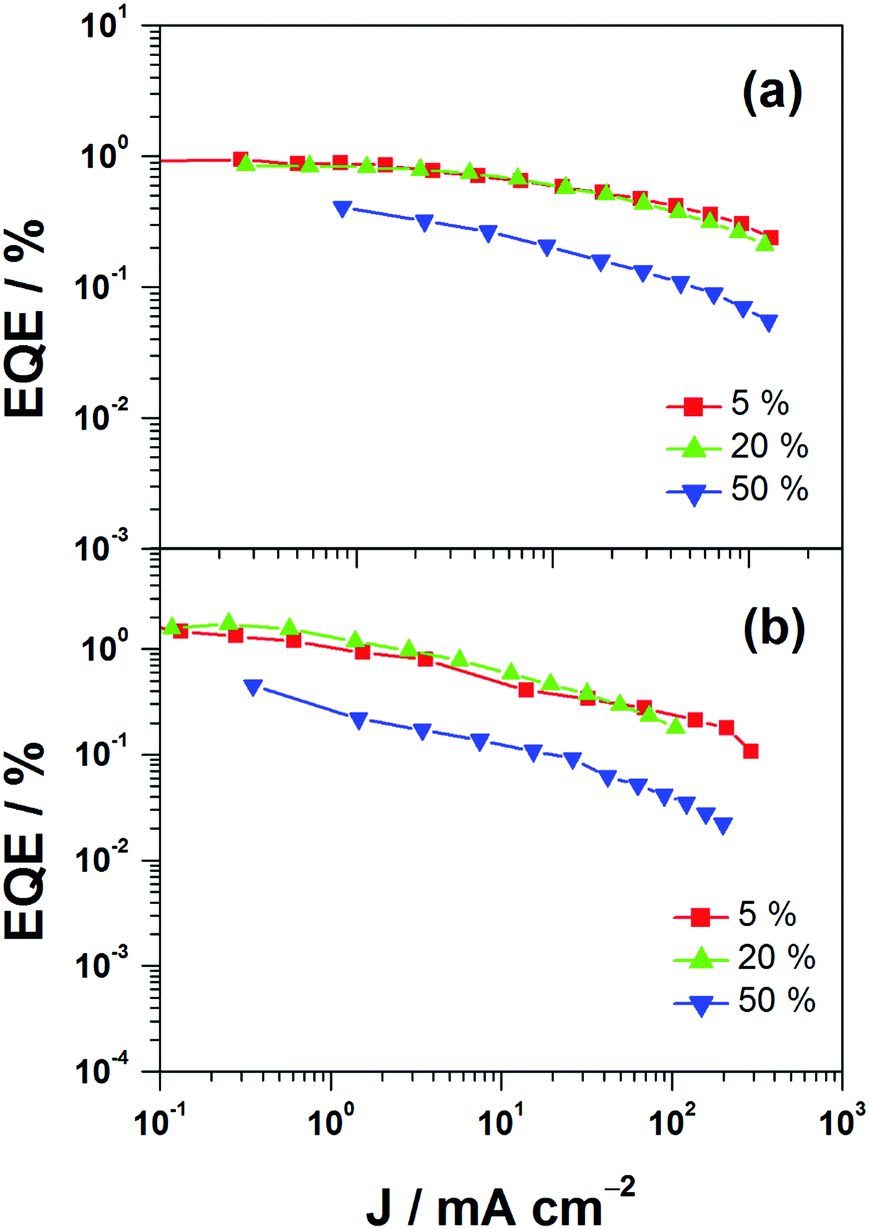
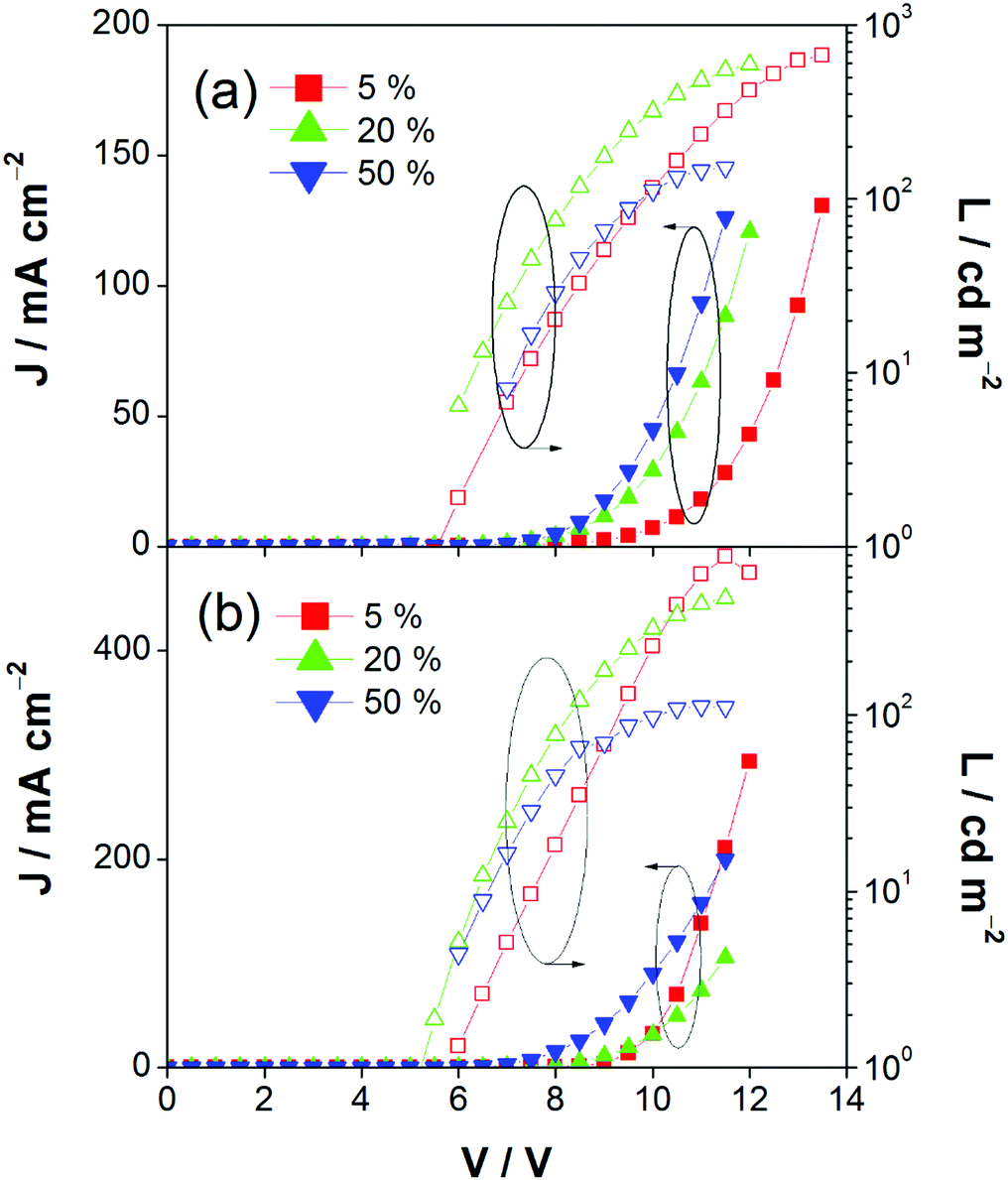
This encapsulation effect of bulky poly(benzyl ether) dendrons can also increase the photoluminescence quantum yields (PLQYs) of the gold(III) complexes in solid-state thin films. As indicated in Table 1, the PLQY of 3 is higher than that of 2. This yields a better performance of the solution-processable OLEDs. In particular, devices based on 2 show a reasonable maximum EQE and current efficiency of 0.9% and 2.5 cd A−1, respectively (Fig. 7a and Table 3). The introduction of more poly(benzyl)ether moieties to yield higher generation complex 3 can improve the device performance. The optimized device exhibits a maximum EQE and current efficiency of 2.4% and 6.7 cd A−1, respectively (Fig. 7b and Table 3). Fig. 8 depicts the current density–voltage–luminance curves of both devices. The performance improvement can be rationalized in terms of a higher PLQY in complex 3 and the minimization of the emission quenching via the suppression of the intermolecular interaction. It should be mentioned that due to the open, floppy structure of the saturated dendron, the performance of OLEDs based on the saturated dendrimer (i.e. maximum EQE of 2.4%) is comparatively lower than those of devices made from the previously reported conjugated dendrimer with the same C^N^C ligand (i.e. maximum EQE of 7.8%),35 similar to the cases of the iridium(III) systems.40 Nevertheless, the incorporation of a saturated poly(benzyl ether) dendrimer into the gold(III) metal center can effectively perturb the packing of the molecules and provides a simple means to generate high-energy vibronic emission in the blue-green region.
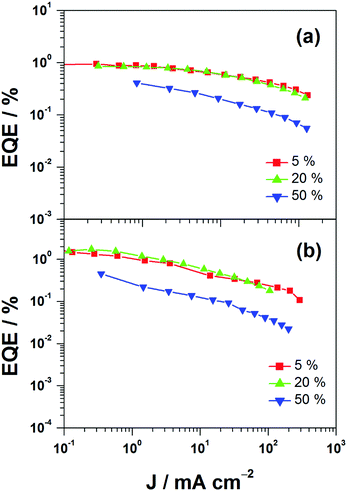 |
| | Fig. 7 EQEs of devices made from 2 and 3. | |
Table 3 Key characteristics of devices made from 2 and 3
| Complex |
Conc./wt% |
Max. current efficiency/cd A−1 |
Max. power efficiency/lm W−1 |
Max. EQE/% |
λ
max/nm (CIE (x, y))a |
|
Data were collected at a current density of 20 mA cm−2.
|
|
2
|
5 |
2.5 |
1.1 |
0.9 |
488 (0.21, 0.35) |
| 20 |
2.3 |
1.2 |
0.8 |
492 (0.23, 0.45) |
| 50 |
0.9 |
0.4 |
0.4 |
496 (0.30, 0.43) |
|
|
|
3
|
5 |
6.7 |
3.5 |
2.4 |
492 (0.24, 0.51) |
| 20 |
4.8 |
2.3 |
1.7 |
492 (0.21, 0.39) |
| 50 |
1.2 |
0.6 |
0.4 |
492 (0.22, 0.38) |
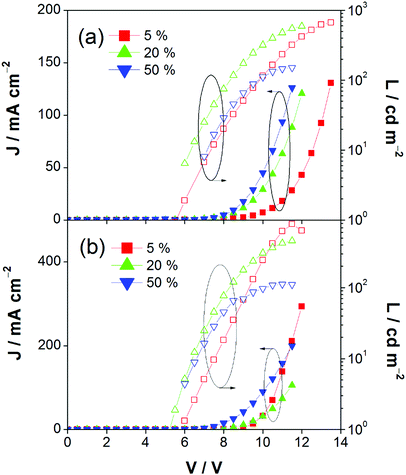 |
| | Fig. 8 Current density–voltage–luminance (J–V–L) of devices made from 2 and 3. | |
Conclusions
A new class of saturated poly(benzyl ether) dendritic gold(III) complexes has been successfully designed and synthesized. The incorporation of the saturated poly(benzyl ether) dendrimer into the gold(III) metal center can effectively perturb the packing of the molecules, suppressing the undesirable excimeric emission that degrades the color purity. By utilizing the second-generation poly(benzyl)ether dendritic gold(III) complex as the phosphorescent dopant, blue-green-emitting PHOLEDs with maximum current efficiency of 6.7 cd A−1 and EQE of 2.4% have been achieved. Taking advantage of the diverse synthetic routes of the saturated poly(benzyl ether) dendrimers that have been well reported in the literature, this work opens up a simple method for the design and synthesis of a new class of saturated poly(benzyl ether) dendritic gold(III) complexes with blue emission in the visible spectrum. These results demonstrate that the alkynylgold(III) complexes with saturated dendrons are promising candidates as blue-emitting phosphorescent dopants for solution-processable OLEDs.
Materials and reagents
Potassium tetrachloroaurate(III) was purchased from Strem Chemicals. All solvents were purified and distilled using standard procedures before use. All other reagents were of analytical grade and were used as received. Tetra-n-butylammonium hexafluorophosphate (Aldrich, 98%) was recrystallized no less than three times from hot absolute ethanol prior to use.
Physical measurements and instrumentation
The UV-vis absorption spectra were recorded on a Cary 50 (Varian) spectrophotometer equipped with a Xenon flash lamp. 1H NMR spectra were recorded on a Bruker DPX-300 (300 MHz) or Bruker DPX-400 (400 MHz) Fourier-transform NMR spectrometer with chemical shifts reported relative to tetramethylsilane. Positive FAB mass spectra were recorded on a Thermo Scientific DFS High Resolution Magnetic Sector Mass Spectrometer. IR spectra were recorded as KBr disks on a Bio-Rad FTS-7 FTIR spectrometer (4000–400 cm−1). Elemental analyses for the metal complexes were performed on the Carlo Erba 1106 elemental analyzer at the Institute of Chemistry, Chinese Academy of Sciences in Beijing. Steady-state excitation and emission spectra were recorded on a Spex Fluorolog-3 Model FL3–211 fluorescence spectrofluorometer equipped with an R2658P PMT detector. Solid-state photophysical measurements were carried out with the solid sample loaded in a quartz tube inside a quartz-walled Dewar flask. Liquid nitrogen was placed into the optical Dewar flask for low temperature (77 K) photophysical measurements. Excited-state lifetimes of solution and glass samples were measured using a conventional laser system. The excitation source used was the 355 nm output (third harmonic, 8 ns) of a Spectra-Physics Quanta-Ray Q-switched GCR-150 pulsed Nd:YAG laser (10 Hz). Luminescence decay signals were detected by a Hamamatsu R928 photomultiplier tube, recorded on a Tektronix Model TDS-620A (500 MHz, 2 GS s−1) digital oscilloscope, and analyzed by using a program for exponential fits. Relative luminescence quantum yields in degassed solutions were measured by the optical dilution method reported by Demas and Crosby.45 A degassed solution of [Ru(bpy)3]Cl2 in an aqueous state (Φlum = 0.042, excitation wavelength at 436 nm) was used as the ref. 46, while those of the thin films were measured on a Hamamatsu C9920-03 Absolute PLQY Measurement System. Cyclic voltammetric measurements were performed by using a CH Instruments, Inc. model CHI 600A electrochemical analyzer. All solutions for electrochemical studies were deaerated with pre-purified argon gas just before measurements. Thermal analyses were performed with a Perkin-Elmer TAC7/DX thermal analysis controller, in which the decomposition temperature, Td, is defined as the temperature at which the material shows a 5% weight loss.
OLED fabrication and characterization
OLEDs were fabricated on patterned ITO glass substrates with a sheet resistance of 30 Ω per square. The substrates were cleaned with Decon 90, rinsed with deionized water and then dried in an oven, and finally treated in an ultraviolet-ozone chamber. A 70 nm thick PEDOT:PSS layer was spin-coated onto the ITO coated glass substrates as a hole-transporting layer. After that, the emissive layer was formed by mixing the dendrimer with MCP to prepare a 10 mg cm−3 solution in chloroform and spin-coating onto the PEDOT:PSS layer to give uniform thin films of 60 nm thickness. Onto this, a 30 nm thick BAlq layer was evaporated as a hole-blocking layer and an electron-transporting layer; while a LiF/Al was used as the metal cathode. All films were sequentially deposited at a rate of 0.1–0.2 nm s−1 without vacuum break. A shadow mask was used to define the cathode and to make four 0.1 cm2 devices on each substrate. Current density–voltage–luminance characteristics and electroluminescence spectra were measured simultaneously with a programmable Keithley model 2420 power source and a PhotoResearch PR-655 spectrometer.
Synthesis and characterization of the poly(benzyl)ether dendritic ligands and poly(benzyl)ether based dendritic alkynylgold(III) complexes
3,5-Dimethoxybenzyl azide (G1-CH2N3), 3,5-bis(3,5-dimethoxybenzyloxy)benzylazide (G2-CH2N3) and [3,5-bis{3,5-bis(3,5-dimethoxybenzyloxy)benzyloxy}]benzylazide (G3-CH2N3) were synthesized according to the published procedures.47 Benzyl ether dendritic ligands were synthesized from their respective precursors by the reaction of different polyarylether azide ligands with 1,4-diethynylbenzene in the presence of a catalytic amount of sodium ascorbate and CuSO4·5H2O. 2,6-Diphenyl-4-(2,5-difluorophenyl)pyridine (2,5-F2C6H3–C^N^C) and the chlorogold(III) precursor, [Au{2,5-F2C6H3–C^N^C}Cl], were prepared by slight modifications of the literature procedures.24,33,48 All the complexes were synthesized by reacting the corresponding alkynes with the chlorogold(III) precursor in the presence of a catalytic amount of copper(I) iodide in triethylamine and dichloromethane.
G1-C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) CH.
A mixture of 1,4-diethynylbenzene (0.15 g, 0.75 mmol) and G1-CH2N3 (0.13 g, 0.90 mmol) in the presence of 5 mol% CuSO4·5H2O (7.50 mg, 0.03 mmol) and 10 mol% sodium ascorbate (11.88 mg, 0.06 mmol) was dissolved in degassed DMF–H2O (8:2 v/v, 10 mL) and stirred for 12 h under a nitrogen atmosphere at 50–60 °C. The crude product was cooled to room temperature and then extracted with diethyl ether (20 mL). The desired product was purified by column chromatography on silica gel using ethyl acetate as the eluent. The removal of the solvent and subsequent recrystallization by diffusion of pentane vapor into a concentrated solution of the product gave ligand G1-CCH as a pale white solid. Yield: 170 mg, 70%. 1H NMR (400 MHz, DMSO-d6, 298 K, relative to Me4Si, δ/ppm): δ 3.11 (s, 1H, –CCH), 3.71 (s, 6H, –OMe), 5.55 (s, 2H, –CH2–), 6.47 (t, J = 2.0 Hz, 1H, –C6H3-OMe-3,5), 6.51 (d, J = 2.0 Hz, 2H, –C6H3-OMe-3,5), 7.54 (d, J = 8.4 Hz, 2H, –Ar), 7.87 (d, J = 8.4 Hz, 2H, –Ar), 8.67 (s, 1H, –C2N3H–). Positive FAB-MS: m/z 319 [M]+. Elemental analyses: found (%): C, 71.34; H, 5.55; N, 13.11. Calcd for C19H17N3O2: C, 71.45; H, 5.36; N, 13.15.
CH.
A mixture of 1,4-diethynylbenzene (0.15 g, 0.75 mmol) and G1-CH2N3 (0.13 g, 0.90 mmol) in the presence of 5 mol% CuSO4·5H2O (7.50 mg, 0.03 mmol) and 10 mol% sodium ascorbate (11.88 mg, 0.06 mmol) was dissolved in degassed DMF–H2O (8:2 v/v, 10 mL) and stirred for 12 h under a nitrogen atmosphere at 50–60 °C. The crude product was cooled to room temperature and then extracted with diethyl ether (20 mL). The desired product was purified by column chromatography on silica gel using ethyl acetate as the eluent. The removal of the solvent and subsequent recrystallization by diffusion of pentane vapor into a concentrated solution of the product gave ligand G1-CCH as a pale white solid. Yield: 170 mg, 70%. 1H NMR (400 MHz, DMSO-d6, 298 K, relative to Me4Si, δ/ppm): δ 3.11 (s, 1H, –CCH), 3.71 (s, 6H, –OMe), 5.55 (s, 2H, –CH2–), 6.47 (t, J = 2.0 Hz, 1H, –C6H3-OMe-3,5), 6.51 (d, J = 2.0 Hz, 2H, –C6H3-OMe-3,5), 7.54 (d, J = 8.4 Hz, 2H, –Ar), 7.87 (d, J = 8.4 Hz, 2H, –Ar), 8.67 (s, 1H, –C2N3H–). Positive FAB-MS: m/z 319 [M]+. Elemental analyses: found (%): C, 71.34; H, 5.55; N, 13.11. Calcd for C19H17N3O2: C, 71.45; H, 5.36; N, 13.15.
G2-CCH.
This was synthesized according to a procedure similar to that of G1-CCH except that G2-CH2N3 (0.23 g, 0.05 mmol) was used in place of G1-CH2N3. A white solid of G2-CCH was obtained. Yield: 230 mg, 78%. 1H NMR (400 MHz, DMSO-d6, 298 K, relative to Me4Si, δ/ppm): δ 3.11 (s, 1H, –CCH), 3.71 (s, 12H, –OMe), 5.00 (s, 4H, –CH2–), 5.55 (s, 2H, –CH2–), 6.47 (t, J = 2.0 Hz, 2H, –C6H3-OMe-3,5), 6.56–6.62 (m, 7H, –C6H3-OMe-3,5 and –C6H3–(OCH2)2-3,5–), 7.54 (d, J = 8.4 Hz, 2H, –Ar), 7.85 (d, J = 8.4 Hz, 2H, –Ar), 8.65 (s, 1H, –C2N3H–). Positive FAB-MS: m/z 592 [M]+. Elemental analyses: found (%): C, 68.91; H, 5.97; N, 6.67. Calcd for C35H33N3O6·H2O: C, 68.95; H, 5.78; N, 6.89.
G3-CCH.
This was synthesized according to a procedure similar to that of G1-CCH except that precursor G3-CH2N3 (0.30 g, 0.03 mmol) was used in place of precursor CH2N3-G1. A white solid of G3-CCH was obtained. Yield: 175 mg, 78%. 1H NMR (400 MHz, DMSO-d6, 298 K, relative to Me4Si, δ/ppm): δ 3.11 (s, 1H, –CCH), 3.71 (s, 24H, –OMe), 4.96 (s, 12H, –CH2–), 5.53 (s, 2H, –CH2–), 6.41 (t, J = 2.0 Hz, 2H, –C6H3-OMe-3,5), 6.56–6.68 (m, 19H, –C6H3-OMe-3,5 and –C6H3-(OCH2)2-3,5–), 7.50 (d, J = 8.4 Hz, 2H, –Ar), 7.83 (d, J = 8.4 Hz, 2H, –Ar), 8.61 (s, 1H, –C2N3H–). Positive FAB-MS: m/z 1137 [M]+. Elemental analyses: found (%): C, 70.51; H, 5.81; N, 3.44. Calcd for C67H64N3O14·1/2H2O: C, 70.32; H, 5.72; N, 3.67.
[Au(2,5-F2C6H3–C^N^C)(CC-G1)] (1).
A mixture of [Au(2,5-F2C6H3–C^N^C)Cl] (54 mg, 0.10 mmol), copper(I) iodide (3.80 mg, 0.02 mmol), triethylamine (2 mL) and G1-CCH (58 mg, 0.10 mmol) was stirred in degassed dichloromethane solution (30 mL) for 12 h under a nitrogen atmosphere at room temperature. After removing the solvent, the crude product was purified by column chromatography on silica gel using ethyl acetate as the eluent. Subsequent recrystallization by diffusion of diethyl ether vapor into a concentrated solution of the product gave [Au(2,5-F2C6H3–C^N^C)(CC-G1)] as a pale yellow solid. Yield: 40 mg, 46%. 1H NMR (400 MHz, DMSO-d6, 298 K, relative to Me4Si, δ/ppm): δ 3.71 (s, 6H, –OMe), 5.55 (s, 2H, –CH2–), 6.47 (d, J = 2.0 Hz, 1H, –C6H3-OMe-3,5), 6.51 (t, J = 2.0 Hz, 2H, –C6H3-OMe-3,5), 7.34 (dt, J = 7.4 and 1.0 Hz, 2H, –C6H4– of C^N^C), 7.44 (dt, J = 7.4 and 1.0 Hz, 2H, –C6H4– of C^N^C), 7.48–7.56 (m, 2H, 2,5-F2C6H3– of C^N^C), 7.58 (d, J = 8.0 Hz, 2H, –Ar), 7.74–7.82 (m, 1H, 2,5-F2C6H3– of C^N^C), 7.86 (d, J = 8.0 Hz, 2H, –Ar), 7.95 (dd, J = 7.8 and 1.0 Hz, 2H, –C6H4– of C^N^C) 8.02 (d, J = 7.8 Hz, 2H, –C6H4– of C^N^C), 8.18 (s, 2H, pyridyl of C^N^C), 8.67 (s, 1H, –C2N3H–). Positive FAB-MS: m/z 856 [M]+. IR (KBr disk): 2150 cm−1ν(C≡C). Elemental analyses: found (%): C, 58.33; H, 3.54; N, 6.43. Calcd for C42H29F2N4O2Au·1/2H2O: C, 58.27; H, 3.49; N, 6.47.
[Au(2,5-F2C6H3–C^N^C)(CC-G2)] (2).
This was synthesized by a method according to a procedure similar to that of [Au(2,5-F2C6H3–C^N^C)(CC-G1)] except that G2-CCH (130 mg, 0.20 mmol) was used in place of G1-CCH. A yellow solid of [Au(2,5-F2C6H3–C^N^C)(CC-G2)] was obtained. Yield: 40 mg, 35%. 1H NMR (400 MHz, DMSO-d6, 298 K, relative to Me4Si, δ/ppm): δ 3.70 (s, 12H, –OMe), 5.00 (s, 4H, –CH2–), 5.55 (s, 2H, –CH2–), 6.41 (t, J = 4.4 Hz, 2H, –C6H3-OMe-3,5), 6.56–6.64 (m, 7H, –C6H3-OMe-3,5 and –C6H3-(OCH2)2-3,5–), 7.34 (dt, J = 7.6 and 1.0 Hz, 2H, –C6H4– of C^N^C), 7.44 (dt, J = 7.8 and 1.0 Hz, 2H, –C6H4– of C^N^C), 7.48–7.55 (m, 2H, 2,5-F2C6H3– of C^N^C), 7.58 (d, J = 7.8 Hz, 2H, –Ar), 7.77–7.81 (m, 1H, 2,5-F2–C6H3– of C^N^C), 7.82 (d, J = 7.8 Hz, 2H, –Ar), 7.94 (dd, J = 7.8 and 1.0 Hz, 2H, –C6H4– of C^N^C), 8.02 (d, J = 7.8 Hz, 2H, –C6H4– of C^N^C), 8.18 (s, 2H, pyridyl of C^N^C), 8.65 (s, 1H, –C2N3H–). Positive FAB-MS: m/z 1128 [M]+. IR (KBr disk): 2146 cm−1ν(C≡C). Elemental analyses: found (%): C, 61.26; H, 4.08; N, 4.88. Calcd for C58H45F2N4O6Au·1/2H2O: C, 61.21; H, 4.07; N, 4.92.
[Au(2,5-F2C6H3–C^N^C)(CC-G3)] (3).
This was synthesized by a method according to a procedure similar to that of [Au(2,5-F2C6H3–C^N^C)(CC-G1)] except that G3-CCH (250 mg, 0.20 mmol) was used in place of G1-CCH. A yellow solid of [Au(2,5-F2C6H3–C^N^C)(CC-G3)] was obtained. Yield: 57 mg, 26%. 1H NMR (400 MHz, DMSO-d6, 298 K, relative to Me4Si, δ/ppm): δ 3.70 (s, 24H, –OMe), 5.00 (s, 12H, –CH2–), 5.54 (s, 2H, –CH2–), 6.41 (t, J = 2.0 Hz, 2H, –C6H3-OMe-3,5), 6.56–6.66 (m, 19H, –C6H3-OMe-3,5 and –C6H3–(OCH2)2-3,5–), 7.33 (dt, J = 8.0 and 1.0 Hz, 2H, –C6H4– of C^N^C), 7.43 (dt, J = 8.0 and 1.0 Hz, 2H, –C6H4– of C^N^C), 7.48–7.55 (m, 2H, 2,5-F2C6H3– of C^N^C), 7.56 (d, J = 8.2 Hz, 2H, –Ar), 7.77–7.82 (m, 1H, 2,5-F2C6H3– of C^N^C), 7.84 (d, J = 8.2 Hz, 2H, –Ar), 7.93 (dd, J = 8.0 and 1.0 Hz, 2H, –C6H4– of C^N^C) 8.03 (d, J = 8.0 Hz, 2H, –C6H4– of C^N^C), 8.18 (s, 2H, pyridyl of C^N^C), 8.61 (s, 1H, –C2N3H–). Positive FAB-MS: m/z 1672 [M]+. IR (KBr disk): 2150 cm−1ν(C≡C). Elemental analyses: found (%): C, 64.76; H, 4.75; N, 3.51. Calcd for C90H77F2N4O14Au: C, 64.59; H, 4.63; N, 3.34.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
V. W.-W. Y. acknowledges support from The University of Hong Kong and the URC Strategic Research Theme on New Materials. The work described in this paper was supported by the Theme-based Research Scheme (TRS) (Project No. T23-713/11) and a General Research Fund from the Research Grants Council of the Hong Kong Special Administrative Region, China (Project No. HKU17304715).
References
- B. W. D'Andrade and S. R. Forrest, Adv. Mater., 2004, 16, 1585–1595 CrossRef.
- W.-Y. Wong and C.-L. Ho, Coord. Chem. Rev., 2009, 253, 1709–1758 CrossRef CAS.
- B. W. D'Andrade, M. A. Baldo, C. Adachi, J. Brooks, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 2001, 79, 1045–1047 CrossRef.
- C. Adachi, M. A. Baldo, S. R. Forrest, S. Lamansky, M. E. Thompson and R. C. Kwong, Appl. Phys. Lett., 2001, 78, 1622–1624 CrossRef CAS.
- C. Adachi, R. C. Kwong, P. Djurovich, V. Adamovich, M. A. Baldo, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 2001, 79, 2082–2084 CrossRef CAS.
- M. A. Baldo, S. Lamansky, P. E. Burrows, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 1999, 75, 4–6 CrossRef CAS.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H.-E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304–4312 CrossRef CAS PubMed.
- J. Brooks, Y. Babayan, S. Lamansky, P. I. Djurovich, I. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2002, 41, 3055–3066 CrossRef CAS PubMed.
- W. Lu, B.-X. Mi, M. C. W. Chan, Z. Hui, C.-M. Che, N. Zhu and S.-T. Lee, J. Am. Chem. Soc., 2004, 126, 4958–4971 CrossRef CAS PubMed.
- S. J. Farley, D. L. Rochester, A. L. Thompson, J. A. K. Howard and J. A. G. Williams, Inorg. Chem., 2005, 44, 9690–9703 CrossRef CAS PubMed.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- C. Cebrian, M. Mauro, D. Kourkoulos, P. Mercandelli, D. Hertel, K. Meerholz, C. A. Strassert and L. De Cola, Adv. Mater., 2013, 25, 437–442 CrossRef CAS PubMed.
- H. Sasabe, H. Nakanishi, Y. Watanabe, S. Yano, M. Hirasawa, Y.-J. Pu and J. Kido, Adv. Funct. Mater., 2013, 23, 5550–5555 CrossRef CAS.
- G. Zhou, W.-Y. Wong and X. Yang, Chem. – Asian. J., 2011, 6, 1706–1727 CrossRef CAS PubMed.
- C.-L. Ho, H. Li and W.-Y. Wong, J. Organomet. Chem., 2014, 751, 261–285 CrossRef CAS.
- X. Yang, G. Zhou and W.-Y. Wong, Chem. Soc. Rev., 2015, 44, 8484–8575 RSC.
- K.-H. Kim, J.-L. Liao, S. W. Lee, B. Sim, C.-K. Moon, G.-H. Lee, H. J. Kim, Y. Chi and J.-J. Kim, Adv. Mater., 2016, 28, 2526–2532 CrossRef CAS PubMed.
- Z.-Q. Zhu, K. Klimes, S. Holloway and J. Li, Adv. Mater., 2017, 29, 1605002 CrossRef PubMed.
- Y. Chi and P.-T. Chou, Chem. Soc. Rev., 2007, 36, 1421–1431 RSC.
- H. Yersin, Top. Curr. Chem., 2004, 241, 1–26 CrossRef CAS.
- V. W.-W. Yam, S. W.-K. Choi, T.-F. Lai and W.-K. Lee, J. Chem. Soc., Dalton Trans., 1993, 1001–1002 RSC.
- K. M.-C. Wong, X. Zhu, L.-L. Hung, N. Zhu, V. W.-W. Yam and H.-S. Kwok, Chem. Commun., 2005, 2906–2908 RSC.
- V. W.-W. Yam, K. M.-C. Wong, L.-L. Hung and N. Zhu, Angew. Chem., Int. Ed., 2005, 44, 3107–3110 CrossRef CAS PubMed.
- K. M.-C. Wong, L.-L. Hung, W. H. Lam, N. Zhu and V. W.-W. Yam, J. Am. Chem. Soc., 2007, 129, 4350–4365 CrossRef CAS PubMed.
- V. K.-M. Au, K. M.-C. Wong, N. Zhu and V. W.-W. Yam, Chem. – Eur. J., 2011, 17, 130–142 CrossRef CAS PubMed.
- A. Szentkuti, M. Bachmann, J. A. Garg, O. Blacque and K. Venkatesan, Chem. – Eur. J., 2014, 20, 2585–2596 CrossRef CAS PubMed.
- J. Fernandez-Cestau, B. Bertrand, M. Blaya, G. A. Jones, T. J. Penfold and M. Bochmann, Chem. Commun., 2015, 51, 16629–16632 RSC.
- M. Bachmann, J. Terreni, O. Blacque and K. Venkatesan, Chem. – Eur. J., 2017, 23, 3837 CrossRef CAS PubMed.
- K. M.-C. Wong, M. M.-Y. Chan and V. W.-W. Yam, Adv. Mater., 2014, 26, 5558–5568 CrossRef CAS PubMed.
- R. Kumar and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 1994–2015 CrossRef CAS PubMed.
- T. von Arx, A. Szentkuti, T. N. Zehnder, O. Blacque and K. Venkatesan, J. Mater. Chem. C, 2017, 3765–3769 RSC.
- M.-C. Tang, C.-H. Lee, S.-L. Lai, M. Ng, M.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2017, 139, 9341–9349 CrossRef CAS PubMed.
- V. K.-M. Au, K. M.-C. Wong, D. P.-K. Tsang, M.-Y. Chan, N. Zhu and V. W.-W. Yam, J. Am. Chem. Soc., 2010, 132, 14273–14278 CrossRef CAS PubMed.
- V. K.-M. Au, D. P.-K. Tsang, K. M.-C. Wong, M.-Y. Chan, N. Zhu and V. W.-W. Yam, Inorg. Chem., 2013, 52, 12713–12725 CrossRef CAS PubMed.
- M.-C. Tang, D. P.-K. Tsang, M. M.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, Angew. Chem., Int. Ed., 2013, 52, 446–449 CrossRef CAS PubMed.
- M.-C. Tang, C. K.-M. Chan, D. P.-K. Tsang, Y.-C. Wong, M. M.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, Chem. – Eur. J., 2014, 20, 15233–15241 CrossRef CAS PubMed.
- M.-C. Tang, D. P.-K. Tsang, Y.-C. Wong, M.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2014, 136, 17861–17868 CrossRef CAS PubMed.
- N. D. McClenaghan, R. Passalacqua, F. Loiseau, S. Campagna, B. Verheyde, A. Hameurlaine and W. Dehaen, J. Am. Chem. Soc., 2003, 125, 5356–5365 CrossRef PubMed.
- G. Zhou, W.-Y. Wong, B. Yao, Z. Xie and L. Wang, Angew. Chem., Int. Ed., 2007, 46, 1149–1151 CrossRef CAS PubMed.
- P. L. Burn, S. C. Lo and I. D. W. Samuel, Adv. Mater., 2007, 19, 1675–1688 CrossRef CAS.
- T. Qin, J. Ding, L. Wang, M. Baumgarten, G. Zhou and K. Müllen, J. Am. Chem. Soc., 2009, 131, 14329–14336 CrossRef CAS PubMed.
- J. Ding, B. Wang, Z. Yue, B. Yao, Z. Xie, Y. Cheng, L. Wang, X. Jing and F. Wang, Angew. Chem., Int. Ed., 2009, 48, 6664–6666 CrossRef CAS PubMed.
- F. K.-W. Kong, M.-C. Tang, Y.-C. Wong, M.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2016, 138, 6281–6291 CrossRef CAS PubMed.
- X. Li, Z. Chen, Q. Zhao, L. Shen, F. Li, T. Yi, Y. Cao and C. Huang, Inorg. Chem., 2007, 46, 5518–5527 CrossRef CAS PubMed.
- G. A. Crosby and J. N. Demas, J. Phys. Chem., 1971, 75, 991–1024 CrossRef CAS.
- J. Van Houten and R. J. Watts, J. Am. Chem. Soc., 1976, 98, 4853–4858 CrossRef CAS.
- J.-W. Lee, B.-K. Kim, J.-H. Kim, W.-S. Shin and S.-H. Jin, Bull. Korean Chem. Soc., 2005, 26, 1790 CrossRef CAS.
- K.-H. Wong, K.-K. Cheung, M. C.-W. Chan and C.-M. Che, Organometallics, 1998, 17, 3505–3511 CrossRef CAS.
|
| This journal is © the Partner Organisations 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 and
Vivian Wing-Wah
Yam
and
Vivian Wing-Wah
Yam