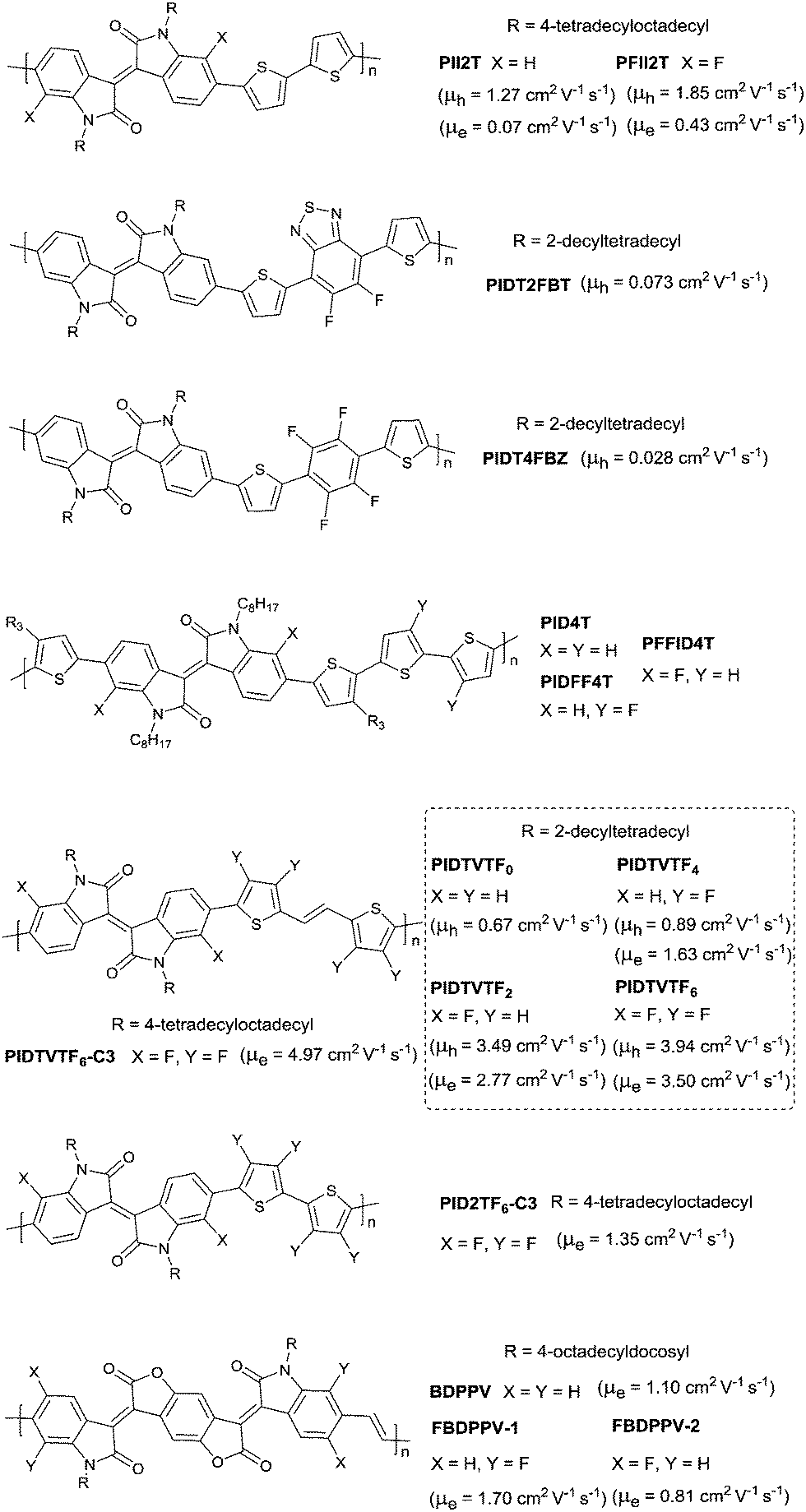
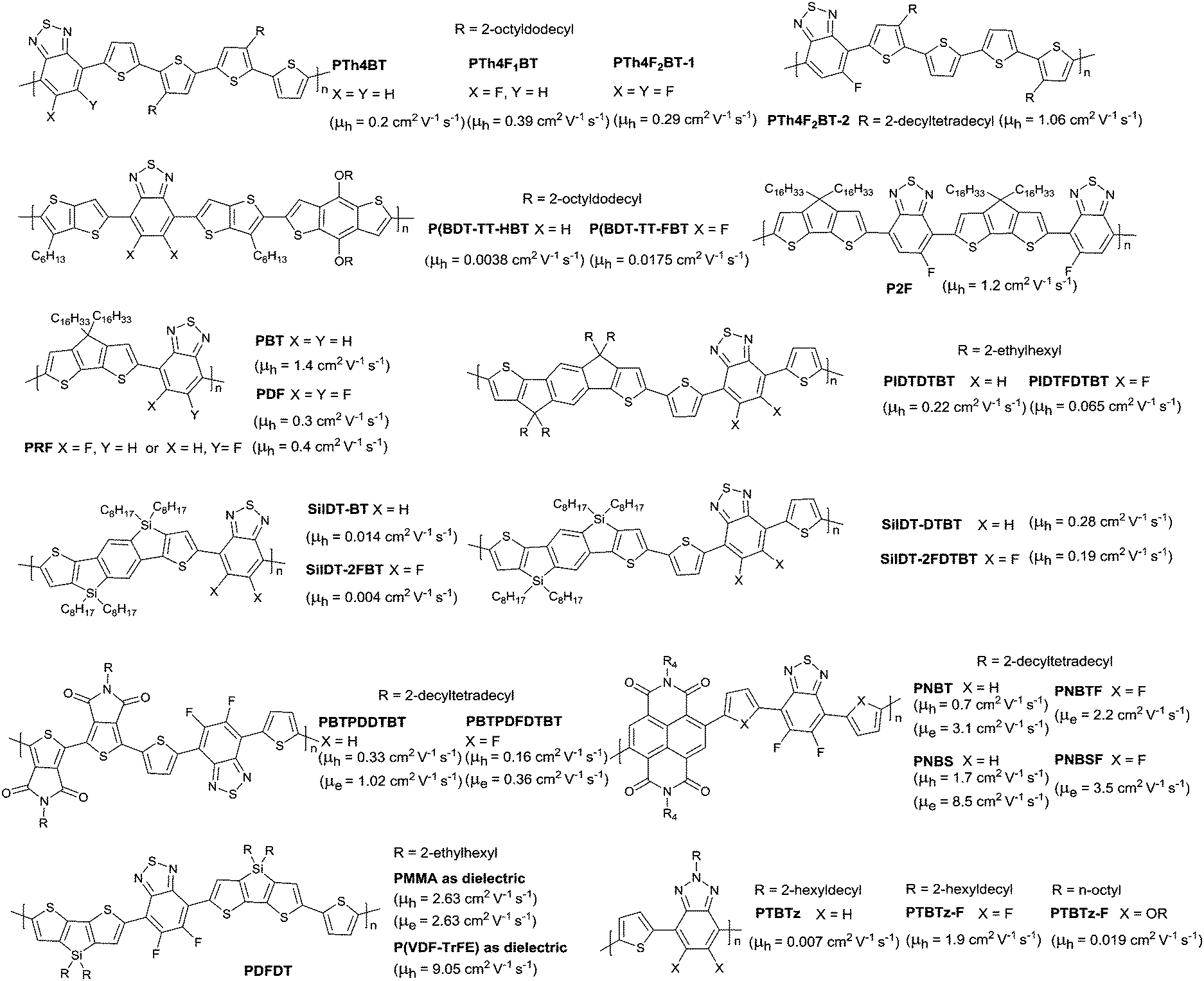
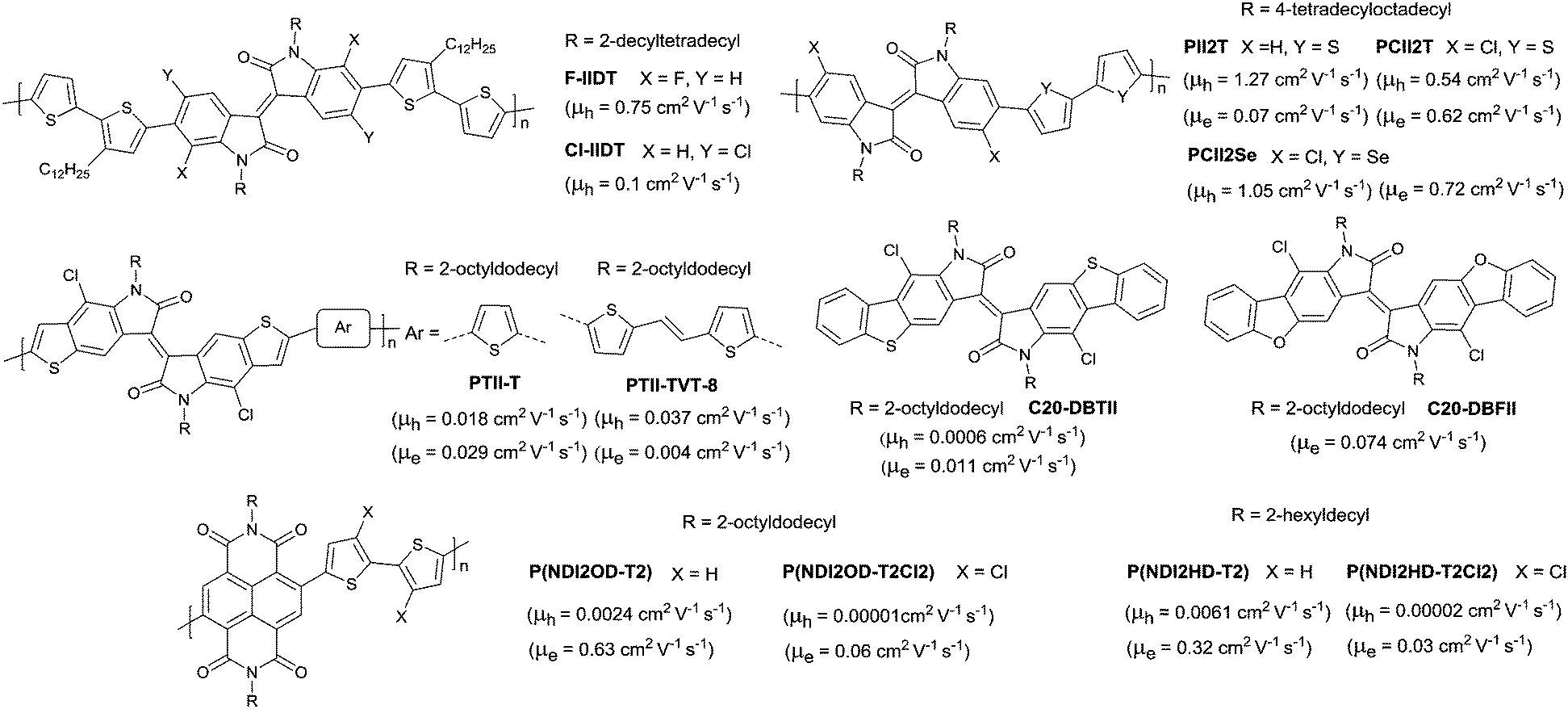
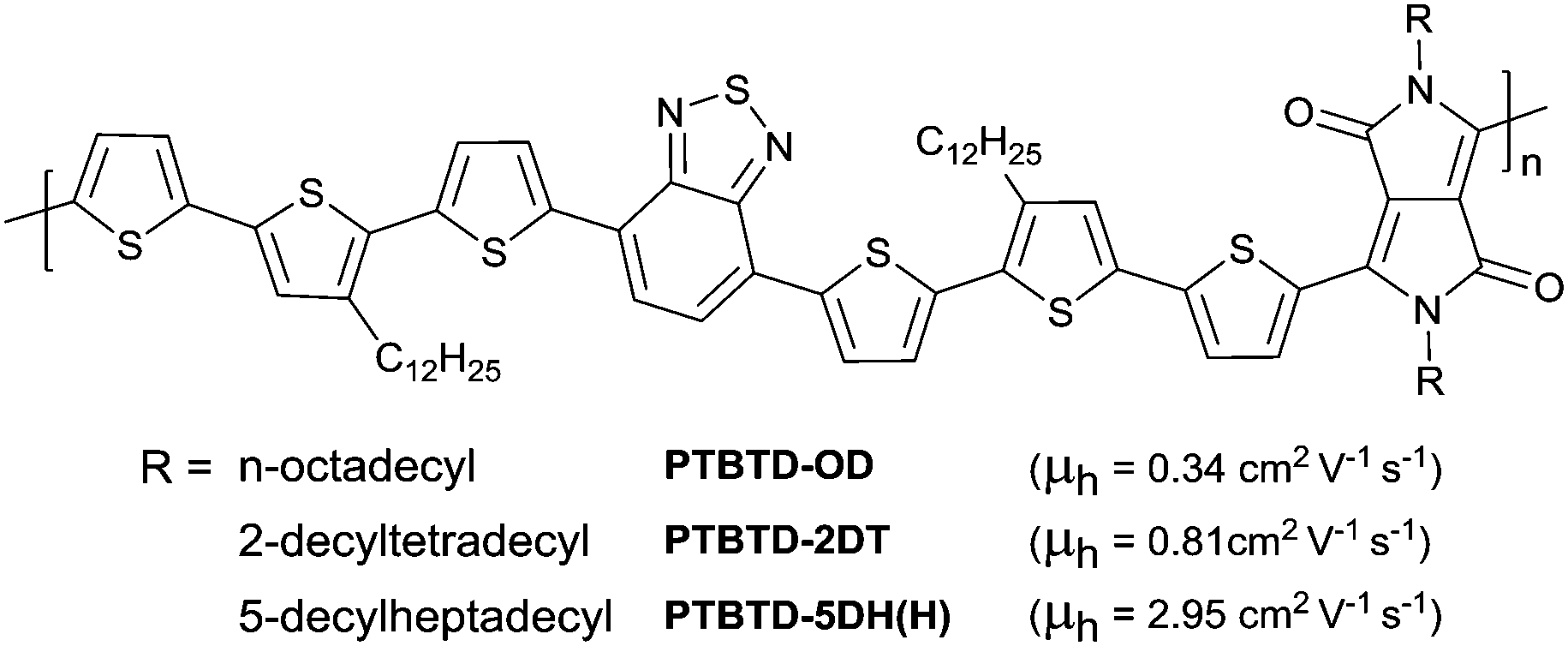
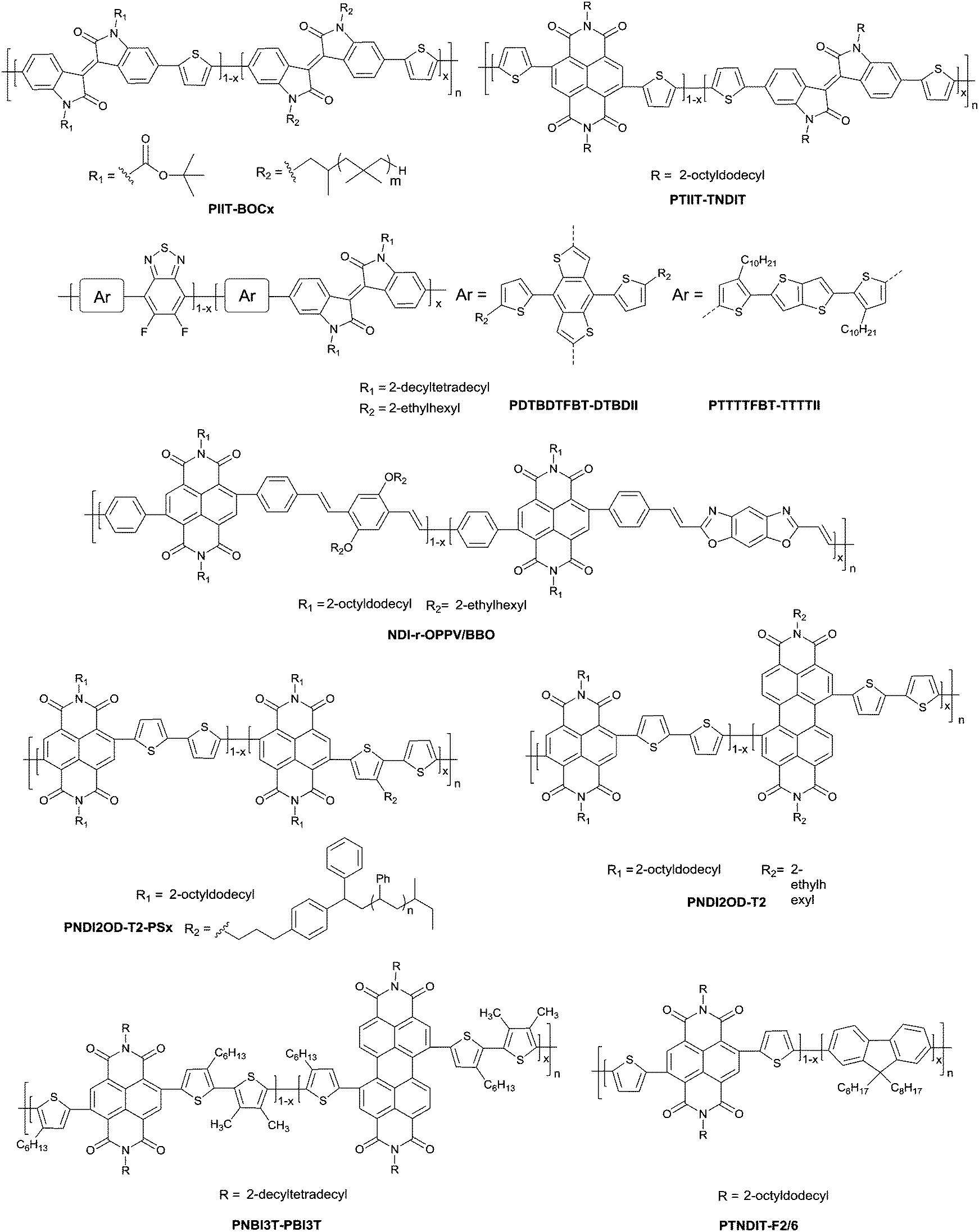
DOI:
10.1039/C7QM00169J
(Review Article)
Mater. Chem. Front., 2017,
1, 2423-2456
Design and effective synthesis methods for high-performance polymer semiconductors in organic field-effect transistors
Received
17th April 2017
, Accepted 3rd July 2017
First published on 7th July 2017
Abstract
To date, versatile polymer semiconductors have been reported for field-effect transistors (FETs). And the third-generation donor–acceptor (D–A) polymers have been among the most intensively studied semiconductors. Meanwhile, there are a variety of methods adopted for the enhancement of performance. To the best of our knowledge, a p-type polymer semiconductor with a highest hole mobility of 52.7 cm2 V−1 s−1, an n-type polymer semiconductor with a highest electron mobility of 8.5 cm2 V−1 s−1 and a balanced ambipolar semiconductor with the highest both hole and electron mobility of over 4 cm2 V−1 s−1 have been achieved. This review describes building block selection, backbone halogenation, side chain engineering and random copolymerization, which are the effective synthesis approaches applied in this field, affording assistance for developing high-performance polymer semiconductors in the future.

Longxian Shi
| Longxian Shi received his BS degree from Wuhan Textile University in 2014. Since September 2015, he has been a master’s student in the Institute of Chemistry, Chinese Academy of Sciences (ICCAS). His research interests focus on molecular materials and devices for organic field-effect transistors. |

Yunlong Guo
| Yunlong Guo received his PhD degree in physical chemistry from the Institute of Chemistry, Chinese Academy of Sciences (ICCAS) in 2010. In April 2016, he became a project associate professor at the Department of Chemistry, University of Tokyo. Now, he is a professor at ICCAS. His research interests include organic–inorganic hybrid perovskite electronics and functional organic field-effect transistors. |

Wenping Hu
| Wenping Hu is a professor of Tianjin University and the Institute of Chemistry, Chinese Academy of Sciences (ICCAS). He received his PhD from ICCAS in 1999. He then joined Osaka University as a research fellow of the Japan Society for the Promotion of Sciences and Stuttgart University as an Alexander von Humboldt fellow. In 2003 he worked for Nippon Telephone and Telegraph, and then returned to ICCAS. In 2016, he moved to Tianjin University. His research focuses on molecular electronics, and he has more than 390 refereed publications with over 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 citations. 000 citations. |

Yunqi Liu
| Yunqi Liu is a professor at the Institute of Chemistry, Chinese Academy of Sciences (ICCAS). He graduated from Nanjing University in 1975, and received a doctorate from the Tokyo Institute of Technology, Japan, in 1991. He was selected as an Academician of CAS in 2015. His research interests include molecular materials and devices, the synthesis and applications of carbon nanomaterials, and organic electronics. He has more than 500 refereed publications with over 22000 citations. |
1. Introduction
Since Tsumura1,2 and his co-workers fabricated the first polythiophene-based organic field-effect transistor (OFET), polymer semiconductors have been widely studied in organic field-effect transistors, as they are flexible, lightweight, transparent, solution-processable and modifiable. Compared to the inorganic semiconductors, the solution-processable polymer semiconductors have been recognized as promising candidates for commercial applications due to their low cost. Among the polymer semiconductors, the third-generation D–A conjugated polymers are more attractive than all donor and all acceptor conjugated polymers because of their relatively favourable charge transport with a low bandgap. In addition, several designs and synthesis means of these materials have been proposed, and these polymers showed excellent OFET behaviour when combined with device architectures (top-gate bottom-contact (TGBC), top-gate top-contact (TGTC), bottom-gate bottom-contact (BGBC) and bottom-gate bottom-contact (BGBC)). This review emphasizes the discussion of design and effective synthesis methods including building block selection, backbone halogenation, side chain engineering and random copolymerization, aiming to provide a valuable message for the development of high-performance polymer semiconductors. For building blocks, diketopyrrolopyrroles (DPPs) and isoindigo (IID) and other typically reported structures will be fully discussed. Then, the section on backbone halogenation will summarize the effect of fluorination and chlorination on the properties of the polymer including energy levels, molecular packing order and coplanarity. After that, the function of side chain engineering for controlling the solubility and condensed state structure of a rigid conjugated polymer will be unfolded. Finally, random copolymerization as an effective way to adjust the various properties of polymer semiconductors will be mentioned.
2. Building blocks
Generally, building blocks in a conjugated polymer can be classified as acceptors and donors according to their electron-donating or electron-withdrawing role in the conjugated system. Obviously, the rational design and introduction of building blocks is a basic and important step for the exploration of high-performance semiconductors in OFETs. Up to now, various kinds of molecules have been used as acceptors in conjugated polymers, such as DPPs, IID, imides, etc. Different acceptors energy matched by suitable donors can generate polymers with a favorable behavior. In this section, a series of common acceptors accompanied by different donors will be discussed.
2.1 Diketopyrrolopyrroles
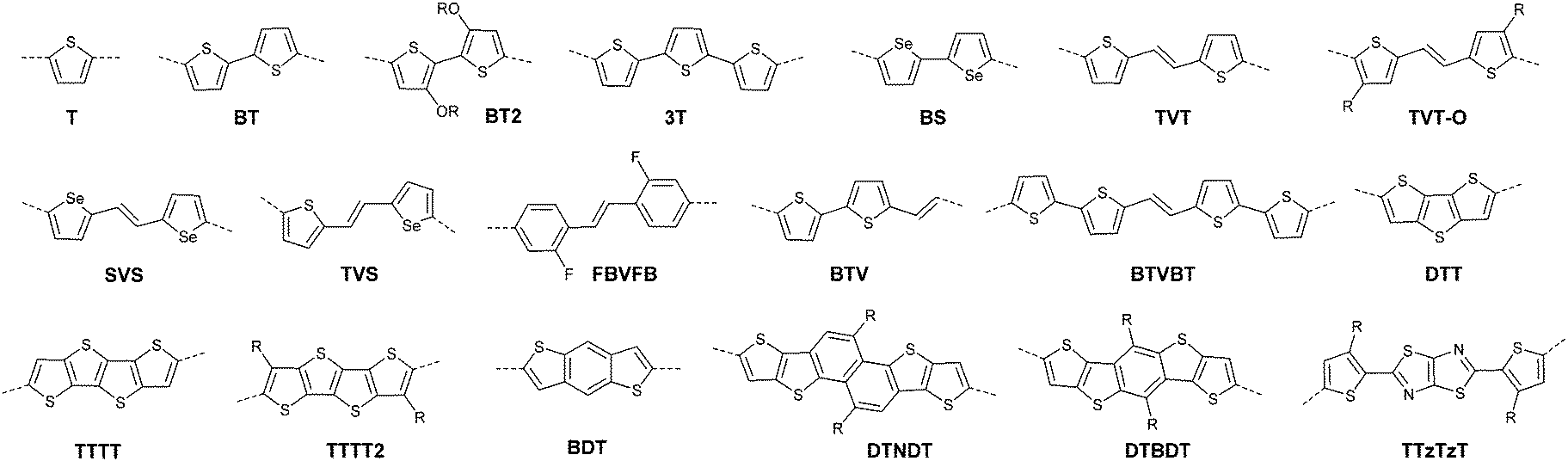
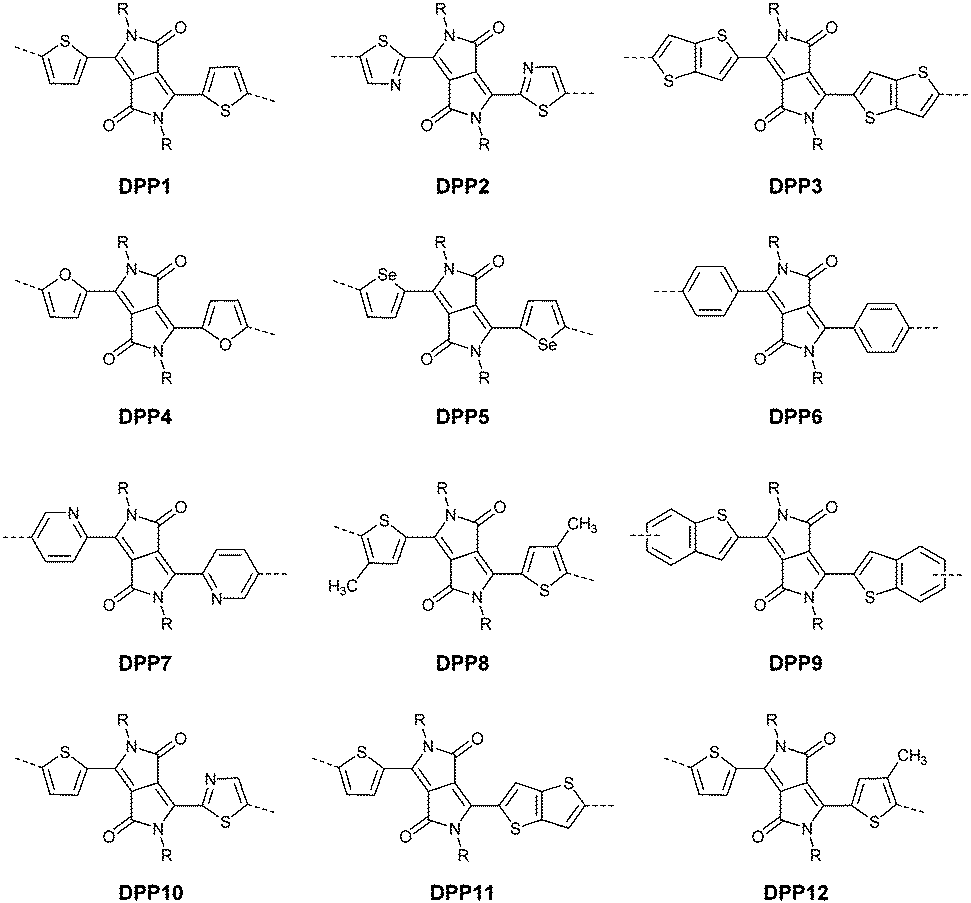
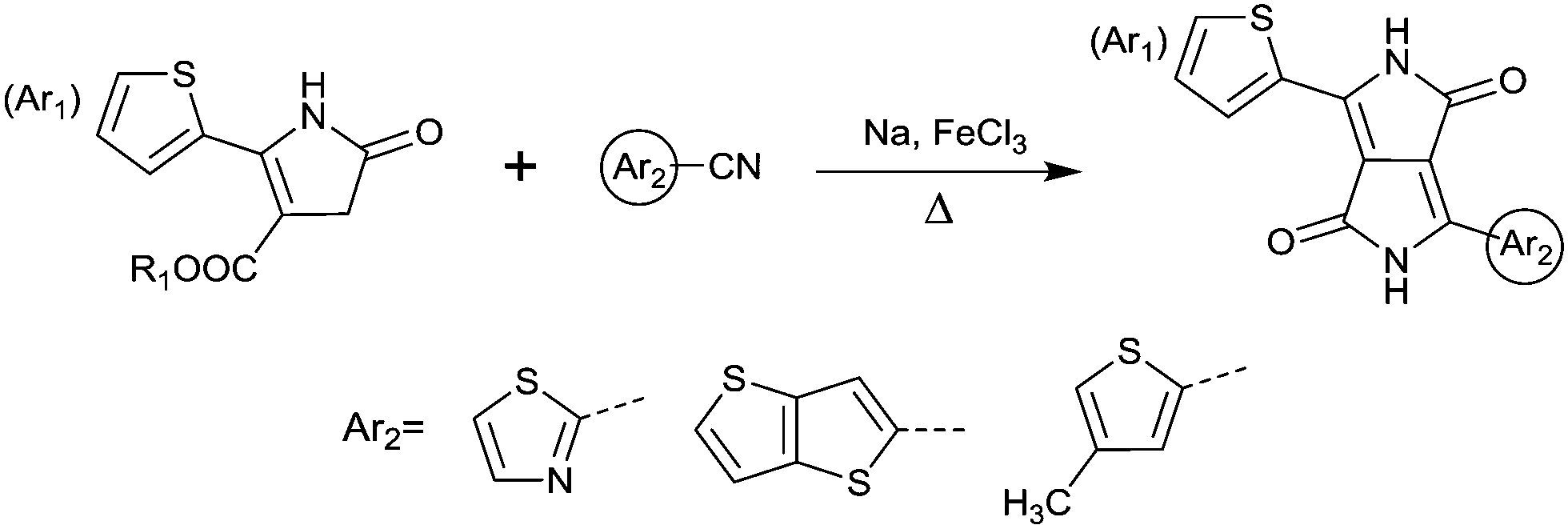
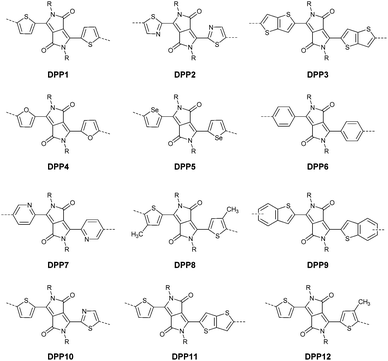
DPPs are excellent acceptors, which possess a planar conjugated bicyclic lactam unit that induces low highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) levels as it is of high electron-withdrawing ability.3Scheme 1 shows a common route for DPP synthesis. It is worth mentioning that the reaction rate could be improved by enhancing the concentration of succinate and temperature. In addition, the elevated temperature and slow addition of succinic ester is needed.4 Different aromatic nitrile original materials give a DPP core with different adjacent aromatic structures, which could be a way to adjust the steric hindrance between the DPP core and the adjacent aromatic unit, the planarity of the molecule and the energy levels. Besides, the different steric hindrances also affect the yields.4
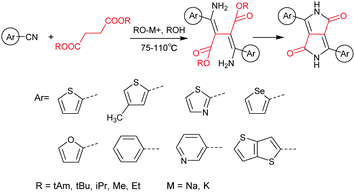 |
| | Scheme 1 A general method for symmetric DPP synthesis. | |
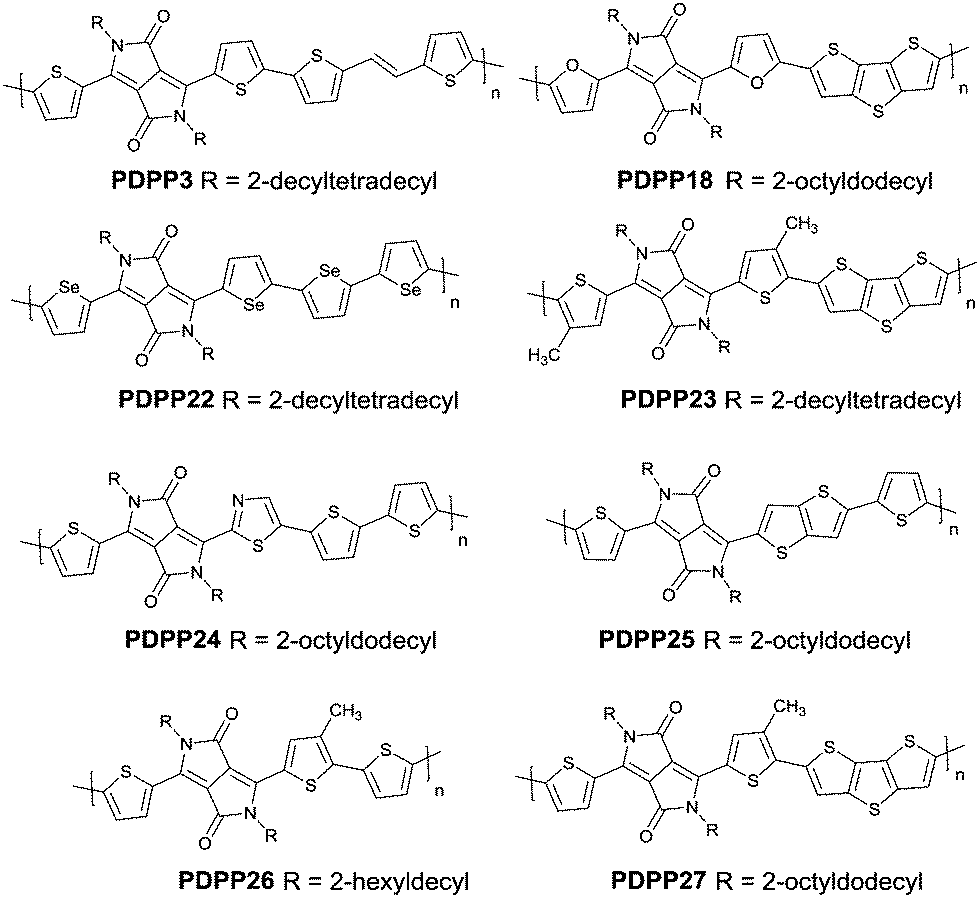
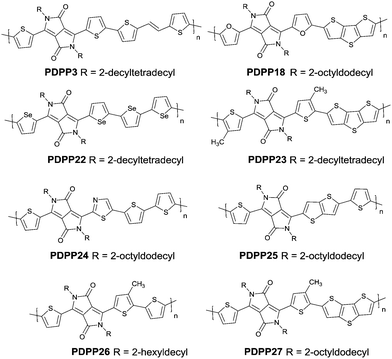
Studies of the DPP-based semiconductors were mainly focused on DPP1 (Fig. 1). As listed in Tables 1 and 2, most high-performance p-type (μh > 1 cm2 V−1 s−1), and even n-type and ambipolar DPP-based polymer semiconductors use DPP1 as the charge acceptor, because DPP1 not only has the fine properties of the core that DPP derivatives possess but also the fine coplanarity between the core and flanked aryl units resulting from the existence of hydrogen⋯oxygen (H⋯O) non-covalent interactions. To the best of our knowledge, the first effective DPP-based polymer for OFETs was reported by Li25 and co-workers in 2010 via DPP1 as the acceptor and thieno[3,2-b]thiophene (TT) as the donor. This fused ring structure polymer showed a p-type semiconductor behaviour with a hole mobility of 0.94 cm2 V−1 s−1. Later on, Chen7et al. utilized (E)-2-(2-(thiophen-2-yl)vinyl)thiophene (TVT) units as the donor monomer and DPP1 as the acceptor monomer to obtain the polymer PDPP3 (Fig. 2 and Table 1) with a high hole mobility of up to 8.20 cm2 V−1 s−1.
Table 1 Electronic parameters for high-performance p-type (μh > 1 cm2 V−1 s−1) DPP-based polymer semiconductors
|
|
| Polymer |
Acceptor |
Donor |
M
n (kDa)/PDI |
Device structure |
HOMO/LUMO (eV) |
Max. μh (cm2 V−1 s−1) |
I
on/Ioff |
Ref. |
| PDPP1 |
DPP1 |
BT2 |
63.7/2.36 |
BGBC |
−5.02/−3.72 |
2.01 |
104–105 |
5
|
| PDPP2 |
DPP1 |
3T |
44.0/1.92 |
BGTC |
−5.12/−3.55 |
3.46 |
108 |
6
|
| PDPP3 |
DPP1 |
TVT |
73.5/2.49 |
BGBC |
−5.28/— |
8.20 |
105–107 |
7
|
| PDPP4 |
DPP1 |
SVS |
35.8/1.62 |
BGTC |
−5.27/— |
12.04 |
>106 |
8
|
| PDPP5 |
DPP1 |
TVS |
91.0/1.61 |
BGTC |
— |
7.60 |
— |
9
|
| PDPP6 |
DPP1 |
FBVFB |
80.2/2.46 |
BGBC |
−5.40/−3.59 |
1.48 |
106–107 |
10
|
| PDPP7 |
DPP1 |
BTVBT |
39.0/2.10 |
BGTC |
−5.07/−3.83 |
1.40 |
>107 |
11
|
| PDPP8 |
DPP1 |
DTT |
29.3/N/A |
BGBC |
−5.27/−3.88 |
3.16 |
3 × 104 |
12
|
| PDPP9 |
DPP1 |
TTTT |
58.1/2.95 |
BGTC |
−5.21/−3.91 |
3.20 |
>106 |
13
|
| PDPP10 |
DPP1 |
TTTT2 |
20.5/3.25 |
BGTC |
−5.3/−3.95 |
2.10 |
3 × 106 |
14
|
| PDPP11 |
DPP1 |
BDT |
56.8/2.41 |
BGTC |
−5.36/−4.02 |
1.31 |
>106 |
13
|
| PDPP12 |
DPP1 |
DTNDT |
56.9/2.93 |
BGTC |
−5.15/−3.48 |
1.80 |
105 |
15
|
| PDPP13 |
DPP1 |
DTBDT |
28.9/2.60 |
BGTC |
−5.39/−3.38 |
2.70 |
107 |
16
|
| PDPP14 |
DPP1 |
TTZTZT |
17.0/4.00 |
BGTC |
−5.14/−3.50 |
1.23 |
105–106 |
17
|
| PDPP15 |
DPP1 |
BS |
19.3/3.25 |
BGTC |
— |
1.50 |
106 |
18
|
| PDPP16 |
DPP1 |
TVT-O |
10.1/2.54 |
BGTC |
−5.10/−3.30 |
1.69 |
— |
19
|
| PDPP17 |
DPP4 |
TVT |
63.8/3.26 |
BGBC |
−5.34/−3.96 |
1.90 |
>106 |
20
|
| PDPP18 |
DPP4 |
DTT |
24.0/2.29 |
BGTC |
— |
4.10 |
102–103 |
21
|
| PDPP19 |
DPP5 |
BT |
114.0/2.41 |
BGBC |
−5.18/−3.86 |
1.79 |
>107 |
22
|
| PDPP20 |
DPP5 |
BS |
217.3/3.21 |
BGBC |
−5.16/−3.90 |
2.08 |
>106 |
22
|
| PDPP21 |
DPP5 |
TVT |
140.7/2.88 |
BGBC |
−5.12/−3.79 |
4.15 |
>107 |
22
|
| PDPP22 |
DPP5 |
SVS |
347.0/3.10 |
BGBC |
−5.10/−3.83 |
5.23 |
>107 |
22
|
| PDPP23 |
DPP8 |
DTT |
29.3/1.38 |
BGBC |
−5.19/−3.85 |
5.27 |
4 × 103 |
12
|
| PDPP24 |
DPP10 |
BT |
75.4/1.57 |
BGBC |
−5.48/−4.03 |
3.05 |
8 × 106 |
23
|
| PDPP25 |
DPP11 |
T |
205.9/1.93 |
BGBC |
— |
5.87 |
2 × 105 |
24
|
| PDPP26 |
DPP12 |
T |
138.8/1.65 |
BGBC |
— |
12.5 |
7 × 104 |
24
|
| PDPP27 |
DPP12 |
DTT |
34.4/1.41 |
BGBC |
−5.25/−3.87 |
5.32 |
7 × 104 |
12
|
 |
| | Fig. 1 Chemical structures of DPP-based acceptors. | |
 |
| | Fig. 2 Chemical structures of PDPP3, PDPP18, PDPP22, PDPP23, PDPP24, PDPP25, PDPP26 and PDPP27. | |
Table 2 Electronic parameters for high-performance n-type and ambipolar DPP-based polymer semiconductors
|
|
| Polymer |
Acceptor |
Donor |
M
n (kDa)/PDI |
Device structure |
HOMO/LUMO (eV) |
Max. μh (cm2 V−1 s−1) |
Max. μe (cm2 V−1 s−1) |
Ref. |
| PDPP28 |
DPP1 |
2CNT |
23.3/2.52 |
BGTC |
−5.48/−3.80 |
— |
0.09 |
47
|
| PDPP29 |
DPP1 |
2CNB |
67.5/1.80 |
BGTC |
−5.63/−3.71 |
— |
0.18 |
47
|
| PDPP30 |
DPP1 |
2Tz |
64.0/3.60 |
BGTC |
−5.88/−2.14 |
— |
0.31 |
48
|
| PDPP31 |
DPP2 |
T |
93.3/3.05 |
TGBC |
−5.63/−4.00 |
— |
0.13 |
26
|
| PDPP32 |
DPP1 |
V |
48.4/2.00 |
TGBC |
−5.28/−3.55 |
0.37 |
0.78 |
50
|
| PDPP33 |
DPP1 |
TV |
49.2/1.90 |
TGBC |
−5.18/−3.39 |
2.96 |
0.36 |
50
|
| PDPP34 |
DPP1 |
BTV |
23.5/2.90 |
TGBC |
−5.13/−3.23 |
1.88 |
0.04 |
50
|
| PDPP35 |
DPP1 |
T |
33.7/6.08 |
BGTC |
−5.30/−4.00 |
1.57 |
0.18 |
51
|
| PDPP36 |
DPP1 |
2FT |
40.8/3.97 |
BGBC |
— |
0.13 |
0.12 |
39
|
| PDPP37 |
DPP1 |
S |
23.3/— |
BGTC |
−5.10/−3.49 |
8.84 |
4.34 |
52 and 53
|
| PDPP38 |
DPP1 |
BTz |
42.4/1.42 |
BGTC |
−5.20/−4.00 |
0.35 |
0.40 |
54
|
| PDPP39 |
DPP1 |
FBT |
24.0/2.01 |
BGTC |
−5.38/−4.18 |
0.21 |
0.42 |
55
|
| PDPP40 |
DPP1 |
2FBT |
25.0/2.34 |
BGTC |
−5.48/−4.22 |
0.10 |
0.30 |
55
|
| PDPP41 |
DPP1 |
PHI |
25.5/2.15 |
BGTC |
−5.30/−3.75 |
0.13 |
0.52 |
56
|
| PDPP42 |
DPP1 |
V |
56.2/2.74 |
BGBC |
— |
0.17 |
0.017 |
40
|
| PDPP43 |
DPP1 |
BF |
102/4.30 |
TGBC |
−5.67/−4.24 |
0.36 |
0.41 |
57
|
| PDPP44 |
DPP1 |
CNTVT |
125.2/1.51 |
BGTC |
−5.77/−3.92 |
0.749 |
7.00 |
58
|
| PDPP45 |
DPP1 |
4FTVT |
59.9/4.91 |
BGTC |
−5.36/−3.50 |
3.40 |
5.86 |
59
|
| PDPP46 |
DPP1 |
BTzVBTz |
20.6/4.99 |
BGTC |
−5.34/−3.43 |
0.32 |
0.13 |
60
|
| PDPP47 |
DPP1 |
TAT |
82.1/2.30 |
BGTC |
−5.39/−3.89 |
2.19 |
0.38 |
49
|
| PDPP48 |
DPP1 |
BTzABTz |
11.0/1.81 |
BGTC |
−5.31/−3.53 |
0.24 |
0.15 |
60
|
| PDPP49 |
DPP1 |
TT |
50/3.87 |
TGBC |
−5.33/−4.07 |
1.60 |
2.00 |
61
|
| PDPP50 |
BBT |
DPP1 |
8.8/1.80 |
BGBC |
−4.55/−3.90 |
1.17 |
1.32 |
62
|
| PDPP51 |
DPP1 |
BTP |
32.8/2.77 |
BGBC |
−5.48/−3.98 |
1.43 |
0.13 |
63
|
| PDPP52 |
DPP3 |
T |
14/5.40 |
TGBC |
−5.06/−3.68 |
1.95 |
0.071 |
29 and 30
|
| PDPP53 |
DPP4 |
2FT |
16.1/3.13 |
BGTC |
−5.40/−4.03 |
0.26 |
0.12 |
35
|
| PDPP54 |
DPP4 |
BTz |
204.6/2.20 |
BGTC |
−5.37/−3.74 |
0.20 |
0.56 |
33
|
| PDPP55 |
DPP4 |
T4FBT |
12.08/1.71 |
BGTC |
−5.48/−4.11 |
0.40 |
0.12 |
34
|
| PDPP56 |
DPP5 |
S |
70/3.00 |
BGBC |
−5.20/−4.02 |
0.1 |
0.1 |
38
|
| PDPP57 |
DPP5 |
TT |
100/2.50 |
BGBC |
−5.10/−3.92 |
1.1 |
0.15 |
38
|
| PDPP58 |
DPP7 |
BT |
26.3/3.56 |
TGBC |
−5.69/−4.33 |
2.78 |
6.30 |
41
|
| PDPP59 |
DPP7 |
T2FBTT |
58.4/1.64 |
BGBC |
−5.66/−4.02 |
0.24 |
0.65 |
43
|
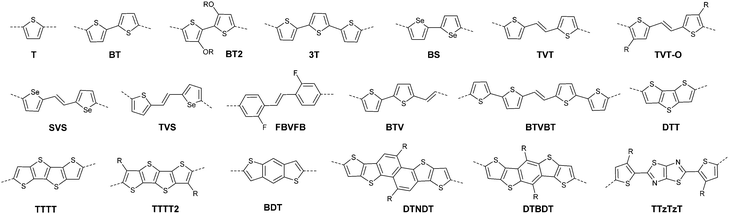
After that, DPP derivatives DPP2,26,27 DPP3,28–30 DPP4,20,21,31–35 DPP5,22,36–38 DPP6,36,39,40 DPP7,39,41–43 DPP8,12 DPP9,44 DPP10,23 DPP11,24 and DPP1212,24 were also intensively investigated in a previous study and are shown in Fig. 2. Among them, DPP1–DPP9 are all symmetric units with thiophene, thiazole, TT, furan, selenophene, benzene, pyridine, 3-methylthiophene and benzothiophene, respectively, while DPP10–DPP12 are asymmetric units. The HOMO and LUMO levels changed following variation of the flanking aryl units. Besides, the HOMO delocalization and coplanarity of a molecule could also be affected by the flanking aryl units, eventually resulting in good or bad charge-carrier hopping between the molecules.3 DPP2 is not a good structure for charge transport as the structure of the two thiazole subunits destroyed the H⋯O non-covalent interaction. And, there are few reports regarding its use for OFETs except organic solar cells. For DPP3, the stronger electron-donating properties of TT than thiophene give DPP3 higher HOMO and LUMO levels, which is not beneficial for n-type and ambipolar performance of semiconductors. Additionally, the larger size of TT than thiophene also has a negative impact on the solubility of the polymer, which causes poorer molecular packing during the device fabrication process and eventually leads to undesirable electrical properties. Therefore, few DPP3-based conjugated polymers exhibit high OFET performance. It is worth mentioning that researchers have proved that DPP4 flanked by furan possesses better solubility in chlorine-free solution than analogous thiophene-based DPP1.45 Chang21 and co-workers also synthesized a high-performance polymer PDPP18 (Fig. 2 and Table 1) with a hole mobility of up to 4.10 cm2 V−1 s−1 based on the acceptor DPP4. Selenium, as an element in the same group as sulfur, possesses more electrons and is of a larger size than sulfur. Based on this, the corresponding HOMO–LUMO levels of DPP5 flanked by selenophene are also higher than DPP1 flanked by thiophene, which has also been confirmed by comparison of the energy levels of the analogues between the DPP1- and DPP5-based polymers listed in Tables 1 and 2. High OFET performance DPP5-based polymer semiconductors also emerge in an endless stream. For example, in 2015, Choi et al.22 obtained a high p-type polymer PDPP22 with a hole mobility of up to 5.23 cm2 V−1 s−1. From the theoretical calculations, it was revealed that DPP6 flanked by two phenyl substituents had a larger band gap and a less coplanar structure, which is detrimental to its amibipolar properties and intermolecular charge transfer.41 So almost no high-performance DPP6-based polymer semiconductors have been reported. However, when the two pyridine units were adjacent to the DPP core instead of benzene or thiophene, low-lying HOMO–LUMO levels and fine coplanarity were observed from calculations and experiments,41 which benefited the electrical performance. More detailed discussion on such a structure will be displayed in the later content. For DPP8 and DPP9, the HOMO–LUMO levels are adjusted by methyl substitution or phenyl fusion. As they are new DPP-based building blocks, their OFET performance still needs to be investigated.
Different from polymers based on symmetric units, the asymmetric units DPP10–DPP12 can provide a conjugated polymer with some special properties such as good solubility. As the two different flanking aromatic substituents are adjacent to the DPP core, the synthesis method of asymmetric DPPs is different from the methods used for symmetric DPPs as shown in Scheme 1, which required two steps to obtain a high yield by linking two different kinds of aromatic nitriles adjacent to the molecular backbone (Scheme 2). Li’s group12,23,24 found that the asymmetric conjugated structure of the DPP-based molecule imparted a less preferential order and good solubility in non-chlorinated solvents for the resulting polymer. For example, DPP12, as an acceptor monomer, with adjacent aromatic substituents of thiophene and 3-methylthiophene on each side, copolymerized with thiophene contributes a high hole mobility to the PDPP26 polymer (Fig. 2 and Table 1) at 12.5 cm2 V−1 s−1. In addition, the hole mobility of asymmetric polymers PDPP2423 (Fig. 2 and Table 1), PDPP2524 (Fig. 2 and Table 1), and PDPP2712 (Fig. 2 and Table 1) was lower than that of PDPP28, with the data of 3.05, 5.87 and 5.32 cm2 V−1 s−1, respectively.
 |
| | Scheme 2 Syntheses of asymmetric DPPs (DPP10, DPP11, and DPP12). | |
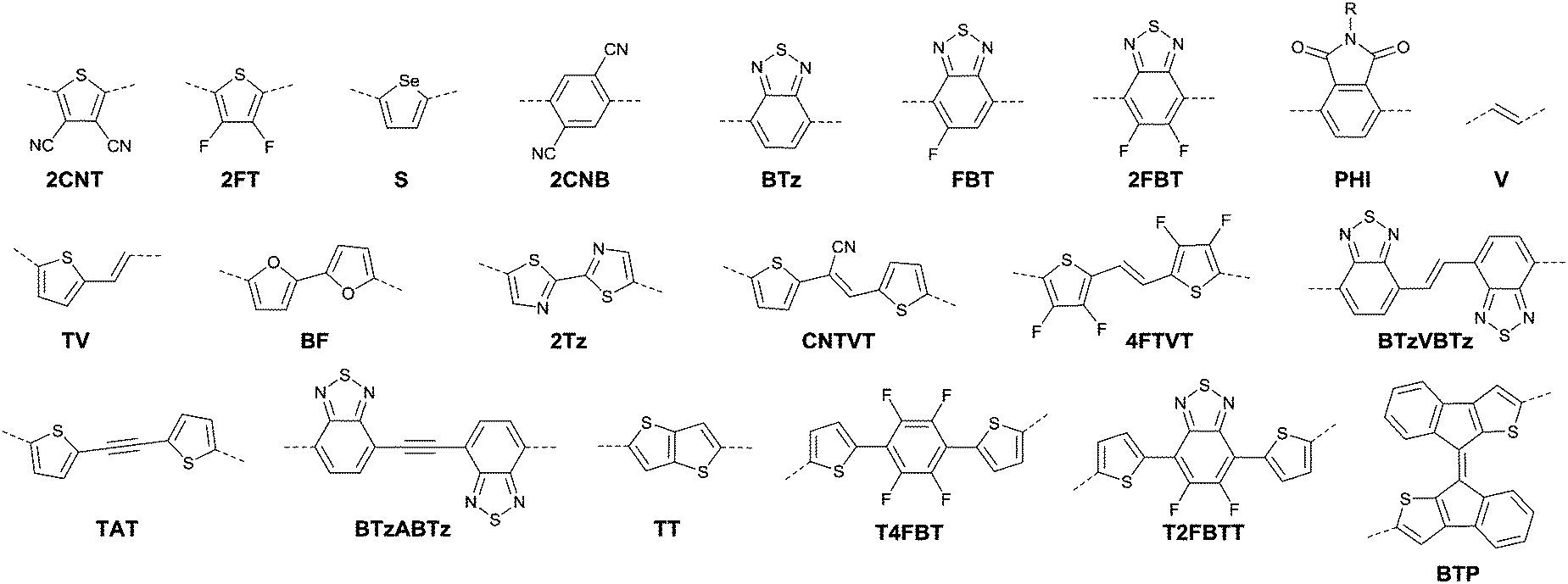
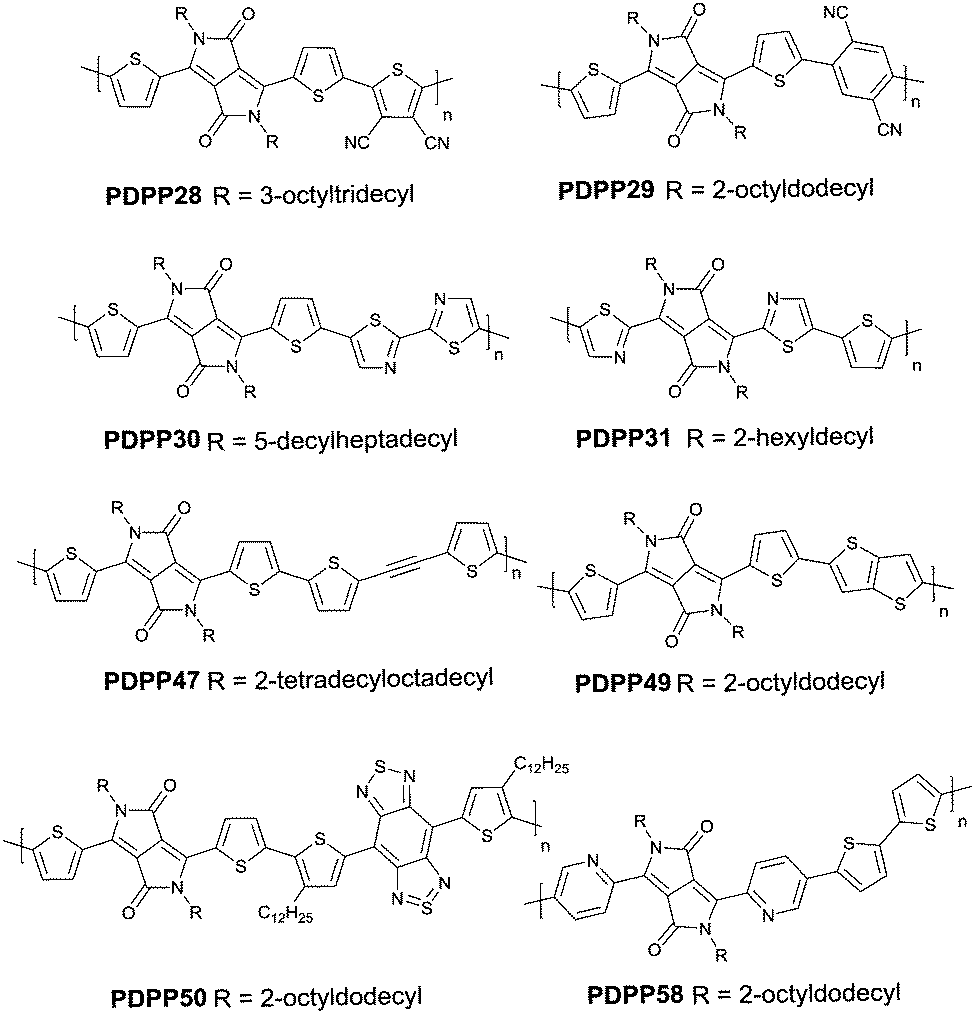
Compared with the p-type polymers, the high performance of n-type and ambipolar behaviour is less reported, resulting from the limited electron acceptors for polymer semiconductors to be of a LUMO energy level lower than −4 eV for the facile injection and efficient transport of electrons.41,46 On the other hand, the present knowledge of n-type and ambipolar polymers constructed from DPP acceptors is still in its infancy. As is shown in Table 2, PDPP28–PDPP3126,47,48 are n-type DPP-based polymers with a mobility lower than 1 cm2 V−1 s−1. In one of the early studies, Sun et al.41 reported a DPP-based ambipolar polymer PDPP58 (Fig. 3 and Table 2) with a relatively high hole mobility (μh = 2.78 cm2 V−1 s−1) and the highest electron mobility (μe = 6.30 cm2 V−1 s−1). Furthermore, this is the first time that DPP7 as the acceptor monomer of PDPP58 was introduced into a polymer semiconductor. Notably, DPP7 has a highly coplanar structure because of the weak steric hindrance between the 2-pyridinyl substituents and the DPP core. Apart from that, pyridine is a relatively electron-deficient structure, as a result of which, the LUMO energy level could be reduced. Thus, PDPP59 (Table 2) reached a high electron mobility and excellent ambipolar properties. Besides, Yun49 and co-workers introduced the donor 2-[2-(thiophen-2-yl)ethynyl]thiophene (TAT) containing a structure of acetylene into the polymer PDPP47 (Fig. 3 and Table 2). Compared with the polymer PDPP3 whose linkage is vinylene, PDPP47 showed ambipolar behaviour caused by the more electron-withdrawing property of acetylene and conformation-insensitive charge transport. Chen et al.61 optimized the device architecture, charge injection and processing of the polymer PDPP49 (Fig. 3 and Table 2) reported for the first time by Li et al.25 After optimization, PDPP49 showed ambipolar behaviour with a hole mobility of up to 1.60 cm2 V−1 s−1 and an electron mobility of up to 2.00 cm2 V−1 s−1. Jonathan D. Yuen62 prepared the polymer PDPP50 (Fig. 3 and Table 2), the benzobisthiadiazole unit of which possesses a high electron-withdrawing ability that is even higher than that of DPP. As a result, DPP was a donor and benzobisthiadiazole was an acceptor in the polymer PDPP50, which presented an excellent fine ambipolar property with a hole mobility of 1.17 cm2 V−1 s−1 and an electron mobility of 1.32 cm2 V−1 s−1.
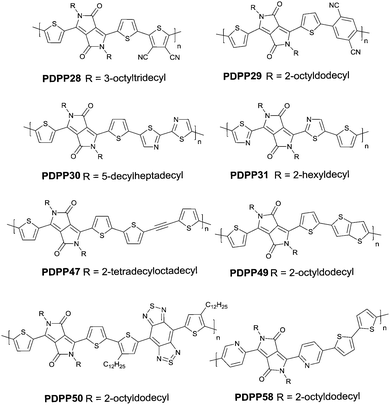 |
| | Fig. 3 Chemical structures of PDPP28, PDPP29, PDPP30, PDPP31, PDPP47, PDPP49, PDPP50 and PDPP58. | |
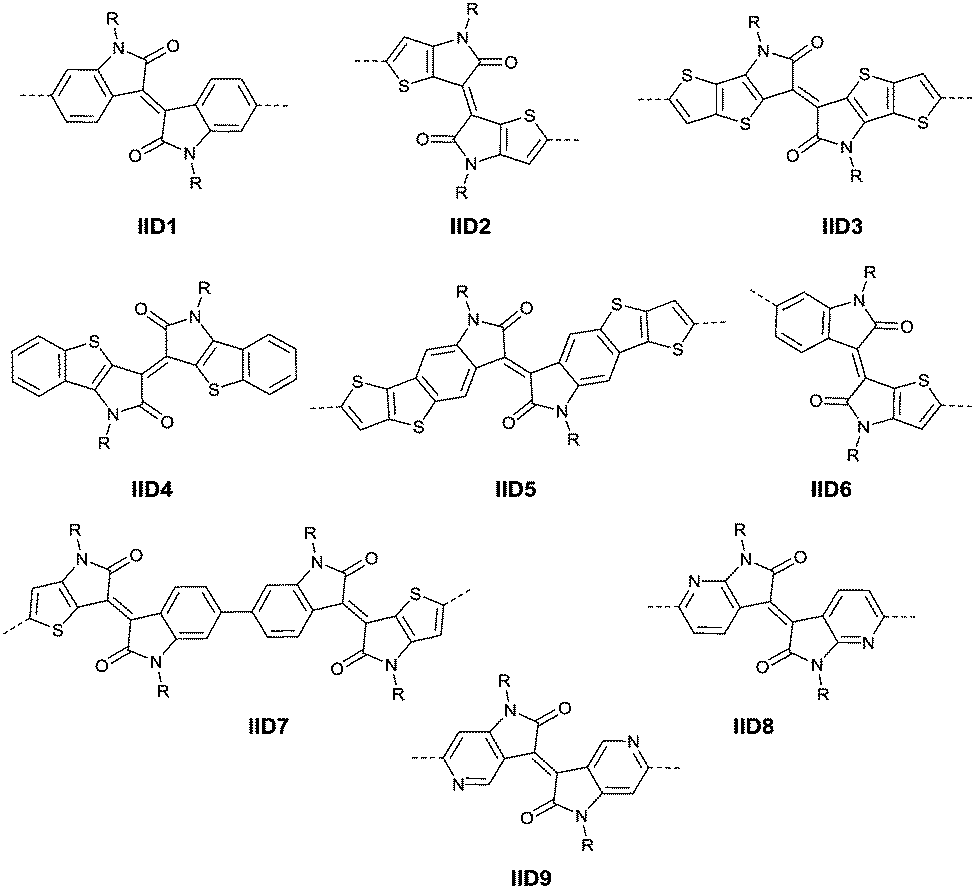
2.2 Isoindigo
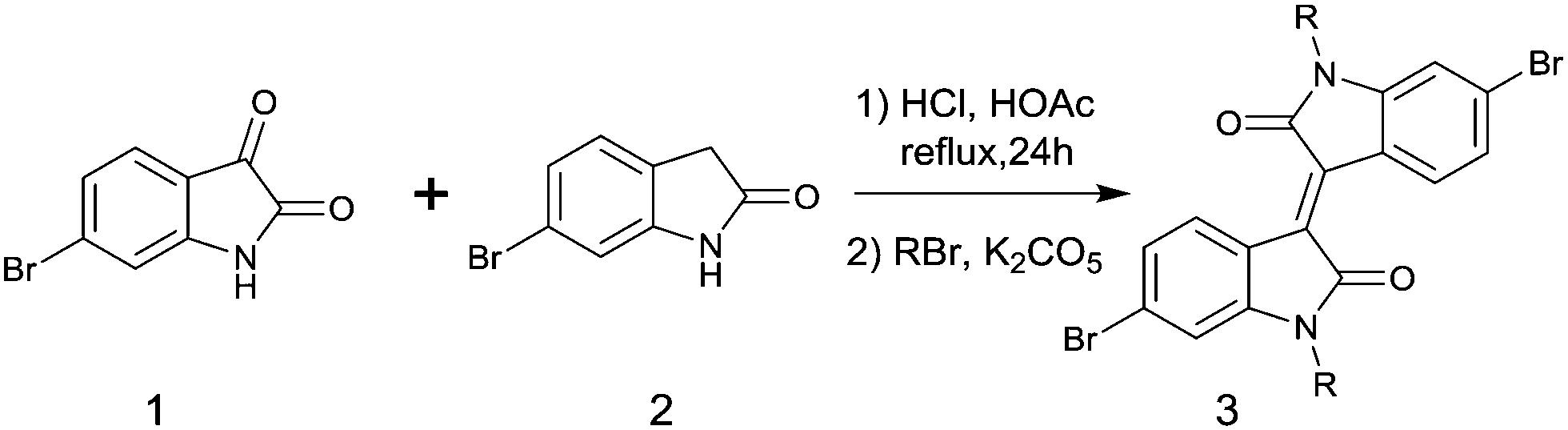
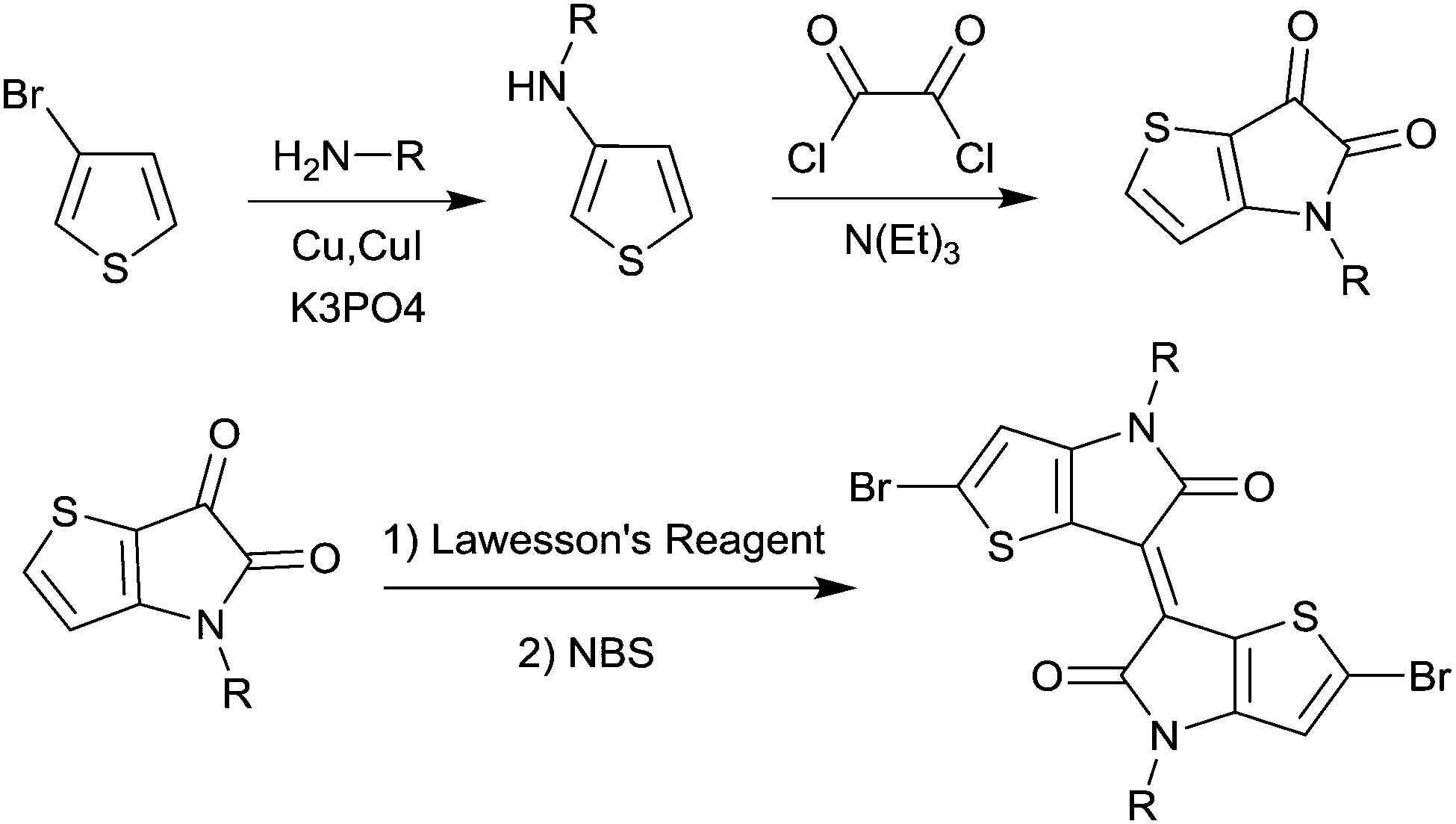
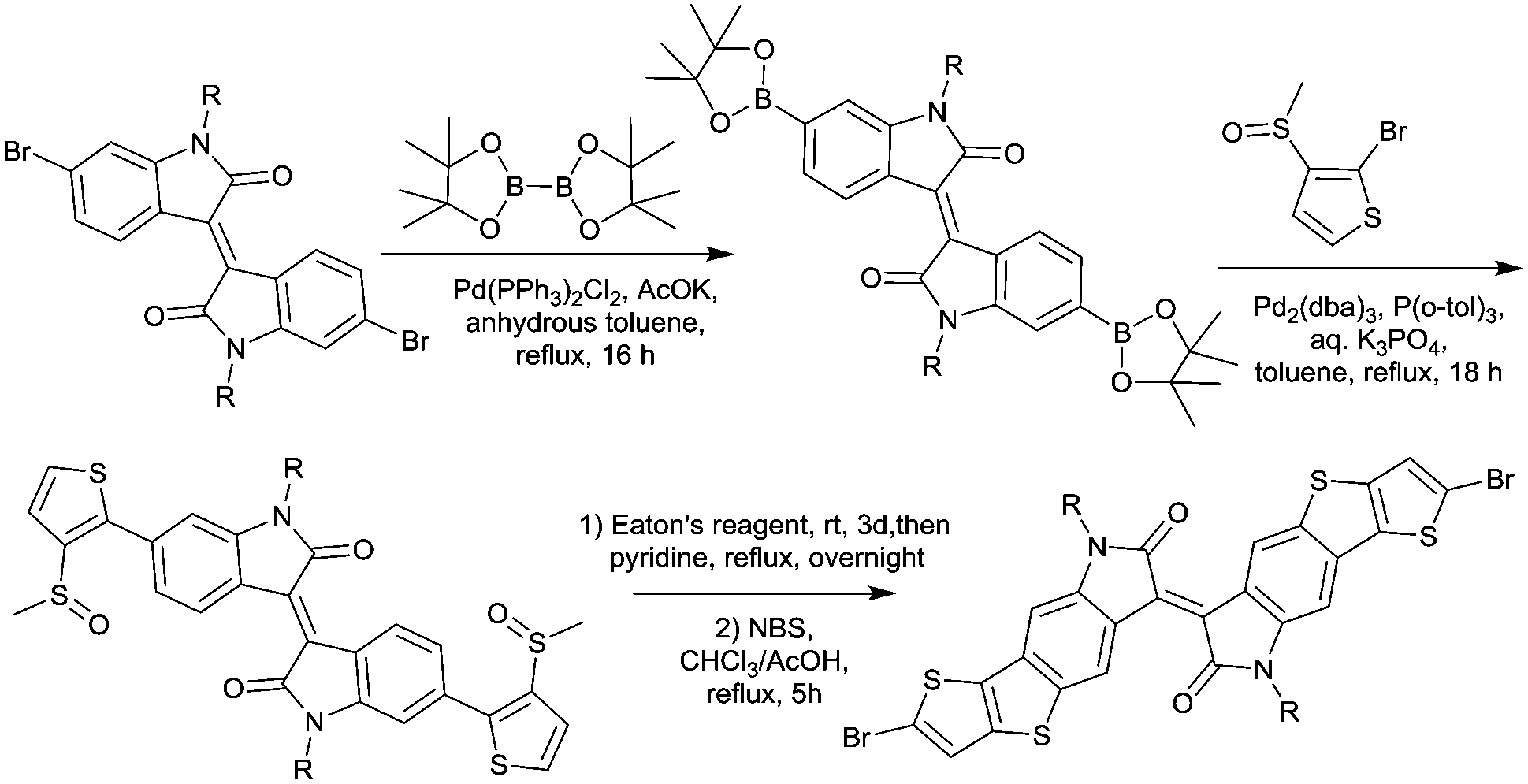
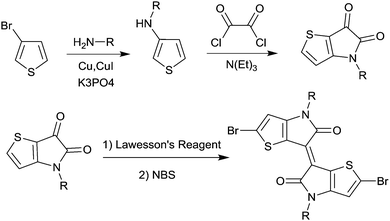
In 2011, IID-based polymer semiconductors were first studied in OFETs.64 They also exhibit a strong electron efficiency owing to the two lactam units similar to DPP in the molecular structure. As shown in Scheme 3, this is a common method for the synthesis of IID as 1 and 2 are commercially available. Such a reaction could afford a brominated IID monomer (3) in 86% yield.64
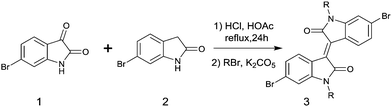 |
| | Scheme 3 General method for the synthesis of a brominated IID monomer. | |
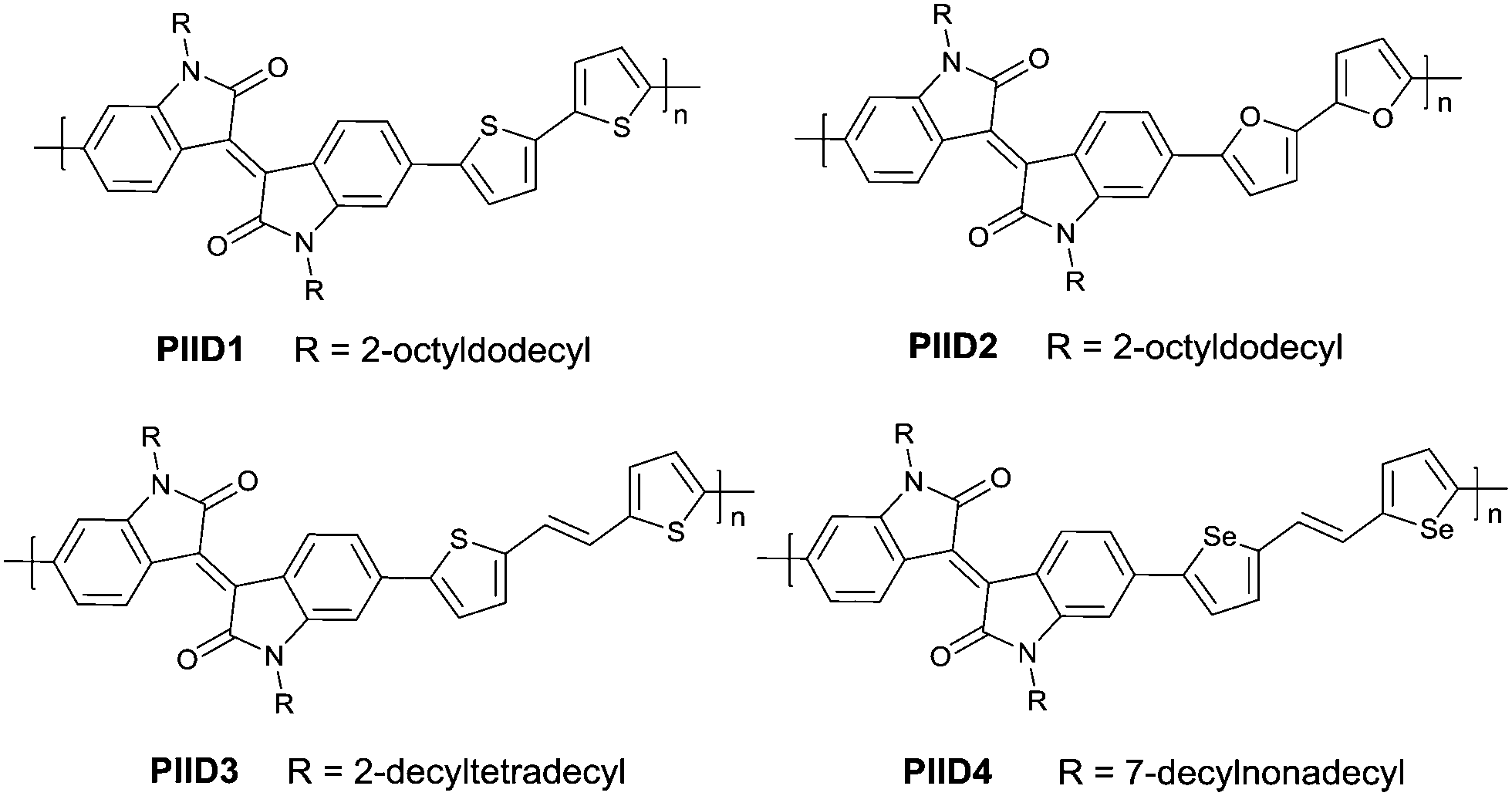
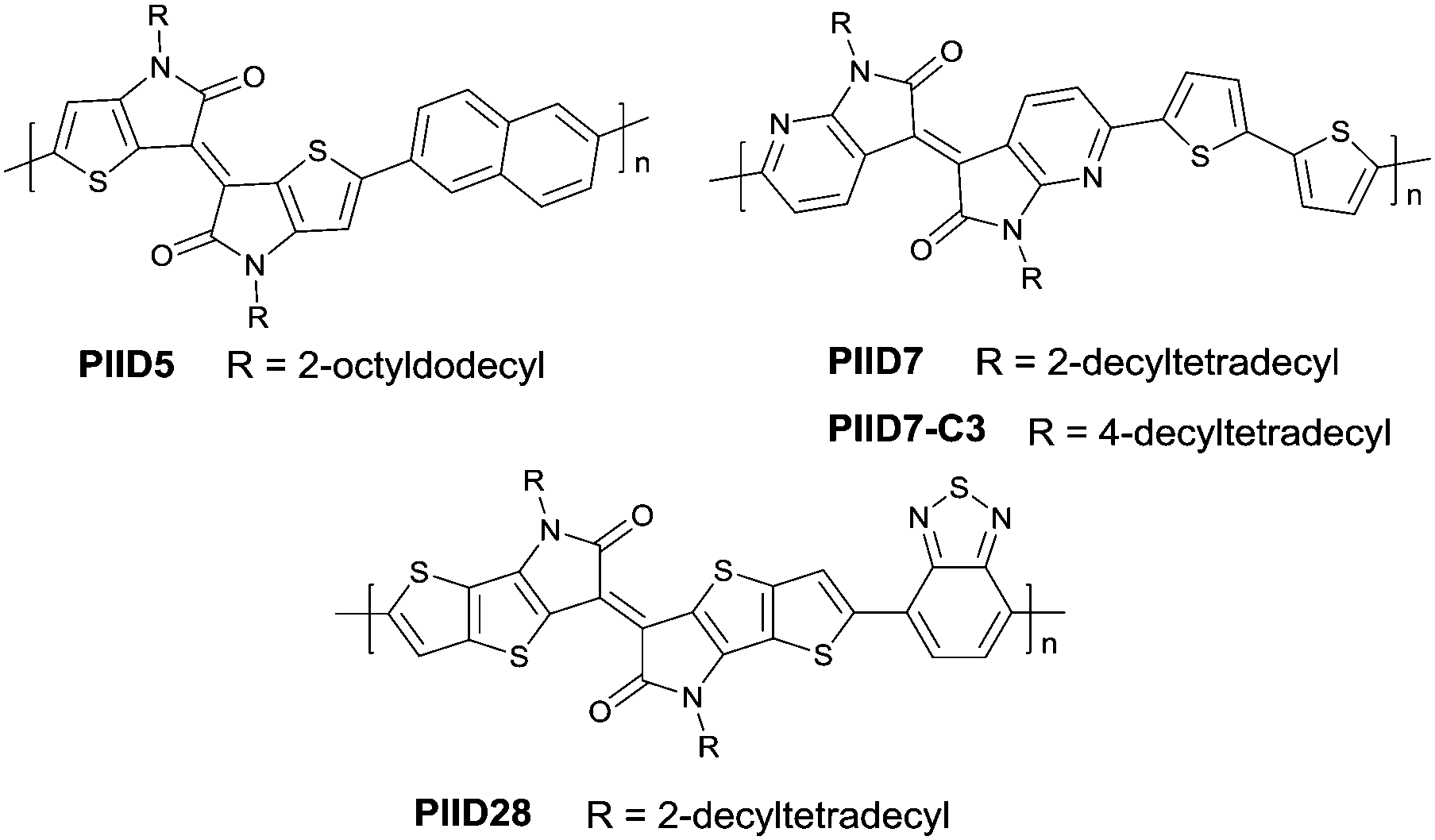
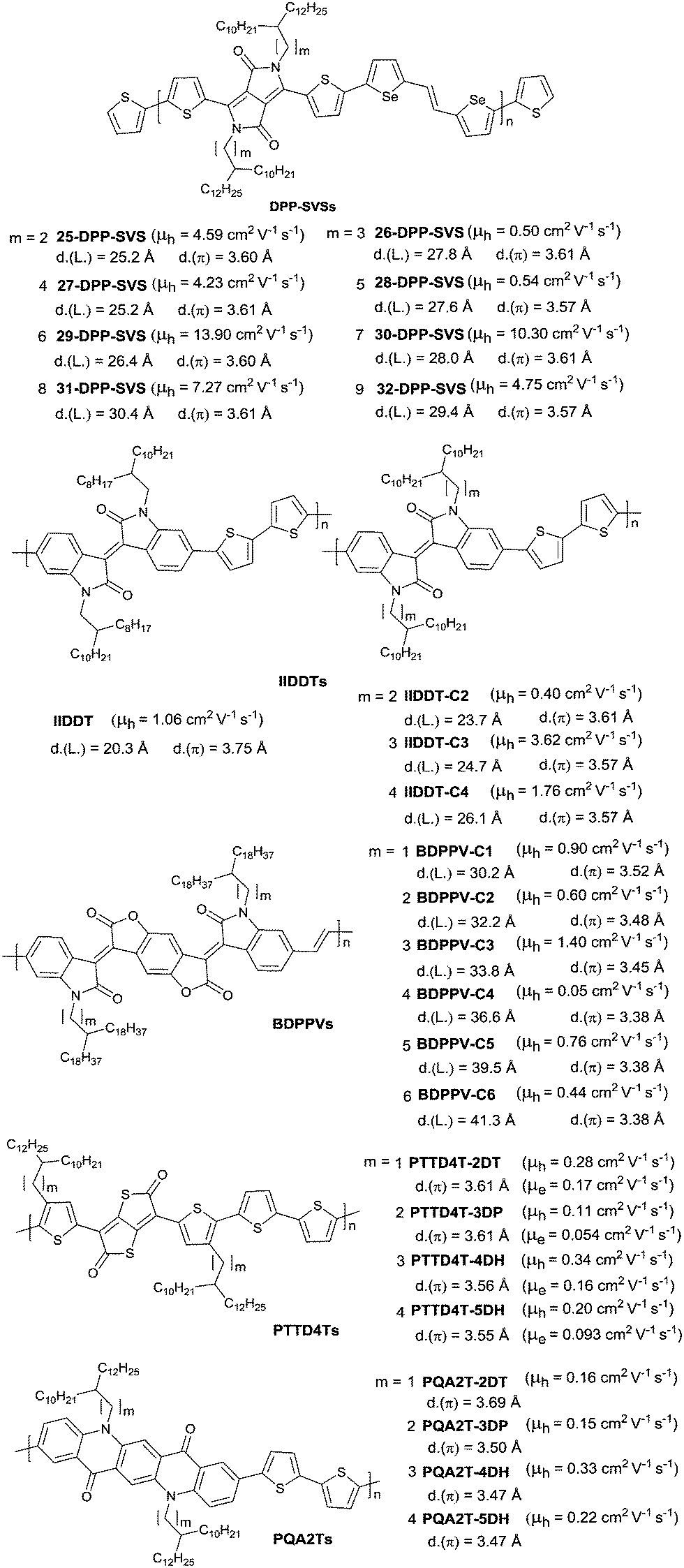
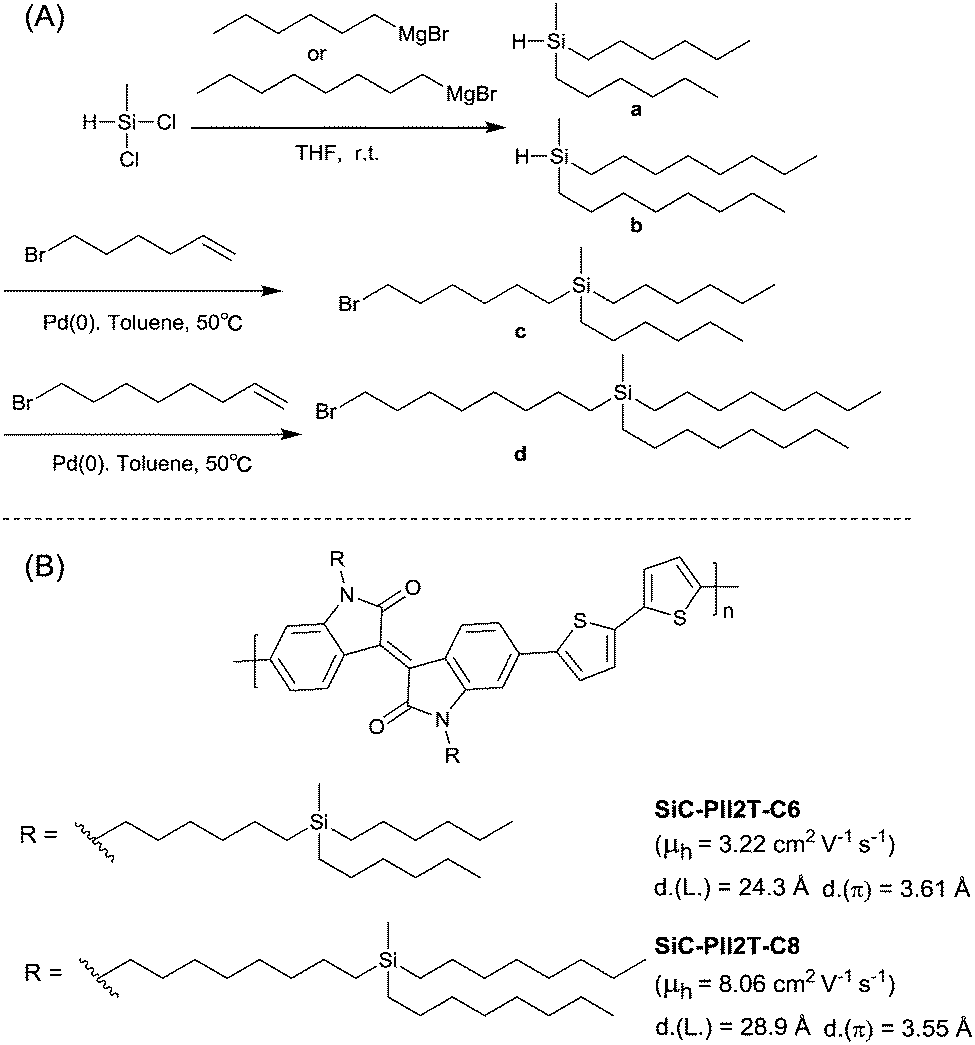
PIID165 (Fig. 4 and Table 3) is one of the IID-based polymers reported for OFETs with a hole mobility of up to 1.06 cm2 V−1 s−1. A series of polymers PIID1 (PIID1–PIID-C6Si, Table 3), PIID2 (PIID2–PIID2-C11Si; Fig. 4 and Table 3), and PIID3 (Fig. 4 and Table 3) are high-performance p-type IID-based polymer semiconductors with a hole mobility over 1 cm2 V−1 s−1. The PIID1 and PIID2 series exhibit side chain engineering, which will be discussed in detail later in this review. It is worth mentioning that the PIID4 polymer (Fig. 4 and Table 3) based on IID1 (Fig. 5) and (E)-2-(2-(selenoph-2-yl)vinyl)selenophene (SVS) exhibited a high hole mobility up to 5.83 cm2 V−1 s−1,66 to the best of our knowledge, which was the highest one based on IID1.
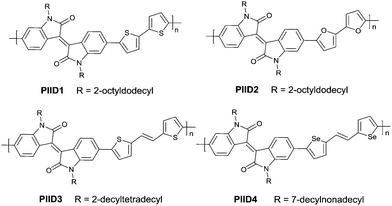 |
| | Fig. 4 Chemical structures of PIID1, PIID2, PIID3, and PIID4. | |
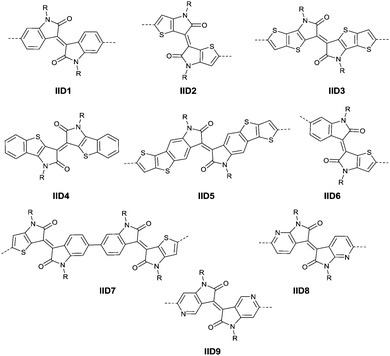 |
| | Fig. 5 Chemical structures of IID-based acceptors. | |
Table 3 Electronic parameters for high-performance p-type (μh > 1 cm2 V−1 s−1) IID- and IID-derivative-based polymer semiconductors
|
|
| Polymer |
Acceptor |
Donor |
M
n (kDa)/PDI |
Device structure |
HOMO/LUMO (eV) |
Max. μh (cm2 V−1 s−1) |
I
on/Ioff |
Ref. |
| PIID1 |
IID1 |
BT |
20.4/2.0 |
BGTC |
−5.70/−3.70 |
1.06 |
>106 |
65
|
| PIID1-C3 |
IID1 |
BT |
39.2/3.2 |
BGTC |
−5.52/−3.74 |
3.62 |
>106 |
65
|
| PIID1-C4 |
IID1 |
BT |
37.3/2.3 |
BGTC |
−5.50/−3.74 |
1.76 |
>106 |
65
|
| PIID1-C6Si |
IID1 |
BT |
138/3.29 |
BGTC |
−5.20/−1.61 |
2.48 |
>106 |
70
|
| PIID2 |
IID1 |
BF |
55.9/2.08 |
BGTC |
−5.32/— |
1.1 |
≈105 |
71 and 72
|
| PIID2-C5Si |
IID1 |
BF |
54.7/1.58 |
BGTC |
−5.35/— |
1.9 |
≈105 |
72
|
| PIID2-C6Si |
IID1 |
BF |
50.4/1.74 |
BGTC |
−5.30/— |
3.2 |
≈106 |
72
|
| PIID2-C7Si |
IID1 |
BF |
56.2/1.91 |
BGTC |
−5.25/— |
3.2 |
≈106 |
72
|
| PIID2-C8Si |
IID1 |
BF |
63.2/1.81 |
BGTC |
−5.25/— |
3.0 |
≈106 |
72
|
| PIID2-C9Si |
IID1 |
BF |
48.3/2.10 |
BGTC |
−5.25/— |
4.8 |
≈106 |
72
|
| PIID2-C10Si |
IID1 |
BF |
34.1/1.37 |
BGTC |
−5.25/— |
2.0 |
≈105 |
72
|
| PIID2-C11Si |
IID1 |
BF |
34.0/1.42 |
BGTC |
−5.35/— |
1.8 |
≈106 |
72
|
| PIID3 |
IID1 |
TVT |
98.8/3.31 |
BGTC |
−5.54/−3.65 |
2.20 |
>106 |
66 and 73
|
| PIID4 |
IID1 |
SVS |
295/2.36 |
BGTC |
−5.22/— |
5.83 |
105 |
66
|
| PIID5 |
IID2 |
D61 |
21.0/4.87 |
TGBC |
−5.12/−3.49 |
14.4 |
— |
68
|
| PIID6 |
IID7 |
BT |
— |
TGBC |
−5.40/−3.73 |
1.1 |
— |
74
|
| PIID7 |
IID8 |
TVT |
61.1/3.00 |
BGBC |
−5.66/−2.04 |
3.63 |
— |
75
|
| PIID7-C3 |
IID8 |
TVT |
50.6/3.87 |
BGBC |
−5.67/−3.64 |
7.28 |
— |
75
|
| PIID8 |
IID13 |
BF |
37.6/2.38 |
BGTC |
−5.32/−4.05 |
1.92 |
— |
76
|
As shown in Scheme 4, thienoisoindigo (IID2, Fig. 5) as a derivative of IID was synthesized for the first time in 2012.67 The dihedral angle between the two subunits is minimized, and the planarity of the molecular backbone is also enhanced when compared with that of IID. The IID2-based polymer PIID568 (Fig. 6) showed a favourable hole mobility up to 5.8 cm2 V−1 s−1 owing to its fine coplanarity and an ultrahigh mobility of 14.4 cm2 V−1 s−1 when high-k gate dielectric poly(vinylidenefluoride-trifluoroethylene) (P(VDF-TrFE)) worked as an insulator for device fabrication.
 |
| | Scheme 4 Synthesis of a brominated thienoisoindigo monomer. | |
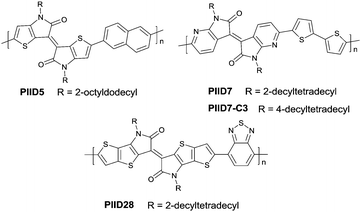 |
| | Fig. 6 Chemical structures of PIID5, PIID7, PIID-C3 and PIID28. | |
IID3, IID4 and IID5 (Fig. 5) also exhibited a smaller dihedral angle than IID1. Polymer PIID2869 (Fig. 6 and Table 4) based on IID3 exhibits a hole mobility of 0.4 cm2 V−1 s−1 and an electron mobility of 0.7 cm2 V−1 s−1. For IID3, the incorporation of thienothiophene extended the fused nature of the IID core and resulted in the increment of the π-orbital overlap, which is good for charge transport. As described in Meager's work,69 the synthesis procedure of IID3 is similar to that of IID2. Besides, as a similar structure to IID3, IID4 has also been mentioned for a small molecule semiconductor but not a polymer. The OFETs based on it showed a hole mobility of 0.18 cm2 V−1 s−1 when annealed under a temperature of 80 °C.77 In order to reduce the electron density of thienoisoindigo and keep the fine planarity of its fine backbone, Yue et al.78 combined the structure of IID and thienoisoindigo and synthesized IID5 (Scheme 5 and Fig. 5). The fusion of phenyl with TT not only lowered the dihedral angle between the linking repeat units to enhance the π–π stacking interaction between the polymer backbones but also increased the polaron delocalization, resulting in the promotion of efficient charge transport. But the experimental result was not good with a poor hole mobility below 1 cm2 V−1 s−1, but with a fine organic photovoltaic (OPV) performance for the polymer based on the acceptor IID5 and the donor thiophene. Such a phenomenon may be caused by the molecular orientation packing, which will be discussed in detail in the later section. The author also demonstrated that a polymer with a high molecular weight has better charge transporting properties than that of a polymer with a low molecular weight.
Table 4 Electronic parameters for high-performance n-type and ambipolar IID- and IID-derivative-based polymer semiconductors
|

|
| Polymer |
Acceptor |
Donor |
M
n (kDa)/PDI |
Device structure |
HOMO/LUMO (eV) |
Max. μh (cm2 V−1 s−1) |
Max. μe (cm2 V−1 s−1) |
Ref. |
| PIID9 |
IID1 |
BTz |
25.0/1.32 |
BGTC |
−5.68/−3.54 |
— |
0.22 |
88
|
| PIID10 |
IID12 |
V |
37.6/2.38 |
TGBC |
−6.21/−4.24 |
— |
1.1 |
83
|
| PIID11 |
IID12 |
BT |
77.2/3.00 |
TGBC |
−5.72/−4.15 |
— |
1.74 |
89
|
| PIID12 |
IID12 |
TVT |
28.07/1.72 |
BGTC |
−5.43/−4.09 |
— |
0.55 |
90
|
| PIID13 |
IID12 |
TAT |
23.95/1.83 |
BGTC |
−5.60/−4.11 |
— |
0.13 |
90
|
| PIID14 |
IID12 |
TT |
26.5/1.65 |
BGTC |
−5.57/−4.10 |
— |
0.65 |
90
|
| PIID15 |
IID14 |
BT |
51.6/2.62 |
TGBC |
−5.80/−4.37 |
— |
3.22 |
87
|
| PIID16 |
IID19 |
BT |
25.9/2.7 |
BGBC |
−5.78/−4.09 |
— |
0.18 |
91
|
| PIID17 |
IID19 |
2Tz |
14.3.2.5 |
BGBC |
−5.99/−4.18 |
— |
0.017 |
91
|
| PIID18 |
IID19 |
TT |
23.5/2.15 |
BGBC |
−5.92/−3.98 |
— |
0.14 |
92
|
| PIID19 |
IID1 |
T |
26/2.1 |
BGBC |
−5.57/−3.86 |
0.04 |
0.1 |
93
|
| PIID1 |
IID1 |
BT |
72.8/1.95 |
TGBC |
−5.31/−3.74 |
1.27 |
0.07 |
94
|
| PIID20 |
IID1 |
BTzVBTz |
49.6/2.53 |
BGTC |
−5.72/−3.51 |
0.076 |
0.04 |
60
|
| PIID21 |
IID1 |
BTzABTz |
13.5/2.09 |
BGTC |
−5.68/−3.49 |
0.03 |
0.086 |
60
|
| PIID22 |
IID1 |
2Tz |
27.2/2.4 |
TGBC |
−5.86/−4.14 |
0.03 |
0.022 |
95
|
| PIID23 |
IID2 |
BTz |
40/2.25 |
TGBC |
−4.86/−3.73 |
0.16 |
0.14 |
67
|
| PIID24 |
IID2 |
TVT |
23/1.84 |
BGTC |
−5.38/−3.76 |
0.12 |
0.0015 |
96
|
| PIID25 |
IID2 |
CNTVT |
38.02/1.87 |
BGTC |
−5.62/−3.82 |
0.07 |
0.19 |
96
|
| PIID26 |
IID2 |
TAT |
33.02/1.67 |
BGTC |
−5.65/−3.78 |
0.43 |
0.03 |
96
|
| PIID27 |
IID3 |
T |
30/2.23 |
TGBC |
−4.8/−3.7 |
0.2 |
0.2 |
69
|
| PIID28 |
IID3 |
BTz |
17/1.59 |
TGBC |
−4.9/−3.9 |
0.4 |
0.7 |
69
|
| PIID29 |
IID3 |
BT |
20/2.05 |
TGBC |
−4.8/−3.6 |
0.4 |
0.1 |
69
|
| PIID7 |
IID8 |
BT |
61.1/3.00 |
TGBC |
−5.66/−2.04 |
0.45 |
0.47 |
75
|
| PIID7-C3 |
IID8 |
BT |
50.6/3.87 |
TGBC |
−5.67/−3.64 |
2.33 |
0.78 |
75
|
| PIID30 |
IID11 |
BT |
49.8/1.76 |
BGBC |
−5.6/−3.7 |
0.11 |
0.035 |
82
|
| PIID31 |
IID12 |
BT3 |
17.4/1.85 |
BGTC |
−5.63/−4.06 |
0.37 |
1.23 |
84 and 97
|
| PIID32 |
IID12 |
BT4 |
19.4/1.98 |
BGTC |
−5.73/−4.01 |
0.17 |
0.70 |
84
|
| PIID33 |
IID12 |
BT5 |
10.2/1.48 |
BGTC |
−5.68/−4.05 |
0.0018 |
0.175 |
84
|
| PIID34 |
IID12 |
TT |
20.9/2.86 |
TGBC |
−5.70/−3.83 |
1.70 |
1.37 |
98
|
| PIID35 |
IID12 |
BDT |
13.9/2.14 |
TGBC |
−5.78/−3.81 |
0.12 |
0.11 |
98
|
| PIID36 |
IID12 |
BDS |
19.2/2.56 |
TGBC |
−5.74/−3.81 |
0.10 |
0.16 |
98
|
| PIID37 |
IID13 |
BT3 |
10.98/1.86 |
BGTC |
−5.18/−3.94 |
0.61 |
0.22 |
85
|
| PIID38 |
IID13 |
TVT2 |
21.96/2.55 |
BGTC |
−4.96/−3.83 |
0.45 |
0.11 |
86
|
| PIID39 |
IID13 |
TVT3 |
13.69/2.11 |
BGTC |
−5.10/−3.70 |
0.86 |
0.19 |
86
|
| PIID40 |
IID15 |
BT |
43.1/4.8 |
BGBC |
−5.65/−3.84 |
0.51 |
0.50 |
99
|
| PIID41 |
IID15 |
TT |
27.6/4.9 |
BGBC |
−5.76/−3.79 |
0.12 |
0.14 |
99
|
| PIID42 |
IID16 |
BT |
128/2.89 |
BGBC |
−5.60/−3.71 |
0.19 |
0.088 |
100
|
| PIID43 |
IID16 |
TVT |
158/1.59 |
BGBC |
−5.71/−3.70 |
0.10 |
0.075 |
100
|
| PIID44 |
IID17 |
BT |
82.1/2.07 |
TGBC |
−5.16/−3.58 |
0.41 |
0.18 |
101
|
| PIID45 |
IID17 |
TVT |
46.2/5.31 |
TGBC |
−5.24/−3.58 |
0.45 |
0.16 |
101
|
| PIID45-C3 |
IID17 |
TVT |
35.5/7.18 |
TGBC |
−5.38/−3.71 |
0.32 |
0.071 |
101
|
| PIID46 |
IID18 |
BT |
21.0/2.3 |
BGBC |
−5.60/−4.06 |
0.10 |
0.14 |
102
|
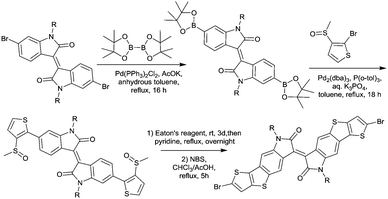 |
| | Scheme 5 Synthesis of a brominated IID5 monomer. | |
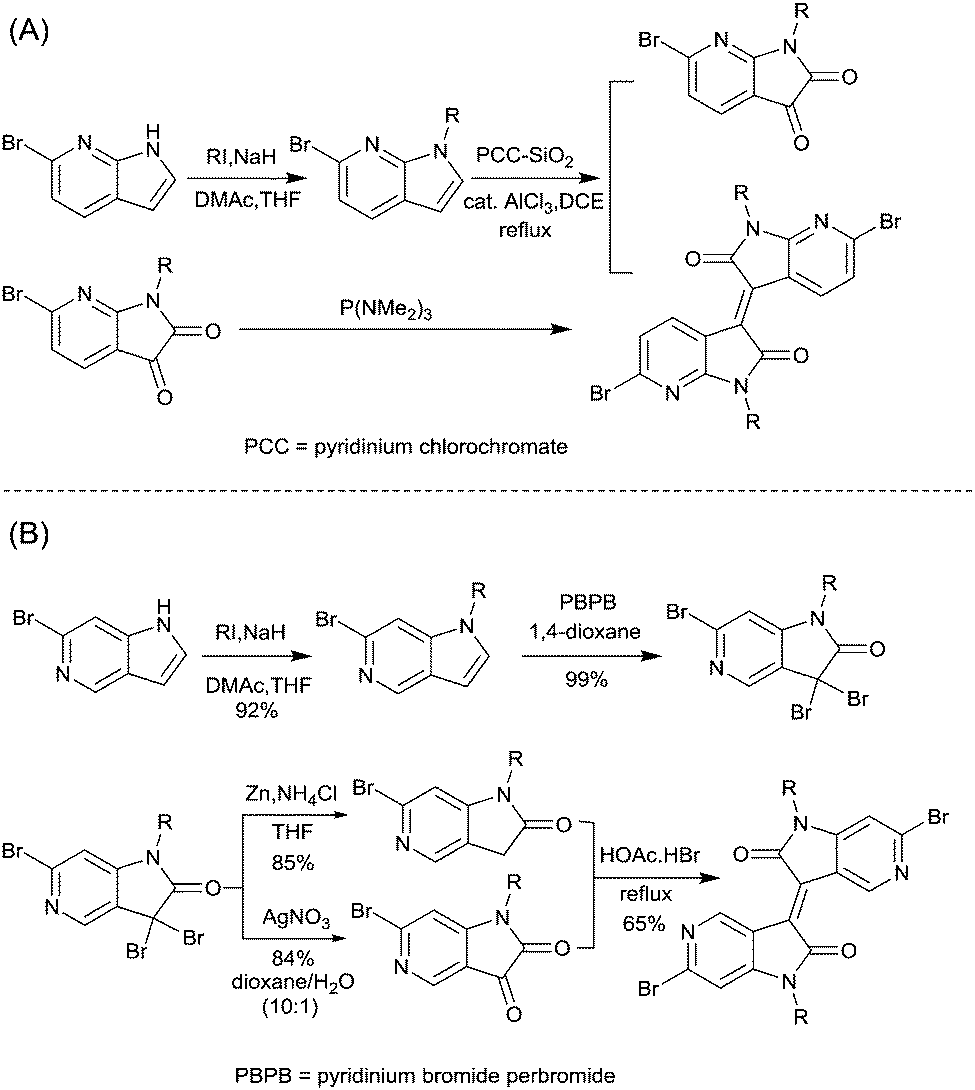
The brominated monomer of IID6 (Fig. 5), as an asymmetric IID-derivative acceptor, was prepared by the acid-promoted condensation of 6-bromo-oxindole and thieno-dioxypyrrole, followed by alkylation and bromination.79 The polymer based on the acceptor IID6 and the donor bithiophene (BT) or 2,5-bis(thiophen-2-yl)thiophene (3T) exhibited a hole mobility below 1 cm2 V−1 s−1, totally different from the high-performance behaviour of polymers based on the asymmetric DPP acceptors, which can be explained by the different packing orientation as well.
James et al.74 coupled two IID6 units via benzene-coupling to obtain the new symmetric acceptor unit IID7 (Fig. 5). In other words, IID7 was a building block for the donor–acceptor based conjugated polymer, which is of a lower LUMO level than IID6. But the corresponding polymer PIID6 (Table 3) formed by the acceptor IID7 and a BT-based donor showed an unexceptional high hole mobility of 1.1 cm2 V−1 s−1 when poly(trifluoroethylene) (PTrFE) acted as a dielectric layer for the OFET.
As shown in Scheme 6, 7,7′-diazaisoindigo75 (IID8, Fig. 5) and 5,5′-diazaisoindigo80 (IID9, Fig. 5) monomers were also synthesized for the acceptor unit of polymer semiconductors in OFETs. Replacement of the aromatic benzene ring with pyridine results in a lower lying LUMO to afford a more electron-deficient acceptor owing to the electron-deficient feature of the pyridinic nitrogen. In addition, the coplanarity of the backbone was enhanced as the dihedral angles between the indolone subunits of 7,7′-diazaisoindigo and 5,5′-diazaisoindigo were reduced. Such effects were proved in theoretical calculations and experiments.75,80 The polymer PIID7-C3 (Fig. 6) based on the 7,7′-diazaisoindigo acceptor exhibited a favourable hole mobility up to 7.28 cm2 V−1 s−1 since the ordered packing of the polymer was caused by the aforementioned fine coplanarity. However, the charge transfer performance of a polymer made from the 5,5′-diazaisoindigo acceptor (IID9) was poor (μh = 1.27 × 10−3 cm2 V−1 s−1)80 and even poorer than the IID-based polymer when copolymerized with the same donor, which still needs to be further studied.
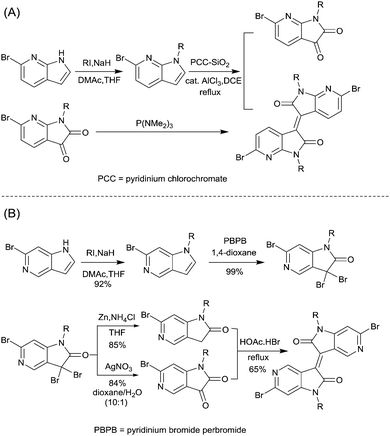 |
| | Scheme 6 Syntheses of brominated (A) 7,7′-diazaisoindigo and (B) 5,5′-diazaisoindigo monomers. | |
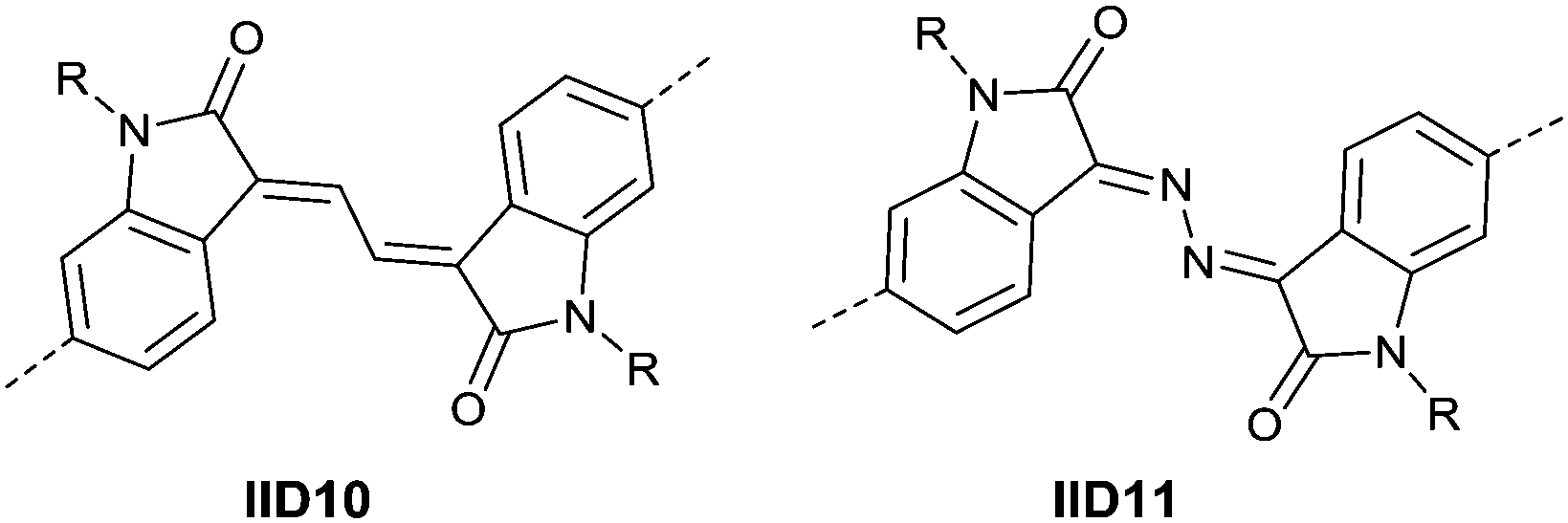
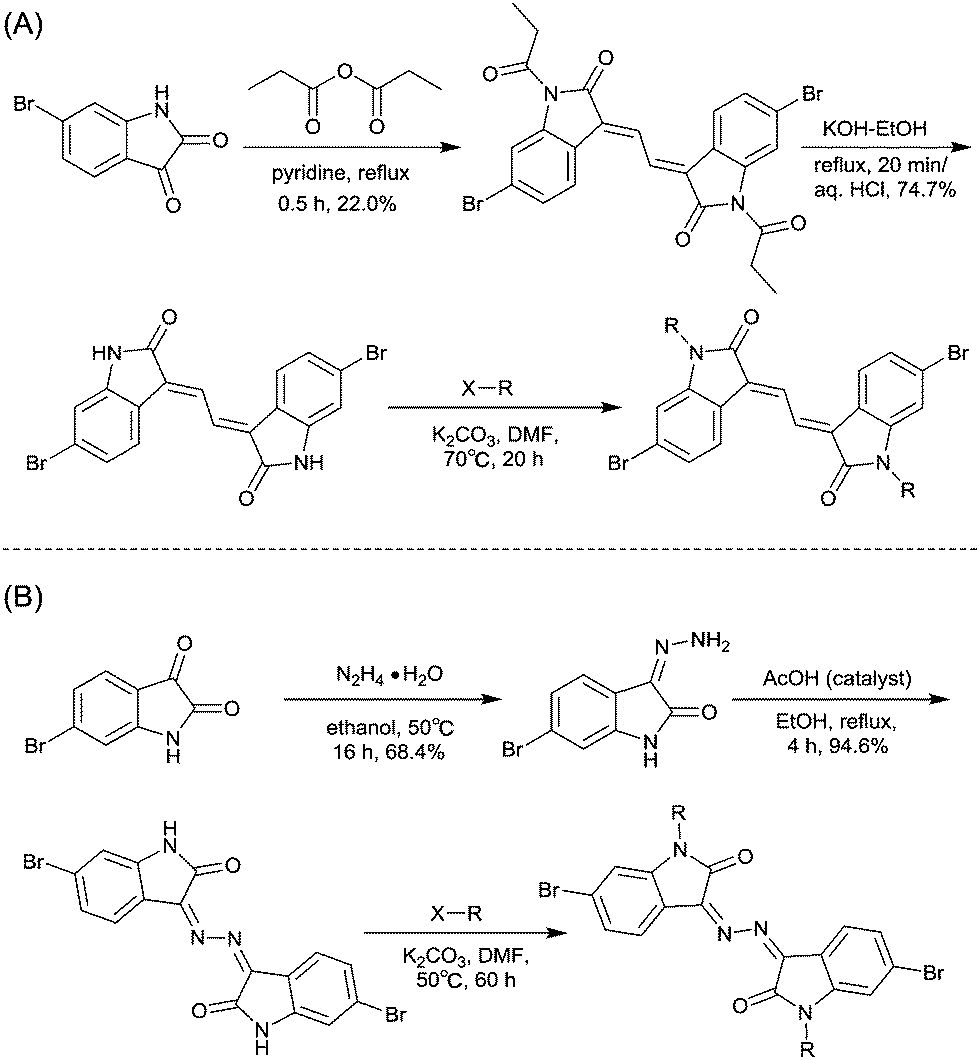
Li’s research group creatively incorporated the ethane-1,2-diylidene moiety and the hydrazine-1,2-diylidene moiety into the two indolin-2-one subunits of IID.81,82 The chemical structure and synthesis steps of them are shown in Fig. 7 (IID10 and IID11) and Scheme 7, respectively. The polymer based on IID10 and BT showed a p-type performance but not with a high charge mobility. However, the polymer based on IID11 and BT showed an ambipolar performance since the azine is a strong electron-withdrawing moiety to afford low LUMO levels.
 |
| | Fig. 7 Chemical structures of IID10 and IID11. | |
 |
| | Scheme 7 Syntheses of brominated (A) IID10 and (B) IID11 monomers. | |
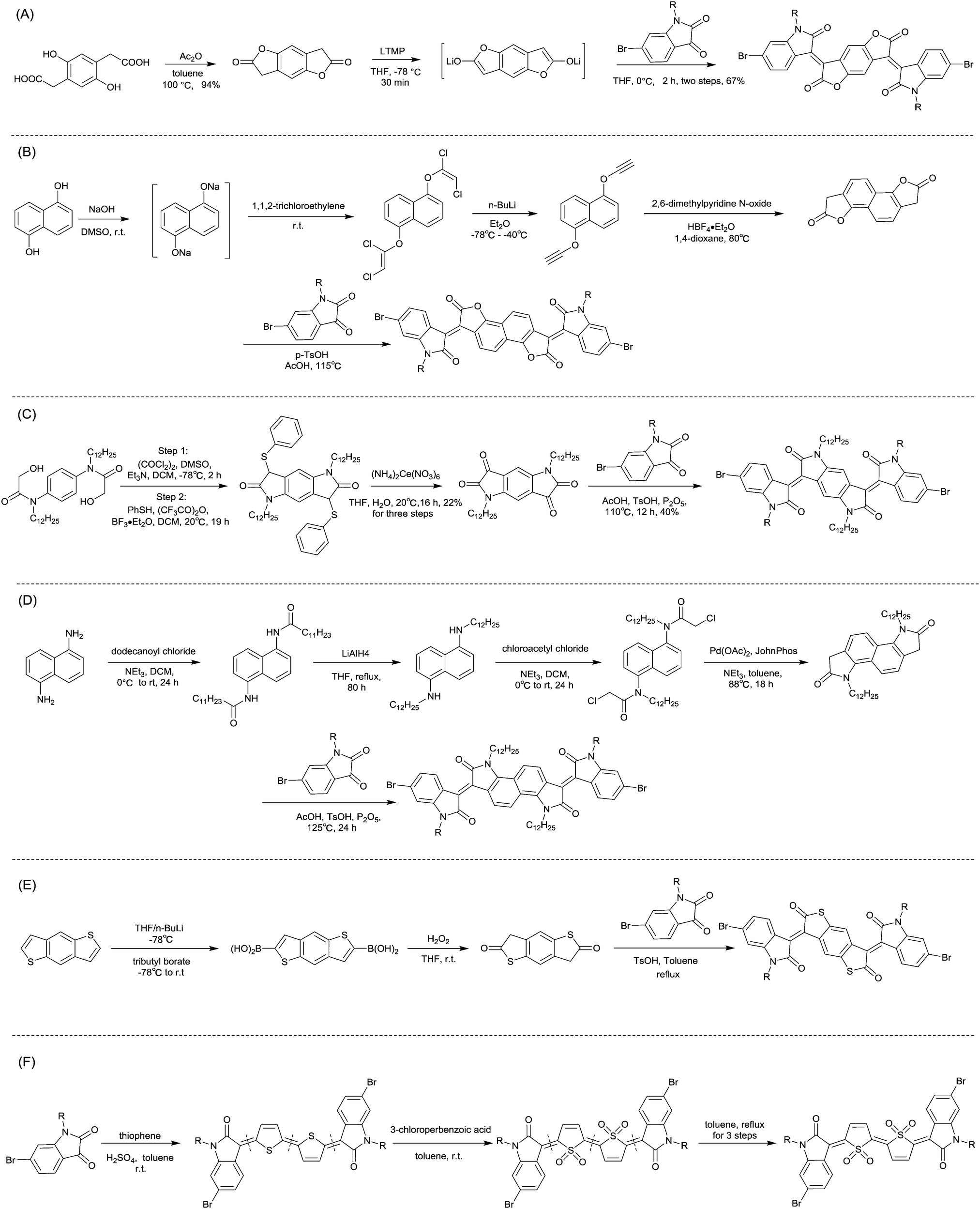
In 2013, Jian Pei83 incorporated the benzodifurandione moiety into the IID molecular backbone to obtain IID12 (Fig. 8, synthesis steps as shown in Scheme 8(A)), which was seen as a poly(p-phenylene vinylene) derivative. The introduction of carbonyl groups reduced the LUMO level of IID12, and more intramolecular hydrogen bonds were formed to “lock” the polymer backbone and ensure the fine molecular packing and crystallinity. As a result, an electron mobility over 1 cm2 V−1 s−1 (PIID10 listed in Table 4) was firstly achieved under ambient conditions for polymer semiconductors. Besides, PIID32–PIID36 (Table 4) based on IID12 exhibited ambipolar properties and PIID3184 (Fig. 9 and Table 4) exhibited a hole mobility of 0.37 cm2 V−1 s−1 and an electron mobility of 1.23 cm2 V−1 s−1 among them. Similar to IID12, IID1385,86 (Fig. 8) and IID1487 (Fig. 8) were also reported. The IID13 based polymers PIID37–PIID3985,86 (Table 4) exhibited ambipolar characteristics. The IID14 based polymer PIID1587 (Fig. 9) exhibited a unipolar n-type performance with a high electron mobility of 3.22 cm2 V−1 s−1 under ambient conditions owing to the introduction of an electron-deficient sp2-nitrogen. Such an effect was proved in the aforementioned polymers PIID7 and PIID7-C3.
 |
| | Fig. 8 Chemical structures of IID12−IID19. | |
 |
| | Scheme 8 Syntheses of brominated (A) IID12, (B) IID15, (C) IID16, (D) IID17, (E) IID18 and (F) IID19 monomers. | |
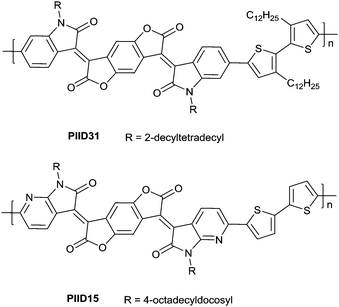 |
| | Fig. 9 Chemical structures of PIID31 and PIID15. | |
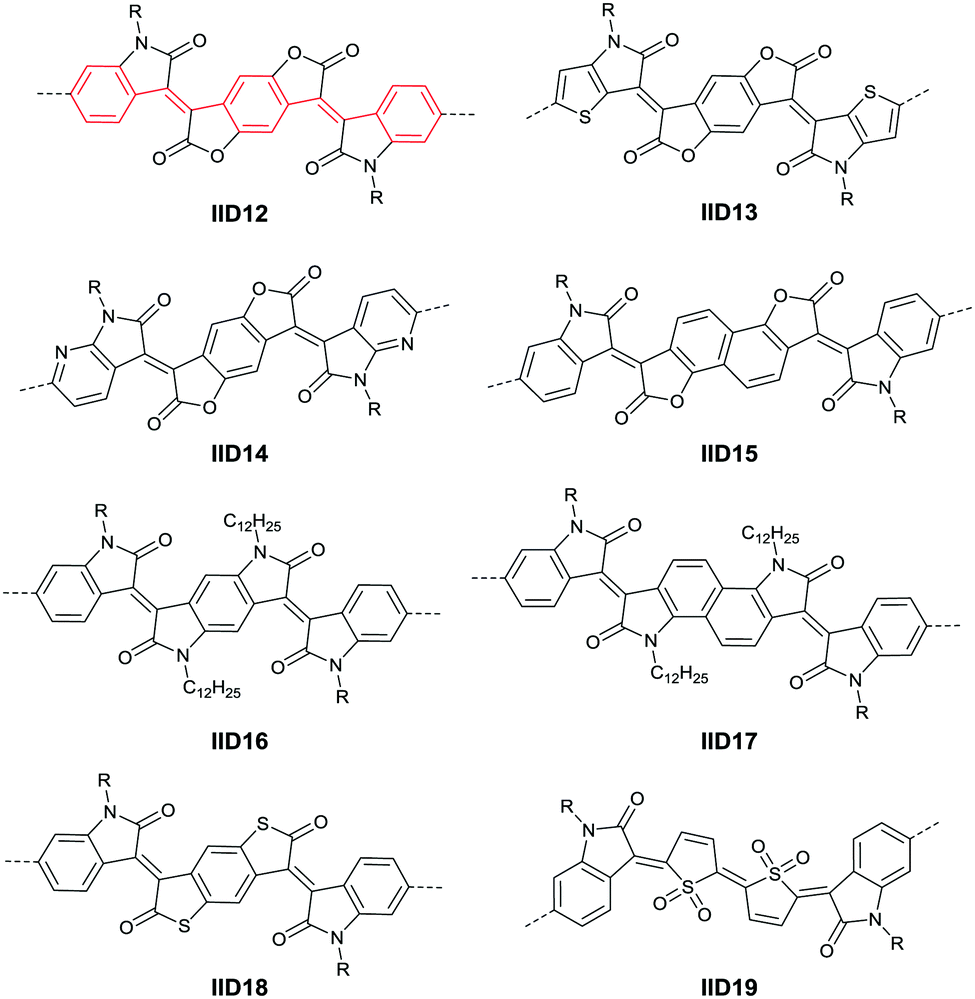
Li et al.99 substituted the naphthalene ring for a central benzene ring to obtain IID15 (Fig. 8, synthesis steps shown in Scheme 8(B)). The large size of naphthalene endows IID15 with a weaker electron-accepting ability than IID12, which is beneficial for the highly balanced ambipolar properties of the IID15-based polymers PIID40 (Fig. 10 and Table 4) and PIID41 (Fig. 10 and Table 4). Replacement of the central benzodifurandione ring by benzodipyrrole-dione76,100 (IID16 as in Fig. 8), naphthodipyrroledione101 (IID17 as in Fig. 8) and benzodithiophenedione102 (IID18 as in Fig. 8) was also studied, and the synthesis steps are displayed in Scheme 8(C), (D) and (E), respectively. For IID16 and IID17 based polymers, the additional N-substituted alkyl chains afford the improvement of solubility of such conjugated polymers in solvent which is beneficial to the processing of the polymers. In 2016, the Li92 research group reported a quinoid-type IID derivative (IID19 as in Fig. 8, synthesis methods are shown in Scheme 8(F)). The IID19-based polymers PIID16–PIID1891,92 (Table 4) exhibited a unipolar n-type semiconductor performance.
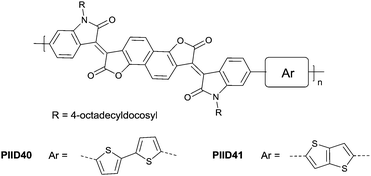 |
| | Fig. 10 Chemical structures of PIID40 and PIID41. | |
2.3 Others
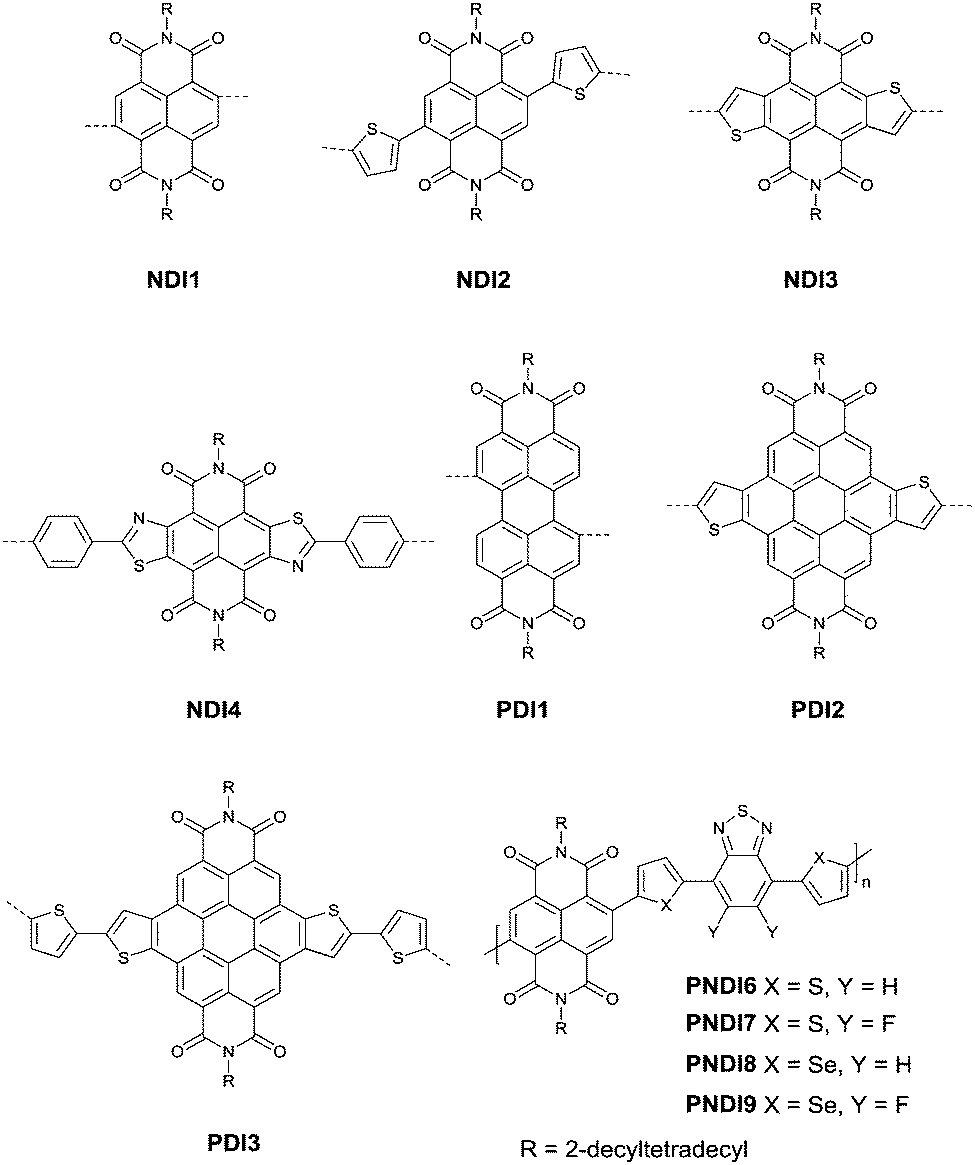
Naphthalenedicarboximide (NDI) and perylenedicarboximide (PDI) based polymers were also studied for OFETs. NDI1,46,103–107 NDI2,108 NDI3,109–111 NDI4,112 PDI1,103,104,106,113 PDI2114 and PDI3115 (Fig. 11) have been reported as the acceptors. The six-membered dicarboxylic imide rings endow the rylene diimides with electron-withdrawing properties. Therefore, the polymers based on NDI and PDI generally show p-channel or ambipolar behaviour. The polymers based on NDI and PDI and their OFET performance are listed in Table 5. The record high electron mobility of 8.5 cm2 V−1 s−1 was achieved for PNDI8105 (Fig. 11).
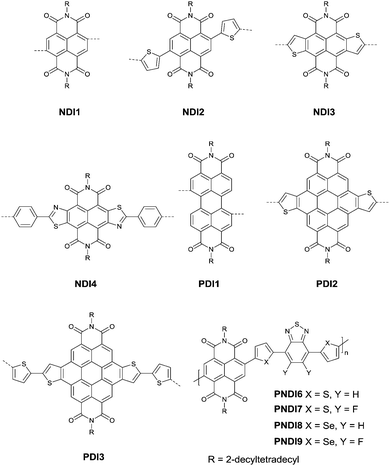 |
| | Fig. 11 Chemical structures of NDI1–NDI4, PDI1–PDI3 and PNDI6–PNDI9. | |
Table 5 Electronic parameters for high-performance polymer (except DPPs and IID- and IID-derivative-based polymers) semiconductors
|
|
| Polymer |
Acceptor |
Donor |
M
n (kDa)/PDI |
Device structure |
HOMO/LUMO (eV) |
Max. μh (cm2 V−1 s−1) |
Max. μe (cm2 V−1 s−1) |
Ref. |
| PNDI1 |
NDI1 |
B |
6.81/1.99 |
BGTC |
−5.173/−3.15 |
— |
0.8 |
103
|
| PNDI2 |
NDI1 |
BTz |
5.17/1.28 |
BGTC |
−5.14/−3.03 |
— |
0.2 |
103
|
| PNDI3 |
NDI1 |
BT |
47.8/5.53 |
BGTC |
−5.36/−3.91 |
— |
0.06 |
104
|
| PNDI4 |
NDI1 |
BT |
52.5/5.51 |
TGBC |
— |
— |
0.45–0.85 |
46
|
| PNDI5 |
NDI1 |
TVT |
23.7/2.46 |
TGBC |
−5.62/−3.90 |
0.3 |
1.57 |
107
|
| PNDI6 |
NDI1 |
BTz5 |
36.7/4.05 |
TGBC |
−5.90/−3.77 |
0.7 |
3.1 |
105
|
| PNDI7 |
NDI1 |
T2FBTT |
48.5/4.57 |
TGBC |
−6.24/−3.85 |
— |
2.2 |
105
|
| PNDI8 |
NDI1 |
SBTzS |
39.7/3.72 |
TGBC |
−5.84/−3.81 |
1.7 |
8.5 |
105
|
| PNDI9 |
NDI1 |
2FSBTzS |
56.1/4.25 |
TGBC |
−6.20/−3.88 |
— |
3.5 |
105
|
| PNDI10 |
NDI1 |
PTZ |
9.97/1.53 |
BGTC |
−5.85/−3.75 |
— |
0.05 |
106
|
| PNDI11 |
NDI2 |
2FT |
13.6/1.53 |
BGTC |
−5.86/−3.93 |
0.22 |
0.05 |
108
|
| PNDI12 |
NDI3 |
BTz |
14.4/2.93 |
BGTC |
—/−4.1 |
— |
0.1 |
109
|
| PNDI13 |
NDI3 |
BT |
27.1/3.3 |
BGTC |
−5.6/−4.4 |
0.1 |
0.27 |
110
|
| PNDI14 |
NDI3 |
TT |
19.0/2.6 |
BGTC |
−5.7/−4.1 |
0.046 |
0.26 |
111
|
| PNDI15 |
NDI3 |
NTz |
15.7/1.73 |
BGTC |
−5.6/−4.2 |
— |
0.21 |
109
|
| PNDI16 |
NDI4 |
V |
88.8/2.47 |
BGTC |
−5.73/−3.86 |
— |
0.015 |
112
|
| PPDI1 |
PDI1 |
B |
16.08/1.53 |
BGTC |
−5.89/−3.49 |
— |
0.04 |
103
|
| PPDI2 |
PDI1 |
BTz |
30.62/1.17 |
BGTC |
−5.49/−3.08 |
— |
0.032 |
103
|
| PPDI3 |
PDI1 |
BT |
11.0/2.9 |
BGTC |
−5.61/−3.96 |
— |
0.002 |
104
|
| PPDI4 |
PDI1 |
DTT |
— |
BGBC |
−5.57/−3.59 |
— |
0.034 |
113
|
| PPDI5 |
PDI1 |
ATTTA |
15/1.1 |
BGBC |
−5.73/−3.53 |
— |
0.075 |
113
|
| PPDI6 |
PDI1 |
PTZ |
11.82/1.57 |
BGTC |
−5.83/−3.79 |
— |
0.05 |
106
|
| PPDI7 |
PDI2 |
T |
10.7/1.6 |
TGBC |
−3.70/−5.56 |
0.04 |
0.30 |
114
|
| PPDI8 |
PDI3 |
BTz5 |
5.3/3.4 |
BGTC |
−5.54/−3.72 |
0.018 |
0.019 |
115
|
| PTPD |
TPD |
2TTT |
16/2.06 |
BGTC |
−5.05/— |
1.29 |
— |
121
|
| PBTPD1 |
BTPD |
TVT |
17.7/3.13 |
BGTC |
−5.65/−3.90 |
0.087 |
— |
123
|
| PBTPD2 |
BTPD |
TVS |
14.5/3.74 |
BGTC |
−5.61/−3.95 |
0.17 |
— |
123
|
| PBTPD3 |
BTPD |
BTz5 |
62.5/2.89 |
BGTC |
−5.72/−4.01 |
0.33 |
1.02 |
122
|
| PBTPD4 |
BTPD |
T2FBTT |
61.3/4.38 |
BGTC |
−5.85/−4.10 |
0.16 |
0.36 |
122
|
| PBTI1 |
BTI |
BTI |
3.59/2.2 |
BG |
−6.28/−3.47 |
— |
>0.01 |
124
|
| PBTI2 |
BTI |
BT |
1.81/1.38 |
BG |
−5.88/−3.04 |
0.01 |
— |
124
|
| PPHI |
PHI |
BT2 |
207.5/— |
BGBC |
— |
0.28 |
— |
126
|
| PNDI |
NTDI |
BTz5 |
20.1/2.9 |
BGBC |
−5.58/−3.58 |
0.82 |
— |
128
|
| PBTz1 |
BTz1 |
TTTT |
75.6/2.59 |
TGBC |
−5.41/−3.7 |
8.7 |
— |
131
|
| PBTz2 |
BTz1 |
CDT |
10.1/2.63 |
BC |
— |
0.17 |
— |
159
|
| PBTz3 |
BTz1 |
4TR |
35/1.63 |
BGTC |
−5.18/— |
0.13–0.20 |
— |
160
|
| PBTz4 |
BTz2 |
T |
27/1.5 |
BGTC |
−5.55/−3.60 |
0.02 |
— |
161
|
| PBTz5 |
BTz2 |
TT |
13/2.1 |
BGTC |
−5.37/−3.60 |
0.26 |
— |
161
|
| PBTz6 |
BTz5 |
SiIDT |
22.3/2.54 |
TGBC |
−5.2/−3.4 |
0.28 |
— |
162
|
| PBTz7 |
BTz6(SF) |
BT4 |
37.0/1.3 |
BGBC |
−5.23/−3.50 |
0.39 |
— |
163
|
| PBTz8 |
BTz7(DF) |
SiIDT |
26.4/2.69 |
TGBC |
−5.2/−3.2 |
0.19 |
— |
162
|
| PBTz9 |
BTz8 |
T |
7/2.0 |
BGTC |
−5.24/−3.37 |
0.007 |
— |
164
|
| PBTz10 |
BTz9 |
T |
35/2.21 |
BGTC |
−5.20/−3.23 |
0.019 |
— |
164
|
| PBTz11 |
BTz10(F) |
T |
7.5/1.19 |
BGTC |
−5.36/−3.44 |
1.9 |
— |
164
|
| PNCz1-1 |
NCz1 |
BT |
52.9/2.1 |
BGTC |
−5.14/−3.46 |
0.23 |
— |
132
|
| PNCz1-2 |
NCz1 |
TNTR |
67.3/1.3 |
BGBC |
−5.40/−3.64 |
0.214 |
— |
133
|
| PNCz2 |
NCz2 |
BT |
28.7/2.1 |
BGTC |
−5.22/−3.55 |
0.040 |
— |
132
|
| PNCz3 |
NCz3 |
BT |
49.7/4.8 |
BGTC |
−5.48/−3.65 |
0.27 |
0.17 |
132
|
| PCDTPT |
TPT |
CDT |
50/— |
BGBC |
— |
52.7 |
— |
134
|
| PBAI1 |
BAI1 |
BDT2 |
41.2/2.47 |
BGBC |
−4.91/−3.63 |
1.5 |
0.41 |
135
|
| PBAI2 |
BAI1 |
CZ |
68.2/4.13 |
BGBC |
−5.03/−3.65 |
0.14 |
0.09 |
135
|
| PBAI3 |
BAI2 |
T |
15.7/3.15 |
TGBC |
−4.24/−3.02 |
0.23 |
0.48 |
165
|
| PBAI4 |
BAI2 |
S |
25/3.32 |
TGBC |
−4.99/−3.77 |
0.20 |
0.99 |
166
|
| PBAI5 |
BAI2 |
B |
29/3.55 |
TGBC |
−5.14/−3.74 |
0.002 |
0.028 |
166
|
| PBAI6 |
BAI2 |
BTz |
40/2.98 |
TGBC |
−4.97/−3.74 |
0.52 |
3.11 |
166
|
| PBPD1 |
BPD |
T |
37/2.4 |
BGBC |
−5.06/−3.62 |
0.22 |
0.16 |
167
|
| PBPD1-Si |
BPD |
T |
28/2.4 |
BGBC |
−5.01/−3.65 |
0.74 |
0.87 |
167
|
| PBPD2 |
BPD |
S |
52/3.1 |
BGBC |
−4.88/−3.65 |
0.22 |
0.05 |
167
|
| PBPD2-Si |
BPD |
S |
13/2.2 |
BGBC |
−4.82/−3.69 |
0.52 |
0.35 |
167
|
| PBPD3 |
BPD |
BT |
7.2/2.22 |
BGBC |
−5.0/−3.7 |
1.24 |
0.82 |
137
|
| PBPD4 |
BPD |
TT |
4.8/2.08 |
BGBC |
−5.0/−3.6 |
1.37 |
— |
137
|
| PBPD5 |
BPD |
TVT |
7.4/2.32 |
BGBC |
−3.62/1.14 |
0.245 |
0.095 |
168
|
| PBPD6 |
BPD |
TVT2 |
12.4/2.36 |
BGBC |
−3.65/1.26 |
0.109 |
0.081 |
168
|
| PP661 |
P661 |
V |
35/2.68 |
TGBC |
−5.74/−3.45 |
— |
0.34 |
140
|
| PP662 |
P662 |
V |
20.4/2.62 |
TGBC |
−5.80/−3.52 |
— |
0.51 |
140
|
| PNTDT |
NTDT |
BDT2 |
60.7/3.15 |
BGTC |
−5.33/−3.67 |
0.0058 |
— |
141
|
| PBFIT |
BFI |
T |
47.5/3.68 |
BGTC |
−3.80/−5.45 |
— |
0.3 |
130
|
| PBFIBT |
BFI |
BT |
— |
BGTC |
−3.74/−5.14 |
— |
0.09 |
130
|
| PBBT1 |
BBT |
TT |
15.1/1.94 |
BGBC |
−4.36/−3.80 |
1.0 |
0.7 |
142
|
| PBBT2 |
BBT |
CDT |
17/2.8 |
BGBC |
−4.8/−4.0 |
0.13 |
0.1 |
143
|
| PBBT3 |
BBT |
SiDT |
18/2.8 |
BGBC |
−4.8/−4.1 |
0.0025 |
0.016 |
143
|
| PBBT4 |
BBT |
FL |
11/2.8 |
BGBC |
−5.1/−3.9 |
0.0017 |
0.007 |
143
|
| PBBT5 |
BBT |
PD |
14/2.8 |
BGBC |
−4.8/−4.2 |
0.011 |
0.011 |
143
|
| PBBT2-C6 |
BBT |
DPP1 |
8.7/1.5 |
BGBC |
−4.55/−3.9 |
0.83 |
1.36 |
62
|
| PDPP50 |
BBT |
DPP1 |
8.8/1.8 |
BGBC |
−4.55/−3.9 |
1.17 |
1.32 |
62
|
| PSeS |
SeS |
TzFLTz |
20.3/3.53 |
BGTC |
−5.34/−3.86 |
0.0073 |
0.087 |
144
|
| PSN |
SN |
TzFLTz |
30.0/2.38 |
BGTC |
−5.21/−3.48 |
0.65 |
— |
144
|
| PSeN |
SeN |
TzFLTz |
12.6/2.21 |
BGTC |
−5.17/−3.53 |
0.0017 |
0.0078 |
144
|
| PBDTTQ-1 |
BDTTQ-1 |
BT3 |
37.3/2.43 |
BGTC |
−5.50/−3.86 |
— |
— |
145
|
| PBDTTQ-2 |
BDTTQ-2 |
BT3 |
11.8/1.66 |
BGTC |
−5.48/−4.01 |
0.0012 |
0.0006 |
145
|
| PBDTTQ-3 |
BDTTQ-3 |
BT |
76.3/3.63 |
BGBC |
— |
0.22 |
0.21 |
148
|
| PTTQ |
TTQ |
D110 |
17.7/2.21 |
BGBC |
−5.40/−3.91 |
0.0018 |
0.0016 |
146
|
| PPTQ-1 |
PTQ-1 |
D110 |
18.3/3.56 |
BGBC |
−5.50/−3.96 |
0.028 |
0.042 |
146
|
| PPTQ-2 |
PTQ-2 |
T |
274/6.3 |
BGTC |
−5.17/−4.21 |
0.03 |
0.014 |
147
|
| PPTQ-3 |
PTQ-3 |
T |
45/6.3 |
BGTC |
−5.21/−4.29 |
0.0011 |
0.02 |
147
|
| PAPhTQ |
APhTQ |
BT |
100.9/6.49 |
BGBC |
— |
0.11 |
0.02 |
148
|
| PBPP1 |
BPP |
T |
37.0/1.13 |
TGBC |
−3.58/−5.34 |
0.012 |
0.03 |
169
|
| PBPP2 |
BPP |
BT |
17/2.47 |
TGBC |
−5.63/−4.05 |
— |
0.001 |
169
|
| PBPP3 |
BPP |
TT |
15/3.41 |
TGBC |
−5.68/−4.07 |
— |
0.001 |
169
|
| PBPP3 |
BPP |
TVT |
20/2.28 |
TGBC |
−5.55/−4.06 |
— |
0.002 |
169
|
| PBPP4 |
BPP |
CDT |
21/1.96 |
TGBC |
−5.44/−3.95 |
— |
0.01 |
169
|
| PBPP5 |
BPP |
BTz |
54/1.25 |
TGBC |
−5.84/−3.99 |
— |
0.01 |
169
|
| PBPP6 |
BPP |
S |
74.0/1.74 |
TGBC |
−3.59/−5.32 |
0.017 |
0.022 |
170
|
| PBPP7 |
BPP |
Fu |
47.0/1.64 |
TGBC |
−3.60/−5.37 |
0.0048 |
0.00058 |
170
|
| PBPP8 |
BPP |
B |
20/2.08 |
BGTC |
−5.39/−3.35 |
— |
0.0024 |
149
|
| PBPT1 |
BPT |
T |
34/1.67 |
TGBC |
−5.27/−4.24 |
0.2 |
0.1 |
150
|
| PBPT2 |
BPT |
BT |
5/3.30 |
TGBC |
−5.33/−4.16 |
0.08 |
0.01 |
150
|
| PDPIDT-T |
DPIDT |
T |
25/1.6 |
TGBC |
−5.4/−3.8 |
0.05 |
0.04 |
152
|
| PDPIDT-P |
DPIDT |
B |
106/1.7 |
TGBC |
−5.2/−3.5 |
0.08 |
— |
152
|
| PDPIDT-2T |
DPIDT |
BT |
10/1.9 |
TGBC |
−5.3/−3.7 |
0.09 |
0.04 |
152
|
| PTPTQD1 |
TPTQD |
BT |
74.7/4.97 |
BGTC |
−5.29/−3.55 |
0.15 |
— |
153
|
| PTPTQD2 |
TPTQD |
TVT |
76.9/2.28 |
BGTC |
−5.14/−3.57 |
0.58 |
— |
153
|
| PPQ2TBT |
PQ2T |
BT |
38.9/3.68 |
BGBC |
— |
0.0064 |
— |
154
|
| PQA2T |
QA |
BT |
12/2.5 |
BGTC |
−5.2/— |
0.27 |
— |
155
|
| PQA3T |
QA |
3T |
11/1.9 |
BGTC |
−5.2/— |
0.24 |
— |
155
|
| PQTE |
QA |
TVT |
20.60/1.15 |
BGTC |
−5.26/−3.38 |
0.67 |
— |
156
|
| PQB |
QA |
BTz5 |
16.2/2.5 |
BGTC |
−5.26/−3.31 |
0.00577 |
— |
157
|
| PQBOC8 |
QA |
BTz5-OR |
40.4/1.8 |
BGTC |
−5.31/−3.06 |
0.30 |
— |
157
|
| PEPD2T |
EPD |
BT |
10.7/— |
BGTC |
−5.50/−3.42 |
0.40 |
— |
158
|
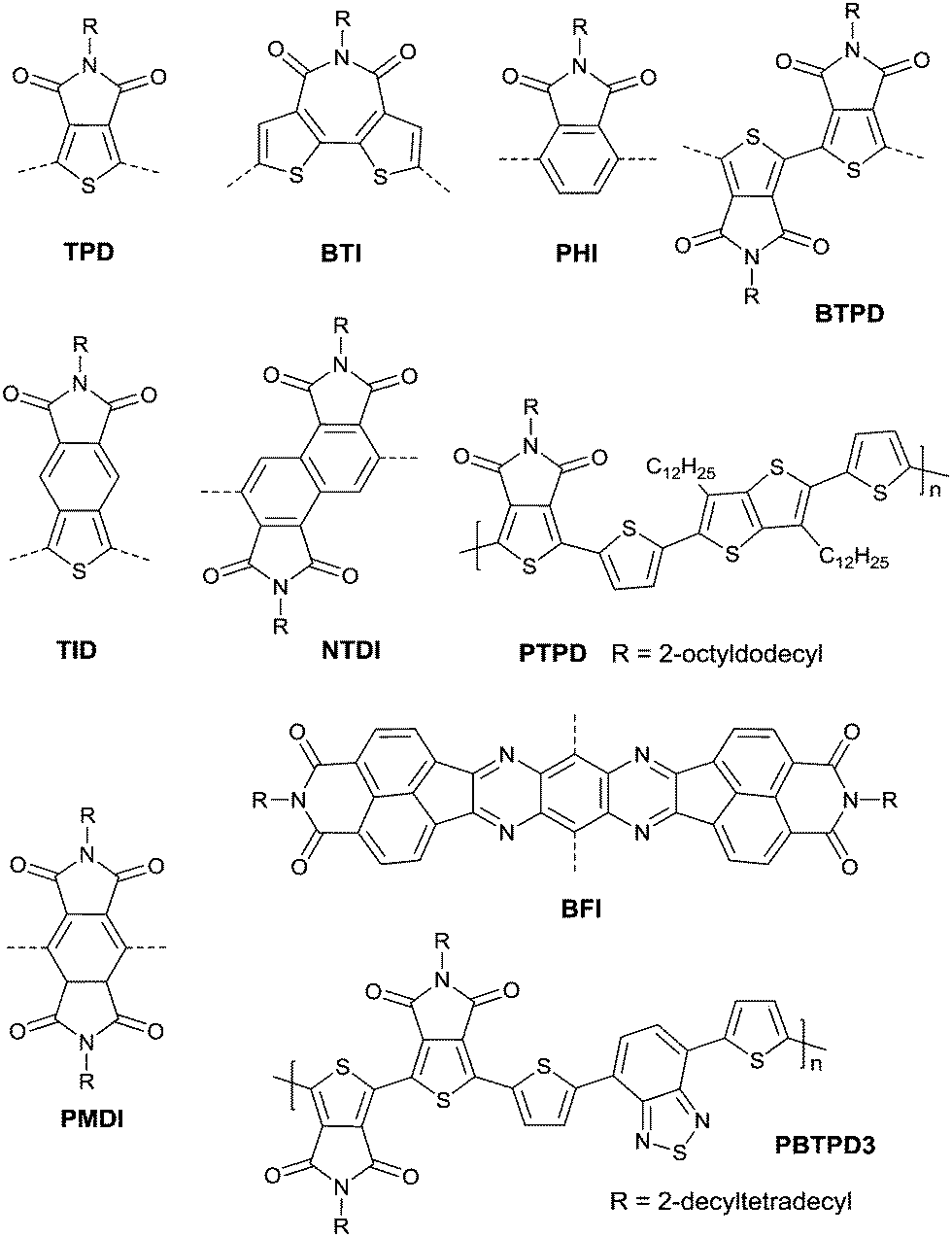
Thieno[3,4-c]pyrrole-4,6-dione (TPD),116–121 1,1′-bithieno[3,4-c]pyrrole-4,4′,6,6′-tetraone (BTPD),122,123 bithiopheneimide (BTI),116,124 thienoisoindoledione (TID),125 phthalimide (PHI),118,126 pyromellitic diimide (PMDI),127 and naphthalene tetracarboxylic diimide128 (NTDI or iso-NDI) were also reported as the arylene imides for polymer semiconductors. PMDI has the same carboxylic group numbers as NDI and PDI, but the OFET performances of the polymers based on it and a different number of thiophene rings were poor.127 Meanwhile, PDI, TPD, BTI, PHI and TID have two fewer carboxylic groups resulting in lower electron-deficiency when compared with NDI and PDI, but they exhibit fine symmetry and planarity, and are rigidly fused, which is beneficial to the enhancement of intermolecular interactions.118 Polymer PTPD (Fig. 12 and Table 5) exhibited a hole mobility of 1.29 cm2 V−1 s−1, while polymers based on BTI (PBTI1-2, Table 5) and PHI (PPHI, Table 5) did not show high mobility. TID-based polymer semiconductors have only been mentioned in organic solar cells.125 BTPD and NTDI, as the dimeric forms of TPD and PHI, exhibit higher electron-withdrawing characteristics. In addition, the strong O⋯S non-covalent interaction between the TPD subunits endowed the anti-coplanar structure of TPD.123 Among the polymers based on BTPD (PBTPD1–4, Table 5), the highest performance was achieved by PBTPD3 (Fig. 12) with an electron mobility of 1.02 cm2 V−1 s−1 and a hole mobility of 0.33 cm2 V−1 s−1, when the charge donor was benzothiadiazole (BTz). Tetraazabenzodifluoranthene diimides (BFI, Fig. 12), as the first examples of heterocyclic diimides, were synthesized by Li et al.129 Later on, they also constructed the polymers PBFIT (Table 5) and PBFIBT (Table 5) based on BFI, among which, PBFIT exhibited a high electron mobility of 0.3 cm2 V−1 s−1 (the highest value for unipolar n-type conjugated polymers before 2013) based on BGTC transistors.130
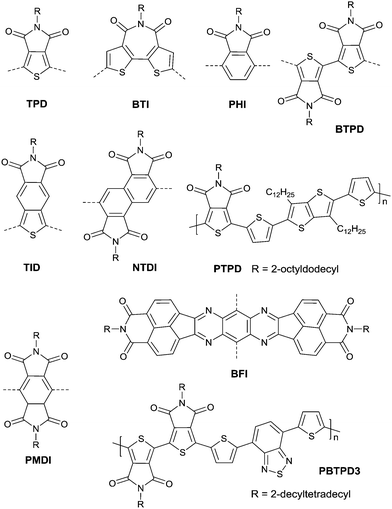 |
| | Fig. 12 Chemical structures of TPD, BTI, PHI, BTPD, TID, NTDI, PTPD, BFI, PMDI and PBTPD3. | |
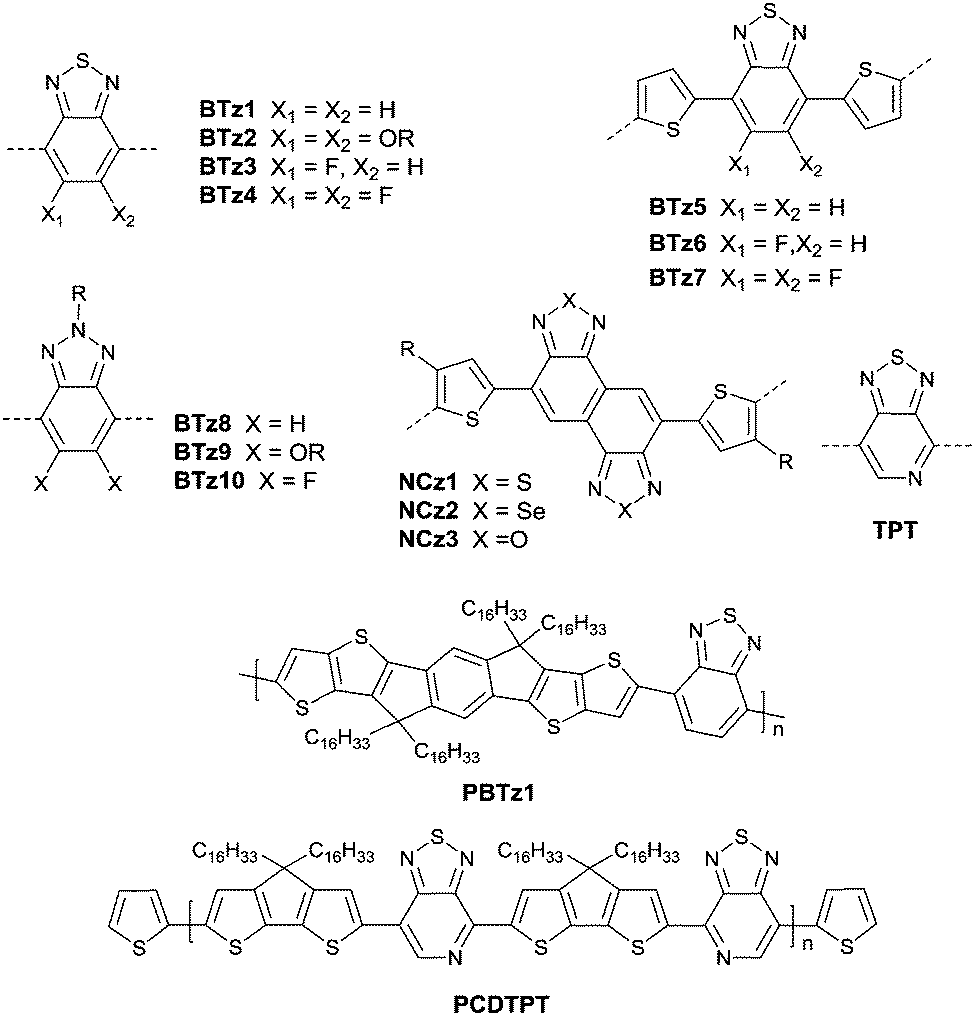
Benzothiadiazole also played the acceptor role in donor–acceptor polymers when a suitable co-monomer was chosen. BTz1–BTz10 (Fig. 13) are BTz derivatives. Zhang et al.131 prepared the BTz-based polymer PBTz1. Indacenodithieno-[3,2-b]thiophene of a largely conjugated structure and fine planarity was copolymerized with BTz. Linear alkyl chains connected to indacenodithieno[3,2-b]thiophene gave the polymer PBTz1 (Fig. 13 and Table 5) good solubility in tetrahydrofuran (THF), chloroform, toluene and chlorobenzene. Therefore, PBTz1-based OFETs showed a high-performance with a hole mobility of up to 6.6 cm2 V−1 s−1 when annealed at 270 °C and the mobility further increased to 8.7 cm2 V−1 s−1 when CuSCN was solution processed to be the electrode. Naphthobischalcogenadiazole (NCz)132,133 was also studied. Kawashima et al.132 investigated the effect of the chalcogen atom on NCz-based conjugated polymers. The results showed that NCz2 (Fig. 13)-based polymer PNCz2 (Table 5) had a poor p-channel behavior caused by the defect of the polymer backbone (the repulsion between the large size of the selenium atom and the adjacent thiophene ring), while the NCz3 (Fig. 13)-based polymer PNCz3 (Table 5) exhibited a good ambipolar property which was even better than that of the naphthobisthiadiazole (NCz1 as in Fig. 13)-based polymer PNCz1 (Table 5). The polymer PCDTPT (Fig. 13 and Table 5) based on TPT (the benzene ring of BTz was replaced by pyridine, Fig. 13) was synthesized as the semiconductor layer for OFETs as reported by Luo and Heeger et al.134 They adopted a novel processing method that regulates the self-assembly and arrangement of the polymer chain on nanogrooved substrates through capillary action. The fabricated corresponding PCDTPT based OFETs (BGBC) via such a method showed a hole mobility of 36.3 cm2 V−1 s−1 for a channel length of 140 μm, while 52.7 cm2 V−1 s−1 for a channel length of 160 μm.
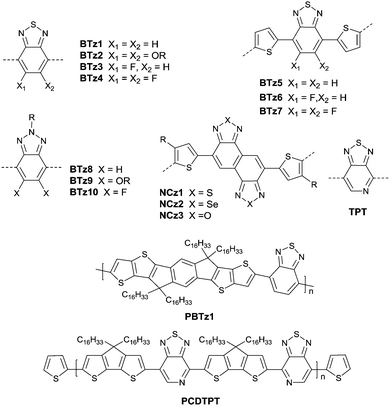 |
| | Fig. 13 Chemical structures of BTz1–BTz10, NTz1–NTz3, TPT, PBTz1 and PCDTPT. | |
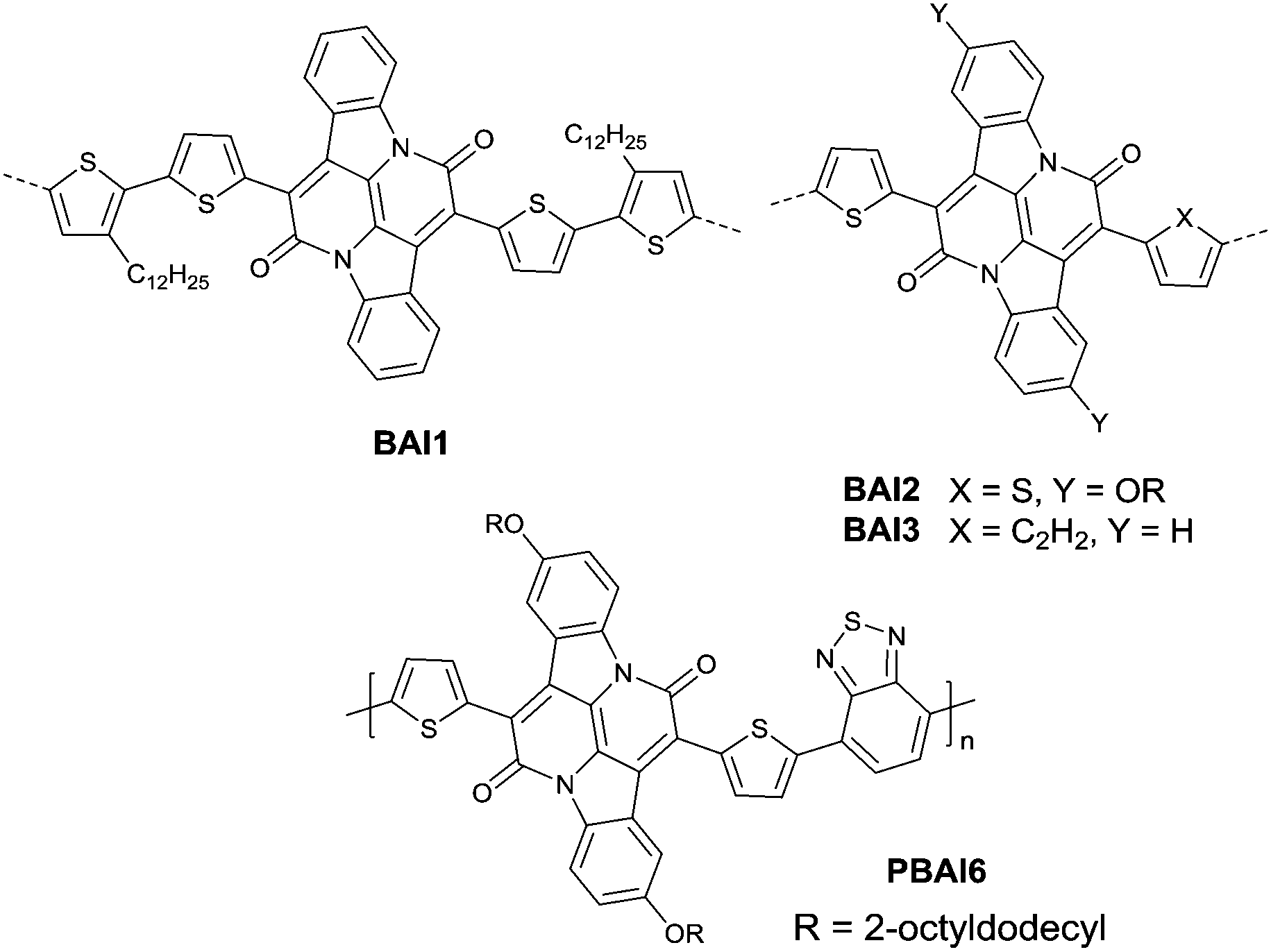
Recently, bay-annulated indigo (BAI), an old natural dye, was also studied as the electron-deficient building block. He et al.135 firstly synthesized the polymers PBAI1 (Table 5) and PBAI2 (Table 5) based on BAI1 (Fig. 14), and a hole mobility of 1.5 cm2 V−1 s−1 and an electron mobility of 0.41 cm2 V−1 s−1 were shown for PBAI1 (Table 5). PBAI2–PBAI6 (Table 5) are also polymers based on BAI. Among them, PBAI6 (Fig. 14 and Table 5) exhibited a high electron mobility of up to 3.11 cm2 V−1 s−1. Besides, Matthew A. Kolaczkowski et al.136 also synthesized the desymmetrized BAI derivative BAI3 (Fig. 14), which was flanked by thiophene on one side and benzene on the other side. But polymers based on BAI3 or other desymmetrized BAI derivatives still have not been reported. As a side note, BAI may be a potential electron-deficient unit for high-performance polymer semiconductors.
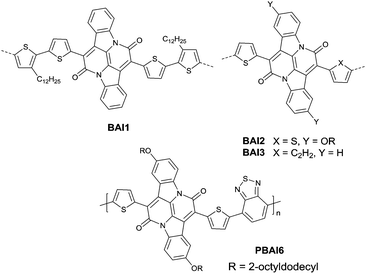 |
| | Fig. 14 Chemical structures of BAI1–BAI3 and PBAI6. | |
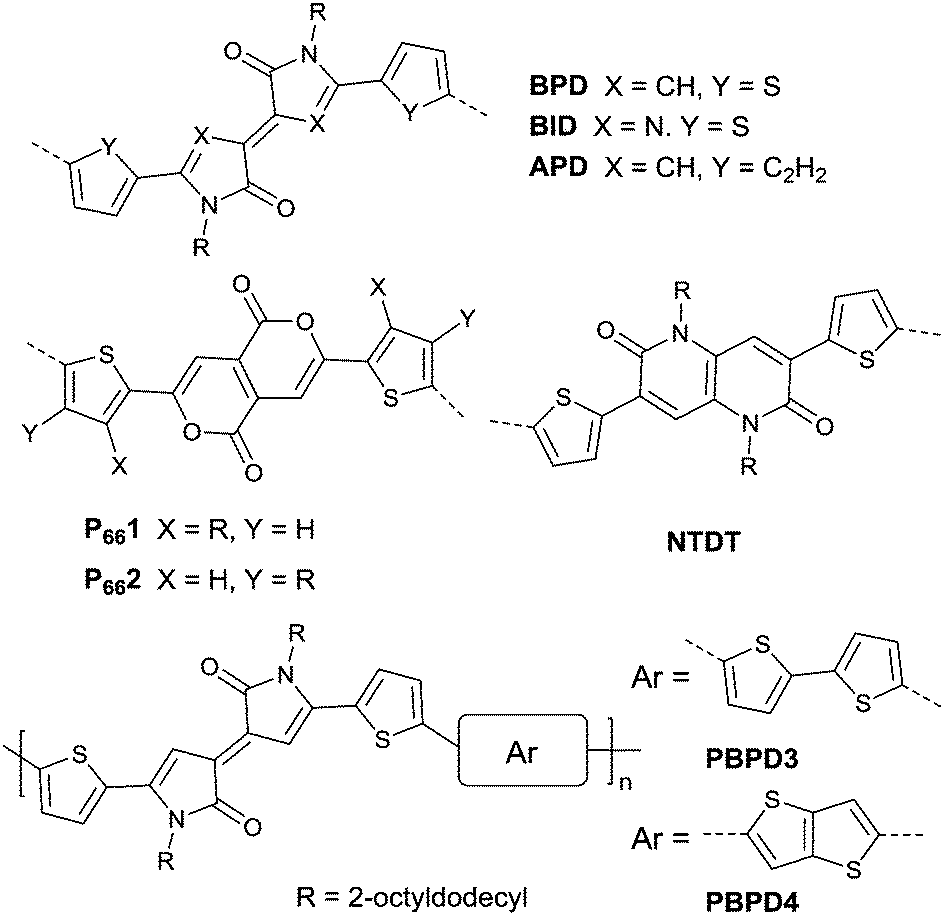
Pechmann dye, as a planar and polar electron-deficient unit, was firstly incorporated into D–A conjugated polymers in 2014 by Zhang’s group.137 The bipyrrolylidene-2,2′(1H,1′H)-dione (BPD, a Pechmann dye derivative) based polymer PBPD3 (Fig. 15 and Table 5) exhibited a narrow bandgap of 1.1 eV and ambipolar behaviour with a hole mobility of 1.24 cm2 V−1 s−1 and an electron mobility of 0.82 cm2 V−1 s−1. In addition, the polymer PBPD4 (Fig. 15 and Table 5) showed p-channel properties with mobility reaching 1.37 cm2 V−1 s−1. Zhang’s group also synthesized polymers PBPD1, PBPD-SiPBPD2, PBPD2-Si, PBPD5, and PBPD6 (Table 5) for polymer FETs, which exhibit a mobility below 1 cm2 V−1 s−1. Similar to BPD, BID138 and APD139 (Fig. 15) were investigated as well but still have not been studied for polymer FETs yet. Finally, Pei140 and co-workers calculated the HOMO and LUMO levels of different kinds of Pechmann dyes via DFT. According to the calculation result that 6,6-endo-dilactones (P66) is of a lower LUMO level than 5,5-exo-dilactams (N–P55, the central part of BPD), they prepared charge acceptor monomers P661 (Fig. 15) and P662 (Fig. 15) and their corresponding polymers PP661 (Table 5) and PP662 (Table 5) successfully. Theory calculations were proved by the OFET characterization with a unipolar electron-transporting behaviour (μe(PP661) = 0.34 cm2 V−1 s−1 and μe(PP662) = 0.51 cm2 V−1 s−1). 3,7-Dithiophen-2-yl-1,5-dialkyl-1,5-naphthyridine-2,6-dione (NTDT as in Fig. 15), which is of a tightly similar structure to P661 and P662, was synthesized by Yoon et al.141 But the performance of the polymer PNTDT (Table 5) based on it was very poor. The synthesis steps of the brominated P661 monomer and the NTDT monomer are shown in Scheme 9.
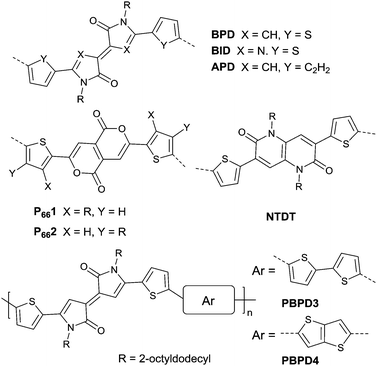 |
| | Fig. 15 Chemical structures of BPD, BID, APD, P661, P662, NTDT, PBPD3 and PBPD4. | |
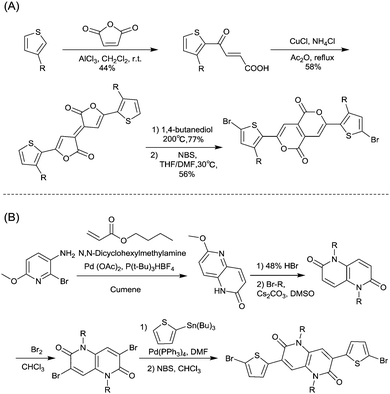 |
| | Scheme 9 Syntheses of brominated (A) P661 and (B) NTDT monomers. | |
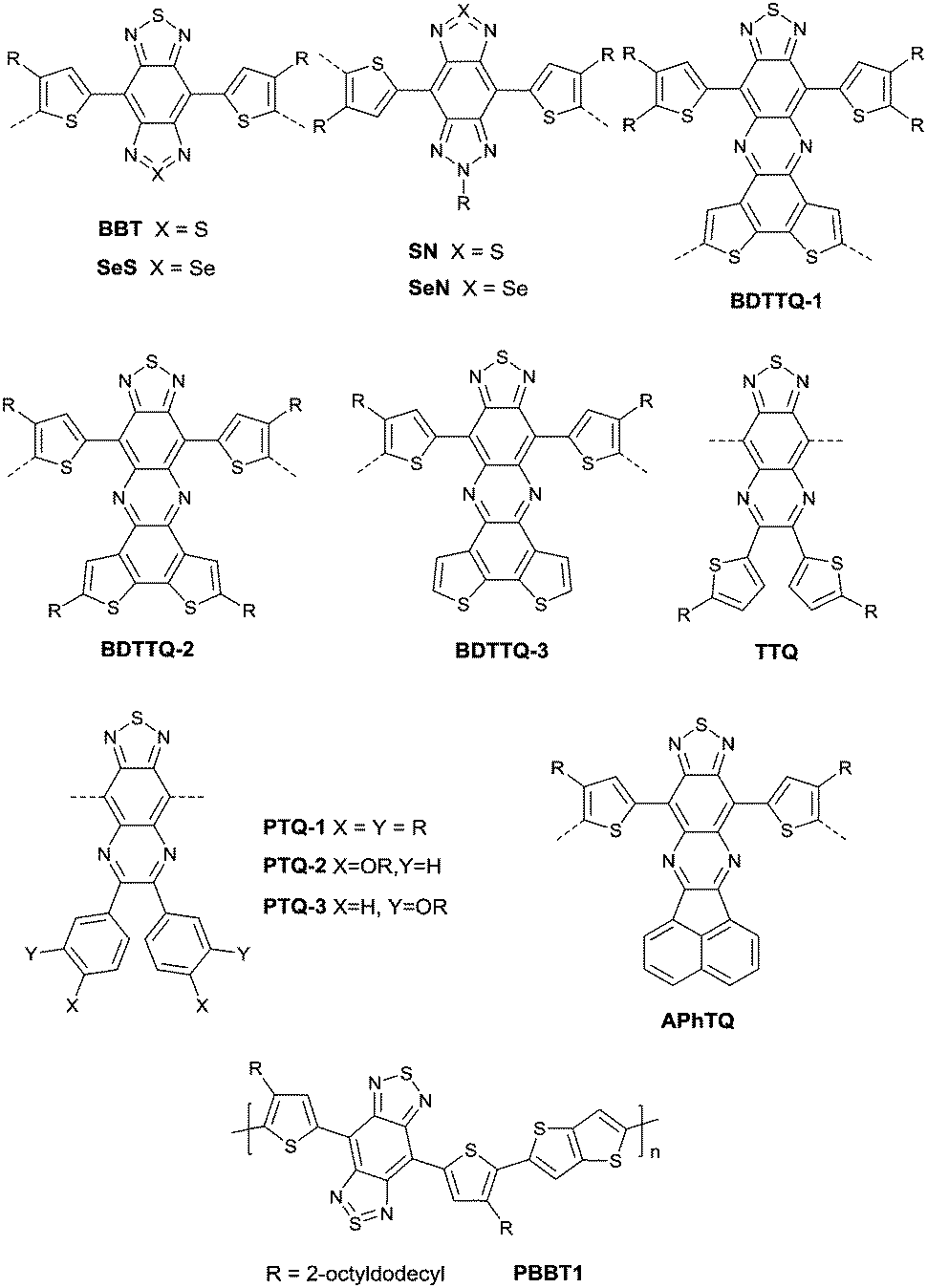
As shown in Fig. 16, benzobisthiadiazole62,142,143 (BBT) was studied as the charge acceptor of polymer semiconductors as well. As aforementioned, BBT is of strong electron-withdrawing ability, and it even acted as a charge acceptor when copolymerized with DPP (PBBT2-C6 and PDDP50 listed in Table 5). The BBT-based polymer PBBT1142 (Fig. 16 and Table 5) showed a nearly balanced ambipolar behaviour with a hole mobility of 1.0 cm2 V−1 s−1 and an electron mobility of 0.7 cm2 V−1 s−1 owing to the low bandgap caused by the strong D–A interaction. Besides, as the side alkyl chain added to neighboring BT heterocycles endows benzobisthiadiazole with processable properties, many other ambipolar polymers based on BBT were synthesized (PBBT2–PBBT5 as listed in Table 5). But their OFET performance still needs to be improved. Subsequently, Wang et al.144 prepared a series of benzobisthiadiazole analogues including selenadiazolo-benzothiadiazole (SeS as in Fig. 16), thiadiazolobenzotriazole (SN as in Fig. 16) and selenadiazolobenzotriazole (SeN as in Fig. 16) via heteroatom substitution and copolymerized them with the same donor (TzFLTz listed in Table 5), respectively. Such polymers did not show promising OFET performance, but the charge polarity of the polymers was proved to be influenced as the polymers PseS (Table 5) and PseN (Table 5) exhibit ambipolar properties while PSN (Table 5) showed p-channel charge transporting behaviour.
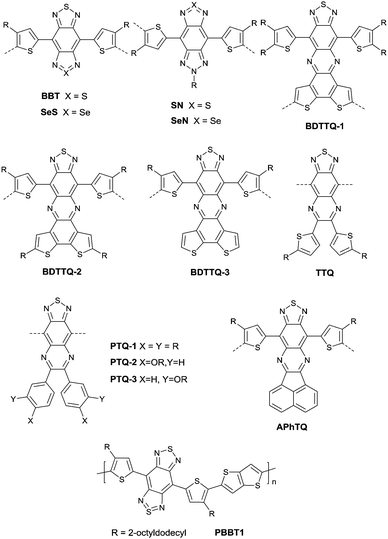 |
| | Fig. 16 Chemical structures of BBT, SeS, SN, SeN, BDTTQ-1–BDTTQ-3, TTQ, PTQ-1–PTQ-3, APhTQ, and PBBT1. | |
Thiadizoloquinoxaline (TQ), as another structure containing thiadiazole, was also investigated. Compared to the BBT acceptor, TQ acceptors have more sites to be alkylated, which will influence the solubility, structure, and packing of the corresponding polymers. As shown in Fig. 16, BDTTQ-1, BDTTQ-2 and BDTTQ-3 are of the same core structures but of different side chain distributions and linking pathways for polymers. PBDTTQ-2145 (Table 5) based on BDTTQ-2 exhibited ambipolar properties while PBDTTQ-1145 (Table 5) based on BDTTQ-1 did not show any field-effect response. Besides, such a phenomenon was also observed for PTQ1–PTQ3 (Fig. 16) based polymers. More recent studies found that the polymer PPTQ1146(Table 5) based on PTQ1 and the polymer PPTQ2147 (Table 5) based on PTQ2 exhibited a balanced ambipolar property while PPTQ3147 showed an accessible n-type behavior.
Baumgarten et al.146 also made a comparison between the TTQ acceptor (Fig. 16) and the PTQ-1 acceptor (Fig. 16) when they copolymerized them with the same donor (thiophene derivatives). The results showed that the polymer PPTQ-1 (Table 5) based on PTQ-1 exhibited ambipolar properties, which were an order of magnitude larger than those of the polymer PTTQ (Table 5) based on TTQ. Apart from that, researchers also compared the polymer PBDTTQ-3148 (Table 5) based on BDTTQ-3 (Fig. 16) with the polymer PAPhTQ (Table 5) based on AphTQ (Fig. 16). The results suggested that PBDTTQ-3 had the best OFET performance of the AQ-based polymers with a hole mobility up to 0.22 cm2 V−1 s−1 and an electron mobility of 0.21 cm2 V−1 s−1.
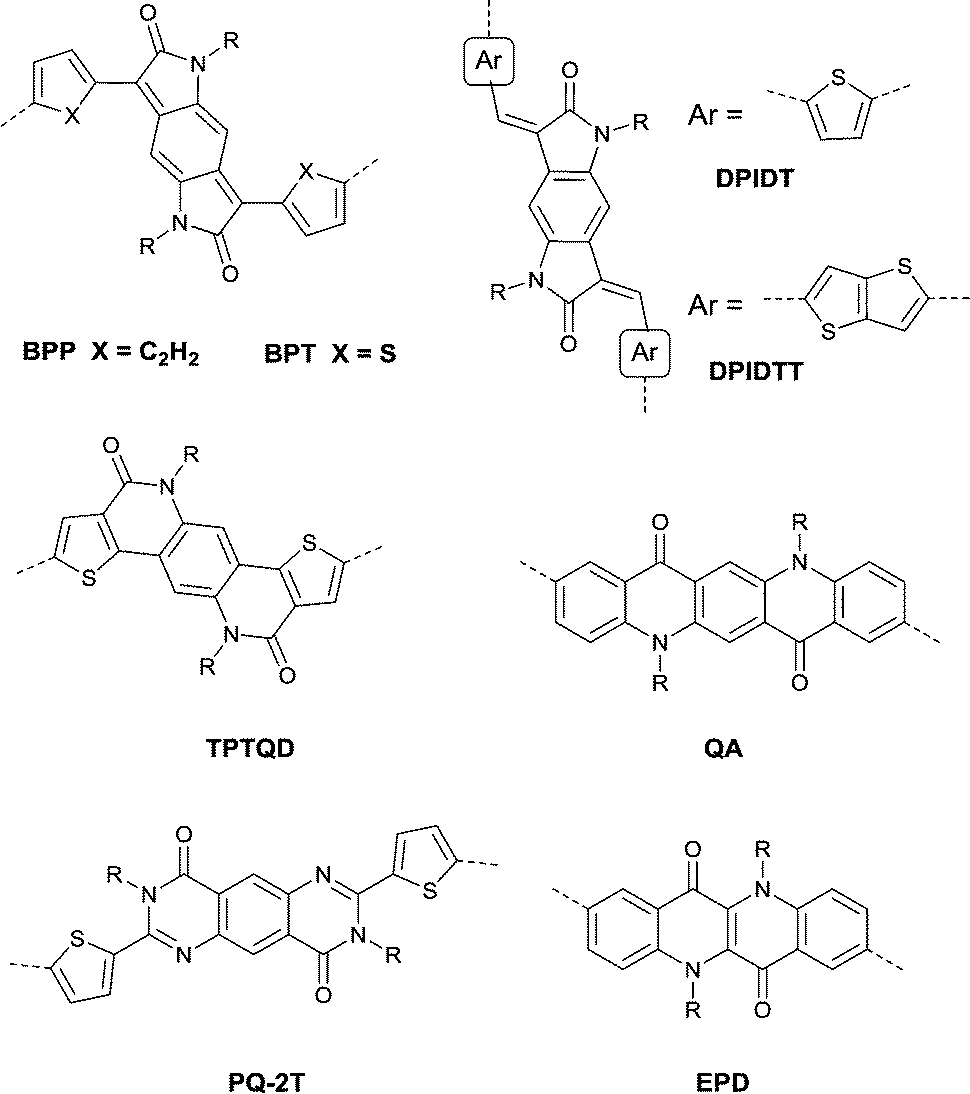
Benzodipyrrolidone (BPD), as the elongated DPP, has also been explored. In 2011, a phenyl-flanked benzodipyrrolidone (BPP in Fig. 17) based polymer for OFETs was synthesized by Cui et al.149 Later on, a series of polymers (PBPP1–PBPP8, as listed in Table 5) based on BPP were investigated, but all of them exhibited poor performance. Similar to the studies in DPP, the polymers PBPT1 (Table 5) and PBPT2 (Table 5) based on thiophene-flanked benzodipyrrolidone (BPT, Fig. 17) were firstly introduced by Rumer et al.150 Both of them showed ambipolar behaviour, and a hole mobility of 0.2 cm2 V−1 s−1 and an electron mobility of 0.1 cm2 V−1 s−1 were observed for the OFETs based on PBPT1. Meanwhile, dihydropyrroloindoledione (DPID), as another unit structurally related to DPP, was explored by Rumer et al.151,152 as well. Thiophene-flanked DPID (DPIDT as in Fig. 17)-based polymers PDPIDT-T (Table 5) and PDPIDT-2T (Table 5) possessed ambipolar behaviour while the polymer PDPIDT-P (Table 5) showed p-type semiconductor characteristics. However, the TT-flanked DPID (DPIDTT, Fig. 17) based polymer (copolymerized with thiophene) was too insoluble to be further studied. Pei et al.153 also synthesized a pentacyclic aromatic lactam, thieno[2′,3′:4,5]pyrido[2,3-g]-thieno[3,2-c]quinoline-4,10(5H,11H)-dione (TPTQD, Fig. 17), via Beckmann rearrangement. The polymers PTPTQD1 (Table 5) and PTPTQD2 (Table 5) exhibited p-channel semiconductor behaviour with a mobility of 0.15 cm2 V−1 s−1 and 0.58 cm2 V−1 s−1, respectively. Similarly, Li154 and co-workers reported polymers consisting of a kind of aromatic lactam unit – pyrimido[4,5-g]quinazoline-4,9-dione (PQ). But the polymer PPQ2TBT (Table 5) based on PQ2T (Fig. 17) exhibited poor p-type behaviour with the mobility at a magnitude of 10−3 cm2 V−1 s−1.
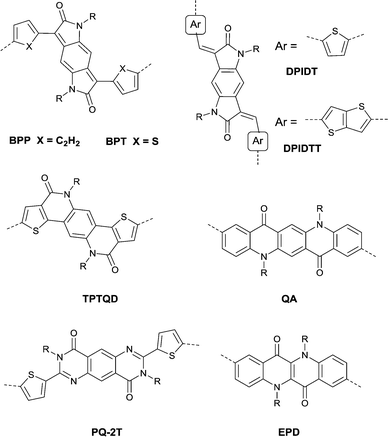 |
| | Fig. 17 Chemical structures of BPP, BPT, DPIDT, DPIDTT, TPTQD, QA, PQ-2T and EPQ. | |
Quinacridones (QAs, Fig. 17), as red-violet pigments, were investigated as well. Osaka155 and co-workers firstly incorporated the quinacridone unit into polymers for OFETs. Researchers found that the orientational order of the polymer PQA2T was improved by the enhancement of molecular weight and a small π–π stacking distance of 3.6 Å was achieved. Nevertheless, the hole mobility of the polymer was not sensitive to the varying molecular weights, which may be caused by the relatively limited delocalization of the HOMO and π-conjugation. The polymer PQA2T (Table 5) exhibited a hole mobility of up to 0.27 cm2 V−1 s−1 and PQA3T (Table 5) showed a hole mobility of up to 0.24 cm2 V−1 s−1. Then, the polymers PQTE,156 PQB157 and PQBOC8,157 as listed in Table 5, were also further studied. Among them, PQTE exhibited the highest hole mobility of 0.67 cm2 V−1 s−1.
Epindolidione (EPD, Fig. 17), as an isomer of IID and of a similar structure to quinacridone, was firstly incorporated into conjugated polymers for OFETs by Pei et al.158via the effective synthesis method (Scheme 10). The corresponding polymer PEPD2T (Table 5) showed p-type semiconductor features with a mobility of up to 0.40 cm2 V−1 s−1.
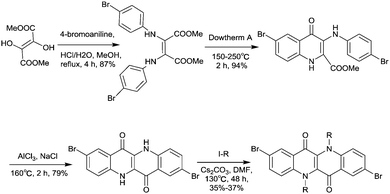 |
| | Scheme 10 Syntheses of the brominated EPD monomer. | |
3. Backbone halogenation
3.1 Fluorination
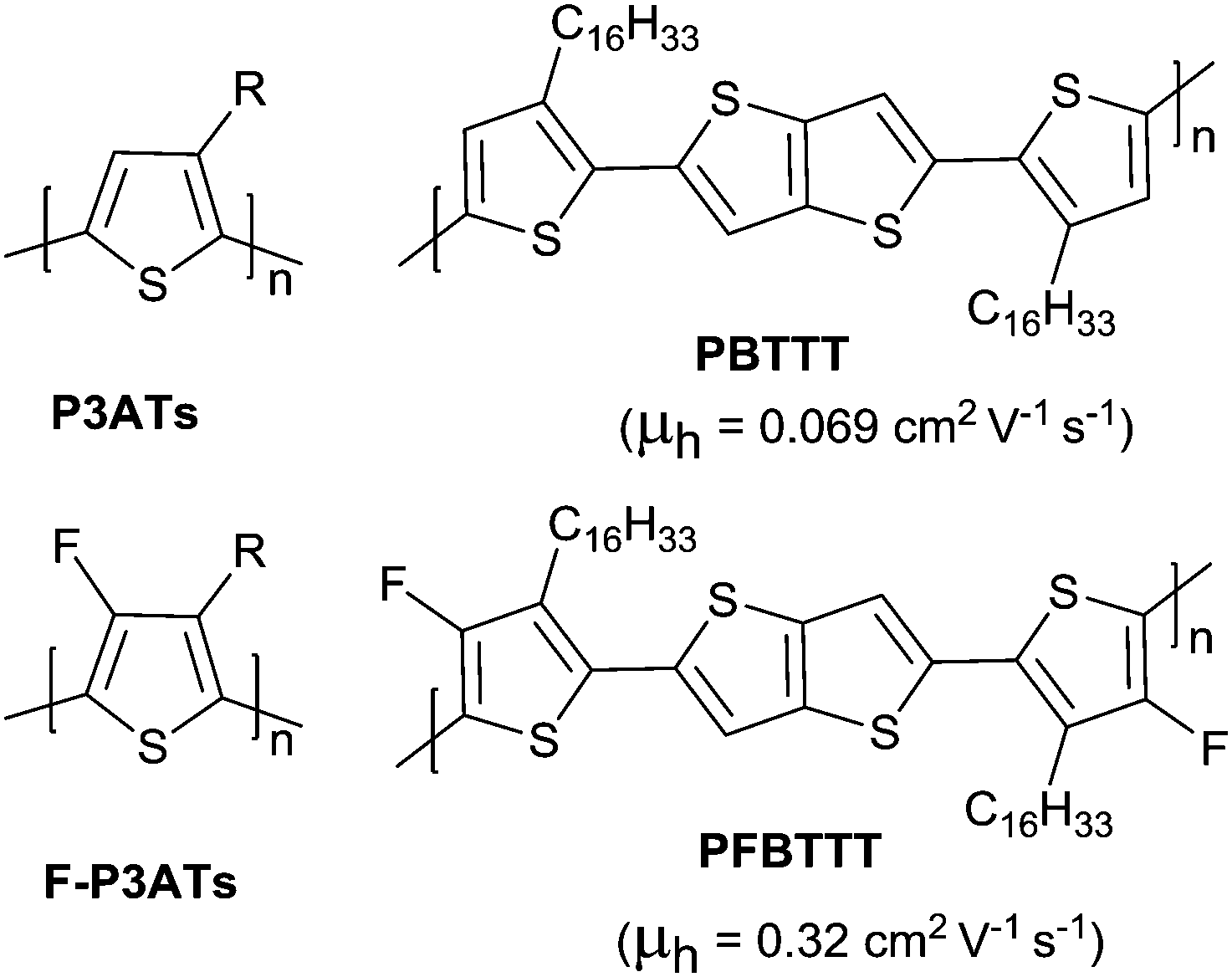
Halogen, cyanogroup and diimide moieties are the commonly electron-deficient groups used for the n-type semiconductors for OFETs. Among them, fluorine substitution on the conjugated polymer has attracted more attention because of the special properties of the fluorine atom, e.g. a small atom size slightly larger than hydrogen (rvanderWaals(F) = 1.35 Å, rvanderWaals(H) = 1.2 Å) and the highest Pauling electronegativity of 4.0.171 Due to its small size, it does not cause steric hindrance enhancement but does affect the crystallinity and orientation of the polymer. As shown in Fig. 18, through comparing poly(3-alkyl-4-fluoro)thiophenes (F-P3ATs) and poly(3-alkyl)-thiophenes (P3ATs), Fei et al.172 found that the ionization potential of the polymer could be enhanced without a significant change in the optical band gap by backbone fluorination. Besides, because the polymer had a more co-planar backbone after fluorination, the average charge carrier mobility of the fluorinated polymer F-P3ATs was 5 orders of magnitude higher than the analogous non-fluorinated polymers P3ATs. Boufflet et al.173 also theoretically and experimentally studied the effect of fluorination on the molecule backbone planarity and OFET performance via a comparison between poly(2,5-bis(3-alkylthiophen-2-yl)thieno[3,2-b]thiophene) (PBTTT as in Fig. 18) and poly(2,5-bis(4-fluoro-3-hexadecyl-thiophen-2-yl)thieno[3,2-b]thiophene) (PFBTTT as in Fig. 18). They illustrated that the physical properties of the polymer were strongly affected by backbone fluorination, including a significant enhancement of the aggregation degree and melting temperature owing to the F⋯S interactions leading to an increase in backbone rigidity and planarity. In addition, the hole mobility in the OFET devices and ambient stability were also improved through backbone fluorination.
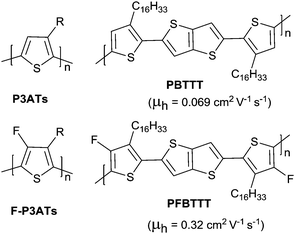 |
| | Fig. 18 Chemical structures of P3ATs, PBTTT, F-P3ATs and PFBTTT. | |
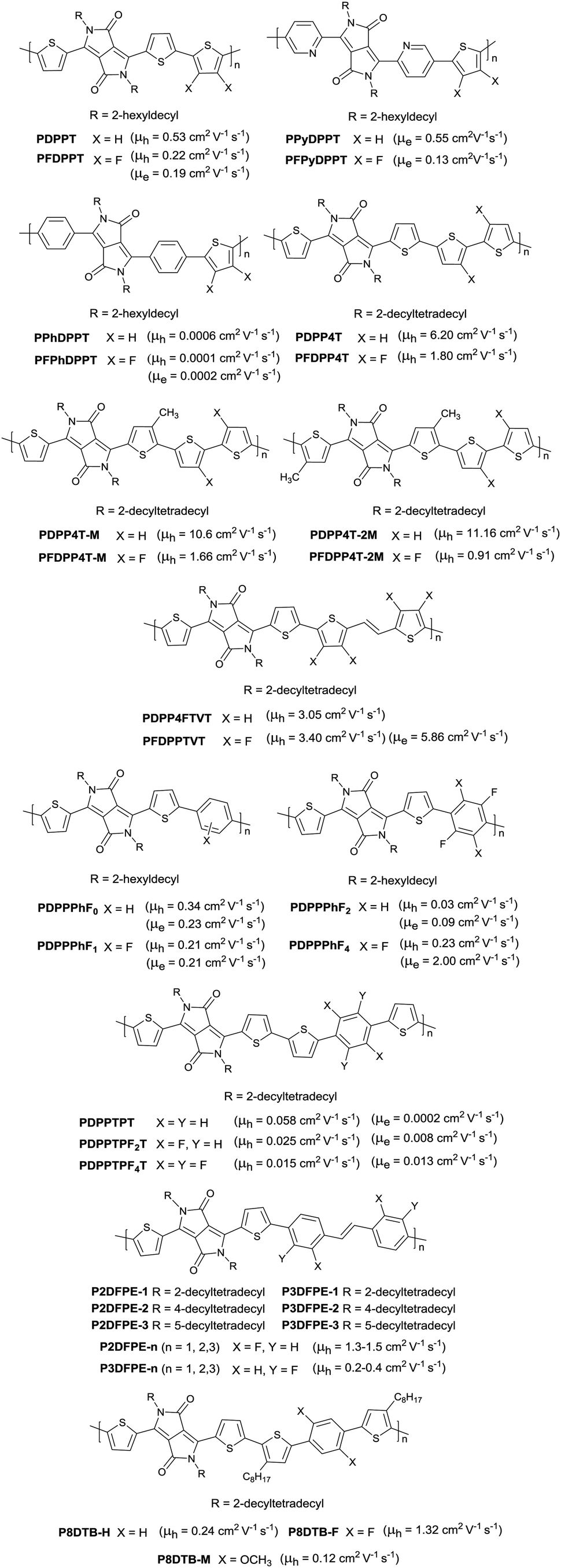
Following the above-mentioned fluorothiophene studies, difluorothiophene was also used as the building block for fluorinated conjugated polymers. In 2015, in order to explore the effect of fluorination of the co-monomer on the charge carrier mobility, optical properties, and crystallinity of different aryl-flanked DPP based polymers, Mueller et al.39 prepared six polymers (PDPPT, PFDPPT, PPyDPPT, PFPyDPPT, PPhDPPT and PFPhDPPT, as shown in Fig. 19) based on DPP having varying flanking aryl units and thiophene or difluorothiophene as the co-monomer. They found that the crystallinity of the DPP-based polymers could be improved by fluorination irrespective of the different flanking aryl units. As shown in Fig. 19, it was also realized that electron transport of such polymers was deeply affected by fluorination while the bulk hole mobility was insensitive to such structural variations. This conclusion was also proved by Gao et al.59 The fluorinated polymer PFDPPTVT (Fig. 19) exhibited a hole mobility of 3.4 cm2 V−1 s−1 and an electron mobility of 5.86 cm2 V−1 s−1, while the nonfluorinated analogue PDPPTVT only showed p-channel transport with a hole mobility of 3.05 cm2 V−1 s−1. This can be explained by the fact that the LUMO level of polymer semiconductors can be reduced by fluorination contributing to easier electron injection from the electrode in OFET devices.171 To further check whether the effect of fluorination on DPP based polymers is positive or negative, Li et al.174 synthesized another six polymers (PDPP4T, PFDPP4T, PDPP4T-M, PFDPP4T-M, PDPP4T-2M and PFDPP4T-2M, as shown in Fig. 19) with variation of the flanking aryl units and BT or difluorobithiophene as the co-monomer. The results showed that the fluorinated DPP polymers exhibit a much lower hole mobility than the nonfluorinated analogues caused by the more “face-on” orientation. Therefore, the influence of fluorination on DPP-based polymers still needs to be deeply studied.
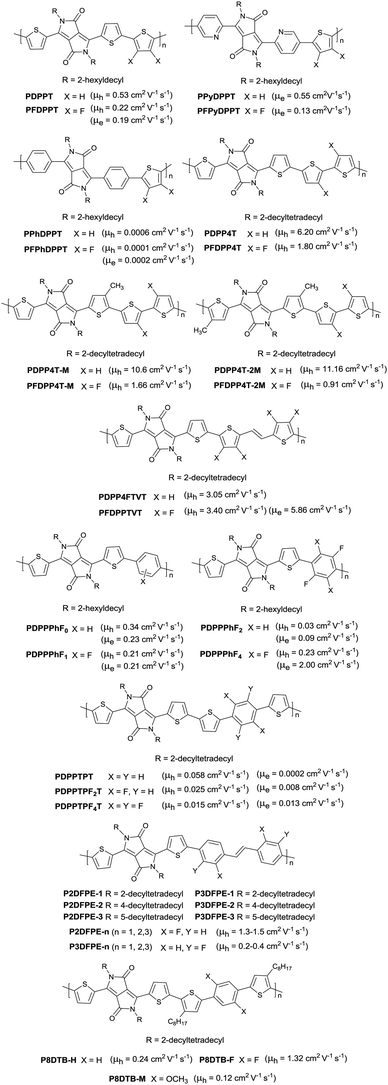 |
| | Fig. 19 Chemical structures of DPP-based fluorinated polymers. | |
Prior to difluorothiophene as the fluorinated building block for polymer semiconductors, a fluorinated phenyl unit was also incorporated into the DPP-based polymer backbone. In 2013, Park et al.171 prepared fluorinated polymers using a fluorinated phenyl unit for n-type semiconductors, for the first time. As shown in Fig. 19 (PDPPPhF0–PDPPPhF4), as the number of fluorine substitutions on the phenylene varies from zero to four, the LUMO level of the corresponding polymers reduced from −3.56 eV to −3.64 eV and the charge transport behaviour slightly changed from p-channel to n-channel. Then PDPPPhF4 showed a high n-type performance with an electron mobility of up to 2.00 cm2 V−1 s−1, resulting from the effective charge hopping induced by the unique structural and electrochemical properties. Such a phenomenon was also observed in polymers PDPPTPT, PDPPTPF2T and PDPPTPF4T by Mueller et al.175 Zhang et al.10 made a comparison between two series of DPP based polymers (P2DFEP-n and P3DFEP-n as in Fig. 19) with two different kinds of difluorodiphenylethene. The results showed that the diverse fluorination substitution positions on diphenylethene caused a different OFET performance of the polymers because of the distinct intra- and intermolecular interactions and backbone conformations. P2DFPE-n containing the F⋯H–C non-covalent interaction (conformation locks) showed a higher hole mobility than P3DFPE-n containing both F⋯H–C and F⋯S non-covalent interactions (conformation locks). There are also many other non-covalent interactions existing in polymers. Besides, P3DFPE-n exhibited a higher HOMO level than P2DFPE-n, which had a non-negligible impact on hole injection of the devices as well. Kim et al.176 investigated the different effect of the O⋯S and F⋯S interactions on the DPP-based polymers through the introduction of the fluorine atoms or methoxy groups into the backbone. It was found that the fluorinated polymer P8DTB-F (Fig. 19) containing the F⋯S interaction exhibited a predominantly edge-on orientation with high crystallinity and a hole mobility of 1.32 cm2 V−1 s−1 while P8DTB-M (Fig. 19) (contain the O⋯S interaction) showed principally a face-on orientation with low hole mobility which is even lower than the non-functionalized polymer P8DTB-H (Fig. 19) of bimodal orientation.
The above-mentioned fluorination process belongs to the charge donor fluorination. Besides, charge acceptor fluorination has also been reported. Compared to the charge donor fluorination, it may be more difficult to fluorinate the charge acceptors in terms of their complicated chemical structures. Fluorinated IID-based polymers were investigated in 2012. Ting Lei94 observed the ambipolar transport behaviour of the IID-based polymer semiconductors for the first time via fluorinating the IID core to reduce the bandgap and HOMO–LUMO levels. The electron mobility of the IID-based polymers was improved from 0.07 cm2 V−1 s−1 to 0.43 cm2 V−1 s−1, while the hole mobility was maintained at the same level after fluorination (PII2T and PFII2T in Fig. 20). Characterization by grazing incident X-ray diffraction (GIXRD) and tapping-mode atomic force microscopy (AFM) also revealed the different interchain interactions and stronger crystalline tendencies caused by fluorination of the IID core. Additionally, there are also some donor fluorinations reported for IID-based polymers. Jo et al.177 prepared two kinds of IID-based polymers with the fluorinated donor of difluorobenzothiadiazole (PIDT2FBT as in Fig. 20) and tetrafluorobenzene (PIDT4FBZ as in Fig. 20). But both of them did not exhibit high performance. Then, Hu et al.178 made a comparison between fluorination on the donor unit and fluorination on the acceptor unit for the IID-quarterthiophene-based D–A semiconductor polymers (PID4T, PIDFF4T and PFFID4T as in Fig. 20). It was shown that fluorination on the donor unit resulted in stronger temperature-dependent aggregation than fluorination on the acceptor unit. But the OFET performance was not mentioned. Subsequently, Gao et al.179 obtained six donor–acceptor conjugated polymers (PIDTVTF0–PIDTVTF6, PIDTVTF6–C3 and PID2TF6–C3 as in Fig. 20) based on the IID core through direct arylation polycondensation (DAP) and checked the impact of multifluorination on the molecular properties. It was demonstrated that more fluorine atoms were introduced into the backbone, and the IID-based polymers showed deeper HOMO (from −4.88 eV to −5.25 eV) and LUMO (from −2.96 eV to −3.36 eV) levels, which resulted in high-performance ambipolar (PIDTVTF6: μh = 3.9 cm2 V−1 s−1, μe = 3.5 cm2 V−1 s−1) and unipolar n-type (PIDTVTF6–C3: μe = 4.97 cm2 V−1 s−1) semiconductors. Isoindigo-based polymers with different backbone conformations were also studied via different fluorination positions. As shown in Fig. 20, FBDPPV-1 and FBDPPV-2 of different backbone conformations were synthesized by Lei et al.180 Both of them showed n-type ambient-stable behaviour owing to the low-lying LUMO levels. In comparison, FBDPPV-2 (EHOMO = −6.22 eV, ELUMO = −4.30 eV) showed slightly lower HOMO and LUMO levels than FBDPPV-1 (EHOMO = −6.19 eV, ELUMO = −4.26 eV). The electron mobility of FBDPPV-1 was higher than that of the nonfluorinated analogue FBDPPV while FBDPPV-2 exhibited a lower electron mobility than FBDPPV. Such a phenomenon could be attributed to the different backbone conformations leading to the different interchain interactions and film microstructures.
 |
| | Fig. 20 Chemical structures of IID- and IID-derivative-based fluorinated polymers. | |
Other fluorinated polymers based on monofluorobenzothiadiazole (FBT) and difluorobenzothiadiazole (2FBT) were reported in numerous studies. As shown in Fig. 21, the FBT-based polymer PTh4F1BT181 and the 2FBT-based polymers PTh4F2BT-1182 and PTh4F2BT-2183 and the nonfluorinated analogue PTh4BT160 with the same donor were explored. The results showed that the non-covalent interaction and the solid-state order were enhanced by fluorine substitution, eventually resulting in an improvement in OFET performance. Such an improvement was also observed in P(BDT-TT-FBT) (Fig. 21) when compared with the nonfluorinated analogue (P(BDT-TT-HBT) as in Fig. 21).184 However, when the co-monomer was cyclopentadithiophene185 (CDT, the corresponding polymers PBT, PDF, PRF and P2F as in Fig. 21), indacenodithiophene186 (IDT, the corresponding polymers PIDTDTBT and PIDTFDTBT as in Fig. 21) or silaindacenodithiophene162 (SiIDT, the corresponding polymers SiIDT-BT, SiIDT-2FBT, SiIDT-DTBT and SiIDT-2FDTBT as in Fig. 21), the 2FBT-based polymer exhibited a lower hole mobility than the nonoflurinated analogues. This opposite phenomenon was due to the solid-state order enhancement and HOMO–LUMO level reduction caused by fluorination. Supposedly, for polymer fluorination, when the effect of the solid-state order enhancement is greater than the effect of the HOMO–LUMO level reduction, the hole mobility will be increased. In contrast, the hole transport is restricted as the reduction effect of the HOMO–LUMO levels is greater than the solid-state order enhancement effect. Additionally, a decrease in both hole mobility and electron mobility was also characterized for the fluorination of some polymers. As shown in Fig. 21, the hole mobility and electron mobility of the polymer PBTPDFDTBT122 were 0.16 and 0.36 cm2 V−1 s−1, respectively, which were lower than the nonfluorinated polymer PBTPDDTBT122 with a hole mobility of 0.33 cm2 V−1 s−1 and an electron mobility of 1.02 cm2 V−1 s−1. And the high-performance polymers PNBT (Fig. 21) and PNBS (Fig. 21) based on NDI and BTz aforementioned exhibited a much lower performance after fluorination. In addition, high-performance with a hole mobility of 9.05 cm2 V−1 s−1 (PDFDT, Fig. 21) and high bias-stress stability were approached when P(VDF-TrFE) containing fluorine was used as the dielectric layer of OFETs (TGBC) for 2FBT-based polymers.187 This favourable hole mobility resulted from the manipulation of energy levels via fluorinated ferroelectric dielectrics and the fluorinated semiconductor PDFDT, which could reduce the Fermi level and the gap between the Fermi level and the HOMO.
 |
| | Fig. 21 Chemical structures of fluorinated polymers. | |
3.2 Chlorination
In addition to fluorination, chlorination was also introduced into the polymer semiconductor backbone. Compared with the fluorine atom, the chlorine atom has a larger size which may cause steric hindrance effects severely. Thus, reports on the chlorination of the polymer semiconductor backbone are rarer than those on fluorination. In 2015, Pei et al.188 compared the effects of fluorination and chlorination in IID-based polymers on the phase separation and molecular orientation. The results showed that the polymers containing the fluorinated IID core (FIIDT, Fig. 22) exhibited a higher OFET performance but a lower OPV performance than the polymers containing the chlorinated IID core as less crystallinity and face-on orientation were induced by the large size of chlorine and the large torsional angle of the backbone. Before that, Pei and co-workers first synthesized two polymers (PCII2T and PCII2Se as in Fig. 22) based on chlorinated IID in 2013.189 They found that the phenylthienyl dihedral angles were larger after chlorination, whereas OFETs based on these types of polymers exhibit an ambipolar behaviour with an increased electron mobility and ambient stability. Later on, chlorinated polymers (PTIIT-T and PTII-TVT-8 as in Fig. 22) and small molecules (C20-DBTII and C20-DBFII as in Fig. 22) based on the IID derivative were also reported by Wan et al.190,191 But their electrical properties were not good. Following the chlorination of polymers based on IID or its derivative, chlorinated polymers composed of NDI via the incorporation of dichlorothiophene were studied.192 As shown in Fig. 22 (P(NDI2OD-T2), P(NDI2HD-T2), P(NDI2OD-T2Cl2) and P(NDI2HD-T2Cl2)), chlorination of NDI-thiophene based polymers was detrimental to the OFET performance with a reduction in charge mobility. In summary, since few examples of chlorination of conjugated polymers have been investigated, the detailed effects of the introduction of chlorine still need to be further studied.
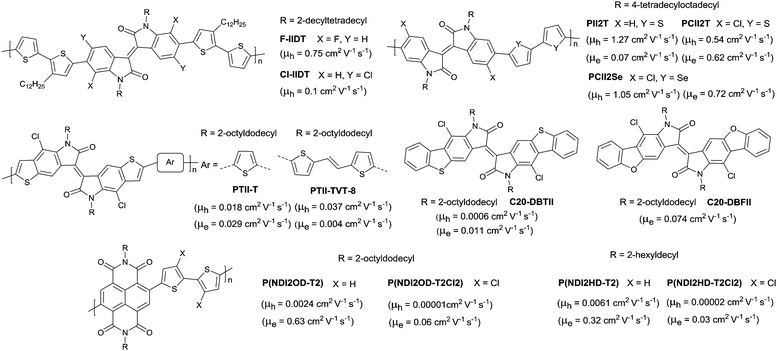 |
| | Fig. 22 Chemical structures of chlorinated polymers and small molecules C20-DBTII and C20-DBFII. | |
4. Side chain engineering
Due to the poor solubility following the rigid backbone of π-conjugated polymers, flexible side chains are always introduced into the polymers. Different side chains consisting of alkyl chains, oligo(ethylene glycol) chains, siloxane-terminated side chains, carbosilane chains, fluoroalkyl chains and so on lead to the different molecular packing distances, crystallinities and interchain interactions, eventually resulting in different OFET performances. Additionally, the form, length, density, distribution and branching position of side chains also affects the property of the polymers.
4.1 Alkyl chains
Alkyl chains are the most widely used side chains to improve the solubility of the conjugated polymers, which have two forms including linear chains and branched chains. In 2006, Park et al.193 investigated the effect of different linear alkyl side chain lengths (–(CH2)nCH3, n = 3, 5, and 7) on the molecular ordering and OFET behaviour of P3AT. According to the Fourier transform infrared (FT-IR) spectra, they found that more ordered side chains were observed following an increase in length, whereas the charge transport behaviour decreased as the alkyl side chain length was increased. Poly(3-butylthiophene) (P3BT) showed 20 times higher hole mobility than poly (3-octylthiophene) (P3OT). However, such a trend was suitable for untreated SiO2 but not for octyltrichlorosilane-treated (OTS) SiO2 transistor dielectric layers. For the latter, the mobility of P3AT with longer side chains could still maintain high values because of the weak interaction between the dielectric surface and side chains and better molecular order, which was confirmed by McCullough et al.194 They demonstrated that the competing processes between backbone packing (π-stacking) and side-chain packing during conjugated polymer self-assembly would result in polymorphic behaviour and disorder. And the mobility of P3ATs with longer side chains was more easily affected by surface treatment than that of P3ATs with shorter side chains.
Compared with linear side chains, branched side chains have attracted more attention from researchers as they can provide better solubility for polymers. Reichmanis et al.195 compared the properties of DPP-based polymers with linear chains and branched chains. As shown in Fig. 23, the polymers PTBTD-2DT and PTBTD-5DH(H) with branched side chains exhibited higher mobility than PTBTD-OD with a linear side chain because of the improved solubility.
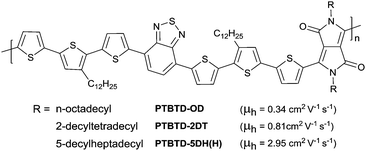 |
| | Fig. 23 Chemical structures of PTBTD-OD, PTBTD-2DT, and PTBTD-5DH(H). | |
In 2013, Kim et al.8 firstly extended the branching point of branched side chains in the DPP-based polymer to achieve an unprecedented record high hole mobility of 12 cm2 V−1 s−1 at that time. Subsequently, they systematically investigated the branched side chain effect on the OFET performance of DPP-SVS polymers (Fig. 24).196 According to the result, the electrical performance of such polymers was sensitive to the branching point of the side chains (or lengths of spacer groups) and its odd–even characteristics. Polymers (25-DPP-SVS, 27-DPP-SVS and 29-DPP-SVS as in Fig. 24) with an even number of carbon atoms in the spacer group showed one order of magnitude higher mobility than the polymers (26-DPP-SVS and 28-DPP-SVS as in Fig. 24) with an odd number of carbon atoms in the spacer group when the number was below six. The highest mobility was observed for the 29-DPP-SVS with a hole mobility up to 13.90 cm2 V−1 s−1, a lamellar distance of 26.4 Å and a π-stacking distance of 3.60 Å, whose carbon number of the spacer group was six. As shown in Fig. 24, similar research studies have been reported for polymers IIDTs,65 BDPPVs,197 PTTD4Ts198 and PQA2Ts.198 Bao et al.70 firstly proved that reduction of the π-stacking distance could increase the charge mobility in conjugated polymers. And the π-stacking distance can be adjusted by the length of the spacer group. For IID and its derivative based polymers IIDTs and BDPPVs, the highest charge transport as well as the low π-stacking distance was observed when the number of carbon atoms in the spacer group was three. For thienothiophenedione- and QA-based polymers PTTD4Ts and PQA2Ts, the π-stacking distance reduced as the spacer group became longer, leading to suppressed steric hindrance. Therefore, there was a clear trend observed that the hole mobility of PQA2Ts increased with a longer group.
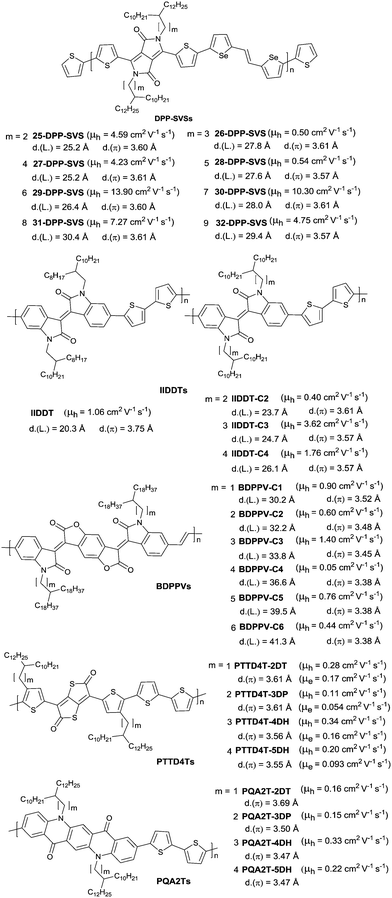 |
| | Fig. 24 Chemical structures of DPP-SVSs, IIDDTs, BDPPVs, PTTD4Ts and PQA2Ts with different branching position alkyl chains. | |
4.2 Oligo(ethylene glycol) chains
Differing from hydrophobic alkyl chains, oligo(ethylene glycol) (OEG) chains are hydrophilic and more flexible due to the ether moieties. They have also been reported for semiconductors as the side chains. In 2015, Wang et al.199 compared the properties of polymers with alkyl side chains (PFDTOBT as in Fig. 25) and OEG side chains (PFDTOBT-O2, PFDTOBT-O3 and PFDTOBT-O4 as in Fig. 25). It was revealed that the π-stacking distance became shorter when OEG chains substituted for alkyl chains. Thus, PFDTOBT-O2, PFDTOBT-O3 and PFDTOBT-O4 showed a higher charge mobility, a narrower bandgap and a red-shifted absorption spectrum in a thin film than PFDTOBT. But the mobility decreased when the length of the OEG side chains was increased, whereas the π-stacking distance did not change. Such mobility reduction resulted from a more inert component following the ORG chain length growth. Meanwhile, the hybrid linear chain consisting of an alkyl chain segment and an OEG chain segment were explored by Cho and co-workers.200 As shown in Fig. 25, PNDI-OR and PNDI-RO were with different adjacent chain structures. The polymer PNDI-RO whose backbone was adjacent to the OEG moiety exhibited a high unipolar n-type property with a mobility up to 1.64 cm2 V−1 s−1. They also demonstrated that the order of the microstructure was dependent on the adjacent side chain, but not the rigid conjugated backbone. Before that, Patil et al.201 adopted another manner of hybrid chains. Two adjacent DPP units, among which one unit was attached to the alkyl side chains and the other unit was attached to the OEG side chains, were used for the synthesis of the conjugated polymer N-CS2DPP-OD-TEG (Fig. 25). Such a polymer showed a wider adsorption behavior (up to ∼1100 nm) and an n-channel semiconductor property with a mobility up to 3 cm2 V−1 s−1. The introduction of OEG side chains not only afforded good solubility of the polymer but also induced the chain to be crystallized spontaneously.
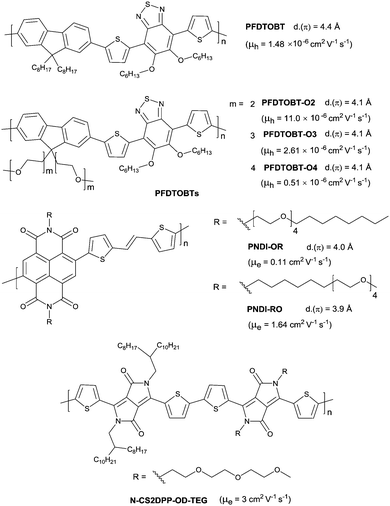 |
| | Fig. 25 Chemical structures of PFDTOBTs, PNDI-OR, PNDI-RO and N-CS2DPP-OD-TEG. | |
4.3 Siloxane-terminated side chains
Siloxane-terminated side chain, as a novel hybrid solubilizing chain, was introduced into a conjugated polymer by Bao et al.70 for the first time. The synthesis of a brominated IID monomer with siloxane-terminated side chains is shown in Fig. 26(A). Siloxane-terminated side chains are more flexible chains because of the longer length of the Si–O bond (LSi–O = 1.64 Å) and the larger angle of the Si–O–Si bond (ASi–O–Si = 143°) compared with the C–C bond (LC–C = 1.53 Å) and the usual tetrahedral angle (AC–C–C ≈ 110°).70,202 As a result, the IID-based polymer PII2T-Si (Fig. 26(B)) with siloxane-terminated side chains was provided with better solubility, a lower π-stacking distance and a larger crystalline coherence length, eventually leading to a higher charge mobility than that of the polymer PII2T (Fig. 26(B)) with branched alkyl side chains. After that, Yang et al.53 introduced siloxane-terminated side chains via N-alkyl. As shown in Fig. 26(B), the polymer PTDPPSe-Si with siloxane-terminated side chains was also accompanied by a closer π-stacking distance of 3.6 Å relative to the polymer PTDPPSe with the alkyl chain. The solution-sheared thin film of PTDPPSe-Si also exhibited a higher ambipolar behaviour with a hole mobility of 3.97 cm2 V−1 s−1 and an electron mobility of 2.20 cm2 V−1 s−1 in comparison with PTDPPSe. Yang et al.203 also synthesized a polymer with siloxane-terminated side chains based on DPP and TT, and a remarkably high stability and hole mobility of over 4.5 cm2 V−1 s−1 were approached. Besides, the introduction of siloxane-terminated side chains into the BPD-based polymer resulted in an improved OFET performance.167
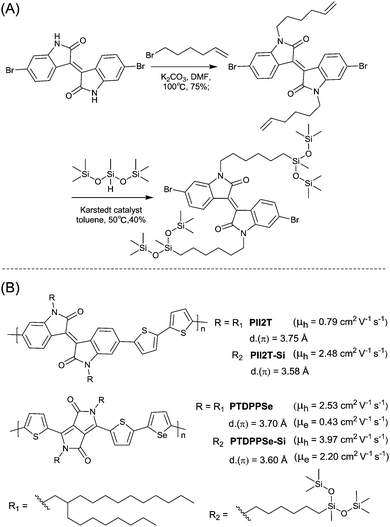 |
| | Fig. 26 (A) Synthesis of the brominated IID monomer with siloxane-terminated side chains. (B) Chemical structures of PII2T, PII2T-Si, PTDPPSe and PTDPPSe-Si. | |
The length of the alkyl spacer between the bulky siloxane group and the conjugated backbone was studied as well for DPP-based polymers (PTDPPSe-Sis as in Fig. 27) and IID-based polymers (PII2F-Sis as in Fig. 27). Yang et al.52 suggested that PTDPPSe-SiC5, whose carbon number of the alkyl spacer was five, exhibited the best ambipolar performance with a hole mobility of up to 8.84 cm2 V−1 s−1 and an electron mobility of up to 4.34 cm2 V−1 s−1 through a solution-shearing method.
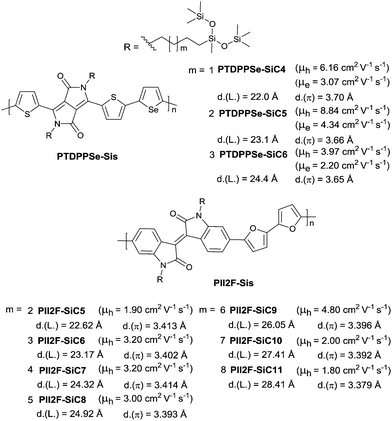 |
| | Fig. 27 Chemical structures of PTDPPSe-Sis and PII2F-Sis. | |
Bao et al.72 demonstrated that the polymer PII2F-SiC9, whose carbon number of the alkyl spacer was nine, showed the highest value for the bulky siloxane group for good solubility (better than PTDPPSe-SiC4) as well as dense microstructures (denser than PTDPPSe-SiC6). Additionally, for IID-based polymers, the thin-film microstructure, crystallinity and domain size but not the length of the spacers played a more important role for thin film charge transport characteristics when the carbon number of the alkyl spacer was three and over three.
4.4 Carbosilane side chains
Compared with siloxane-terminated side chains, carbosilane chains, as chains also containing the silicon element, have been rarely reported in conjugated polymers for OFETs. In 2016, Chen et al.204 synthesized high mobility, stretchable, and mechanically stable polymer semiconductors via introduction of carbosilane side chains into a IID-based polymer (Fig. 28). When the length of the carbosilane chains increased, the lamellar spacing of the polymer increased and the π-stacking distance decreased. Thus, the polymer SiC-PII2T-C8 achieved a high hole mobility of up to 8.06 cm2 V−1 s−1. In addition, it still had a mobility over 1 cm2 V−1 s−1 under 60% strain and could be simultaneously operated over 400 stretching/releasing cycles. Generally, these novel carbosilane side chains may be a potential choice for high-performance semiconductors in future studies.
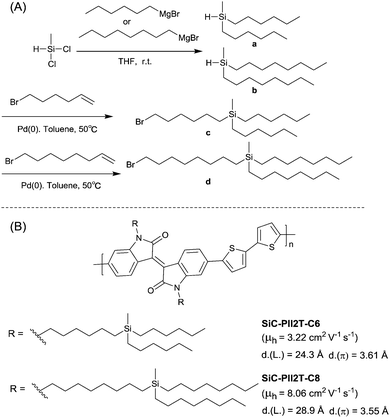 |
| | Fig. 28 (A) Synthesis of carbosilane chains. (B) Chemical structures of SiC-PII2T-C6 and SiC-PII2T-C8. | |
4.5 Fluoroalkyl chains
As aforementioned, the fluorine element was positioned adjacent to the conjugated backbone directly with the aim of adjusting the conformation of the backbone and the energy levels. Since fluoroalkyl chains possess many special properties including rigidity, thermal stability, hydrophobicity, chemical and oxidative resistance and self-assembly ability, such chains were also studied as soluble side chains especially for n-type organic semiconductors.205 In 2013, Kim et al.206 utilized semifluorinated alkyl chains for the substitution of hydrocarbon alkyl chains of PQT12 (Fig. 29), which resulted in the self-assembly of the polymer SFA-PQT (Fig. 29) via the fluorophobic effect accompanied by the improvement of mobility without a thermal annealing process. Cho and co-workers205 introduced a semifluorinated alkyl chain into NDI-based polymers. The polymers PNDIF-2T (Fig. 29) and PNDIF-TVT (Fig. 29) with the semifluorinated alkyl side chain exhibited a well-ordered microstructure better than the analogues with hydrocarbon alkyl side chains as semifluorinated side chains induced rigid backbone organization of the conjugated polymer due to their strong self-organization. The results obtained from 1-chloronaphthalene(CN)-processed polymers PNDIF-2T and PNDIF-TVT exhibited a high electron mobility of up to 6.50 cm2 V−1 s−1 and 5.64 cm2 V−1 s−1, respectively, with a high on–off current ratio of 105. Later on, the DPP unit with semifluorinated side chains was also investigated. As shown in Fig. 29, the polymer PFDPP-BT with semifluorinated side chains showed 3 times higher hole mobility than PDPP-BT with hydrocarbon alkyl chains. AFM and X-ray diffraction (XRD) characterization also confirmed the positive function of semifluorinated alky chains for the thin film morphology and crystallinity. As there are still few reports focused on semifluorinated alkyl chains or even on per-fluorinated side chains,207 which have been mentioned in OPVs, the effect of fluoroalkyl side chains on polymer semiconductors for OFETs stills needs to be further explored.
 |
| | Fig. 29 Chemical structures of PQT12, SFA-PQT, PNDIF-2T, PNDIF-TVT, PDPP-BT and PFDPP-BT. | |
5. Random copolymers
Generally, Stille polycondensation,208 Suzuki polycondensation209 and DAP210 are common methods used for the polymerization process of conjugated polymers (Scheme 11). In earlier studies, investigators concentrated on regular conjugated polymers with the aim to release the high charge transporting property. For regular conjugated copolymers, the HOMO and LUMO levels are often controlled by selecting different pairs of charge donors and acceptor monomers, which may be limited by the complexity and difficulty of designing monomers with different structures. Therefore, random copolymerization, as a facile method, has been utilized in the synthesis of polymer semiconductors to regulate the HOMO–LUMO energy levels, crystallinity, solubility, molecular packing, functionality, etc.
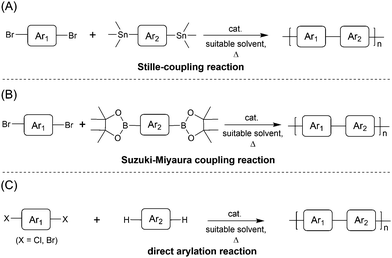 |
| | Scheme 11 Paths for polymerization of conjugated polymers including (A) Stille coupling reaction, (B) Suzuki–Miyaura coupling reaction, and (C) direct arylation reaction. | |
In 2011, Jenekhe et al.40 synthesized the regular copolymers HD-PPTV (Fig. 30) and HD-PPPV (Fig. 30) and the random copolymer PPTPV (Fig. 30). The results showed that HD-PPTV exhibited an ambipolar behaviour and HD-PPPV exhibited a unipolar p-type behaviour. Besides, the random copolymer PPTPV exhibited ambipolar behaviour between HD-PPTV and HD-PPPV when it was composed of 50% HD-PPTV and 50% HD-PPPV. In addition, the optical absorption spectra and XRD patterns of PPTPV also showed a photophysical property and crystallinity behaviour between HD-PPTV and HD-PPPV. Later on, Kim et al.211 systematically explored the effect of the ratio between two monomers on the property of the random copolymer PDPP-Th-Se (Fig. 31). It was suggested that the crystallinity of PDPP-Th-Se could be systematically manipulated by adjusting the ratio between two different electron donors – selenophene and thiophene. The melting and crystallization temperatures as well as the crystallinity of PPTPV were improved following the enhancement of the Se content, which resulted in the enhancement of the OFET performance. Based on such an adjustment effect of random copolymers, their solubility could also be finely tuned. Kwon and co-workers achieved good solubility of the random copolymer PDPP-BTT-SVS (Fig. 31) in a non-chlorinated solvent and high OFET performance (μh = 6.51 cm2 V−1 s−1) when 10% bithienothiophene (BTT) and 90% SVS were present.212 After that, a series of random copolymers (PDPP-BT-SVS,213 PDPP-2T-TVT,214 PDPP-TVT-SVS215 and PDPP-CNTVT-SVS216 as in Fig. 31) based on the thiophene-flanked DPP as the charge acceptor and two different charge donors were also reported. Among them, Kim and co-workers realized the modulation of PDPP-CNTVT-SVS from p-type (μh = 6.23 ± 0.4 cm2 V−1 s−1, CNTVT/SVS = 1:9) to n-type (μe = 6.88 ± 1.01 cm2 V−1 s−1, CNTVT/SVS = 9:1) as well as a balanced ambipolar (μh = 3.15 ± 0.2 cm2 V−1 s−1, μe = 3.03 ± 0.15 cm2 V−1 s−1, CNTVT/SVS = 1:1) semiconductor via precisely regulating the copolymerization ratio of 2,3-di(thiophen-2-yl)acrylonitrile (CNTVT)/SVS.216
 |
| | Fig. 30 Chemical structures of regular copolymers HD-PPTV and HD-PPPV and the random copolymer PPTPV. | |
 |
| | Fig. 31 Chemical structures of PDPP-Th-Se, PDPP-BTT-SVS, PDPP-BT-SVS, PDPP-2T-TVT, PDPP-TVT-SVS and PDPP-CNTVT-SVS. | |
Some special polymer semiconductors based on random copolymers have also been reported. Bao et al.217 incorporated the non-conjugated segment – 2,6-pyridine dicarboxamide (PDCA) – into a DPP and TVT-based polymer via random copolymerization (PDPP-TVT-TPDCAT, Fig. 32(A)). The introduction of PDCA provided enhanced mechanical properties via the formation of intra- and intermolecular hydrogen-bonding or variation of the film morphology (Fig. 32(B)). This kind of polymer exhibited intrinsically stretchable and healable properties. When the OFETs based on PDPP-TVT-TPDCAT were strained up to 100%, their charge mobility decreased linearly and slowly. And the mobility value could return to the initial one when the strain was released. Besides, the tolerance measurement of common movements (Fig. 32(C)) via mounting OFETs on human limbs revealed that the devices could maintain an average hole mobility over 0.1 cm2 V−1 s−1 as well.
 |
| | Fig. 32 (A) Chemical structure of a stretchable and healable semiconducting polymer based on DPP (renamed as PDPP-TVT-TPDCAT here); (B) description of variation of the polymer structure during stretching and releasing; (C) photographs of folded, twisted and stretched OFETs on the human skin. ((B and C) were reproduced with permission from Macmillan Publishers Limited, part of Springer Nature. Copyright 2016.) | |
Zhang et al.218 also rationally utilized the random copolymerization strategy to control the hydrogen bonds. Different from Bao's work, Zhang et al. introduced the functional groups via the side chains instead of the main backbone. As is shown in Fig. 33, DPP-quaterthiophene-based units containing alkyl chains with urea groups were randomly copolymerized with those containing branching alkyl chains without urea groups. IR and 1H NMR characterizations verified the existence of inter-chain hydrogen bonds. And the polymer PDPP4T-U (Fig. 33) exhibited the highest hole mobility of up to 13.1 cm2 V−1 s−1 after thermal annealing at just 100 °C when (1 − x)/x = 1:10, which resulted from the ordered lamellar packing of alkyl chains and inter-chain π–π stacking.
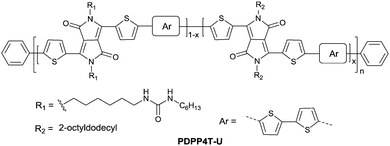 |
| | Fig. 33 Chemical structure of a random copolymer containing a urea group (renamed as PDPP4T-U here). | |
In addition to random copolymerization occurring between two DPP-based monomers, other monomers were also reported for random copolymers. As shown in Fig. 34, Ajayaghosh et al.219 utilized the random compolymerization method to explore how the fused chalcogenophene affected the molecular packing and charge carrier transport by varying the percentage composition of the fused chalcogenophene in the polymers TDPBTBT and BTDPBTBT. The results showed that the charge carrier mobility could be enhanced via increasing the percentage composition of fused chalcogenophene. Chen and co-workers prepared a novel charge donor di(thiophen-2-yl)thieno[3,2-b]thiophene (DTTT) and randomly copolymerized it with the DPP-based unit.220 It was indicated that the random copolymer PDTTT-T-DPP (Fig. 34) exhibited a higher hole mobility than the regular analogues formed by the direct polymerization between DTTT and DPP without thiophene as the linkage. Besides, as shown in Fig. 34, other random copolymers PDPP-BTD,221 PBPD-T-DPP222 and PDPP-T-ID223 containing commonly studied electron deficient units (BT, IID, and BPD) were also reported. Among them, the properties of PDPP-BTD and PBPD-T-DPP could be finely tuned to be suitable for either OPV devices or OFETs via adjusting the random copolymerization ratio ((1 − x)/x) of monomers. While the polymer PDPP-T-ID was only tested for OPV devices.
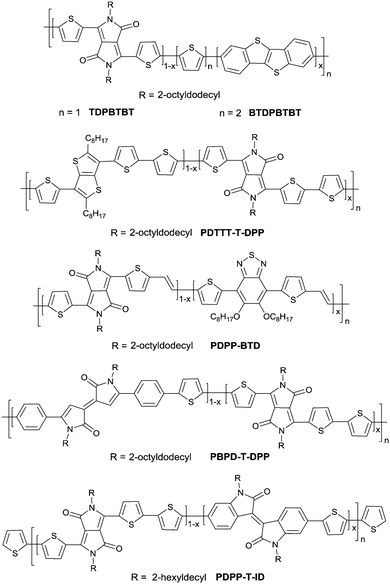 |
| | Fig. 34 Chemical structures of random copolymers TDPBTBT, BTDPBTBT, PDTTT-T-DPP, PDPP-BTD, PBPD-T-DPP and PDPP-T-ID. | |
In 2016, Fang and co-workers combined random copolymerization with side chain engineering for the synthesis of the IID-based polymer PIIT-BOCx (Fig. 35).224 The long polyisobutylene side-chains provided solubility for the polymer while t-Boc groups, as a thermal cleavable group, afforded a latent hydrogen bond for cross-linking when thermally treated. The crosslinked polymer PIIT-BOCx exhibited a favourable solvent-resistant property which was beneficial for application in solution-processable multiple-layer electronic devices. In addition to the traditional synthesis methods used for polycondensation of the IID-based polymer, Kiriy et al.225 adopted a novel method via zinc-activating the monomers. As shown in Fig. 35, the low-bandgap random copolymer PTIIT-TNDIT was prepared by the copolymerization of zinc-activated IID- and NDI-based monomers. Such a polymerization approach proceeds quickly at room temperature, which is an advantage compared to traditional polymerization methods. However, the OFET performance of the polymer PTIIT-TNDIT was not discussed in this article, and such a synthesis method still needs to be deeply studied for more semiconducting polymers. Other random copolymers PDTBDTFBT-DTBDII (Fig. 35) and PTTTTFBT-TTTTII (Fig. 35) based on IID and 2FBT were also reported but with poor OFET performance.226
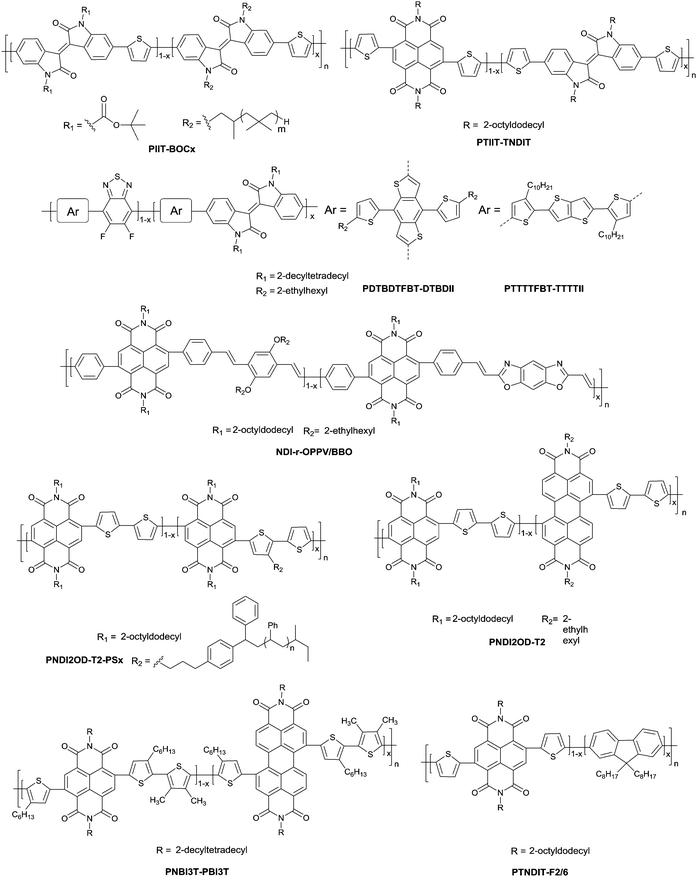 |
| | Fig. 35 Chemical structures of random copolymers PIIT-BOCx, PTIlT-TNDIT, PDTBDTFBT-DTBDII (renamed here), PTTTTFBT-TTTTII (renamed here), NDI-r-OPPV/BBO, PNDI2OD-T2-PSx, PNDI2OD-T2, PNBI3T-PBI3T and the gradient copolymer PTNDIT-F2/6. | |
Furthermore, as shown in Fig. 35, imide-based random copolymers were reported as well. For NDI-r-OPPV/BBO227 (Fig. 35), oligo(p-phenylenevinylene) (OPPV) with side chains served to improve the solubility of the NDI-benzobisoxazole (BBO) based segment. NDI-r-OPPV/BBO was synthesized by the Horner–Wadsworth–Emmons polymerization method. OFETs of NDI-r-OPPV/BBO showed dominant n-type properties different from the balanced ambipolar performance of the regular polymer based on NDI and OPPV. In 2016, Bao et al.228 introduced polystyrene (PS) oligomer side chains into the NDI-based polymer and investigated their effect by means of tuning the monomer ratio in the random copolymer (PNDI2OD-T2-PSx, Fig. 35). The results showed that the ambient stability of the NDI-based polymer was obviously improved while keeping the favourable electron transporting performance when 20 mol% of PS side chains were present in the polymer. They assumed that PS side chains worked as the molecular encapsulation layer around the conjugated polymer backbone to relieve the electron traps. Additionally, PNDI2OD-T2229 (Fig. 35) and PNBI3T–PBI3T230 (Fig. 35), as random copolymers consisting of NDI and PDI, were also reported. But their OFET performance was not mentioned.
Gradient or even block-like copolymer PTNDIT-F2/6 (Fig. 35), distinguished from random copolymers, was also synthesized by the zinc-activating approach aforementioned.231 Such a gradient or even block-like behaviour resulted from the faster polymerization of the fluorine segment than the NDI based segment, and was confirmed by NMR, gel permeation chromatography (GPC), AFM, and fluorescence quenching experiments. But the OFET properties of such a novel polymer were not measured in the literature.
6. Conclusions and outlook
This review summarized the versatile chemical strategies, consisting of building block selection, backbone halogenation, side chain engineering and random copolymerization, for high-performance semiconductor polymers in OFETs. Suitable building block selection for charge donor and acceptor energy level matching as well as facile charge carrier injection can increase the semiconductor performance of the polymer. Halogenation including fluorination and chlorination can subtly modify the energy levels, molecular packing order and coplanarity of polymers via electron-withdrawing properties and non-covalent bonds. Compared with chlorination, fluorination is more effective due to the smaller size of the fluorine atom. Side chain engineering is a favourable method to tune the solubility and condensed state structure of the rigid conjugated polymer backbone for increasing molecular weight and coplanarity, which will eventually result in the enhancement of OFET performance. What's more, random copolymerization is a facile and efficient method that can creatively adjust the energy levels, molecular orientations, solubilities, interchain interactions, charge transport properties, and so on.
To some extent, findings on new building blocks are limited, and they still need to be deeply studied. In addition, some new building blocks with high OFET performance as mentioned above, such as BAI, should be paid more attention in future research works. On the other hand, halogenation, side chain engineering and random copolymerization as remarkable tools for favorable and functional (e.g. stretchable and healable) polymer semiconductors may be the research focus in future works. Additionally, the device architecture is also essential for high-performance OFETs, which is not discussed in detail here. In summary, high-performance polymer based OFETs are expected when the chemical approaches mentioned above are combined with rational device architectures.
Acknowledgements
This work was financially supported by the National Key R&D Program of “Strategic Advanced Electronic Materials” (No. 2016YFB0401100), the National Natural Science Foundation of China (51233006 and 21633012), and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB12030100).
Notes and references
- A. Tsumura, H. Koezuka and T. Ando, Appl. Phys. Lett., 1986, 49, 1210 CrossRef CAS.
- X. Guo, R. P. Ortiz, Y. Zheng, M. G. Kim, S. Zhang, Y. Hu, G. Lu, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2011, 133, 13685–13697 CrossRef CAS PubMed.
- D. Chandran and K.-S. Lee, Macromol. Res., 2013, 21, 272–283 CrossRef CAS.
- M. Grzybowski and D. T. Gryko, Adv. Opt. Mater., 2015, 3, 280–320 CrossRef CAS.
- Z. Mao, W. Zhang, J. Huang, K. Shi, D. Gao, Z. Chen and G. Yu, Polym. Chem., 2015, 6, 6457–6464 RSC.
- Z. Yi, X. Sun, Y. Zhao, Y. Guo, X. Chen, J. Qin, G. Yu and Y. Liu, Chem. Mater., 2012, 24, 4350–4356 CrossRef CAS.
- H. Chen, Y. Guo, G. Yu, Y. Zhao, J. Zhang, D. Gao, H. Liu and Y. Liu, Adv. Mater., 2012, 24, 4618–4622 CrossRef CAS PubMed.
- I. Kang, H. J. Yun, D. S. Chung, S. K. Kwon and Y. H. Kim, J. Am. Chem. Soc., 2013, 135, 14896–14899 CrossRef CAS PubMed.
- H. H. Choi, J. Y. Baek, E. Song, B. Kang, K. Cho, S. K. Kwon and Y. H. Kim, Adv. Mater., 2015, 27, 3626–3631 CrossRef CAS PubMed.
- W. Zhang, K. Shi, J. Huang, D. Gao, Z. Mao, D. Li and G. Yu, Macromolecules, 2016, 49, 2582–2591 CrossRef CAS.
- M. Jang, J. H. Kim, D. H. Hwang and H. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 12781–12788 CAS.
- F. Yang, C. Li, J. Zhang, G. Feng, Z. Wei and W. Li, Org. Electron., 2016, 37, 366–370 CrossRef CAS.
- G. E. Park, J. Shin, D. H. Lee, T. W. Lee, H. Shim, M. J. Cho, S. Pyo and D. H. Choi, Macromolecules, 2014, 47, 3747–3754 CrossRef CAS.
- J. R. Matthews, W. Niu, A. Tandia, A. L. Wallace, J. Hu, W.-Y. Lee, G. Giri, S. C. B. Mannsfeld, Y. Xie, S. Cai, H. H. Fong, Z. Bao and M. He, Chem. Mater., 2013, 25, 782–789 CrossRef CAS.
- M. He, W. Li, Y. Gao, H. Tian, J. Zhang, H. Tong, D. Yan, Y. Geng and F. Wang, Macromolecules, 2016, 49, 825–832 CrossRef CAS.
- S. Park, B. T. Lim, B. Kim, H. J. Son and D. S. Chung, Sci. Rep., 2014, 4, 5482 CrossRef CAS PubMed.
- S. Subramaniyan, F. S. Kim, G. Ren, H. Li and S. A. Jenekhe, Macromolecules, 2012, 45, 9029–9037 CrossRef CAS.
- J. S. Ha, K. H. Kim and D. H. Choi, J. Am. Chem. Soc., 2011, 133, 10364–10367 CrossRef CAS PubMed.
- S.-H. Kang, H. R. Lee, G. K. Dutta, J. Lee, J. H. Oh and C. Yang, Macromolecules, 2017, 50, 884–890 CrossRef CAS.
- H. Chen, Y. Guo, Z. Mao, D. Gao and G. Yu, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1970–1977 CrossRef CAS.
- J. Chang, P. Sonar, Z. Lin, C. Zhang, J. Zhang, Y. Hao and J. Wu, Org. Electron., 2016, 36, 113–119 CrossRef CAS.
- T. W. Lee, D. H. Lee, J. Shin, M. J. Cho and D. H. Choi, Polym. Chem., 2015, 6, 1777–1785 RSC.
- Y. Yu, Y. Wu, A. Zhang, C. Li, Z. Tang, W. Ma, Y. Wu and W. Li, ACS Appl. Mater. Interfaces, 2016, 8, 30328–30335 CAS.
- Y. Ji, C. Xiao, Q. Wang, J. Zhang, C. Li, Y. Wu, Z. Wei, X. Zhan, W. Hu, Z. Wang, R. A. Janssen and W. Li, Adv. Mater., 2016, 28, 943–950 CrossRef CAS PubMed.
- Y. Li, S. P. Singh and P. Sonar, Adv. Mater., 2010, 22, 4862–4866 CrossRef CAS PubMed.
- W. Li, W. S. Roelofs, M. Turbiez, M. M. Wienk and R. A. Janssen, Adv. Mater., 2014, 26, 3304–3309 CrossRef CAS PubMed.
- B. Carsten, J. M. Szarko, L. Lu, H. J. Son, F. He, Y. Y. Botros, L. X. Chen and L. Yu, Macromolecules, 2012, 45, 6390–6395 CrossRef CAS.
- X. Liu, J. Huang, J. Xu, D. Gao, W. Zhang, K. Shi and G. Yu, RSC Adv., 2016, 6, 35394–35401 RSC.
- J. Shin, G. E. Park, D. H. Lee, H. A. Um, T. W. Lee, M. J. Cho and D. H. Choi, ACS Appl. Mater. Interfaces, 2015, 7, 3280–3288 CAS.
- H. Bronstein, Z. Chen, R. S. Ashraf, W. Zhang, J. Du, J. R. Durrant, P. S. Tuladhar, K. Song, S. E. Watkins, Y. Geerts, M. M. Wienk, R. A. Janssen, T. Anthopoulos, H. Sirringhaus, M. Heeney and I. McCulloch, J. Am. Chem. Soc., 2011, 133, 3272–3275 CrossRef CAS PubMed.
- P. Sonar, J. Chang, Z. Shi, E. Gann, J. Li, J. Wu and C. R. McNeill, J. Mater. Chem. C, 2015, 3, 9299–9305 RSC.
- J. Yuan, X. Huang, F. Zhang, J. Lu, Z. Zhai, C. Di, Z. Jiang and W. Ma, J. Mater. Chem., 2012, 22, 22734 RSC.
- P. Sonar, T. R. Foong, S. P. Singh, Y. Li and A. Dodabalapur, Chem. Commun., 2012, 48, 8383–8385 RSC.
- P. Sonar, J. Chang, Z. Shi, J. Wu and J. Li, J. Mater. Chem. C, 2015, 3, 2080–2085 RSC.
- P. Sonar, J. Chang, J. H. Kim, K. H. Ong, E. Gann, S. Manzhos, J. Wu and C. R. McNeill, ACS Appl. Mater. Interfaces, 2016, 8, 24325–24330 CAS.
- J. Dhar, T. Mukhopadhay, N. Yaacobi-Gross, T. D. Anthopoulos, U. Salzner, S. Swaraj and S. Patil, J. Phys. Chem. B, 2015, 119, 11307–11316 CrossRef CAS PubMed.
- M. Shahid, R. S. Ashraf, Z. Huang, A. J. Kronemeijer, T. McCarthy-Ward, I. McCulloch, J. R. Durrant, H. Sirringhaus and M. Heeney, J. Mater. Chem., 2012, 22, 12817 RSC.
- M. Shahid, T. McCarthy-Ward, J. Labram, S. Rossbauer, E. B. Domingo, S. E. Watkins, N. Stingelin, T. D. Anthopoulos and M. Heeney, Chem. Sci., 2012, 3, 181–185 RSC.
- C. J. Mueller, C. R. Singh, M. Fried, S. Huettner and M. Thelakkat, Adv. Funct. Mater., 2015, 25, 2725–2736 CrossRef CAS.
- P.-T. Wu, F. S. Kim and S. A. Jenekhe, Chem. Mater., 2011, 23, 4618–4624 CrossRef CAS.
- B. Sun, W. Hong, Z. Yan, H. Aziz and Y. Li, Adv. Mater., 2014, 26, 2636–2642 CrossRef CAS PubMed.
- X. Zhang, C. Xiao, A. Zhang, F. Yang, H. Dong, Z. Wang, X. Zhan, W. Li and W. Hu, Polym. Chem., 2015, 6, 4775–4783 RSC.
- P. Li, L. Xu, H. Shen, X. Duan, J. Zhang, Z. Wei, Z. Yi, C.-a. Di and S. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 8620–8626 CAS.
- J. Huang, X. Liu, D. Gao, C. Wei, W. Zhang and G. Yu, RSC Adv., 2016, 6, 83448–83455 RSC.
- S. M. Lee, H. R. Lee, A.-R. Han, J. Lee, J. H. Oh and C. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 15652–15661 CAS.
- H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dotz, M. Kastler and A. Facchetti, Nature, 2009, 457, 679–686 CrossRef CAS PubMed.
- Y. Xiong, X. Qiao and H. Li, Polym. Chem., 2015, 6, 6579–6584 RSC.
- B. Fu, C.-Y. Wang, B. D. Rose, Y. Jiang, M. Chang, P.-H. Chu, Z. Yuan, C. Fuentes-Hernandez, B. Kippelen, J.-L. Brédas, D. M. Collard and E. Reichmanis, Chem. Mater., 2015, 27, 2928–2937 CrossRef CAS.
- H.-J. Yun, H. H. Choi, S.-K. Kwon, Y.-H. Kim and K. Cho, Chem. Mater., 2014, 26, 3928–3937 CrossRef CAS.
- D. Yoo, B. Nketia-Yawson, S.-J. Kang, H. Ahn, T. J. Shin, Y.-Y. Noh and C. Yang, Adv. Funct. Mater., 2015, 25, 586–596 CrossRef CAS.
- J. S. Lee, S. K. Son, S. Song, H. Kim, D. R. Lee, K. Kim, M. J. Ko, D. H. Choi, B. Kim and J. H. Cho, Chem. Mater., 2012, 24, 1316–1323 CrossRef CAS.
- J. Lee, A. R. Han, H. Yu, T. J. Shin, C. Yang and J. H. Oh, J. Am. Chem. Soc., 2013, 135, 9540–9547 CrossRef CAS PubMed.
- J. Lee, A. R. Han, J. Kim, Y. Kim, J. H. Oh and C. Yang, J. Am. Chem. Soc., 2012, 134, 20713–20721 CrossRef CAS PubMed.
- P. Sonar, S. P. Singh, Y. Li, M. S. Soh and A. Dodabalapur, Adv. Mater., 2010, 22, 5409–5413 CrossRef CAS PubMed.
- J. Lee, M. Jang, S. M. Lee, D. Yoo, T. J. Shin, J. H. Oh and C. Yang, ACS Appl. Mater. Interfaces, 2014, 6, 20390–20399 CAS.
- G. Zhang, J. Guo, J. Zhang, P. Li, J. Ma, X. Wang, H. Lu and L. Qiu, Polym. Chem., 2015, 6, 418–425 RSC.
- W. Hong, B. Sun, H. Aziz, W. T. Park, Y. Y. Noh and Y. Li, Chem. Commun., 2012, 48, 8413–8415 RSC.
- H. J. Yun, S. J. Kang, Y. Xu, S. O. Kim, Y. H. Kim, Y. Y. Noh and S. K. Kwon, Adv. Mater., 2014, 26, 7300–7307 CrossRef CAS PubMed.
- Y. Gao, X. Zhang, H. Tian, J. Zhang, D. Yan, Y. Geng and F. Wang, Adv. Mater., 2015, 27, 6753–6759 CrossRef CAS PubMed.
- J. Kim, A. R. Han, J. Hong, G. Kim, J. Lee, T. J. Shin, J. H. Oh and C. Yang, Chem. Mater., 2014, 26, 4933–4942 CrossRef CAS.
- Z. Chen, M. J. Lee, R. Shahid Ashraf, Y. Gu, S. Albert-Seifried, M. Meedom Nielsen, B. Schroeder, T. D. Anthopoulos, M. Heeney, I. McCulloch and H. Sirringhaus, Adv. Mater., 2012, 24, 647–652 CrossRef CAS PubMed.
- J. D. Yuen, J. Fan, J. Seifter, B. Lim, R. Hufschmid, A. J. Heeger and F. Wudl, J. Am. Chem. Soc., 2011, 133, 20799–20807 CrossRef CAS PubMed.
- C. Li, Z. Mao, H. Chen, L. Zheng, J. Huang, B. Zhao, S. Tan and G. Yu, Macromolecules, 2015, 48, 2444–2453 CrossRef CAS.
- T. Lei, Y. Cao, Y. Fan, C. J. Liu, S. C. Yuan and J. Pei, J. Am. Chem. Soc., 2011, 133, 6099–6101 CrossRef CAS PubMed.
- T. Lei, J. H. Dou and J. Pei, Adv. Mater., 2012, 24, 6457–6461 CrossRef CAS PubMed.
- K. H. Park, K. H. Cheon, Y. J. Lee, D. S. Chung, S. K. Kwon and Y. H. Kim, Chem. Commun., 2015, 51, 8120–8122 RSC.
- R. S. Ashraf, A. J. Kronemeijer, D. I. James, H. Sirringhaus and I. McCulloch, Chem. Commun., 2012, 48, 3939–3941 RSC.
- G. Kim, S. J. Kang, G. K. Dutta, Y. K. Han, T. J. Shin, Y. Y. Noh and C. Yang, J. Am. Chem. Soc., 2014, 136, 9477–9483 CrossRef CAS PubMed.
- I. Meager, M. Nikolka, B. C. Schroeder, C. B. Nielsen, M. Planells, H. Bronstein, J. W. Rumer, D. I. James, R. S. Ashraf, A. Sadhanala, P. Hayoz, J. C. Flores, H. Sirringhaus and I. McCulloch, Adv. Funct. Mater., 2014, 24, 7109–7115 CAS.
- J. Mei, D. H. Kim, A. L. Ayzner, M. F. Toney and Z. Bao, J. Am. Chem. Soc., 2011, 133, 20130–20133 CrossRef CAS PubMed.
- Y. Zhou, T. Kurosawa, W. Ma, Y. Guo, L. Fang, K. Vandewal, Y. Diao, C. Wang, Q. Yan, J. Reinspach, J. Mei, A. L. Appleton, G. I. Koleilat, Y. Gao, S. C. Mannsfeld, A. Salleo, H. Ade, D. Zhao and Z. Bao, Adv. Mater., 2014, 26, 3767–3772 CrossRef CAS PubMed.
- J. Mei, H.-C. Wu, Y. Diao, A. Appleton, H. Wang, Y. Zhou, W.-Y. Lee, T. Kurosawa, W.-C. Chen and Z. Bao, Adv. Funct. Mater., 2015, 25, 3455–3462 CrossRef CAS.
- J. Shin, H. A. Um, D. H. Lee, T. W. Lee, M. J. Cho and D. H. Choi, Polym. Chem., 2013, 4, 5688 RSC.
- D. I. James, S. Wang, W. Ma, S. Hedström, X. Meng, P. Persson, S. Fabiano, X. Crispin, M. R. Andersson, M. Berggren and E. Wang, Adv. Electron. Mater., 2016, 2, 1500313 CrossRef.
- J. Huang, Z. Mao, Z. Chen, D. Gao, C. Wei, W. Zhang and G. Yu, Chem. Mater., 2016, 28, 2209–2218 CrossRef CAS.
- Y. Cao, J. S. Yuan, X. Zhou, X. Y. Wang, F. D. Zhuang, J. Y. Wang and J. Pei, Chem. Commun., 2015, 51, 10514–10516 RSC.
- T. Hasegawa, M. Ashizawa and H. Matsumoto, RSC Adv., 2015, 5, 61035–61043 RSC.
- W. Yue, R. S. Ashraf, C. B. Nielsen, E. Collado-Fregoso, M. R. Niazi, S. A. Yousaf, M. Kirkus, H. Y. Chen, A. Amassian, J. R. Durrant and I. McCulloch, Adv. Mater., 2015, 27, 4702–4707 CrossRef CAS PubMed.
- M. S. Chen, J. R. Niskala, D. A. Unruh, C. K. Chu, O. P. Lee and J. M. J. Fréchet, Chem. Mater., 2013, 25, 4088–4096 CrossRef CAS.
- Y. Lu, Y. Liu, Y. Z. Dai, C. Y. Yang, H. I. Un, S. W. Liu, K. Shi, J. Y. Wang and J. Pei, Chem. – Asian J., 2017, 12, 302–307 CrossRef CAS PubMed.
- S. Chen, B. Sun, C. Guo, W. Hong, Y. Meng and Y. Li, Chem. Commun., 2014, 50, 6509–6512 RSC.
- W. Hong, C. Guo, B. Sun and Y. Li, J. Mater. Chem. C, 2015, 3, 4464–4470 RSC.
- T. Lei, J. H. Dou, X. Y. Cao, J. Y. Wang and J. Pei, J. Am. Chem. Soc., 2013, 135, 12168–12171 CrossRef CAS PubMed.
- X. Wang, H. H. Choi, G. Zhang, Y. Ding, H. Lu, K. Cho and L. Qiu, J. Mater. Chem. C, 2016, 4, 6391–6400 RSC.
- G. Zhang, Z. Ye, P. Li, J. Guo, Q. Wang, L. Tang, H. Lu and L. Qiu, Polym. Chem., 2015, 6, 3970–3978 RSC.
- G. Zhang, J. Chen, Y. Dai, S. Song, Z. Ye, H. Lu, L. Qiu and K. Cho, Dyes Pigm., 2017, 137, 221–228 CrossRef CAS.
- Y.-Z. Dai, N. Ai, Y. Lu, Y.-Q. Zheng, J.-H. Dou, K. Shi, T. Lei, J.-Y. Wang and J. Pei, Chem. Sci., 2016, 7, 5753–5757 RSC.
- G. Kim, A. R. Han, H. R. Lee, J. Lee, J. H. Oh and C. Yang, Chem. Commun., 2014, 50, 2180–2183 RSC.
- T. Lei, J. H. Dou, X. Y. Cao, J. Y. Wang and J. Pei, Adv. Mater., 2013, 25, 6589–6593 CrossRef CAS PubMed.
- G. Zhang, J. Guo, M. Zhu, P. Li, H. Lu, K. Cho and L. Qiu, Polym. Chem., 2015, 6, 2531–2540 RSC.
- Y. Deng, J. Quinn, B. Sun, Y. He, J. Ellard and Y. Li, RSC Adv., 2016, 6, 34849–34854 RSC.
- Y. Deng, B. Sun, Y. He, J. Quinn, C. Guo and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 3459–3462 CrossRef CAS PubMed.
- R. Stalder, S. R. Puniredd, M. R. Hansen, U. Koldemir, C. Grand, W. Zajaczkowski, K. Müllen, W. Pisula and J. R. Reynolds, Chem. Mater., 2016, 28, 1286–1297 CrossRef CAS.
- T. Lei, J.-H. Dou, Z.-J. Ma, C.-H. Yao, C.-J. Liu, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2012, 134, 20025–20028 CrossRef CAS PubMed.
- P. Li, H. Wang, L. Ma, L. Xu, F. Xiao, Z. Yi, Y. Liu and S. Wang, Sci. China: Chem., 2016, 59, 679–683 CrossRef CAS.
- H. J. Yun, H. H. Choi, S. K. Kwon, Y. H. Kim and K. Cho, ACS Appl. Mater. Interfaces, 2015, 7, 5898–5906 CAS.
- G. Zhang, P. Li, L. Tang, J. Ma, X. Wang, H. Lu, B. Kang, K. Cho and L. Qiu, Chem. Commun., 2014, 50, 3180–3183 RSC.
- X. Zhou, N. Ai, Z.-H. Guo, F.-D. Zhuang, Y.-S. Jiang, J.-Y. Wang and J. Pei, Chem. Mater., 2015, 27, 1815–1820 CrossRef CAS.
- Y. Deng, B. Sun, Y. He, J. Quinn, C. Guo and Y. Li, Chem. Commun., 2015, 51, 13515–13518 RSC.
- Y. He, C. Guo, B. Sun, J. Quinn and Y. Li, Chem. Commun., 2015, 51, 8093–8096 RSC.
- Y. Jiang, Y. Gao, H. Tian, J. Ding, D. Yan, Y. Geng and F. Wang, Macromolecules, 2016, 49, 2135–2144 CrossRef CAS.
- Y. He, J. Quinn, Y. Deng and Y. Li, Org. Electron., 2016, 35, 41–46 CrossRef CAS.
- S. Vasimalla, S. P. Senanayak, M. Sharma, K. S. Narayan and P. K. Iyer, Chem. Mater., 2014, 26, 4030–4037 CrossRef CAS.
- Z. Chen, Y. Zheng, H. Yan and A. Facchetti, J. Am. Chem. Soc., 2008, 131, 8–9 CrossRef PubMed.
- Z. Zhao, Z. Yin, H. Chen, L. Zheng, C. Zhu, L. Zhang, S. Tan, H. Wang, Y. Guo, Q. Tang and Y. Liu, Adv. Mater., 2017, 29, 1602410 CrossRef PubMed.
- W. Zhou, Y. Wen, L. Ma, Y. Liu and X. Zhan, Macromolecules, 2012, 45, 4115–4121 CrossRef CAS.
- H. Chen, Y. Guo, Z. Mao, G. Yu, J. Huang, Y. Zhao and Y. Liu, Chem. Mater., 2013, 25, 3589–3596 CrossRef CAS.
- S. Park, J. Cho, M. J. Ko, D. S. Chung and H. J. Son, Macromolecules, 2015, 48, 3883–3889 CrossRef CAS.
- M. Nakano, I. Osaka and K. Takimiya, Macromolecules, 2015, 48, 576–584 CrossRef CAS.
- Y. Fukutomi, M. Nakano, J. Y. Hu, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2013, 135, 11445–11448 CrossRef CAS PubMed.
- K. Nakano, M. Nakano, B. Xiao, E. Zhou, K. Suzuki, I. Osaka, K. Takimiya and K. Tajima, Macromolecules, 2016, 49, 1752–1760 CrossRef CAS.
- S. Subramaniyan, T. Earmme, N. M. Murari and S. A. Jenekhe, Polym. Chem., 2014, 5, 5707 RSC.
- X. Zhao, L. Ma, L. Zhang, Y. Wen, J. Chen, Z. Shuai, Y. Liu and X. Zhan, Macromolecules, 2013, 46, 2152–2158 CrossRef CAS.
- H. Usta, C. Newman, Z. Chen and A. Facchetti, Adv. Mater., 2012, 24, 3678–3684 CrossRef CAS PubMed.
- C. Zhang, G. Zhao, W. Zeng, K. Tian, H. Dong, W. Hu, J. Qin and C. Yang, Org. Electron., 2015, 16, 101–108 CrossRef CAS.
- X. Guo, N. Zhou, S. J. Lou, J. W. Hennek, R. Ponce Ortiz, M. R. Butler, P. L. Boudreault, J. Strzalka, P. O. Morin, M. Leclerc, J. T. Lopez Navarrete, M. A. Ratner, L. X. Chen, R. P. Chang, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2012, 134, 18427–18439 CrossRef CAS PubMed.
- T. Y. Chu, J. Lu, S. Beaupre, Y. Zhang, J. R. Pouliot, S. Wakim, J. Zhou, M. Leclerc, Z. Li, J. Ding and Y. Tao, J. Am. Chem. Soc., 2011, 133, 4250–4253 CrossRef CAS PubMed.
- D. Chen, Y. Zhao, C. Zhong, S. Gao, G. Yu, Y. Liu and J. Qin, J. Mater. Chem., 2012, 22, 14639 RSC.
- T.-Y. Chu, J. Lu, S. Beaupré, Y. Zhang, J.-R. Pouliot, J. Zhou, A. Najari, M. Leclerc and Y. Tao, Adv. Funct. Mater., 2012, 22, 2345–2351 CrossRef CAS.
- M.-C. Yuan, M.-Y. Chiu, S.-P. Liu, C.-M. Chen and K.-H. Wei, Macromolecules, 2010, 43, 6936–6938 CrossRef CAS.
- Q. Wu, M. Wang, X. Qiao, Y. Xiong, Y. Huang, X. Gao and H. Li, Macromolecules, 2013, 46, 3887–3894 CrossRef CAS.
- X. Qiao, Q. Wu, H. Wu, J. Zhang and H. Li, Adv. Funct. Mater., 2017, 27, 1604286 CrossRef.
- D. H. Lee, K. H. Kim, J. Shin, M. J. Cho and D. H. Choi, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1228–1235 CrossRef CAS.
- J. A. Letizia, M. R. Salata, C. M. Tribout, A. Facchetti, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2008, 130, 9679–9694 CrossRef CAS PubMed.
- H. Li, S. Sun, T. Salim, S. Bomma, A. C. Grimsdale and Y. M. Lam, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 250–260 CrossRef CAS.
- X. Guo, F. S. Kim, S. A. Jenekhe and M. D. Watson, J. Am. Chem. Soc., 2009, 131, 7206–7207 CrossRef CAS PubMed.
- X. Zhou, Y. Cao, X.-Y. Wang, Z.-H. Guo, J.-Y. Wang and J. Pei, Asian J. Org. Chem., 2014, 3, 209–215 CrossRef CAS.
- Z. Zhao, Z. Wang, C. Ge, X. Zhang, X. Yang and X. Gao, Polym. Chem., 2016, 7, 573–579 RSC.
- H. Li, F. S. Kim, G. Ren, E. C. Hollenbeck, S. Subramaniyan and S. A. Jenekhe, Angew. Chem., Int. Ed., 2013, 52, 5513–5517 CrossRef CAS PubMed.
- H. Li, F. S. Kim, G. Ren and S. A. Jenekhe, J. Am. Chem. Soc., 2013, 135, 14920–14923 CrossRef CAS PubMed.
- W. Zhang, Y. Han, X. Zhu, Z. Fei, Y. Feng, N. D. Treat, H. Faber, N. Stingelin, I. McCulloch, T. D. Anthopoulos and M. Heeney, Adv. Mater., 2016, 28, 3922–3927 CrossRef CAS PubMed.
- K. Kawashima, I. Osaka and K. Takimiya, Chem. Mater., 2015, 27, 6558–6570 CrossRef CAS.
- S.-W. Cheng, D.-Y. Chiou, C.-E. Tsai, W.-W. Liang, Y.-Y. Lai, J.-Y. Hsu, C.-S. Hsu, I. Osaka, K. Takimiya and Y.-J. Cheng, Adv. Funct. Mater., 2015, 25, 6131–6143 CrossRef CAS.
- C. Luo, A. K. Kyaw, L. A. Perez, S. Patel, M. Wang, B. Grimm, G. C. Bazan, E. J. Kramer and A. J. Heeger, Nano Lett., 2014, 14, 2764–2771 CrossRef CAS PubMed.
- B. He, A. B. Pun, D. Zherebetskyy, Y. Liu, F. Liu, L. M. Klivansky, A. M. McGough, B. A. Zhang, K. Lo, T. P. Russell, L. Wang and Y. Liu, J. Am. Chem. Soc., 2014, 136, 15093–15101 CrossRef CAS PubMed.
- M. A. Kolaczkowski, B. He and Y. Liu, Org. Lett., 2016, 18, 5224–5227 CrossRef CAS PubMed.
- Z. Cai, H. Luo, P. Qi, J. Wang, G. Zhang, Z. Liu and D. Zhang, Macromolecules, 2014, 47, 2899–2906 CrossRef CAS.
- J. Wang, X. Chen, G. Zhang, Z. Liu and D. Zhang, J. Mater. Chem. C, 2014, 2, 1149–1157 RSC.
- A. D. Thilanga Liyanage, B. Milián-Medina, B. Zhang, J. Gierschner and M. D. Watson, Macromol. Chem. Phys., 2016, 217, 2068–2073 CrossRef CAS.
- X.-Y. Wang, M.-W. Zhang, F.-D. Zhuang, J.-Y. Wang and J. Pei, Polym. Chem., 2016, 7, 2264–2271 RSC.
- W. S. Yoon, D. W. Kim, J.-M. Park, I. Cho, O. K. Kwon, D. R. Whang, J. H. Kim, J.-H. Park and S. Y. Park, Macromolecules, 2016, 49, 8489–8497 CrossRef CAS.
- J. Fan, J. D. Yuen, M. Wang, J. Seifter, J. H. Seo, A. R. Mohebbi, D. Zakhidov, A. Heeger and F. Wudl, Adv. Mater., 2012, 24, 2186–2190 CrossRef CAS PubMed.
- J. D. Yuen, R. Kumar, D. Zakhidov, J. Seifter, B. Lim, A. J. Heeger and F. Wudl, Adv. Mater., 2011, 23, 3780–3785 CAS.
- Y. Wang, H. Masunaga, T. Hikima, H. Matsumoto, T. Mori and T. Michinobu, Macromolecules, 2015, 48, 4012–4023 CrossRef CAS.
- C. An, S. R. Puniredd, X. Guo, T. Stelzig, Y. Zhao, W. Pisula and M. Baumgarten, Macromolecules, 2014, 47, 979–986 CrossRef CAS.
- T. Dallos, D. Beckmann, G. Brunklaus and M. Baumgarten, J. Am. Chem. Soc., 2011, 133, 13898–13901 CrossRef CAS PubMed.
- T. T. Steckler, P. Henriksson, S. Mollinger, A. Lundin, A. Salleo and M. R. Andersson, J. Am. Chem. Soc., 2014, 136, 1190–1193 CrossRef CAS PubMed.
- C. An, M. Li, T. Marszalek, D. Li, R. Berger, W. Pisula and M. Baumgarten, Chem. Mater., 2014, 26, 5923–5929 CrossRef CAS.
- W. Cui, J. Yuen and F. Wudl, Macromolecules, 2011, 44, 7869–7873 CrossRef CAS.
- J. W. Rumer, M. Levick, S. Y. Dai, S. Rossbauer, Z. Huang, L. Biniek, T. D. Anthopoulos, J. R. Durrant, D. J. Procter and I. McCulloch, Chem. Commun., 2013, 49, 4465–4467 RSC.
- J. W. Rumer, S.-Y. Dai, M. Levick, L. Biniek, D. J. Procter and I. McCulloch, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1285–1291 CrossRef CAS.
- J. W. Rumer, S.-Y. Dai, M. Levick, Y. Kim, M.-B. Madec, R. S. Ashraf, Z. Huang, S. Rossbauer, B. Schroeder, L. Biniek, S. E. Watkins, T. D. Anthopoulos, R. A. J. Janssen, J. R. Durrant, D. J. Procter and I. McCulloch, J. Mater. Chem. C, 2013, 1, 2711 RSC.
- Y. Cao, Z.-H. Guo, Z.-Y. Chen, J.-S. Yuan, J.-H. Dou, Y.-Q. Zheng, J.-Y. Wang and J. Pei, Polym. Chem., 2014, 5, 5369 RSC.
- J. Quinn, C. Guo, B. Sun, A. Chan, Y. He, E. Jin and Y. Li, J. Mater. Chem. C, 2015, 3, 11937–11944 RSC.
- I. Osaka, M. Akita, T. Koganezawa and K. Takimiya, Chem. Mater., 2012, 24, 1235–1243 CrossRef CAS.
- H. Li, X. Wang, F. Liu and H. Fu, Polym. Chem., 2015, 6, 3283–3289 RSC.
- H. Li, C. Gu, L. Jiang, L. Wei, W. Hu and H. Fu, J. Mater. Chem. C, 2013, 1, 2021 RSC.
- C. Y. Yang, K. Shi, T. Lei, J. Wang, X. Y. Wang, F. D. Zhuang, J. Y. Wang and J. Pei, ACS Appl. Mater. Interfaces, 2016, 8, 3714–3718 CAS.
- M. Zhang, H. N. Tsao, W. Pisula, C. Yang, A. K. Mishra and K. Müllen, J. Am. Chem. Soc., 2007, 129, 3472–3473 CrossRef CAS PubMed.
- K. H. Ong, S. L. Lim, H. S. Tan, H. K. Wong, J. Li, Z. Ma, L. C. Moh, S. H. Lim, J. C. de Mello and Z. K. Chen, Adv. Mater., 2011, 23, 1409–1413 CrossRef CAS PubMed.
- W. Lee, G.-H. Kim, S.-J. Ko, S. Yum, S. Hwang, S. Cho, Y.-H. Shin, J. Y. Kim and H. Y. Woo, Macromolecules, 2014, 47, 1604–1612 CrossRef CAS.
- B. C. Schroeder, Z. Huang, R. S. Ashraf, J. Smith, P. D'Angelo, S. E. Watkins, T. D. Anthopoulos, J. R. Durrant and I. McCulloch, Adv. Funct. Mater., 2012, 22, 1663–1670 CrossRef CAS.
- P.-Y. Chao, H.-C. Wu, C. Lu, C.-W. Hong and W.-C. Chen, Macromolecules, 2015, 48, 5596–5604 CrossRef CAS.
- S. Yum, T. K. An, X. Wang, W. Lee, M. A. Uddin, Y. J. Kim, T. L. Nguyen, S. Xu, S. Hwang, C. E. Park and H. Y. Woo, Chem. Mater., 2014, 26, 2147–2154 CrossRef CAS.
- K. J. Fallon, N. Wijeyasinghe, N. Yaacobi-Gross, R. S. Ashraf, D. M. E. Freeman, R. G. Palgrave, M. Al-Hashimi, T. J. Marks, I. McCulloch, T. D. Anthopoulos and H. Bronstein, Macromolecules, 2015, 48, 5148–5154 CrossRef CAS.
- K. J. Fallon, N. Wijeyasinghe, E. F. Manley, S. D. Dimitrov, S. A. Yousaf, R. S. Ashraf, W. Duffy, A. A. Y. Guilbert, D. M. E. Freeman, M. Al-Hashimi, J. Nelson, J. R. Durrant, L. X. Chen, I. McCulloch, T. J. Marks, T. M. Clarke, T. D. Anthopoulos and H. Bronstein, Chem. Mater., 2016, 28, 8366–8378 CrossRef CAS.
- S.-F. Yang, Z.-T. Liu, Z.-X. Cai, H.-W. Luo, P.-L. Qi, G.-X. Zhang and D.-Q. Zhang, Macromolecules, 2016, 49, 5857–5865 CrossRef CAS.
- Z. Cai, Z. Liu, H. Luo, P. Qi, G. Zhang and D. Zhang, Chin. J. Chem., 2014, 32, 788–796 CrossRef CAS.
- J. W. Rumer, S. Rossbauer, M. Planells, S. E. Watkins, T. D. Anthopoulos and I. McCulloch, J. Mater. Chem. C, 2014, 2, 8822–8828 RSC.
- K. C. Lee, W. T. Park, Y. Y. Noh and C. Yang, ACS Appl. Mater. Interfaces, 2014, 6, 4872–4882 CAS.
- J. H. Park, E. H. Jung, J. W. Jung and W. H. Jo, Adv. Mater., 2013, 25, 2583–2588 CrossRef CAS PubMed.
- Z. Fei, P. Boufflet, S. Wood, J. Wade, J. Moriarty, E. Gann, E. L. Ratcliff, C. R. McNeill, H. Sirringhaus, J. S. Kim and M. Heeney, J. Am. Chem. Soc., 2015, 137, 6866–6879 CrossRef CAS PubMed.
- P. Boufflet, Y. Han, Z. Fei, N. D. Treat, R. Li, D.-M. Smilgies, N. Stingelin, T. D. Anthopoulos and M. Heeney, Adv. Funct. Mater., 2015, 25, 7038–7048 CrossRef CAS.
- A. Zhang, C. Xiao, Y. Wu, C. Li, Y. Ji, L. Li, W. Hu, Z. Wang, W. Ma and W. Li, Macromolecules, 2016, 49, 6431–6438 CrossRef CAS.
- C. J. Mueller, E. Gann, C. R. McNeill and M. Thelakkat, J. Mater. Chem. C, 2015, 3, 8916–8925 RSC.
- H. G. Kim, B. Kang, H. Ko, J. Lee, J. Shin and K. Cho, Chem. Mater., 2015, 27, 829–838 CrossRef CAS.
- J. W. Jo, J. H. Kim and J. W. Jung, Dyes Pigm., 2016, 133, 333–338 CrossRef CAS.
- H. Hu, K. Jiang, J.-H. Kim, G. Yang, Z. Li, T. Ma, G. Lu, Y. Qu, H. Ade and H. Yan, J. Mater. Chem. A, 2016, 4, 5039–5043 CAS.
- Y. Gao, Y. Deng, H. Tian, J. Zhang, D. Yan, Y. Geng and F. Wang, Adv. Mater., 2017, 1606217, DOI:10.1002/adma.201606217.
- T. Lei, X. Xia, J. Y. Wang, C. J. Liu and J. Pei, J. Am. Chem. Soc., 2014, 136, 2135–2141 CrossRef CAS PubMed.
- M. Hao, X. Li, K. Shi, D. Xie, X. Zeng, J. Fang, G. Yu and C. Yang, Polym. Chem., 2015, 6, 6050–6057 RSC.
- J. F. Jheng, Y. Y. Lai, J. S. Wu, Y. H. Chao, C. L. Wang and C. S. Hsu, Adv. Mater., 2013, 25, 2445–2451 CrossRef CAS PubMed.
- Y. Zhu, Z. Chen, Y. Yang, P. Cai, J. Chen, Y. Li, W. Yang, J. Peng and Y. Cao, Org. Electron., 2015, 23, 193–198 CrossRef CAS.
- X. Wang, Z.-G. Zhang, H. Luo, S. Chen, S. Yu, H. Wang, X. Li, G. Yu and Y. Li, Polym. Chem., 2014, 5, 502–511 RSC.
- M. Wang, M. Ford, H. Phan, J. Coughlin, T. Q. Nguyen and G. C. Bazan, Chem. Commun., 2016, 52, 3207–3210 RSC.
- H. Bronstein, J. M. Frost, A. Hadipour, Y. Kim, C. B. Nielsen, R. S. Ashraf, B. P. Rand, S. Watkins and I. McCulloch, Chem. Mater., 2013, 25, 277–285 CrossRef CAS.
- B. Nketia-Yawson, H. S. Lee, D. Seo, Y. Yoon, W. T. Park, K. Kwak, H. J. Son, B. Kim and Y. Y. Noh, Adv. Mater., 2015, 27, 3045–3052 CrossRef CAS PubMed.
- Y.-Q. Zheng, Z. Wang, J.-H. Dou, S.-D. Zhang, X.-Y. Luo, Z.-F. Yao, J.-Y. Wang and J. Pei, Macromolecules, 2015, 48, 5570–5577 CrossRef CAS.
- T. Lei, J.-H. Dou, Z.-J. Ma, C.-J. Liu, J.-Y. Wang and J. Pei, Chem. Sci., 2013, 4, 2447 RSC.
- N. Zhao, N. Ai, M. Cai, X. Wang, J. Pei and X. Wan, Polym. Chem., 2016, 7, 235–243 RSC.
- S. Xu, N. Ai, J. Zheng, N. Zhao, Z. Lan, L. Wen, X. Wang, J. Pei and X. Wan, RSC Adv., 2015, 5, 8340–8344 RSC.
- G.-S. Ryu, Z. Chen, H. Usta, Y.-Y. Noh and A. Facchetti, MRS Commun., 2016, 6, 47–60 CrossRef CAS.
- Y. D. Park, D. H. Kim, Y. Jang, J. H. Cho, M. Hwang, H. S. Lee, J. A. Lim and K. Cho, Org. Electron., 2006, 7, 514–520 CrossRef CAS.
- G. Sauvé, A. E. Javier, R. Zhang, J. Liu, S. A. Sydlik, T. Kowalewski and R. D. McCullough, J. Mater. Chem., 2010, 20, 3195 RSC.
- B. Fu, J. Baltazar, A. R. Sankar, P.-H. Chu, S. Zhang, D. M. Collard and E. Reichmanis, Adv. Funct. Mater., 2014, 24, 3734–3744 CrossRef CAS.
- J. Y. Back, H. Yu, I. Song, I. Kang, H. Ahn, T. J. Shin, S.-K. Kwon, J. H. Oh and Y.-H. Kim, Chem. Mater., 2015, 27, 1732–1739 CrossRef CAS.
- J.-H. Dou, Y.-Q. Zheng, T. Lei, S.-D. Zhang, Z. Wang, W.-B. Zhang, J.-Y. Wang and J. Pei, Adv. Funct. Mater., 2014, 24, 6270–6278 CrossRef CAS.
- G. E. Park, J. Shin, D. H. Lee, M. J. Cho and D. H. Choi, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1226–1234 CrossRef CAS.
- B. Meng, H. Song, X. Chen, Z. Xie, J. Liu and L. Wang, Macromolecules, 2015, 48, 4357–4363 CrossRef CAS.
- R. Kim, B. Kang, D. H. Sin, H. H. Choi, S. K. Kwon, Y. H. Kim and K. Cho, Chem. Commun., 2015, 51, 1524–1527 RSC.
- C. Kanimozhi, N. Yaacobi-Gross, K. W. Chou, A. Amassian, T. D. Anthopoulos and S. Patil, J. Am. Chem. Soc., 2012, 134, 16532–16535 CrossRef CAS PubMed.
- J. E. Mark, Acc. Chem. Res., 2004, 37, 946–953 CrossRef CAS PubMed.
- A. R. Han, J. Lee, H. R. Lee, J. Lee, S.-H. Kang, H. Ahn, T. J. Shin, J. H. Oh and C. Yang, Macromolecules, 2016, 49, 3739–3748 CrossRef CAS.
- H.-C. Wu, C.-C. Hung, C.-W. Hong, H.-S. Sun, J.-T. Wang, G. Yamashita, T. Higashihara and W.-C. Chen, Macromolecules, 2016, 49, 8540–8548 CrossRef CAS.
- B. Kang, R. Kim, S. B. Lee, S. K. Kwon, Y. H. Kim and K. Cho, J. Am. Chem. Soc., 2016, 138, 3679–3686 CrossRef CAS PubMed.
- H. G. Jeong, B. Lim, D. Khim, M. Han, J. Lee, J. Kim, J. M. Yun, K. Cho, J. W. Park and D. Y. Kim, Adv. Mater., 2013, 25, 6416–6422 CrossRef CAS PubMed.
- P. Homyak, Y. Liu, S. Ferdous, F. Liu, T. P. Russell and E. B. Coughlin, Chem. Mater., 2015, 27, 443–449 CrossRef CAS.
- B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Chem. Rev., 2011, 111, 1493–1528 CrossRef CAS PubMed.
- J. Sakamoto, M. Rehahn, G. Wegner and A. D. Schluter, Macromol. Rapid Commun., 2009, 30, 653–687 CrossRef CAS PubMed.
- T. Bura, J. T. Blaskovits and M. Leclerc, J. Am. Chem. Soc., 2016, 138, 10056–10071 CrossRef CAS PubMed.
- K.-H. Kim, S. Park, H. Yu, H. Kang, I. Song, J. H. Oh and B. J. Kim, Chem. Mater., 2014, 26, 6963–6970 CrossRef CAS.
- H. J. Yun, G. B. Lee, D. S. Chung, Y. H. Kim and S. K. Kwon, Adv. Mater., 2014, 26, 6612–6616 CrossRef CAS PubMed.
- H.-J. Yun, J. Cho, D. S. Chung, Y.-H. Kim and S.-K. Kwon, Macromolecules, 2014, 47, 7030–7035 CrossRef CAS.
- E. Y. Ko, G. E. Park, D. H. Lee, H. A. Um, J. Shin, M. J. Cho and D. H. Choi, ACS Appl. Mater. Interfaces, 2015, 7, 28303–28310 CAS.
- J. Cho, S. J. Park, S. M. Lee, J. U. Ha, E. S. Ahn, S. T. Chang, S. K. Kwon, D. S. Chung and Y. H. Kim, Macromol. Rapid Commun., 2016, 37, 2057–2063 CrossRef CAS PubMed.
- D. Khim, Y. R. Cheon, Y. Xu, W.-T. Park, S.-K. Kwon, Y.-Y. Noh and Y.-H. Kim, Chem. Mater., 2016, 28, 2287–2294 CrossRef CAS.
- W.-T. Park, G. Kim, C. Yang, C. Liu and Y.-Y. Noh, Adv. Funct. Mater., 2016, 26, 4695–4703 CrossRef CAS.
- J. Yao, C. Yu, Z. Liu, H. Luo, Y. Yang, G. Zhang and D. Zhang, J. Am. Chem. Soc., 2016, 138, 173–185 CrossRef CAS PubMed.
- V. S. Nair, J. Sun, P. Qi, S. Yang, Z. Liu, D. Zhang and A. Ajayaghosh, Macromolecules, 2016, 49, 6334–6342 CrossRef CAS.
- L. Chen, F. Wu, Z. Deng, L. Feng, P. Gu, H. Dong, W. Hu and Y. Chen, Polym. Chem., 2015, 6, 7684–7692 RSC.
- H. Li, F. Liu, X. Wang, C. Gu, P. Wang and H. Fu, Macromolecules, 2013, 46, 9211–9219 CrossRef CAS.
- P. Qi, Z. Wang, Z. Liu, S. Yang, Y. Yang, J. Yao, G. Zhang and D. Zhang, Polym. Chem., 2016, 7, 3838–3847 RSC.
- J. W. Jung, F. Liu, T. P. Russell and W. H. Jo, Energy Environ. Sci., 2013, 6, 3301 CAS.
- Z.-H. Guo, N. Ai, C. R. McBroom, T. Yuan, Y.-H. Lin, M. Roders, C. Zhu, A. L. Ayzner, J. Pei and L. Fang, Polym. Chem., 2016, 7, 648–655 RSC.
- Y. Karpov, J. Maiti, R. Tkachov, T. Beryozkina, V. Bakulev, W. Liu, H. Komber, U. Lappan, M. Al-Hussein, M. Stamm, B. Voit and A. Kiriy, Polym. Chem., 2016, 7, 2691–2697 RSC.
- P. Deng, Y. Lei, B. Wu, X. Zheng, Y. Lu, F. Zhu and B. S. Ong, Dyes Pigm., 2016, 134, 251–257 CrossRef CAS.
- N. B. Kolhe, A. Z. Ashar, K. S. Narayan and S. K. Asha, Macromolecules, 2014, 47, 2296–2305 CrossRef CAS.
- T. Kurosawa, Y.-C. Chiu, Y. Zhou, X. Gu, W.-C. Chen and Z. Bao, Adv. Funct. Mater., 2016, 26, 1261–1270 CrossRef CAS.
- S. Sharma, N. B. Kolhe, V. Gupta, V. Bharti, A. Sharma, R. Datt, S. Chand and S. K. Asha, Macromolecules, 2016, 49, 8113–8125 CrossRef CAS.
- K. Nakabayashi, M. Yamada and H. Mori, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3151–3158 CrossRef CAS.
- R. Tkachov, H. Komber, S. Rauch, A. Lederer, U. Oertel, L. Häußler, B. Voit and A. Kiriy, Macromolecules, 2014, 47, 4994–5001 CrossRef CAS.
|
| This journal is © the Partner Organisations 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Wenping
Hu
*ac and
Yunqi
Liu
*a,
Wenping
Hu
*ac and
Yunqi
Liu