
Advancements on the molecular design of nanoantibiotics: current level of development and future challenges
Roxana
Jijie
a,
Alexandre
Barras
 a,
Florina
Teodorescu
b,
Rabah
Boukherroub
*a and
Sabine
Szunerits
*a
a,
Florina
Teodorescu
b,
Rabah
Boukherroub
*a and
Sabine
Szunerits
*a
aUniv. Lille, CNRS, Centrale Lille, ISEN, Univ. Valenciennes, UMR 8520, IEMN, F-59000 Lille, France. E-mail: sabine.szunerits@univ-lille1.fr
bInstitute of Physical Chemistry “Ilie Murgulescu”, Romanian Academy, 060021 Bucharest, Romania
First published on 10th July 2017
Abstract
Numerous antimicrobial drugs have been developed and commercialized to kill and inhibit the growth of pathogenic microbes. The therapeutic efficiency of these drugs has however become often inadequate for the treatment of microbial infections, the reasons being multiple. Oral administration results often in only partial absorption and up to 50% of the active compound can be excreted in the urine. Furthermore, antibiotic therapy is also frequently accompanied by gastrointestinal complications, including vomiting, nausea and diarrhea. From a therapeutic point of view, the low solubility of antibiotics in lipid membranes presents a limitation for rapid and efficient treatment of infections. The preferred route for the delivery of antimicrobial drugs is the oral one; oral formulations capable of extending the duration of drug delivery and minimizing side effects have received much attention. The combination of antimicrobial agents with nanomaterials has been seen as a particularly promising strategy to overcome these limitations. Particulate formulations have proven their efficiency in achieving better pharmacokinetic profiles and improving the bio availability of several antibiotics. Antibiotic modified particles have shown their capability to protect some of the antimicrobial drugs from stomach acid and first-pass metabolisms in the gastrointestinal tract. Likewise, particle formulations can increase the circulation times of a drug and the local concentration gradient across absorptive cells. The present review describes the current status of different types of antibiotic loaded nano-systems. Key principles such as antibiotic loading, the release mechanism and the mode of action involved in pathogen killing or inhibition will be discussed in more detail. It is hoped that the general knowledge obtained here will help in the generation of advanced nanomaterials loaded with antimicrobials agents.
Design, System, ApplicationThe development of antibiotic formulations capable of extending the duration of their delivery and minimizing side effects has received much attention. Integration of antibiotics with nanostructures is a particularly promising strategy as the newly formed nanoantibiotics, also known as nanobiotics, have proven more efficient against the eradication of pathogenic bacteria and associated infectious diseases. This review outlines the recent advances made in the design and use of different types of antibiotic loaded nano-systems and the strategies employed for optimizing the design of nanobiotics. How to translate this exciting technology into clinical applications for the treatment of infectious diseases is still one of the main obstacles; any advances in the understanding of the mode of action of nanobiotics and their toxicity will help in fine tuning their molecular design. This review might help in getting a better global understanding of the current nanobiotic design strategies employed. |
1. Introduction
Infectious microorganisms spread through the body via the circulatory system. They are normally removed from the blood stream by macrophages, which are present in all major organs such as the liver, spleen, bone marrow, etc. However, many microorganisms are able to escape the macrophage digestion process, making intracellular infections difficult to eradicate. Treatment with antimicrobial agents, in most cases antibiotics, is currently the only viable strategy to kill and inhibit further growth of pathogenic microorganisms. Typically, antimicrobials kill bacteria by binding to some vital compounds of bacterial metabolism or impeding normal cellular activities. For instance, β-lactams such as penicillin and cephalosporin inhibit bacterial cell wall synthesis; tetracyclines, macrolides, clindalycin, metronidazole and quinolones inhibit protein synthesis; sulphonamides and trimethoprim have an inhibitory effect on enzyme synthesis. Despite the great progress made in antimicrobial development, many infectious diseases remain difficult to treat. One major reason is that many antimicrobials are difficult to transport though cell membranes and have low activity inside the cells. Toxicity to healthy tissues and solubility issues are additional limitations for their use in large quantities. Another current issue when using antibiotics stems from acquired resistance from infectious microbes towards these drugs. To address these issues, alternative antimicrobial drug systems have been proposed, one being antibiotic loaded nanoparticles. Such “nanoantibiotics” allow altering and improving the pharmacokinetics and therapeutic levels of antimicrobial agents in contrast to the free counterparts. Many advantages of nanoparticle based drug delivery have been recognized, including improved solubility of drugs, prolonged circulation, release of drugs in a controlled and sustained manner, targeted delivery to tissues and cells of interest, as well as concurring delivery of multiple agents for combined therapy. This is also applicable to antimicrobial nanosystems. Moreover, antimicrobial loaded nanoparticles can enter host cells through endocytosis and then release the antibiotic load to treat intracellular infections. As a result, a number (<40) of nanoparticle-based systems are approved for clinical trials and are under different stages of clinical tests. Most of the clinically approved intravenous nanoparticle therapies are targeted for the treatment of various cancer types, including solid malignancies, ovarian, breast, leukemia, prostate, liver, lymphomas, bladder, pancreatic, etc. When it comes to the treatment of bacterial infections using nanoantibiotics, the only clinically approved system is AmBisome by Gildeas Science for the treatment of serious fungal infections and leishmaniasis (Table 1).1 CAL02, with sphingomyelin and cholesterol loaded on liposomes for toxin neutralization, is under Ph I clinical trial against pneumonia infections. No antibiotic encapsulated nanosystem is under trial currently.| Particles | Active agent | Name | Application |
|---|---|---|---|
| Liposomes | Sphingomyelin and cholesterol | CAL02 (Combioxin SA) | Ph I, pneumonia infections |
| Liposomes | Amphotericin B | AmBisome (Gildeas Science) | Clinically approved treatment of invasive fungal infections |
While numerous studies for tumor-enhanced drug delivery have been performed, the corresponding research on the treatment of bacterial infections has just really started. One sufficient challenge in nanomedicine translation is the contraction between prolonged circulation time achieved by the integration of PEG and other hydrophilic polymers, and targeted delivery to pathogens mostly requiring nanoparticles with a hydrophobic and positively charged surface. The negatively charged walls of many types of bacteria interact with the positively charged nanostructures through electrostatic interactions and results in permeability changes or even the complete destruction of the cell wall and thus the pathogen itself.
This review focuses on more recent developments of antibiotic-loaded nanostructures including liposomes, polymeric nanoparticles and dendrimers, metallic and magnetic nanoparticles, carbon-based nanostructures and mesoporous silica nanostructures (Fig. 1). Since liposomes were proposed as nanostructures for antibiotic delivery about forty years ago, the different nanotechnologies employed for increasing the bioavailability of antibiotics, thus allowing targeted antibiotic therapy, have been reviewed in several reports.2–10 The increase in public awareness of the emergence of antibiotic resistance and unsolved problems in treating bacterial infections makes a more timely review of importance. This feature article focuses in addition not only on the road map of antimicrobial lipid and polymer nanoparticles, which have been greatly covered by two recent review articles,4,9 but presents our vision of the progress in the use of metallic, magnetic and carbon-based nanostructures such as C-dots and graphene for antibiotic delivery and bacterial treatment. The characteristic features of different carbon-based nanostructures such as structural, optical, thermal and mechanical features, make them promising nanostructures for future antibacterial applications.
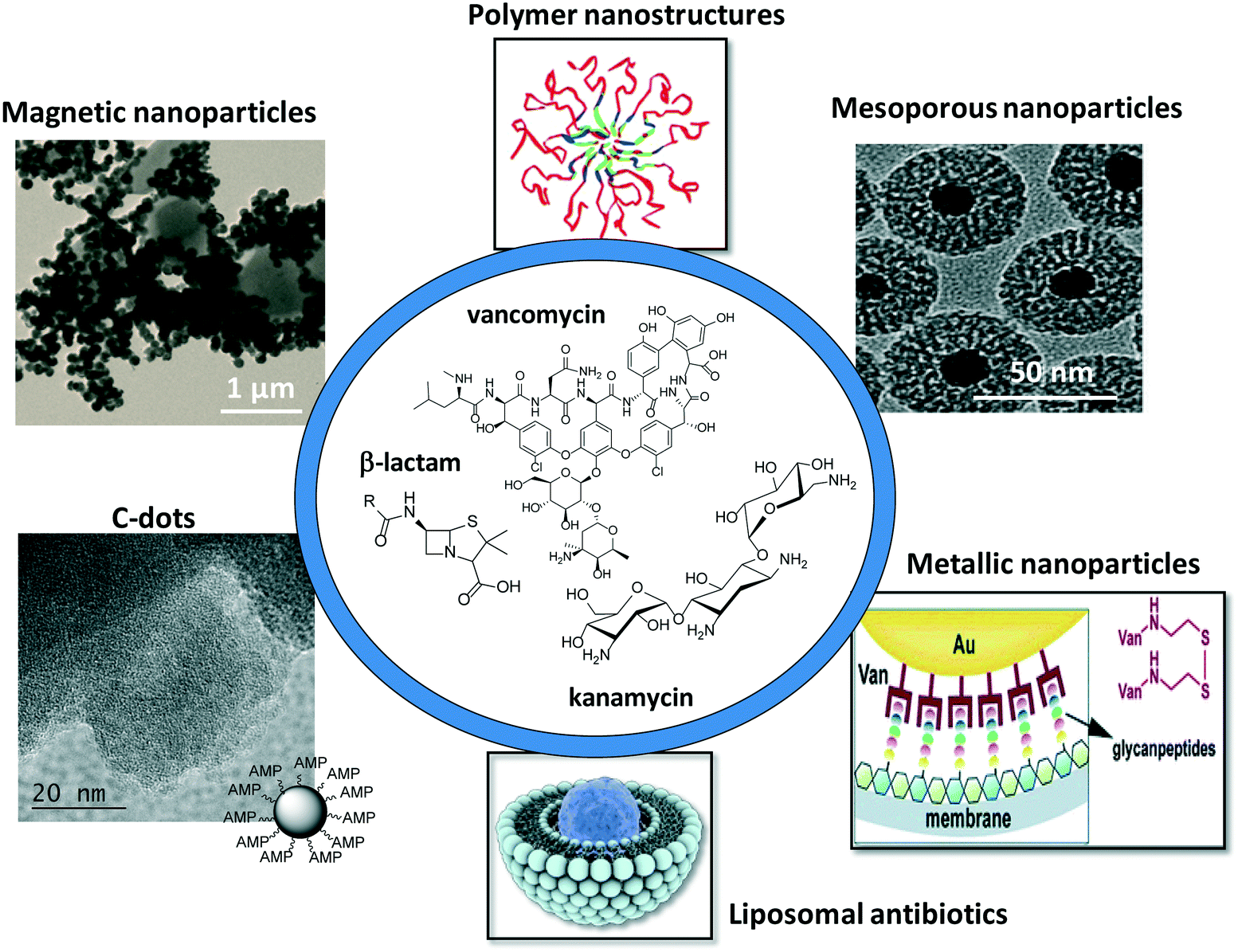 | ||
| Fig. 1 Schematic illustration of the different nanoparticle platforms for antimicrobial drug loading and release. | ||
2. Antibiotics: one of many antimicrobial agents
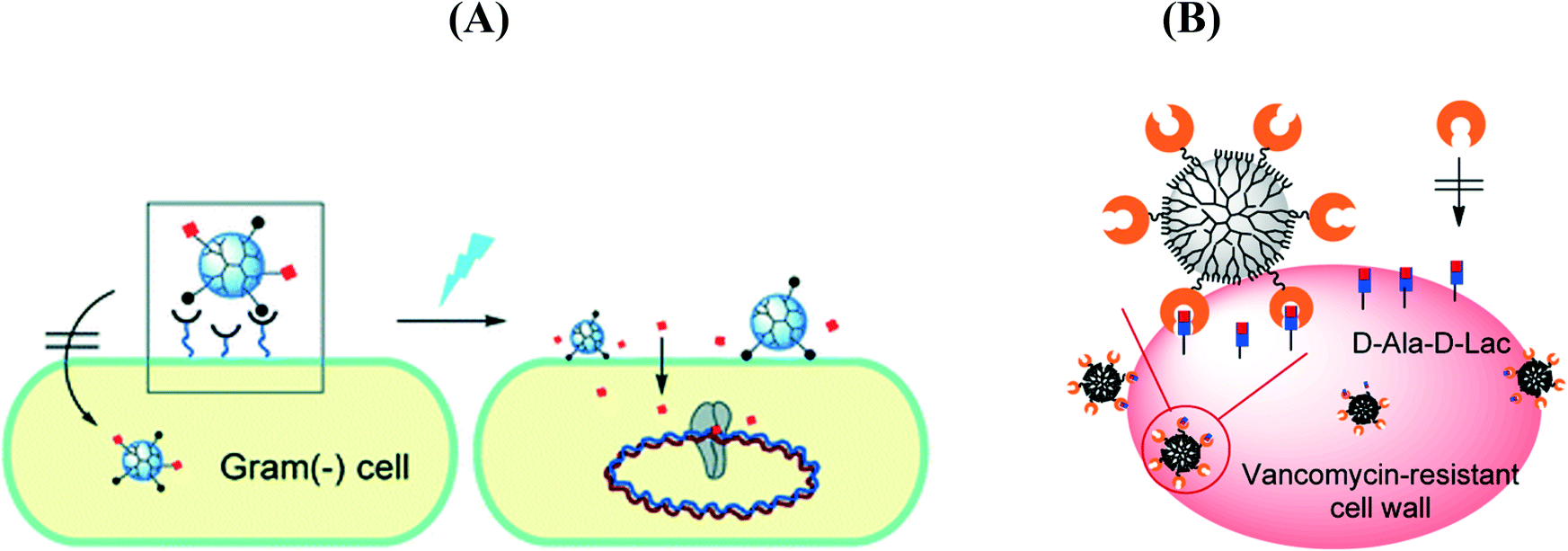
Antibiotics, a class of antimicrobial agents, used in the treatment and prevention of bacterial infections, have been considered as one of the wonder discoveries of the 20th century. Although there are well over 100 antibiotics on the market, the majority come from only a few types of drugs (Table 2). Their mode of action (Fig. 2A) differs due to the nature of their structure and degree of affinity to certain target sites within bacterial cells.11 Penicillin, cephalosporin, bacitracin and vancomycin are four antibiotics targeting the cell wall of bacteria by inhibiting cell wall synthesis. Cationic peptides such as colistin, on the other hand, are inhibitors of cell membrane functions. Membranes are important barriers that segregate and regulate the intra- and extracellular flow of substances. A disruption or damage to this structure results in leakage of important solutes essential for the cell's survival. Because this structure is found in both eukaryotic and prokaryotic cells, the action of this class of antibiotics is often poorly selective and can often be toxic for systemic use in the mammalian host. Most clinical usage is therefore limited to topical applications. Aminoglycosides, macrolides, and tetracyclines are inhibitors of protein synthesis, an essential process necessary for the multiplication and survival of all bacterial cells. These antibacterial agents target bacterial protein synthesis by binding to either the 30S or 50S subunits of the intracellular ribosomes. This activity then results in the disruption of the normal cellular metabolism of the bacteria, and consequently leads to the death of the organism or the inhibition of its growth and multiplication. Another mode of action is based on the inhibition of nucleic acid synthesis with quinolones, metronidazole and rifampin as examples. Other antibiotics act on selected cellular processes essential for the survival of the bacterial pathogens: sulfonamides and trimethoprim disrupt the folic acid pathway, which is a necessary step for bacteria to produce precursors important for DNA synthesis. Sulfonamides target and bind further to dihydropteroate synthase, while trimethophrim inhibit dihydrofolate reductase; both enzymes are essential for the production of folic acid, a vitamin synthesized by bacteria.| Antibiotic class | Examples | Target/mode of action |
|---|---|---|
| β-Lactams | Penicillin (ampicillin), cephalosporins (cephamycin), penemes (meropenem), monobactams (aztreonam) | Inhibiting cell wall synthesis |
| Aminoglycosides | Gentamicin, streptomycin, spectinomycin, and tobramycin | Inhibitors of protein synthesis |
| Glycopeptides | Vancomycin, teicoplanin | Inhibiting cell wall synthesis |
| Macrolides | Erythromycin, clarithromycin, azithromycin | Inhibitors of protein synthesis |
| Fluoroquinolones | Ciprofloxacin, levofloxacin, ofloxacin | Inhibition of nucleic acid synthesis |
| Sulfonamides | Co-trimoxazole and trimethoprim, sulfamethoxazole | Act on selected cellular processes and metabolism |
| Tetracyclines | Tetracycline and doxycycline | Inhibitors of protein synthesis |
| Cationic peptides | Colistin | Cell membrane functions |
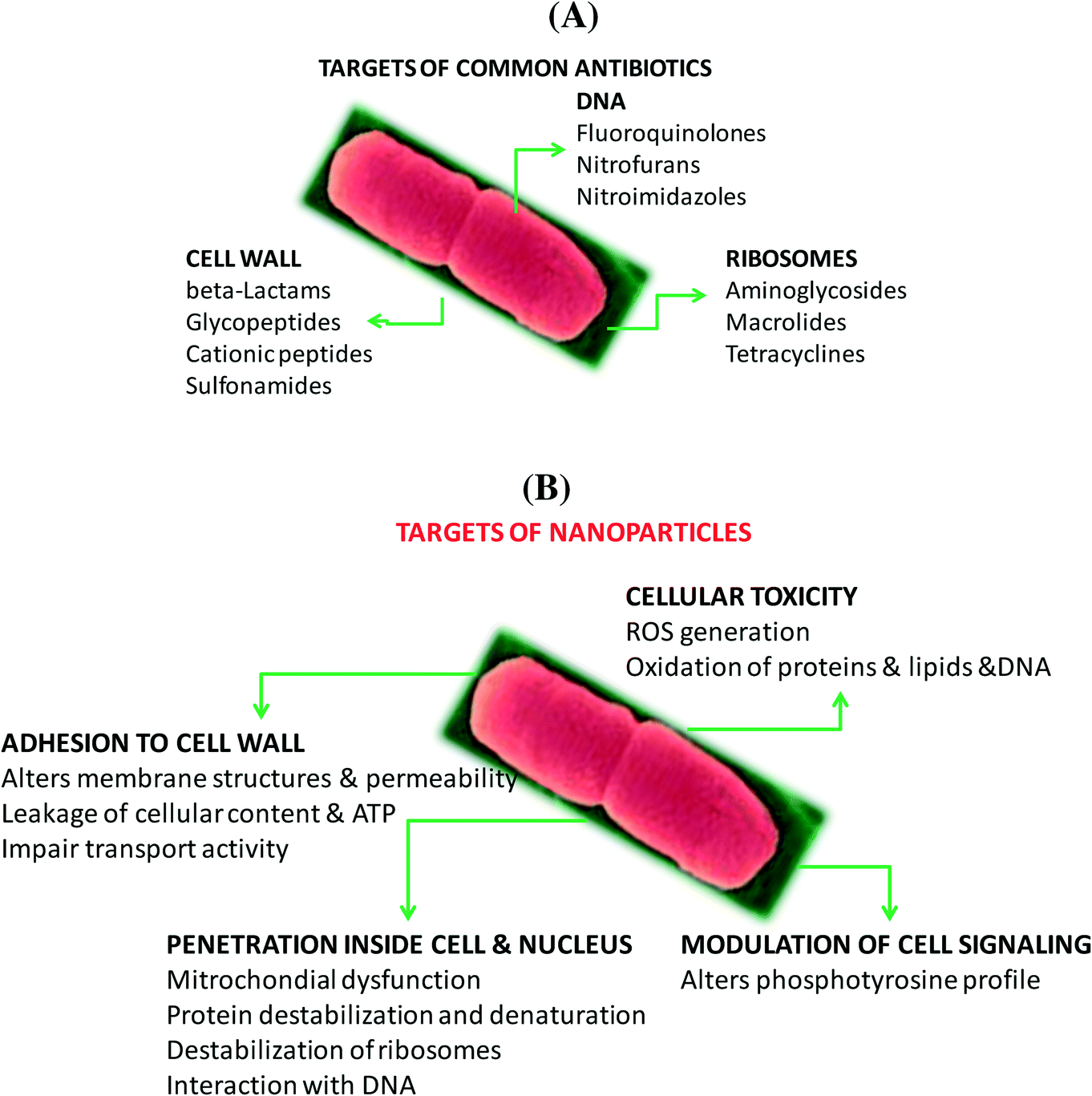 | ||
| Fig. 2 Major targets of common antimicrobial agents (A) and nanoparticles (B). | ||
Regrettably, the over and misuse of most of these drugs have been accompanied by the rapid appearance of resistant strains. A wide range of biochemical and physiological mechanisms may be responsible for resistance and are well described in some review articles; the reader is referred to them.12,13 Resistant strains of Gram-positive bacteria (such as methicillin-resistant Staphylococcus aureus, MRSA) pose a continuing threat. A range of antibiotics available to combat them remains comparatively robust. It is the emergence of resistant Gram-negative strains that currently presents the greater threat, in particular Pseudomonas aeruginosa, Enterobacter cloacae and E. coli. These bacteria can be deadly, causing infections from urinary tract infections (UTI) to life threatening pneumonias and bloodstream infections. As it is harder for antibiotics to penetrate Gram-negative bacterial cell walls, these pathogens are particularly difficult to eradicate and a very few antibiotics can currently combat them. This situation might be different when using nanostructures.
In the case of nanostructures, a wide range of pathogen inactivation mechanisms is possible depending greatly on the composition of the nanoparticles, their size, as well as surface charge and surface function (Fig. 2B). Next to cellular dysfunctions due to the interaction of the particles with the microbial cell membrane, changing permeability and transport activity, particles may also penetrate inside the cells and affect vital cellular functioning.14 Electrostatic interactions of metallic nanoparticles with sulfur-containing proteins, present in the cell wall, can cause irreversible changes in the cell wall structure, resulting in its disruption which in turn affects the integrity of the lipid bilayer and permeability of the cell membrane. An increase in membrane permeability affects a cell's ability to properly regulate transport activity through the plasma membrane and results in the leakage of cellular contents, including ions, proteins, reducing sugars and sometimes the cellular energy reservoir, ATP. The thickness and composition of the cell wall of the microorganisms play an important role in the way nanoparticles interact (Fig. 2C). Gram-negative bacteria, such as E. coli, are more susceptible to cell membrane damage than Gram-positive bacteria, such as S. aureus due to the difference in the organization of a key component of the cell membrane, the peptidoglycans. In Gram-positive bacteria, the cell wall is composed of a negatively charged peptidoglycan layer of about 30 nm in thickness, while Gram-negative bacteria exhibit peptidoglycan layers of ∼3–4 nm in thickness. Gram-negative bacteria contain on the other hand lipopolysaccharides (LPS) in the cell membrane, which contribute to the structural integrity of the membrane as well as membrane protection from chemical attacks. The negative charge of LPS together with the thinner peptidoglycan layer promotes adhesion of Ag NPs and makes bacteria more susceptible to antimicrobial therapy. Other modes of action of particles are based on blocking the sugar metabolism or increasing the cellular oxidative stress in the microbes. Indeed, oxidative stress is an indication of toxic effects caused by heavy metal ions, such as Ag+. Increased concentrations of metal ions are expected to cause an increase in cellular oxidative stress with the ability to produce reactive oxygen species (ROS) such as hydrogen peroxide (H2O2), the superoxide anion (O2−), hydroxyl radical (OH˙), and singlet oxygen (1O2). Reactive oxygen species can also be generated intracellularly during mitochondrial oxidative phosphorylation. As the cycle of phosphorylation and dephosphorylation is essential for microbial growth and cellular activity, examining the phosphotyrosine profile of bacterial proteins from both Gram-positive and Gram-negative bacteria offers a useful way to study the effect of particles on their mode of action.
While a variety of nanostructures displaying intrinsic antibacterial properties have been identified (Ag NPs, Cu NPs, ZnO NPs, etc.) using one or several of the targeting mechanisms outlined in Fig. 2B, the focus of this review is directed only on the effect of antibiotic loaded nanostructures. While the development of bacterial resistance is common to all antibiotics, some previous studies showed that the activity of aminoglycosidic antibiotics (streptomycin, gentamycin, neomycin) is protected when immobilized on gold nanoparticles.15 One reason for this result might be due to the multivalent presentation of antibiotic ligands on the nanoparticles. As demonstrated for carbohydrate modified nanoparticles,16–21 a single particle with a large surface to volume ratio is ready for the attachment of multiple ligands, providing an easy and powerful possibility for enhancing the affinity and/or increasing locally the concentration of the ligand.
The formulation of different antibiotic modified nanostructures will thus be the focus of the rest of the review.
3. Different nanoantibiotic formulations
3.1. Liposomes
Liposomes are spherical vesicles made of one or more phospholipid bilayers with a diameter varying between 20 nm and 10 μm depending on the type of phospholipid used.22 Hydrophobic substances can be loaded inside the lipid bilayer, while hydrophilic compounds are entrapped in the aqueous core. For the entrapment of hydrophilic elements, large unilamellar vesicles (LUVs) are mainly used as the volume of encapsulated water is high. Multilamellar vesicles (MLVs) and in particular small unilamellar vesicles (SUVs) of ≈100 nm are more adapted for the integration of hydrophobic compounds such as antibiotics. The basic steps involved in the preparation of antibiotic loaded liposomes consist of drying down the lipids using an organic solvent, dispersing the lipid in aqueous media, down-sizing the hydrated suspension by sonication or extrusion followed by purification.23Liposomes have become universal drug carrier systems due to the easy manner of manipulating their physico-chemical properties, enabling the design of nanocarriers with the desired pharmacokinetic properties.7 Numerous studies have focused on the entrapment of antibiotics in liposomes with the aim to enhance their antibacterial activity and to reduce toxic side effects during antibiotic therapy. The advantages of liposomal antibiotics include an improved plasma circulation time of the antibiotic when encapsulated in polyethylene glycol (PEG)-coated (STEALTH) formulations. The PEG layer protects the vesicles and allows a long liposome circulation in the blood system.24 The possibility of adding targeting ligands to these nanocarriers allowed not only targeted treatment, but also improved the activity of antibiotics against extracellular pathogens and eradication of bacterial structures such as biofilms. In the area of antimicrobial resistance with a limited number of new antibiotics coming onto the market, improving currently available antibiotics upon the design of liposomal formulations is one way to overcome some of these resistance related issues. Promising results were for example reported for the eradication of drug-resistant Pseudomonas aeruginosa due to a direct interaction/fusion between liposomes and their bacterial cells.25,26
Most studies regarding liposomal antibiotics deal with aminoglycosides, quinolones, polypeptides, and betalactams. Indeed, the first antibiotic to be entrapped was penicillin.27 It was shown that entrapment extends indeed the rate of penicillin elimination from the plasma. Some years later, the capacity of dihydrostreptomycin (DHS) sulfate (a derivative of streptomycin, a semisynthetic aminoglycoside antibiotic) loaded liposomes formed from a mixture of egg phosphatidylcholine, cholesterol and phosphatidic acid in a 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 molar ratio to enhance the antimicrobial action towards S. aureus compared to the free drug was shown.28 Multilamellar negatively charged liposomes (lipid bilayer vesicles) composed of phosphatidylcholine, cholesterol, and phosphatidylserine (molar ratio, 6:3:1) were produced by Desiderio and Campbell to entrap an aqueous solution of cephalothin.29 This formulation was effective in the elimination of Salmonella typhimurium in infected macrophages. The possibility of entrapping more than one antibiotic was demonstrated in 1983 by Stevenson et al.,30 who showed a 10-fold increase in intracellular antibacterial activity by loading streptomycin and chloramphenicol, within neutral or anionic unilamellar liposomes composed of egg phosphatidylcholine and cholesterol. The encapsulation of piperacillin, a β-lactam, into liposomes prepared with phosphatidylcholine and cholesterol (1:1) allowed the protection of the antibiotic from enzymatic hydrolysis by staphylococcal β-lactamase, resulting in enhanced activity against Staphylococcus aureus.31 Indeed, the literature is rich and some excellent recent reviews have been devoted to this topic.4,5,7Table 3 lists some more recent examples of liposomal antibiotics. The formulation of chloramphenicol in liposomes was recently proposed as an easy manner to overcome problems associated with the toxicity of the antibiotic.32 Li et al. showed that a liposomal co-delivery of daptomycin and clarithromycin is highly effective and less toxic for the treatment of methicillin-resistant S. aureus infections in the presence of only one-thirtieth of the concentration required in liposomes containing daptomycin only (Fig. 3A).33 A liposomal complex containing the phospholipid cardiolipin co-entrapped with levofloxacin was formulated by Gaidukevich et al. to suppress the growth of Mycobacterium tuberculosis and reduce the minimum inhibitory concentration of cardiolipin (33.5 μM) and levofloxacin up to 2 μg ml−1.34 Antimicrobial activities of herbal extracts with lysozyme were evaluated against two Gram-positive (B. subtilis and M. luteus) and two Gram-negative (E. coli and S. marcescens) bacteria by Matouskova et al.35 The integration of selective antimicrobial peptides such as S-thanatin onto liposomes with encapsulated levofloxacin via a lipid PEG linker was recently reported by Fan et al.36 (Fig. 3B). The positively charged nanostructures showed a strong interaction with bacterial cells with an increased uptake of levofloxacin. Indeed, the TS–liposome–levofloxacin (TS-LPs-LEV) complex exhibited excellent activity against Klebsiella pneumoniae ATCC 700603 and restored the susceptibility of multidrug-resistant K. pneumoniae clinical isolates to levofloxacin in vitro.
:2:1 molar ratio to enhance the antimicrobial action towards S. aureus compared to the free drug was shown.28 Multilamellar negatively charged liposomes (lipid bilayer vesicles) composed of phosphatidylcholine, cholesterol, and phosphatidylserine (molar ratio, 6:3:1) were produced by Desiderio and Campbell to entrap an aqueous solution of cephalothin.29 This formulation was effective in the elimination of Salmonella typhimurium in infected macrophages. The possibility of entrapping more than one antibiotic was demonstrated in 1983 by Stevenson et al.,30 who showed a 10-fold increase in intracellular antibacterial activity by loading streptomycin and chloramphenicol, within neutral or anionic unilamellar liposomes composed of egg phosphatidylcholine and cholesterol. The encapsulation of piperacillin, a β-lactam, into liposomes prepared with phosphatidylcholine and cholesterol (1:1) allowed the protection of the antibiotic from enzymatic hydrolysis by staphylococcal β-lactamase, resulting in enhanced activity against Staphylococcus aureus.31 Indeed, the literature is rich and some excellent recent reviews have been devoted to this topic.4,5,7Table 3 lists some more recent examples of liposomal antibiotics. The formulation of chloramphenicol in liposomes was recently proposed as an easy manner to overcome problems associated with the toxicity of the antibiotic.32 Li et al. showed that a liposomal co-delivery of daptomycin and clarithromycin is highly effective and less toxic for the treatment of methicillin-resistant S. aureus infections in the presence of only one-thirtieth of the concentration required in liposomes containing daptomycin only (Fig. 3A).33 A liposomal complex containing the phospholipid cardiolipin co-entrapped with levofloxacin was formulated by Gaidukevich et al. to suppress the growth of Mycobacterium tuberculosis and reduce the minimum inhibitory concentration of cardiolipin (33.5 μM) and levofloxacin up to 2 μg ml−1.34 Antimicrobial activities of herbal extracts with lysozyme were evaluated against two Gram-positive (B. subtilis and M. luteus) and two Gram-negative (E. coli and S. marcescens) bacteria by Matouskova et al.35 The integration of selective antimicrobial peptides such as S-thanatin onto liposomes with encapsulated levofloxacin via a lipid PEG linker was recently reported by Fan et al.36 (Fig. 3B). The positively charged nanostructures showed a strong interaction with bacterial cells with an increased uptake of levofloxacin. Indeed, the TS–liposome–levofloxacin (TS-LPs-LEV) complex exhibited excellent activity against Klebsiella pneumoniae ATCC 700603 and restored the susceptibility of multidrug-resistant K. pneumoniae clinical isolates to levofloxacin in vitro.
| Active lipids | Antibiotics | Bacteria | Ref. |
|---|---|---|---|
| Chol: cholesterol; DC-Col: 3b-[N-(N′,N′-dimethylaminoethane)-carbamoyl]cholesterol; DCP: dicetyl phosphate; DDAB: didecyldimethylammonium bromide; DMPG: 1,2-dimyristoyl-sn-glycero-3-phosphorylglycerol sodium salt; DOPC: dioleoylphosphatidylcholine; DPPC: dipalmitoylphosphatidylcholine; DPPG: 1,2-dipalmitoyl-sn-glycero-3-phosphorylglycerol sodium salt; DOPA: 1,2-di-(9Z-octadecenoyl)-sn-glycero-3-phosphate; DOPE: 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine; DSPC: 1,2-distearoyl-sn-glycero-3-phosphocholine; DSPE: 1,2-distearoyl-sn-glycero-3-phosphoethanolamine; DSPG: 1,2-distearoyl-sn-glycero-3-phosphoglycerol; HSPC: hydrogenated soy phosphatidylcholine; Mal: maleimide; PC: phosphatidylcholine; PHEPC: partially hydrogenated egg phosphatidylcholine; POPC: 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine. | |||
| DC-Chol | Ciprofloxacin | P. aeruginosa | 37 and 38 |
| DC-Chol, Chol, DOPC, DOPE | Ampicillin, penicillin G | E. coli XL1 Blue strain | 39 |
| Soybean lecithin, Chol (chitosan) | Vancomycin hydrochloride | — | 40 |
| Chol, HSPC (mPEG2000–DSPE and chitosan) | Daptomycin, clarithromycin | MRSA | 33 |
| Commercial Lipoquin (ARD-3100) | Ciprofloxacin | Francisella tularensis strain Schu S4 | 41 |
| Egg PC, Chol | Gentamicin | MRSA | 42 |
| DSPC, DSPG or Egg PC, DOPA | Doxycycline | H. pylori | 43 and 44 |
| DPPC, DOPC, DPPG | Tobramycin | Burkholderia cepacia complex | 45 |
| DPPC, DDAB, DCP, Chol | Clarithromycin | P. aeruginosa, B. subtilis | 46 |
| DPPC | Polymyxin B | P. aeruginosa | 47 |
| DPPC, POPC, Chol | Polymyxin B | B. bronchiseptica, P. aeruginosa, E. coli, K. pneumoniae, A. lwoffii, A. baumannii | 48 |
| DOPC, Chol | Colistin | — | 49 |
| DSPC, DCP, DMPG | Vancomycin | MRSA | 50 and 51 |
| PHEPC, Chol PEG-DSPE | Clarithromycin | M. avium | 52 |
| DPPC, Chol, cholesteryl tetraethyleneglycol fucose | Ampicillin, metronidazole | H. pylori | 53 |
| DPPC, Chol, (DSPE–PEG–Mal) | Gentamicin | E. coli, P. aeruginosa, S. aureus | 54 |
| PC/Chol + chitosan modified Au NPs | Vancomycin | MRSA | 55 |
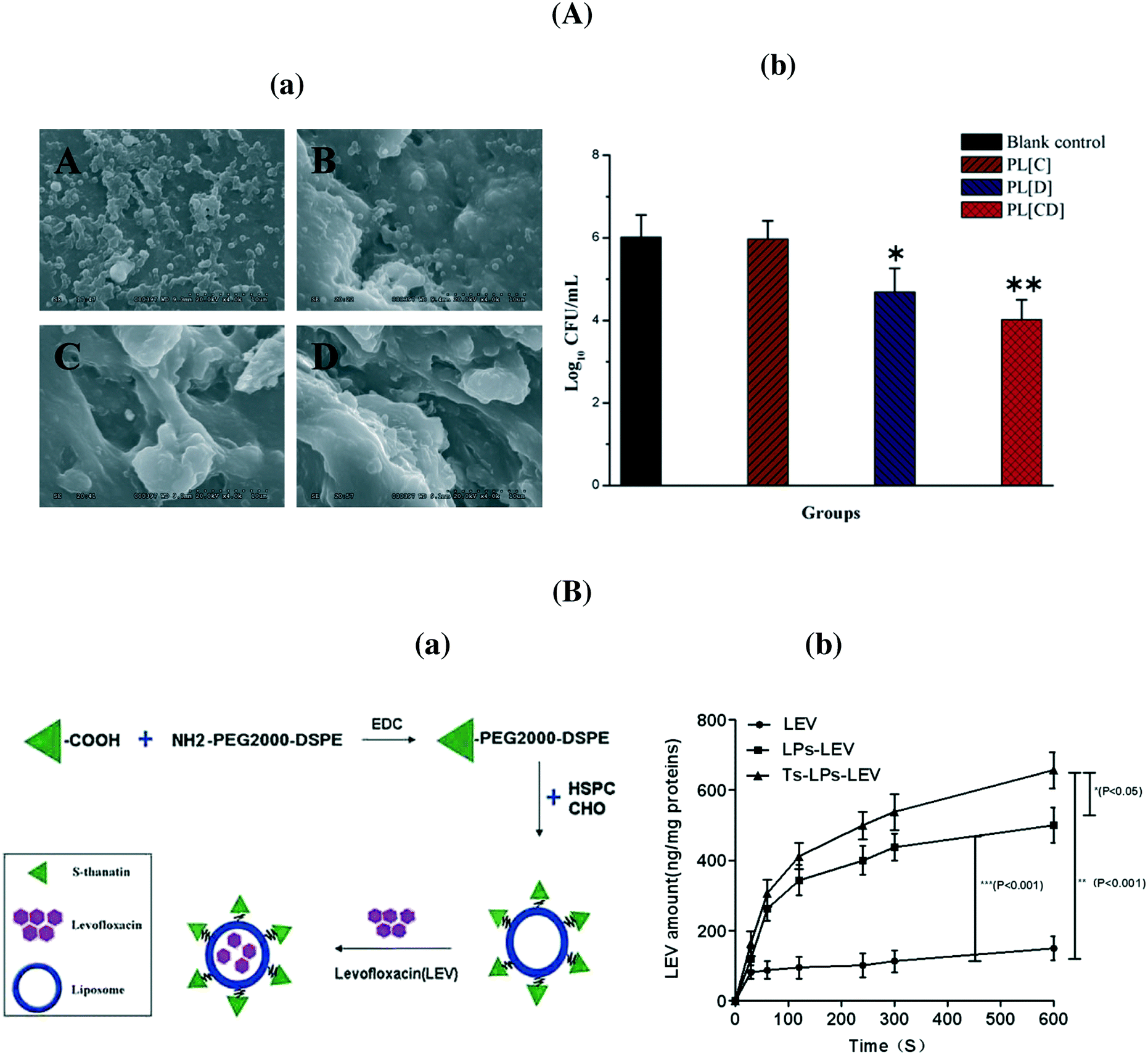 | ||
| Fig. 3 (A) (a) MRSA 252 biofilm survival and morphology examined after seven days. (A) No treatment; (B) PEGylated liposome with clarithromycin, (C) PEGylated liposome with daptomycin; (D) PEGylated liposome with clarithromycin + daptomycin; (b) a comparison of bacterial survival in the presence of the indicated formulations; (B) (a) formulation of S-thanatin antibiotic liposomal formulation, (b) intra-bacterial levofloxacin accumulation of levofloxacin in K. pneumoniae ATCC 700603 (reprinted with permission from ref. 36). | ||
Despite their common use, one of the main drawbacks of liposomal antibiotics is the short shelf-life of lipid vesicles which limits drug stability. This is mainly due to the hydrolysis of ester bonds or the oxidation of unsaturated acyl chains of the lipids used in their formulation. Passive drug leakage is another issue to be controlled. Furthermore, the encapsulation process has an important influence on the therapeutic utility of the nanocarriers and liposomal antibiotics are only useful if a therapeutic amount of drug and a reasonable amount of lipids is used, as high doses of lipids can be toxic.56
3.2. Polymer nanoparticles and hydrogels
To overcome some of the problems related to liposomal antibiotics, polymer particles loaded with antibiotics were developed. Polymer-based nanoparticles are submicron-sized colloidal particles in which the antibiotic can be embedded or encapsulated within their polymeric matrix or adsorbed or conjugated onto their surface.57 Such particles are composed of biodegradable or biocompatible materials such as poly(lactic acid) (PLA), poly(lactic-co-glycolic acid) (PLGA), chitosan, gelatin, alginic acid, etc. and have been extensively explored as means for drug solubilization, stabilization and targeting.4,58 Compared to the free drug, polymeric particles have many advantages including improved drug bioavailability, higher carrier capacity, protection of the antibiotic against enzymatic and hydrolytic degradation, the ability to release the antibiotic in a controlled behavior and the capability to adapt to different routes of administration. By virtue of their enhanced permeability, they can concentrate in inflamed and infected locations. Furthermore, the advantages of polymeric particles are their high stability, controlled synthesis (size, surface charge, the presence of functional groups), and drug release profiles that can be accurately tuned by selecting the appropriate polymer. Some excellent review articles about polymeric nanoparticles have been published recently.4,9,59,60Table 4 summarizes some of the recent developments in antibiotic loaded polymeric nanoparticles, including hydrogels to fight infections.| Particles | Antibiotics | Bacterial strain | Ref. |
|---|---|---|---|
| BMA: butyl methacrylate; CAMP: cyclic adenosine monophosphate; DMAEMA = 2-(dimethylamino)ethyl methacrylate, PAA: 2-propylacrylic acid; PAE: poly(beta-amino ester); PCL: poly-caprolactone; PLGA: poly(lactic-co-glycolic acid); PLH: poly(L-histidine); PC: phenyl cephalosporin, PP: phenylacetic acid, PEG: poly(ethylene glycol); PEO: poly(ethylene oxide); POEGMA = poly(oligo(ethylene glycol) methacrylate); PQDMA: poly(2-dimethylamino)ethyl methacrylate; PVA: poly(vinyl alcohol). | |||
| PLGA | Cefazolin, ciprofloxacin, colistin clindamycin, doxycycline, vancomycin | S. aureus, P. aeruginosa, E. coli | 61 |
| PLGA | Amikacin | P. aeruginosa | 62 |
| PLGA | Ciproflaxin | P. aeruginosa, S. aureus | 63 |
| PLGA/CAMP | Colistin | P. aeruginosa | 64 |
| PLGA/polypyrrole | Vancomycin | S. aureus | 65, 66 |
| PLGA/PCL | Vancomycin, rifampin, linezolid, daptomycin | S. aureus | |
| POEGMA-b-PVA | Gentamicin | P. aeruginosa | 67 |
| PEG/PAA | Amikacin, colistin | S. aureus, E. coli | 68 |
| PEG-PCL-PAE | Vancomycin | S. aureus | 69 |
| p(DMAEMA-BMA-PAA) | Farnesol | Streptococcus mutans | 70 |
| Alginate/chitosan | Tobramycin | P. aeruginosa | 71 |
| Carboxymethyl-chitosan | Fentamycin sulfate (GS) | S. aureus | 72 |
| Core–shell supramolecular gelatin nanoparticles (SGNPs) | Vancomycin | S. epidermidis, S. aureus, E. coli, P. vulgaris, S. marcescens, P. aeruginosa | 73 |
| Polymeric triple-layered nanogel | Vancomycin | S. aureus | 74 |
| Mannose-modified hydrogel | Vancomycin | MRSA | 75 |
| Dendritic-based hydrogels | Amoxicillin | — | 76 |
| Chitosan-based hydrogel | Gentamycin sulfate | S. aureus | 72 |
| PEG/PAA | Amikacin, colistin | S. aureus, E. coli | 68 |
| PLGA-PLH-PEG | Vancomycin | S. aureus | 77 |
| PEG-b-PP, PEG-b-PC | Vancomycin, gentamicin | S. aureus | 78 |
| PAA/gelatin-based hydrogel | Gentamycin sulfate, vancomycin hydrochloride | — | 79 |
PLGA particles containing colistin as well as cationic antimicrobial peptides were prepared by d'Angelo et al. using an emulsion/solvent diffusion technique.64 Post-coating with chitosan and PVA allowed modulating surface properties and improving the transport of the nanosystem through artificial mucus; the particles displayed excellent efficacy in biofilm eradication compared to free colistin. Co-delivery of nitric oxide and gentamicin from polymeric nanoparticles was proposed recently by Nguyen et al. as efficient means for reducing the viability of P. aeruginosa biofilms by more than 90%.67 The group of Scott developed tobramycin encapsulated polymer nanostructures, which showed the capability to penetrate the thick DNA-rich mucus on people with a P. aeruginosa pulmonary infection.71 Ciproflaxin loaded poly(lactic-co-glycolic acid) nanoparticles modified with DNase I were investigated by Torrents and co-workers for the eradication of established biofilms.63 While the particles were not able to prevent biofilm formation, from planktonic bacteria, they successfully reduced biofilm mass and size (Fig. 4A). Repeated administration over 3 days reduced 95% and eradicated more than 99.8% of established biofilms.63 Polymeric nanofibers, composed of PLGA and PCL, were developed by Ashbaugher et al. for local co-delivery of a combination of antibiotics.80 The release can be adjusted by varying the PLGA/PCL polymer ratio and proved to be highly effective in preventing medical device infections in patients. Benoit and co-workers synthesized positively charged nanoparticles from diblock copolymers composed of 2-(dimethylamino)ethyl methacrylate (DMAEMA), butyl methacrylate (BMA) and 2-propylacrylic acid (PAA) for loading with farnesil, a hydrophobic antibacterial drug.70 A pH dependent release was observed, resulting in the disruption of Streptococcus mutans biofilms 4-fold more efficiently than free farnesol (Fig. 4B). These particles also efficiently attenuated biofilm virulence in vivo using a clinically relevant topical treatment regime in a rodent dental carrier disease model.70
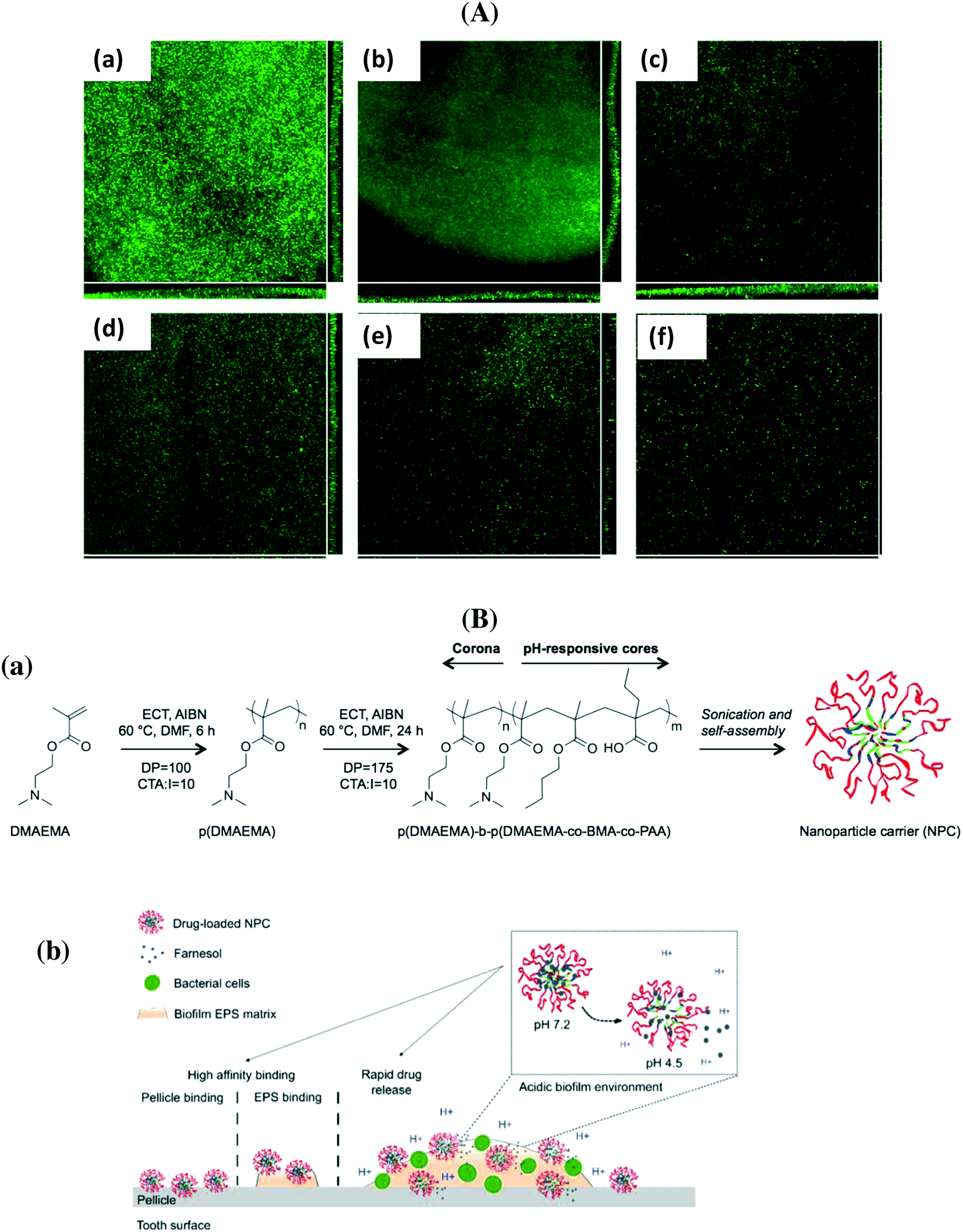 | ||
| Fig. 4 (A) Flow cell analysis of P. aeruginosa biofilm formation in the absence (a) and when treated with free ciproflaxin (b), free ciproflaxin + DNase I (c), PLGA–ciproflaxin (d), PLGA–ciproflaxin + PLGA–DNase I (e) and PLGA–ciproflaxin–DNAse I (f) (reprinted with permission from ref. 63); (B) (a) structure of diblock copolymers used in two-step RAFT polymerizations, (b) proposed mode of action of pH-responsive polymeric nanoparticles for prevention and/or treatment of biofilms (reprinted with permission from ref. 70). | ||
In order to eliminate the adverse side effects due to premature and nonspecific antibiotic release, considerable efforts have been devoted to the development of ‘on-demand’ release strategies. In this context, hydrogels have attracted a great deal of attention as a matrix for the controlled delivery of antibiotics. Li et al. developed an efficient and specific antibiotic delivery system based on core–shell supramolecular gelatin nanoparticles (SGNPs).73 The surface of the SGNPs was decorated with red blood cell (RBC) membranes and then vancomycin (Van) was encapsulated in the hybrid matrix (Van⊂SGNPs@RBC). After reaching the infection sites, the RBC membranes on the nanosystem reduce the clearance by the immune system and act as detoxifiers by absorbing the exotoxins produced by bacteria, while the gelatin core is degraded by the gelatinase and the encapsulated vancomycin is released and kills the local bacteria. In vitro studies proved that Van⊂SGNPs@RBC were internalized by macrophage cells and displayed negligible toxicity towards human embryonic kidney and human hepatocyte cell lines. The confocal microscopy images (Fig. 5A) revealed that the Van⊂SGNPs@RBC exhibited higher antibacterial activity against S. aureus than toward S. epidermis with a MIC value of 3 ± 0.8 μg mL−1 and no inhibition effect for Gram-negative bacteria.73
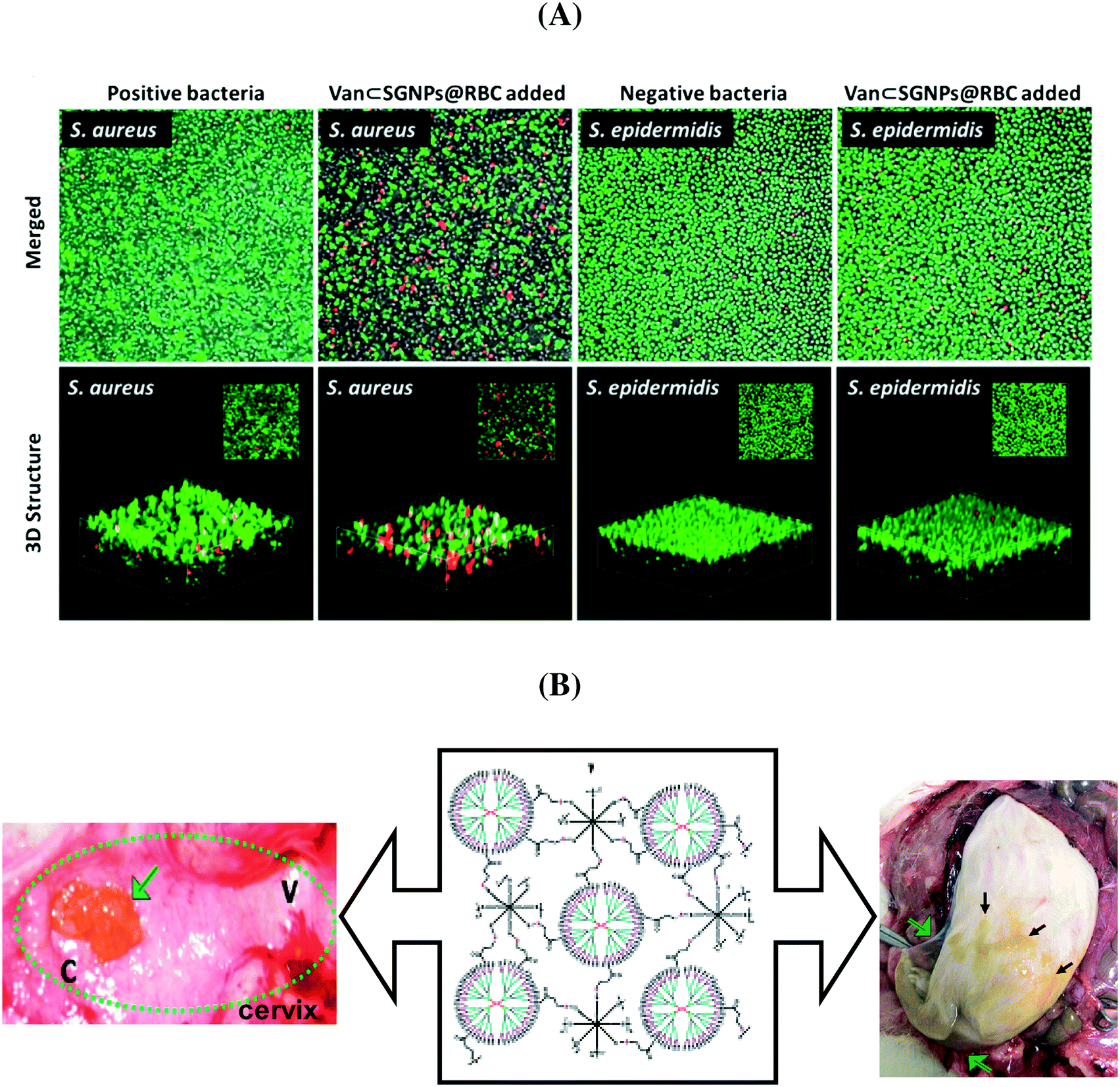 | ||
| Fig. 5 (A) Antibacterial efficiency of Van⊂SGNPs@RBC against S. aureus and S. epidermidis (reprinted with permission from ref. 73); (B) dendritic-based hydrogels (10 wt%) after intravaginal (left) and intracervical (right) application in pregnant guinea pigs (reprinted with permission from ref. 76). | ||
A bacterial-lipase sensitive polymeric triple-layered nanogel (TLN) as a nanocarrier for vancomycin delivery to bacterial infection sites was proposed by Xiong et al.74 They demonstrated that lipase-secreting S. aureus bacteria triggered rapid drug release and the released drug was able to kill the intracellular bacterial cells. The same group investigated mannosyl modified nanogels for targeted and activatable delivery of vancomycin to infection sites.75 The phosphatase and phospholipase produced by the bacteria can degrade the polyphosphoester core of the nanogel, triggering antibiotic release. The mannosyl-modified gel exhibited excellent inhibition capacity against intracellular and in vivo bacterial infections, using Raw264.7 cells and zebrafish embryo infected with MRSA as models.75
Recently, dendritic-based hydrogels showed great potential in the loading and release of therapeutic moieties. Navath and co-workers reported that amoxicillin entrapped into injectable and biodegradable hydrogels obtained by cross-linking of a thiopyridyl functionalized PAMAM dendrimer [(NH2)49-G4-(NH-PDP)15] with 8-arm PEG (MW ∼ 20 kDa) is a viable therapeutic option to treat ascending genital infections during pregnancy.76 The results confirmed that the dendritic-based hydrogels ensured a sustained intravaginal delivery of amoxicillin; additionally, the gel components were located on the epithelial surface and did not cross the fetal membrane (Fig. 5B). A gentamycin sulfate loaded genipin cross-linked carboxymethyl-chitosan hydrogel was proposed as a simple approach to achieve combined antibacterial efficiency and enhanced osteoblastic cell response, with significant inhibition of bacteria growth and biofilm formation of S. aureus. The antibacterial activity of the hydrogel was linked to the disruption of the bacterial wall and membrane.72 The design of materials that exhibit antibacterial properties and biocompatibility is of great impact in the biomedical field because biomaterial related infections remain a serious biomedical problem and are expensive and complex to treat.
Recently, dendrimeric macromolecules have been reported to be effective platforms for targeted drug delivery to treat bacterial infections (Table 5).
| Particles | Antibiotics | Bacteria | Ref. |
|---|---|---|---|
| Dendrimers | Ciprofloxacin | E. coli | 81 |
| Dendrimers | Vancomycin | S. aureus | 82 |
| Lipid-dendrimer NPs | Vancomycin | S. aureus, MRSA | 83 |
| Dendrimers | Tobramycin | P. aeruginosa | 84 |
| Dendrimers | Ciprofloxacin | S. aureus, E. coli | 85 |
| Dendrimers | Erythromycin, tobramycin | S. aureus, E. faecalis, E. coli, A. baumanni, K. pneumoniae, E. cloacae | 86 |
| Dendrimers | Norfloxacin | S. aureus, E. coli | 87 |
| Dendrimers | Nadifloxacin | E. coli, P. aeruginosa and P. hauseri | 88 |
| Dendrimers | Rifampicin | — | 89 |
| Dendrimers | Azythromycin | C. trachomatis | 90 |
Choi et al. prepared ciprofloxacin attached to a lipopolysaccharide-binding poly(amidoamine) dendrimer nanoconjugate for bacterial cell wall-targeted delivery of the antibiotic under light irradiation (Fig. 6A)81 and a vancomycin-conjugated poly(amidoamine) dendrimer for pathogen detection and isolation (Fig. 6B).82 A lipid–dendrimer hybrid nanoparticle (LDHN) system conjugated with vancomycin was synthesized by Sonawane et al.83 to fight methicillin-resistant S. aureus (MRSA) infections. Due to the sustained release of vancomycin from LDHNs, the nanosystem showed prolonged and enhanced antibacterial activity. Reymond et al. reported the synergistic effect of glycopeptide dendrimers (FD2) with tobramycin against P. aeruginosa biofilm. The complex nanostructures displayed an effective biofilm inhibition and degradation properties at a sub-inhibitory concentration of both compounds, 4-fold lower than the FD2 minimal biofilm inhibitory concentration (MBIC) and 5-fold lower than the antibiotic MBIC. More recently, VanKoten et al. reported the enhanced antibacterial activity of fourth generation poly(amidoamine) dendrimers decorated with a quaternary ammonium compound against Gram-positive and Gram-negative bacteria.91
 | ||
| Fig. 6 (A) Light controlled release of ciprofloxacin by a cell wall-targeted dendrimer nanoconjugate (reprinted with permission from ref. 81); (B) vancomycin-conjugated poly(amidoamine) dendrimer as a nanoplatform for targeting Gram-positive bacteria (reprinted with permission from ref. 82). | ||
3.3. Gold and silver nanostructures
Metallic nanoparticles (NPs) have generated huge interest in many biomedical fields due to their ease of synthesis and functionalization, and their plasmonic features, allowing the development of theranostic applications.92,93 Due to their small size, large surface area to volume ratio, stability over high temperature and translocation into the cells, inorganic nanoparticles are considered as a viable alternative for carrying a large amount of antibiotics and the efficacy of metallic NPs conjugated with antibiotics has thus been the subject of intense investigations. These studies showed that gold nanostructures94,95 used in combination with antibiotics exhibit enhanced bactericidal activity on a range of Gram-positive and Gram-negative bacteria and higher stability over an extended storage compared to the free antibiotics (Table 6). While silver nanoparticles (Ag NPs) are bactericidal per se and have proven to be the most effective against bacteria among the metallic nanoparticles,96,97 it has been reported that the combined use of Ag NPs with antibiotics results in enhanced and synergetic antimicrobial effects.98| Particle | Antibiotics | Comments | Bacteria | Ref. |
|---|---|---|---|---|
| PVP: polyvinylpyrrolidone. | ||||
| Au NPs | Gentamicin | d = 16 nm, citrate capped | E. coli K12 | 95 |
| Au NPs | Vancomycin | d = 4–14 nm, biomass coated Au NPs of negative surface charge electrostatic interaction | E. coli, VRSA, VSSA | 99 |
| Au NPs | Vancomycin | d < 20 nm, interaction with bis(vancomycin) cystamide via Au–S bonds | E. coli, VRE | 100 |
| Au NPs | Cefaclor | d = 22–52 nm, primary amine group of cefaclor acted as both the reducing and the capping agent | S. aureus, E. coli | 102 |
| Au NPs | Gentamicin | d = 10–12 nm, electrostatic interaction | S. aureus | 105 |
| Au3+ Au NPs | Penicillin G, amoxicillin, methicillin, cephalexin, cefixime, erythromycin, gentamicin, amikacin, tetracycline, ciprofloxacin, clindamycin, nitrofurantoin, nalidixic acid, vancomycin | d = 1.2–17.5 nm, tannin-free black tea reduction, Au3+ coordination complex with available donor groups such as nitrogen, sulfur and phosphorus in antibiotics | P. aeruginosa, E. coli, S. aureus | 108 |
| Au NPs | Streptomycin, gentamycin, neomycin | d = 12–15 nm, citrate capped, electrostatic interactions | S. aureus, M. luteus, E. coli, P. aeruginosa | 109 |
| Au NPs | Ampicillin, streptomycin, kanamycin | d = 14 nm, NaBH4 reduction in the presence of antibiotics | E. coli DH5α; M. luteus, S. aureus | 110 |
| Au NPs | Ciprofloxacin | d = 4 and 20 nm, nitrogen atom of the NH moiety of piperazine group binds on Au NPs | — | 111 |
| Ag NPs | Ampicillin, penicillin G, isoniazid | d = 20–50 nm, Ag NPs embedded on silica shells functionalized with several antibiotics | E. coli, P. aeruginosa S. aureus, B. subtilis, C. albicans | 104 |
| Ag NPs | Ampicillin, chloramphenicol, erythromycin, gentamicin, tetracycline, vancomycin | d = 3–20 nm, Allophylus cobbe leaf extract acts as a reducing as well as stabilizing/capping agent, mixed with antibiotics | P. aeruginosa, S. flexneri, S. aureus, K. pneumoniae | 112 |
| Ag NPs | Amoxicillin | d = 4 and 20 nm, mixed with antibiotics | E. coli | 113 |
| Ag NPs | Streptomycin, ampicillin, tetracycline |
d = 38.3 (citrate), 19.3 (SDS), 16.0nm (PVP), mixed with antibiotics |
E. coli, S. aureus | 114 |
Mainly two strategies are employed to conjugate antibiotics onto the surface of metallic NPs: (i) adsorption mediated by intermolecular forces such ionic interactions between the amino group of vancomycin and the negative surface charge of Au NPs99 or via Au–S bonds100,101 and (ii) covalent binding.94,102–104 However, the exact mechanism governing the enhancement of the antibacterial properties of antibiotic modified metallic NPs is still highly speculative. In general, the enhanced antibacterial effects of metallic NP/antibiotic complexes have been attributed to the ability of the antibiotic to cause structural change and degradation together with the high affinity of metallic NPs to bind to the bacterial cell wall ensuring a local increase of the antibiotic concentration. Perry and co-workers102 reported a one-pot synthesis of spherical 52–22 nm Au NPs capped with cefaclor, a second-generation antibiotic where the primary amine groups of cefaclor acted as both the reducing and the capping agent for the synthesis of Au NPs leaving the β-lactam ring of cefaclor available for activity against microbes (Fig. 7A). The cefaclor reduced Au NPs showed a minimum inhibition concentration (MIC) for S. aureus and E. coli of 10 and 100 μg mL−1, respectively. Coating the particles into a poly(ethylenimine) (PEI) modified glass surface resulted in an antimicrobial coating suitable for biomedical applications. AFM images suggested that the action of these particles takes place through the combination of cefaclor inhibiting the synthesis of peptidoglycan layers and the particles generating holes in the bacterial cell walls, thereby increasing the permeability of the cell wall, resulting in the leakage of cell contents and eventually cell death. In addition, the Au NPs increase the long-term activity of cefaclor, which maintains 100% antibacterial activity after 5 days, while cefaclor has a half-life of ca. 9 h with 11% activity in solution.
 | ||
| Fig. 7 (A) The antibacterial effect of cefaclor reduced spherical gold nanoparticles as shown by AFM images after incubation for 6 h at 37 °C (reprinted permission from ref. 102); (B) TEM micrographs of (a) vancomycin-resistant Staphylococcus aureus (VRSA) and (b) E. coli bacteria cells treated with vancomycin-modified Au NPs (reprinted with permission from ref. 99), (c) streptomycin-modified Au NPs interacting with E. coli (reprinted with permission from ref. 94). | ||
Ahangari et al.105 reported that gentamicin (Gm) can be successfully conjugated onto Au NPs via ionic interactions with 347 gentamicin molecules per gold nanosphere. Bio-distribution studies on BALB/c mice infected with Staphylococcus aureus illustrated a good retention of gentamicin conjugated Au NPs in the bacterial infection site. Similar results were reported by Gu et al.100 and Fayaz et al.99 using vancomycin coated Au NPs, exhibiting enhanced activities against vancomycin-resistant enterococci (VRE)100 and vancomycin-resistant Staphylococcus aureus (VRSA), vancomycin-sensitive S. aureus (VSSA) and E. coli.99 The vancomycin modified Au NPs interact with the building elements of the outer membrane causing structural changes, degradation and finally cell wall lysis. Indeed as seen from the TEM images,99 the presence of vancomycin modified Au NPs is seen in abundance on the cell wall surface (Fig. 7B). Similar results were found by Saha et al.94 using streptomycin-modified Au NPs (Fig. 7B). Importantly, the antibiotic-conjugated Au NPs are more stable than the free antibiotic withstanding even harsh storage conditions.
Shahverdi et al.106 investigated the effect of trivalent gold ions (Au3+) and gold nanoparticles (Au NPs) modified with 14 different antibiotics against P. aeruginosa, S. aureus and E. coli. The Au NPs showed no increase in the antibacterial effect of the antibiotics; the susceptibility of P. aeruginosa increased however in the presence of Au3+ and some antibiotics (methicillin, erythromycin, vancomycin, penicillin G, clindamycin, nalidic acid) up to 147%. Independently, Burygin et al.95 demonstrated that Au NPs had no enhancing effect on the antibacterial activity of gentamicin. They suggested that Au NPs enhanced the antibacterial activity of the antibiotic only when the drug was covalently attached onto the nanoparticles' surface, leading to a stable conjugate. In a different work, the same group demonstrated enhanced activity of ciprofloxacin in the presence of ZnO nanoparticles107 and of different antibiotics in the presence of Ag NPs. The increased activity in the presence of ZnO nanoparticles against S. aureus was associated with the pumping activity of the NorA protein and enhanced adsorption of antibiotics into cells.
3.4. Carbon based materials
The use of carbon based materials for the removal of antibiotics by adsorption with high concentration has been well studied.115–119 Surprisingly the literature on the integration of antibiotics onto carbon based structures to ablate bacteria is rather limited (Table 7). In the case of fullerenes and in particular C60, photodynamic inactivation of bacteria was reported120 and the effect proved to be more pronounced on Gram-positive than on Gram-negative bacteria, indicating that the bactericidal action was dependent on the fullerene insertion into the cell wall of the bacteria. Carbon nanotubes modified with amphotericin B, an antibiotic for the treatment of coronial fungal infections, were investigated by Bianco and co-workers for the treatment of Candida parapsilosis and Candida albicans based infections.121| Particles | Antibiotics | Comments | Bacteria | Ref. |
|---|---|---|---|---|
| CDs | Ciprofloxacin | d = 2–5 nm, polyethylene glycol diamine capped CDs conjugated with glutamic acid ciprofloxacin | E. coli | 128 |
| CDs | Vancomycin | d = 2.2 nm, formation using citric acid and urea covalently linked with Van | S. aureus, B. subtilis, L. monocutogenes | 126 |
| CDs | Ferry ciprofloxacin hydrochloride | d = 2–5 nm, synthesis from gum arabic | E. coli, P. aeruginosa, B. subtilis, S. aureus | 129 |
| CDs | Tetracycline alone and associated with β-cyclodextrin | d = 2–5 nm, carbon dot coated alginate beads | — | 130 |
| CDs | Ampicillin | d = 40 nm, covalent linking to amine-functionalized C-dots formed from citric acid and ethylenediamine | E. coli | 131 |
| prGO | Ampicillin | Electrophoretically deposited prGO/PEI films modified covalently | E. coli | 133 |
| Graphene nanosheets | Gentamicin sulfate | Non-covalent loading of methanol derived graphene nanosheets | E. coli | 132 |
| Carbon nanotubes | Amphotericin B | Covalent linking via carboxylated sites along sidewalls of CNTs | Candida parapsilosis, Candida albicans | 134 |
Functional carbon dots (CDs) have shown to open promising avenues for bacteria imaging, detection and inactivation.122–129 However, very few studies have used CDs as a carrier platform for antibiotics to control pathogenic infections. Krishna et al.128 employed polyethylene glycol diamine capped CDs conjugated with glutamic acid and ciprofloxacin for simultaneous crack visualization and drug delivery to control bacterial infection (Fig. 8A). Thakur et al.129 found that CDs can act as efficient nanosinks for delivery of ferry ciprofloxacin hydrochloride. The release of antibiotic was sustained and constant under physiological conditions over 24 h and the nanocomposites displayed the highest antibacterial activity against P. aeruginosa. Gogoi et al.130 found that the decoration of calcium alginate beads with carbon dots (CA-CDs) improved the stability of the CA beads, in addition to swelling and the antibiotic's loading capacity. Zhong et al.126 synthesized CDs modified with vancomycin for fast and accurate sensing of Gram positive bacteria, including S. aureus, B. subtilis and L. monocytogenes. Our group reported that ampicillin loaded CDs are highly effective in inactivating the growth of E. coli (Fig. 8B).131 The antibacterial efficiency can be further increased upon visible light illumination where moderate quantities of reactive oxygen species were generated, resulting in additional photodynamic inactivation of the pathogens.
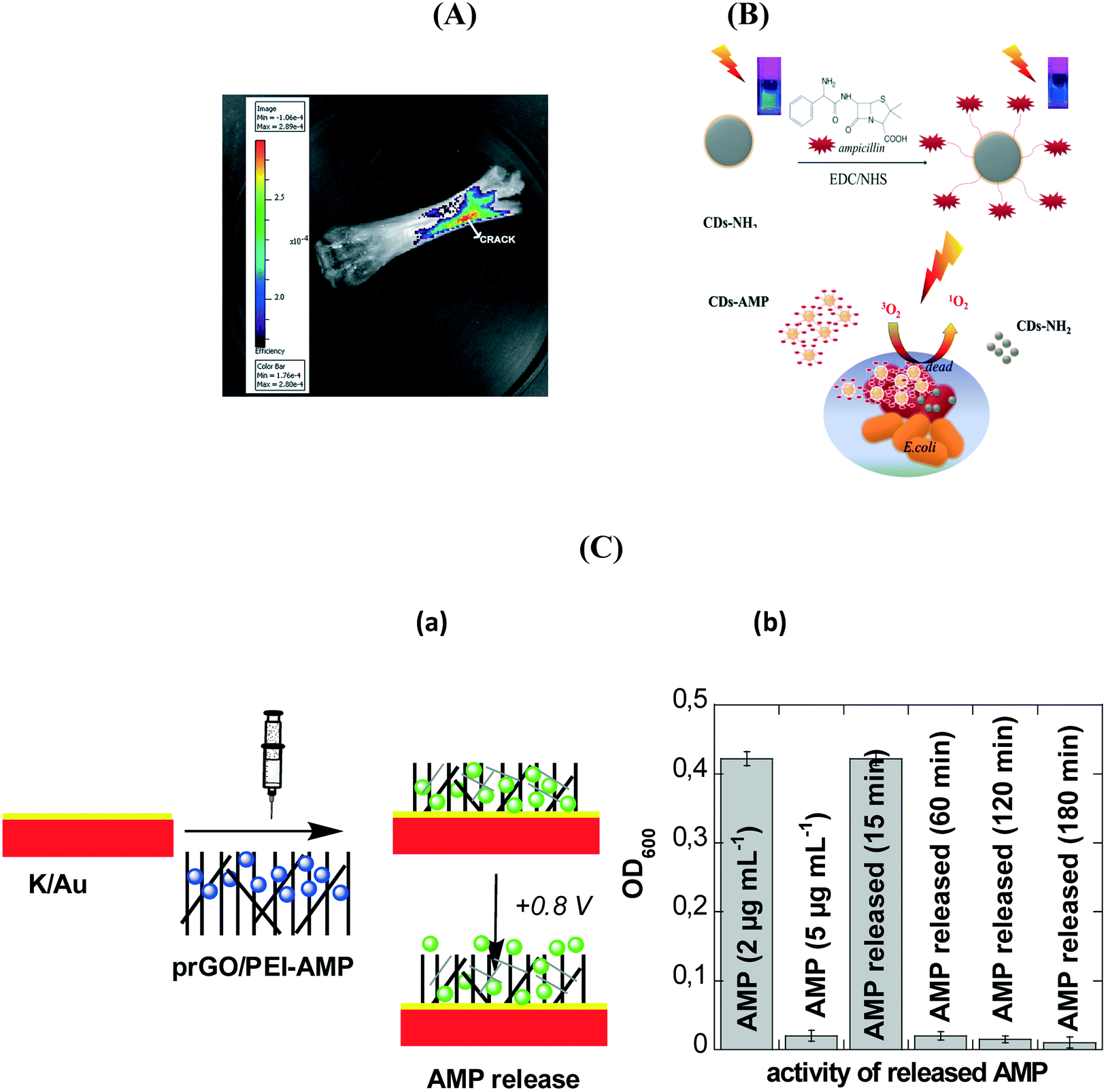 | ||
| Fig. 8 (A) Bone crack detection by CDGAC imaged with the Xenogen (Caliper Life Sciences) IVIS system (reprinted with permission from ref. 128); (B) antibacterial activity of ampicillin (AMP) modified CDs functionalized with ampicillin together with the formation of single oxygen under visible light illumination;131 (C) (a) formation of an electrochemically activatable ampicillin (AMP) loaded interface based on the coating of flexible electrodes with prGO/PEI-AMP, (b) comparison of the action of free AMP (2 μg mL−1; negative control), AMP (5 μg mL−1; positive control) and electrochemically released AMP at different time intervals when incubated with E. coli (1 × 108 cfu mL−1) for 2 h.133 | ||
2D materials such as graphene based nanostructures also attracted huge interest in the loading and release of antibiotics. Pandey et al.132 investigated the loading and release behaviour of gentamicin sulphate on methanol derived graphene. An efficient drug release of 62.75% was obtained at pH 3. Moreover, the graphene nano-platform loaded with antibiotic inhibited bacterial growth in an enhanced manner. Porous reduced graphene oxide (prGO) modified with polyethyleneimine (PEI) was shown to be highly interesting for ampicillin loading and for on-demand drug release using electrochemical bias.133 The interaction of ampicillin with prGO was believed to occur through π–π stacking and/or electrostatic interactions, resulting in the preservation of the amino group upon integration into prGO and its antibacterial function against Gram-negative bacteria. Application of +0.8 V resulted in a significant ampicillin release over time (Fig. 8C). The positively charged interface seems to be favorable for zwitterionic molecules such as ampicillin. Following the electrochemical release, the activity of the released ampicillin to kill pathogens was evaluated. The minimum inhibitory concentration of ampicillin against E. coli C43 isolates was MIC50 = 4.9 ± 0.5 μg mL−1. Application of +0.8 V for 15 min resulted in a release of 2 μg mL−1, below the MIC50 value, while after 1 h the critical 5 μg mL−1 was released from the matrix.
3.5. Magnetic nanoparticles
Different pathogen related issues such as the fast and efficient insulation and elimination of bacteria for water disinfection, food decontamination, infectious disease detection and identification, etc. can be addressed by using magnetic nanoparticles (MPs). MP based drug delivery systems retain also the ability to track the movement of the particles through the body135 as they can interact with external magnetic fields, offering different opportunities in nanomedicine such as a contrast agent in MRI, magnetic hyperthermia or magnetically triggerable drug delivery systems.136 While MPs are not bactericidal, loading of antibiotics on their surface makes them able to interact specifically with pathogens and might result in bacterial ablation (Table 8).| Particles | Antibiotics | Comments | Bacteria | Ref. |
|---|---|---|---|---|
| MPs | Vancomycin | d = 8–10 nm, surface thiol exchange with thiol of vancomycin | S. aureus, S. saprophyticus | 137 |
| MPs | Vancomycin | d = 50–2800 nm, covalent linkage | Gram-positive and Gram-negative | 138 |
| MPs | Methicillin | d = 83 nm, polymers on encapsulated MPs | S. epidermidis biofilm penetration | 139 |
A series of vancomycin-modified MPs were prepared as the COOH groups of vancomycin can be easily activated and further integrated onto the nanostructures.137,138,140,141 Vancomycin, a glycopeptide antibiotic, interacts strongly with a broad range of Gram-positive bacteria through a simple five-hydrogen bond motif between the heptapeptide backbone of vancomycin and the D-alanyl-D-alanine-dipeptide extending from the cell wall. The interaction is strong with respect to small molecule–bimolecule interactions with a dissociation constant of 1–4 μM at pH 7.142,143 It was shown that the time required to efficiently magnetically confine bacteria decreases when the size of the MP and the vancomycin surface coverage increase.138 Recently, a biocompatible multi-compartment nanocarrier containing hydrophobic MPs combined with mPEG-PDLLA (poly(D)-(L)-lactic acid) and the hydrophilic antibiotic methicillin was investigated for the treatment of medical-device associated infections (Fig. 9A).139 The MP polymersomes penetrated 20 μm thick S. epidermidis biofilms with high efficiency following the application of an external magnetic field with complete biofilm eradication using an optimized MP formulation containing 40 μg mL−1 MP and 20 μg mL−1 methicillin. The evaluation of the capture capability of vancomycin loaded MPs was recently performed by Zhu et al. using the classical plate counting method.141 Excellent enrichment capabilities were observed once the MP/vancomycin particles were PEGylated (Fig. 9B) with an enrichment >60% even at ultralow bacteria concentrations of 1 × 101 cfu mL−1. Our group demonstrated that magnetic PEGylated graphene oxide nanosheets loaded with 17 μg mL−1 of ampicillin allows highly efficient magnetic separation and bacteria inactivation (Fig. 9C).
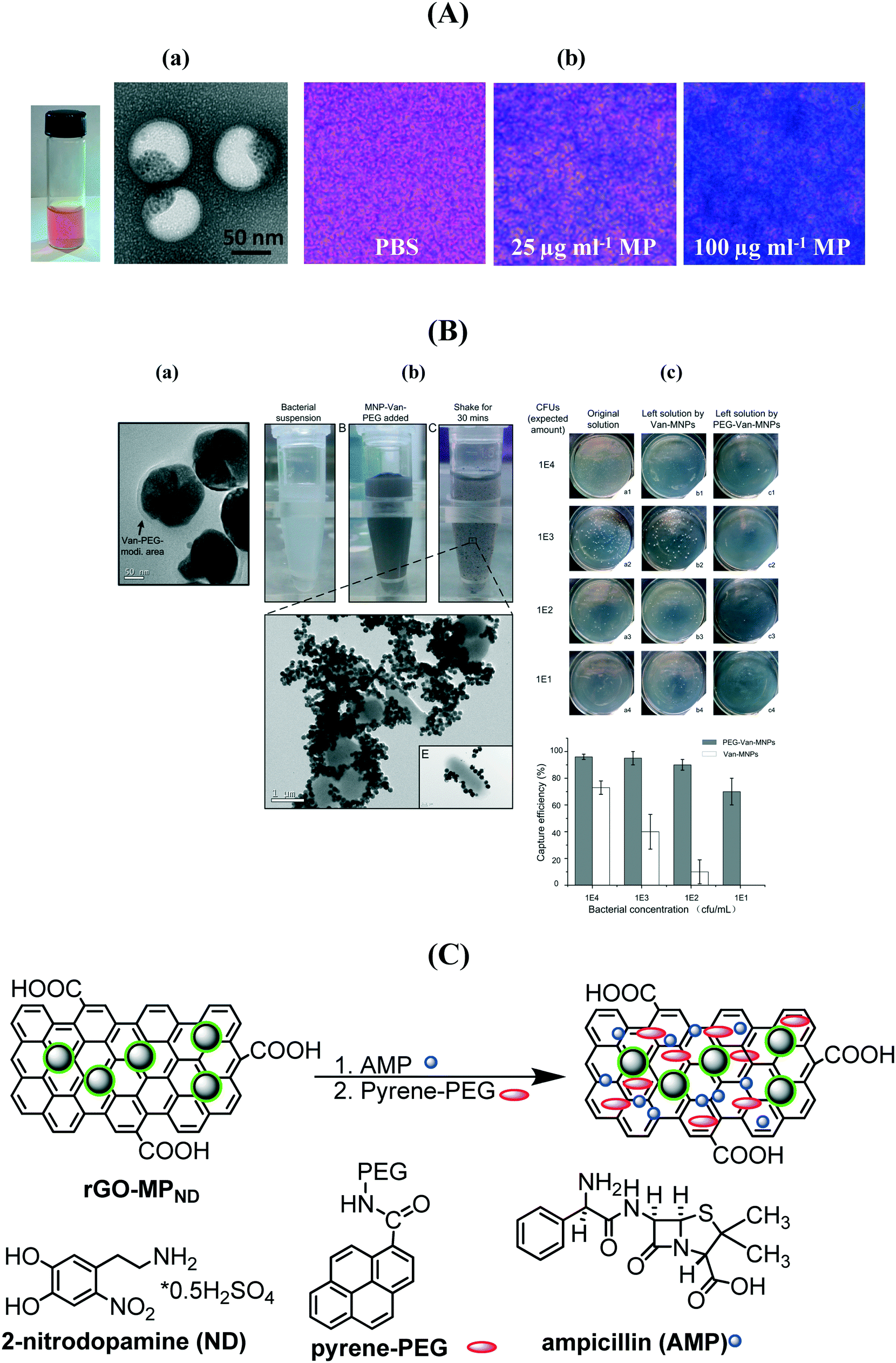 | ||
| Fig. 9 (A) (a) Photograph of a magnetic/methicillin polymersome solution together with TEM images; (b) Prussian blue staining of biofilms treated with and without iron oxide-encapsulated polymersomes (reprinted with permission from ref. 139); (B) (a) TEM image of MP–vancomycin–PEG nanocarriers, (b) photographs of bacterial suspensions before and after shaking for 30 min with MP/vancomycin/PEG and TEM image showing the phenomenon of bacterial aggregation, (c) enrichment capability of MP/vancomycin and MP/van/PEG (reprinted with permission from ref. 141); (C) formulation of ampicillin (AMP) modified PEGylated magnetic reduced graphene oxide. | ||
3.6. Other nanoparticles
The design of gated mesoporous supports able to deliver drugs is a fast-growing area that has attracted much attention of many reserchers.144 The high porosity of mesoporous silica nanoparticles (MSNs) ensuring a high drug-loading capacity and a sustained release of antibiotics at the target sites has been employed to maximize the antibacterial effects of antibiotics (Table 9). After the pioneering work by Vallet-Regi et al.145 on ordered mesoporous silica as a matrix for drug delivery, loading with amoxicillin was performed.146 The release of amoxicillin was observed to be dependent on the different physical states of the material with a faster release rate measured from powder than the disk form. This is in contrast to the gentamicin loaded highly ordered hexagonal mesoporous silica structure (SBA-15) where no significant difference was observed.147 Nairi et al. postulated that ampicillin release under physiological conditions is strongly affected by the surface charge and not the pore size.148 Similarly, Savimli and Yilmaz reported that the functionalization of MSNs with various groups, including amine, thiol and methyl can modulate the amount of amoxicillin loaded and released.149 The introduction of ammonic groups greatly retarded the amoxicillin release from MCM-41.150 Ciprofloxacin incorporated inside a silica shell (cip@SiO2) was found to be active against both Gram-positive and Gram-negative bacteria, with a slight decrease in the MIC for E. coli DH5α compared to the free antibiotic.151 The difference between the free drug and the nanoconjugate was believed to be due to the difference in hydrophobicity. Most interestingly, silica-based porous materials were applied as a scaffold in bone repair (Fig. 10A)152 or as a coating material to incorporate antibiotics into metallic implantable materials to prevent bacterial infections (Fig. 10B).153| Particles | Antibiotics | Ref. |
|---|---|---|
| SBA-15: highly ordered hexagonal mesoporous silica structure; MSNs: mesoporous silica nanostructures; mSiNTs: mesoporous silica nanotubes. | ||
| SBA-15 | Amoxicillin | 146 |
| SBA-15 | Gentamicin | 147 |
| mSiNTs | Ampicillin | 154 |
| MSNs | Ampicillin | 148 |
| MSNs | Amoxicillin | 149 |
| SBA-15 | Erythromycin | 155 |
| MSNs | Amoxicillin | 150 |
| SiO2 | Ciprofloxacin | 151 |
| MSNs | Gentamicin sulfate | 152 |
| Silica xerogel loaded into a porous TiO2 | Tetracycline-hydrochloride | 153 |
| MCM-41 capped with ε-polylysine | Vancomycin | 156 |
 | ||
| Fig. 10 (A) SEM images of (a) gentamycin sulfate (50 wt%) loaded mesoporous silica–hydroxyapatite particles (HMS–HA) embedded in poly(lactide-co-glycolide) (PLGA) proposed as a biodegradable drug delivery composite for bone replacement (reprinted with permission from ref. 152); (B) SEM images of porous TiO2 before (a) and after (b) silica xerogel containing tetracycline-hydrochloride filtration (reprinted with permission from ref. 153). | ||
4. Conclusion and perspectives
The use of nanoparticles as delivery vehicles for antimicrobial agents represents a new and promising approach in the design of effective therapies against pathogenic and antibiotic resistant bacteria. Such structures possess several clinical advantages due to the intrinsic physico-chemical properties of the nanostructures. Due to the large surface-to-volume ratio, several antibiotics together with targeting molecules can be loaded on the nanostructures. Co-delivery of two or more antibiotics allows the development of a therapeutic modality for combination therapy. This might help in targeting specific pathogens but might be also a long-term strategy to overcome antibiotic resistance. The use of antibiotic loaded nanocarriers overcomes in addition solubility and stability issues of current antibiotics and minimizes drug-induced side effects. The administration of PEG-modified antibiotic-loaded particles increases in addition the drug circulation time and has a strong impact on the overall pharmacokinetics. While, currently, most research is focused on the use of these nanostructures via intravenous administration, nanoparticles' administration can be extended to oral, nasal, pulmonary, parenteral, intra-ocular, or transdermal routes. The numerous benefits of antibiotic-loaded nanostructures over free antimicrobial agents are thus opening a wide range of opportunities. Especially, the possibility of stimulus-controlled delivery is a promising strategy for treating localized infections, assuring a high local concentration of therapeutic molecules and the conjugation of nanoparticles with specific ligands allows a targeted delivery.The choice of the nanoparticle based platform might depend on the looked for application. While all of the different nanostructures showed improved antibacterial properties when compared to the free antibiotic in solution, differences in synthetic effort, toxicity and mechanical and chemical properties make these nanoantibiotics rather different from each other. Table 10 tries to point out the advantages and disadvantages of each nanosystem.
| Particles | Advantages | Disadvantages |
|---|---|---|
| Liposomes | Improved pharmacokinetics | Short shelf-life which limits drug stability |
| Better biodistribution | Drug leakage over time | |
| Decreased toxicity | Strong influence of the encapsulation process on therapeutic utility | |
| Target selective | Complex preparation on laboratory scale | |
| Expensive | ||
| Polymers | Good stability | Presence of a reactive group can influence the rate and yield of the conjugation reaction and stability of the polymer |
| Good encapsulation efficiency | ||
| Controllable release rate/duration | Nondegradable backbones must have a molecular weight below the renal threshold, which is typically around 50 kDa to be safe | |
| Wide availability of polymers | ||
| Access to antimicrobial polymers (chitosan) | ||
| Target selective | ||
| Hydrogels | High drug loading | Low elastic force of many hydrogels limits their use in load-bearing applications as these can decompose and release drug |
| Humidity controlled environment favorable to drug stability | ||
| Responsive structures for drug release | ||
| Target selective | ||
| Metallic particles | Intrinsic antibacterial activity | Aggregation issues resulting in decreased antibacterial effects |
| Easy to synthesise with different shapes, forms | Release of metal ions into the medium which can be potentially toxic | |
| Improved stability of the drug | Accumulation in the body after administratio | |
| Combination with other antibacterial approaches (PTT, PDT) | ||
| Target selective | ||
| Magnetic structures | Ability to track their movement through the body | Corrosion related issues |
| Magnetic forces might not be strong enough to overcome the force of blood to help in the accumulation to the target site | ||
| Useful for imaging applications (MRI) | ||
| Carbon-based materials | Easy and cheap fabrication | Diversity in C-based materials making commercialization impossible |
| Covalently and non-covalently linked depending on carbon structures | Presence of heavy metal ion impurities from the synthetic procedure | |
| Tuneable chemical and physical properties allowing imaging | ||
| Mesoporous silica | Sustained and controlled release of drug if protected | Burst release |
| Inhibition of release due to blocking of pores | ||
| Non-toxic | Difficulties in controlling the pore size on large scale synthesis | |
| Good biocompatibility and elimination in the body due to degradation | ||
| Variable surface/volume ratio for improved loading | ||
| Facile surface modification using silanol groups |
One key obstacle in treating infectious diseases with these nanoantibiotics is in translating this exciting technology into clinical applications. Currently, only one particle formulation for the treatment of fungal diseases is clinically approved and another for the treatment of Pseudomonas is undergoing Ph1 clinical trials. Profound knowledge about the potential toxicity of these novel nanoantibiotics is greatly needed to guarantee successful clinical translation. The toxic effects of antimicrobial nanosystems on the central nervous system are still not known and the interaction of particles with cells and tissues remains poorly understood. Clinical translation and integration rely on a consistent and reproducible product. The size-specific properties of these nanosystems demand, in addition, the development and use of synthetic methods with high reproducibility on a large scale. With few exceptions, nanoparticles used in pre-clinical studies are almost exclusively synthesized in small batches. One of the main challenges facing clinical translation of nanoparticles is controlling their biological fate. Rapid clearance by immune cells can be overcome using PEG, sugar, serum protein modified particles, or concepts which have been successfully used in clinically approved products like Doxil and PEGylated doxorubicin loaded liposomes. Overall, the outlook for nanoparticle based drug delivery is promising and will hopefully in the future lead to promising clinical products for antibiotic-modified carriers. Unfortunately, the results presented to date seem to represent evolutionary improvements over existing options rather than major breakthroughs; further work will clearly be required to reach the goal of more general and powerful treatments and cures.
Acknowledgements
Financial support from the Centre National de la Recherche Scientifique (CNRS), the University Lille 1, the Hauts-de-France region, the CPER “Photonics for Society”, the Agence National de la Recherche (ANR) and the EU through FLAG-ERA JTC 2015-Graphtivity, and the Marie Sklodowska-Curie Action (H2020-MSCA-RISE-2015, PANG-690836) is acknowledged.References
- A. C. Anselmo and S. Mitragotri, Bioeng. Transl. Med., 2016, 1, 10–29 Search PubMed.
- H.-J. Choi, D. Wendell and C. D. Montemagno, NanoBiotechnology, 2007, 3, 66–75 CrossRef CAS.
- R. P. Rennie, Handb. Exp. Pharmacol., 2012, 211, 45–65 CAS.
- K. Forier, K. Raemdonck, S. C. De Smedt, J. Demeester, T. Coenye and K. Braeckmans, J. Controlled Release, 2014, 190, 607–623 CrossRef CAS PubMed.
- H. Pinto-Alphandary, A. Andremont and P. Couvreur, Int. J. Antimicrob. Agents, 2000, 13, 155–168 CrossRef CAS PubMed.
- M. Veerapandian and K. Yun, Appl. Microbiol. Biotechnol., 2011, 90, 1655–1667 CrossRef CAS PubMed.
- Z. Drulis-Kawa and A. Dorotkiewicz-Jach, Int. J. Pharm., 2010, 387, 187–198 CrossRef CAS PubMed.
- L. Zhang, D. Pornpattananangkul, C.-M. J. Hu and C.-M. Huang, Curr. Med. Chem., 2010, 17, 585–594 CrossRef CAS PubMed.
- A. Muñoz-Bonilla and M. Fernández-García, Eur. Polym. J., 2015, 65, 46–62 CrossRef.
- Y. Zhao and X. Jiang, Nanoscale, 2013, 5, 8340–8350 RSC.
- M. Morar and G. D. Wright, Annu. Rev. Genet., 2010, 44, 25–51 CrossRef CAS PubMed.
- S. B. Levy and B. Marshall, Nat. Med., 2004, 10, S122–S129 CrossRef CAS PubMed.
- J. Davies and D. Davies, Microbiol. Mol. Biol. Rev., 2010, 74, 417–433 CrossRef CAS PubMed.
- M. B. Habash, A. J. Park, E. C. Vis, R. J. Harris and C. M. Khursigara, Antimicrob. Agents Chemother., 2014, 58, 5818–5830 CrossRef PubMed.
- A. Nirmala Grace and K. Pandian, Colloids Surf., A, 2007, 297, 63–70 CrossRef.
- Y. C. Lee and R. T. Lee, Acc. Chem. Res., 1995, 28, 321–327 CrossRef CAS.
- M. Mammen, S.-K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 CrossRef.
- C.-C. Lin, Y.-C. Yeh, C.-Y. Yang, C.-L. Chen, G.-F. Chen, C.-C. Chen and Y.-C. Wu, J. Am. Chem. Soc., 2002, 124, 3508–3509 CrossRef CAS PubMed.
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 CrossRef CAS PubMed.
- L. L. Kiessling, J. E. Gestwicki and L. E. Strong, Curr. Opin. Chem. Biol., 2000, 4, 696–703 CrossRef CAS PubMed.
- M. Durka, K. Buffet, J. Iehl, M. Holler, J.-F. Nierengarten, J. Taganna, J. Bouckaert and S. P. Vincent, Chem. Commun., 2011, 47, 1321–1323 RSC.
- A. Bangham, M. M. Standish and G. Weissmann, J. Mol. Biol., 1965, 13, 253–259 CrossRef CAS PubMed.
- A. Akbarzadeh, R. Rezaei-Sadabady, S. Davaran, S. W. Joo, N. Zarghami, Y. Hanifehpour, M. Samiei, M. Kouhi and K. Nejati-Koshki, Nanoscale Res. Lett., 2013, 8, 102 CrossRef PubMed.
- S. Labana, R. Pandey, S. Sharma and G. Khuller, Int. J. Antimicrob. Agents, 2002, 20, 301–304 CrossRef CAS PubMed.
- Z. Drulis-Kawa, A. Dorotkiewicz-Jach, J. Gubernator, G. Gula, T. Bocer and W. Doroszkiewicz, Int. J. Pharm., 2009, 367, 211–219 CrossRef CAS PubMed.
- R. E. Hancock and F. S. Brinkman, Annu. Rev. Microbiol., 2002, 56, 17–38 CrossRef CAS PubMed.
- G. Gregoriadis, FEBS Lett., 1973, 36, 292–296 CrossRef CAS PubMed.
- P. F. Bonventre and G. Gregoriadis, Antimicrob. Agents Chemother., 1978, 13, 1049 CrossRef CAS PubMed.
- J. V. Desiderio and S. G. Campbell, J. Infect. Dis., 1983, 148, 563–570 CrossRef CAS PubMed.
- M. Stevenson, A. Baillie and R. Richards, Antimicrob. Agents Chemother., 1983, 24, 742–749 CrossRef CAS PubMed.
- M. Nacucchio, M. Bellora, D. Sordelli and M. D'aquino, Antimicrob. Agents Chemother., 1985, 27, 137–139 CrossRef CAS PubMed.
- S. G. Ingebrigtsen, A. Didriksen, M. Johannessen, N. Škalko-Basnet and A. M. Holsæter, Int. J. Pharm., 2017, 526, 538–546 CrossRef CAS PubMed.
- Y. Li, T. Su, Y. Zhang, X. Huang, J. Li and C. Li, Drug Delivery, 2015, 22, 627–637 CrossRef CAS PubMed.
- S. Gaidukevich, Y. L. Mikulovich, T. Smirnova, S. Andreevskaya, G. Sorokoumova, L. Chernousova, A. Selishcheva and V. Shvets, Bull. Exp. Biol. Med., 2016, 160, 675 CrossRef CAS PubMed.
- P. Matouskova, I. Marova, J. Bokrova and P. Benesova, Food Technol. Biotechnol., 2016, 54, 304 Search PubMed.
- X. Fan, J. Fan, X. Wang, P. Wu and G. Wu, Frontiers in pharmacology, 2015, 6, 249 CrossRef PubMed.
- C. Liu, J. Shi, Q. Dai, X. Yin, X. Zhang and A. Zheng, Drug Dev. Ind. Pharm., 2015, 41, 272–278 CrossRef CAS PubMed.
- S. Antonela Antoniu, Expert Rev. Anti-Infect. Ther., 2012, 10, 1439–1446 CrossRef CAS PubMed.
- I. Colzi, A. N. Troyan, B. Perito, E. Casalone, R. Romoli, G. Pieraccini, N. Škalko-Basnet, A. Adessi, F. Rossi and C. Gonnelli, Eur. J. Pharm. Biopharm., 2015, 94, 411–418 CrossRef CAS PubMed.
- Z. Yang, J. Liu, J. Gao, S. Chen and G. Huang, Int. J. Pharm., 2015, 495, 508–515 CrossRef CAS PubMed.
- K. A. Hamblin, S. J. Armstrong, K. B. Barnes, C. Davies, J. P. Wong, J. D. Blanchard, S. V. Harding, A. J. Simpson and H. S. Atkins, Antimicrob. Agents Chemother., 2014, 58, 3053–3059 CrossRef PubMed.
- D. G. Atashbeyk, B. Khameneh, M. Tafaghodi and B. S. Fazly Bazzaz, Pharm. Biol., 2014, 52, 1423–1428 CrossRef CAS PubMed.
- S. Thamphiwatana, W. Gao, D. Pornpattananangkul, Q. Zhang, V. Fu, J. Li, J. Li, M. Obonyo and L. Zhang, J. Mater. Chem. B, 2014, 2, 8201–8207 RSC.
- S. Thamphiwatana, V. Fu, J. Zhu, D. Lu, W. Gao and L. Zhang, Langmuir, 2013, 29, 12228–12233 CrossRef CAS PubMed.
- A.-S. Messiaen, K. Forier, H. Nelis, K. Braeckmans and T. Coenye, PLoS One, 2013, 8, e79220 Search PubMed.
- M. Alhajlan, M. Alhariri and A. Omri, Antimicrob. Agents Chemother., 2013, 57, 2694–2704 CrossRef CAS PubMed.
- J. He, K. Abdelraouf, K. R. Ledesma, D. S.-L. Chow and V. H. Tam, Int. J. Antimicrob. Agents, 2013, 42, 559–564 CrossRef CAS PubMed.
- M. Alipour, M. Halwani, A. Omri and Z. E. Suntres, Int. J. Pharm., 2008, 355, 293–298 CrossRef CAS PubMed.
- S. J. Wallace, J. Li, R. L. Nation, R. J. Prankerd and B. J. Boyd, J. Pharm. Sci., 2012, 101, 3347–3359 CrossRef CAS PubMed.
- L. Sande, M. Sanchez, J. Montes, A. J. Wolf, M. A. Morgan, A. Omri and G. Y. Liu, J. Antimicrob. Chemother., 2012, 67, 2191–2194 CrossRef CAS PubMed.
- A. Pumerantz, K. Muppidi, S. Agnihotri, C. Guerra, V. Venketaraman, J. Wang and G. Betageri, Int. J. Antimicrob. Agents, 2011, 37, 140–144 CrossRef CAS PubMed.
- J. E. de Steenwinkel, W. van Vianen, T. Marian, H. A. Verbrugh, M. A. van Agtmael, R. M. Schiffelers and I. A. Bakker-Woudenberg, J. Antimicrob. Chemother., 2007, 60, 1064–1073 CrossRef CAS PubMed.
- P.-L. Bardonnet, V. Faivre, P. Boullanger, J.-C. Piffaretti and F. Falson, Eur. J. Pharm. Biopharm., 2008, 69, 908–922 CrossRef CAS PubMed.
- N. Monteiro, M. Martins, A. Martins, N. A. Fonseca, J. N. Moreira, R. L. Reis and N. M. Neves, Acta Biomater., 2015, 18, 196–205 CrossRef CAS PubMed.
- D. Pornpattananangkul, L. Zhang, S. Olson, S. Aryal, M. Obonyo, K. Vecchio, C.-M. Huang and L. Zhang, J. Am. Chem. Soc., 2011, 133, 4132–4139 CrossRef CAS PubMed.
- A. Sharma and U. S. Sharma, Int. J. Pharm., 1997, 154, 123–140 CrossRef CAS.
- K. M. El-Say and H. S. El-Sawy, Int. J. Pharm., 2017, 528, 675–691 CrossRef CAS PubMed.
- B. Bharatwaj, L. Wu, J. A. Whittum-Hudson and S. R. da Rocha, Biomaterials, 2010, 31, 7376–7385 CrossRef CAS PubMed.
- W. S. Cheow and K. Hadinoto, Microb. Biofilms, 2014, 227–238 CAS.
- W. Gao, R. H. Fang, S. Thamphiwatana, B. T. Luk, J. Li, P. Angsantikul, Q. Zhang, C.-M. J. Hu and L. Zhang, Nano Lett., 2015, 15, 1403–1409 CrossRef CAS PubMed.
- S. R. Shah, A. M. Henslee, P. P. Spicer, S. Yokota, S. Petrichenko, S. Allahabadi, G. N. Bennett, M. E. Wong, F. K. Kasper and A. G. Mikos, Pharm. Res., 2014, 31, 3379–3389 CrossRef CAS PubMed.
- P. Sabaeifard, A. Abdi-Ali, M. R. Soudi, C. Gamazo and J. M. Irache, Eur. J. Pharm. Sci., 2016, 93, 392–398 CrossRef CAS PubMed.
- A. Baelo, R. Levato, E. Julián, A. Crespo, J. Astola, J. Gavaldà, E. Engel, M. A. Mateos-Timoneda and E. Torrents, J. Controlled Release, 2015, 209, 150–158 CrossRef CAS PubMed.
- I. d'Angelo, B. Casciaro, A. Miro, F. Quaglia, M. L. Mangoni and F. Ungaro, Colloids Surf., B, 2015, 135, 717–725 CrossRef PubMed.
- W.-L. Chiang, T.-T. Lin, R. Sureshbabu, W.-T. Chia, H.-C. Hsiao, H.-Y. Liu, C.-M. Yang and H.-W. Sung, J. Controlled Release, 2015, 199, 53–62 CrossRef CAS PubMed.
- D. Das and S. Pal, RSC Adv., 2015, 5, 25014–25050 RSC.
- T.-K. Nguyen, R. Selvanayagam, K. K. Ho, R. Chen, S. K. Kutty, S. A. Rice, N. Kumar, N. Barraud, H. T. Duong and C. Boyer, Chem. Sci., 2016, 7, 1016–1027 RSC.
- Y. Wu, J. Liang, F. Horkay and M. Libera, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 64–72 CrossRef CAS.
- L. Chu, H. Gao, T. Cheng, Y. Zhang, J. Liu, F. Huang, C. Yang, L. Shi and J. Liu, Chem. Commun., 2016, 52, 6265–6268 RSC.
- B. Horev, M. I. Klein, G. Hwang, Y. Li, D. Kim, H. Koo and D. S. Benoit, ACS Nano, 2015, 9, 2390–2404 CrossRef CAS PubMed.
- J. Deacon, S. M. Abdelghany, D. J. Quinn, D. Schmid, J. Megaw, R. F. Donnelly, D. S. Jones, A. Kissenpfennig, J. S. Elborn and B. F. Gilmore, J. Controlled Release, 2015, 198, 55–61 CrossRef CAS PubMed.
- F. Wu, G. Meng, J. He, Y. Wu, F. Wu and Z. Gu, ACS Appl. Mater. Interfaces, 2014, 6, 10005–10013 CAS.
- L.-L. Li, J.-H. Xu, G.-B. Qi, X. Zhao, F. Yu and H. Wang, ACS Nano, 2014, 8, 4975–4983 CrossRef CAS PubMed.
- M.-H. Xiong, Y. Bao, X.-Z. Yang, Y.-C. Wang, B. Sun and J. Wang, J. Am. Chem. Soc., 2012, 134, 4355–4362 CrossRef CAS PubMed.
- M. H. Xiong, Y. J. Li, Y. Bao, X. Z. Yang, B. Hu and J. Wang, Adv. Mater., 2012, 24, 6175–6180 CrossRef CAS PubMed.
- R. S. Navath, A. R. Menjoge, H. Dai, R. Romero, S. Kannan and R. M. Kannan, Mol. Pharmaceutics, 2011, 8, 1209–1223 CrossRef CAS PubMed.
- A. F. Radovic-Moreno, T. K. Lu, V. A. Puscasu, C. J. Yoon, R. Langer and O. C. Farokhzad, ACS Nano, 2012, 6, 279–4287 CrossRef PubMed.
- Y. Li, G. Liu, X. Wang, J. Hu and S. Liu, Angew. Chem., Int. Ed., 2016, 55, 1760–1764 CrossRef CAS PubMed.
- M. Changez, V. Koul and A. K. Dinda, Biomaterials, 2005, 26, 2095–2104 CrossRef CAS PubMed.
- A. G. Ashbaugh, X. Jiang, J. Zheng, A. S. Tsai, W.-S. Kim, J. M. Thompson, R. J. Miller, J. H. Shahbazian, Y. Wang and C. A. Dillen, Proc. Natl. Acad. Sci. U. S. A., 2016, 201613722 Search PubMed.
- P. T. Wong, S. Tang, J. Mukherjee, K. Tang, K. Gam, D. Isham, C. Murat, R. Sun, J. R. Baker and S. K. Choi, Chem. Commun., 2016, 52, 10357–10360 RSC.
- S. K. Choi, A. Myc, J. E. Silpe, M. Sumit, P. T. Wong, K. McCarthy, A. M. Desai, T. P. Thomas, A. Kotlyar, M. M. B. Holl, B. G. Orr and J. R. Baker, ACS Nano, 2013, 7, 214–228 CrossRef CAS PubMed.
- S. J. Sonawane, R. S. Kalhapure, S. Rambharose, C. Mocktar, S. B. Vepuri, M. Soliman and T. Govender, Int. J. Pharm., 2016, 504, 1–10 CrossRef CAS PubMed.
- G. Michaud, R. Visini, M. Bergmann, G. Salerno, R. Bosco, E. Gillon, B. Richichi, C. Nativi, A. Imberty and A. Stocker, Chem. Sci., 2016, 7, 166–182 RSC.
- N. Wrońska, A. Felczak, K. Zawadzka, A. Janaszewska, B. Klajnert, M. Bryszewska and K. Lisowska, New J. Chem., 2014, 38, 2987–2992 RSC.
- K. Winnicka, M. Wroblewska, P. Wieczorek, P. T. Sacha and E. A. Tryniszewska, Molecules, 2013, 18, 8607–8617 CrossRef CAS PubMed.
- Y.-J. Zhou, M.-X. Zhang, R. C. Hider and T. Zhou, FEMS Microbiol. Lett., 2014, 355, 124–130 CrossRef CAS PubMed.
- A. Felczak, K. Zawadzka, N. Wrońska, A. Janaszewska, B. Klajnert, M. Bryszewska, D. Appelhans, B. Voit and K. Lisowska, New J. Chem., 2013, 37, 4156–4162 RSC.
- P. V. Kumar, A. Asthana, T. Dutta and N. K. Jain, J. Drug Targeting, 2006, 14, 546–556 CrossRef CAS PubMed.
- M. K. Mishra, K. Kotta, M. Hali, S. Wykes, H. C. Gerard, A. P. Hudson, J. A. Whittum-Hudson and R. M. Kannan, Nanomedicine, 2011, 7, 935–944 CrossRef CAS PubMed.
- H. W. VanKoten, W. M. Dlakic, R. Engel and M. J. Cloninger, Mol. Pharmaceutics, 2016, 13, 3827–3834 CrossRef CAS PubMed.
- S. Szunerits and R. Boukherroub, Chem. Commun., 2012, 48, 8999–9010 RSC.
- N. Khlebtsov, V. Bogatyrev, L. Dykman, B. Khlebtsov, S. Staroverov, A. Shirokov, L. Matora, V. Khanadeev, T. Pylaev and N. Tsyganova, Theranostics, 2013, 3, 167–180 CrossRef CAS PubMed.
- B. Saha, J. Bhattacharya, A. Mukherjee, A. Ghosh, C. Santra, A. K. Dasgupta and P. Karmakar, Nanoscale Res. Lett., 2007, 2, 614 CrossRef CAS.
- G. Burygin, B. Khlebtsov, A. Shantrokha, L. Dykman, V. Bogatyrev and N. Khlebtsov, Nanoscale Res. Lett., 2009, 4, 794 CrossRef CAS PubMed.
- Y.-H. Hsueh, K.-S. Lin, W.-J. Ke, C.-T. Hsieh, C.-L. Chiang, D.-Y. Tzou and S.-T. Liu, PLoS One, 2015, 10, e0144306 Search PubMed.
- M. A. Raza, Z. Kanwal, A. Rauf, A. N. Sabri, S. Riaz and S. Naseem, Nanomaterials, 2016, 6, 74 CrossRef PubMed.
- M. Rai, A. Yadav and A. Gade, Biotechnol. Adv., 2009, 27, 76–83 CrossRef CAS PubMed.
- A. M. Fayaz, M. Girilal, S. A. Mahdy, S. Somsundar, R. Venkatesan and P. Kalaichelvan, Process Biochem., 2011, 46, 636–641 CrossRef.
- H. Gu, P. Ho, E. Tong, L. Wang and B. Xu, Nano Lett., 2003, 3, 1261–1263 CrossRef CAS.
- A. N. Brown, K. Smith, T. A. Samuels, J. Lu, S. O. Obare and M. E. Scott, Appl. Environ. Microbiol., 2012, 78, 2768–2774 CrossRef CAS PubMed.
- A. Rai, A. Prabhune and C. C. Perry, J. Mater. Chem., 2010, 20, 6789–6798 RSC.
- Y. E. Hur, S. Kim, J.-H. Kim, S.-H. Cha, M.-J. Choi, S. Cho and Y. Park, Mater. Lett., 2014, 129, 185–190 CrossRef CAS.
- M. Tudose, D. C. Culita, C. Munteanu, J. Pandele, E. Hristea, P. Ionita, I. Zarafu and M. C. Chifiriuc, J. Inorg. Organomet. Polym. Mater., 2015, 25, 869–878 CrossRef CAS.
- A. Ahangari, M. Salouti, Z. Heidari, A. R. Kazemizadeh and A. A. Safari, Drug Delivery, 2013, 20, 34–39 CrossRef CAS PubMed.
- A. R. Shahverdi, A. Fakhimi, H. R. Shahverdi and S. Minaian, Nanomedicine, 2007, 3, 168–171 CrossRef CAS PubMed.
- M. Banoee, S. Seif, Z. E. Nazari, P. Jafari-Fesharaki, H. R. Shahverdi, A. Moballegh, K. M. Moghaddam and A. R. Shahverdi, J. Biomed. Mater. Res., Part B, 2010, 93, 557–561 CrossRef PubMed.
- Z. E. Nazari, M. Banoee, A. A. Sepahi, F. Rafii and A. R. Shahverdi, Gold Bull., 2012, 45, 53–59 CrossRef CAS.
- A. N. Grace and K. Pandian, Colloids Surf., A, 2007, 297, 63–70 CrossRef CAS.
- D. Bhattacharya, B. Saha, A. Mukherjee, C. R. Santra and P. Karmakar, Nanosci. Nanotechnol., 2012, 2, 14–21 CrossRef.
- R. T. Tom, V. Suryanarayanan, P. G. Reddy, S. Baskaran and T. Pradeep, Langmuir, 2004, 20, 1909–1914 CrossRef CAS PubMed.
- S. Gurunathan, J. W. Han, D.-N. Kwon and J.-H. Kim, Nanoscale Res. Lett., 2014, 9, 373 CrossRef PubMed.
- P. Li, J. Li, C. Wu, Q. Wu and J. Li, Nanotechnology, 2005, 16, 1912 CrossRef CAS.
- A. J. Kora and L. Rastogi, Bioinorg. Chem. Appl., 2013, 2013, 871097 Search PubMed.
- O. Akhavan, E. Ghaderi and A. Esfandiar, J. Phys. Chem. B, 2011, 115, 6279–6288 CrossRef CAS PubMed.
- M. Bottini, N. Rosato and N. Bottini, Biomacromolecules, 2011, 12, 3381–3393 CrossRef CAS PubMed.
- B. Peng, L. Chen, C. Que, K. Yang, F. Deng, X. Deng, G. Shi, G. Xu and M. Wu, Sci. Rep., 2016, 6, 31920 CrossRef CAS PubMed.
- X. Zhang, J. Shen, N. Zhuo, Z. Tian, P. Xu, Z. Yang and W. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 24273–24280 CAS.
- Y. Gao, J. Wu, X. Ren, X. Tan, T. Hayat, A. Alsaedi, C. Cheng and C. Chen, Environ. Sci.: Nano, 2017, 4, 1016–1024 RSC.
- P. Mroz, G. P. Tegos, H. Gali, T. Wharton, T. Sarna and M. R. Hamblin, Photochem. Photobiol. Sci., 2007, 6, 1139–1149 CAS.
- W. Wu, S. Wieckowski, G. Pastorin, M. Benincasa, C. Klumpp, J. P. Briand, R. Gennaro, M. Prato and A. Bianco, Angew. Chem., Int. Ed., 2005, 44, 6358–6362 CrossRef CAS PubMed.
- Y.-P. Sun, B. Zhou, Y. Lin, W. Wang, K. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang and H. Wang, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef CAS PubMed.
- L. Cao, S.-T. Yang, X. Wang, P. G. Luo, J.-H. Liu, S. Sahu, Y. Liu and Y.-P. Sun, Theranostics, 2012, 2, 295–301 CrossRef CAS PubMed.
- Q. Wang, X. Huang, Y. Long, X. Wang, H. Zhang, R. Zhu, L. Liang, P. Teng and H. Zheng, Carbon, 2013, 59, 192–199 CrossRef CAS.
- C.-I. Weng, H.-T. Chang, C.-H. Lin, Y.-W. Shen, B. Unnikrishnan, Y.-J. Li and C.-C. Huang, Biosens. Bioelectron., 2015, 68, 1–6 CrossRef CAS PubMed.
- D. Zhong, Y. Zhuo, Y. Feng and X. Yang, Biosens. Bioelectron., 2015, 74, 546–553 CrossRef CAS PubMed.
- M. J. Meziani, X. Dong, L. Zhu, L. P. Jones, G. E. LeCroy, F. Yang, S. Wang, P. Wang, Y. Zhao and L. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 10761–10766 CAS.
- A. S. Krishna, C. Radhakumary, M. Antony and K. Sreenivasan, J. Mater. Chem. B, 2014, 2, 8626–8632 RSC.
- M. Thakur, S. Pandey, A. Mewada, V. Patil, M. Khade, E. Goshi and M. Sharon, J. Drug Delivery, 2014, 2014, 282193 Search PubMed.
- N. Gogoi and D. Chowdhury, J. Mater. Chem. B, 2014, 2, 4089–4099 RSC.
- R. Jijie, A. Barras, J. Bouckaert, N. Dumitrascu, S. Szunerits and R. Boukherroub, J. Mater. Chem., 2017 Search PubMed , submitted.
- H. Pandey, V. Parashar, R. Parashar, R. Prakash, P. W. Ramteke and A. C. Pandey, Nanoscale, 2011, 3, 4104–4108 RSC.
- S. Boulahneche, R. Jijie, A. Barras, F. Chekin, S. K. Singh, J. Bouckaert, M. S. Medjram, S. Kurungot, R. Boukherroub and S. Szunerits, J. Mater. Chem. B, 2017 10.1039/C7TB00687J.
- W. Wu, S. Wieckowski, G. Pastorin, M. Benincasa, C. Klumpp, J.-P. Briand, R. Gennaro, M. Prato and A. Bianoc, Angew. Chem., Int. Ed., 2005, 44, 6358–6362 CrossRef CAS PubMed.
- O. Veiseh, J. W. Gunn and M. Zhang, Adv. Drug Delivery Rev., 2010, 62, 284–304 CrossRef CAS PubMed.
- H. Mok and M. Zhang, Expert Opin. Drug Delivery, 2013, 10, 73–87 CrossRef CAS PubMed.
- H. Gu, K. Xu, C. Xu and B. Xu, Chem. Commun., 2006, 941–949 RSC.
- A. J. Kell, G. Stewart, S. Ryan, R. Peytavi, M. Boissinot, A. Huletsky, M. G. Bergeron and B. Simard, ACS Nano, 2008, 2, 1777–1788 CrossRef CAS PubMed.
- B. M. Geilich, I. Gelfat, S. Sridhar, A. L. van de Ven and T. J. Webster, Biomaterials, 2017, 119, 78–85 CrossRef CAS PubMed.
- H. Gu, P.-L. Ho, K. W. Tsang, L. Wang and B. Xu, J. Am. Chem. Soc., 2003, 125, 15702–15703 CrossRef CAS PubMed.
- M. Zhu, W. Liu, H. Liu, Y. Liao, J. Wei, X. Zhou and D. Xing, ACS Appl. Mater. Interfaces, 2015, 7, 12873–12881 CAS.
- J. Rao, I. J. Colton and G. M. Whitesides, J. Am. Chem. Soc., 1997, 119, 9336–9340 CrossRef CAS.
- B. K. Hubbard and C. T. Walsh, Angew. Chem., Int. Ed., 2003, 42, 730–765 CrossRef CAS PubMed.
- A. P. A. P. Wight and M. E. Davis, Chem. Rev., 2002, 102, 3589–3614 CrossRef CAS PubMed.
- M. Vallet-Regi, A. Ramila, R. Del Real and J. Pérez-Pariente, Chem. Mater., 2001, 13, 308–311 CrossRef CAS.
- M. Vallet-Regı, J. Doadrio, A. Doadrio, I. Izquierdo-Barba and J. Pérez-Pariente, Solid State Ionics, 2004, 172, 435–439 CrossRef.
- A. Doadrio, E. Sousa, J. Doadrio, J. P. Pariente, I. Izquierdo-Barba and M. Vallet-Regı, J. Controlled Release, 2004, 97, 125–132 CrossRef CAS PubMed.
- V. Nairi, L. Medda, M. Monduzzi and A. Salis, J. Colloid Interface Sci., 2017, 497, 217–225 CrossRef CAS PubMed.
- F. Sevimli and A. Yılmaz, Microporous Mesoporous Mater., 2012, 158, 281–291 CrossRef CAS.
- Z. Li, K. Su, B. Cheng and Y. Deng, J. Colloid Interface Sci., 2010, 342, 607–613 CrossRef CAS PubMed.
- M. Rosemary, I. MacLaren and T. Pradeep, Langmuir, 2006, 22, 10125–10129 CrossRef CAS PubMed.
- X. Shi, Y. Wang, L. Ren, N. Zhao, Y. Gong and D.-A. Wang, Acta Biomaterial., 2009, 5, 1697–1707 CrossRef CAS PubMed.
- C.-M. Han, E.-J. Lee, H.-E. Kim, Y.-H. Koh and J.-H. Jang, Thin Solid Films, 2011, 519, 8074–8076 CrossRef CAS.
- R. K. Singh, T.-H. Kim, J.-J. Kim, E.-J. Lee, J. C. Knowles and H.-W. Kim, RSC Adv., 2013, 3, 8692–8704 RSC.
- J. C. Doadrio, E. M. Sousa, I. Izquierdo-Barba, A. L. Doadrio, J. Perez-Pariente and M. Vallet-Regí, J. Mater. Chem., 2006, 16, 462–466 RSC.
- N. Mas, I. Galiana, L. Mondragon, E. Aznar, E. Climent, N. Cabedo, F. Sancenon, J. Ramano Murguia, R. Martinez-Manez, M. D. Marcos and P. Amoros, Chem. – Eur. J., 2013, 19, 11167–11171 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |