
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlasma source mass spectrometry for radioactive waste characterisation in support of nuclear decommissioning: a review
Ian W.
Croudace
 *a,
Ben C.
Russell
ab and
Phil W.
Warwick
a
*a,
Ben C.
Russell
ab and
Phil W.
Warwick
a
aGAU-Radioanalytical, University of Southampton, NOCS, Southampton, SO14 3ZH, UK. E-mail: iwc@noc.soton.ac.uk
bNational Physical Laboratory, Teddington, TW11 0LW, UK
First published on 20th December 2016
Abstract
The efficient characterization of nuclear waste materials represents a significant challenge during nuclear site decommissioning, with a range of radionuclides requiring measurement in varied and often complex sample matrices. Of the available measurement techniques, inductively coupled plasma mass spectrometry (ICP-MS) has traditionally been applied to long-lived radionuclides, particularly in the actinide series. With recent advances in the technique, both the sensitivities achievable and number of radionuclides potentially measurable has expanded, with the reduced procedural time offering significant economic benefits to nuclear site waste characterization compared with traditional radiometric (typically alpha and beta spectrometry) techniques. This review provides a broad assessment of recent developments, improvements in capability and describes the advantages and drawbacks of ICP-MS with regards to sample introduction and instrument design. The review will be of interest to international agencies concerned with nuclear decommissioning as well as nuclear site laboratories, project managers and sites involved in environmental monitoring and nuclear forensics.
1 Introduction
Over the last 30 years, the application of ICP-MS for radionuclide quantification has grown significantly. Initially, ICP-MS techniques focused on longer lived radionuclides where their low specific activities favored atom-counting over radiometric techniques (typically alpha and beta spectrometry). For example, U, Th, Pu, 99Tc, and 237Np have been measured using ICP-MS since its early days. With the shift to decommissioning, other long lived but less abundant radionuclides such as 93Zr have also been quantified using ICP-MS. In addition, improvements in instrument sensitivity achieved through advances in sample introduction, mass spectrometry configuration and design and vacuum pump technologies, has opened up the possibility of using ICP-MS for quantification of shorter-lived radionuclides such as 90Sr, significantly reducing the analytical time for such analyses (Fig. 1).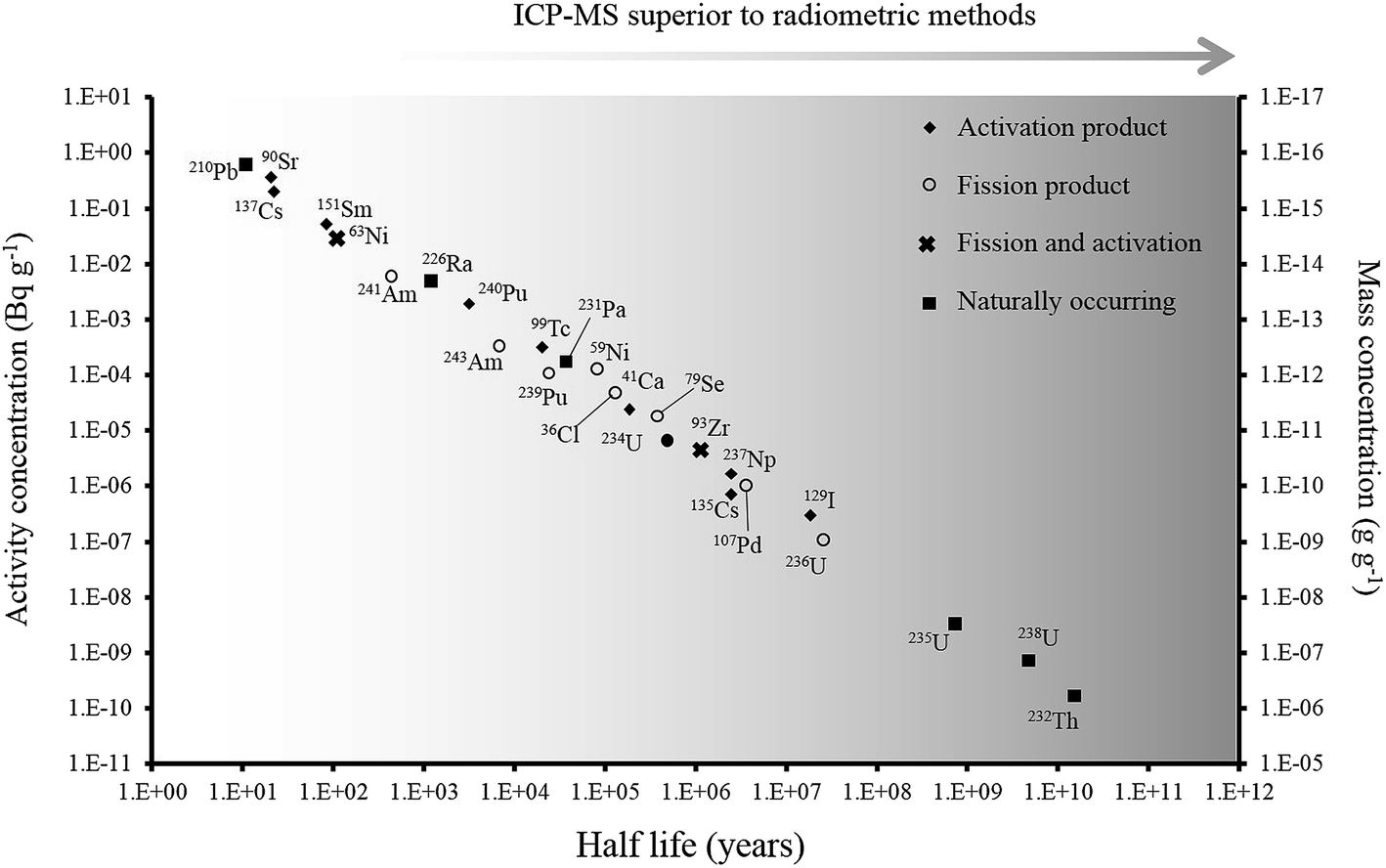 | ||
| Fig. 1 Half-life versus minimum detectable activity (activity that gives count rate that is >3 times standard deviation of the background count rate) and concentration of selected radionuclides, labelled according to their method of production. Adapted from Russell et al. (2014).1 | ||
Historically, radionuclide analysis within the nuclear sector has supported environmental monitoring programs, health physics, process control, effluent and waste characterization and personnel monitoring. Analytical programs tended to focus on relatively short-lived radionuclides that were likely to contribute significantly to personnel and public doses or contamination of the workplace, or which provided information on reactor performance. However, in recent years, many first and second generation nuclear facilities worldwide have either entered or are approaching shutdown and decommissioning phases. This has led to a rapidly increasing demand for radionuclide analysis to characterize the wastes arising from site decommissioning programs (e.g. plant contamination assessments, radioactively contaminated land etc.)
As well as focusing on radionuclides that contribute to radiological worker dose and waste activity inventories in the short term, analytical strategies are now also required to quantify the low-abundance long lived radionuclides that will impact on waste repository safety cases over 103 to 106 years. The change in emphasis to decommissioning has resulted in a number of analytical challenges. These include the need for rapid radionuclide characterization of wastes prior to sentencing, the provision of techniques capable of measuring low-abundance long-lived radionuclides in the presence of other significantly higher abundance radionuclides and the requirement to analyze diverse and complex matrices. In all these cases, mass spectrometric techniques, and particularly ICP-MS, offer some unique capabilities, which help to address these challenges. Given the increasing expectations facing radioanalytical science arising from decommissioning and the expanding programme of decommissioning worldwide, it is timely to review the state-of-the-art regarding ICP-MS analysis of radionuclides and to explore how the technique could be more widely applied in the future.
2 History
There have been numerous developments in the field of mass spectrometry.2–20 In 1983 a significant advance was the introduction of the first commercial ICP-QMS,21,22 an instrument that allowed elements and isotope rations to be measured at high sensitivity. The ICP-QMS uses an inductively coupled argon plasma as an excitation source to ionize the sample and a quadrupole mass spectrometer as an analyser to separate and selectively transmit analyte ions of a single mass-to-charge ratio (m/z) to the detector. During this process, sample ions rapidly undergo large temperature (6000 K-to-room temp.) and pressure (760 to 10−6 Torr) reductions. In essence, ions are systematically transferred from the plasma to the detector in a highly controlled electrostatic field within a dynamically increasing vacuum which was followed several years later by early measurements of radionuclides.22–27 The rapidity of ICP-QMS and ability to simultaneously measure multiple radionuclides were established as major advantages compared to alpha and beta counting techniques,28–30 whilst the robustness of the technique better-suited to routine analysis compared to alternative mass spectrometric techniques, specifically thermal ionization mass spectrometry (TIMS).30,31 Additionally, sample introduction into ICP-QMS could be achieved from a solid, liquid or gas.32Early studies were critical in establishing the uncertainties associated with radionuclide detection by ICP-QMS and considerations for instrumental setup. This included sample pre-treatment prior to sample introduction to improve detection limits,23 the impact of sample introduction on sensitivity and interference removal,27,29,33–35 the importance of abundance sensitivity in removing peak tailing,26 and the use of internal standardization to account for matrix effects.36 As well as advances in quadrupole instrument design, the development of other ICP-MS setups including sector field (ICP-SFMS), collision/reaction cell instruments and multiple detector systems (MC-ICP-MS) has increased the sensitivity, interference-removal capability, and the number of nuclides measurable (Fig. 2). This has significantly expanded the toolbox for the radioanalytical chemist with regards to nuclear waste characterization and decommissioning.
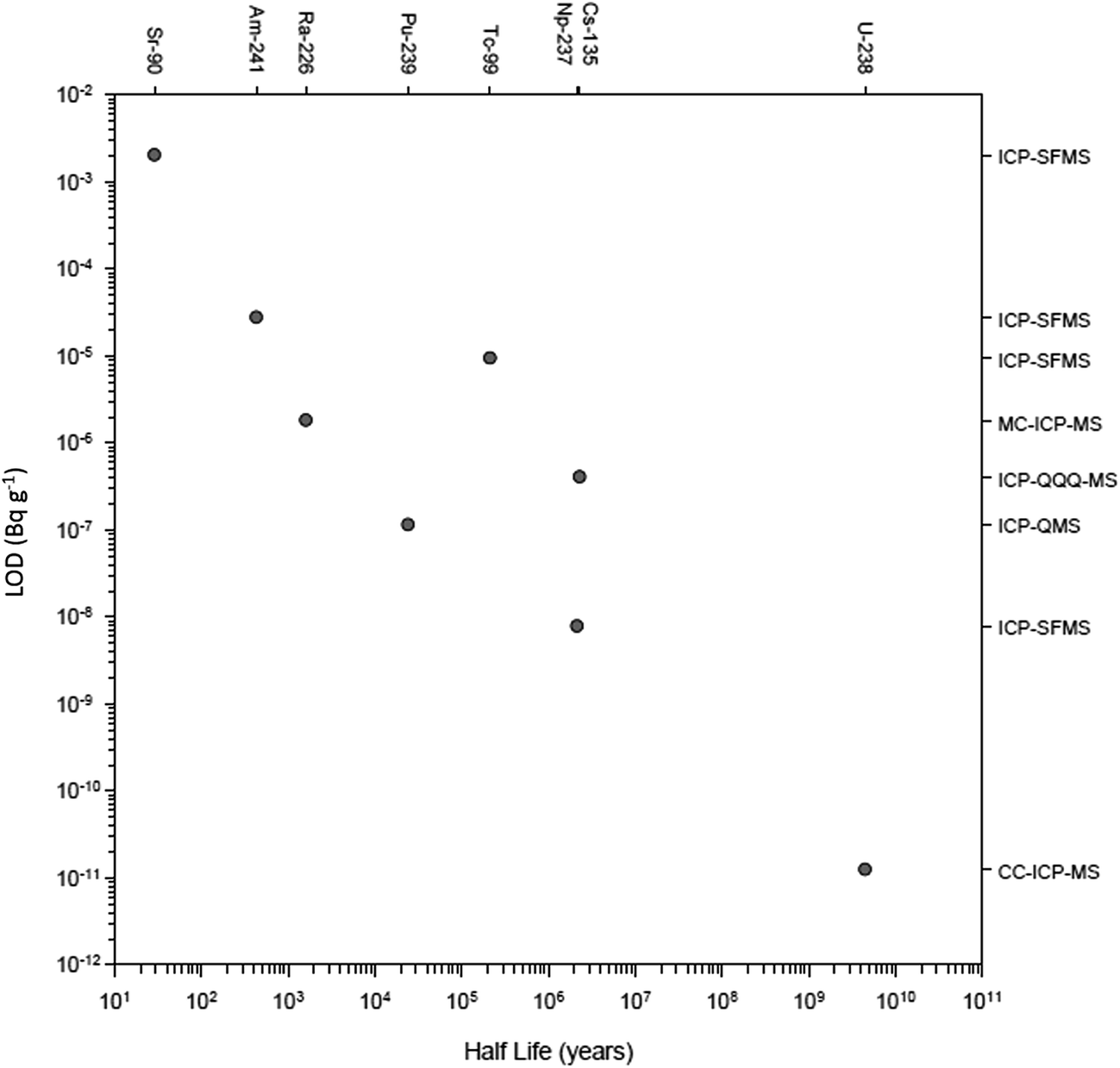 | ||
| Fig. 2 Recent detection limits achieved for multiple radionuclides as a function of their half-life. | ||
3 Mass spectrometry vs. radiometric analysis
The specific activity (the rate of radioactive decay for a given mass of isotope) for a radionuclide is inversely proportional to the half-life and hence, for long lived radionuclides, more sensitive measurements can potentially be achieved by determining the concentration rather than the activity of the radionuclide (Fig. 1). More recently, there has been a growing interest in quantifying long-lived, low abundance radionuclides (e.g.41Ca, 59Ni, 63Ni, 93Zr, 135Cs, 151Sm) formed through fission or neutron activation. These radionuclides were not considered significant during operational phases as their contribution to operator dose was significantly lower than for the short lived radionuclides such as 90Sr and 137Cs (Fig. 3). However, such radionuclides contribute significantly to the long-term nuclear waste repository dose estimates. Measurement of these radionuclides radiometrically is challenging as their emissions are often masked by the more abundant short-lived isotopes. For example, in fresh fission wastes the 135Cs![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :137Cs atomic ratio is approximately 1:1 whereas the activity ratio is 1:80000. In such instances, mass spectrometric techniques are the best analytical approach and bring significant benefits through analytical cost saving and sample throughput.
:137Cs atomic ratio is approximately 1:1 whereas the activity ratio is 1:80000. In such instances, mass spectrometric techniques are the best analytical approach and bring significant benefits through analytical cost saving and sample throughput.
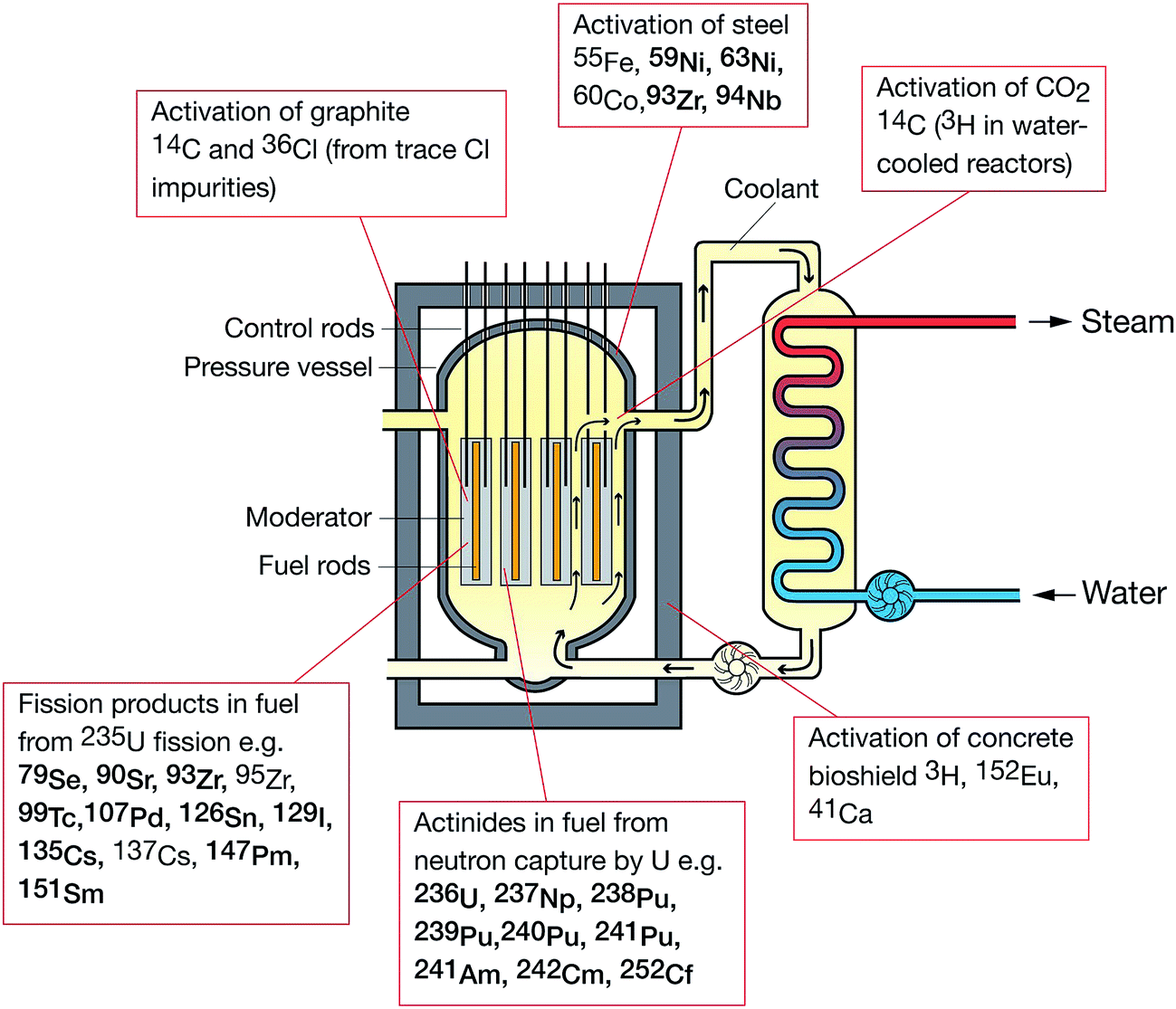 | ||
| Fig. 3 Formation route of medium to long-lived radionuclides in a nuclear reactor. | ||
In some cases, radiometric techniques are limited to measurement of radionuclides with relatively short half-lives. Consequently, isotope ratio measurements are limited to monitoring of nuclear incidents shortly after the event (e.g.134Cs/137Cs measurements at Fukushima), and are no longer applicable to samples affected by atmospheric weapons test fallout or Chernobyl. Advances in ICP-MS have put this technique in a position where 135Cs (2.3 million year half-life) is measurable, enabling determination of the 135Cs/137Cs ratio, expanding measurement options over longer timescales. In other cases, radiometric techniques are unable to separate isotopes with similar decay energies e.g.239Pu and 240Pu, whereas ICP-MS is capable of accurate measurement of the 239Pu/240Pu ratio, which can vary significantly depending on the source of contamination, and therefore represents a significant advance with regards to routine monitoring and nuclear forensics.
The significant reduction in counting time compared to radiometric techniques was identified as an early advantage of ICP-MS for particular radionuclides. As the technique has advanced, there has been a focus on reducing the sample preparation time using techniques such as online chemical separation coupled directly to the instrument, interference separation using an integrated collision or reaction cell, and improvements in sensitivity and isotope ratio accuracies using sector-field and multi-collector instruments, respectively.
Accurate and low uncertainty measurements of the decay properties of radionuclides is of importance in numerous fields including decay heat calculations in the nuclear industry and calibration of instruments.37,38 ICP-MS can achieve low uncertainty measurement of the number of atoms in a sample, which combined with radiometric activity measurements allow determination of the decay constant and half-life. This information is used as part of development of technical standards, which improves measurement with regards to measurement quality, reproducibility and comparison between studies39,40
4 Sample introduction
Development of high sensitivity sample introduction has been a vital part of improving detection limits, and is potentially a key factor in reducing isobaric, polyatomic and tailing interferences depending on the instrumental setup.41,42 A number of studies have compared the performance of different sample introduction techniques41,43,44 and these are summarised in Table 1.| Technique | Comments | Radionuclide applications ref. |
|---|---|---|
| Solution nebulization | • Range of designs, some with high tolerance to solid content | 45–52 |
| • Sample uptake rate as low as 50 μL min−1 | ||
| • Relatively high oxide and hydride formation | ||
| • Sample to plasma transfer efficiency can be 1–2% | ||
| Desolvating sample introduction | • Reduced solvent loading, low oxide and hydride formation | 53–57 |
| • Ultrasonic nebulisers have high sample uptake rate (∼1 mL min−1) | ||
| Direct injection | • 100% sample to plasma efficiency | 34, 41, 48 and 58 |
| • Increased solvent loading into plasma increases oxide and hydride formation | ||
| Flow injection (FI) | • Direct, real-time measurements | 59–62 |
| • Reduced sample preparation compared to offline separation | ||
| • 100% sample to plasma efficiency | ||
| • High maintenance | ||
| • Ultra-trace measurement difficult | ||
| Sequential injection (SI) | • Evolution of flow injection | 63 |
| • Delivery of eluents, washing solutions and standards without reconfiguring the manifold | ||
| • Potential cross-contamination using a single manifold | ||
| Lab on chip | • Downscaling of sequential injection | 64 |
| • Reagent-based assay to sub-μL levels | ||
| Laser ablation (LA-ICP-MS) | • Direct measurement of solid samples | 65–76 |
| • Surface and depth profiling, or measurement of single particles | ||
| • Reduced hydride and oxide formation because of ‘dry’ plasma | ||
| • Solution nebulization preferable for bulk sample composition | ||
| • Lack of reference materials | ||
| Electro thermal vaporization (ETV) | • High analyte transport efficiency (20–80%) | 77–81 |
| • Low oxide and hydride formation | ||
| • Can handle complex sample matrices | ||
| • Inferior detection limit compared to solution nebulization | ||
| Glow discharge | • Complete material characterization | 82 and 83 |
| • Interferences can arise from discharge gas and sample matrix | ||
| • Isobaric interferences prevents direct determination of radionuclides | ||
| High performance liquid chromatography (HPLC) | • Rapid separation compared to offline chemical separation | 48, 74, 84 and 85 |
| • Separation and detection of multiple radionuclides from the same sample | ||
| Cold plasma | • Rapid isobaric interference separation compared to offline chemical separation | 86 and 87 |
| • Sensitivity dependent on sample matrix | ||
| • Several potential applications e.g.79Se/79Br, 126Sn/126Te, 129I/129Xe | ||
| Capillary electrophoresis | • Low sample volumes (nL to μL) | 88–90 |
| • Rapid separation compared to offline chemical separation |
5 Quadrupole ICP-MS (ICP-QMS)
ICP-QMS achieved the first successful measurements of radionuclides by ICP-MS. Early instruments were unable to achieve separation of isobaric and polyatomic interferences because of their limited abundance sensitivity. Their use was originally focused on higher mass radionuclides such as 238U that do not suffer from the same number of interferences as lower mass radionuclides. However, advances in instrumental sensitivity, versatility with regards to sample introduction, and equipping certain instruments with a collision/reaction cell has improved detection limits and expanded applications over time.MicroMass Ltd. (Wilmslow, UK) introduced an early version of a collision cell instrument (Platform ICP-MS), with the aim of thermalizing ions and dissociating disturbing molecular ions such as argides. Compared to operating without a collision cell, ion transmission, sensitivity and isotope ratio precision can be improved.74 However, there are conflicting views on the impact of ICP-CC-MS on abundance sensitivity. On the one hand, collisions in the cell can reduce the ion kinetic energy, potentially improving the abundance sensitivity by increasing the residence time of ions in the mass analyser which results in better mass separation.49 However, the collision gas increases pressure in the mass analyser, with residual gas ions leading to scattering of ions in the quadrupole, which at higher gas flow rates can have a negative impact on the abundance sensitivity.
The Perkin Elmer Elan 6100 Dynamic Reaction Cell (DRC) (based on Elan 6000 ICP-QMS) is an early example of a reaction cell instrument, capable of operating with multiple gases including NH3, CH4, H2 and He. DRC instruments are equipped with a Bandpass Tuning feature, which offers mass discrimination against interfering by-products formed in the cell, whilst allowing analyte transmission. This compares to a collision cell setup that often operates with an energy filter to prevent newly formed interferences from leaving the cell. This can lead to an energy overlap between the analyte and interferences that may ultimately increase instrument backgrounds, decrease analyte signal and adversely impact detection limits.91 Bandura et al. (2005) extensively studied cell-based separation of radionuclides from overlapping isotopes using a Perkin Elmer Elan DRC, with the results outlining the cell gas used and the likelihood of the reaction occurring.92 The DRC series has been updated with the NexION series, which is equipped with a quadrupole ion deflector that turns the ion beam 900 prior to the entrance to the collision/reaction cell. The NexION series is also equipped with an additional hyper skimmer cone to improve the removal of unionized material.
The Agilent 8800 Triple Quadrupole ICP-MS (ICP-QQQ-MS) consists of two quadrupoles positioned either side of a collision–reaction cell (termed the Octopole Reaction System, ORS). It is in effect an ICP-MS/MS or tandem mass spectrometer. Positioning a quadrupole before the entrance to the cell means the ion beam can be mass filtered prior to the cell entrance, enabling greater control over the ions entering the cell, preventing undesirable secondary polyatomic ions from forming in the cell. Secondly, the additional quadrupole improves the abundance sensitivity, with a theoretical value of 10−14. This is advantageous for radionuclides affected by tailing from a stable isotope of the same element e.g.88Sr on 90Sr, and 127I on 129I. The 8800 has recently been superseded by the 8900, offering improvements including more rapid sample acquisition, measurement at higher masses (beneficial for actinide-based cell products) and introduction of samples with up to 25% total dissolved solid content.
6 Sector field ICP-MS (ICP-SFMS)
The introduction of ICP-SFMS offered lower background and higher sensitivity compared with ICP-QMS, enabling lower limits of detection for radionuclide measurement. The counting efficiency of ICP-SFMS is generally on the order of ∼0.1%, with low background signals of <1 counts per second, compared to typical ICP-QMS values of 0.01% counting efficiency and several counts per second background.93,94 ICP-SFMS can therefore theoretically analyze smaller bulk samples with lower analyte concentrations.95 Operating at higher mass resolution can be used to reduce or remove isobaric and polyatomic interferences, however, this is at the expense of ion transmission and therefore sensitivity.94 Reproducibility can also be affected, with typical values at low resolution of <0.02%, compared to <0.1% at medium and high resolution.96 Additionally, overlapping peaks from stable isobars affecting radionuclide detection cannot be resolved, even at high resolution. Therefore, ICP-SFMS is generally operated at low mass resolution to maximize instrumental sensitivity,97,98 with interference removal dependent on sample introduction and/or chemical separation.Multi-collector instruments (MC-ICP-MS) are fitted with multiple ion counting detectors, which eliminates the need to cycle a number of small ion beams through a single detector. In addition, the effects of ion beam instability are eliminated, and flat-topped peaks and high precision isotope ratios (∼0.001%) are achievable, which is not possible using single detector instruments.51,54,99 For example, the Thermo Neptune (equipped with eight Faraday cups that are interchangeable with ion counting detectors) are capable of flat-topped peaks at medium mass resolution (4000), unlike single detector ICP-SFMS instruments. Multiple collectors also increase the signal in proportion to the number of available ion counting channels. For example, Taylor et al. (2003) demonstrated that with two ion-counting channels, the dwell time of 240Pu/239Pu is doubled, or only half the amount of plutonium is needed to give similar counting statistics to the peak jumping method.51
Finally, the Spectro-MS is a fully simultaneous, double focusing ICP-SFMS consisting of an entrance slit, ESA, energy slit, magnetic sector field and a solid state detector split into 4800 channels, which is capable of measuring the entire mass system.100 For every analysis, the entire mass spectrum is captured, rather than focusing over a single mass unit. A summary of commercially available ICP-MS is given in Table 2.
| Instrument type | Thermo Scientific | Agilent | Perkin Elmer | Nu | Spectro |
|---|---|---|---|---|---|
| Collision/reaction cell | iCap Q | 7800 | NexION 300 series | ||
| 7900 | |||||
| 8900 | |||||
| Sector field | Element 2 | Attom | Spectro MS | ||
| Element XR | |||||
| Multi-collector | Neptune Plus | Plasma II | |||
| Plasma 1700 |
7 Future developments in hardware
7.1 Miniaturization
Current applications of miniature instruments include drug screening of people, packages, luggage and vehicles, as well as forensics and environmental analysis.101–103 The instruments developed can be self-contained with volumes of approximately 50 cm3, although this may exclude ionization sources and compressed gas cylinders.103 There are a number of ionization techniques (Table 3) and commercially available instruments.104 The pump and sample introduction system have a significant influence on instrument size and weight, whilst miniaturization can lead to a reduction in performance. Instruments typically have detection limits in the ppm to ppb range,105,106 and to date there have been no applications for nuclear decommissioning. However, this approach potentially offers no sample preparation, and on-site, real-time analysis without having to return samples to an off-site laboratory.| Abbreviation | Full name |
|---|---|
| DBDI | Dielectric barrier discharge ion source |
| DESI | Desorption electrospray ionization |
| DART | Direct analysis in real time |
| LEMS | Laser electrospray mass spectrometry |
| FAPA | Flowing atmospheric pressure afterglow |
| LTP | Low temperature plasma |
| LP-DBDI | Low pressure dielectric barrier discharge ionization |
7.2 Extreme environments
There have been a limited number of studies into instrument contamination following handling of active samples. Oak Ridge National Laboratory investigated contamination of ICP-QMS from 90Sr and 137Cs in the pump oil, and swabs of solid components.107 No activity was measured on the detector, with the majority in the interface region. Whilst any activity on the sample cone was burned off by the torch, some was detected on the skimmer cone. Two separate studies found contamination on the quadrupoles.108,109There are a number of variables that must be considered, including the ion transmission, which is likely to vary depending on the age of the instrument. Contamination is also likely to vary with sample introduction system, instrument design, radionuclide and activity analyzed, and the sample throughput.110 It is therefore good practice to monitor solid components such as cones, detectors and quadrupoles when they are replaced, prior to disposal. Some instruments have been modified in order to handle higher activity samples, with the nebulizer, torch and sliding interface situated inside a glovebox.97,111–116
8 Application to radionuclide measurement
The following radionuclides, of direct interest to the nuclear decommissioning, waste characterisation and repository safety cases, have been considered as part of this review: 36Cl, 41Ca, 59Ni and 63Ni, 79Se, 90Sr, 93Zr, 99Tc, 107Pd, 129I, 135Cs and 137Cs, 151Sm, 210Pb, 226Ra and 228Ra, 231Pa, Th and U isotopes, 237Np, Pu isotopes, 241Am, and isotopes of Cm and Cf. The generalised origin of these radionuclides is indicated in Fig. 3.8.1 Sample preparation
A range of possible dissolution methods exist (Table 4), with the choice of technique guided by the sample matrix and radionuclide(s) of interest. Some materials that will dissolve easily or with persistence in mineral acids (or in some cases using microwave-induced heating in pressurised vessels) followed that approach. Up to the mid-1990s, radioanalytical practitioners encountering samples that contained components resistant to acid digestion procedures would have followed a fusion-based approach where the sample matrix was completely opened-out using fluxes such as alkali carbonates and fluorides. In 1996, Croudace and co-workers,117 effectively demonstrated the significant benefits of using lithium borate fusion as a rapid and effective digestion method to the radioanalytical community. Prior to this, borate fusion was almost exclusively employed for elemental analysis (X-ray fluorescence analysis and ICP-OES) where it was established for its broad sample dissolution capability in an effective, safe, rapid manner and where the glasses formed could be readily dissolved in mineral acids. Up to the 1996 study referred to, radioanalytical practitioners tended to use classical (and less effective) methods such as mineral acid leaching or fusions with alkali carbonates, fluorides or hydroxides. Croudace et al. (1998) noted that the chemical nature of the radionuclides and their location (lattice-bound or adsorbed) needed to be considered and that some actinides such as Pu can often be digested using mineral acids, whereas high fired Pu was more intransigent and required a total dissolution techniques e.g. borate fusion.51,117 Similarly, whilst 135Cs and 137Cs have been recovered by acid leaching for a range of sample matrices, it was proven that complete recovery in clay-rich sediments was only achievable using lithium borate fusion.1 Additionally, certain radionuclides (99Tc and 129I) suffer from losses due to volatility, which must be considered during sample preparation. A summary of sample digestion methods available for radionuclides has recently been published.119| Digestion or analyte extraction method | Problems/comments | Silicates | Oxides | Sulphates | Carbonates | Borates | Phosphates | Metals, carbides, silicidesa | References | |
|---|---|---|---|---|---|---|---|---|---|---|
| a Some samples (metals, carbides and disilicides may require an oxidative pretreatment prior to borate fusion. Adapted from Croudace et al. 2016.119 | ||||||||||
| Acids & alkalis | HCl and/or HNO3 | Microwave digestion, heating in PTFE or PFA pressure vessels may be effective. Full recovery of analytes potentially low. Oxidation of sample may be required to prevent volatilization. Difficult to achieve full dissolution. Possible volatility issues with: As, Ge, Po, S, Sb, Se, Tc | ✗ | ✗ | ✗ | 120 | ||||
| HF/HClO4 acid mix | Only small sample masses readily treatable. HF needs to be removed prior to analysis. Insoluble fluoride precipitates in large sample volumes. Perchlorates potentially explosive. Frequently requires the use of HCl and/or HNO3. Possible volatility issues with: As, B, Ge, Po, Sb, Tc | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | 121 | ||
| HF/H2SO4 acid mix | Small sample volumes treatable. HF needs to be removed prior to analysis. Many evaporation stages | ✗ | 120 | |||||||
| Caustic digests | Widely applied to dissolve halogens, Tc | |||||||||
| Traditional alkali fusions | NaOH fusion | Opens out mineral lattices but requires lengthy post fusion treatment. Dissolution of Pt hardware possible | ✗ | ✗ | ✗ | 122 | ||||
| NaCO3 fusion | Opens out mineral lattices but requires lengthy treatment. Dissolution of Pt hardware possible. Elevated Pb or Fe(II) will alloy with Pt hardware. Possible volatility issues with: As, Hg, Po, Tc, Tl, Se | ✗ | ✗ | ✗ | ✗ | 123 | ||||
| Na2O2 fusion or sinter with acid digestion | Attack of Pt hardware possible. Typical fusion temperature of 250–500 °C. Small sample volumes treatable. Time intensive procedure to dissolve the alkaline fusion cake. Possible volatility issues with Au & Ru | ✗ | ✗ | 124 | ||||||
| Alkali fluoride followed by pyrosulfate | Hazardous as HF produced; requires treatment with pyrosulfate to remove fluorides. Will attack Pt hardware | ✗ | ✗ | 125 | ||||||
| Borate fusions | Borate fusion ± acid digestion | Flexible method with no problems. Effectively digests most materials (some may require an oxidant pretreatment) and is ideal for many elemental and isotopic analysis purposes. High purity lithium borate fluxes used to ensure low analytical blanks. Sample size can vary from 0.1–10 g. Sample: flux ratios from 1:1 upward. Pt–Au crucibles used which are easily cleaned. Typical fusion temperature <1000–1200 °C. Possible volatility issues with: Hg, Pb Po & Tl |
✗ | ✗ | ✗ | ✗ | ✗ | 117 | ||
| Fluxless fusion | Flux free fusion ± acid digestion | Small sample volumes treatable. Conducted in inert Ar atmosphere. Typical fusion temperature >1300 °C. May require addition of SiO2 and MgO if silicate poor to help glass formation. Possible volatility issues with Hg, Pb & Tl | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | 126 |
| Thermal oxidation | Analyte(s) trapped in bubbler or condenser | Used to liberate volatile radionuclides e.g. pyrolyser, HBO2 | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| Laser ablation | Direct ablation, Viridiscan, LIBS/ICP-MS | Effective in liberating small quantities of elemental material in a controlled manner. Ablation can occur in carrier gas, by trapping in a filter medium or under acid to reduce later digestion problems | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | 127–129 |
8.2 Chlorine-36
Chlorine-36 (half-life 3.02 × 105 years) is formed by neutron activation of stable 35Cl, which is present as an impurity in concrete and other reactor components.130,131 Concrete and graphite wastes generated from decommissioning are the main sample matrices of interest, with mg kg−1 concentrations of 35Cl in concrete, combined with the high neutron capture cross-section (43 barns) and high volume of waste concrete that must be characterised.130 Chlorine-36 is sometimes measured alongside 129I, and the volatility of both nuclides must be considered during sample preparation. Past separation techniques include leaching from concrete, followed by addition of oxidants to convert Cl and I to halides, which are then trapped in sodium hydroxide.130 Alternatively, precipitation of Cl as AgCl followed by ion exchange chromatography has been applied to concrete, aluminium and graphite samples,132 whilst an extraction chromatographic material (CL resin) was developed by Triskem International that is applicable to separation of 36Cl and 129I from decommissioning samples.133Chlorine-36 is a beta emitting radionuclide (Emax = 0.71 MeV) that can be measured by liquid scintillation counting (LSC), with Ashton et al. (1999) achieving a minimum detectable activity of 9.7 mBq g−1 in concrete samples when measured in combination with 129I.130 In a separate study, Hou et al. (2007) achieved a LOD of 14 mBq in various decommissioning samples, with the Cl chemical yield assessed by ICP-SFMS (MicroMass Plasma Trace 2).132 The LOD for ICP-MS measurement for stable Cl was 0.01 mg kg−1, equivalent to 12.2 Bq kg−1 36Cl. The specific activity of 36Cl (1.07 × 109 Bq g−1) makes it well suited to mass spectrometric measurement, however, there is no known ICP-MS procedure for measurement of 36Cl. Aside from isobaric 36S (36.0% abundance), peak tailing from 35Cl, and isobaric overlap 36Ar in the plasma gas must be overcome, which will be dependent on the abundance sensitivity of the instrument and the use of a collision or reaction cell, respectively. AMS is capable of separating 36Cl from the 36S isobar, and has achieved a detection limit of 0.1 Bq kg−1 in food for sample sizes of 3–4 g,131 with good reproducibility (<2%) for samples with a 36Cl/Cl ratio >10−12.134
8.3 Calcium-41
Calcium-41 (t1/2 = 1.03 × 105 years) is formed through thermal neutron capture of stable 40Ca (natural abundance 96.94%), and also naturally through cosmic ray spallation. The latter has led to interest in measurement in the fields of cosmochemistry and geomorphology, whilst the high bioavailability is of interest in biomedical tracing in the progress of bone disease.135–141 Calcium-41 is present in reactor shield concrete, and is therefore a key radionuclide with regards to high-volume low and intermediate level waste originating from decommissioning, as well as in long-term monitoring of nuclear waste repositories.140,142–144Decay of 41Ca to 41K ground state by electron capture emits low energy X-rays and Auger electrons (0.3–0.6 keV), and is therefore measurable by X-ray spectrometry or LSC.139,142 X-ray spectrometry is straightforward but the low counting efficiency and low abundance of X-rays (11.4% for 3.31 keV) results in low sensitivity.142 This also impacts measurement by LSC, which requires separation of other radionuclides prior to measurement.142,143 A detection limit of 0.02 Bq g−1 was achieved for a 5 g concrete sample following a 60 minute count time.142 Calcium-41 has been successfully separated from the bulk matrix and radiometric interferences by numerous techniques including ion exchange, liquid–liquid extraction, liquid membrane separation, and precipitations including calcium fluoride, carbonate and oxalate.139,143,145
The long half-life of 41Ca and low energy X-ray and Auger electrons makes it well-suited to mass spectrometric determination, with an additional interest in measurement of the 41Ca:40Ca ratio in nuclear samples. There is no known ICP-MS application for detection of 41Ca, due to significant tailing effects from 40Ca, as well as 40Ar (99.60% natural abundance) in the plasma gas, with additional interferences from isobaric 41K (natural abundance 6.73%) and instrument-generated 40CaH.136 Even in highly contaminated samples, a 41Ca isotopic abundance of 10−9 relative to 40Ca must be determined118 (compared to natural terrestrial ratios of ∼10−14 to 10−15).137,139 A half-life value of 9.94 ± 0.15 × 104 years was recently determined using multiple techniques including TIMS.144 A 42Ca–48Ca double spike as well as two NIST reference materials of known isotopic composition were used to correct for isotopic fractionation. A mathematical correction was applied to account for tailing from 40Ca, and isobaric 40K using 39K (natural abundance 93.26%) by monitoring at mass 41 when processing natural Ca samples.
RIMS is a highly sensitive technique for isotopic 41Ca/40Ca measurements in the 10−10 to 10−11 range,136,139 whilst AMS is capable of isotope ratio measurements as low as 10−15.139,145 Given the difficulty in routinely measuring 41Ca, Nottoli et al. (2013) estimated the activity from measurement of 60Co in ion exchange resins used for primary fluid purification in pressurized water reactors (PWR's).145 AMS has been compared to LSC for detection of 41Ca in concrete and other solid samples including sediment and soil.139,143 Given the fundamental difference in the two techniques, there was good agreement between results in both studies, however Hampe et al. noted significant deviations at lower activities.139
8.4 Nickel-59 and nickel-63
Neutron irradiation of Ni, Ni alloys and stainless steels result in the formation of two long-lived Ni radioisotopes, 59Ni (t1/2 = 7.6 × 104 years) and 63Ni (t1/2 = 99 years). Irradiation of 63Cu can also result in the formation of 63Ni via63Cu(n,p)63Ni reaction. Typical 59Ni/63Ni activity ratios are ∼1:100 and most published methodologies have focused on the quantification of 63Ni as the dominant radionuclide in recently irradiated materials. However, the long-lived 59Ni will have a more long-term impact on waste radionuclide inventories and its characterization is becoming of greater interest. Measurement of 63Ni is relatively straightforward through measurement of the 66.95 keV beta emissions by liquid scintillation analysis, with detection limits of typically 14 mBq.146 Thin window beta counting has also been used, achieving limits of detection of 1 mBq for a 3000 min count time.147 Measurement of 59Ni is more complicated. Theoretically, it should be possible to detect the associated X-ray emissions (5.88–6.49 keV) using liquid scintillation analysis, but in practice these emissions are indistinguishable from the more abundant beta emissions arising from the associated 63Ni. X-ray spectrometry using LEGe of Si(Li) detectors has therefore typically been employed for 59Ni measurement although such approaches are relatively insensitive with detection limits of 1–2 Bq being reported.148 In all cases, chemical purification of the Ni fraction is required prior to measurement of the Ni radioisotopes. Purification of Ni is most commonly achieved using dimethylglyoxime (DMG) at pH 8–9, either via precipitation or solvent extraction of the Ni–DMG complex, or via adsorption of Ni onto an extraction chromatographic material incorporating DMG loaded onto an inert support. Co-adsorption of other transition metals (notably Co, Fe and Cu) can be prevented by including ammonium acetate in the load solution. Ni/Co decontamination factors of 103 have been reported using such separations.146 Further purification of Ni from Co can be achieved using anion exchange chromatography, with Co being retained from 9 M HCl solutions, with Ni/Co decontamination factors approaching 104.149 Purification of Ni prior to AMS measurement through the formation of volatile Ni(CO)4 has also been described and this could have significance in the development of robust Ni/Co separation procedures prior to ICP-MS measurement.150
Given the long half-life of 59Ni, the radionuclide should be measurable with reasonable sensitivity using mass spectrometric techniques. In practice, 59Ni is always associated with stable Ni with a maximum 59Ni/Ni ratio of 10−7. In addition, mass spectrometric measurement of 59Ni is impacted by an isobaric interference from the only stable isotope of Co, 59Co. Measurement of 59Ni and 63Ni by AMS following chemical separation of the Co interference has been reported.150,151 AMS was considered in preference to ICP-MS for the measurement of 59Ni to detect nuclear waste container leaks in the Kara Sea152 with proposed detection limits of 2–5 fg (6–15 μBq). Although less sensitive, ICP-MS should still be of use for general waste characterization, yet application of ICP-MS to 59Ni measurement has not been reported to date. This most likely reflects the challenges in measuring the low abundance 59Ni in the presence of a significantly higher signal of 58Ni and the associated abundance sensitivity interference along with the isobaric interference from 59Co. The extremely low abundance sensitivities (theoretically 10−10) associated with ICP-QQQ-MS may ultimately permit measurement of 59Ni by ICP-MS. Isobaric interference from 59Co cannot be resolved using high resolution mass spectrometry as a resolution of >50000 would be required to achieve this. Effective separation of Ni and Co must therefore be achieved either through chemical separation prior to ICP-MS measurement or via reaction gas technologies to remove the 59Co interference. To date, effective separation of Ni and Co using dynamic reaction cell-based techniques has not been extensively investigated. Bandura et al. (2006) suggested N2O as a reaction gas for Co/Ni separation. Co+ reacts at a faster rate with N2O compared with Ni+.92 However, formation of NiO+ was still observed and a Co/Ni separation factor of only 6 was determined. Efficient off-line chemical separation of Ni and Co therefore appears critical to the application of ICP-MS for 59Ni determination.
8.5 Selenium-79
Selenium-79 is a long lived (t1/2 = 6.0 × 105 years) fission product with a low energy beta emission (Emax = 0.151 MeV). The radionuclide is of interest for post-closure repository performance assessments given its potentially mobile nature. Measurement of 79Se is potentially well suited to ICP-MS, however this is problematic given its low abundance in nuclear wastes, its relatively high ionisation potential (9.75 eV) and isobaric interference arising from 79Br (50.6% abundance), as well as polyatomic interferences arising from 39K40Ar and 38Ar40ArH. Aguerre and Frechou (2006) developed a scheme to effectively separate Se from Br and associated fission products (although 125Sb was co-extracted with the Se) with final Se measurement by ICP-QMS (Perkin Elmer Elan 6000).153 Plasma power was increased to 1300 W to maximise sensitivity, with a detection limit of 0.15 μg L−1, equivalent to 50 Bq L−1 79Se. There is also the potential to use cold plasma conditions (reduced RF power) in combination with chemical separation to separate 79Se from 79Br, although there is no known application of this approach. Compte et al. (1995) developed a method for determination of 79Se in high activity fission product solutions (1010 Bq L−1) using ETV-ICP-MS, following single stage chemical separation, with 79Se measured at a concentration of 0.43 mg L−1 (120.01 Bq L−1).154 ETV-ICP-MS has also been used in combination with LSC for determining the half-life of 79Se, following separation from a reprocessing solution using liquid–liquid extraction and ion exchange chromatography.1558.6 Strontium-90
Strontium-90 is a beta-emitting radionuclide (decay energy 0.546 MeV) with a half-life of 28.8 years that decays to 90Y (half-life 2.67 days) and then on to stable 90Zr. Strontium-90 is of critical importance in nuclear waste management, environmental monitoring and radiation protection. Additionally, 90Sr is a mobile element that can accumulate in soils and plants via precipitation and ion exchange mechanisms,62,77,86 as well as in bones and teeth if inhaled or ingested, because of its similar chemical properties to calcium. This increases the risk of leukemia and bone cancer.86,87,156 An extensive review of sample preparation and measurement techniques for 90Sr is presented elsewhere,62 with a recent review paper focusing on mass spectrometric measurement of 90Sr, along with 135Cs and 137Cs.157The primary limitation affecting ICP-MS measurement of 90Sr is an isobaric interference from stable 90Zr (natural abundance 51.45%). The similarity in mass requires a resolution of ∼30000 for effective separation, which is beyond the capabilities of ICP-SFMS.86 Even in highly 90Sr-contaminated soils surrounding the Chernobyl Nuclear Power Plant, a 90Zr decontamination factor of ∼106 is still required.77,158 An additional interference is peak tailing from stable 88Sr (natural abundance 82.6%), which is present at high concentrations in environmental samples (20–300 mg kg−1 in soils, 7–9 mg L−1 in seawater).87 Finally, multiple polyatomic interferences can arise from reactions of elements in the plasma.54
Beta-counting techniques are applicable to highly sensitive detection of 90Sr, either through direct measurement of 90Sr, or via90Y.62 The overlapping spectra of these two radionuclides can be resolved by calculating an ingrowth of 90Y, or more commonly waiting for 2–3 weeks for the establishment of secular equilibrium, followed by long count times, depending on the detection limits required. The short measurement time for ICP-MS means the concentration of 90Y daughter product is negligible (∼0.02% of the total intensity at m/z = 90 in nuclear fuel samples) and will not interfere with 90Sr determination.158
Chemical separation is commonly based on extraction chromatography using commercially available Sr-resin (Triskem International). For more complex sample matrices, this is commonly preceded by a pre-concentration stage such as calcium oxalate precipitation.77,86,158–160 Online chemical separation is advantageous when a rapid response is required, for example in the prompt assessment of contamination following the Fukushima accident in 2011.160,161 A lab-on-valve based setup successfully removed 99.8% Zr using Sr-resin prior to measurement with a Perkin Elmer Elan II DRC-ICP-MS, whilst an automated solid phase elution (SPE) system coupled to the same instrument reduced the chemical separation and analysis time by ∼50% compared to offline separation.160 Grinberg et al. (2007) investigated ETV-based separation of Sr from Zr and Y using a commercial graphite tube in combination with DRC-ICP-MS.77 In Zr-free samples, ETV using a graphite tube achieved a Sr detection limit of 1.8 ng kg−1 (9.2 × 103 Bq kg−1), compared to 0.1 ng kg−1 (513.2 Bq kg−1) using solution nebulization. However, in the presence of 100 mg kg−1 Zr, the detection limit for solution nebulization increased to 89 ng kg−1 (4.5 × 105 Bq kg−1) compared to 2.9 ng kg−1 (1.4 × 104 Bq kg−1) using ETV.
A Perkin Elmer Elan II DRC-ICP-MS is the most commonly applied instrument to 90Sr measurement (Table 5). With O2 as the reaction gas, 90Zr is oxidized to 90Zr16O, while the same oxidation reaction with 90Sr is not energetically favorable and would require a stronger oxidant such as N2O, or a higher O2 flow rate.87,160 DRC-ICP-MS has been applied to detection of 90Sr in soil samples in the vicinity of the Chernobyl nuclear power plant.158 A Zr decontamination factor of >107 was achieved by a combination of Sr-resin and DRC-ICP-MS. The abundance sensitivity was calculated as ∼3 × 10−9, with a detection limit of 4 pg L−1 in Zr-free solutions, increasing to 200 pg L−1 (1026.5 Bq L−1) in digested soils. Strontium-90 has also been measured in soil contaminated by the accident at the Fukushima nuclear power plant, using on-line Sr-resin separation combined with DRC-ICP-MS, with a detection limit of 0.77 pg L−1 (4.0 Bq L−1).160 Taylor et al. (2007) measured 90Sr by DRC-ICP-MS in samples collected from Perch Lake, Ontario, with detection limits of 0.1 ng kg−1 (513.2 Bq kg−1) 0.04 ng kg−1 (205.3 Bq kg−1) and 0.003 ng L−1 (15.4 Bq L−1) for sediments, plants and water, respectively.159 There was a good agreement in the values measured for a standard reference material by Cerenkov counting and DRC-ICP-MS, with detection limits for the two techniques of 13.7 pg kg−1 (70.3 Bq kg−1) and 97.7 pg kg−1 (501.4 Bq kg−1), respectively. A recent study by Amr et al. (2016) measured 90Sr (along with 137Cs and Pu isotopes) by triple quadrupole ICP-MS (Agilent 8800) in Qatar soils and sediments.162 Following acid leaching and extraction chromatography using Sr-resin (Triskem International), 90Sr was separated from 90Zr using oxygen in the collision–reaction cell, with 90Sr measured at an average concentration of 0.61 fg g−1 (3.1 Bq g−1).
| Reference | Instrument | Model | Matrix | Chemical separation | LOD, pg L−1 (Bq L−1) |
|---|---|---|---|---|---|
| 86 | SFMS | Element | Urine | CaPO4 pptate, Sr-resin, medium res, cold plasma | 0.4 (2.1) |
| 64 | SFMS | Element | Groundwater | Medium res, cold plasma | 11 (56.5) |
| 161 | DRC-MS | Perkin Elmer Elan DRC II | Soils | On-line solid phase elution, reaction cell | 600 (3079.4) |
| 160 | DRC-MS | Perkin Elmer Elan DRC II | Soils | Sr resin, reaction cell | 0.8 (3.9) |
| 77 | DRC-MS | Environmental samples | Sr resin, ETV sample introduction, reaction cell | 1800 (9238.3) | |
| 158 | DRC-MS | Perkin Elmer Elan DRC II | Soils | Sr resin, reaction cell | 200 (1026.5) |
| CC-QMS | Collision cell | 900 (4619.1) | |||
| 86 | CC-QMS | Platform, Micromass Ltd | Urine | CaPO4 pptate, Sr-resin, collision cell | 2000 (1.0 × 104) |
A less common approach to 90Sr measurement is ICP-SFMS operating with cold plasma conditions (forward power = 650–750 W) and medium mass resolution (R = 4000–4500).86,87 Cold plasma conditions are applicable to separation of Sr from Zr based on the difference in ionization potential (5.7 eV and 6.8 eV, respectively), although sensitivity becomes more dependent on the sample matrix under these conditions. Medium mass resolution reduces instrument sensitivity compared to low resolution, but also reduces tailing from stable 88Sr.132 When a combination of medium mass resolution and cold plasma are applied, peak tailing of 88Sr is considered the critical factor affecting accurate detection of 90Sr. Abundance sensitivity values of 6 × 10−7 and 0.8 × 10−7 have been recorded at medium resolution, which is an improvement of 2 orders of magnitude compared to low mass resolution,76 but is lower than that of DRC-ICP-MS. A detection limit of 0.4 pg L−1 (2.1 Bq L−1) was achieved for urine samples by ICP-SFMS, compared to 2 ng L−1 (10.5 Bq L−1) for ICP-QMS operating with a collision cell (Platform, Micromass Ltd).86
8.7 Zirconium-93
Zirconium-93 is a fission product (6.35% thermal neutron fission yield) and is also produced via the neutron irradiation of 92Zr (natural abundance 17.1% σtherm – 0.2 barns). Although theoretically predicted previously, 93Zr was first positively identified in irradiated uranium by Steinberg and Glendenin in 1950.163 The long half-life (t1/2 = 1.53 × 106 years) and corresponding low specific activity (9.3 × 107 Bq g−1) means that the nuclide is ideally suited to mass spectrometric detection. In addition, the low beta energy (Emax = 56 keV) and lack of gamma emission and, in recently irradiated materials, potential interference from 95Zr makes radiometric detection less attractive. Given the low ionisation efficiency of Zr, ICP-MS-based techniques are preferable to TIMS-based approaches. Measurement of atomic percentages of a range of fission products, including 93Zr, in spent nuclear fuel dissolver solutions by direct measurement has been reported using a Perkin Elmer Elan 250 ICP-MS,164 although isobaric interferences were identified as an issue. In general, isobaric interferences arising from 93Nb (100% abundance), 93mNb and 93Mo (both activation products) and polyatomic 92MoH+ must be mitigated by chemically separating the interfering species from 93Zr prior to measurement. Chemical separations of 93Zr/93Nb have been reported based on solvent extraction into benzoylphenylhydroxylamine–trichloromethane–HCl165 and ion-exchange chromatography in HCl/HF media.166 Isobaric interference suppression on-line using ICP-DRC-MS has not been employed. Bandura et al. (2006) noted that no reactions had been identified that would permit separation of 93Nb/93Mo interferences in a reaction cell.92Chartier et al. (2008) reported an isotope-dilution MC-ICP-MS (Isoprobe) approach using a 96Zr spike.165 A mass bias of 3% amu−1 was observed for Zr and corrected for through measurement of 94Zr/90Zr ratios (reference value 0.3378 ± 0.0002) in a natural Zr standard.165 The relative uncertainty of measured [93Zr] was estimated as 0.6% (k = 2). The procedure was subsequently applied to the determination of the half-life of 93Zr.167
8.8 Technetium-99
Technetium-99 is a pure-beta emitting radionuclide with a half-life of 2.13 × 105 years, and is a high yield fission product (6.1% and 5.9% fission yield from thermal neutron fission of 235U and 239Pu, respectively) of significant interest with regards to nuclear waste characterization and decommissioning. The relatively high mobility is a concern with regards to waste storage and disposal, whilst the high solubility in seawater (as TcO4−) makes it an important seawater tracer.168 A comprehensive review of analytical methods for 99Tc is published elsewhere.169Low level gas flow GM counter or LSC are applicable to 99Tc measurement. The long half-life and low decay energy (Emax = 292 keV) means long count times for LSC if detection limits in the pg g−1 range are required,169 whilst interferences from 103Ru, 106Ru and 90Sr must be removed prior to measurement. ICP-MS measurement of 99Tc is impacted by an isobaric interference from stable 99Ru (natural abundance 12.76%), which can be corrected for by chemical separation, or monitoring 101Ru (natural abundance 17.06%). Several isotopes have been used for drift correction, including 103Rh and 115In. Issues with 103Rh are memory effects, and instability in dilute HNO3 for long analytical runs.168,170 By comparison, 115In does not suffer from memory effects and has minimal disturbance from 99Tc16O.169 There are no stable isotopes of Tc, although long-lived 97Tc and 98Tc (both with 4.2 × 106 year half-lives) can be used as both a yield tracer and internal standard.169
Solvent extraction, ion exchange chromatography and extraction chromatography are all effective approaches to separation of 99Tc, with extraction chromatography using TEVA resin the most commonly used.169 The volatility of Tc must be considered during sample preparation, with losses noted when the temperature of a HNO3 solution (up to 8 M concentration) increased from 100 °C to 150 °C.169 Such losses must also be considered when dry ashing samples to decompose organic matter in solid samples.171 No significant losses were observed when ashing at 800 °C for less than 6 hours for a seaweed sample. Technetium-99 has been measured in a range of environmental and biological samples by ETV-ICP-MS (Perkin Elmer HGA-600 MS), using NH4OH as matrix modifier to form an alkaline solution and minimize loss of Tc during drying and ashing. Depending on the daily performance, the detection limit ranged from 0.5–1.0 pg mL−1 (3.2 × 10−4 to 6.3 × 10−4 Bq mL−1) compared to 5–50 pg mL−1 (3.2 × 10−4 to 3.2 × 10−2 Bq mL−1) using radiochemical techniques.
A modular automated separator (MARS Tc-99; Northstar Technologies, Madison WI, USA) was developed for rapid analysis of groundwater samples from boreholes at a low and intermediate waste repository. The system was coupled to ICP-QMS (Varian 810), with an activity of 1658 ± 7 mBq L−1 (2.6 ng mL−1) measured for a method standard, which was in good agreement with the certified value of 1598 ± 40 mBq L−1 (2.5 ng mL−1).172 The detection limit of the procedure developed was 0.12 mBq mL−1 (0.19 pg mL−1), whilst the system developed was potentially applicable to other radionuclides. In a separate study, large volume water samples were measured by ICP-QMS (Thermo X-series II) following chemical separation by ferrous hydroxide co-precipitation, and online SI using 2 TEVA columns.168 Decontamination factors of 1 × 106 and 5 × 105 were achieved for Ru and Mo, with a detection limit of 11.5 pg m−3 (7.5 mBq m−3) for a 200 L sample. Slightly lower decontamination factors of 4 × 104 and 1 × 105 for Mo and Ru, respectively, were recorded following 2 TEVA column-separations of environmental samples prior to ICP-QMS (Thermo X-series II) measurement, with a detection limit of 0.15 mBq g−1 (0.24 pg mL−1).171 Samples were left for 1 week following separation to allow for decay of 99mTc (half-life 6.0 hours).
Measurement of 99Tc in waste stream samples was carried out at the Lan-Yu low level waste storage site, Taiwan, where Tc was previously estimated from a scaling factor based on the activity of 137Cs.173 Following acid digestion and TEVA resin separation, samples were measured by ICP-QMS (Agilent 7500a), with a detection limit of 0.021 ng g−1 (0.013 Bq g−1). Accurate quantification meant a revision of the scaling factors being used was recommended, to the extent that reclassification of historical waste should also be carried out. The performance of ICP-QMS (Thermo X-series II) and ICP-SFMS (Thermo Element 2) was compared for low-level radioactive waste following digestion and separation using TEVA extraction chromatography resin.170 Instrumental detection limits of 0.024 ng L−1 (0.015 Bq L−1) and 0.015 ng L−1 (0.0095 Bq L−1) were measured for ICP-QMS and ICP-SFMS, respectively. The minimum detectable activity for LLW samples was <8.5 mBq g−1 (13.4 pg g−1) for ICP-QMS, <5.9 mBq g−1 (9.3 pg g−1) for ICP-SFMS and <414–511 mBq g−1 (806.8 pg g−1) for a gas proportional counter, whilst the method detection limit (for a 0.1 g waste sample) was 13.6 ng L−1 (8.5 Bq L−1) for ICP-QMS, and 1.19 ng g−1 (745 mBq g−1) for LSC (no value was given for ICP-SFMS).
8.9 Palladium-107
Palladium-107 is a long lived (t1/2 = 6.5 × 106 years) low energy beta emitting radionuclide (Emax = 33 keV). The radionuclide is produced with a thermal neutron fission yield of 0.15%. The specific activity of 1.9 × 107 Bq g−1 indicates that mass spectrometric measurement would be favorable. Isobaric interferences would arise from natural 107Ag (51.8% abundance), as well as a potential polyatomic interference from 91ZrO+. There are few published procedures based on ICP-MS for measurement of 107Pd. Bibler et al. (1997) used a ICP-QMS (Fisons/VG Plasmaquad) to measure fission products including 107Pd in HLW sludge's and glass.174 It was noted that direct measurement of 107Pd was not possible due to isobaric interference from silver which is used to scavenge iodine and is therefore present in the waste stream. Bannochie et al. (2009) noted a similar problem and calculated 107Pd activities via measurement of 105Pd and correcting for the relative fission yield of the two isotopes.175 Interference from Ag could also be overcome through chemical separation, with Bienvenu et al. (1998) noting that Pd could be separated from Ag using liquid–liquid extraction into triphenyl phosphine in nitro-2-phenylpentyl ether from 2 M HNO3 with Pd being back-extracted into 4 M ammonia.176 However, decontamination factors were not sufficient for high Ag containing samples, and an electrothermal vaporization approach was subsequently developed whereby Ag was volatilized at <1000 °C and Pd volatilized at 1500 °C.8.10 Iodine-129
Iodine-129 is a fission product (0.6% yield), and is also produced naturally via cosmic-ray-induced spallation of Xe, spontaneous fission of 238U, neutron bombardment of Te in the geosphere, and via thermal neutron-induced fission of stable I in the lithosphere.177–180 Iodine-129 decays with a half-life of 1.57 × 107 years to stable 129Xe, and is a weak beta-gamma emitter with maximum beta decay energy of 29.78 keV (37.2%) and gamma energy of 39.58 keV (7.42%).Iodine is a redox-sensitive element, which controls speciation and exchange between the ocean/atmosphere terrestrial environment.177,181 The high concentration and solubility in water makes 129I a useful oceanographic tracer,179,182 and has also been used in distinguishing the discharge pathways from European reprocessing facilities at Sellafield and La Hague,182 and more recently following the Fukushima accident.183 Measurement of 129I in Fukushima-contaminated samples has also been applied as an analogue for reconstruction of the distribution and activity of shorter-lived 131I (half-life 8.02 days).184
Traditionally, radiochemical neutron activation analysis (RNAA) was the only method applied to 129I detection in environmental samples for a number of years.185 Beta, gamma and X-ray-based techniques are limited by interferences and shielding issues, as well as long count times for low activity samples.191 For mass spectrometric determination, the primary concern is accurate detection of 129I in the presence of high concentrations of stable 127I (natural abundance 100%). Isotopic 129I/127I ratios range from 10−13 to 10−12 prior to anthropogenic nuclear activities, compared to 10−11 to 10−9 in the marine and terrestrial environment, with ratios as high as 10−8 to 10−3 at reprocessing sites.178,180,182,186–188 The 129I/127I ratio measurable by the instrument is therefore a critical factor. Decomposition of solid samples and chemical separation prior to quantification are additional challenges, with losses due to volatization and/or transformation to an undesirable chemical form.197 Separation techniques include ion chromatography, and phyrohydrolysis and solvent extraction for soil samples.179,184,189
Until recently, ICP-MS was generally used for detection of stable 127I, while AMS was the favored technique for 129I detection, because it can achieve instrumental background ratios of 10−15 to 10−13 (Table 6).181,182,184–186 ICP-MS was considered not to be capable of distinguishing between iodine isotopes, with applications limited to more contaminated samples with 129I/127I ratios greater than ∼1010.177,180,187,189 Additional interferences include the low ionization efficiency of I (33.9%) owing to its high first ionization energy (10.45 eV),190 isobaric 129Xe (natural abundance 26.40%) present as an impurity in the plasma gas, and polyatomic interferences (particularly from collision or reaction cell instruments) including 127IH2, 97MoO2, 113CdO, 115In14N and 89Y40Ar.179,191 An additional challenge is selection of an appropriate internal standard, with possible options including Cs, Re and Te.179 One study selected Te owing to the similarity in ionization energy with I (9.01 eV),190 whilst another did not use an internal standard owing to limitations with the elements listed.179
| Reference | Iodine isotope | Instrument | Model | Matrix | Chemical separation | LOD, pg L−1 (Bq L−1) |
|---|---|---|---|---|---|---|
| 192 | 127 | QMS | X series II | Lake sediments | ||
| 129 | AMS | |||||
| 181 | 127 | AMS | Aerosol particles | |||
| 129 | ||||||
| 193 | 127 | AMS | Environmental samples | |||
| 129 | ||||||
| 177 | 127 | QMS | X series II | Scandinavian soil and sediment | Sequential extraction into soil/sediment fractions | |
| 129 | AMS | |||||
| 178 | 127 | QMS | X series II | Baltic sea seawater | ||
| 129 | AMS | |||||
| 187 | 127 | DRC-MS | Elan DRCe | Soil | O2-based extraction with heating, extraction from activated carbon | |
| 129 | ||||||
| 188 | 127 | CC-QMS | Agilent 7500ce/cx | Baltic sea, lake and rain waters from Finland | Anion exchange, solvent extraction | |
| 129 | AMS | |||||
| 182 | 127 | QMS | Seawater and aerosol filters | Liquid–liquid exchange for air filters | ||
| 129 | AMS | Seawater samples anion exchange | ||||
| Matrix adjustment for I-127, but no matrix separation | ||||||
| 194 | 127 | AMS | Pacific ocean and Japan seawater | Solvent extraction | ||
| 129 | ||||||
| 190 | 127 | SFMS | Element 2 | Japanese coastal seawater (pre-Fukushima) | 2300 (0.015) | |
| 129 | ||||||
| 183 | 127 | QMS | X series II | Fukushima seawater samples | Anion exchange | |
| 129 | AMS | |||||
| 189 | 127 | QQQ-MS | Agilent 8800 | Fukushima soil samples | Pyrohydrolysis and solvent extraction | |
| 129 | ||||||
| 189 | 127 | ORS-QMS | Agilent 7700X | Fukushima soil samples | Pyrohydrolysis and solvent extraction | 1500 (0.0098) |
| 129 | ||||||
| 179 | 127 | SFMS | Element 2 | Snow samples from Greenland (Fukushima contaminated) | Online IC separation (anion exchange) | 700 (0.0046) |
| 129 | ||||||
| 184 | 127 | ORS-QMS | Agilent 7700 | Fukushima soil samples | Pyrohydrolysis | |
| QQQ-MS | Agilent 8800 | |||||
| 129 | AMS | |||||
| 180 | 127 | AMS | Woodward iodine corporation | |||
| 129 |
Iodine-129 was detected in vegetation to a detection limit of 1.4 ng kg−1 (9.2 mBq kg−1) using a Plasma Quad PQ2 ICP-QMS,191 with the 129Xe interference corrected for using 131Xe (natural abundance 21.23%). The volatility of iodine was seen as an advantage that partially overcame the low transport efficiency (2–4% with pneumatic nebulization), as the formation of droplets in the spray chamber increased surface area and rate of vaporization, with vapors then taken up into the ICP. More recently, a Perkin Elmer Elan DRCe was applied to measurement of soils in land surrounding a fuel reprocessing site in Tokai, Japan.187 Using oxygen as a reactive gas, the 129Xe interference was reduced, however ion transport was generally suppressed, and the instrument 129I/127I background of ∼10−8 was not improved in DRC mode due to 127IH2 formation with trace impurities in the reaction gas. The background was improved to 10−10 using Axial Field Technology, which applies an accelerating axial field to ions in the DRC, improving instrument stability and reducing matrix effects. Fukushima-contaminated soil samples were measured using an Agilent 8800 ICP-QQQ-MS,189 with O2 used for 129Xe removal, and in-cell polyatomic interferences reduced by the additional quadrupole mass filter positioned before the entrance to the cell. A sample standard bracketing technique was used to correct for mass discrimination and improve reproducibility, with good agreement between ICP-MS and AMS.
ICP-SFMS (Thermo Element 2) detection of 129I was achieved in Fukushima-contaminated Greenland snow samples.179 Clean laboratory conditions (class 10–100) were used for sample preparation, whilst separation of Mo, In and Cd using online ion chromatography reduced polyatomic interference formation. A desolvating sample introduction system (Apex-Q (ESI)) tripled the sensitivity compared a cyclonic Peltier-cooled spray chamber, however the nitrogen carrier gas increased Xe background by an order of magnitude. The formation of 127I2H was controlled by the instrumental setup e.g. sample uptake and gas flow rate, and plasma temperature. This is compared to an ‘on-peak’ baseline subtraction by Ohno et al. (2013) when using ICP-QQQ-MS, with a 127IH2/127I ratio of 3 × 10−8 calculated from aspiration of a 100 mg L−1 stable I solution.189 The inability of ICP-SFMS to eliminate 129Xe was resolved by monitoring changes in the background Xe signal in Ar plasma gas to determine whether a background correction was necessary.
8.11 Caesium 135 and 137
High yield fission products 135Cs and 137Cs (6.58% and 6.22%, respectively) are present in environmental samples as a result of releases from nuclear power plants and reprocessing sites, nuclear accidents, and fallout from atmospheric weapons testing. Caesium-137 (t1/2 = 30.07 years) is established as an important radionuclide in radiation protection, environmental monitoring and waste disposal. By comparison, 135Cs is a long-lived radioisotope (t1/2 = 2.3 × 106 years) with a comparatively low radiation risk; however it is a significant contributor to the long term radiological risk associated with deep geological disposal. Furthermore, the 135Cs/137Cs ratio varies with reactor, weapon and fuel type, and recent studies have achieved precise determination of 135Cs/137Cs ratios as a forensic tool to identify the source of radioactive contamination.195–200Caesium-137 decays by beta emission to short-lived metastable isomer 137mBa, with maximum energies of 514 keV (94.4% yield) and 1175 keV (5.4% yield). This is immediately accompanied by gamma ray emission of 661.7 keV (85.1% yield) to form 137Ba. Gamma spectrometry is generally favored because it exploits the good gamma yield of the 661.6 keV energy that is not susceptible to significant absorption, whilst the ability to directly count most samples without any chemical separation is also beneficial. Caesium-135 decays with a maximum beta particle energy of 269 keV, however, measurement by beta counting is restricted by any 137Cs also present in the sample with a radioactivity concentration that is typically 5 orders of magnitude higher. Measurement of 135Cs by mass spectrometry offers a considerable advantage because of the larger atom population compared with radioactive decays. A review of measurement of 135Cs and 137Cs and 135Cs/137Cs isotope ratios in environmental samples has been published recently,201 and Table 7 gives a summary of recent procedures for 135Cs/137Cs measurement.
| Reference | Instrument | Model | Matrix | Chemical separation | LOD, pg L−1 (Bq L−1 135Cs/137Cs) |
|---|---|---|---|---|---|
| 214 | QMS | Spent fuel pellets and solutions | Cation exchange | ||
| 215 | QMS | VG Elemental Plasma Quad 2 | Waste tank sludge and supernatants | Ba(OH)2 precipitation | 209000 (8.53/6.7 × 105) |
| 216 | QMS | Perkin Elmer Elan 5000 | High activity waste | Cation exchange | 16000 (0.65/5.12 × 105) |
| 79 | QMS | Perkin Elmer Elan 5000 | Cs-137 standard | ETV sample introduction | 200 (0.01/640.1) |
| 217 | QMS | PQ Excell | Groundwater | On-line cation exchange | 2 (8.2 × 10−5/6.4) |
| 207 and 209 | MC-ICP-MS | GV Isoprobe | Spent fuel pellets | Anion exchange and HPLC, reaction cell | |
| 1 | SFMS | Element 2XR | Fission product standard | AMP, cation exchange, extraction chromatography | 50 (2.0 × 10−3/160.0) |
| 208 | SFMS | Perkin Elmer Elan 5000 | Simulated spent fuel | Capillary electrophoresis | 4000 (0.16/1.3 × 104) |
| 195 | DRC-MS | Perkin Elmer Elan DRC II | Soil and sediment | AMP, anion exchange, online cation exchange, reaction cell | 200 (0.01/640.1) |
| 198 | QQQ-MS | Agilent 8800 | Rainwater | Reaction cell | 270 (0.01/864.2) |
| 199 | QQQ-MS | Agilent 8800 | Environmental samples | AMP, anion exchange, cation exchange, reaction cell | |
| 200 | QQQ-MS | Agilent 8800 | Environmental samples | AMP, anion exchange, cation exchange, reaction cell | 10/6 (4.1 × 10−4/19.2) |
A key challenge for mass spectrometric measurement is removal of isobaric interferences from naturally occurring 135Ba and 137Ba (isotopic abundances 6.6% and 11.2%, respectively), peak tailing from stable 133Cs (isotopic abundance 100%), and polyatomic interferences including 95Mo40Ar, 97Mo40Ar, 119Sn16O and 121Sb16O. For mass spectrometric measurement, Cs separation has commonly been achieved using ammonium molybdophosphate (AMP) followed by anion exchange and/or cation exchange chromatography, most commonly Dowex AG1 and Dowex AG50W (both Bio-Rad, USA), respectively.1,195,197,198,200,202 Other techniques include extraction chromatography using calixarenes,203 and potassium nickel hexacyanoferrate (KNiFC).200 Barium-138 (natural abundance 71.7%) has been used to assess the effectiveness of separation at masses 135 and 137, however any interference correction will reduce the accuracy of 135Cs/137Cs values. Caesium-137 values can be verified by gamma spectrometry, however if a significant correction is required this will not validate the 135Cs result.204–206
Online separation has been applied to Cs and Ba separation prior to ICP-MS analysis. For example, Isnard et al. (2009) achieved a 135Cs and 137Cs detection limit of 2 pg L−1 (8.2 × 10−8 Bq L−1) and 1 pg L−1 (3.2 × 10−3 Bq L−1), respectively, using on-line chromatographic separation with a CG3 IonPac guard column (Dionex, USA) for a spiked groundwater matrix.207 Following AMP and anion exchange separation, Taylor et al. (2008) achieved online Cs/Ba separation in sediment samples by attaching a Dionex CS12A 3 × 150 mm cation exchange column onto a high performance liquid chromatography (HPLC) module, and coupling this system with ICP-MS.195
Sample introduction-based separation has also been applied to Cs/Ba separation. Song et al. used ETV with potassium thiocyanate (KSCN) as a chemical modifier.79 At a volatilization temperature of 1100 °C, Cs was vaporized in a 104 excess of Ba, with a Ba signal increase at mass 135 of only 1%. The detection limit of 135Cs using QMS (Perkin Elmer Elan 5000) was calculated as 0.2 ng L−1 (0.01 Bq L−1) An alternative online separation technique is capillary electrophoresis (CE-ICP-MS), which was effectively applied to 135Cs/137Cs measurement in plutonium uranium redox extraction (PUREX) and mixed oxide fuel (MOX) samples.208 The procedure achieved 133Cs detection limits of 6000 ng L−1 (245.0 Bq L−1) and 4 ng L−1 (0.2 Bq L−1) in ICP-QMS (Perkin Elmer Elan 5000) and ICP-SFMS (Thermo Element 2), respectively.
A reaction cell has been applied to Cs separation, with N2O, and CH3Cl both effectively used as reactive gases.195,209 For example, kinetic data from Lavrov et al. (2004) indicates the reaction for Ba with N2O has an efficiency of 32%, compared to 0.01% for Cs.210 An Agilent 8800 ICP-QQQ-MS has been used for 135Cs/137Cs detection in rainwater and environmental samples contaminated following the accident at the Fukushima Daiichi NPP.198–200 Using N2O as a reactive gas, a 135Cs and 137Cs detection limit of 10 pg L−1 (equivalent to 4.1 × 10−7 Bq L−1 and 3.2 × 10−2 Bq L−1, respectively) was achieved in a Ba-free solution, compared to 100 pg L−1 (4.1 × 10−6 Bq L−1) and 270 pg L−1 (8.6 × 10−1 Bq L−1) for 135Cs and 137Cs, respectively, in solutions containing 100 μg L−1 Ba. These values are higher than the instrumental and method detection limits achievable using ICP-SFMS.1 However, the ICP-QQQ-MS offers the combination of chemical and instrument-based separation, whilst the two quadrupoles have measured a 133Cs/135Cs abundance sensitivity of ∼10−10.
A procedure using a Thermo Element 2XR ICP-SFMS achieved an instrumental detection limit of 1 pg L−1 for stable 133Cs. The absence of instrument-based separation meant highly efficient chemical separation techniques using high purity reagents and clean laboratory conditions was required to minimize Ba contamination prior to sample introduction. Following chemical separation, a higher method detection limit of 20 pg L−1 was calculated (equivalent to 8.2 × 10−7 Bq L−1 and 6.4 × 10−2 Bq L−1 for 135Cs and 137Cs, respectively).1 Time independent fractionation is an issue affecting accurate isotope ratio measurements by ICP-MS. One cause of this is space charge effects, as lighter ions are deflected to a greater extent than heavier ions during ion extraction.207 The absence of a 135Cs/137Cs certified reference material makes accurate mass bias correction challenging, and limits the ability to compare results between studies. Isnard et al. (2009) applied a sample standard bracketing technique using natural 121Sb, 123Sb and 151Eu, 153Eu to correct for mass bias, as the average mass of these isotopes is close to that of 137Cs.207 The isotopic fractionation affecting TIMS and ICP-MS, as well as the absence of a certified reference material to verify accuracy of 135Cs/137Cs measurements, led to a comparison of the two techniques for uranium oxide and mixed oxide fuel samples.
Of the alternative mass spectrometric techniques, Lee et al. (1993) were the first to develop a procedure for measuring 135Cs/137Cs in environmental samples using TIMS.211 There have been multiple recent TIMS applications including sediment core samples to highlight fractionation of 135Cs and 137Cs following aboveground nuclear tests at the Nevada Nuclear test site,196 and environmental samples contaminated by the Fukushima Daiichi accident.202 TIMS is affected by time-dependent isotopic fractionation, as lighter isotopes are preferentially evaporated from the filament. The lack of an available certified Cs isotopic standard makes it difficult to measure mass fractionation, however has been anticipated to be small relative to estimated uncertainties.196 Alternatively, mass fractionation can be corrected by measuring 133Cs/137Cs and 135Cs/137Cs ratios during the analysis.207
Pibida et al. used both RIMS and TIMS to determine the chronological age of nuclear fuel burn up samples through measurement of 135Cs/137Cs.212 For low-level detection in environmental samples, improvements in overall efficiency and chemical separation were required, whilst the absence of a commercially available instrument and time-consuming optimization of instrument efficiency and chemical preparation are the main limitations to this approach. AMS measurement is limited by the high energies required to separate Cs and Ba isobars, which makes separation a complex and costly process.206 A method to produce a beam of Cs anions, and production of suitable yield tracers to determine the efficiency of the process are also required.213
8.12 Samarium-151
Samarium-151 is a lanthanide element fission product, with a 235U thermal neutron fission yield of 0.53%. The radionuclide has a half-life of 94.7 years giving a specific activity of 9.25 × 1011 Bq g−1. Quantification of 151Sm is typically required in determining nuclear fuel burn up and in nuclear waste characterization. Samarium-151 is a pure beta emitting radionuclide (Emax = 76 keV), which is typically analyzed using liquid scintillation counting following separation of 151Sm from other rare earth radionuclides and 241Am. Separation of Sm from other lanthanides is usually achieved using either extraction chromatographic materials such as HDEHP-based resins,218 ion exchange chromatography, or HPLC219,220 based techniques. More recently, isotachophoretic separation coupled with ICP-MS has been developed for lanthanide quantification of spent nuclear fuel.221There are only a limited number of papers relating to measurement of 151Sm by ICP-MS, although ICP-MS has also been used in nuclear fuel burn up assessments to measure the variation in stable Sm isotopic composition.85 Isobaric interferences arise from stable 151Eu (47.81%) and polyatomic species 119Sn 16O2, 133Cs 18O, 134Ba 17O, 135Ba 16O and 150Nd 1H. Due to the potential for interference from Eu and Nd and their chemical similarity to Sm, reliable 151Sm measurements by ICP-MS rely on robust chemical separation of the rare earth elements.
Alonso et al. (1995) measured actinides and fission products in spent nuclear fuel using HPLC coupled to an Elan 5000 ICP-QMS.214 Lanthanide separation was performed using CG5/CS5 guard/separation columns and isocratic elution with 0.1 M oxalic acid and 0.19 M LiOH. The HPLC eluent was introduced into the ICP-MS via a cross flow nebulizer and Scott type, double pass spray chamber. Plasma RF power was optimized at 1050 W. Spent nuclear fuel 151Sm/150Sm values of 0.0119–0.0175 were reported. The same experimental set-up was used to measure fission products, including 151Sm, in UO2 and MOX fuel pellets.204
Wolf et al. (2004) coupled HPLC and ICP-MS to separate rare earth elements prior to measurement.84 HPLC separation was achieved using an Amberlite CG50 (2 × 50 mm2) guard column coupled with a Dionex CS5A (2 × 250 mm2) analytical column, a linear gradient elution of 0.040.0.26 M hydroxyisobutyric acid (HIBA) with a 0.30 mL min−1 flow rate. The eluent from the HPLC was introduced directly into the ICP-MS via a CETAC MCN-100, Model M-2 pneumatic microconcentric nebulizer. Measurements were performed using a Fison PlasmaQuad 2+ quadrupole ICP-MS. The within-sample precision of 151Sm measurement was quoted as 6%.
Isnard et al. (2005) separated Sm and Gd off-line using HPLC, followed by MC-ICP-MS measurement of Sm isotopes, including 151Sm, using a GV Isoprobe-N MC-ICP-MS.222 Sample introduction was via a microconcentric nebulizer and cyclonic spray chamber. Plasma RF power was set at 1350 W, with measurements performed via static Faraday detectors. Analysis of MOX spent nuclear fuel gave mean 151Sm/150Sm atom ratios of 0.04319 with a standard deviation of 0.2%. Pitois et al. (2008) coupled capillary electrophoretic separation with both ICP-QMS (PerkinElmer ELAN 5000) and ICP-SFMS (Thermo Element 2) to measure rare earth fission products although not 151Sm.208 Detection limits of 8 ng mL−1 (ICP-QMS) and 7 pg mL−1 (ICP-SFMS) were reported for rare earth elements, which would be equivalent to 6500 Bq mL−1 and 6.5 Bq mL−1 of 151Sm.
8.13 Lead-210
The longest lived radioisotopes of Pb are 202Pb (t1/2 = 5.3 × 104 years), 205Pb (t1/2 = 1.5 × 107 years), and 210Pb (t1/2 = 22.23 years). Lead-210 is naturally occurring as part of the 238U decay series, and has been measured in the fields of geochronology, air flux measurements of radon223,224 radiation protection associated with uranium mining, and environmental contamination from the iron and steel industry.225 Lead-210 is a beta emitting radionuclide (Emax 63.5 keV, 100% intensity), and is also a weak alpha (decay energy 3.72 MeV, 1.9 × 10−6% intensity) and gamma emitter (12.56 keV, 22.0% intensity). Measurement is therefore potentially achievable by multiple radiometric techniques. Gamma spectrometry measurement is affected by limited sample throughput, and prior knowledge of the sample geometry and composition is required. The low energy gamma ray, self-absorption and interferences from other gamma emitters and X-rays must also be considered.224 Lead-210 measurement by beta counting using a GM counter is achieved through measuring the 210Bi daughter (1.2 MeV Emax), following an ingrowth period of 8–9 days. Finally, measurement by alpha spectrometry is achievable through measurement of 210Po, however an ingrowth time of greater than 3 months is required.224Mass spectrometric measurement of 210Pb is affected by peak tailing from stable 208Pb (52.4% abundance), and polyatomic interferences associated with Pt, Hg, Bi and Pb.226 Polyatomic interferences combined with the relatively short half-life of 210Pb have been highlighted as the causes for measurement precision being inferior to that of alpha spectrometry.223 Pre-concentration of 210Pb prior to measurement is achievable by co-precipitation with cobalt-ammonium pyrrolidine dithiocarbonate,226 or as lead sulphate,225 whilst chemical separation from interferences can be performed by extraction chromatography using Sr-resin.226 Given the Pt-based interferences, Pt sampler and skimmer cones should be avoided, as high backgrounds have been reported at m/z = 210.225
Larivière et al. compared ICP-QMS (Perkin Elmer Elan 5000), ICP-DRC-MS (Perkin Elmer Elan 6100) and ICP-SFMS (Thermo Element 2) for measurement of 210Pb in water.226 All instruments were equipped with an Apex-Q sample introduction system, and a detection limit of 90 mBq L−1 (0.03 pg L−1) was recorded for final measurements. The tailing removal was greatest using ICP-DRC-MS (>3 × 109), compared to >2 × 106 for QMS, and 1 × 105 for SFMS. In a separate study, CC-ICP-MS (Agilent 7500ce) was applied to measurement of standards and solutions spiked with 210Pb, and sediment samples from waste associated with an on oil and gas production field in Egypt.224 The recovery was assessed using isotope dilution with 206Pb. The results showed good agreement with those obtained by gamma spectrometry, with a detection limit of 0.21 ng L−1 (593.24 Bq L−1). The formation of polyatomic interferences was reduced using krypton as a collision gas at a flow rate of 0.4 mL min−1, which was determined by assessing the 208Pb/206Pb ratio with the change in gas flow rate. It was also noted that potential lead hydride formation could be removed using a desolvating sample introduction system. Technologically enhanced 210Pb as a result of iron and steel manufacture has been measured along with 210Po using a combination of ICP-QMS (Perkin Elmer Elan 9000), gamma spectrometry and alpha spectrometry,225 with a Pb activity range of <7 Bq kg−1 (2.5 pg kg−1) to 4240 Bq kg−1 (1.5 ng kg−1).
8.14 Radium-226 and 228
Radium-226 and 228 are naturally occurring radioactive materials, with large amounts produced from mining and extraction of fossil fuels, as well as from the phosphate industry and uranium mine tailings.227,228 Radium-228 is also continually produced from the decay of 232Th in shelf sediments.229 The natural occurrence of these nuclides has led to application as a tracer for ocean circulation, seawater sediment fluxes and particulate residence times.229 Radium-226 decays with a half-life of 1599 years, with maximum alpha decay energy of 4.79 MeV (94.6%) and gamma emission of 186 keV (32.8%). The half-life of 228Ra is comparatively short (5.76 years), with a maximum beta decay energy of 39 keV.Radiometric detection is achievable by alpha or gamma spectrometry, however both require a lengthy ingrowth period for secular equilibrium to be established between 228Ra and 228Th.229,230 Whilst ICP-MS measurement needs lengthy sample treatment to remove multiple potential polyatomic interferences prior to sample introduction (including 88Sr138Ba, 87Sr139La, 86Sr140Ce, 208Pb18O, and 186W40Ar), no ingrowth time is required, and the sample analysis time is also significantly lower than radiometric measurements.228,229 Chemical separation is generally achieved by diffusive gradients in thin films (DGT) or manganese dioxide (MnO2) combined with ion exchange and/or extraction chromatography.227,230–233
Measurement of the 228Ra is challenging given the significantly shorter half-life and lower concentration in environmental samples compared to 226Ra. Recent applications have taken advantage of the high instrumental sensitivity of ICP-SFMS for 226Ra/228Ra detection, using either single227,231–233 or multi-collector instruments229,230,233 (Table 8). Instruments have been successfully operated at both low resolution,231–233 and medium resolution,227 with the latter setup improving polyatomic interference removal at the expense of one order of magnitude sensitivity loss when analysing seawater samples (1200 cps per ng L−1 at low resolution compared to 100 cps per ng L−1 at medium resolution). A recent study of 226Ra in high salinity seawater using a Perkin Elmer NexION 300x achieved a detection limit of 100 pg L−1 (3.7 Bq L−1),228 which is several orders of magnitude higher than that of ICP-SFMS. Multiple elements have been applied for mass bias correction, given the absence of a certified 226Ra/228Ra reference material, including 207Pb/208Pb, 238U/235U, and 229Th/232Th.229,230
| Reference | Instrument | Model | Matrix | Chemical separation | LOD pg L−1 (Bq L−1) |
|---|---|---|---|---|---|
| 228 | CC-QMS | Perkin Elmer NexION 300x | High salinity wastewater | AG50 (IE) | 100 (3.7) |
| 227 | SFMS | Element II | Seawater | ||
| 231 | SFMS | Element II | Water and sediment pore water | DGT | 0.5 (0.02) |
| 232 | SFMS | Element II | Water and sediments | DGT, MnO2 pptate, Sr resin (EC) | 0.5 (0.02) |
| 233 | MC-ICP-MS | Nu plasma | Natural waters | MnO2, AG50 (IE), Sr resin (EC) | 0.05 (0.002) |
| 0.05 (0.002) | |||||
| 230 | MC-ICP-MS | Neptune | Seawater and suspended particles | MnO2, AG50, AG1, Sr resin | 0.091 (0.003) |
| 229 | MC-ICP-MS | Nu instruments | Seawater |
8.15 Protactinium-231
Protactinium-231 is the longest lived intermediate daughter of 235U, with a half-life of 3.28 × 104 years, and can also be produced as a by-product in the thorium fuel cycle via a fast neutron reaction with 232Th or 232U. Protactinium-231 is an alpha emitting radionuclide (5.149 MeV), decaying to 227Ac. A major application of 231Pa is in precise isotope ratio calculations for uranium-series age dating, and as an indicator of climate change, and is commonly measured in combination with Th and U isotopes. This required high precision measurements of isotopic ratios, which can be achieved using TIMS (typically 2–3% precision).234–236 However, extensive matrix separation, and challenges associated with sample loading limit sample throughput.234 Alpha spectrometry has been used for measurement of 231Pa in seawater samples, however cubic-metre levels of sample are required, followed by extensive chemical separation, with low final measurement precision (5–10%).234 ICP-MS measurement can rival the precision of TIMS, whilst achieving a significant improvement in sample throughput. The main interference is peak tailing from 232Th, which can be removed by chemical separation prior to measurement if necessary. Protactinium-234 (t1/2 = 26.97 days) has been used a spike in some studies,234,237 however, consideration must be given to decay to 233U, leading to changes in the 231Pa/233Pa ratio over time, with losses of Pa to beaker walls also highlighted as an issue.Applications include a fossil coral fragment measured by MC-ICP-MS (FISONS PLASMA 54), with samples introduced by a CETAC MCN6000 desolvating nebulizer.238 The reduced sample handling, sample size and increased throughput compared to TIMS were highlighted as advantages. Choi et al. (2001) measured 231Pa along with 230Th in seawater by ICP-SFMS (Element) equipped with a desolvating nebulizer (Cetac MCN-6000), with a detection limit of 0.4 fg, corresponding to 0.02 fg g−1 (3.5 × 10−8 Bq g−1) in 20 L seawater samples.234 In a separate study, silicate rock samples were measured following anion exchange and extraction chromatography (TRU (Triskem International)) separation by MC-ICP-MS (Thermo Neptune) equipped with a desolvating nebulizer (Cetac Aridus), with a detection limit of 200 fg L−1 (3.5 × 10−6 Bq L−1).237
8.16 Thorium isotopes
Thorium is a naturally occurring element, with an abundance in the earth's crust 3–4 times higher than uranium, and has also been used as an alternative fuel for nuclear fission. The most abundant isotope is 232Th with a half-life of 14.02 × 109 years, and undergoes alpha decay to 228Ra. Additional radioisotopes with long half-lives measurable by ICP-MS are 229Th (t1/2 = 7340 years) and 230Th (t1/2 = 7.5 × 104 years). Thorium is commonly measured as 232Th along with isotopes of U and Pu in bioassay samples, as well as with uranium in chronology, paleoclimatology, archaeology, hydrology, geochemistry and oceanography.239,240 Alternative mass spectrometric measurement techniques include TIMS and SIMS.241 When Th is measured by ICP-MS in combination with U, the 232Th tailing can impact measurement of 233U, as can the formation of 232Th1H.242 A summary of recent measurement of Th isotopes by ICP-MS is given in Table 9, with a review of mass spectrometric determination of Th published elsewhere.243| Reference | Instrument | Model | Matrix | Chemical separation | LODa, pg L−1 (Bq L−1) |
|---|---|---|---|---|---|
| a LOD for 232Th unless indicated otherwise. | |||||
| 246 | QMS | X-series | Urine | MICROTENE-TOPO chromatography | |
| 250 | QMS | X-series II | River water | Stacked TEVA and DGA | 229Th: 4490 (34.9) |
| 230Th: 4490 (3.4) | |||||
|
232Th: 694000 (2.8 × 10−3) |
|||||
| 249 | QMS | Agilent 7500 ce/cx | Ores and mill tailings | ||
| 245 | QMS | Perkin Elmer Elan 6000 | Urine | TiCl4 precipitation and Dowex 1-x8 | QMS-400 (1.6 × 10−6) |
| SFMS | Element | SFMS-200 (8.1 × 10−7) | |||
| 242 | SFMS | Element 2XR | Urine, decommissioning samples including paint and tape | ||
| 247 | DRC-MS | Perkin Elmer DRC-e | Natural waters | 500 (2.0 × 10−6) | |
| 248 | DRC-MS | Perkin Elmer DRC-e | Environmental samples | FIA, UTEVA | 2800 (1.1 × 10−5) |
As instrument sensitivity has improved, the low-level measurement of Th is potentially impacted by environmental contamination during sample preparation. The concentrations of Th, U and their progenies in the reagents and labware used has become an increasingly important issue, with clean laboratory conditions and cleaning of materials prior to use required for some applications. Hoppe et al. (2013) investigated the use of low background materials and maintenance of low background levels of Th and U.244 It was concluded that sample preparation is the limiting factor affecting sensitivity for very low level measurements, and thorough cleaning and acid leaching of these materials has made very sensitive measurements possible.
Several studies have measured Th in combination with U and/or Pu in urine.242,245,246 Becker et al. (2004) measured Th and U using LA-ICP-MS, which minimized sample preparation, whilst quantification issues associated with laser ablation were resolved with matrix-matched standards.245 Measurements were performed by both ICP-QMS (Perkin Elmer Elan 6000) and ICP-SFMS (Thermo Element) coupled to laser ablation (CETAC LSX 200). The Th recovery ranged from 97–104% at doping concentrations of 0.52–2.49 ng L−1 (2.1 × 10−6 Bq L−1 to 1.0 × 10−5 Bq L−1) with detection limits of 0.4 ng L−1 (3.5 × 10−8 Bq g−1) and 0.2 ng L−1 (8.1 × 10−7 Bq g−1) for ICP-QMS and ICP-SFMS, respectively. The study highlighted the advantages of ICP-MS for actinide measurement over alpha spectrometry, which was a less sensitive approach that required more extensive sample preparation. In a separate study, Shi et al. (2013) measured Th, U and Pu in urine samples with regards to dose assessment, as well as measuring decommissioning samples including tape and paint.242 Samples were measured by ICP-SFMS (Thermo Element XR) with an Elemental Scientific Apex desolvating sample introduction system, which reduced the 232Th1H formation rate to 1.3 × 10−5, compared to 6.6 × 10−5 for solution nebulization using a Micromist nebulizer. Cozzella and Pelttirossi (2013) measured Th, U and Pu in urine by ICP-MS rather than alpha spectrometry, highlighting the ability to process a large number of samples whilst maintaining an acceptable measurement uncertainty.246 Samples were measured by ICP-QMS (Thermo X-series) with a Burgener nebulizer. Prior to sample introduction, Th was stabilized in urine with Triton X-100 and then mixed with MICROTENE-TOPO, shaken vigorously and then loaded onto a column prior to elution. Thorium recovery ranged from 93 ± 0.2% to 120 ± 1.2% over a spiked concentration range of 0.5–2 μg L−1 (2.0 × 10−3 to 8.1 × 10−3 Bq g−1).
Thorium and U were measured along with multiple rare earth elements in natural spring water samples in Brazil, in relation to supplying safe potable water to nearby towns.247 Water samples were measured by ICP-DRC-MS (Perkin Elmer ELAN DRC-e) equipped with a sea spray nebulizer and cyclonic spray chamber. The method detection limit was 0.5 ng L−1 (2.0 × 10−6 Bq g−1) Avivar et al. (2011) measured Th and U in multiple environmental samples (different water samples, a phosphogypsum sample, and a channel sediment reference material) using multi-syringe FIA (MSFIA), using UTEVA resin (Triskem International) for online separation and pre-concentration.248 The system was coupled to ICP-DRC-MS (Perkin Elmer Elan DRC-e) equipped with a Scott spray chamber and cross-flow nebulizer. Combination of LOV with MSFIA achieved a 232Th detection limit of 2.8 ng L−1 (1.1 × 10−5 Bq L−1), compared to 120 ng L−1 (4.9 × 10−4 Bq L−1) using FIA only. Tuovinen (2015) compared ICP-MS to XRF, ERD, EPMA, gamma spectrometry and alpha spectrometry for determination of Th and U in ore and mill tailing samples collected in Finland.249 Thorium was measured over a concentration range of 7–157 mg L−1 (2.8–637.2 Bq L−1) by ICP-QMS (Agilent 7500 ce/cx), with the results showing generally good agreement with other techniques. The lowest values were measured by ICP-MS compared to other techniques, which was the result of challenges with sample preparation.
8.17 Uranium isotopes
Uranium is naturally occurring, with an average concentration of ∼4 μg g−1 in the terrestrial crust, 3 μg L−1 in seawater (uniformly distributed in the world's oceans) and ranging from 0.5–500 μg L−1 in surface freshwater depending on the extent of contamination.78 The environmental occurrence of uranium is mostly in hexavalent form, associated with oxygen in nature as the uranyl ion UO22+. Under strongly reducing conditions, uranium is present in tetravalent form in strongly reducing medium such as high organic material (UO2). Uranium-238 is the major isotope (99.27%), with additional minor isotopes of 234U (0.006%) and 235U (0.72%).Uranium was one of the early nuclides to be measured by ICP-MS, with studies initially focusing on the elemental concentration of 238U. As instrumental performance improved, there was an increasing focus on isotopic ratio measurements, initially 235U/238U, and more recently 234U/238U, and 236U/238U, with isotopic analysis enabling distinction between exposure to natural and anthropogenic U sources. Reviews of uranium determination using atomic spectrometric techniques, and in environmental samples, are published elsewhere,78,251 and Table 10 gives a summary of recent ICP-MS procedures for U isotopic measurements.
| Reference | Instrument | Model | Sample matrix | Chemical separation | LODa, pg L−1 (Bq L−1) |
|---|---|---|---|---|---|
| a LOD for 238U unless indicated otherwise. | |||||
| 78 | QMS | Plasma Quad 2+ | Environmental samples (oyster tissue and pine needles) | Pneumatic nebulizer: 5400 (6.7 × 10−5) to 48000 (6.0 × 10−4) |
|
| ETV: 900 (1.1 × 10−5) to 21000 (2.6 × 10−4) |
|||||
| 255 | Ion trap MS | LTQ-XL | Water, U ore, soil | EESI | |
| 97 | QMS | Elan 5000 | Uranium oxide leachate | Anion exchange | 30000 (3.7 × 10−4) |
| 258 | QMS | Yokogawa PMS-2000 | Environmental samples | TEVA resin | 100 (1.2 × 10−6) |
| 259 | QMS | Perkin Elmer Elan 6000 | Urine | 4000 (5.0 × 10−5) | |
| 250 | QMS | X-series II | River water | Stacked TEVA and DGA resins | 233U: 6.5 (2.3 × 10−3) |
| 234U: 6.5 (1.5 × 10−3) | |||||
| 235U: 23.8 (1.9 × 10−6) | |||||
| 236U: 6.5 (1.6 × 10−5) | |||||
| 238U: 2820 (3.5 × 10−5) | |||||
| 260 | QMS | Agilent 7500 | Urine | Ca phosphate, stacked TEVA, TRU, DGA resin | |
| 261 | DRC-MS | Perkin Elmer Elan 6100 DRC | Urine | Instrument LOD: 4 (5.0 × 10−8) | |
| Method LOD: 22 (2.2 × 10−11) | |||||
| 262 | SFMS | Element 2 | Urine | Sample dilution only | 200 (2.5 × 10−6) |
| 263 | SFMS | Element | Standard solutions | ||
| 49 | SFMS | Element | Soil samples 4–20 km north and west of Chernobyl | Anion exchange | Soil: 40 (5.0 × 10−7) |
| Water: 0.2 (2.5 × 10−9) | |||||
| 259 | SFMS | Element | Urine | Instrument LOD: 60 (7.5 × 10−7) | |
| Method LOD: <3000 (3.7 × 10−5) | |||||
| 264 | SFMS | Plasma Trace 2 | Urine | Phosphate precipitation | 5 (6.2 × 10−8) |
| CC-MC-ICP-MS | Micromass Isoprobe | 2× TRU | 1 (1.2 × 10−8) | ||
| MC-ICP-MS | VG Elemental P54 | UTEVA resin | 200 (2.5 × 10−6) | ||
| 265 | SFMS | Element XR | Swab samples | Stacked ion exchange and extraction chromatography | |
| 265 | SFMS | Element 2 | Urine | Ca and Mg co-precipitation | |
| 266 | SFMS | Element 2 | Urine | UTEVA resin | 235U: 0.8 (6.4 × 10−8) |
| 236U: 0.05 (1.2 × 10−7) | |||||
| 238U: 100 (1.2 × 10−9) | |||||
Several spectral interferences must be considered for uranium analysis, with Ir, Bi, Th or Ru effectively used for interference correction.78 Potential platinum-argide interferences from the use of platinum cones can be avoided by using nickel cones,252 whilst chloride-based interferences with elements including Au and Hg must also be considered.78 Uranium-236 detection is potentially affected by both 235U1H formation, and tailing from 238U. Sector field instruments are a popular choice for uranium determination due to low background, high sensitivity at low resolution, and ability to remove interferences at high resolution.78 Additionally, the simultaneous data acquisition of MC-ICP-MS combined with sector field sensitivity leads to more precise isotope ratio determination.253 Determination of uranium concentration and/or isotopic ratios in bioassay samples is commonly carried out through measurement of urine samples, as it is non-invasive, whilst the complexity of the sample matrix represents a challenge for isotope ratio determination.252–254 For example, uranium was measured as part of the Baltimore VA Depleted Uranium Clinical Follow-up Program by ICP-SFMS (Thermo Element 2).252 Measurement of 235U/238U and 236U/238U was compared for a quartz concentric nebulizer with a cyclonic spray chamber, and APEX Q (Elemental Scientific) sample introduction system. Superior sensitivity was recorded using the Apex Q, which was preferable for low U concentrations, whilst the concentric nebulizer and cyclonic spray chamber was more robust and suitable for higher U concentrations. The limited abundance sensitivity and 235UH formation meant the background equivalent concentration of 236U was 25 times higher than for 235U and 238U, with more scattered 236U/238U ratios at total uranium concentrations <10 ng L−1. Additionally, the accuracy of 235U measurements was poor when total U concentrations were less than 5 ng L−1.
In a separate study, Arnason et al. (2015) carried out an inter-laboratory comparison for uranium concentration and multiple isotope ratio values in urine, with results from sites using ICP-QMS or ICP-SFMS.254 As the total uranium concentration decreased, the concentration measured by isotope dilution only were higher than those that used chemical separation or digestion prior to measurement. At a concentration of 50 ng L−1, the predicted 234U/238U value was 0.000053. Sites that carried out digestion and chemical separation prior to measurement yielded values from 5.2 × 10−5 to 7.2 × 10−5, compared to 1.5 × 10−3 to 1.8 × 10−2 for isotope-dilution-only methods. A significant positive bias was also seen for 235U/238U for dilution only-methods, accounted for by polyatomic interference at m/z = 235. Finally, for 236U/238U, only studies that applied chemical separation were considered, and, of these five studies, four either did not report a result or reported a non-detect result. The conclusion from this study was that accurate and precise isotope ratio measurements was more dependent on the analytical methodology and instrument capability than measurement of total U concentration. Arnason et al. also measured uranium isotopes in urine samples at volumes from 1–8 mL by ICP-SFMS (Element 2) equipped with a Cetac Aridus desolvating sample introduction system, following chemical separation using UTEVA extraction chromatography separation.253 At spike concentrations of 2.5–25 ng L−1, 235U/238U was successfully detected, with an improvement in precision as U concentration increased. By comparison, 234U/238U was only detected in samples spiked with 25 ng L−1. It was also realized that whilst a higher urine volume achieved better precision, the uranium recovery was lower.
Liu et al. (2011) applied extractive electrospray ionization mass spectrometry (EESI-MS) to determination of 235U/238U in uranyl nitrate solutions, prepared from samples including natural water, uranium ore and soil.255 EESI-MS is typically used for organic compounds, and in this study samples were directly measured without pre-concentration or separation. The resulting mass spectrum looked for detection of uranyl nitrate at m/z 456 [238UO2(NO3)3−], as well as peaks for 234 and 235. There was a relative error in isotope ratios of 0.21–0.25%, and RSD of 1.54–1.81%. This approach overcomes the extensive sample preparation that can lead to U losses, and also offers fast analysis speed (∼5 minutes per sample). Results were comparable between EESI-MS and ICP-QMS (Agilent 7500ce) over a U concentration range of ∼2.6 μg L−1 to ∼3.1 mg L−1. Results from EESI-MS returned a RSD of 1.25–3.26%, compared to 0.71–1.46% for ICP-MS. The EESI used in this study was home-made, and it was suggested that improvements in RSD would be realized using a commercial EESI source.
Uranium-236 was measured in soil samples in the vicinity of Chernobyl by ICP-SFMS (Thermo Element), ICP-QMS (Perkin Elmer Elan 6000) and ICP-CC-MS (Micromass Platform ICP), with high precision isotope ratio measurements were performed using a MC-ICP-MS (Nu Instruments).256 Multiple sample introduction systems (micro-concentric Micromist nebulizer, Q-DIHEN or MCN equipped with an Aridus desolvating sample introduction (CETAC)) were investigated. An abundance sensitivity for 236U/238U of 5 × 10−6, 3 × 10−7, 6 × 10−7 and 3 × 10−7 was measured for ICP-SFMS, MC-ICP-MS, ICP-QMS and ICP-CC-MS, respectively. The precision ranged from 0.28–0.34% for 236U/238U for single collector instruments, improving to up to 0.07% for MC-ICP-MS equipped with USN or MCN with an Aridus. Ultra-low 236U/238U ratios were recently measured at isotopic ratios of <10−7 by ICP-QQQ-MS (Agilent 8800).257 The two mass filters effectively removed tailing from the peak at 238U, whilst the 235U1H interference was removed by reacting UH with O2 in the collision–reaction cell. The interference-removal capability meant that accurate isotopic ratios could be measured without the need for spectral interference correction.
8.18 Neptunium-237
Neptunium-237 is an alpha emitting radionuclide (maximum decay energy 4.78 MeV (47.6%) with a half-life of 2.1 million years. Neptunium is present in the environment as a result of atmospheric weapons test fallout and nuclear fuel reprocessing. A review of 237Np measurement in nuclear and environmental samples was recently published.266 Samples investigated include soils, sediment, groundwater, seawater and various other environmental samples.82,267–277Neptunium occurs in both tetra- and pentavalent states, and the variation in oxidation state depending on the reagents and conditions used makes sample preparation challenging.278 Bulk separation from the sample matrix has been achieved by techniques including iron hydroxide precipitation272 and lanthanum hydroxide precipitation,275 followed by ion exchange or extraction chromatography.82,268–272,275,278 Neptunium is often measured along with Pu following chemical separation, given the similar chemical behavior of the two elements in HNO3 and HCl on anion exchange and extraction chromatography resins.63,268–271,274–276,279 A micro-flow injection sample introduction system combined with membrane desolvation improved the instrument sensitivity, and the detection limit by concentrating the analyte into a smaller volume.276 In a separate study, Qiao et al. incorporated TEVA resin into a sequential injection (SI) system for simultaneous Np and Pu measurement in environmental samples, or Bio-RAD AG MP-1M for soil, sediment and seaweed.279
The major interference impacting 237Np detection is tailing from 238U, which must be removed by chemical separation, or by the instrument if abundance sensitivity is sufficient.276 The 237Np detection limit has been noted to increase with increasing U concentration (0.32 fg (8.3 × 10−9 Bq) with no uranium, compared to 10 fg (2.6 × 10−7 Bq) with 2500 pg of U, with a negative impact on detection limit once the U concentration exceeded 30 pg).
The lack of a suitable tracer for 237Np has been identified as a limitation,266,276 particularly for isotope dilution applications.274,280 Neptunium-236 (154000 year half-life) is potentially suitable,269,274 but suffers from a lack of commercial availability and production difficulties.280,281 Alternative isotopes of neptunium (239Np and 235Np) are less well suited given their short half-lives (2.35 days and 396 days, respectively) and suffer from potential isobaric interferences from 239Pu and 235U, respectively.63 Plutonium-242 (half-life 3.75 × 105 years) has been used as a tracer for determination of both 237Np and Pu isotopes,276 given its similar chemical behavior and commercial availability.269,270,274,275,280 An issue with this is variation in chemical fractionation between Np and Pu during analysis, which will increase uncertainty,280 and an isotope of the same element is desirable.
Neptunium-237 has frequently been determined using ICP-SFMS268,270–274 (Table 11). Kim et al. (2004) simultaneously measured 237Np with 239Pu and 240Pu in environmental samples by ICP-SF-MS (Micromass Plasma Trace 2) equipped with an Aridus desolvating sample introduction system and T1-H micro-concentric nebulizer.270 Chemical separation of Np and Pu was achieved using an automated SI system using TEVA extraction chromatography resin, with a 237Np detection limit of 2.5 pg L−1 (6.5 × 10−5 Bq L−1). In a separate study, an Agilent 7500 QMS equipped with an Octopole Reaction System was operated in ‘no gas’ mode for Np measurement.275 However, there was evidence that Np would behave similarly to Pu with CO2 as the reactive gas, which could be used as a basis for separation of Np and Pu from U.282 It was noted that applying chemical separation prior to ICP-SFMS measurement could lower the detection limit.
| Reference | Instrument | Model | Matrix | Chemical separation | LOD, pg L−1 (Bq L−1) |
|---|---|---|---|---|---|
| 283 | QMS | Perkin Elmer Elan 5000 | Natural groundwater | Capillary electrophoresis | 50000000 (1.3 × 103) |
| 260 | QMS | Agilent 7500 | Urine | Ca phosphate, stacked TEVA, TRU, DGA resin | |
| 275 | QMS | Agilent 7500 | Large soil samples | 8700 (0.2) | |
| 63 | QMS | X-series II | Environmental samples | Hydroxide precipitate, sequential injection (TEVA resin) | |
| 279 | QMS | X-series II | Soil, sediment, seaweed | Iron hydroxide precipitation, sequential injection (anion exchange) | 1.5 (3.9 × 10−5) |
| 267 | QMS | VG Elemental PlasmaQuad PQ2+ | Environmental samples | TOA extraction chromatography | 460 (1.2 × 10−2) |
| 268 | SFMS | Finnigan MAT Element | Environmental samples | TRU resin | |
| 82 | SFMS | Not given | Irish sea sediments | Glow discharge | 80000 (2.1) |
| 269 | SFMS | Finnigan-MAT Element | Environmental samples | La(OH)3 co-precipitation, TEVA resin | 0.5 (1.3 × 10−5) |
| 270 | SFMS | Micromass PlasmaTrace 2 | Environmental samples | Automated sequential injection (TEVA resin) | 2.5 (6.5 × 10−5) |
| 272 | SFMS | Finnigan MAT Element 2 | Soil | TEVA resin | 0.2–0.4 (5.2 × 10−6 to 1.0 × 10−5) |
| 284 | SFMS | Element 2 | Soils and sediments from river Yenisei | TEVA resin |
Sediments have been measured by glow discharge mass spectrometry.82 The instrument was operated at a mass resolution of around 6000, resulting in high transmission (>75%) and removal of tailing interferences, as well as reduced sample preparation time compared to chemical separation. Irish Sea sediment samples were measured after being compacted into pellets. Neptunium was determined in groundwater at the Nevada National Security Site (NNSS) by ICP-SFMS (NuPlasma) at concentrations ranging from <4 × 10−4 to 2.6 mBq L−1 (0.015–99.7 pg L−1) with all values below the US Environment Protection Agency drinking limits of 560 mBq L−1.277 The outcome was used to evaluate retardation of Np relative to other radionuclides in NNSS groundwater.
8.19 Plutonium isotopes
Plutonium is present in the environment as a result of nuclear weapons tests, reactor accidents, and discharges from reprocessing facilities, and is arguably the most frequently studied transuranic element. Isotopes of particular interest with regards to ICP-MS measurement are 238Pu (t1/2 = 87.7 years), 239Pu (t1/2 = 24110 years) and 240Pu (t1/2 = 6561 years) (Table 13). Several reviews on the determination of plutonium isotopes are given elsewhere.95,120,285
Isotope ratio measurements by ICP-MS (most commonly 240Pu/239Pu) have been effectively used to determine the source of nuclear contamination286 (Table 12). This is a significant advance over traditional alpha spectrometry detection, which is unable to resolve the similar alpha energies of 239Pu and 240Pu (5.24 and 5.25 MeV, respectively). Measurement of Pu isotopes by alpha spectrometry also requires extensive chemical separation and counting times on the order of days to weeks.
:239Pu atom ratios for different sources286,299
| Source | 240Pu/239Pu |
|---|---|
| a After fuel burn up (the amount of energy extracted from the primary fuel source).48,287 | |
| Integrated weapon test fallout | 0.18 |
| Weapon production | 0.01–0.07 |
| Chernobyl accident | 0.40 |
| MAGNOX reactor | 0.23a |
| Pressurized heavy water reactor | 0.41a |
| Advanced gas-cooled reactor (AGR) | 0.57a |
| Pressure tube boiling water reactor | 0.67a |
| Boiling water reactor (BWR) | 0.40a |
| Pressurized water reactor (PWR) | 0.43a |
| Fukushima prefecture coast sediments | 0.19–0.26 |
| Reference | Instrument | Model | Matrix | Chemical separation | LOD, pg L−1 (Bq L−1)a |
|---|---|---|---|---|---|
| a LOD for 239Pu unless indicated otherwise. | |||||
| 42 | SFMS | Plasma Trace 2 | Environmental samples | Sr resin and TEVA resin | 3 (6.5 × 10−5 Bq L−1) |
| 283 | QMS | Perkin Elmer Elan 5000 | Natural groundwater | Capillary electrophoresis | 50000000 (1.2 × 105) |
| 300 | QMS | PlasmaQuad2+ | Environmental samples | 239/240Pu: 10 (2.3 × 10−2/8.4 × 10−2) | |
| 289 | QMS | PQ Excell-s | Soil | Flow injection (UTEVA resin) | 4.3 (9.9 × 10−3) |
| 301 | QMS | Varian 810 MS | Environmental samples | Flow injection (TEVA resin) | 3 (6.9 × 10−3) |
| 302 | QMS | Perkin Elmer Elan 6000 | Environmental samples | TEVA resin | |
| 260 | QMS | Agilent 7500 | Urine | Ca phosphate precipitation, stacked TEVA, TRU, DGA resins | |
| 275 | QMS | Agilent 7500 | Large soil samples | 700 (1.6) | |
| 63 | QMS | X-series II | Environmental samples, sequential injection | Hydroxide precipitation, sequential injection (TEVA resin) | |
| 279 | QMS | X-series II | Soil, sediment, seaweed, sequential injection | Iron hydroxide precipitation, SI-based AG1 | 1.5 (3.5 × 10−3) |
| 250 | QMS | X-series II | River water | Stacked TEVA and DGA resin | 239/240/242Pu: 0.3 (6.2 × 10−4, 2.3 × 10−3, 4.0 × 10−5) |
| 97 | QMS | Elan 5000 | Uranium oxide leachate | Anion exchange | 30000 (68) |
| 283 | QMS | Perkin Elmer Elan 5000 | Natural groundwater | Capillary electrophoresis | 50000000 (1.2 × 105) |
| 282 | CC-QMS | GV Isoprobe | U-235 target | ||
| 268 | SFMS | Finnigan MAT Element | Environmental samples | TRU resin | |
| 49 | SFMS | Element | Soil samples 4–20 km north and west of Chernobyl | Anion exchange | |
| 291 | SFMS | Element 2 | Urine | None | 4.7 (1.1 × 10−2) |
| 270 | SFMS | Micromass PlasmaTrace 2 | Environmental samples | Automated sequential injection (TEVA resin) | 2.1 (4.8 × 10−3) |
| 240Pu: 0.42 (9.7 × 10−4) | |||||
| 300 | SFMS | Axiom SC | Environmental samples | SFMS: 239/240Pu: 0.1 (2.3 × 10−4/8.4 × 10−4) | |
| 241Pu: 0.05 (0.2) | |||||
| 303 | SFMS | Thermo Element | Urine | Ca phosphate precipitation and TEVA resin | 1.0 × 10−3 (2.3 × 10−6) |
| 57 | SFMS | Element 2 | Environmental samples | CaF2 precipitation, UTEVA and TRU resin | 9.2 (2.1 × 10−2) to 15 (3.5 × 10−2) |
| 304 | SFMS | Micromass PlasmaTrace 2 | Marine sediments | Calcination, anion exchange chromatography | 240/241/242Pu: 3–4 (2.5 × 10−2 to 3.4 × 10−2/11.4–15.2/4.4 × 10−4 to 5.9 × 10−4) |
| 239/240Pu: 8–9 (1.8 × 10−2 to 2.1 × 10−2/6.7 × 10−2 to 7.6 × 10−2) | |||||
| 284 | SFMS | Element 2 | Soils and sediments from river Yenisei | TEVA resin | |
Measurement of 239Pu is affected by tailing from 238U, with decontamination factors of 108 or higher required depending on the sample matrix.285,286 A detection limit of 1.0 fg (2.3 × 10−6 Bq) was achieved for 239Pu at U concentrations from 0–30 pg using ICP-SFMS (Thermo Element XR), compared to 8.6 fg (2.0 × 10−5 Bq) when U concentration was increased to 2500 pg.276 There is an additional polyatomic interference from 238U1H, which can be removed by U/Pu separation, or by reducing hydride formation using a desolvating sample introduction system. Plutonium-238 measurement is challenging owing to low concentrations, and an isobaric interference from 238U, whilst 241Pu can be affected by isobaric 241Am. Chemical separation of Pu prior to sample introduction is generally a 2 stage process-bulk matrix co-precipitation, followed by actinide pre-concentration and separation using extraction chromatography.63,275,282,288–290 More rapid FI or SI techniques have been effectively applied to urine,291 soils,275,289 and environmental samples.63 A number of Pu isotopes including 236Pu, 242Pu and 244Pu have been effectively used as tracers,63 with isotope dilution a common technique that can also address issues with plasma instability and ion intensity drift.95
Carbon dioxide has been used as a reactive gas for U separation from Pu for a 235U target using CC-ICP-MS (GV Isoprobe MC-ICP-MS).282 At CO2 gas flow rates >0.5 mL min−1, U is present as UO+ (∼95%) and UO2+ (∼5%), compared to Pu, which was present as Pu+ (40%), PuO+ (∼60%), and PuO2+ (∼2%). A recent study used lab-on-valve separation of Pu using bead injection extraction chromatography micro-flow system was coupled to ICP-QMS (Thermo X-series II) equipped with Xs skimmer cone and Burgener nebulizer.292 The design enabled processing of large urine volumes (1 L), Pu chemical yields >90%, and completion of the analytical procedure in less than 3 hours, compared to 1–2 days for manual processing. A detection limit of 1.0–1.5 pg L−1 (equivalent to 2.3 × 10−3 to 3.4 × 10−3 Bq L−1 for 239Pu and 1.5 × 10−4 to 2.2 × 10−4 Bq L−1 for 242Pu) was achieved for 239Pu and 242Pu, however, potential improvements to the procedure were suggested, including overcoming the limited flow rate (<1 mL min−1) due to back-pressure build up and settling of beads within the system, which could lead to leakage and/or malfunctioning for long-term operations.
Plutonium isotope ratios (239Pu/240Pu) were determined from alpha planchettes originally prepared from samples from the Mayak nuclear facility, Russia.293 The aim was to show that direct analysis of samples was possible without the time-consuming dissolution and chemical separation associated with solution ICP-MS. A potential advantage is that this approach can be applied to planchette samples round the world, to generate a large amount of new data from existing samples. Samples had previously been separated from the sample matrix, and were measured by ICP-QMS (Agilent 7700X). If the 239+240Pu activity on the planchette was <1 Bq, the statistical variation in isotope ratios was too high and reliable determination of 240Pu was not possible. It was noted that the likelihood of distinguishing between sources of Pu could be improved by chemical separation to remove U from the sample prior to measurement.
Varga et al. investigated rapid and direct measurement of U and Pu, 235U/239Pu and 236U/240Pu chronometers without chemical separation using a Thermo Element 2 ICP-SFMS equipped in a glovebox.294 The instrument was run in low resolution mode with a low-flow micro concentric nebulizer, and a quartz glass spray chamber. A second method incorporating extraction chromatography prior to measurement was also tested, with multiple U/Pu ratios measured. Results from two Pu CRM's were in good agreement with archive purification dates.
Bu et al. measured Pu isotopes in lichen, kelp, moss and horse mussel collected from Alaska by ICP-SF-MS (Thermo Element XR).295 The instrument was operated in low resolution, equipped with an Apex (Elemental Scientific) sample introduction system and PFA nebulizer. A 242Pu tracer was used, and a correction factor was applied to correct for uranium hydride interference, however this was expected to be low as chemical separation was applied prior to sample introduction. Mass discrimination was determined from measurement of 235U/238U in a natural U solution, with the overall procedure validated by measuring 239+240Pu in a CRM. A measured value of 5.18 ± 0.10 Bq kg−1 was in good agreement with the certified value of 5.30 ± 0.16 Bq kg−1. The total 239+240Pu content in sample measured ranged from 3.8 to 573 Bq kg−1 dry weight. The difference in isotope ratio values measured showed isotopically heavier 240Pu/239Pu values in marine samples compared to terrestrial samples, accounted for as being a large input of Pu into the Pacific Ocean, most likely Marshall Islands high yield tests.
A comparison of liquid scintillation and ICP-SFMS for 241Pu measurement in nuclear waste slurries was reported by Jäggi et al. (2012).296 ICP-MS measurements were performed using a double focusing ICP-SFMS (Thermo Element 2). Samples were introduced via an Apex nebulizing system connected to an ACM desolvation system and a self-aspirating PFA-ST nebulizer. Correction for tailing and hydride formation were performed following measurement of a 238U standard. Correction factors for mass 239, 240 and 241 arising from 238U were typically 9 × 10−6, 1 × 10−6 and 3 × 10−7. A detection limit of 0.27 Bq g−1 (117.6 pg g−1) for a 0.1 g slurry sample was achieved.
Isotope ratios were measured from multiple environmental samples collected in Finland by ICP-SFMS (Thermo Element 2) operating in low resolution, fitted with a coaxial nebulizer and cyclonic spray chamber.297 Instrument performance was carried out using a certified reference material containing natural U, and quality of 240Pu/239Pu measurements was validated from a CRM. For surface air samples collected in 1963, values mostly fell in the range 0.15–0.25, indicative of global weapons fallout. Results from environmental samples showed a wider range in values from 0.13 ± 0.1 to 0.53 ± 0.03, indicative of both global fallout and the Chernobyl accident. As well as digestion and chemical separation, old alpha planchettes were wet-ashed and purified by extraction chromatography prior to measurement.
Both airborne Pu and U originating from the Fukushima Daiichi NPP were identified in the atmosphere 120 km from the site through measurement of aerosol samples, with a sampling time of approximately one week.298 Following ashing and chemical separation, samples were analyzed by a combination of AMS and ICP-SFMS. Whilst the amount of environmental U and Pu increased, the dose from airborne Pu was negligible. ICP-SFMS was used for analysis of 234U/238U and 235U/238U, however AMS was advantageous for Pu measurement due to excellent suppression of hydride interferences. No Pu spike was added because of the low levels being investigated, and the potential negative impact on detection limit that adding a spike could have. Zheng et al. presented the first data on distribution of Pu isotopes in surface sediments 30 km off Fukushima.287 The 240Pu/239Pu ratios were measured using ICP-SFMS with Apex (Elemental Scientific) sample introduction. Chemical separation was performed using anion exchange chromatography, and several reference materials were used for method validation. Isotope ratio results were compared to background data in Japanese estuaries and the western North Pacific, and it was found that no Pu contamination was detected outside the 30 km zone around the plant. A more comprehensive assessment of Pu isotope measurements in seawater and sediments within the 30 km zone was recommended, to improve understanding of marine environmental behavior.
Detection limits in the attogram range in sediment and seaweed reference materials was achieved using ICP-SFMS (Element XR) equipped with the Jet interface,56 comparable to sensitivities achievable by AMS. Despite the larger orifice of the Jet sample cone, the Jet interface did not increase the hydride formation rate when operating with a desolvating sample introduction system. Without the Jet interface, the precision of 240Pu/239Pu was 20.5% and accuracy −3.3%, compared to 5.0% and 0.83% when equipped with the Jet interface and Cetac Aridus II desolvating sample introduction system. Plutonium has also been determined in urine samples by multiple techniques-alpha spectrometry, ICP-SFMS (Thermo Element XR) and AMS.299 The minimum detectable activity for SF-ICP-MS (23 fg) was two times better than alpha spectrometry (50 fg), but inferior to AMS (0.44 fg).
8.20 Americium-241
Americium-241 is produced as an activation product, and from beta decay of 241Pu, with a half-life of 432.2 years. Americium-241 is an alpha emitter (5.44 MeV, 12.8% yield and 5.49 MeV, 85.2% yield), and gamma emitter (59.5 keV, 36% abundance), with gamma spectrometry considered inadequate for low-level 241Am determination.62 Alpha spectrometry is the most sensitive and frequently used approach, and measurement is generally combined with chemical yield tracer 243Am (t1/2 = 7.4 × 103 years), which decays by alpha emission (5.28 MeV, 87.4% yield and 5.23 MeV, 11% yield), and gamma emission (74.7 keV, 62.8% abundance). A comprehensive review of analytical methods for americium determination is published elsewhere.62,305Measurement of 241Am is affected by an isobaric interference from 241Pu, and from 240PuH+, with Pu typically present at comparable or higher concentrations than that of 241Am in materials of reactor origin. There are also multiple polyatomic interferences, and 243Am (half-life 3730 years) has been applied as an internal standard,57,306 with 242Pu used to assess 241Pu contamination.98 Alternatively, a mixed elemental standard can be used for external calibration of the instrument.116 Chemical separation techniques prior to sample introduction include precipitation/co-precipitation and/or liquid–liquid extraction, ion exchange chromatography and extraction chromatography.57,306 Providing these interferences can be effectively removed, ICP-SFMS can match the detection limits of alpha spectrometry.62
Krachler et al. (2014) noted the absence of matrix-matched reference materials, leading to determination of Am in spent nuclear fuels using both high resolution ICP-OES and ICP-SFMS (Thermo Element 2) installed in a glovebox.116 The absence of a certified reference material meant instrumental mass bias was estimated based on the difference in sensitivity between 232Th and 238U in the internal standard, and assuming the same instrument sensitivity for all actinides. The mean concentration differed by a maximum of 4% between ICP-OES and ICP-MS.
Isotope-dilution ICP-SFMS has been used for the determination of pg kg−1 concentrations of 241Am in sediments (Agarande et al., 2001).307 Measurements of 241Am relative to a 243Am spike were performed using a VG Elemental Axiom ICP-MS with single collector and MCN6000 desolvating microconcentric nebulizer. Plutonium-242 was also added to confirm effective separation of Pu and Am prior to measurement. Measurements were performed in low resolution mode and mass bias was corrected using the U isotopic standard IRMM-72/1. Limits of detection of 0.2 Bq kg−1 (1.6 pg kg−1) were reported. Americium was analyzed along with Pu in Chernobyl-contaminated samples by ICP-SFMS (Thermo Element 2) in comparison to alpha spectrometry.62 Good agreement was observed between the two techniques for Certified Reference Materials (IAEA-384 and IAEA-385). The measurement time for ICP-SFMS was several minutes, compared to count times of days by alpha spectrometry. ICP-SFMS achieved a detection limit of 13.2 μBq g−1 (0.1 fg g−1) for ICP-SFMS, with a precision of 0.8–3%, compared to 4 μBq g−1 (0.03 fg g−1) and a precision of 1–5% by alpha spectrometry. Mass discrimination was again determined by applying a linear correction using a natural uranium solution. The combined measurement of Pu and Am allowed calculation of date of contamination based on the 241Pu/241Am ratio, assuming release of radionuclides was over a short time rather than continuous, as 241Am is also produced by decay of 241Pu. In a separate study, Varga measured 241Am in two environmental reference materials (sediment and seaweed) by ICP-SFMS (Thermo Element),98 with alpha spectrometry used to validate the procedure. The removal of polyatomic interferences was tested using stable element standards of Bi, Pb, Hg and Tl, whilst isobaric 241Pu removal was monitored using a 242Pu isotopic tracer. A detection limit of 0.86 fg g−1 (0.11 mBq g−1) was achieved, which was comparable to that of alpha spectrometry 0.79 fg (0.10 mBq).
A method for 241Am determination in urine was developed and validated at the Centre for Disease Control and Prevention (CDC).306 Measurements were performed using ICP-SFMS (Thermo Element XR) equipped with a CETAC Aridus desolvating sample introduction system, with a detection limit of 0.22 pg L−1 (0.028 Bq L−1). These values were in good agreement with liquid scintillation counting (LSC), and NIST CRM target values. The analytical bias from −0.3 to 1.7% for observed values compared to target values, and 2.1–3% for samples run in an internal comparison with LSC. The previous procedure using LSC returned a detection limit of 32.3 pg L−1 (4.2 Bq L−1), higher than the CDC action level of 0.73 pg L−1 (0.09 Bq L−1). It was noted that if U concentrations exceeded 10 μg L−1, more aggressive rinsing was required to eliminate U from solution, with analytical bias increasing to between −0.62 and −5.61% compared to NIST target values.
On-line extraction chromatographic separation of actinides, including 241Am, from urine matrices coupled with ICP-QMS has also been evaluated (Hang et al., 2004).308 Detection limits of 0.15 pg mL−1 (0.02 Bq mL−1) were reported for a 25 mL urine sample volume. Coupling of flow injection analysis with ICP-SFMS has also been reported for the analysis of environmental samples, with detection limits down to 0.6 fg being reported.268
Wang et al. recently developed a method for 241Am measurement in large (2–20 g) soil samples using ICP-SFMS (Thermo Element XR) equipped with a high efficiency nebulizer (HEN).309 Multiple chemical separation techniques were investigated for separation of Am from soil matrix components, rare earth elements, and ICP-MS interferences (Bi, Tl, Hg, Pb, Hf and Pt). A good chemical recovery of Am (76–82%) and detection limit of 0.012 mBq g−1 was achieved, whilst decontamination factors of 7 × 105 for Pu was the highest reported for 241Am studies, enabling measurement in Fukushima sourced soils contaminated with 241Pu.
8.21 Curium/californium
Curium isotopes are produced via successive neutron capture by Am and subsequent beta decay. The main Cm isotopes produced in nuclear reactors are 242Cm (t1/2 = 162.9 days), 243Cm (t1/2 = 28.9 years), 244Cm (t1/2 = 18.2 years), 245Cm (t1/2 = 8480 years), 246Cm (t1/2 = 4760 years) and 248Cm (t1/2 = 3.48 × 105 years). For Cf, the main isotopes are 249Cf (t1/2 = 351 years), 250Cf (t1/2 = 13.1 years), 251Cf (t1/2 = 898 years) and 252Cf (t1/2 = 2.6 years). To date, there are no reported applications of ICP-MS for Cm or Cf measurement in nuclear wastes. Kurosaki et al. (2014) reported the measurement of 243Cm, 244Cm, 245Cm, 246Cm and 240Pu using a Thermo Finigan Element 2 for nuclear forensics applications.310 Gourgiotis et al. (2010) used ICP-QMS (Thermo Electron X-series) for Cm and Cf isotope measurement in transmutation studies.311 Sample introduction was via a quartz Peltier-cooled impact bead spray chamber (natural aspiration) and micro concentric nebulizer. Two peak jump routines were employed to separately measure high abundance (248Cm/246Cm) and low abundance (245Cm/246Cm, 247Cm/246Cm, 249(Bk + Cf)/251Cf, 250Cf/251Cf, and 252Cf/251Cf) isotope ratios. High precision isotope ratio measurement was achieved using corrected peak centering (3 points per peak with the central point associated with the maximum count rate). Dead time and mass bias corrections (using sample standard bracketing) were also applied. Peak tailing and hydride interferences were corrected by measuring the 237/238U ratio and 238UH/238U: ratio for a U010 standard. Abundance sensitivities of 1.97 ± 0.02 ppm and hydride formation of (35.7 ± 0.1) × 10−6 were reported. Chromatographic separation of 249Bk and 249Cf using Dionex HPLC was developed to permit separate determination of 249Bk/248Cm and 249Cf/248Cm ratios.3129 Conclusions
Advances in the capability and sensitivity of recent ICP-MS instruments have stimulated new interest from the nuclear sector. This stems from the potential of these instruments to routinely and swiftly measure a range of key radionuclides considered important in nuclear decommissioning programmes. ICP-MS has proven itself as a versatile technique with regards to sample introduction and instrument setup, both of which can be used to improve sensitivity and/or interference removal. For the majority of radionuclides suitable for ICP-MS measurement, interference removal is the critical aspect affecting optimised detection limits. Effective measurement must be combined with robust, effective sample digestion and chemical separation (either through wet chemistry techniques, sample introduction and/or using an instrument with collision/reaction cell capabilities).For long-lived radionuclides, ICP-MS offers significant benefits in sample throughput compared to the traditional radiometric methods (alpha and beta counting techniques). These benefits arise from reduced measurement time per sample, and reduced preparation time with the use of on-line separation and collision/reaction cell instruments and provide significant potential economic benefits to nuclear sites. The improvements in ICP-MS means that a number of long-lived radionuclides are now measureable that are very challenging or even not measurable by radiometric techniques e.g.93Zr, 107Pd, 135Cs, 41Ca and 59Ni. These radionuclides are of increasing interest to regulatory agencies concerned with long-term nuclear waste storage and disposal. Additionally, ICP-MS (particularly multi-collector instruments) offers the ability to accurately measure isotopic ratios e.g.239Pu/240Pu and 236U/238U, enabling the determination of the source of contamination, rather than the activity concentration alone.
Acknowledgements
The authors warmly thank Bob Nesbitt, Rex Taylor and Andy Milton for ICP-MS collaborations over the years. Ben Russell thanks the NDA for partial support via an NDA Bursary (awarded to Profs Croudace and Warwick), and funding through the National Measurement System by the Department of Business, Energy and Industrial Strategy.References
- B. C. Russell, I. W. Croudace, P. E. Warwick and J. A. Milton, Anal. Chem., 2014, 86, 8719–8726 CrossRef CAS PubMed.
- W. Wien, Ann. Phys., 1898, 65, 440 CrossRef.
- J. J. Thomson, Philos. Mag., 1897, 44, 29 Search PubMed.
- A. J. Dempster, Phys. Rev., 1918, 11, 316 CrossRef CAS.
- F. W. Aston, Philos. Mag., 1919, 38, 709 Search PubMed.
- F. W. Aston, Philos. Mag., 1920, 39, 449 CrossRef CAS.
- F. W. Aston, Proc. R. Soc. A, 1927, 115, 487 CrossRef CAS.
- W. Bartky and A. J. Dempster, Phys. Rev., 1929, 33, 1019 CrossRef CAS.
- J. Mattauch and R. Hertzog, Z. Phys., 1934, 89, 786 CrossRef CAS.
- K. T. Bainbridge and E. B. Jordan, Phys. Rev., 1936, 50, 282 CrossRef CAS.
- A. O. Nier, Rev. Sci. Instrum., 1940, 11, 212 CrossRef CAS.
- A. O. Nier, Rev. Sci. Instrum., 1947, 18, 39 CrossRef.
- J. A. Hipple, J. Appl. Phys., 1942, 13, 551 CrossRef CAS.
- H. Ewald and H. Hintenberger, Methoden und Anwendungen der Massenspektroskopie, Verlag Chemie GmbH, Weinheim, Germany, 1953 Search PubMed.
- S. Penner, Rev. Sci. Instrum., 1961, 32, 150 CrossRef.
- H. Ewald, E. Konecny, H. Opower and H. Rösler, Z. Naturforsch., C: Biochem., Biophys., Biol., Virol., 1964, 19, 194 Search PubMed.
- S. Fukumoto, T. Matsuo and H. Matsuda, J. Phys. Soc. Jpn., 1968, 25, 946 CrossRef CAS.
- J. Frantzen, Jahrbuch der Wittheit zu Bremen 2006/2007, Hauschild Verlag, Bremen, Germany, 2008, vol. 181, ISBN 978-3-89757-529-5 Search PubMed.
- A History of European Mass Spectrometry, ed. K. R. Jennings, IM Publications LLP, Chichester, UK, 2012, ISBN 978-1-906715-04-5, pp. 285 Search PubMed.
- K. S. Sharma, Int. J. Mass Spectrom., 2013, 349–350, 3–8 CrossRef CAS.
- G. P. Russ and J. M. Bazan, Spectrochim. Acta, Part B, 1987, 42, 49–62 CrossRef.
- D. W. Boomer and M. J. Powell, Anal. Chem., 1987, 59, 2810–2813 CrossRef CAS PubMed.
- R. M. Brown, S. E. Long and C. J. Pickford, Sci. Total Environ., 1988, 70, 265–274 CrossRef CAS.
- G. L. Beck and O. T. Farmer, J. Anal. At. Spectrom., 1988, 3, 771–773 RSC.
- C. K. Kim, A. Takaku, M. Yamamoto, H. Kawamura, K. Shiraishi, Y. Igarashi, S. Igarashi, H. Takayama and N. Ikeda, J. Radioanal. Nucl. Chem., 1989, 132(1), 131–137 CrossRef CAS.
- A. S. Hursthouse, M. S. Baxter, K. McKay and F. R. Livens, J. Radioanal. Nucl. Chem., 1992, 157(2), 281–294 CrossRef CAS.
- C. K. Kim, R. Seki and S. Morita, J. Anal. At. Spectrom., 1991, 6, 205–209 RSC.
- J. S. Crain and B. L. Mikesell, Appl. Spectrosc., 1992, 46(10), 1498–1502 CrossRef CAS.
- C. Riglet, O. Provitina, J. L. Dautheribes and D. Revy, J. Anal. At. Spectrom., 1992, 7, 923–927 RSC.
- M. R. Smith, E. J. Wyse and D. W. Koppenaal, J. Radioanal. Nucl. Chem., 1992, 160(2), 341–354 CrossRef CAS.
- R. J. Rosenberg, J. Radioanal. Nucl. Chem., 1993, 171(2), 465–482 CrossRef CAS.
- J. S. Crain, J. S. Yaeger, F. P. Smith, J. A. Alvarado, L. L. Smith, J. T. Kiely and D. G. Graczyk, Argonne National Laboratory 4th CIRMS Meeting, NIST, Gaithersberg, 28-30 Nov 1995 Search PubMed.
- J. S. Crain and J. Alvarado, J. Anal. At. Spectrom., 1994, 9, 1223–1227 RSC.
- E. J. Wyse and D. R. Fisher, Radiat. Prot. Dosim., 1994, 55(3), 199–206 CAS.
- J. S. Crain, L. L. Smith, J. S. Yaeger and J. A. Alvarado, J. Radioanal. Nucl. Chem., 1995, 194(1), 133–139 CrossRef CAS.
- Y. Igarashi, C. K. Kim, Y. Takaku, K. Shiraishi, M. Yamamooto and N. Ikeda, Anal. Sci., 1990, 6, 157–164 CrossRef CAS.
- S. Collins, S. Pommé, S. M. Jerome and A. K. Pearce, Appl. Radiat. Isot., 2015, 104, 203–211 CrossRef CAS PubMed.
- S. M. Collins, A. K. Pearce, K. Ferreira, A. Fenwick, P. H. Regan and J. Keightley, Appl. Radiat. Isot., 2015, 99, 46–53 CrossRef CAS PubMed.
- NPL Radioactivity Group, http://www.npl.co.uk/science-technology/radioactivity/products-and-services/primary-and-secondary-standards, accessed 17/08/16.
- NIST Radionuclide Half-Life measurements, http://www.nist.gov/pml/data/halflife.cfm, accessed 17/08/16.
- J. S. Becker and H. J. Dietz, Fresenius' J. Anal. Chem., 1999, 364, 482–488 CrossRef CAS.
- C. S. Kim, C. U. Kim, J. I. Lee and K. J. Lee, J. Anal. At. Spectrom., 2000, 15, 247–255 RSC.
- S. F. Boulyga and J. S. Becker, Fresenius' J. Anal. Chem., 2001, 370, 612–617 CrossRef CAS PubMed.
- K. K. Falkner, G. P. Klinkhammer, C. Ungerer and D. M. Christie, Annu. Rev. Earth Planet. Sci., 1995, 23, 409–449 CrossRef CAS.
- C. M. Barshick, D. C. Duckworth and D. H. Smith, Inorganic Mass Spectrometry Fundamentals and Applications, Oak Ridge National Laboratory, Marcel Dekker Inc., Oak Ridge, Tennessee, 2000 Search PubMed.
- R. Thomas, A Beginner's Guide to ICP-MS, Part II: The Sample Introduction System, Spectroscopy, 2001, 16(5), 56–60 Search PubMed.
- W. Kerl, J. S. Becker, H.-J. Dietze and W. Dannecker, Fresenius' J. Anal. Chem., 1997, 359, 407–409 CrossRef CAS.
- J. S. Becker and H. J. Dietz, J. Anal. At. Spectrom., 1999, 14, 1493–1500 RSC.
- S. F. Boulyga and J. S. Becker, J. Anal. At. Spectrom., 2002, 17, 1143–1147 RSC.
- N. Jakubowski, L. Moens and F. Vanhaecke, Spectrochim. Acta, Part B, 1998, 53(12), 1739–1763 CrossRef.
- R. N. Taylor, T. Warneke, J. A. Milton, I. W. Croudace, P. E. Warwick and R. W. Nesbitt, J. Anal. At. Spectrom., 2003, 18, 480–484 RSC.
- J. A. McLean, J. S. Becker, S. F. Boulyga, H. J. Dietze and A. Montaser, Int. J. Mass Spectrom., 2001, 208, 193 CrossRef CAS.
- S. F. Boulyga, C. Testa, D. Desideri and J. S. Becker, J. Anal. At. Spectrom., 2001, 16, 1283–1289 RSC.
- D. Larivière, V. F. Taylor, R. D. Evans and R. J. Cornett, Spectrochim. Acta, Part B, 2006, 61(8), 877–904 CrossRef.
- M. G. Minnich and R. S. Houk, J. Anal. At. Spectrom., 1998, 13, 167–174 RSC.
- J. Zheng, J. Nucl. Radiochem. Sci., 2015, 15(1), 7–13 CAS.
- Z. Varga, G. Surányi, N. Vajda and Z. Stefánka, Microchem. J., 2006, 85, 39–45 CrossRef.
- J. S. Becker, Spectrochim. Acta, Part B, 2003, 58, 1757–1784 CrossRef.
- M. Hollenbach, J. Grohs, S. Mamich and M. Kroft, J. Anal. At. Spectrom., 1994, 9, 927–933 RSC.
- J. H. Aldstadt, J. M. Kuo, L. L. Smith and M. D. Erickson, Anal. Chim. Acta, 1996, 319, 135–143 CrossRef CAS.
- E. H. Hansen and M. Miró, Trends Anal. Chem., 2007, 26(1), 18–26 CrossRef CAS.
- N. Vajda and C.-K. Kim, J. Radioanal. Chem., 2010, 284, 341–366 CrossRef CAS.
- J. Qiao, X. Hou, P. Roos and M. Miró, J. Anal. At. Spectrom., 2010, 25, 1769–1779 RSC.
- J. Ruzicka, Analyst, 2000, 125, 1053–1060 RSC.
- S. F. Boulyga, D. Desideri, M. A. Meli, C. Testa and J. S. Becker, Int. J. Mass Spectrom., 2003, 226, 329–339 CrossRef CAS.
- M. D. Seltzer, Appl. Spectrosc., 2003, 57(9), 1173–1177 CrossRef CAS PubMed.
- J. S. Becker, Int. J. Mass Spectrom., 2005, 242, 183–195 CrossRef CAS.
- L.-S. Chen, T. Wang, Y.-K. Hsieh, C.-H. Hsu, J. C.-T. Lin and C.-F. Wang, J. Membr. Sci., 2014, 456, 202–208 CrossRef CAS.
- S. Bürger and L. R. Riciputi, J. Environ. Radioact., 2009, 100(11), 970–976 CrossRef PubMed.
- Z. Varga, Anal. Chim. Acta, 2008, 625, 1–7 CrossRef CAS PubMed.
- F. Pointurier, A. Hubert and A. C. Pottin, J. Radioanal. Nucl. Chem., 2013, 296, 609–616 CrossRef CAS.
- J. V. Cizdziel, M. E. Ketterer, D. Farmer, S. H. Faller and V. F. Hodge, Anal. Bioanal. Chem., 2008, 390, 521–530 CrossRef CAS PubMed.
- J. S. Becker and H. J. Dietz, Fresenius' J. Anal. Chem., 2000, 368, 23–30 CrossRef CAS PubMed.
- J. S. Becker, C. Pickhardt and H.-J. Dietze, Int. J. Mass Spectrom., 2000, 202, 283–297 CrossRef CAS.
- Viridiscan, http://www.viridian-tc.co.uk/viridiscan.html, accessed 17/08/16.
- R. C. Marin, J. E. S. Sarkis and M. R. L. Nascimento, J. Radioanal. Nucl. Chem., 2013, 295, 99–104 CrossRef CAS.
- P. Grinberg, S. Willie and R. E. Sturgeon, J. Anal. At. Spectrom., 2007, 22(11), 1409–1414 RSC.
- J. S. Santos, L. S. G. Teixeira, W. N. L. dos Santos, V. A. Lemos, J. M. Godoy and S. L. C. Ferreira, Anal. Chim. Acta, 2010, 674, 143–156 CrossRef CAS PubMed.
- M. Song, T. U. Probst and N. G. Berryman, Fresenius' J. Anal. Chem., 2001, 370(6), 744–751 CrossRef CAS PubMed.
- J. B. Truscott, L. Bromley, P. Jones, E. Hywel Evans, J. Turner and B. Fairman, J. Anal. At. Spectrom., 1999, 14, 627–631 RSC.
- P. Grinberg, S. Willie and R. E. Sturgeon, J. Anal. At. Spectrom., 2005, 20, 717–723 RSC.
- L. A. De las Heras, E. Hrnecek, O. Bildstein and M. Betti, J. Anal. At. Spectrom., 2002, 17, 1011–1014 RSC.
- M. Betti and L. A. de las Heras, Spectrochim. Acta, Part B, 2004, 59(9), 1359–1376 CrossRef.
- S. F. Wolf, D. L. Bowers and J. C. Cunnane, J. Radioanal. Nucl. Chem., 2005, 263(3), 581–586 CrossRef CAS.
- I. Günther-Leopold, N. Kivel, J. K. Waldes and B. Wernli, Anal. Bioanal. Chem., 2008, 309, 503–510 CrossRef PubMed.
- A. P. Vonderheide, M. V. Zoriy, A. V. Izmer, C. Pickhardt, J. A. Caruso, P. Ostapczuk, R. Hille and J. S. Becker, J. Anal. At. Spectrom., 2004, 19(5), 675–680 RSC.
- M. V. Zoriy, P. Ostapczuk, L. Halicz, R. Hille and J. S. Becker, Int. J. Mass Spectrom., 2005, 242(2–3), 203–209 CrossRef CAS.
- A. R. Timerbaev and R. M. Timerbeav, Trends Anal. Chem., 2013, 51, 44–50 CrossRef CAS.
- C. Möser, R. Kautenburger and H. P. Beck, Electrophoresis, 2012, 33(9–10), 1482–1487 CrossRef PubMed.
- C. Bresson, E. Ansoborlo and C. Vidaud, J. Anal. At. Spectrom., 2011, 26, 593–601 RSC.
- Perkin Elmer, Elan II DRC brochure, http://www.esc.cam.ac.uk/esc/files/Department/facilities/icp-ms/drcii-b.pdf, accessed 18/05/2016.
- D. R. Bandura, V. I. Baranov, A. E. Litherland and S. D. Tanner, Int. J. Mass Spectrom., 2006, 235–236, 312–327 CrossRef.
- U. Nygren, I. Rodushkin, C. Nilsson and D. C. Baxter, J. Anal. At. Spectrom., 2003, 18(12), 1426–1434 RSC.
- M. Moldovan, E. M. Krupp, A. E. Holliday and O. F. X. Donard, J. Anal. At. Spectrom., 2004, 1(9), 815–822 RSC.
- M. E. Ketterer and S. C. Szechenyi, Spectrochim. Acta, Part B, 2008, 63, 719–737 CrossRef.
- M. Hamester, D. Wiederin, J. Wills, W. Kerl and C. B. Douthitt, Fresenius' J. Anal. Chem., 1999, 364(5), 495–498 CrossRef CAS.
- D. Solatie, P. Carbol, M. Betti, F. Bocci, T. Hiernaut, V. V. Rondinella and J. Cobos, Fresenius' J. Anal. Chem., 2000, 368, 88–94 CrossRef CAS PubMed.
- Z. Varga, Anal. Chim. Acta, 2007, 587, 165–169 CrossRef CAS PubMed.
- M. E. Wieser and J. B. Schwieters, Int. J. Mass Spectrom., 2005, 242(2–3), 97–115 CrossRef CAS.
- Spectro, http://www.spectro.com/products/icp-ms-spectrometers, Overview of ICP-MS spectrometers, accessed 18/05/2016.
- N. L. Sanders, S. Kothari, G. Huang, G. Salazar and R. G. Cooks, Anal. Chem., 2010, 82, 5313–5316 CrossRef CAS PubMed.
- J. K. Dalgleish, K. Hou, Z. Ouyang and R. G. Cooks, Anal. Lett., 2012, 45, 1440–1446 CrossRef CAS.
- J. M. Wells, M. J. Roth, A. D. Keil, J. W. Grossenbacher, D. R. Justes, G. E. Patterson and D. J. Barket Jr, J. Am. Soc. Mass Spectrom., 2008, 19(10), 1419–1424 CrossRef CAS PubMed.
- A. Malcolm, S. Wright, R. R. A. Syms, N. Dash, M.-A. Schwab and A. Finlay, Anal. Chem., 2010, 82, 1751–1758 CrossRef CAS PubMed.
- P. I. Hendricks, J. K. Dalgleish, J. T. Shelley, M. A. Kirleis, M. T. McNicholas, L. Li, T.-C. Chen, C.-H. Chen, J. S. Duncan, F. Boudreau, R. J. Noll, J. P. Denton, T. A. Roach, Z. Ouyang and R. G. Cooks, Anal. Chem., 2014, 86, 2900–2908 CrossRef CAS PubMed.
- S. Kumano, M. Sugiyama, M. Yamada, K. Nishimura, H. Hasegawa, H. Morokuma, H. Inoue§ and Y. Hashimoto, Anal. Chem., 2013, 85(10), 5033–5039 CrossRef CAS PubMed.
- J. M. Giaquinto, J. M. Keller and A. M. Meeks, J. Radioanal. Nucl. Chem., 1998, 234(1–2), 137–141 CrossRef CAS.
- F. Pilon, S. Lorthioir, J. Birolleau and S. Lafontan, J. Anal. At. Spectrom., 1996, 11(9), 759–764 RSC.
- J. I. G. Alonso, D. Thoby-Schultzendorff, B. Giovanonne, L. Koch and H. Wiesmann, J. Anal. At. Spectrom., 1993, 8, 673–679 RSC.
- J. D. Burraston, PhD thesis, GAU-Radioanalytical, Ocean and Earth Science, University of Southampton, National Oceanography Centre, 2014 Search PubMed.
- R. Forsyth and U.-B. Eklund, Spent nuclear fuel corrosion: SKB Technical Report 95-04, Swedish Nuclear Fuel and Waste Management Company, 1995 Search PubMed.
- M. Betti, G. Rasmussen, T. Hiernaut and L. Koch, J. Anal. At. Spectrom., 1994, 9, 385–391 RSC.
- M. R. Smith, O. T. Farmer III, J. H. Reeves and D. W. Koppenaal, J. Radioanal. Nucl. Chem., 1995, 194(1), 7–13 CrossRef CAS.
- J. M. Barrero Moreno, M. Betti and J. I. Garcia Alonso, J. Anal. At. Spectrom., 1997, 12, 355–361 RSC.
- O. B. Egorov, M. J. O'Hara, O. T. Farmer III and J. W. Grate, Analyst, 2000, 126, 1594–1601 RSC.
- M. Krachler, R. Alvarez-Sarandes and S. V. Winckel, J. Anal. At. Spectrom., 2014, 29, 817–824 RSC.
- I. W. Croudace, P. E. Warwick, R. Taylor and S. Dee, Anal. Chim. Acta, 1998, 371, 217–225 CrossRef CAS.
- C. S. Kim, C.-K. Kim, P. Martin and U. Sansone, J. Anal. At. Spectrom., 2007, 22, 827–841 RSC.
- I. W. Croudace, P. E. Warwick, D. G. Reading and B. C. Russell, Trends Anal. Chem., 2016, 85, 120–129 CrossRef CAS.
- R. Bock, A Handbook of Decomposition Methods in Analytical Chemistry, International Textbook Company Limited, London, 1979 Search PubMed.
- B. Parsa, J. Radioanal. Nucl. Chem., 1992, 157, 65–73 CrossRef CAS.
- U.S. Environmental Protection Agency, Rapid Method for Sodium Hydroxide/Sodium Peroxide Fusion of Radioisotope Thermoelectric Generator Materials in Water and Air Filter Matrices Prior to Plutonium Analyses for Environmental Remediation Following Radiological Incidents, 2014, EPA/600/F-14/284 Search PubMed.
- S. Fisher and R. Kunin, Anal. Chem., 1957, 29, 400–402 CrossRef CAS.
- C. Galindo, L. Mougin and A. Nourreddine, Appl. Radiat. Isot., 2007, 65, 9–16 CrossRef CAS PubMed.
- C. W. Sill, K. W. Puphal and F. D. Hindman, Anal. Chem., 1974, 46, 1725–1737 CrossRef CAS PubMed.
- J. S. Fedorowich, J. P. Richards, J. C. Cain, R. Kerrich and J. Fan, Chem. Geol., 1993, 106, 229–249 CrossRef CAS.
- M. Dong, D. Oropeza, J. Chirinos, J. J. González, J. Lu, X. Mao and R. E. Russo, Spectrochim. Acta, Part B, 2015, 109, 44–50 CrossRef CAS.
- K. Subedi, T. Trjos and J. Almirall, Spectrochim. Acta, Part B, 2015, 103–104, 76–83 CrossRef CAS.
- Y. Lee, S.-H. Nam, K.-S. Ham, J. Gonzalez, D. Oropeza, D. Quarles Jr, J. Yoo and R. E. Rosso, Spectrochim. Acta, Part B, 2016, 118, 102–111 CrossRef CAS.
- L. Ashton, P. Warwick and D. Giddings, Analyst, 1999, 124, 627–632 RSC.
- M. Baxter, L. Castle, H. M. Crews, M. Rose, C. Garner, G. Lappin and D. Leong, Food Addit. Contam., Part A, 2009, 26, 139–144 CrossRef CAS PubMed.
- X. Hou, L. F. Ostergaard and S. P. Nielsen, Anal. Chem., 2007, 79, 3126–3134 CrossRef CAS PubMed.
- A. Zulaf, S. Happel, P. Warwick, B. M. Mokili, A. Bombard and H. Junglas, http://www.triskem-international.com/fr/iso_album/poster_rrmc_2011_cl_resin.pdf, accessed 17/08/16.
- M. Martschini, P. Andersson, O. Fortsner, R. Golser, D. Hanstorp, A. O. Lindahl, W. Kutschera, S. Pavetich, A. Priller, J. Rohlén, P. Steier, M. Suter and A. Wallner, Nucl. Instrum. Methods Phys. Res., Sect. B, 2013, 294, 115–120 CrossRef CAS.
- P. W. Kubik, D. Elmore, N. J. Conard, K. Nishiizumi and J. R. Arnold, Nature, 1986, 319, 568–570 CrossRef CAS.
- P. Müller, B. A. Bushaw, K. Blaum, S. Diel, C. Geppert, A. Nähler, N. Trautmann, W. Nörtershäuser and K. Wendt, Anal. Chem., 2001, 370, 508–512 CrossRef.
- K. D. A. Wendt, C. Geppert, M. Miyabe, P. Müller, W. Nörtershäuser and N. Trautmann, J. Nucl. Sci. Technol., 2002, 39(4), 303–307 CrossRef CAS.
- C. Hennessy, M. Berglund, M. Ostermann, T. Walczyk, H.-A. Synal, C. Geppert, K. Wendt and P. D. P. Taylor, Nucl. Instrum. Methods Phys. Res., Sect. B, 2005, 299, 281–292 CrossRef.
- D. Hampe, B. Gleisberg, S. Akhmadaliev, G. Rugel and S. Merchel, J. Radioanal. Nucl. Chem., 2013, 296, 617–624 CrossRef CAS.
- G. S. Jackson, D. J. Hillegonds, P. Muzikar and B. Goehring, Nucl. Instrum. Methods Phys. Res., Sect. B, 2013, 313, 14–20 CrossRef CAS PubMed.
- S. Merchel, L. Benedetti, D. L. Bourlès, R. Braucher, A. Deqald, T. Faestermann, R. C. Finkel, G. Korschinek, J. Masarik, M. Poutivtsev, P. Rochette, G. Rugel and K.-O. Zell, Nucl. Instrum. Methods Phys. Res., Sect. B, 2010, 268, 1179–1184 CrossRef CAS.
- X. Hou, Radiochim. Acta, 2005, 93, 611–617 CrossRef CAS.
- P. E. Warwick, I. W. Croudace and D. J. Hillegonds, Anal. Chem., 2009, 81, 1901–1906 CrossRef CAS PubMed.
- G. Jörg, Y. Amelin, K. Kossert, C. Lierse and L. V. Gostomski, Geochim. Cosmochim. Acta, 2012, 88, 51–65 CrossRef.
- E. Nottoli, D. Bourlès, P. Bienvenu, A. Labet, M. Arnold and M. Bertaux, Appl. Radiat. Isot., 2013, 82, 340–346 CrossRef CAS PubMed.
- X. Hou, L. F. Østergaard and S. P. Nielsen, Anal. Chim. Acta, 2005, 535(1–2), 297–307 CrossRef CAS.
- E. Holm, P. Roos and B. Skwarzec, Appl. Radiat. Isot., 1992, 43(1–2), 371–376 CrossRef CAS.
- I. Gresits and S. Tölgyesi, J. Radioanal. Nucl. Chem., 2003, 258(1), 107–122 CrossRef CAS.
- P. E. Warwick and I. W. Croudace, Anal. Chim. Acta, 2006, 567, 277–285 CrossRef CAS.
- A. A. Marchetti, L. J. Hainsworth, J. E. McAninch, M. R. Leivers, P. R. Jones, I. D. Proctor and T. Straume, Nucl. Instrum. Methods Phys. Res., Sect. B, 1997, 123(1–4), 230–234 CrossRef CAS.
- P. Persson, M. Kiisk, B. Erlandsson, M. Faarinen, R. Hellborg, G. Skog and K. Stenström, Nucl. Instrum. Methods Phys. Res., Sect. B, 2000, 172(1–4), 188–192 CrossRef CAS.
- M. E. Mount, D. W. Layton, N. M. Lynn and T. F. Hamilton, Report UCRL-JC-130412, Lawrence Livermore National Laboratory, USA, 1998 Search PubMed.
- S. Aguerre and C. Frechou, Talanta, 2006, 69(3), 565–571 CrossRef CAS PubMed.
- J. Compte, P. Bienvenu, E. Brochard, J.-M. Fernandez and G. Andreoletti, J. Anal. At. Spectrom., 2003, 18, 702–707 RSC.
- P. Bienvenu, P. Cassette, G. Andreoletti, M.-M. Be, J. Comte and M.-C. Lépy, Appl. Radiat. Isot., 2007, 65(3), 355–364 CrossRef CAS PubMed.
- S. Brun, S. Bessac, D. Uridat and B. Boursier, J. Radioanal. Nucl. Chem., 2002, 253(2), 191–197 CrossRef CAS.
- W. Bu, J. Zheng, X. Liu, K. Long, S. Hu and S. Uchida, Spectrochim. Acta, Part B, 2016, 119, 65–75 CrossRef CAS.
- J. Feuerstein, S. F. Boulyga, P. Galler, G. Stingeder and T. Prohaska, J. Environ. Radioact., 2008, 99(11), 1764–1769 CrossRef CAS PubMed.
- V. Taylor, R. D. Evans and R. J. Cornett, Anal. Bioanal. Chem., 2007, 387(1), 343–350 CrossRef CAS PubMed.
- Y. Takagai, M. Furukawa, Y. Kameo and K. Suzuki, Anal. Methods, 2014, 6(2), 355–362 RSC.
- M. Sakama, Y. Nagano, T. Saze, S. Higaki, T. Kitade, N. Izawa, O. Shikino and S. Nakayama, Appl. Radiat. Isot., 2013, 81, 201–207 CrossRef CAS PubMed.
- M. A. Amr, A.-F. I. Helal, A. T. Al-Kinani and P. Balakrishnan, J. Environ. Radioact., 2016, 153, 73–87 CrossRef CAS PubMed.
- E. P. Steinberg and L. E. Glendenin, Phys. Rev., 1950, 78, 624 CrossRef CAS.
- J. I. G. Alonso, D. Thoby-Schultzendorff, B. Giovanonne, J.-P. Glatz, G. Pagliosa and L. Koch, J. Anal. At. Spectrom., 1994, 9, 1209–1215 RSC.
- F. Chartier, H. Isnard, J. P. Degros, A. L. Faure and C. Fréchou, Int. J. Mass Spectrom., 2008, 270(3), 127–133 CrossRef CAS.
- S. Dulanská, B. Remenec, V. Gardoňová and L. Mátel, J. Radioanal. Nucl. Chem., 2012, 293, 635–640 CrossRef.
- P. Cassette, F. Chartier, H. Isnard, C. Fréchou, I. Laszak, J. P. Degros, M. M. Bé, M. C. Lépy and I. Tartes, Appl. Radiat. Isot., 2010, 68, 122–130 CrossRef CAS PubMed.
- K. Shi, J. Qiao, W. Wu, P. Roos and X. Hou, Anal. Chem., 2012, 84, 6783–6789 CrossRef CAS PubMed.
- K. Shi, X. Hou, P. Roos and W. Wu, Anal. Chim. Acta, 2012, 709, 1–20 CrossRef CAS PubMed.
- T. Y. Su, T. L. Tsay, H. C. Wu and L. C. Men, J. Radioanal. Nucl. Chem., 2015, 303, 1245–1248 CrossRef CAS.
- K. Shi, X. Hou, P. Roos and W. Wu, Anal. Chem., 2012, 84, 2009–2016 CrossRef CAS PubMed.
- K. H. Chung, S. D. Choi, G. S. Choi and M. J. Kang, Appl. Radiat. Isot., 2013, 81, 57–61 CrossRef CAS PubMed.
- L. S. Chen, T. H. Wang, Y. K. Hsieh, L. W. Jian, W. H. Chen, T. L. Tsai and C. F. Wang, J. Radioanal. Nucl. Chem., 2014, 299, 1883–1889 CrossRef CAS.
- N. E. Bibler, W. T. Boyce, C. J. Coleman and W. F. Kinard, Report WSRC-MS-97-0011, Office of Scientific and Technical Information, Oak Ridge, USA, 1997 Search PubMed.
- C. J. Bannochie, N. E. Bibler and D. P. DiPrete, WM2009 conference proceedings, March 1–5 2009, Phoenix, USA, 2009 Search PubMed.
- P. G. Bienvenu, E. A. Brochard and E. A. Excoffier, in Application of inductively coupled plasma mass spectrometry to radionuclide determinations: second volume, ed. R.W. Morrow and J.S. Crain, ASTM, West Conshohocken, USA, 1998 Search PubMed.
- V. Hansen, P. Roos, A. Aldahan, X. Hou and G. Possnert, J. Environ. Radioact., 2011, 102, 1096–1104 CrossRef CAS PubMed.
- V. Hansen, P. Yi, X. Hou, A. Aldahan, P. Roos and G. Possnert, Sci. Total Environ., 2011, 412–413, 296–303 CrossRef CAS PubMed.
- Ž. Ežerinskis, A. Spolaor, T. Kirchgeorg, G. Cozzi, P. Vallelonga, H. A. Kjaer, J. Šapolaité, C. Barbante and R. Druteikiené, J. Anal. At. Spectrom., 2014, 29, 1827–1834 RSC.
- C. Vockenhuber, N. Casacuberta, M. Christl and H.-A. Synal, Nucl. Instrum. Methods Phys. Res., Sect. B, 2015, 361, 510–516 CrossRef.
- E. Englund, A. Aldahan, X. L. Hou, G. Possnert and C. Söderström, Nucl. Instrum. Methods Phys. Res., Sect. B, 2010, 268, 1139–1141 CrossRef CAS.
- R. Michel, A. Daraoui, M. Gorny, D. Jakob, R. Sachse, L. Tosch, H. Nies, I. Goronocy, J. Herrman, H. A. Synal, M. Stocker and V. Alfimov, Sci. Total Environ., 2012, 419, 151–169 CrossRef CAS PubMed.
- X. Hou, P. Povinec, L. Zhang, K. Shi, D. Biddulph, C.-C. Chang, Y. Fan, R. Golser, Y. Hou, M. Ješkovský, A. J. Tim Jull, Q. Liu, M. Luo, P. Steier and W. Zu, Environ. Sci. Technol., 2013, 47(7), 3091–3098 CAS.
- Y. Muramatsu, H. Matsuzaki, C. Toyama and T. Ohno, J. Environ. Radioact., 2015, 139, 344–350 CrossRef CAS PubMed.
- J. M. Gómez-Guzmán, S. M. Enamorado-Báez, A. R. Pinto-Goméz and J. M. Abril-Hernández, Int. J. Mass Spectrom., 2011, 303, 103–108 CrossRef.
- V. Alfimov and H.-A. Synal, Nucl. Instrum. Methods Phys. Res., Sect. B, 2010, 268, 769–772 CrossRef CAS.
- H. Fujiwara, K. Kawabata, J. Suzuki and O. Shikino, J. Anal. At. Spectrom., 2011, 26, 2528–2533 RSC.
- J. Lehto, T. Räty, X. Hou, J. Paatero, A. Aldahan, G. Possnert, J. Flinkman and H. Kankaanpää, Sci. Total Environ., 2012, 419, 60–67 CrossRef CAS PubMed.
- T. Ohno, Y. Muramatsu, Y. Shikamori, C. Toyama, N. Okabe and H. Matsuzaki, J. Anal. At. Spectrom., 2013, 28, 1283–1287 RSC.
- J. Zheng, H. Takata, K. Tagami, T. Aono, K. Fujita and S. Uchida, Microchem. J., 2012, 100, 42–47 CrossRef CAS.
- R. J. Cox, C. J. Pickford and M. Thompson, J. Anal. At. Spectrom., 1992, 7, 635–640 RSC.
- E. Englund, A. Aldahan, X. L. Hou, R. Petersen and G. Possnert, Nucl. Instrum. Methods Phys. Res., Sect. B, 2010, 268, 1102–1105 CrossRef CAS.
- X. Hou, W. Zhou, N. Chen, L. Zhang, Q. Liu, M. Luo, Y. Fan, W. Liang and Y. Fu, Anal. Chem., 2010, 82, 7713–7721 CrossRef CAS PubMed.
- T. Suzuki, S. Otosaka, J. Kubawara, H. Kawamura and T. Kobayashi, Biogeosciences, 2013, 10, 3839–3847 CrossRef.
- V. Taylor, R. Evans and R. Cornett, J. Environ. Radioact., 2008, 99(1), 109–118 CrossRef CAS PubMed.
- J. E. Delmore, D. C. Snyder, T. Tranter and N. R. Mann, J. Environ. Radioact., 2011, 102(11), 1008–1011 CrossRef CAS PubMed.
- D. C. Snyder, J. E. Delmore, T. Tranter, N. R. Mann, M. L. Abbott and J. E. Olson, J. Environ. Radioact., 2012, 110, 46–52 CrossRef CAS PubMed.
- T. Ohno and Y. Muramatsu, J. Anal. At. Spectrom., 2014, 29(2), 347–351 RSC.
- J. Zheng, K. Tagami, W. Bu, S. Uchida, Y. Watanabe, Y. Kubota, S. Fuma and S. Ihara, Environ. Sci. Technol., 2014, 48, 5433–5438 CrossRef CAS PubMed.
- J. Zheng, W. Bu, K. Tagami, Y. Shikamori, K. Nakano, S. Uchida and N. Ishii, Anal. Chem., 2014, 86(14), 7103–7110 CrossRef CAS PubMed.
- B. C. Russell, I. W. Croudace and P. E. Warwick, Anal. Chim. Acta, 2015, 890, 7–20 CrossRef CAS PubMed.
- Y. Shibahara, T. Kubota, T. Fuji, S. Fukutani, T. Ohta, K. Takamiya, R. Okumura, S. Mizuno and H. Yamana, J. Nucl. Sci. Technol., 2014, 51(5), 575–579 CrossRef CAS.
- B. C. Russell, P. E. Warwick and I. W. Croudace, Anal. Chem., 2014, 86(23), 11890–11896 CrossRef CAS PubMed.
- J. M. B. Moreno, J. I. G. Alonso, P. Arbore, G. Nicolaou and L. Koch, J. Anal. At. Spectrom., 1996, 11(10), 929–935 RSC.
- S. Asai, Y. Hanzawa, K. Okamura, N. Shinohara, J. Inagawa, S. Hotoku, K. Suzuki and S. Kaneko, J. Nucl. Sci. Technol., 2011, 48(5), 851–854 CrossRef CAS.
- J. Eliades, X.-L. Zhao, A. E. Litherland and W. E. Kieser, Nucl. Instrum. Methods Phys. Res., Sect. B, 2013, 294, 361–363 CrossRef CAS.
- H. Isnard, M. Granet, C. Caussignac, E. Ducarme, A. Nonell, B. Tran and F. Chartier, Spectrochim. Acta, Part B, 2009, 64(11–12), 1280–1286 CrossRef.
- A. Pitois, L. A. De Las Heras and M. Betti, Int. J. Mass Spectrom., 2008, 270, 118–126 CrossRef CAS.
- M. Granet, A. Nonell, G. Favre, F. Chartier, H. Isnard, J. Moureau, C. Caussignac and B. Tran, Spectrochim. Acta, Part B, 2008, 63(11), 1309–1314 CrossRef.
- V. V. Lavrov, V. Blagojevic, G. K. Koyanagi, G. Orlova and D. K. Bohme, J. Phys. Chem. A, 2004, 108(26), 5610–5624 CrossRef CAS.
- T. Lee, K. The-Lung, L. Hsiao-Ling and C. Ju-Chin, Geochim. Cosmochim. Acta, 1993, 57(14), 3493–3497 CrossRef CAS.
- L. Pibida, C. McMahon and B. A. Bushaw, Appl. Radiat. Isot., 2004, 60(2–4), 567–570 CrossRef CAS PubMed.
- C. Macdonald, C. R. J. Charles, R. J. Cornett, X. L. Zhao, W. E. Kieser and A. E. Litherland, Congress de l'ACP, Contribution ID: 155 Type: oral, Session Classification : (T2–9) Instrumentation-DIMP/Instrumentation-DPIM, 2014 Search PubMed.
- J. I. G. Alonso, F. Sena, P. Arbore, M. Betti and L. Koch, J. Anal. At. Spectrom., 1995, 10, 381–393 RSC.
- A. Meeks, J. Giaquinto and J. Keller, J. Radioanal. Nucl. Chem., 1998, 234(1), 131–136 CrossRef CAS.
- J. M. B. Moreno, M. Betti and G. Nicolaou, J. Anal. At. Spectrom., 1999, 14(5), 875–879 RSC.
- M. Liezers, O. T. Farmer and M. Thomas, J. Radioanal. Nucl. Chem., 2009, 282(1), 309–313 CrossRef CAS.
- J. E. Martin, in Environmental Radiochemical Analysis, ed. G. W. A. Newton, Royal Society of Chemistry, London, UK, 1999 Search PubMed.
- S. Sumiya, N. Hayashi, H. Katagiri and O. Narita, Sci. Total Environ., 1993, 130–131, 303–315 Search PubMed.
- M. Yoshida, S. Sumiya, H. Watanabe and K. Tobit, J. Radioanal. Nucl. Chem., 1995, 197, 219–227 CrossRef CAS.
- L. Vio, G. Crétier, F. Chartier, V. Geertsen, A. Gourgiotis, H. Isnard and J.-L. Rocca, Talanta, 2012, 99, 586–593 CrossRef CAS PubMed.
- H. Isnard, R. Brennetot, C. Caussignac, N. Caussignac and F. Chartier, Int. J. Mass Spectrom., 2005, 246, 66–73 CrossRef CAS.
- M. Baskaran, J. Environ. Radioact., 2011, 102, 500–513 CrossRef CAS PubMed.
- M. A. Amr, K. A. Al-Saad and A. I. Helal, Nucl. Instrum. Methods Phys. Res., Sect. A, 2010, 615, 237–241 CrossRef CAS.
- A. E. M. Khater and W. F. Bakr, J. Environ. Radioact., 2011, 102, 527–530 CrossRef CAS PubMed.
- D. Larivière, K. M. Reiber, R. D. Evans and R. J. Cornett, Anal. Chim. Acta, 2005, 549, 188–196 CrossRef.
- Z. Varga, Anal. Bioanal. Chem., 2008, 390, 511–519 CrossRef CAS PubMed.
- T. Zhang, D. Bain, R. Hammack and R. D. Vidic, Environ. Sci. Technol., 2015, 49(5), 2969–2976 CrossRef CAS PubMed.
- Y.-T. Hsieh and G. M. Henderson, J. Anal. At. Spectrom., 2011, 26, 1338–1346 RSC.
- M. Bourquin, P. Van Beek, J. L. Reyss, J. Riotte and R. Freydier, Mar. Chem., 2011, 126, 132–138 CrossRef CAS.
- M. Leermakers, Y. Gao, J. Navez, A. Poffijn, K. Croes and W. Baeyens, J. Anal. At. Spectrom., 2009, 24, 1115–1117 RSC.
- Y. Gao, W. Baeyens, S. De Galan, A. Poffijn and M. Leermakers, Environ. Pollut., 2010, 158, 2439–2445 CrossRef CAS PubMed.
- G. Sharabi, B. Lazar, Y. Kolodny, N. Teplyakov and L. Halicz, Int. J. Mass Spectrom., 2010, 294, 112–115 CrossRef CAS.
- M. S. Choi, R. Francois, K. Sims, M. P. Bacon, S. Brown-Leger, A. P. Fleer, L. Ball, D. Schneider and S. Pichat, Mar. Chem., 2001, 76, 99–112 CrossRef CAS.
- D. A. Pickett, M. T. Murrell and R. W. Williams, Anal. Chem., 1994, 66(7), 1044–1049 CrossRef CAS.
- C.-C. Shen, H. Cheng, R. L. Edwards, S. B. Moran, H. N. Edmonds, J. A. Hoff and R. B. Thomas, Chem. Geol., 2002, 3–4, 165–178 CrossRef.
- M. Regelous, S. P. Turner, T. R. Elliott, K. Rostami and C. J. Hawkesworth, Anal. Chem., 2004, 76, 3584–3589 CrossRef CAS PubMed.
- R. A. Mortlock, R. G. Fairbanks, T.-C. Chiu and J. Rubenstone, Geochim. Cosmochim. Acta, 2005, 69(3), 649–657 CrossRef CAS.
- C.-C. Shen, R. L. Edwards, H. Cheng, J. A. Dorale, R. B. Thomas, S. B. Moran, S. E. Weinstein and H. N. Edmonds, Chem. Geol., 2002, 185, 165–178 CrossRef CAS.
- H. Cheng, R. L. Edwards, C.-C. Shen, V. J. Polyak, Y. Asmerom, J. Woodhead, J. Hellstrom, Y. Wang, X. Kong, C. Spötl, X. Wang and E. C. Alexander Jr, Earth Planet. Sci. Lett., 2013, 371–372, 82–91 CrossRef CAS.
- H. Cheng, R. L. Edwards, J. Hoff, C. D. Gallup, D. A. Richards and Y. Asmerom, Chem. Geol., 2000, 169, 17–33 CrossRef CAS.
- Y. Shi, R. Collins and C. Broome, J. Radioanal. Nucl. Chem., 2013, 269, 509–515 CrossRef.
- S. K. Aggarwal, Radiochim. Acta, 2016, 104(7), 445–455 CrossRef CAS.
- E. W. Hoppe, N. R. Overman and B. D. LaFerriere, Low Radioactivity Techniques, AIP Conf. Proc., 2013, 1549, 58–65 CrossRef CAS.
- J. S. Becker, M. Burow, M. V. Zoriy, C. Pickhardt, P. Ostapczuk and R. Hille, J. Anal. At. Spectrom., 2004, 25(5), 197–202 CAS.
- M. L. Cozzella and R. Pettirossi, Radiation Protection Institute ENEA, Casaccia, Rome, Italy, 2012 Search PubMed.
- H. E. L. Palmieri, E. A. N. Knupp, L. M. L. A. Auler, L. M. F. Gomes and C. C. Windmöller, International Nuclear Atlantic Conference- INAC, 2011 Search PubMed.
- J. Avivar, L. Ferrer, M. Cass and V. Cerà, J. Anal. At. Spectrom., 2012, 27, 327–334 RSC.
- H. Tuovinen, D. Vesterbacka, E. Pohjolainen, D. Read, D. Solatie and J. Lehto, J. Geochem. Explor., 2015, 148, 174–180 CrossRef CAS.
- A. Habibi, B. Boulet, M. Gleizes, D. Larrriiivière and G. Cote, Anal. Chim. Acta, 2015, 883, 109–116 CrossRef CAS PubMed.
- A. Borylo, J. Radioanal. Nucl. Chem., 2013, 295, 621–631 CrossRef CAS.
- P. J. Gray, L. Zhang, H. Zu, M. McDiarmid, K. Squibb and J. A. Centeno, Microchem. J., 2012, 105, 94–100 CrossRef CAS.
- J. G. Arnason, C. N. Pellegri and P. J. Parsons, J. Anal. At. Spectrom., 2013, 28, 1410–1419 RSC.
- J. G. Arnason, C. N. Pellegri and P. J. Parsons, J. Anal. At. Spectrom., 2015, 30, 126–138 RSC.
- C. Liu, B. Hu, J. Shi, J. Li, X. Zhang and H. Chen, J. Anal. At. Spectrom., 2011, 26, 2045–2051 RSC.
- S. F. Boulyga, J. L. Matusevich, V. P. Mironov, V. P. Kudrjashov, L. Halicz, I. Segal, J. A. McLean, A. Montaser and J. S. Becker, J. Anal. At. Spectrom., 2002, 17, 958–964 RSC.
- M. Tanimizu, N. Sugiyama, E. Ponzevera and G. Bayon, J. Anal. At. Spectrom., 2013, 28, 1372–1376 RSC.
- S. Uchida, R. García-Tenorio, K. Tagami and M. García-León, J. Anal. At. Spectrom., 2000, 15, 889–892 RSC.
- R. S. Pappas, B. G. Ting, J. M. Jarrett, D. C. Paschal, S. P. Caudill and D. T. Miller, J. Anal. At. Spectrom., 2002, 17, 131–134 RSC.
- S. L. Maxwell III and V. D. Jones, Talanta, 2009, 80, 143–150 CrossRef PubMed.
- J. W. Ejnik, T. I. Todorov, F. G. Mullick, K. Squibb, M. A. McDiarmid and J. A. Centeno, Anal. Bioanal. Chem., 2005, 382, 73–79 CrossRef CAS PubMed.
- P. Krystek and R. Ritsema, Anal. Bioanal. Chem., 2002, 374, 226–229 CrossRef CAS PubMed.
- M. Magara, T. Sakakibara, S. Kurosawa, M. Takahashi, S. Sakurai, Y. Hanzawa, F. Esaka, K. Watanabe and S. Usada, J. Anal. At. Spectrom., 2002, 17, 1157–1160 RSC.
- R. R. Parrish, M. F. Thirlwall, C. Pickford, M. Horstwood, A. Gerdes, J. Anderson and D. Coggon, Health Phys., 2006, 90(2), 127–138 CrossRef CAS.
- X. Dai and S. K. Tremblay, J. Radioanal. Nucl. Chem., 2011, 289, 461–466 CrossRef CAS.
- P. Thakur and G. P. Mulholland, Appl. Radiat. Isot., 2012, 70, 1747–1778 CrossRef CAS PubMed.
- Y.-Q. Ji, J.-Y. Li, S.-G. Luo, T. Wu and J.-L. Liu, Fresenius' J. Anal. Chem., 2001, 371, 49–53 CrossRef CAS PubMed.
- J. B. Truscott, P. Jones, B. E. Fairman and E. H. Evans, Anal. Chim. Acta, 2001, 433, 245–253 CrossRef CAS.
- T. C. Kenna, J. Anal. At. Spectrom., 2002, 17, 1471–1479 RSC.
- C. S. Kim, C. K. Kim and K. J. Lee, J. Anal. At. Spectrom., 2004, 19, 743–750 RSC.
- Q. C. Chen, H. Dahlgaard, S. P. Nielsen and A. Aarkrog, J. Radioanal. Nucl. Chem., 2002, 253(3), 451–458 CrossRef CAS.
- M. Ayranov, U. Krähenbühl, H. Sahli, S. Röllin and M. Burger, Radiochim. Acta, 2005, 93, 631–635 CAS.
- P. Lindahl, P. Roos, E. Holm and H. Dahlgaard, J. Environ. Radioact., 2005, 82, 285–301 CrossRef CAS PubMed.
- T. C. Kenna, J. Environ. Radioact., 2009, 100, 547–557 CrossRef CAS PubMed.
- S. L. Maxwell, B. K. Culligan, V. D. Jones, S. T. Nichols, M. A. Bernard and G. W. Noyes, Anal. Chim. Acta, 2010, 682, 130–136 CrossRef CAS PubMed.
- Y. Shi, X. Dai, C. Li, R. Collins, S. Kramer-Tremblay, R. Riopel and C. Broome, J. Anal. At. Spectrom., 2014, 29, 1708–1713 RSC.
- P. Zhao, R. M. Tinnacher, M. Zavarin and A. B. Kersting, J. Environ. Radioact., 2014, 137, 163–172 CrossRef CAS PubMed.
- N. Guérin, M.-A. Langevin, K. Nadeau, C. Labrecque, A. Gagnè and D. Larivière, Appl. Radiat. Isot., 2010, 68, 2132–2139 CrossRef PubMed.
- J. Qiao, X. Hou, P. Roos and M. Miró, Anal. Chem., 2011, 83, 374–381 CrossRef CAS PubMed.
- B. S. Matteson, S. K. Hanson, J. L. Miller and W. J. Oldham Jr, J. Environ. Radioact., 2015, 142, 62–67 CrossRef CAS PubMed.
- S. Jerome, P. Ivanov, C. Larijani, D. H. Parker and P. H. Regan, J. Environ. Radioact., 2014, 138, 315–322 CrossRef CAS PubMed.
- A. Gourgiotis, M. Granet, H. Isnard, A. Nonell, C. Gautier, G. Stadelmann, M. Aubert, D. Durand, S. Legand and F. Chartier, J. Anal. At. Spectrom., 2010, 25, 1939–1945 RSC.
- B. Kuczewski, C. M. Marquardt, A. Seibert, H. Geckeis, J. V. Kratz and N. Trautmann, Anal. Chem., 2003, 75(24), 6769–6774 CrossRef CAS PubMed.
- S. Röllin, H. Sahli, R. Holzer, M. Astner and M. Burger, Appl. Radiat. Isot., 2009, 67, 821–827 CrossRef PubMed.
- J. Qiao, X. Hou, M. Miró and P. Roos, Anal. Chim. Acta, 2009, 652, 66–84 CrossRef CAS PubMed.
- T. Warneke, PhD thesis, GAU-Radioanalytical, Ocean and Earth Science, University of Southampton, 2002 Search PubMed.
- J. Zheng, T. Aono, S. Uchida, J. Zhang and M. C. Honda, Geochem. J., 2012, 46, 361–369 CrossRef CAS.
- N. Erdmann, G. Hermann, G. Huber, S. Köhler, J. V. Kratz, A. Mansel, M. Nunneman, G. Passler, N. Trautmann, A. Turchin and A. Waldek, Fresenius' J. Anal. Chem., 1997, 359, 378–381 CrossRef CAS.
- Y. Ohtsuka, Y. Takaku, K. Nishimura, J. Kimura, S. Hisamatsu and J. Inaba, Anal. Sci., 2006, 22, 309–311 CrossRef CAS PubMed.
- D. Larivière, T. A. Cumming, S. Kiser, C. Li and R. J. Cornett, J. Anal. At. Spectrom., 2007, 22, 1–9 RSC.
- B. G. Ting, R. S. Pappas and D. C. Paschal, J. Anal. At. Spectrom., 2003, 18, 795–797 RSC.
- J. Qiao, X. Hou, P. Roos and M. Miró, Anal. Chem., 2013, 85, 2853–2859 CrossRef CAS PubMed.
- S. Cagno, K. Hellemans, O. C. Lind, L. Skipperud, K. Janssens and B. Salbu, Environ. Sci.: Processes Impacts, 2014, 16, 306–312 CAS.
- Z. Varga, A. Nicholl, M. Wallenius and K. Mayer, J. Radioanal. Nucl. Chem., 2015, 307(3), 1919–1926 CrossRef PubMed.
- K. Bu, J. V. Cizdziel and D. Dasher, J. Environ. Radioact., 2013, 124, 29–36 CrossRef CAS PubMed.
- M. Jäggi, S. Röllin, J. A. C. Alvaredo and J. Eikenberg, Appl. Radiat. Isot., 2012, 70, 360–364 CrossRef PubMed.
- S. Salminen-Paatero, U. Nygren and J. Paatero, J. Environ. Radioact., 2012, 113, 163–170 CrossRef CAS PubMed.
- T. Shinonaga, P. Steier, M. Lagos and T. Ohkura, Environ. Sci. Technol., 2014, 48, 3608–3814 CrossRef PubMed.
- H. Hernández-Mendoza, E. Chamizo, A. Delgado, M. García-León and A. Yllera, J. Anal. At. Spectrom., 2011, 26, 1509–1513 RSC.
- F. N. Pointurier, N. Baglan and P. Hémet, Appl. Radiat. Isot., 2004, 60, 561–566 CrossRef CAS PubMed.
- V. N. Epov, R. D. Evans, J. Zheng, O. F. X. Donard and M. Yamada, J. Anal. At. Spectrom., 2007, 22, 1131–1137 RSC.
- M. L. D. P. Godoy, J. M. Godoy and L. A. Roldão, J. Environ. Radioact., 2007, 97, 124–136 CrossRef CAS PubMed.
- M. V. Zoriy, L. Halicz, M. E. Ketterer, C. Pickhardt, P. Ostapczuk and J. S. Becker, J. Anal. At. Spectrom., 2004, 19, 362 RSC.
- O. F. X. Donard, F. Bruneau, M. Moldovan, H. Garraud, V. N. Epov and D. Boust, Anal. Chim. Acta, 2007, 587, 170–179 CrossRef CAS PubMed.
- S. K. Aggarwal, Mass Spectrom. Rev., 2016 DOI:10.1002/mas.21506.
- G. Xiao, D. Saunders, R. L. Jones and K. L. Caldwell, J. Radioanal. Nucl. Chem., 2014, 301, 285–291 CrossRef CAS PubMed.
- M. Agarande, S. Benzoubir, P. Bouisset and D. Calmet, Appl. Radiat. Isot., 2001, 55(2), 161–165 CrossRef CAS PubMed.
- W. Hang, L. Zhu, W. Zhong and C. Mahan, J. Anal. At. Spectrom., 2004, 19, 966–972 RSC.
- Z. Wang, J. Zheng, L. Cao, K. Tagami and S. Uchida, Anal. Chem., 2016, 88(14), 7387–7394 CrossRef CAS PubMed.
- H. Kurosaki, J. R. Cadieux and S. B. Clark, J. Anal. At. Spectrom., 2014, 29, 2419–2423 RSC.
- A. Gourgiotis, H. Isnard, M. Aubert, E. Dupont, I. AlMahamid, G. Tiang, L. Rao, W. Lukens, P. Cassette, S. Panebianco, A. Letourneau and F. Chartier, Int. J. Mass Spectrom., 2010, 291(3), 101–107 CrossRef CAS.
- A. Gourgiotis, H. Isnard, A. Nonell, M. Aubert, G. Stadelmann, E. Dupont, I. AlMahamid, G. Tiang, L. Rao, W. Lukens, P. Cassette, S. Panebianco, A. Letourneau and F. Chartier, Talanta, 2013, 106, 39–44 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |