
Towards hydroxamic acid linked zirconium metal–organic frameworks†
Carla F.
Pereira
abc,
Ashlee J.
Howarth
c,
Nicolaas A.
Vermeulen
c,
Filipe A.
Almeida Paz
b,
João P. C.
Tomé
ad,
Joseph T.
Hupp
*c and
Omar K.
Farha
*ce
aDepartment of Chemistry & QOPNA, University of Aveiro, 3810-193 Aveiro, Portugal
bDepartment of Chemistry & CICECO – Aveiro Institute of Materials, University of Aveiro, 3810-193 Aveiro, Portugal
cDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, Illinois 60208, USA. E-mail: j-hupp@northwestern.edu; o-farha@northwestern.edu
dCQE, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisboa, Portugal
eDepartment of Chemistry, Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabia
First published on 30th January 2017
Abstract
Solvent-assisted linker exchange (SALE) is performed on the Zr-based metal–organic framework (MOF), UiO-66, to exchange benzene-1,4-dicarboxylate with benzene-1,4-dihydroxamate linkers. Characterization of the material before and after SALE is performed using powder X-ray diffraction (PXRD), scanning electron microscopy (SEM), nitrogen adsorption, diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) and thermogravimetric analysis (TGA). The stability of the parent and daughter material is evaluated at pH 1 and 12 and the structural integrity evaluated by PXRD, mass balance and porosity measurements.
Introduction
Metal–organic frameworks (MOFs) constitute an interesting class of materials that continue to develop and expand both in structure and function.1 In the most elementary sense, these materials are prepared by the coordination of multitopic organic linkers to metal ion or cluster nodes. MOFs can be designed to exhibit specific characteristics including strong coordination bonds, well defined geometries, crystallinity and porosity.1a,2 MOFs have already demonstrated their value as promising materials for a variety of applications including, but not limited to, catalysis,3 photoluminescence,4 gas storage and separation,5 conductivity6 and biomedicine.7The design and preparation of new MOFs constitutes one of the major challenges in the field and it can be tackled in one of two main ways: (1) by designing building blocks (metal nodes, organic linkers and/or SBUs) to drive the formation of specific topologies8 and/or (2) by applying postsynthetic modification (PSM) to known MOFs to obtain frameworks which may be difficult or impossible to obtain using de novo synthetic methods.9 In regards to the latter approach, solvent-assisted linker exchange (SALE) is a PSM technique which can give access to MOFs that are not accessible or challenging to obtain de novo. SALE involves the replacement of structural linkers in a parent MOF with a new linker of choice, offering the possibility to obtain a daughter MOF with the same topology but different functionality.9a,c,f–h This process occurs via a single-crystal-to-single-crystal transformation, but solvent still plays a key role in SALE and needs to be carefully selected in order to ensure complete dissolution and facile diffusion of the new linker to be incorporated in the MOF.9a,c,f–i
Herein, we report the application of SALE to incorporate benzene-1,4-dihydroxamic acid (H2BDHA) linkers into UiO-66 (Fig. 1). If a significant portion of the ditopic carboxylic acid linkers could be replaced with benzene-1,4-dihydroxamic acid, the resulting framework should have increased stability compared to the parent framework, UiO-66, since hydroxamic acids form stronger bonds with Zr(IV) compared to carboxylic acids.10 Hydroxamic acids have features of both hydroxylamines and carboxylic acids and they are made by reacting hydroxylamines or O/N-protected hydroxylamines with an activated acyl group.10a,11 They are very well known as siderophores10a,e and for their therapeutic potential.10a,c,11b,12 In the context of surface functionalization, hydroxamic acids provide some advantages over carboxylic and phosphonic acids in terms of stability10d,13 and bond strength.13c
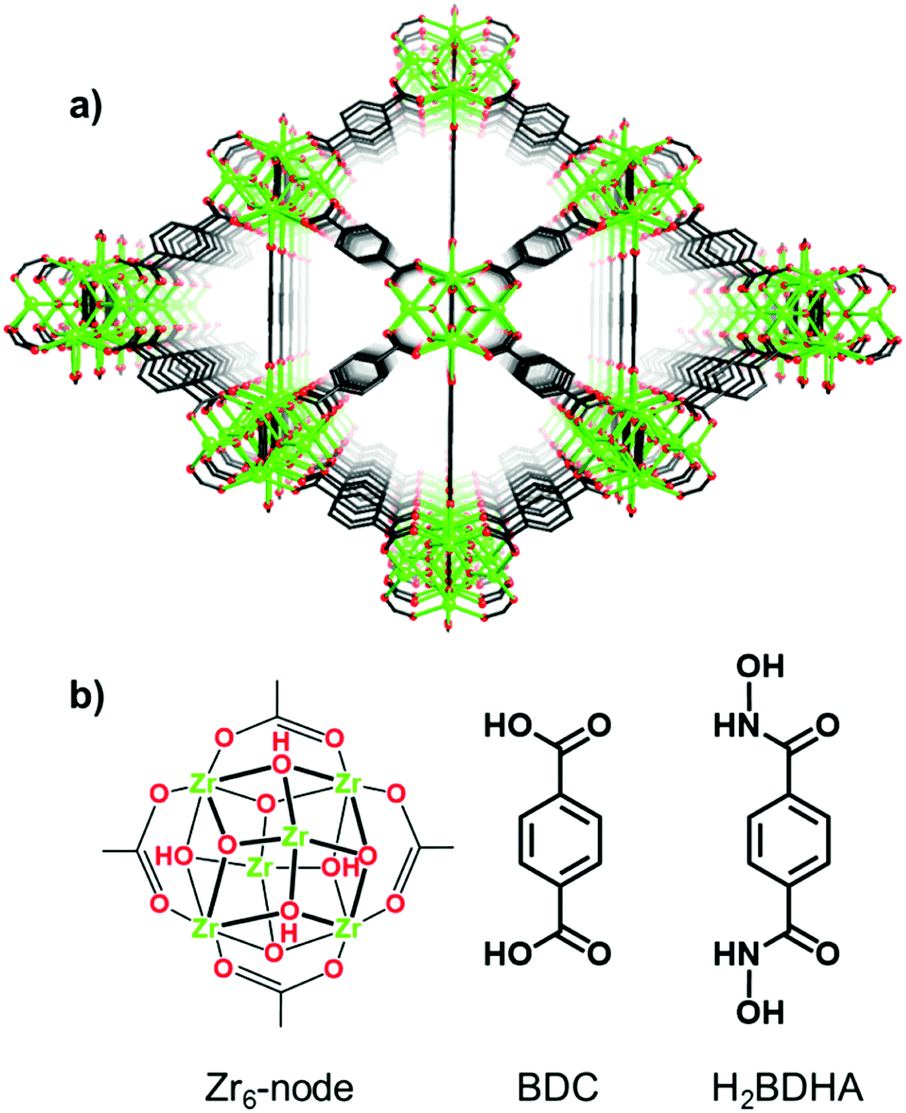 | ||
| Fig. 1 Structure of (a) UiO-66 and (b) Zr6-node and benzene-1,4-dicarboxylic acid (BDC) linker of UiO-66 and benzene-1,4-dihydroxamic acid (H2BDHA) used in SALE. | ||
Given the strength of Zr–hydroxamate bonds, it is expected to be very difficult to obtain a porous, crystalline material by de novo synthesis using a Zr precursor and hydroxamic acid linkers. Indeed, attempts at synthesizing a MOF de novo using ZrCl4 and benzene-1,4-dihydroxamic acid linkers under solvothermal conditions were unsuccessful. We therefore chose to apply SALE to obtain a hydroxamic acid containing Zr-based MOF. In addition to the preparation and characterization of UiO-66-H2BDHA, we also report a comparative study of the stability of both materials (UiO-66 and UiO-66-H2BDHA) by evaluating their structural integrity using powder X-ray diffraction (PXRD) and porosity using nitrogen adsorption isotherms after exposure to aqueous solutions with pH 1 and 12.
Results and discussion
In order to prepare a hydroxamic acid containing MOF, we first synthesized benzene-1,4-dihydroxamic acid (H2BDHA) from the readily available starting materials, dimethyl terephthalate and hydroxylamine hydrochloride (Scheme S1 in the ESI†), with good yield (57%).14 The free linker was then characterized by nuclear magnetic resonance (NMR) spectroscopy, mass spectrometry and by diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) (details in Section 1.1 in the ESI†).In the current work, UiO-66, a fine white powder, was prepared at 80 °C using benzene-1,4-dicarboxylic acid (BDC), ZrCl4 and concentrated HCl as modulator.15 UiO-66 (30 mg; 0.018 mmol) was then subjected to SALE, by soaking the MOF in a solution of H2BDHA (4 equivalents) in N,N′-dimethylformamide (DMF) (5 mL) in an oven at 100 °C for 6 days. The solution of H2BDHA was replaced every 24 hours over the 6 day period. UiO-66-H2BDHA was isolated as a pale yellow solid by centrifugation and washed several times with DMF and ethanol. Finally, the solid was isolated from ethanol and dried in an oven at 80 °C followed by drying under vacuum at 120 °C for 12 hours.
The evolution of SALE in UiO-66 was monitored at 24 hour intervals by 1H NMR spectroscopy (Fig. 2). For each data point, 1–2 mg of the material was isolated, dried and suspended with sonication in 3 drops of D2SO4, before adding 0.6 mL of deuterated dimethyl sulfoxide (DMSO-d6) to dissolve the suspension. The clear yellow solution was sonicated further to ensure complete digestion of the MOF and then transferred to an NMR tube. The 1H NMR spectrum of the digested UiO-66-H2BDHA sample obtained after 6 days is shown in Fig. 3. Due to the presence of D2SO4 the labile protons are not detected.
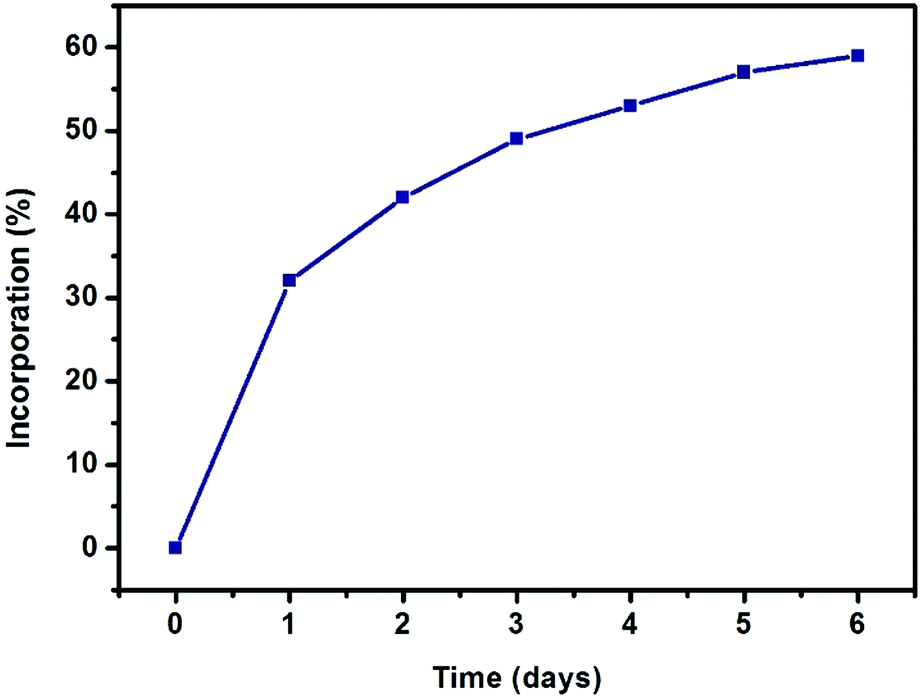 | ||
| Fig. 2 Progress of SALE in UiO-66 expressed as percent incorporation of benzene-1,4-dihydroxamic acid (monitored by 1H NMR spectroscopy). | ||
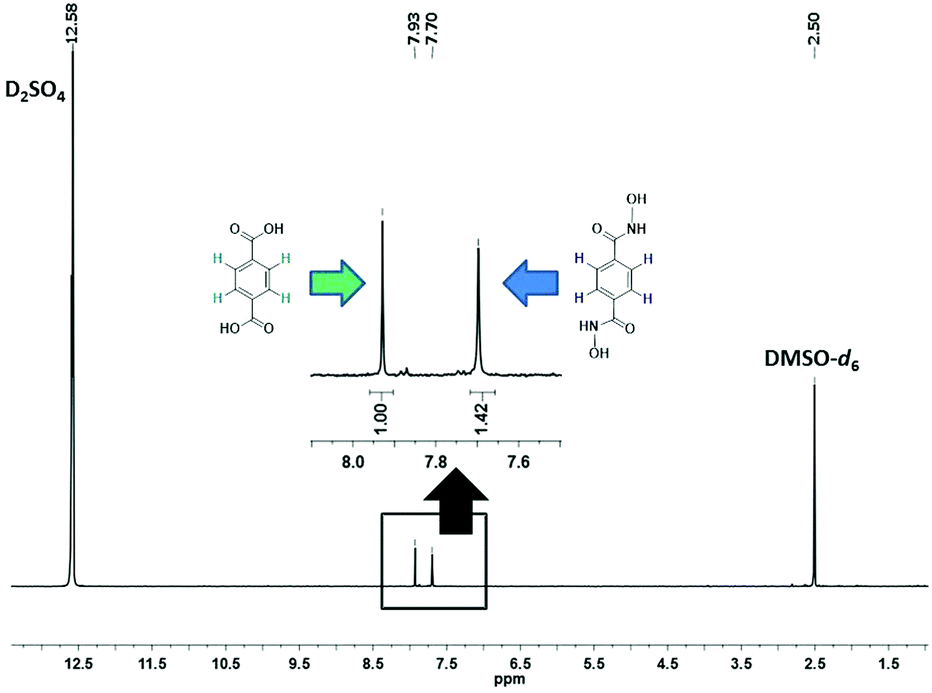 | ||
| Fig. 3 1H NMR (600 MHz) spectrum of UiO-66-H2BDHA in D2SO4 and DMSO-d6 (6 days, after activation). | ||
As depicted in Fig. 2, more than 30% incorporation of H2BDHA is observed in the first day and nearly 60% incorporation is achieved by the 6th day.
Fig. 4 details some structural comparisons between the parent and daughter MOFs. In Fig. 4a, the bulk crystallinity and crystal shape of each material is shown by PXRD and scanning electron microscopy (SEM), respectively. It is clear that the PXRD pattern of UiO-66-H2BDHA resembles that of UiO-66.
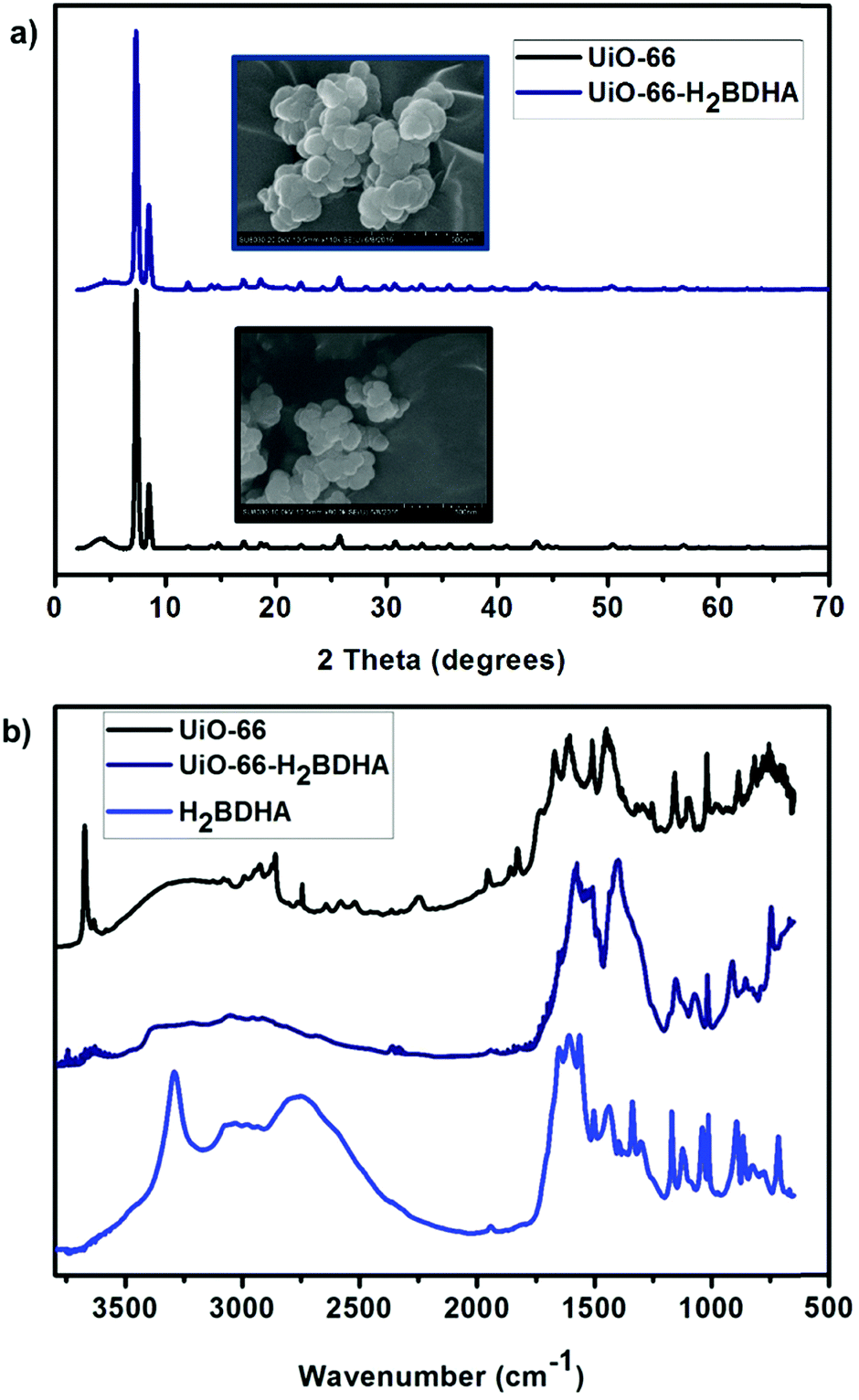 | ||
| Fig. 4 (a) PXRD pattern and SEM images of UiO-66 and UiO-66-H2BDHA. (b) DRIFTS spectrum of UiO-66, UiO-66-H2BDHA and H2BDHA. | ||
The infrared spectrum of a hydroxamic acid commonly exhibits a very strong NH stretching vibration between 3150 and 3350 cm−1 and a broad OH stretching vibration due to intermolecular hydrogen bonding between 2730 and 2930 cm−1. The CH stretching vibrations are commonly visible between 2800 and 3050 cm−1. The characteristic CO stretching vibration near 1630 cm−1 is typically observed as one broad band caused by intramolecular hydrogen bonding, but two sharp stretches corresponding to the symmetric and asymmetric modes can occasionally be observed.11a,14 The DRIFTS spectrum of H2BDHA depicted in Fig. 4b exhibits the features of hydroxamic acids discussed above. The DRIFTS spectrum of the parent UiO-66 material shows a peak at 3636 cm−1 corresponding to terminal –OH groups on the Zr-node of the framework. These Zr–OH sites may be present on the surface of the MOF or a result of structural defects. In the case of the daughter material, UiO-66-H2BDHA, the peak corresponding to terminal –OH groups is decreased significantly, suggesting that a portion of the new H2BDHA linkers are sealing defects in the structure and are likely bound monotopically. The spectrum also shows peaks at 1569 cm−1 and 3353 cm−1 corresponding to the hydroxamato carbonyl and NH vibration of the H2BDHA linker, further confirming the incorporation of hydroxamic acids in the structure of UiO-66. The TGA data for both materials was also collected and is shown in Fig. S4 (ESI†).
The porosity of the parent and daughter MOF was measured after activation by solvent exchange with ethanol, followed by heating under vacuum at 120 °C to remove the solvent. It was discovered that while UiO-66 has a Brunauer–Emmett–Teller (BET) area of 1580 m2 g−1, UiO-66-H2BDHA exhibits a BET area of 1050 m2 g−1 (Fig. 5).
 | ||
| Fig. 5 N2 isotherms of UiO-66 and UiO-66-H2BDHA at 77 K. | ||
In order to test whether the incorporation of hydroxamic acid linkers in UiO-66 leads to an increase in framework stability, we compared the parent and daughter materials after soaking in aqueous solutions with pH 1 and 12. It should be noted that the parent material in this case is UiO-66 made using HCl as a modulator, meaning that the resulting material is expected to contain missing linker and/or missing node defects. The amount of UiO-66-H2BDHA solid obtained at the end of the pH treatment was found to be nearly the same when compared with the initial weight of material used, but in the case of UiO-66, a weight loss of approximately 35% was observed under both pH conditions tested. Both parent and daughter framework materials were found to retain bulk crystallinity (Fig. S5, ESI†). A more significant decrease in porosity was observed however, for UiO-66 compared to UiO-66-H2BDHA after soaking in aqueous solutions of pH 1 and 12 (Fig. 6 and Table S1, ESI†). The modest increase in stability of the daughter framework can be attributed to the replacement of BDC linkers with more strongly coordinating H2BDHA linkers as well as the sealing of defects in UiO-66 parent framework with strongly coordinating H2BDHA linkers bound monotopically. We also compared the stability of the new daughter material UiO-66-H2BDHA with the ‘defect-free’ sample of UiO-66 synthesized using acetic acid as modulator. Here we observed that the ‘defect-free’ material maintained crystallinity after soaking at pH 1 and 12 (Fig. S6, ESI†), but an increase in BET area was observed. The increase in surface area could be attributed to the introduction of defects in the structure (Fig. S7 and Table S1, ESI†). This may further suggest that sealing the parent, defective UiO-66 framework with H2BDHA linkers helps to stabilize the framework from degradation at the surface.
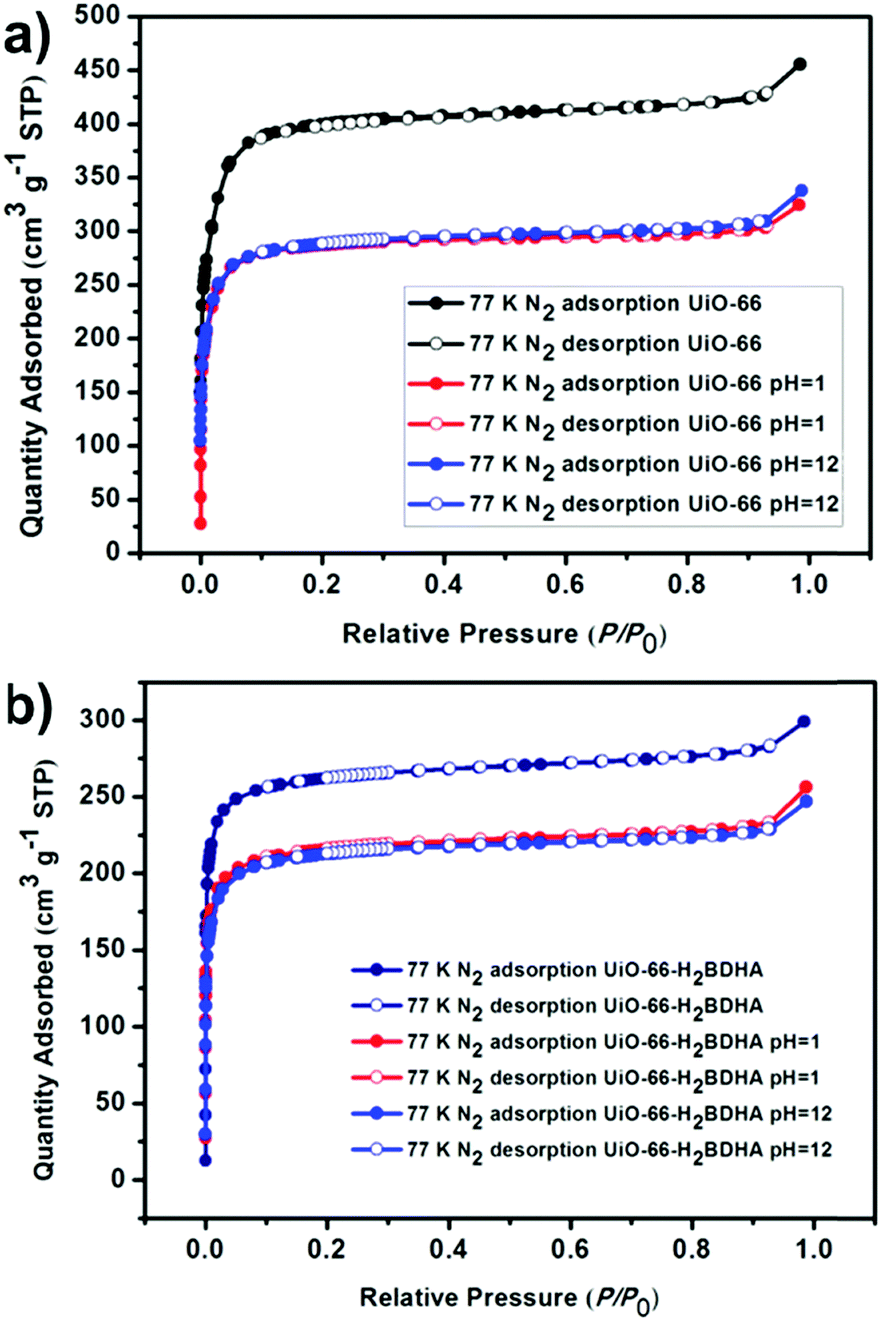 | ||
| Fig. 6 Evaluation of the porosity of UiO-66 (a) and UiO-66-H2BDHA (b) after exposure to aqueous solutions of pH 1 and 12. | ||
Conclusions
In this report, we introduce a hydroxamic acid linker into the Zr-based MOF, UiO-66, via SALE. The synthesis, structural characterization and stability testing of the daughter MOF, UiO-66-H2BDHA, is presented. The daughter MOF was found to have increased stability compared to the defective UiO-66 parent MOF as a result of the replacement of BDC linkers with more strongly coordinating H2BDHA linkers and the sealing of structural defects. The use of hydroxamic acid linkers to construct Zr-based MOFs is very challenging de novo, but is of interest given the strong bonds formed between Zr and hydroxamates. This study shows that the use of post-synthetic techniques, such as SALE, may allow access to robust hydroxamic acid-based MOFs and warrants further exploration to obtain frameworks constructed solely using hydroxamate linkers.Experimental
General experimental
Powder X-ray diffraction (PXRD) data were collected at ambient temperature on an Bruker Kappa Apex 2 (Cu Kα IμS microfocus λ = 1.54178), equipped with a Bruker Apex 2 CCD detector with a detector distance of 150 nm in transmission mode with 180° oscillation (45 kV, 0.65 mA). Intensity data were collected by the step-counting method (step 0.01°) in the range of 2 ≤ 2θ° ≤ 70 range. Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) were recorded on a Nicolet 7600 FTIR spectrometer equipped with an MCT detector. The spectra were collected under Ar atmosphere in a Praying Mantis™ cell (samples loaded in a drybox under Ar). The MOF samples were first activated at 120 °C under high vacuum for 12 h. These activated MOF samples (∼3 wt%) were then combined with anhydrous KBr, ground up thoroughly, and then loaded into the instrument as a powder. The spectra were collected at 1 cm−1 resolution over 64 scans under N2 purge. A sample of powder KBr was utilized as the background. Nitrogen adsorption isotherm measurements were carried out on a Micromeritics Tristar II 3020 (Micromeritics Instrument Corporation) at 77 K. Samples were heated at 120 °C under vacuum for 12 h on a SmartVacPrep instrument (Micromeritics Instrument Corporation, Norcross, GA, USA) prior to measuring the isotherms. SEM (scanning electron microscopy) images were collected using a Hitachi SU-8030 instrument. Mass spectrum was collected on a Bruker AmazonSL instrument configured for electrospray ionization (ESI) using positive mode.1H NMR spectra were collected on a Bruker Avance III 600 MHz spectrometer. Thermogravimetric analysis (TGA) experiments were carried out on a Q500 thermogravimetric analyzer (TA Instruments Inc., New Castle, DE). The samples were heated from room temperature to 800 °C with a gas flow (90 mL min−1) at a heating rate of 5 °C min−1. Sodium hydroxide, dimethyl terephthalate, benzene-1,4-dicarboxylic acid and ZrCl4 were obtained from Sigma-Aldrich. Hydroxylamine hydrochloride was purchased from Fisher. All these chemicals were used as received without further purification.
Synthesis of benzene-1,4-dihydroxamic acid (H2BDHA)
Benzene-1,4-dihydroxamic acid (H2BDHA) was synthesized as previously reported,14 but with some modifications. Hydroxylamine hydrochloride (1.79 g, 27.76 mmol) and sodium hydroxide (2.06 g, 51.50 mmol) were combined in 20 mL methanol and shaken vigorously. The NaCl was then removed via filtration and the filtrate was combined with 1 g (5.15 mmol) of dimethyl terephthalate in 30 mL methanol. The solution was stirred overnight at ambient temperature. The solvent was removed in vacuo and the solid material was dissolved in 20 mL H2O. The solution was acidified to a pH of 5.5 with 5% HCl. The precipitate was collected by filtration and dried in vacuo. Yield = 57%. 1H NMR (600 MHz, DMSO-d6): δ 11.32 (s, 2H, OH), δ 9.14 (s, 2H, NH), δ 7.80 (s, 4H, ArH). 13C NMR (151 MHz, DMSO-d6): δ 127.4, 135.5, 164.2. TOF-MS ESI+: m/z calculated for C8H9N2O4, 197.1; found, 198.1 [M + H]+. Selected DRIFTS data (cm−1): n(NH) = 3296 vs; n(CH) = 3077–2927 b; n(OH) = 2871–2607 b and n(CO) = 1616 (vs – very strong; b – broad).Preparation of UiO-66
UiO-66 was prepared as described previously.15 ZrCl4 (125 mg; 0.54 mmol) in 5 mL of N,N′-dimethylformamide (DMF) was sonicated for 20 minutes in the presence of concentrated HCl (1 mL). Then, benzene-1,4-dicarboxylic acid (123 mg, 0.75 mmol) in 10 mL of DMF was added and the reaction mixture was sonicated for 20 minutes and heated overnight at 80 °C. The obtained materials were washed with DMF (2 × 30 mL) and with ethanol (2 × 30 mL) and dried at 80 °C. UiO-66 was then activated at 120 °C for 12 hours prior to the measurement of the N2 isotherms.SALE of H2BDHA into UiO-66
In a typical experiment, H2BDHA (4 equivalents) was placed in 5 mL of DMF and sonicated for 10 minutes. UiO-66 (30 mg, 0.018 mmol) was then added and the reaction mixture was placed in an oven at 100 °C for 6 days, while replacing the solution of H2BDHA every 24 hours. The material was then washed with DMF and ethanol, and dried at 80 °C, followed by activation at 120 °C for 12 hours.The evolution of SALE was monitored using 1H NMR spectroscopy and the samples were prepared in the following way: 1–2 mg of MOF material was placed in a 1.5-dram vial and then 3 drops of D2SO4 were added and the sample was sonicated until a well-dispersed suspension was formed. 0.6 mL of deuterated dimethyl sulfoxide (DMSO-d6) was then added to dissolve the suspension. The sample was then transferred to an NMR tube and the 1H NMR spectra were obtained by locking on DMSO.
Stability tests
In order to study the stability of the different materials, 40 mg of each material was soaked overnight in 3 mL of aqueous solutions with pH 1 and 12. The materials were then washed with water and ethanol, and dried at 80 °C followed by drying under vacuum at 120 °C for 12 hours. After isolation and drying, the material was re-weighed. At the end of the pH treatment, in the case of UiO-66-H2BDHA, the amount of material recovered was nearly the same when compared with the initial weight, but in the case of UiO-66 a weight loss of about 35% was observed under both pH conditions tested.Acknowledgements
C. F. P., F. A. A. P and J. P. C. gratefully thank Fundação para a Ciência e a Tecnologia (FCT, Portugal), the European Union, QREN, FEDER through Programa Operacional Factores de Competitividade (COMPETE), CICECO – Aveiro Institute of Materials, POCI-01-0145-FEDER-007679 (FCT Ref. UID/CTM/50011/2013), QOPNA (FCT UID/QUI/00062/2013) and CQE (FCT UID/QUI/0100/2013) research units, financed by national funds through the FCT/MEC and when appropriate co-financed by FEDER under the PT2020 Partnership Agreement. FCT is also gratefully acknowledged for the Ph.D. grant No. SFRH/BD/86303/2012 (to CP). Authors gratefully acknowledge funding from the Army Research Office (project number W911NF-13-1-0229). A. J. H. thanks NSERC for a postdoctoral fellowship. We thank Dr. Martino Rimoldi for his help with the NMR analysis and Cornelius Audu for providing the defect-free sample of UiO-66.Notes and references
- (a) O. M. Yaghi and H. L. Li, J. Am. Chem. Soc., 1995, 117, 10401–10402 CrossRef CAS; (b) G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC; (c) P. Silva, S. M. F. Vilela, J. P. C. Tomé and F. A. A. Paz, Chem. Soc. Rev., 2015, 44, 6774–6803 RSC.
- (a) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed; (b) O. K. Farha and J. T. Hupp, Acc. Chem. Res., 2010, 43, 1166–1175 CrossRef CAS PubMed; (c) F. A. A. Paz, J. Klinowski, S. M. F. Vilela, J. P. C. Tomé, J. A. S. Cavaleiro and J. Rocha, Chem. Soc. Rev., 2012, 41, 1088–1110 RSC; (d) A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 15018 CrossRef CAS.
- (a) J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC; (b) K. Leus, Y. Y. Liu and P. Van Der Voort, Catal. Rev.: Sci. Eng., 2014, 56, 1–56 CrossRef CAS; (c) J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- J. Rocha, L. D. Carlos, F. A. A. Paz and D. Ananias, Chem. Soc. Rev., 2011, 40, 926–940 RSC.
- (a) O. K. Farha, A. Ö. Yazaydin, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, Nat. Chem., 2010, 2, 944–948 CrossRef CAS PubMed; (b) J.-R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed; (c) N. M. Padial, E. Q. Procopio, C. Montoro, E. López, J. E. Oltra, V. Colombo, A. Maspero, N. Masciocchi, S. Galli, I. Senkovska, S. Kaskel, E. Barea and J. A. R. Navarro, Angew. Chem., Int. Ed., 2013, 52, 8290–8294 CrossRef CAS PubMed; (d) C. E. Wilmer, O. K. Farha, T. Yildirim, I. Eryazici, V. Krungleviciute, A. A. Sarjeant, R. Q. Snurr and J. T. Hupp, Energy Environ. Sci., 2013, 6, 1158–1163 RSC.
- (a) J. A. Hurd, R. Vaidhyanathan, V. Thangadurai, C. I. Ratcliffe, I. L. Moudrakovski and G. K. H. Shimizu, Nat. Chem., 2009, 1, 705–710 CrossRef CAS PubMed; (b) N. C. Jeong, B. Samanta, C. Y. Lee, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 51–54 CrossRef CAS PubMed; (c) M. Bazaga-Garcia, R. M. P. Colodrero, M. Papadaki, P. Garczarek, J. Zoń, P. Olivera-Pastor, E. R. Losilla, L. Leon-Reina, M. A. G. Aranda, D. Choquesillo-Lazarte, K. D. Demadis and A. Cabeza, J. Am. Chem. Soc., 2014, 136, 5731–5739 CrossRef CAS PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- (a) S. R. Halper, L. Do, J. R. Stork and S. M. Cohen, J. Am. Chem. Soc., 2006, 128, 15255–15268 CrossRef CAS PubMed; (b) X. Yang and A. E. Clark, Inorg. Chem., 2014, 53, 8930–8940 CrossRef CAS PubMed; (c) D. Alezi, A. M. P. Peedikakkal, L. J. Weseliński, V. Guillerm, Y. Belmabkhout, A. J. Cairns, Z. J. Chen, L. Wojtas and M. Eddaoudi, J. Am. Chem. Soc., 2015, 137, 5421–5430 CrossRef CAS PubMed.
- (a) B. J. Burnett, P. M. Barron, C. Hu and W. Choe, J. Am. Chem. Soc., 2011, 133, 9984–9987 CrossRef CAS PubMed; (b) M. Kim, J. F. Cahill, H. Fei, K. A. Prather and S. M. Cohen, J. Am. Chem. Soc., 2012, 134, 18082–18088 CrossRef CAS PubMed; (c) S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed; (d) A. D. Burrows and S. M. Cohen, CrystEngComm, 2012, 14, 4095 RSC; (e) S. Takaishi, E. J. DeMarco, M. J. Pellin, O. K. Farha and J. T. Hupp, Chem. Sci., 2013, 4, 1509–1513 RSC; (f) O. Karagiaridi, W. Bury, E. Tylianakis, A. A. Sarjeant, J. T. Hupp and O. K. Farha, Chem. Mater., 2013, 25, 3499–3503 CrossRef CAS; (g) P. Deria, J. E. Mondloch, O. Karagiaridi, W. Bury, J. T. Hupp and O. K. Farha, Chem. Soc. Rev., 2014, 43, 5896–5912 RSC; (h) O. Karagiaridi, W. Bury, J. E. Mondloch, J. T. Hupp and O. K. Farha, Angew. Chem., Int. Ed., 2014, 53, 4530–4540 CrossRef CAS PubMed; (i) O. Karagiaridi, N. A. Vermeulen, R. C. Klet, T. C. Wang, P. Z. Moghadam, S. S. Al-Juaid, J. F. Stoddart, J. T. Hupp and O. K. Farha, Inorg. Chem., 2015, 54, 1785–1790 CrossRef CAS PubMed; (j) C. J. Stephenson, J. T. Hupp and O. K. Farha, Inorg. Chem., 2016, 55, 1361–1363 CrossRef CAS PubMed.
- (a) M. J. Miller, Chem. Rev., 1989, 89, 1563–1579 CrossRef CAS; (b) M. H. Parker, E. A. Lunney, D. F. Ortwine, A. G. Pavlovsky, C. Humblet and C. G. Brouillette, Biochemistry, 1999, 38, 13592–13601 CrossRef CAS PubMed; (c) R. Codd, Coord. Chem. Rev., 2008, 252, 1387–1408 CrossRef CAS; (d) C. Koenigsmann, T. S. Ripolles, B. J. Brennan, C. F. A. Negre, M. Koepf, A. C. Durrell, R. L. Milot, J. A. Torre, R. H. Crabtree, V. S. Batista, G. W. Brudvig, J. Bisquert and C. A. Schmuttenmaer, Phys. Chem. Chem. Phys., 2014, 16, 16629–16641 RSC; (e) E. Ahmed and S. J. M. Holmström, Microb. Biotechnol., 2014, 7, 196–208 CrossRef CAS PubMed; (f) P. Lassalas, B. Gay, C. Lasfargeas, M. J. James, V. Tran, K. G. Vijayendran, K. R. Brunden, M. C. Kozlowski, C. J. Thomas, A. B. Smith, D. M. Huryn and C. Ballatore, J. Med. Chem., 2016, 59, 3183–3203 CrossRef CAS PubMed.
- (a) D. A. Brown, R. A. Coogan, N. J. Fitzpatrick, W. K. Glass, D. E. Abukshima, L. Shiels, M. Ahlgrén, K. Smolander, T. T. Pakkanen, T. A. Pakkanen and M. Peräkylä, J. Chem. Soc., Perkin Trans. 2, 1996, 2673–2679 RSC; (b) C. J. Marmion, D. Griffith and K. B. Nolan, Eur. J. Inorg. Chem., 2004, 3003–3016 CrossRef CAS; (c) V. Krchnak, Mini-Rev. Med. Chem., 2006, 6, 27–36 CrossRef CAS PubMed; (d) P. Trapencieris, J. Strazdina and P. Bertrand, Chem. Heterocycl. Compd., 2012, 48, 833–855 CrossRef CAS; (e) D. Borovika, P. Bertrand and P. Trapencieris, Chem. Heterocycl. Compd., 2014, 49, 1560–1578 CrossRef CAS.
- (a) E. M. F. Muri, M. J. Nieto, R. D. Sindelar and J. S. Williamson, Curr. Med. Chem., 2002, 9, 1631–1653 CrossRef CAS PubMed; (b) S. Bertrand, J. J. Helesbeux, G. Larcher and O. Duval, Mini-Rev. Med. Chem., 2013, 13, 1311–1326 CrossRef CAS PubMed.
- (a) W. R. McNamara, R. C. Snoeberger III, G. Li, C. Richter, L. J. Allen, R. L. Milot, C. A. Schmuttenmaer, R. H. Crabtree, G. W. Brudvig and V. S. Batista, Energy Environ. Sci., 2009, 2, 1173–1175 RSC; (b) W. R. McNamara, R. L. Milot, H.-E. Song, R. C. Snoeberger III, V. S. Batista, C. A. Schmuttenmaer, G. W. Brudvig and R. H. Crabtree, Energy Environ. Sci., 2010, 3, 917–923 RSC; (c) T. P. Brewster, S. J. Konezny, S. W. Sheehan, L. A. Martini, C. A. Schmuttenmaer, V. S. Batista and R. H. Crabtree, Inorg. Chem., 2013, 52, 6752–6764 CrossRef CAS PubMed.
- D. Griffith, K. Krot, J. Comiskey, K. B. Nolan and C. J. Marmion, Dalton Trans., 2008, 137–147 RSC.
- M. J. Katz, Z. J. Brown, Y. J. Colon, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49, 9449–9451 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qm00364h |
| This journal is © the Partner Organisations 2017 |