
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSubstituent effects on stereoselectivity of dihalocarbene reactions with cyclohexadiene and on the reactivity of bis-dihalocyclopropanes in electrophilic nitrations en route to pyrimidine N-oxides†
Kseniya N.
Sedenkova
ab,
Elena B.
Averina
 *ab,
Yuri K.
Grishin
a,
Julia V.
Kolodyazhnaya
a,
Victor B.
Rybakov
a,
Tamara S.
Kuznetsova
a,
Audrey
Hughes
c,
Gabriel dos Passos
Gomes
c,
Igor V.
Alabugin
c and
Nikolay S.
Zefirov
ab
*ab,
Yuri K.
Grishin
a,
Julia V.
Kolodyazhnaya
a,
Victor B.
Rybakov
a,
Tamara S.
Kuznetsova
a,
Audrey
Hughes
c,
Gabriel dos Passos
Gomes
c,
Igor V.
Alabugin
c and
Nikolay S.
Zefirov
ab
aLomonosov Moscow State University, Department of Chemistry, Leninskie Gory, 1-3, Moscow 119991, Russia. E-mail: elaver@med.chem.msu.ru; alabugin@chem.fsu.edu; Fax: +7-495-939-3969; Tel: +7-495-939-3969
bIPhAc RAS, Severnyi Proezd, 1, Chernogolovka, Moscow Region, 142432, Russia
cDepartment of Chemistry & Biochemistry, Florida State University, 95 Chieftan Way, Tallahassee, FL 32306, USA
First published on 17th October 2017
Abstract
Tricyclic bis-adducts of cyclohexa-1,4-diene with bromofluorocarbene and non-symmetric adducts with both bromofluoro- and dichlorocarbenes were synthesised selectively. The treatment of the bis-adducts with nitrating reagents in acetonitrile affords the products of heterocyclization of a sole dihalogenocyclopropane into 4-fluoropyrimidine N-oxide. The difference in the reactivity of bis-cyclopropanes with different sets of halogen substituents leads to selective heterocyclization of bromofluorocyclopropanes without affecting the dichlorocyclopropane moiety.
Introduction
Pyrimidine and quinazoline rings are frequently encountered in pharmaceuticals with a wide range of activities, including anticonvulsant, antibacterial, antifungal, antiviral and anticancer properties.1 Because halogen- substituted pyrimidines can be readily modified via SNAr reactions, they serve as commonly used synthetic intermediates for the construction of libraries of heterocyclic compounds.2 Nevertheless, no general methods for the construction of a fluoropyrimidine system via cyclization have been described until our recent work.3Previously, we have reported the heterocyclization of gem-bromofluorocyclopropanes I into 4-fluoropyrimidine N-oxides II involving the treatment with nitrosating or nitrating reagents in the presence of organic nitriles (Scheme 1).4,5 This reaction was used to develop efficient preparative approaches to various 4-substituted pyrimidine derivatives III,6,7 including those with antiviral activities.
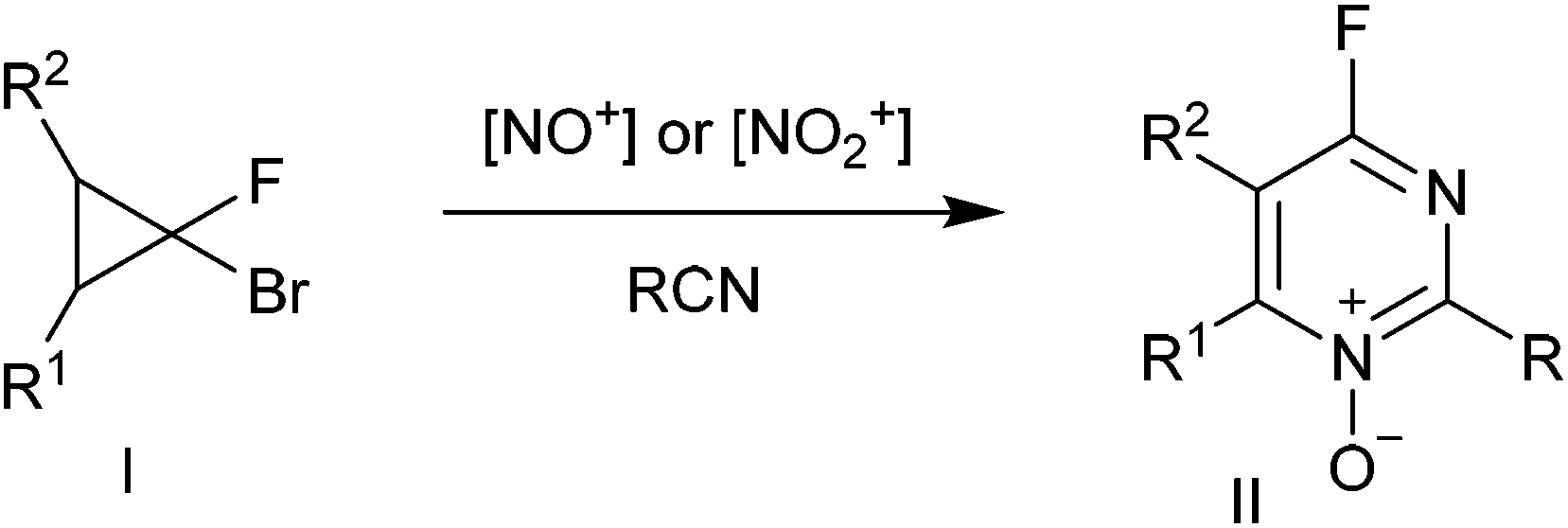 | ||
| Scheme 1 Three-component heterocyclization of gem-bromofluorocyclopropanes. | ||
In order to evaluate further the scope and limitations of the three-component heterocyclization and to investigate the reactivity of the neighboring three-membered rings in this process, we obtained novel bis(gem-dihalocyclopropyl) compounds 1, 2 and involved them in the reaction with nitrating reagents. The heterocyclization of bis(gem-dihalocyclopropanes) would open a direct synthetic pathway to dimeric pyrimidine derivatives. This process is of particular interest because bis(heterocyclic) compounds can act as bivalent ligands with increased bioactivities. For instance, the bivalent positive allosteric modulators of AMPA receptors were shown to be significantly more potent than the respective monovalent analogs.8,9
Results and discussion
The starting bis-cyclopropanes 1, 2 were obtained via a two-stage carbene cycloaddition to cyclohexa-1,4-diene (3) (Scheme 2). In the first stage diene 3 was treated with bromofluorocarbene, generated from 4 eq. of dibromofluoromethane under phase-transfer conditions.10 The intermediate monocyclopropane 4 was isolated in 62% yield and introduced for further cyclopropanation. The alkene moiety of 4 was relatively inactive in the reaction with bromofluorocarbene, and only continuous treatment of 4 with a large excess of dibromofluoromethane afforded the bis-adduct 1 in a good yield. In contrast, the interaction of dichlorocarbene with 4 proceeded smoothly for 24 h leading to compound 2, containing both dichlorocyclopropane and bromofluorocyclopropane moieties.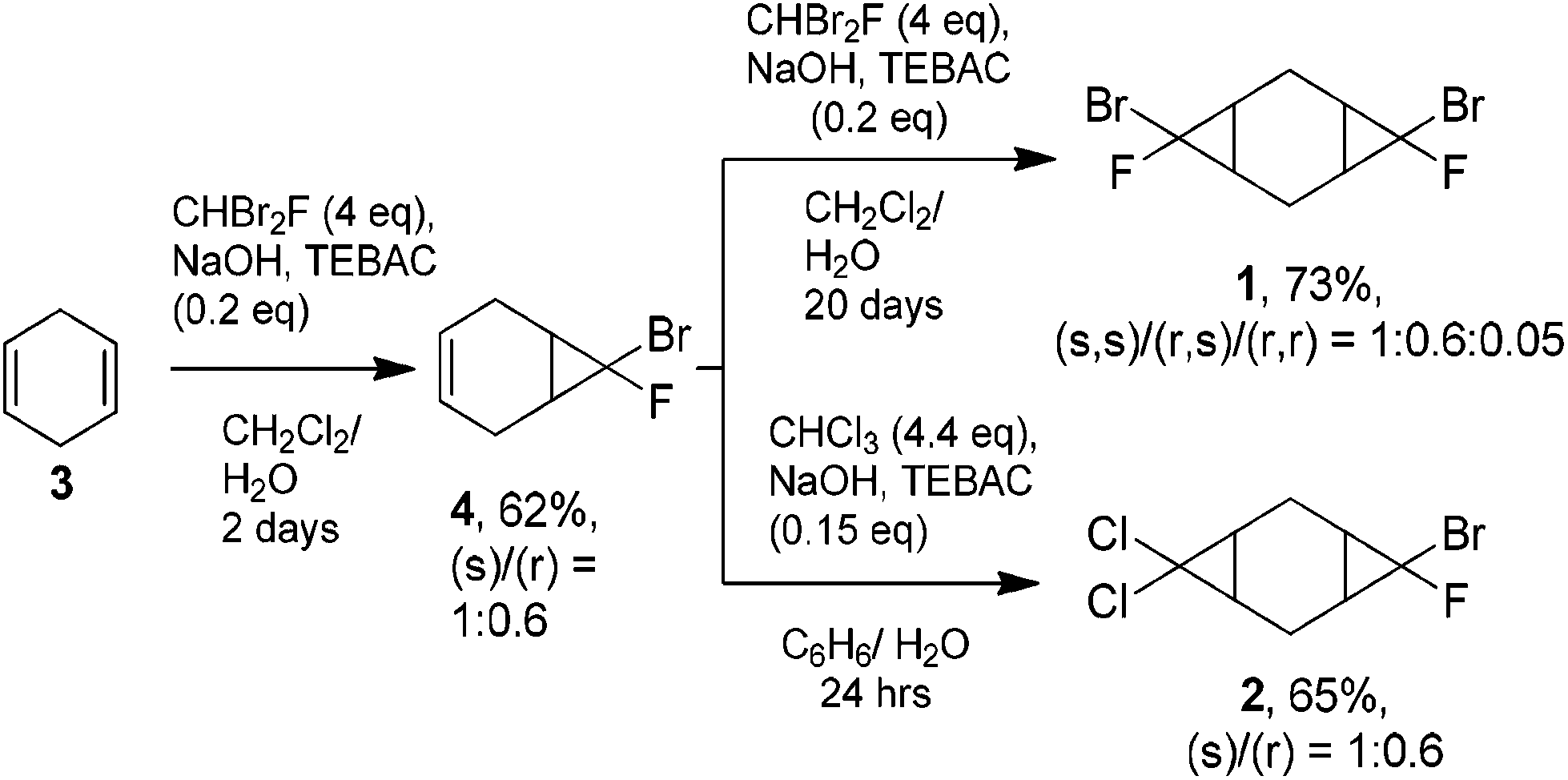 | ||
| Scheme 2 Preparation of tricyclic compounds 1, 2. | ||
The mono-adduct 4 can exist as two diastereomers, depending on the exo/endo-position of halogen atoms in bromofluorocyclopropane (Scheme 3). Adduct 4 was obtained as a mixture of (r) and (s)-isomers,11 (r)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :(s) = 0.6:1. Here and elsewhere the position of the fluorine atom was determined by using 1H and 19F NMR spectra: a large value of spin–spin coupling constant (3JHF = 19–20 Hz) corresponds to the cis-orientation of fluorine and hydrogen atoms in the (s)-isomer, while the (r)-isomer with the trans-orientation of the abovementioned atoms is characterized with a significantly smaller constant (3JHF < 6 Hz) (Scheme 3).
:(s) = 0.6:1. Here and elsewhere the position of the fluorine atom was determined by using 1H and 19F NMR spectra: a large value of spin–spin coupling constant (3JHF = 19–20 Hz) corresponds to the cis-orientation of fluorine and hydrogen atoms in the (s)-isomer, while the (r)-isomer with the trans-orientation of the abovementioned atoms is characterized with a significantly smaller constant (3JHF < 6 Hz) (Scheme 3).
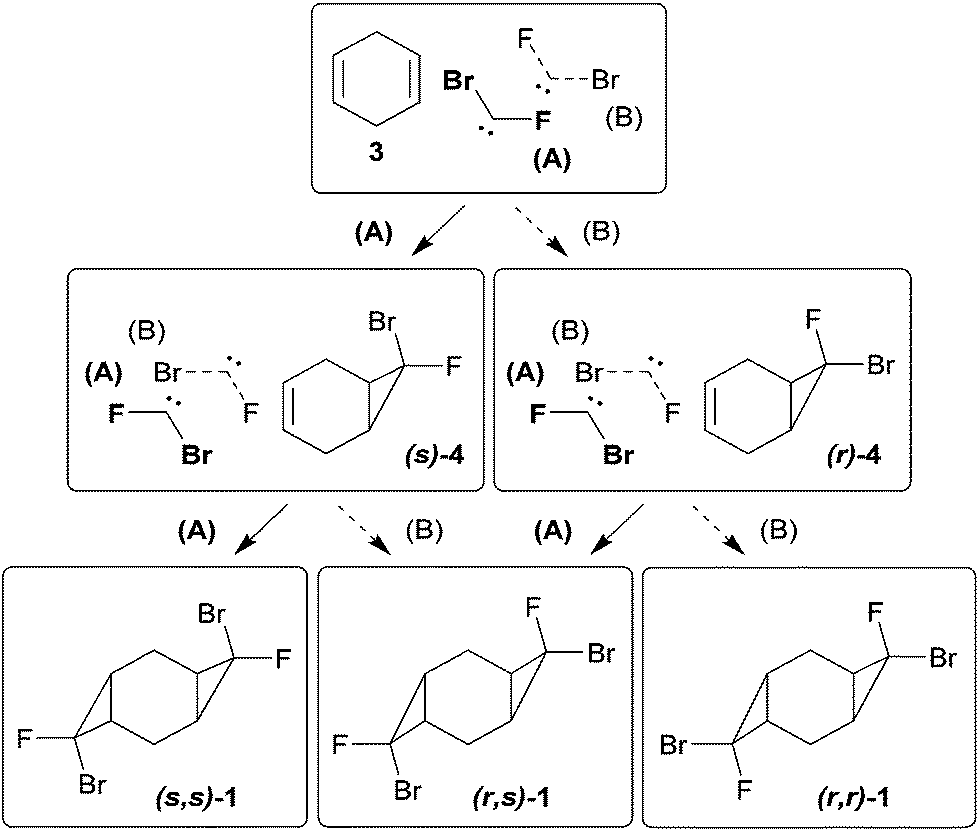 | ||
| Scheme 3 Addition of bromofluorocarbene to diene 3 and alkene 4. | ||
For the bis-adducts 1 and 2, the number of possible diastereomers increases due to the additional choices in the relative positions of halogens and the orientation of cyclopropane rings. Because all of the literature bis-cycloaddition reactions of cyclohexadiene 3 with dihalocarbenes containing two identical halogen atoms were reported to give only the products in which the cyclopropane units are in trans-orientation to each other,12–15 we assume the same trans-preference to be observed for compounds 1, 2.
The observed experimental selectivity for the formation of bis-adduct 1 revealed the formation of isomers (s,s)-1, (r,s)-1 and (r,r)-1 in the ratio of 1:0.6:0.05 (Scheme 3). The major isomer (s,s)-1 can be separated from the mixture of minor isomers (r,s)-1 and (r,r)-1via column chromatography. The structure of the (s,s)-isomer was unambiguously proven with X-ray analysis (Fig. 1).16 Such geometry and isomer ratios of the starting compounds and products indicate the strong preference to the exo-orientation of fluorine during the addition of bromofluorocarbene to alkene 4 (even in comparison to cyclohexadiene 3). The opposite orientation of carbene, leading to compound (r,r)-1, is far less probable (Scheme 3). “Mixed” bis-adduct 2 was isolated as a mixture of (r) and (s)-isomers in the ratio of 0.6:1, that exactly corresponds to the isomer composition of starting monocyclopropane 4.
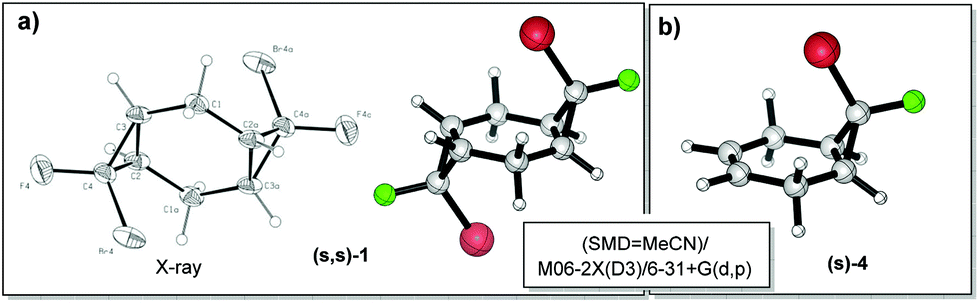 | ||
| Fig. 1 (a) The molecular structure of compound (s,s)-1 according to X-ray crystallography and DFT computations. (b) DFT geometry of compound (s)-4. | ||
An interesting structural effect on the geometry of bis-cyclopropanes stems from the presence of two halogens at the cyclopropyl rings. The central “cyclohexane” ring of halogen-substituted tricyclooctanes 1 and 2 is remarkably close to planarity17 (deviations of <0.3 degree) in a sharp contrast to the twisted geometry of the respective bicyclo[4.1.0]heptane. The monoadduct 4, where a double bond is still present, features an almost flat six-membered ring as well.
The reactivity of bis-adducts 1, 2 was investigated under the conditions of three-component heterocyclization. Individual diastereomer (s,s)-1 appeared to be completely inert toward the treatment with either nitronium triflate or the mixture of nitric and triflic acids in acetonitrile. The isomers (r,s)-1 and (r,r)-1 demonstrated a higher reactivity: their slow interaction with the mixture of fuming nitric and triflic acids afforded tricyclic 4-fluoropyrimidine N-oxide 5, the product of heterocyclization with the involvement of only one dihalocyclopropane moiety (Scheme 4). It should be mentioned that (r)-5 is formed from both (r,s)- and (r,r)-isomers of cyclopropane 1 as the major isomer ((r)-5/(s)-5 = 1/0.5). Presumably, the electrophilic attack of the nitronium cation comes preferably from the side of the six-membered ring opposite to a bulky bromine atom.
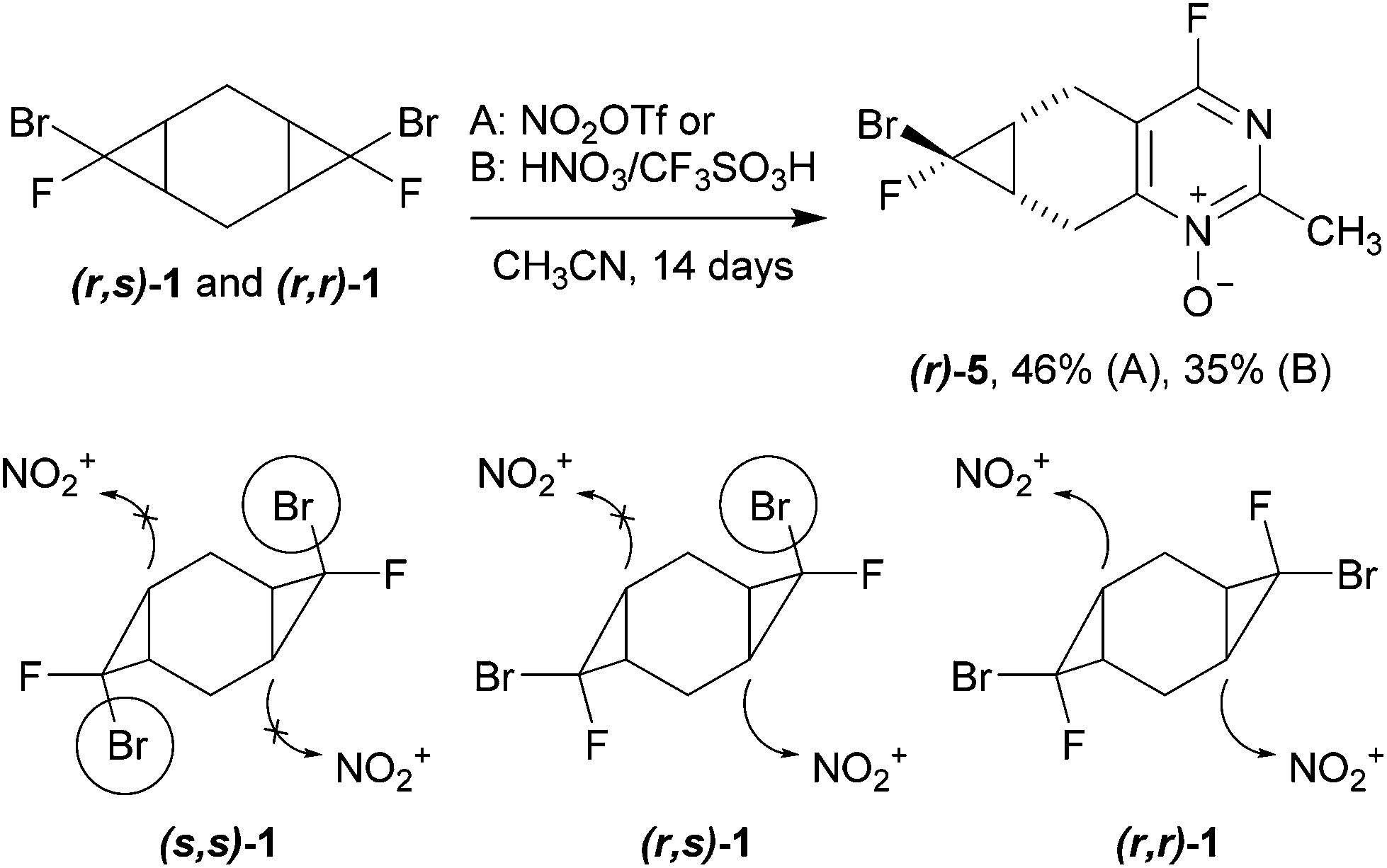 | ||
| Scheme 4 Heterocyclization of compound 1 under the treatment with nitrating agents. | ||
In order to compare the reactivity of dichloro- and bromofluorocyclopropanes, we investigated the heterocyclization of the unsymmetrical compound 2 containing two different dihalogenocyclopropane moieties. The treatment of bis-adduct 2 with the excess of nitrating agents in acetonitrile was performed at room temperature for 14 days. The only product of the reaction was 4-fluoropyrimidine N-oxide 6, resulting from the heterocyclization of the bromofluorocyclopropane moieties (Scheme 5). Attempts to transform the remaining dichlorocyclopropane moieties into heterocycle via the reaction of 6 with nitronium triflate under various conditions led to the decomposition of compound 6.
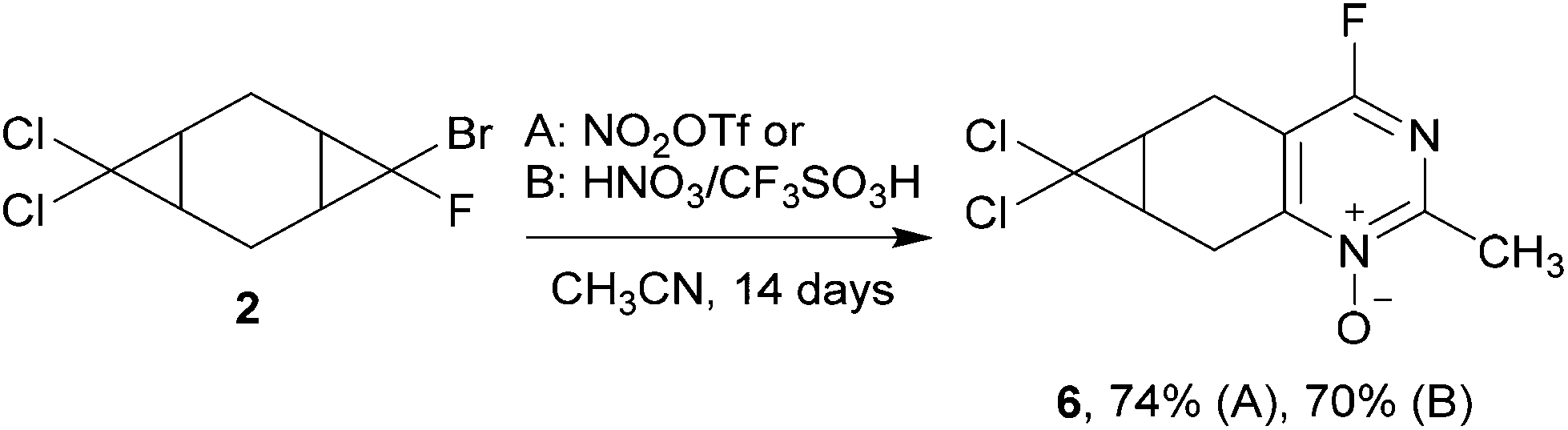 | ||
| Scheme 5 Heterocyclization of compound 2 under the treatment with nitrating agents. | ||
Computational analysis
Methods
Calculations were carried using the meta-hybrid (U)M06-2X functional18 and the 6-31+G(d,p) basis set for all atoms, with an ultrafine integration grid (99590 points). A broken-spin approach was applied when necessary. The implicit SMD19 solvation model was used to simulate the effects of acetonitrile (MeCN) throughout the calculated structures when necessary. Grimme's D3 version (zero damping) for empirical dispersion20 was also included. Frequency calculations were carried out for all structures to confirm them as either a minimum or a TS. Intrinsic Reaction Coordinates (IRC)21 were determined for the TS of interest. Orbital interactions were analyzed by the Natural Bond Orbitals (NBO) method and calculated with the 2nd-order perturbation approach.22 All DFT calculations were performed with Gaussian ‘0923 with the integrated NBO6.24 Three-dimensional structures and orbital plots were produced with CYLView 1.0.125 and Chemcraft 1.8.26
Results and discussion
As the above experimental data illustrate, both the cyclopropanation and the nitration steps reveal interesting substituent effects that are not readily understandable from a qualitative basis. Below, we report additional insights into the origin of the observed trends using computational methods. These methods reveal unexpected complexity for simple reactions and reveal surprisingly large effects of substituents on the stability and reactivity of the cyclopropane units.Cyclopropanation
Interestingly, the introduction of two halogens in the fused cyclopropane ring is thermodynamically favourable (Scheme 6a). The fusion of two dihalocyclopropanes in a cyclohexane ring also leads to significant stabilization of the tricyclic structures (Scheme 6b). We will show below that this stabilization has an impact on reactivity. On the other hand, the relative thermodynamic stabilities of the stereoisomeric fused mono- and bis-cyclopropanes derived from 1,4-cyclohexadiene are similar to each other (Scheme 6c).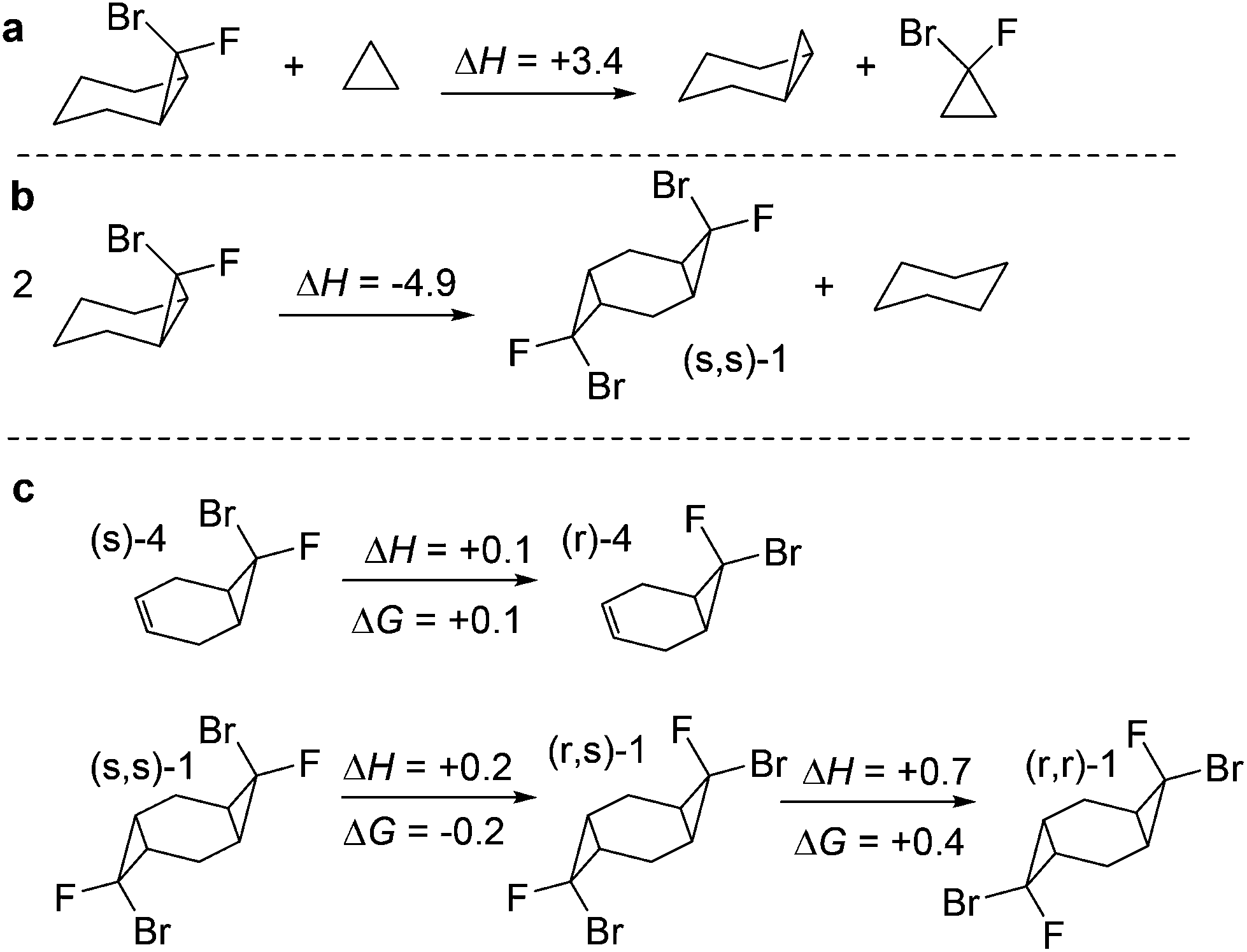 | ||
| Scheme 6 Selected isodesmic equations for evaluating the structural effects on stability of cyclic structures related to this work. (a) Effect of halogen substitution on the stability of fused cyclopropanes. (b) Stabilization of bicyclic structures with the addition of a second fused cyclopropane. (c) Relative stabilities of the stereoisomeric mono- and bis-cyclopropanes. Energies in kcal mol−1. | ||
The cyclopropanation reactions are highly (>60 kcal mol−1) exergonic and irreversible. As a consequence, the experimental selectivities are determined by reaction kinetics and the observed reactivity/selectivity trends should originate from electronic effects that are specific for the TS. The interplay of electronic effects involved in cyclopropanation of alkenes with singlet carbenes is complicated and the sequence of well-choreographed geometric changes along the internal reaction coordinate (IRC) path has been compared to a molecular dance.27 As a four-electron process, the suprafacial reaction is forbidden according to the Woodward–Hoffmann rules. Thus, the initial approach directs the empty orbital of carbene towards the target π-system, masquerading as a two-electron process.28 Only when the formation of the first bond is sufficiently advanced, the second bond starts to form in an asynchronous process with a concomitant rotation of the XCY moiety relative to its target. The IRC scan shows that the cyclopropanation step has no energy barrier in the Electronic (E) PES. The barrier only appears at the Gibbs Free energy (G) surface when the entropy effects are included. Similar “entropic” barriers have been reported earlier for exothermic bimolecular processes.29 Both (r) and (s) stereoisomers are formed through similar highly asynchronous steps. The frequency analysis of structures evolving along the cycloaddition IRC path reveals two barriers associated with structures that have imaginary frequencies. The first structure corresponds to the formation of the first C–C bond and the 2nd one corresponds to the formation of the second C–C bond and the cyclopropane ring. Geometries corresponding to these points (labelled “TS1” and “TS2”) are shown below (Scheme 7).
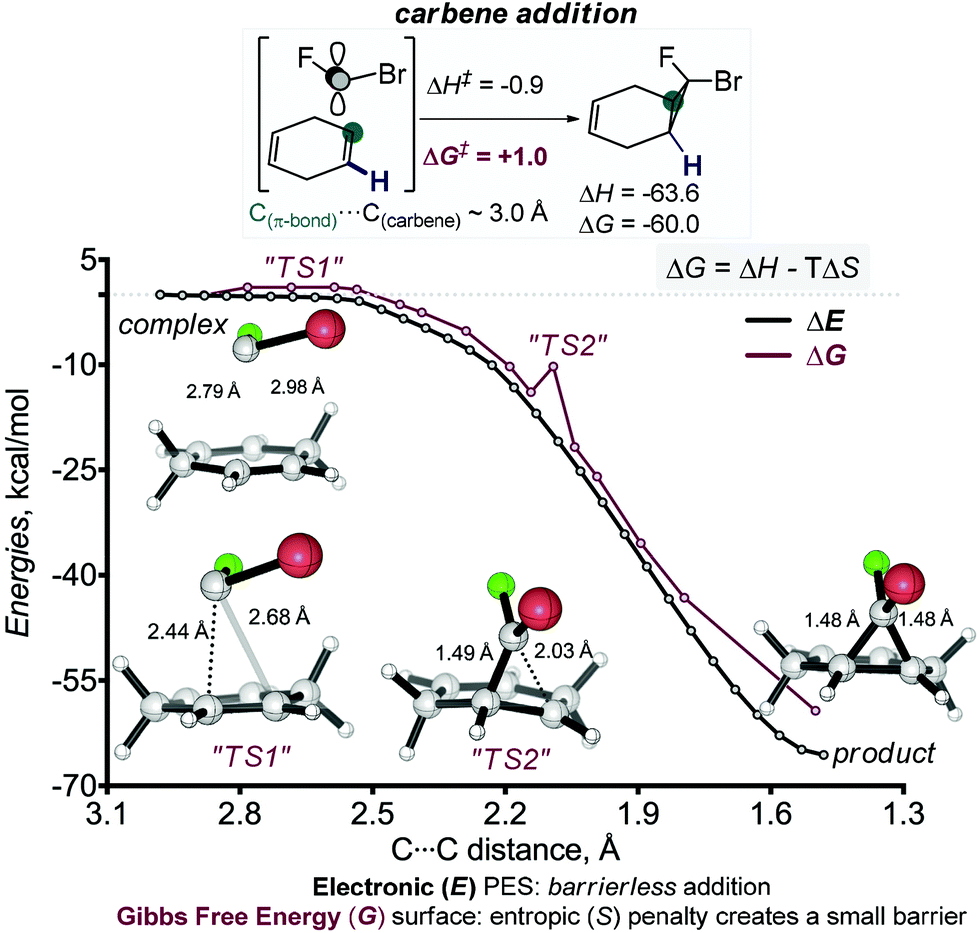 | ||
| Scheme 7 The first cyclopropanation of 1,4-cyclohexadiene is highly asynchronous. Although the process is barrierless at the potential energy surface, inclusion of entropy creates small barriers at the Gibbs Free Energy surface. Two structures are shown from the highly asynchronous bond-forming region. These structures (labelled as “TS1” and “TS2”) display imaginary frequencies corresponding to one of the C–C bonds formation. The distances at the reaction coordinate axis indicate the separation of the carbene carbon from the remote carbon of the double bond. See ESI† for the analogous scan for the 2nd regioisomer.30 | ||
The absence (or near absence) of the energy barriers complicates the application of transition state theory for the analysis of stereoselectivity. However, the inspection of structures and energies along the alternative IRC paths for the formation of the two products suggests a simple model for the observed selectivity. The path towards the experimentally preferred product is ∼1 kcal mol−1 lower at the carbene approach stage. One can trace this energy lowering to electrostatic stabilization created by the favourable Coulombic contacts between the negatively charged fluorine (qNBOF = −0.35e) and the vinyl hydrogen atoms at the attacked alkene moiety.31 This interaction favours the formation of the stereoisomeric product with the exo-F substituent (s)-4 (Scheme 8). The opposite isomer (r)-4 where the exo-Br group is positioned on the top of the vinyl hydrogens does not enjoy the same Coulombic attraction (qNBOBr = −0.05e).
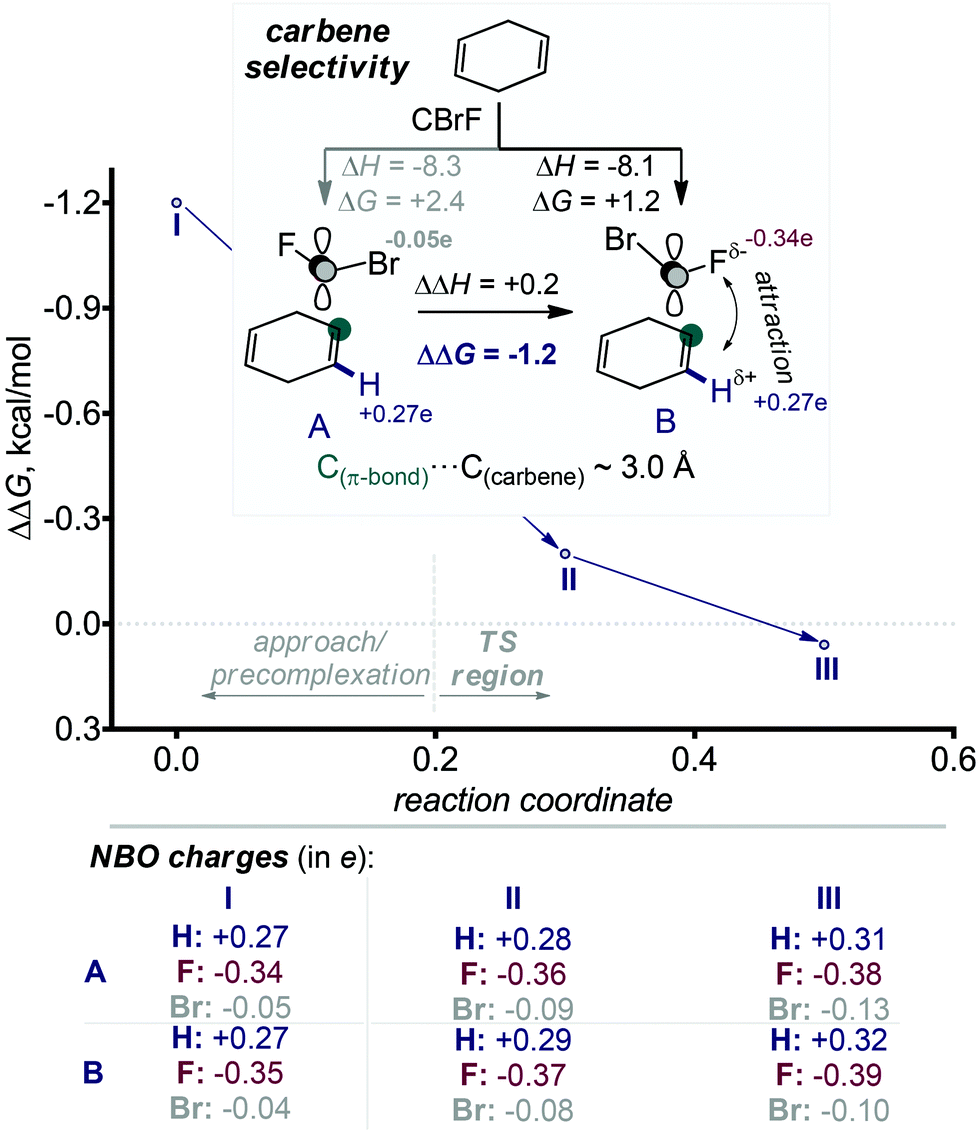 | ||
| Scheme 8 Selectivity in the cyclopropanation step stems from electrostatic interactions between halogen and H on the vinylic carbon at the early stages of the interaction, before the first C–C bond is formed. Selected NBO charges on each point show that the attractive fluorine-hydrogen interaction favours the experimentally observed path B. | ||
On the other hand, computations for the second cyclopropanation steps do reveal the presence of larger (albeit still small) activation barriers. Scheme 9 illustrates the preference for the endo-Br TS that leads to the experimentally preferred stereoisomer. The formation of pre-reaction complexes at this level of theory is strongly exothermic (ΔH = −11 kcal mol−1) but only very weakly exergonic (ΔG = −0.1 kcal mol−1). Gratifyingly, experimental selectivities agree with the difference in the energies of the stereodifferentiating cyclopropanation transition states. The computational analysis finds ∼1 kcal mol−1 preference for the experimentally observed isomer (Scheme 9) which again corresponds to the “exo” orientation of the F substituent.
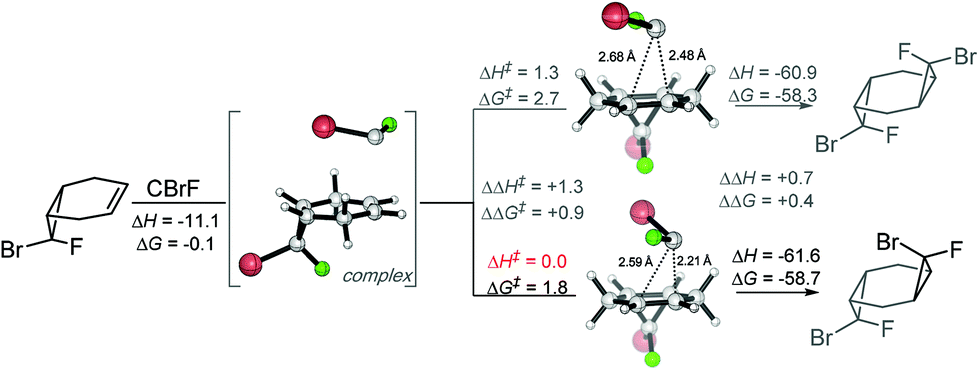 | ||
| Scheme 9 Second cyclopropanation is kinetically resolved towards the experimentally observed products. Energies in kcal mol−1. | ||
“Nitration”
These experimental results bring a number of interesting questions related to the mechanism and selectivity of interactions of functionalized fused cyclopropanes with the nitronium cation. In particular, why is compound (s,s)-1 unreactive whereas its stereoisomers undergo heterocyclization? Why do the cyclopropyl units in compounds 5 and 6 remain intact? Why does the bromofluorocyclopropyl unit react selectively in the presence of a dichlorocyclopropyl moiety? Why are tricyclic bis-cyclopropanes less reactive than their bicyclic mono-cyclopropane analogues?4The proposed mechanism of heterocyclization is described in a previous study.5 A slightly modified version of the mechanism is offered in Scheme 10 but the full discussion of its complexity along the multiple reaction steps is not the purpose of the present discussion.
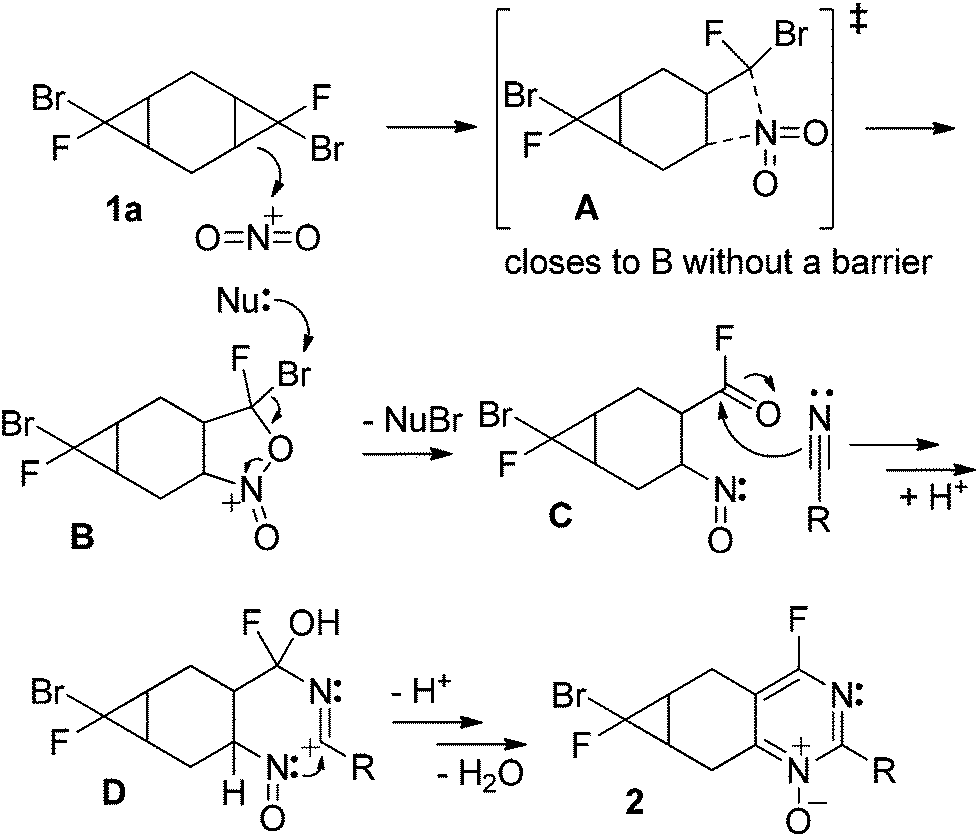 | ||
| Scheme 10 An abridged mechanistic scenario for the heterocyclization. | ||
The key premise of the following analysis is that the ease and selectivity of the cyclopropyl ring opening is determined at the very early stages of the cascade and should correlate with the donor ability of this bond towards the nitronium cation. In this work, we will start with a simple approach that uses thermodynamics of a single-electron transfer (SET) path as the guiding principle. Interestingly, this simple criterion can provide a rationale for some of the experimentally observed trends in the relative reactivity. When necessary, a more accurate analysis can be provided based on the comparison of activation barriers for the direct electrophilic attack of the nitronium cation in the strained C–C bond.
The SET energies readily explain the differences in the reactivity of bicyclic and tricyclic systems. As we discussed above, the halogenated bis-cyclopropane adducts are ∼5 kcal mol−1 stabilized relative to the monocyclopropane adducts. This decreased strain is reflected in their lower reactivity in reaction with the nitronium cation. Whereas SET from bicyclic monocyclopropanes to nitronium is ∼11 kcal mol−1 exothermic for the endo-Br monocyclopropane, the same reaction is only ∼1 kcal mol−1 exothermic for the analogous tricyclic bis-cyclopropane (s,s)-1 at the M06-2X(D3) level of theory in MeCN (Scheme 11). Interestingly, the monocyclic bis-halocyclopropanes are relatively poor donors in comparison with their bi- and tri-cyclic intermediates.
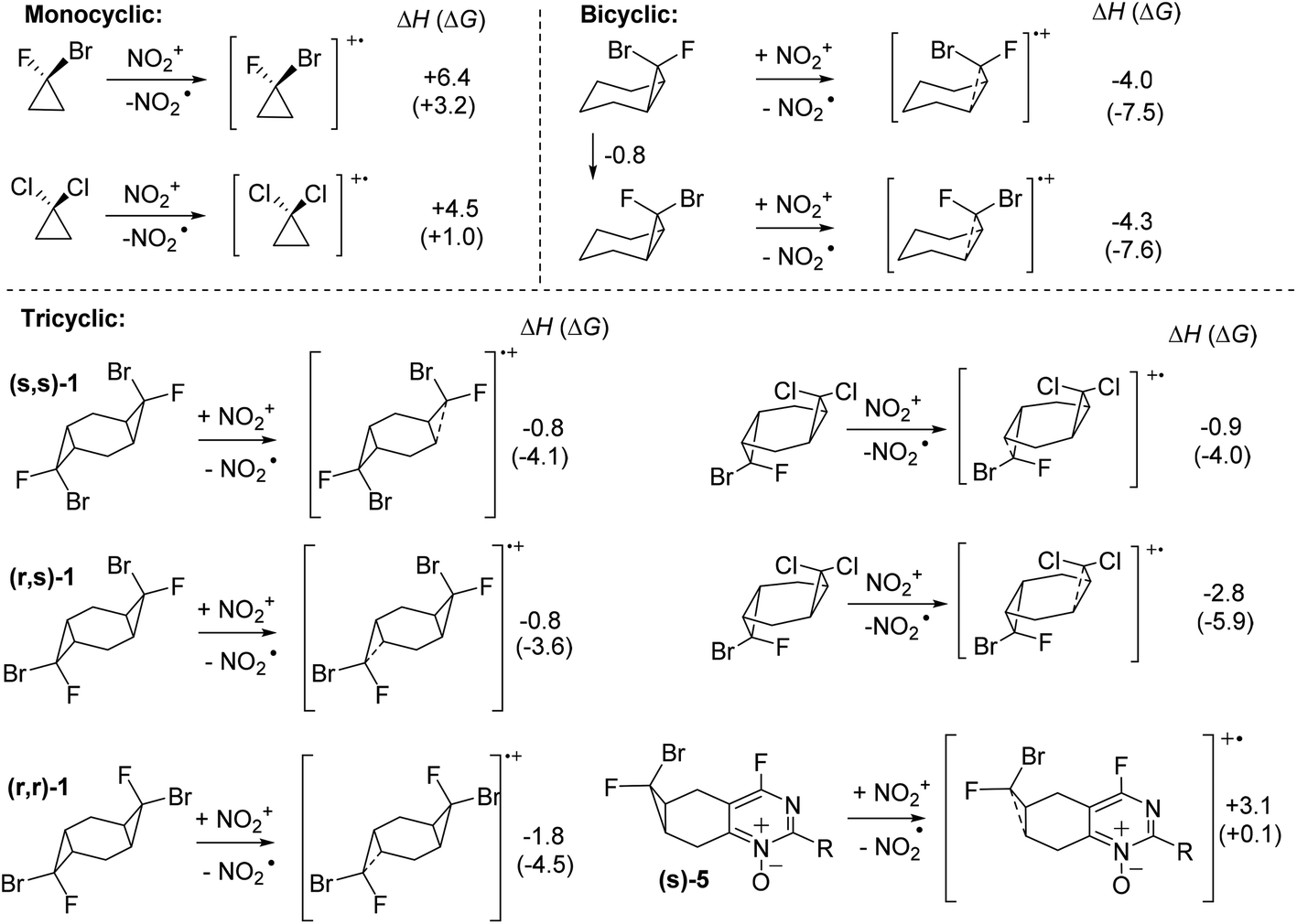 | ||
| Scheme 11 SET selectivity of mono-, bi- and tricyclic dihalocyclopropanes. Enthalpies and free energies (in parenthesis) are at the (SMD = MeCN)/(UBS)M06-2X(D3)/6-31+G(d,p), in kcal mol−1. | ||
The calculated difference in the SET favorability also provides a simple explanation to why the cyclopropyl units in compounds 5 and 6 remain intact. Annealing an acceptor heterocyclic system decreases the donor ability of the cyclopropane unit as illustrated by the endothermicity of electron transfer to nitronium from compound 5 (Scheme 11).
The relative reactivity of (s,s)-1 and (r,s)-1 is more nuanced. Although, the less reactive (s,s)-1 isomer is a ∼1 kcal mol−1 weaker donor in the reaction with nitronium, this relatively small difference cannot fully explain the relatively large difference in reactivity. Furthermore, the calculated energies of the two isomeric radical-cations produced from the non-symmetric bis-cyclopropane suggest that SET from the dichlorosubstituted ring is thermodynamically preferred to SET from the F, Br-subsituted cyclopropane. This trend is not unique to the tricyclic systems – calculations on the parent 1,2-bromo-1-fluoro and 1,1-dichlorocyclopropanes showed a similar trend. Because the relative stabilities of the two radical-cations do not agree with the experimentally observed preference for the selective reaction at the Br, F-substituted ring, we have proceeded one step further and calculated the activation barriers for the electrophilic attack of the nitronium cation in the strained cyclopropane C–C bond in these compounds.
The calculated TS geometries correspond to a non-classic cationic structure originating from the direct attack of the nitronium cation at the C–C bond center. The two C⋯N distances are similar (∼2.1–2.2 Å). Considerable bending at the NO2 moiety indicates a significant electron transfer from cyclopropane (ca. 0.17e, per NBO analysis) along with the formation of incipient C⋯N bonds (Scheme 12).
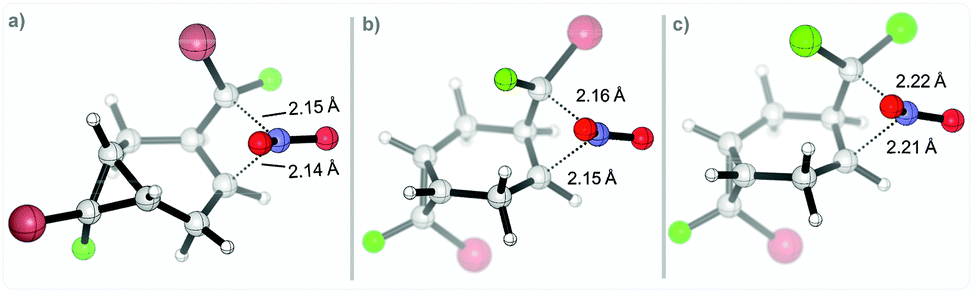 | ||
| Scheme 12 Selected TS for the nitration step on the two different positions for: (r,s)-1 in (a) and (b); 2 in (c). Geometries at the (SMD = MeCN)/(U)M06-2X(D3)/6-31+G(d,p) level of theory. | ||
Interestingly, a further IRC analysis of these NO2+ additions finds a post-TS bifurcation that can potentially lead to two different local minima, depending on which of the cyclopropane carbons makes the full C–N bond first. The formation of the C–N bond in the CH group and the C–O bond in the dihalogenated carbon provides an intermediate that leads to the observed product. The alternative, higher energy, intermediate where the dihalogenated carbon makes a C–N bond does not seem to contribute to the reaction progress (Scheme 13). This system should provide an interesting platform for the future studies of dynamic effects on reaction selectivity.32
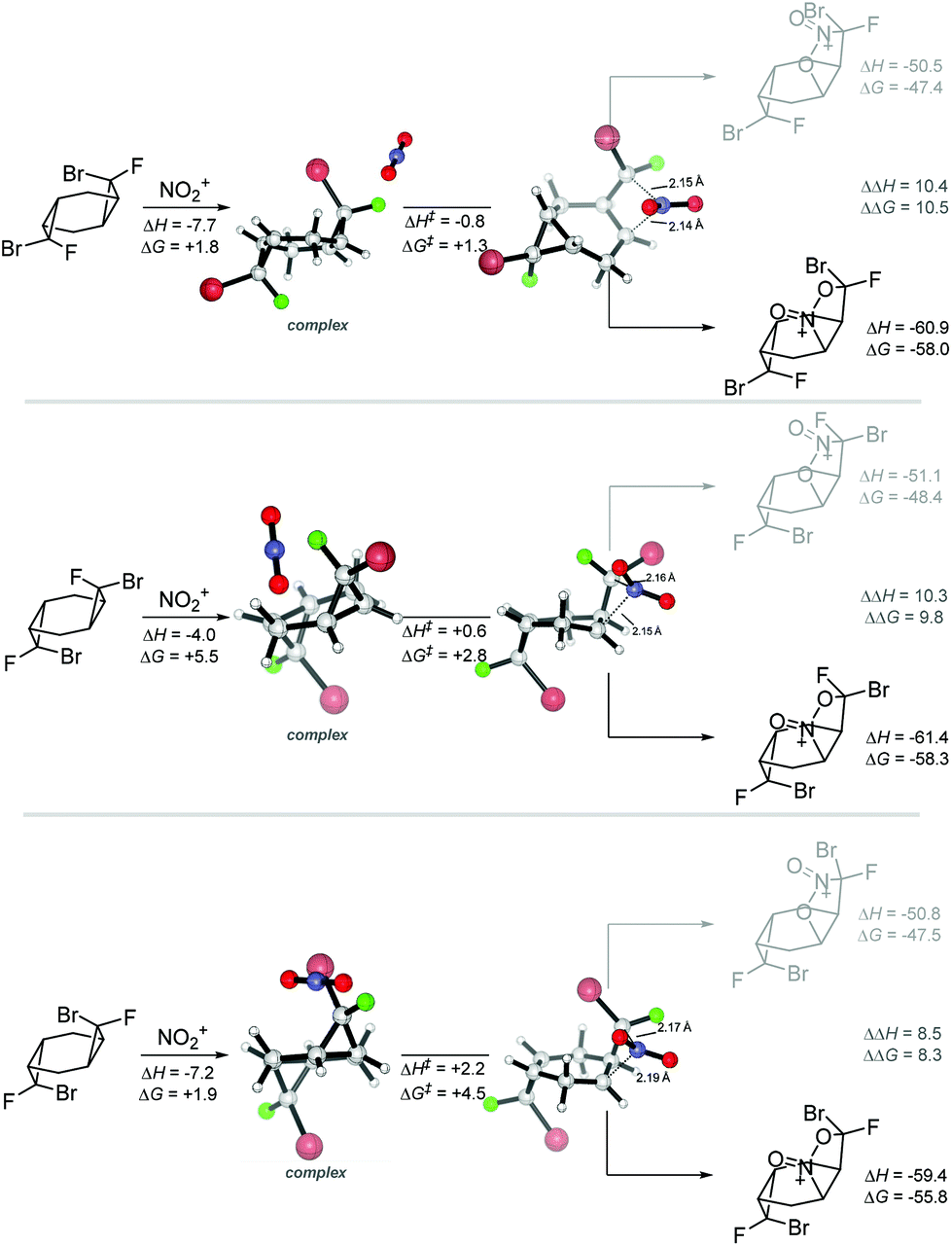 | ||
| Scheme 13 Selected examples for the bifurcating reaction path in the nitration step. The more stable products correspond to the expected intermediaries of the experimentally observed cascade reaction. Energies at the (SMD = MeCN)/(U)M06-2X(D3)/6-31+G(d,p) level of theory, in kcal mol−1. | ||
The comparison of TS energies for the reaction of compounds (r,s)-and2 show that experimentally observed selectivities agree well with the difference in the activation barriers. For example, a cyclopropane opening in (r,s)-1 has ∼5 kcal mol−1 lower barrier when the Br substituent is positioned anti to the attacking nitronium. In a similar way, the activation barrier is ∼6 kcal mol−1 higher for the attack on the dichlorocyclopropane moiety of the non-symmetric substrate 2 (Scheme 14).
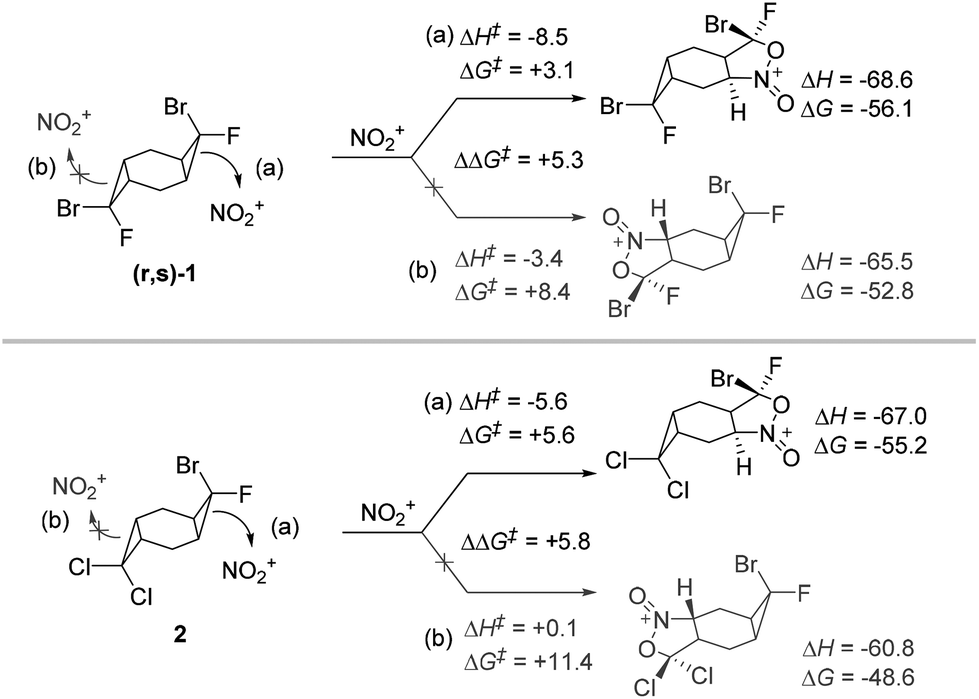 | ||
| Scheme 14 Activation barriers for cyclopropane opening in non-symmetric bis-cyclopropanes. Energies, at the (SMD = MeCN)/(U)M06-2X(D3)/6-31+G(d,p) level of theory, in kcal mol−1. | ||
It is interesting that the dichlorocyclopropane unit remains unreactive under conditions when the bromofluoro cyclopropane undergoes ring opening and heterocyclization (Scheme 14). This behavior is another illustration of the connection between molecular geometry and hybridization in cyclopropanes that leads to a strong dependence of their strain energies from the nature of exocyclic substituents. Cyclopropanes with acceptor groups are “torn” between the necessity to allocate the increased p-character to the endocyclic C–C bonds (to decrease the ring strain) and to the exocyclic C–X bonds (to satisfy the correlation between electronegativity of X and hybridization of carbon orbital in the C–X bond, known as Bent's rule33). It was shown earlier that the strain-inducing destabilizing effects of Cl and Br are similar and noticeably smaller than the effect of F.34
Conclusions
In conclusion, bis-adducts of cyclohexa-1,4-diene with bromofluorocarbene 1 and mixed adducts with the bromofluoro- and dichlorocarbenes 2 were prepared and investigated in reactions with nitrating reagents. Interesting stereoselectivity effects were discovered in cyclopropanations of 1,4-cyclohexadiene (CHD) and related bicyclic alkenes by bromofluoro carbene (CFBr). These effects were proposed to originate from electrostatic F⋯H interactions that favor the (s-) (or F-exo) isomer.Nitration selectively affords products of a one-fold heterocyclization – the remaining three-membered ring is deactivated by the formation of an adjacent electron withdrawing heterocycle. Nitration of a non-symmetric bis-cyclopropane 2 proceeded selectively to involve the bromofluorocyclopropane moiety, leaving the dichloro substituted cyclopropane ring intact. The computational analysis provided the initial insights into the possible origin of the observed selectivity trends by revealing the differences in the donor ability of cyclopropane rings with various substituents. Computed transition state structures and activation barriers for the direct electrophilic insertion of nitronium cation into the strained C–C bonds of cyclopropanes reveal large effects of the exocyclic substituents that are consistent with experimentally observed selectivities.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Russian Science Foundation (Project 17-15-01455) for financial support of this work. The research work was carried out using a diffractometer STADI VARI Pilatus-100K and a NMR spectrometer Agilent 400-MR purchased under the program of MSU development. Computational studies at FSU were supported by the National Science Foundation (CHE-1465142).Notes and references
- (a) G. W. Rewcastle, Pyrimidines and their Benzo Derivatives, in Comprehensive Heterocyclic Chemistry III, ed. A. R. Katritzky, C. A. Ramsden, E. F. V. Scriven, R. J. K. Taylor, Elsevier Ltd., Oxford, 2008, vol. 8, pp. 120–27 Search PubMed; (b) V. Sharma, N. Chitranshi and A. K. Agarwal, Int. J. Med. Chem., 2014, 202784, DOI:10.1155/2014/202784; (c) K. Singh and T. Kaur, MedChemComm, 2016, 7, 749–768 RSC.
- For selected recent examples see: (a) J.-N. Chen, X.-F. Wang, T. Li, D.-W. Wu, X.-B. Fu, G.-J. Zhang, X.-C. Shen and H.-S. Wang, Eur. J. Med. Chem., 2016, 107, 12–25 CrossRef CAS PubMed; (b) S. G. Hammer, S. Gobleder, F. Naporra, H.-J. Wittmann, S. Elz, M. R. Heinrich and A. Strasser, Bioorg. Med. Chem. Lett., 2016, 26, 292–300 CrossRef CAS PubMed; (c) K. M. Khan, S. Iqbal, M. A. Bashir, N. Ambreen, S. Perveen and W. Voelter, Tetrahedron Lett., 2015, 56, 1179–1182 CrossRef CAS; (d) S. Boyd, R. D. M. Davies, S. L. Degorce, S. Groombridge, J. S. Scott and S. Stokes, Tetrahedron Lett., 2016, 57, 152–154 CrossRef CAS.
- D. J. Brown, The Chemistry of Heterocyclic Compounds: The Pyrimidines, ed. E. C. Taylor, A. Weissberger, John Wiley & Sons, Inc., New York, 1994, vol. 52, pp. 329–437 Search PubMed.
- K. N. Sedenkova, E. B. Averina, Y. K. Grishin, A. G. Kutateladze, V. B. Rybakov, T. S. Kuznetsova and N. S. Zefirov, J. Org. Chem., 2012, 77, 9893–9899 CrossRef CAS PubMed.
- K. N. Sedenkova, E. B. Averina, Y. K. Grishin, A. B. Bacunov, S. I. Troyanov, I. V. Morozov, E. B. Deeva, A. V. Merkulova, T. S. Kuznetsova and N. S. Zefirov, Tetrahedron Lett., 2015, 56, 4927–4930 CrossRef CAS.
- K. N. Sedenkova, E. B. Averina, Y. K. Grishin, T. S. Kuznetsova and N. S. Zefirov, Tetrahedron Lett., 2014, 55, 483–485 CrossRef CAS.
- K. N. Sedenkova, E. V. Dueva, E. B. Averina, Y. K. Grishin, D. I. Osolodkin, L. I. Kozlovskaya, V. A. Palyulin, E. N. Savelyev, B. S. Orlinson, I. A. Novakov, G. M. Butov, T. S. Kuznetsova, G. G. Karganova and N. S. N. Zefirov, Org. Biomol. Chem., 2015, 13, 3406–3415 CAS.
- D. S. Karlov, M. I. Lavrov, V. A. Palyulin and N. S. Zefirov, Russ. Chem. Bull., 2016, 65, 58 Search PubMed.
- M. I. Lavrov, V. V. Grigor'ev, S. O. Bachurin, V. A. Palyulin and N. S. Zefirov, Dokl. Biochem. Biophys., 2015, 464, 322 CrossRef CAS PubMed.
- C. Mueller, F. Stier and P. Weyerstahl, Chem. Ber., 1977, 110, 124–137 CrossRef CAS.
- The commonly used “endo/exo” nomenclature is not strictly applicable to such structures according to the IUPAC guidelines (https://goldbook.iupac.org/html/E/E02094.html) that limit it to the bridged molecules of type bicyclo[x.y.z]alkane (x ≥ y > z > 0) where each of bridges has at least one atom. For this reason, we use the IUPAC nomenclature for pseudoasymmetric centers. If the IUPAC nomenclature were expanded to the [4.1.0] systems reported in this work, “r” would correspond to “endo-F” and “s” would correspond to the “endo-Br” isomer.
- W. Kuhn, H. Marschall and P. Weyerstahl, Chem. Ber., 1977, 110, 1564–1575 CrossRef CAS.
- R. L. Danheiser, J. M. Morin and E. J. Salaski, J. Am. Chem. Soc., 1985, 107, 8066–8073 CrossRef CAS.
- W. R. Dolbier Jr., M. Odaniec, E. Gomulka, M. Jaskolski and H. Koroniak, Tetrahedron, 1984, 40, 3945–3948 CrossRef.
- W. R. Dolbier Jr. and M. A. Battiste, Chem. Rev., 2003, 103, 1071–1098 CrossRef PubMed.
- CCDC 984432† contains the supplementary crystallographic data for this paper.
- For structurally similar cyclic systems, see: (a) W. D. Kumler, R. Boikess, P. Bruck and S. Winstein, J. Am. Chem. Soc., 1964, 86, 3126–3130 CrossRef CAS; (b) L. Xu, H. Brinker and W. B. Smith, Magn. Reson. Chem., 1992, 30, 1196–1202 CrossRef CAS.
- The M06-2X functional has demonstrated good thermodynamic data for organic reactions. For more details, see: (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS; (b) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- K. Fukui, Acc. Chem. Res., 1982, 14, 363 CrossRef.
- NBO analysis transforms the canonical delocalized molecular orbitals from DFT calculations into localized orbitals that are closely tied to the chemical bonding concepts. Each of the localized NBO sets is complete and orthonormal. The filled NBOs describe the hypothetical, strictly localized Lewis structure. The interactions between filled and antibonding orbitals represent the deviation from the Lewis structure and can be used to measure delocalization. For example, delocalizing interactions can be treated via the 2nd-order perturbation energy approach as
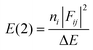 , where ni is the population of a donor orbitals, Fij is the Fock matrix element for the interacting orbitals i and j, and ΔE is the energy gap between these orbitals.
, where ni is the population of a donor orbitals, Fij is the Fock matrix element for the interacting orbitals i and j, and ΔE is the energy gap between these orbitals. - M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, M. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian ‘09 Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) F. Weinhold, C. R. Landis and E. D. Glendening, Int. Rev. Phys. Chem., 2016, 35, 1 CrossRef; A. E. Reed and F. J. Weinhold, Chem. Phys., 1985, 83, 1736 Search PubMed; (b) A. E. Reed and F. Weinhold, Isr. J. Chem., 1991, 31, 277 CrossRef CAS; (c) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS; (d) F. Weinhold, Schleyer P.v.R. Encyclopedia of Computational Chemistry, Wiley, New-York, 1998, pp. 3, 1792 Search PubMed.
- CYLview, 1.0b; Legault, C. Y., Université de Sherbrooke, 2009 (http://www.cylview.org).
- ChemCraft 1.8, http://www.chemcraftprog.com, accessed in February 2016.
- I. V. Alabugin, General Stereoelectronic Trends – Donors, Acceptors, and Chameleons, in Stereoelectronic Effects: A Bridge Between Structure and Reactivity, John Wiley & Sons, Ltd, Chichester, UK, 2016. DOI:10.1002/9781118906378.ch5.
- (a) L. Xu, C. E. Doubleday and K. N. Houk, J. Am. Chem. Soc., 2011, 133, 17848 CrossRef CAS PubMed; (b) E. Kraka and D. Cremer, Acc. Chem. Res., 2010, 43, 591 CrossRef CAS PubMed.
- See, for example: O. M. Gonzalez-James, E. E. Kwan and D. A. Singleton, J. Am. Chem. Soc., 2012, 134, 1914 CrossRef CAS PubMed.
- We use this approach only as a guide for our understanding of the observed selectivity. A more thorough investigation of this step would require dynamics that do not restrain any modes of freedom for this system.
- (a) B. Gold, N. E. Shevchenko, N. Bonus, G. B. Dudley and I. V. Alabugin, J. Org. Chem., 2012, 77, 75 CrossRef CAS PubMed; (b) B. Gold, G. B. Dudley and I. V. Alabugin, J. Am. Chem. Soc., 2013, 135, 1558 CrossRef CAS PubMed; (c) R. Maji and S. E. Wheeler, J. Am. Chem. Soc., 2017, 139, 12441 CrossRef CAS PubMed; (d) C. R. Pitts, M. A. Siegler and T. Lectka, J. Org. Chem., 2017, 82, 3996 CrossRef CAS PubMed.
- (a) B. K. Carpenter, J. Am. Chem. Soc., 1985, 107, 5730 CrossRef CAS; (b) S. L. Debbert, B. K. Carpenter, D. A. Hrovat and W. T. Borden, J. Am. Chem. Soc., 2002, 124, 7896 CrossRef CAS PubMed; (c) B. K. Carpenter, J. N. Harvey and A. J. Orr-Ewing, J. Am. Chem. Soc., 2016, 138, 4695 CrossRef CAS PubMed; (d) C. Doubleday Jr., K. Bolton and W. L. Hase, J. Am. Chem. Soc., 1997, 119, 5251 CrossRef; (e) C. Doubleday, C. P. Suhrada and K. N. Houk, J. Am. Chem. Soc., 2006, 128, 90 CrossRef CAS PubMed; (f) Z. Chen, Y. Nieves-Quinones, J. R. Waas and D. A. Singleton, J. Am. Chem. Soc., 2014, 136, 1312 CrossRef PubMed; (g) Y. Nieves-Quinones and D. A. Singleton, J. Am. Chem. Soc., 2016, 138, 15167 CrossRef CAS PubMed; (h) D. Samanta, A. Rana and M. Schmittel, J. Org. Chem., 2014, 79, 8435 CrossRef CAS PubMed; (i) Y. J. Hong and D. J. Tantillo, J. Am. Chem. Soc., 2014, 136, 2450 CrossRef CAS PubMed; (j) B. Biswas and D. A. Singleton, J. Am. Chem. Soc., 2015, 137, 14244 CrossRef CAS PubMed.
- I. V. Alabugin, S. Bresch and M. Manoharan, J. Phys. Chem. A, 2014, 118, 3663 CrossRef CAS PubMed.
- I. V. Alabugin, S. Bresch and G. P. Gomes, J. Phys. Org. Chem., 2015, 28, 147 CrossRef CAS.
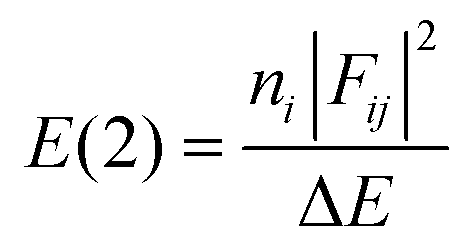
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data for all the obtained compounds, and X-ray data for compound (s,s)-1. CCDC 984432. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ob02463k |
| This journal is © The Royal Society of Chemistry 2017 |