 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Boundary layer friction of solvate ionic liquids as a function of potential
Hua
Li
 a,
Mark W.
Rutland
bc,
Masayoshi
Watanabe
d and
Rob
Atkin†
*a
a,
Mark W.
Rutland
bc,
Masayoshi
Watanabe
d and
Rob
Atkin†
*a
aPriority Research Centre for Advanced Fluids and Interfaces, The University of Newcastle, Callaghan, NSW 2308, Australia. E-mail: rob.atkin@uwa.edu.au
bSchool of Chemical Science and Engineering, KTH Royal Institute of Technology, SE100 44 Sweden
cChemistry, Materials and Surfaces, SP Technical Research Institute of Sweden, SE114 86 Sweden
dDepartment of Chemistry and Biotechnology, Yokohama National University, Yokohama 240-8501, Japan
First published on 3rd January 2017
Abstract
Atomic force microscopy (AFM) has been used to investigate the potential dependent boundary layer friction at solvate ionic liquid (SIL)–highly ordered pyrolytic graphite (HOPG) and SIL–Au(111) interfaces. Friction trace and retrace loops of lithium tetraglyme bis(trifluoromethylsulfonyl)amide (Li(G4) TFSI) at HOPG present clearer stick-slip events at negative potentials than at positive potentials, indicating that a Li+ cation layer adsorbed to the HOPG lattice at negative potentials which enhances stick-slip events. The boundary layer friction data for Li(G4) TFSI shows that at HOPG, friction forces at all potentials are low. The TFSI− anion rich boundary layer at positive potentials is more lubricating than the Li+ cation rich boundary layer at negative potentials. These results suggest that boundary layers at all potentials are smooth and energy is predominantly dissipated via stick-slip events. In contrast, friction at Au(111) for Li(G4) TFSI is significantly higher at positive potentials than at negative potentials, which is comparable to that at HOPG at the same potential. The similarity of boundary layer friction at negatively charged HOPG and Au(111) surfaces indicates that the boundary layer compositions are similar and rich in Li+ cations for both surfaces at negative potentials. However, at Au(111), the TFSI− rich boundary layer is less lubricating than the Li+ rich boundary layer, which implies that anion reorientations rather than stick-slip events are the predominant energy dissipation pathways. This is confirmed by the boundary friction of Li(G4) NO3 at Au(111), which shows similar friction to Li(G4) TFSI at negative potentials due to the same cation rich boundary layer composition, but even higher friction at positive potentials, due to higher energy dissipation in the NO3− rich boundary layer.
Introduction
Ionic liquids (ILs) are salts with melting points below 100 °C.1–4 ILs have attracted significant interest in electrochemistry due to their high ionic conductivity, wide electrochemical window, negligible vapour pressure and non-flammability.1,5–11 Recent growing interest in advanced electrochemical devices such as lithium ion batteries, electric double layer capacitors and fuel cells has led to the development of a new class of ILs known as solvate ionic liquids (SILs).12 SILs are formed by certain alkali metal salts, usually lithium salts, coordinating with a strong ligand (solvate), like polyethylene oxide oligomers (glymes) in equimolar concentration. A Li+ ion is bound by the glyme to form a complex cation, with the anion coming from the alkali metal salt. The resultant SIL has the form Li(glyme) X and has physicochemical properties that are typical of ILs including high thermal stability, wide electrochemical window, high ionic conductivity, and high Li+ transfer number.13The formation of the complex cation in SILs is due to partial donation of lone pair electrons from the glyme to Li+. The definition of a “good” or “poor” SIL relies on the competition between the glyme ligand and the anion for coordination to Li+. For example, Li(G4) TFSI is a good SIL, as the weak basic TFSI− anion coordinates weakly with the Li+ ion, allowing stable formation of the Li(G4)+ complex cation and a negligible amount of free glymes.11 Conversely, Li(G4) NO3 is a poor SIL, due to the highly favourable coordination of Li+ with the strongly basic NO3− anion leading to high concentration of free glymes in the mixture and reduced the ionic properties of the obtained SIL.8
Recently, the bulk structure of SILs has been investigated;14,15 however, ion arrangements of SILs at electrode interfaces have rarely been investigated despite their importance for electrochemical applications.16 Conventional IL ions interact strongly with solid surfaces and present pronounced interfacial structures, which can be divided into three distinct regions: the boundary (surface-adsorbed) layer, the transition zone (near-surface layers) and the bulk phase.17–24 The boundary layer consists of a single layer of ions in direct contact with the solid surface. The composition of this layer strongly depends on the properties of the solid substrate. Adjacent to the boundary layer are several near-surface layers, referred to as the transition zone. Through this transition zone, which is typically a few nanometres across, the pronounced interfacial layer structure decays into the bulk morphology. The boundary layer ion composition can be tuned by applying a potential to the electrode surface; it changes from cation rich to anion rich when the electrode surface switches from negatively charged to positively charged.25–27 The near-surface layers also respond to the potential and propagate further from electrode surfaces at higher potentials.28–30
AFM force curves and computational simulations have been used to investigate the potential dependant interfacial structure of Li(G4) TFSI and Li(G4) NO3 on HOPG and Au(111) electrodes.16 A 0.25 nm final layer is observed at negative potentials on both surfaces, which is likely due to either compression of the complex cation adsorbed structure or desolvation of the tetraglyme from Li+. However, it has not been revealed which possibility is more likely to occur, and thus ion arrangements at negatively charged electrode surfaces are not clear, which limits the application of SILs in electrochemical devices.
Boundary layer friction of ILs is mainly determined by ion arrangements (i.e., composition and orientation) in the boundary layer, therefore investigating the response of boundary layer friction to the electrode potential will provide further information on ion arrangements in the boundary layer.26,27 In this work, AFM is applied to investigate the boundary layer friction of two SILs, Li(G4) TFSI and Li(G4) NO3, at HOPG and Au(111) electrode surfaces as a function of potential. The outcomes will pave the way for estimation of boundary layer structure at different SIL–electrode interfaces and thus underpin the development of new lithium electrolytes for advanced lithium batteries and other electrochemical devices.
Material and methods
All chemicals were purchased from Sigma-Aldrich; SILs were prepared as described previously.16 Water content in the obtained SILs was below 100 ppm, determined using Karl Fischer titration. HOPG surfaces (ZYB grade, NT-MDT) were freshly cleaved before experiments; Au(111) surfaces (a gold film of ∼150 nm thickness on mica, Agilent Technologies) were carefully rinsed with Milli-Q water, dried under nitrogen and irradiated with ultraviolet light for 20 min before use.Lateral (frictional) force measurements were performed using a Bruker Multimode VIII AFM with an EV scanner in contact mode. Three sharp silicon tips (spring constant = 2.0 ± 0.2 N m−1, tip radius 8 nm) from the same batch (model NSC36, Mikromasch, Tallinn, Estonia) were used over the course of the investigation. During the measurements, an AFM electrochemistry fluid cell (MMTMEC, Bruker) was used to hold solvate ILs on HOPG or Au(111) surfaces, which were both the working electrodes and the solid substrates. A 0.25 mm Cu wire was used as the counter electrode, and a 0.25 mm Pt wire was used as the “quasi” reference electrode. The tip and electrochemistry fluid cell were cleaned immediately prior to use by careful rinsing in Milli-Q water, drying under nitrogen and irradiation with ultraviolet light for 20 min. A potential is applied between the surface and the SIL. The open circuit potentials (OCP) of the SILs investigated in this work are all close to 0 V (within ±0.1 V) on both surfaces, thus we just applied 0 V, ±0.5 V and ±1.0 V for all of the systems investigated.
Lateral force measurements were acquired by performing lateral AFM scans with a scan angle of 90° (with respect to the cantilever long axis) with the slow scan axis disabled. At the HOPG surfaces, friction trace and retrace loops were collected with a scan size of 3 nm and a scan rate of 10 Hz at a fixed normal load of 50 nN. At both HOPG and Au(111) surfaces, the lateral force vs. normal load curves were measured with a scan size of 100 nm and a scan rate of 30 Hz. The lateral deflection signal (i.e., cantilever twist) was converted to lateral force using a customized function produced in Matlab 7.0 which takes into account the torsional spring constant and the geometrical dimensions of the cantilever.27
Results and discussion
Boundary layer ion arrangements of ILs can be evaluated by investigation of boundary layer friction. This study first measured the potential dependent friction trace and retrace loops on the boundary layer for Li(G4) TFSI at HOPG at a constant normal load of 50 nN, then examined the variation of boundary layer friction on the normal load at various potentials for Li(G4) TFSI at both HOPG and Au(111), and compared with the friction for Li(G4) NO3 at the Au(111) surface.Friction line trace and retrace loops for Li(G4) TFSI at HOPG
Fig. 1 shows the friction line trace and retrace loops for Li(G4) TFSI at HOPG as a function of applied potential at a constant normal load of 50 nN. Previous studies at HOPG in air have shown that the AFM tip undergoes clear stick-slip motion, which can be described by the Tomlinson model.26,31 Basically, as the AFM tip is dragged over the atomic structure of the surface, it sticks to a certain surface position until the energy stored in the deflected cantilever spring becomes large enough to activate a slip of the tip to the next atomic position. This stick-slip event presents a sawtooth-like modulation of lateral force as a function of tip displacement.32 The area enclosed in this sawtooth-like line trace–retrace loop corresponds to the energy dissipated by stick-slip events and provides a quantitative measure of the energy lost during friction. However, as shown in Fig. 1, when Li(G4) TFSI is present between the HOPG surface and the AFM tip, the magnitude and regularity of stick-slip events in the line trace and retrace loops are less significant for all potentials, except for −1.0 V, which still shows clear stick-slip motion. These results suggest the adsorption of a SIL boundary layer at the HOPG surface, which is consistent with our previous interfacial structure study at SIL–HOPG interfaces.16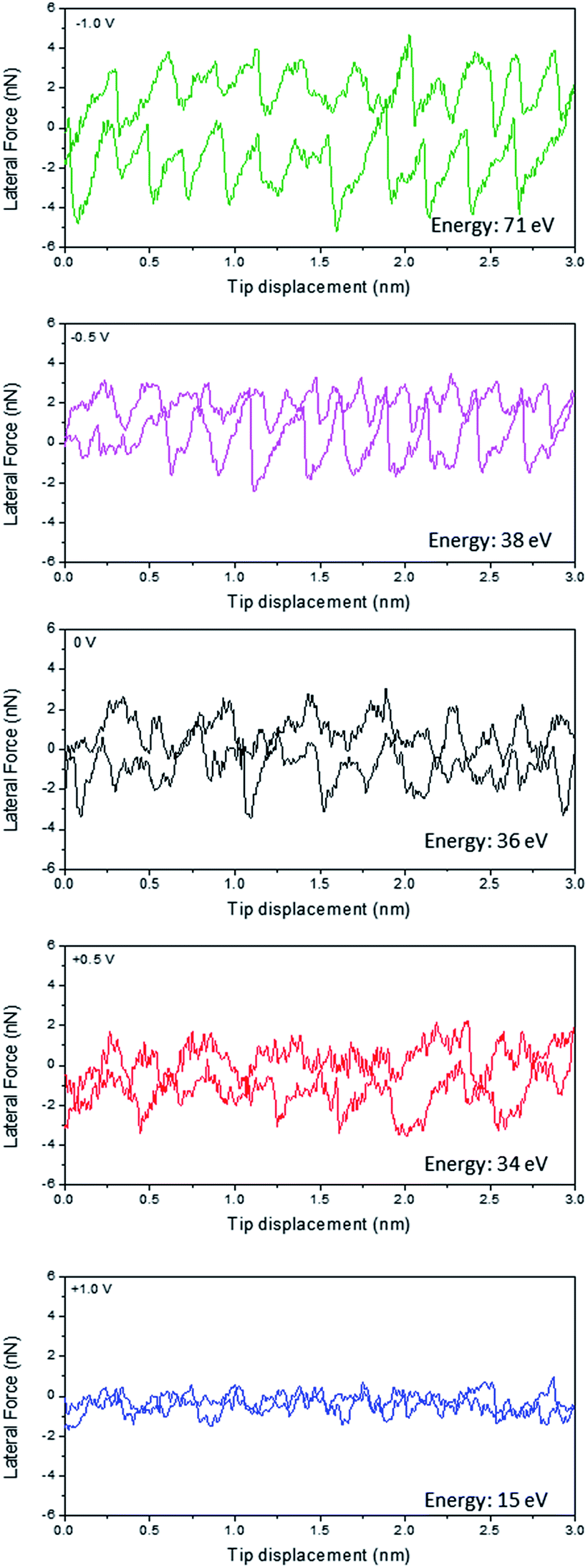 | ||
| Fig. 1 Lateral force versus AFM tip displacement while sliding over an HOPG surface with a scan width of 3 nm at 58.6 nm s−1 at a normal load of 50 nN in Li(G4) TFSI at 0 V, −0.5 V, −1.0 V, +0.5 V, and +1.0 V (versus Pt quasi reference electrode). The energy dissipation is calculated from the area enclosed by the curve. | ||
At 0 V, the energy dissipated (the area enclosed by the line trace and retrace loop) for Li(G4) TFSI reduces significantly compared to that in air.26 The stick-slip events are irregular with jumps ranging from 0.3 nm to 0.4 nm. These jump sizes are generally wider than the dimensions of HOPG lattices (0.25 nm), but are broadly consistent with the molecular dimensions of Li(G4)+ complex cations (0.38 nm), which indicates the presence of a layer containing significant amounts of the Li(G4)+ cation adsorbed at the HOPG interface, as seen in a previous study.16
At positive potentials, the energy dissipation reduces by more than 50% compared to that at 0 V, from 34 eV at 0 V to 15 eV at +1.0 V. The stick-slip widths, varying between 0.3 nm and 0.6 nm, are generally close to the molecular dimensions of TFSI− anions (0.47 nm), and suggest that a TFSI− anion rich boundary layer adsorbs at positive potentials due to electrostatic attractions, in line with a previous interfacial structure study.16 The modulation of the friction line trace reduces when the potential increases from 0 V to +0.5 V; it reduces further and can hardly be discerned at +1.0 V. This indicates that at more positive potentials, a smoother anion rich boundary layer forms due to stronger electrostatic interactions between the anions and the positively charged surface leading to better packing of the adsorbed TFSI− anions; this smoother anion rich boundary layer thus results in lower energy dissipation and friction.
In contrast, at negative potentials more energy is dissipated than at 0 V. The stick-slip events at negative potentials are more obvious, especially at −1.0 V which are as clear and ordered as those detected in air.26 The widths of the jumps (0.2–0.3 nm) are significantly smaller than the dimensions of the Li(G4)+ complex cation (0.38 nm) and TFSI− anion, but are close to the dimensions of the HOPG lattice (0.25 nm). At negative potentials, cations are expected to adsorb at the SIL–HOPG interface so as to balance the negative charges. Our previous AFM force curve study at SIL–HOPG interfaces and other studies have all suggested that it is Li+ cations rather than Li(G4)+ complex cations that lie in the boundary layer and resist the direct contact between the HOPG surface and the AFM tip at negative potentials, because Li+ cations can desolvate from Li(G4)+ complex cations at negative potentials under high pressure and adsorb strongly at the HOPG surface due to high adsorption energy.16,33 The friction line trace and retrace data shown in Fig. 1 further confirms this. As a Li+ ion is spherical and has a slightly smaller dimension (0.15 nm) than the HOPG lattice, it can pack onto the HOPG lattice such that each Li+ cation can occupy an HOPG site. Therefore, the adsorbed Li+ cation boundary layer is strongly templated by the HOPG surface, leading to more obvious stick-slip motions. The stick-slip events are more significant at −1.0 V than at −0.5 V, suggesting a better ordered Li+ cation boundary layer templated from HOPG lattices due to stronger electrostatic interactions.
Boundary layer friction for Li(G4) TFSI at HOPG surfaces
The next set of experiments tested the boundary layer friction as a function of potential for Li(G4) TFSI at HOPG surfaces, as shown in Fig. 2(a). Similar to the results obtained for conventional protic and aprotic IL–HOPG interfaces,26,34 two regimes are present at all potentials investigated. At low normal loads (FN < 8 nN), multiple ion layers remain between the AFM tip and the HOPG surface; lateral force increases sharply with normal load. At higher normal loads (FN > 8 nN), only the strongly adsorbed boundary ion layer remains between the AFM tip and the solid surface, and the lateral force increases more gradually with normal load. The normal load delineating the transition between the multilayer and boundary regimes corresponds to the final step in the normal force curve, as published in a previous nanostructure study.16 The focus of the following discussion is the boundary layer regime, when the normal load is greater than 8 nN and the AFM tip is in contact with a single adsorbed ion layer.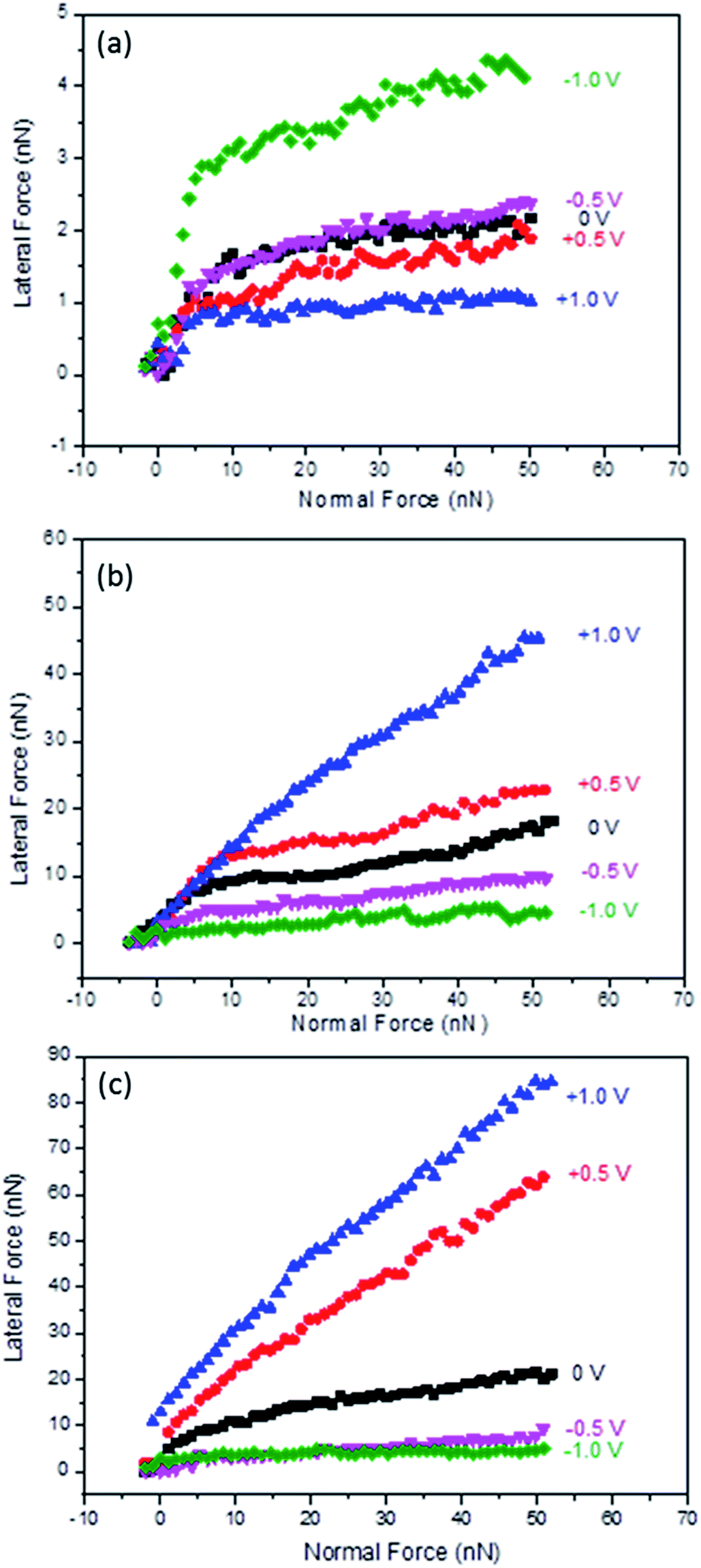 | ||
| Fig. 2 Lateral force versus normal load as a function of applied potential for Li(G4) TFSI at HOPG (a) and Au(111) (b), and Li(G4) NO3 at Au(111) (c). | ||
For the boundary layer friction data at HOPG surfaces (Fig. 2(a)), the lateral force for 0 V lies in the middle. Applying positive potentials decreases the lateral force, while applying negative potentials increases the lateral force compared to 0 V. In the boundary layer regime, energy is mainly dissipated through (1) multilayer expulsion between the AFM tip and the sliding surface, which primarily determines the force at the beginning of the boundary layer regime, and (2) boundary layer ions, which is mainly manifested in the magnitude of the boundary layer friction coefficient.34–36
For the expulsion of multilayers, although all near-surface layers can contribute to energy dissipation, the interactions between the boundary layer and the first near-surface layer are most important as they can cause the greatest disruption of the native structure (and interactions between ions).34 The stronger the attraction between the boundary layer and near-surface layers, the higher the energy dissipation and the higher the lateral force at the beginning of the boundary regime. At negative potentials, especially at −1.0 V, the initial lateral force in the boundary regime is high. This is likely because although Li+ is desolvated from tetraglyme as suggested from Fig. 1 and previous studies, the attractions between the Li+ rich boundary layer and the tetraglyme layer on top of it are still very strong, and thus result in high energy dissipation and high initial lateral force. However, at 0 V and positive potentials, the attractions between the adsorbed boundary layer ions (Li(G4)+ for 0 V and TFSI− for positive potentials) and the first near surface layer (rich in counter ions) are weaker due to more delocalised charges and thus weaker electrostatic forces; therefore, the initial lateral forces at the beginning of the boundary layer regime are lower.
The boundary layer friction coefficients (μ = FL/FN) for Li(G4) TFSI at HOPG were obtained for normal loads between 8 nN and 50 nN for each potential, shown in Table 1. μ is mainly determined by the roughness of the boundary layer, which is significantly influenced by the order and orientation of the ions in the boundary layer. The friction coefficients for −0.5 V and −1.0 V (0.024 and 0.033) are higher than those at 0 V (0.017). This is consistent with more significant stick-slip motions detected at more negative potentials, cf.Fig. 1, due to more desolvated Li+ cations sitting on HOPG lattices. Further, the adsorbed Li+ cations interact weakly with each other laterally due to the spherical shape and lack of solvophobic interactions through carbon chains; they are more easily shaved away by the AFM tip, resulting in a rougher sliding plane. However, at positive potentials the friction coefficients are generally lower than at 0 V. This is because at positive potentials, TFSI− anions enriched in the boundary layer have strong interactions with the HOPG surface and good lateral integrity within the boundary layer through the anion fluorocarbon units, and thus form a smoother and better-defined plane for the AFM tip to slide across. Also TFSI− anions tend to orientate parallel to the HOPG surface in trans position at positive potentials;37,38 this trans position is more energetically favoured and thus shows a lower friction coefficient. At 0 V, the boundary layer is rich in Li(G4)+ cations, but the population of TFSI− anions is also high.16 The sliding plane is rougher than at positive potentials but does not show as obvious stick-slip motion as at negative potentials, thus the friction coefficient at 0 V is higher than at positive potentials but lower than at negative potentials.
| −1.0 V | −0.5 V | 0 V | +0.5 V | +1.0 V | ||
|---|---|---|---|---|---|---|
| HOPG | Li(G4) TFSI | 0.033 | 0.024 | 0.017 | 0.020 | 0.010 |
| Au(111) | Li(G4) TFSI | 0.059 | 0.12 | 0.21 | 0.25 | 0.74 |
| Li(G4) NO3 | 0.048 | 0.12 | 0.24 | 1.0 | 1.3 | |
Effects of surface properties on boundary layer friction
The effects of electrode surface properties were investigated by measuring the lateral forces as a function of potential for Li(G4) TFSI at Au(111) surfaces, shown in Fig. 2(b). Similar to HOPG, a multilayer regime (FN < 8 nN) and a boundary layer regime (FN > 8 nN) are detected at Au(111); what we focus on here is still the boundary regime where only a single ion layer separates the Au(111) substrate and the AFM tip.At Au(111) the response of boundary layer friction to variation in potential is completely different from that at HOPG, with much higher lateral forces and friction coefficients for more positive potentials. At −1.0 V, the lateral force and friction coefficients are lowest and essentially in the same order of magnitude with those at HOPG at the same voltage, cf.Fig. 2 and Table 1. These results suggest that the compositions of the boundary layers at both surfaces are similar and rich in Li+ cations, once again, consistent with the previous AFM force curve study.16 The friction coefficient at Au(111) is ∼80% higher than that at HOPG, possibly due to the restructure of gold at negative potentials increasing surface roughness,39,40 which leads to a rougher Li+ cation layer at Au(111) than at HOPG. However, as Au(111) becomes less negatively charged, the lateral forces of Li(G4) TFSI increases, completely the opposite trend to that observed on HOPG. At +1.0 V, when the interfacial layer is rich in TFSI− anions, the friction coefficient is more than one order of magnitude higher than that found at −1.0 V. These results suggest that in contrast to those found at HOPG, the TFSI− anion rich layer is less lubricating than the Li+ cation rich layer at Au(111) surfaces. One reason is that TFSI− anions may have a lower affinity to Au(111) due to a lack of solvophobic interactions between the anion fluorocarbon units and the gold atoms, so they can be shaved away more easily at high shear forces, resulting in a rougher sliding plane.
Effects of anion structure on boundary layer friction
The impact of anion structure on boundary layer friction was investigated using Li(G4) NO3 at Au(111), cf.Fig. 2(c). Li(G4) NO3 is a poor SIL and has a weaker interfacial nanostructure compared to Li(G4) TFSI according to a previous study.16 Similarly to Li(G4) TFSI at Au(111), Li(G4) NO3 is more lubricating at negative potentials and less lubricating at positive potentials, suggesting that the cation rich layer is more lubricating than the anion rich layer.In fact, at the same negative potential and 0 V, lateral forces are of the same magnitude and the friction coefficients are comparable for Li(G4) NO3 and Li(G4) TFSI, cf.Fig. 2 and Table 1. As the boundary layers are all Li+ cation rich at negative potentials and Li(G4)+ rich at 0 V, these results imply that the influence of the anion structure is not significant, and it is the composition and ordering of cations that control the lubricity at negative potentials and 0 V. The only difference is that for Li(G4) NO3 the lateral forces at −0.5 V are closer to those at −1.0 V, whereas for Li(G4) TFSI the lateral forces are obviously higher at −0.5 V than at −1.0 V. This difference suggests that Li(G4) NO3 can form a better lubricating Li+ cation rich layer at a less negative potential due to weaker coordination between Li+ and tetraglyme in the poor Li(G4) NO3 SIL.
At positive potentials, however, the lateral forces and friction coefficients of Li(G4) NO3 are almost doubled compared to those of Li(G4) TFSI at the same potential. At positive potentials, the boundary layer is rich in NO3− anions for Li(G4) NO3, which has much weaker lateral integrity due to the planar structure and lack of fluorocarbon units compared to TFSI− anions, and thus may have more defects and higher roughness. Also Li(G4) NO3 is a poor SIL, containing a significant population of Li NO3 salts and free glyme molecules. At positive potentials, although NO3− anions are rich in the boundary layer, it is conceivable that a significant amount of Li+ cations are co-located in the layer due to the smaller size of the anions and the ensuing volume considerations – as well as due to non-electrostatic interactions with the (higher concentration of) anions. At high shear forces these positive cations are easily shaved away, leaving defects in the sliding plane, and thus increasing roughness and friction significantly.
Conclusions
The boundary layer frictions of SILs at HOPG and Au(111) interfaces have been investigated as a function of potential using AFM. At HOPG, friction trace and retrace loops reveal that the stick-slip events for Li(G4) TFSI are negligible at +1.0 V, but most significant at −1.0 V. This suggests that the desolvated Li+ cations from Li(G4)+ complexes adsorb on top of HOPG lattices, leading to clearer stick-slip events at negative potentials, supporting an earlier hypothesis.16 The ability of the interface to locally uncomplex the cation is potentially an additional self-assembly tool that can be used to control interfacial sliding.The potential dependent boundary layer friction data of Li(G4) TFSI at HOPG shows that the Li+ cation rich boundary layer at negative potentials is less lubricating than the TFSI− anion rich boundary layer at positive potentials. This suggests that stick-slip events are the predominant energy dissipation pathways for HOPG surfaces; the stick-slip events are more significant in Li+ cation rich boundary layers due to a better templated structure of the HOPG surface. Conversely, at Au(111), Li(G4) TFSI shows much higher friction at positive potentials than at negative potentials, which is similar to that at HOPG. The similar friction at both negatively charged HOPG and Au(111) surfaces means that the boundary layer ion arrangements at both surfaces are comparable at negative potentials, both rich in Li+ cations. At Au(111), the TFSI− anion rich boundary layer at positive potentials is less lubricating than the Li+ cation rich boundary layer at negative potentials, indicating that energy dissipation due to boundary layer anion reorientation is significant and leads to high friction at Au(111) surfaces. This is supported by boundary layer friction data of Li(G4) NO3 at Au(111). Li(G4) NO3 shows similar friction to Li(G4) TFSI at negative potentials, as the cation rich boundary layer composition and orientation are similar, but higher friction at positive potentials due to higher energy dissipation in the NO3− rich boundary layer than in the TFSI− rich boundary layer. The outcomes help to understand ion arrangements and energy dissipation pathways at SIL–electrode interfaces, and thus assist the development of new lithium electrolytes for advanced lithium batteries and other electrochemical devices.
Acknowledgements
This research was supported by an ARC Discovery Project (DP120102708). RA thanks the ARC for a Future Fellowship (FT120100313). MR thanks the Swedish Research Council (VR 621-2011-4361) and the Knut and Alice Wallenberg Foundation (KAW2012.0078) for financial support. Mr Ben McLean is acknowledged for assistance preparing SILs. Mr Peter Cooper and Mr Andre Cook are acknowledged for valuable discussions.References
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis, M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232–250 CAS.
- G. A. Baker, S. N. Baker, S. Pandey and F. V. Bright, Analyst, 2005, 130, 800–808 RSC.
- M. J. Earle and K. R. Seddon, Pure Appl. Chem., 2000, 72, 1391–1398 CrossRef CAS.
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 115, 6357–6426 CrossRef CAS PubMed.
- G. M.-A. Girard, M. Hilder, K. Whitbread, D. Nucciarone, S. Zavorine, M. Moser, M. Forsyth, D. R. MacFarlane and P. C. Howlett, Phys. Chem. Chem. Phys., 2015, 17, 8706–8713 RSC.
- S. A. Mohd Noor, P. C. Howlett, D. R. MacFarlane and M. Forsyth, Electrochim. Acta, 2013, 114, 766–771 CrossRef CAS.
- K. Ueno, J.-W. Park, A. Yamazaki, T. Mandai, N. Tachikawa, K. Dokko and M. Watanabe, J. Phys. Chem. C, 2013, 117, 20509–20516 CAS.
- C. Austen Angell, Y. Ansari and Z. Zhao, Faraday Discuss., 2012, 154, 9–27 RSC.
- H. Ohno, Electrochemical aspects of ionic liquids, John Wiley & Sons, 2011 Search PubMed.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mat., 2009, 8, 621–629 CrossRef CAS PubMed.
- A. M. Smith and S. Perkin, J. Phys. Chem. Lett., 2015, 6, 4857–4861 CrossRef CAS PubMed.
- K. Ueno, K. Yoshida, M. Tsuchiya, N. Tachikawa, K. Dokko and M. Watanabe, J. Phys. Chem. B, 2012, 116, 11323–11331 CrossRef CAS PubMed.
- T. Tamura, K. Yoshida, T. Hachida, M. Tsuchiya, M. Nakamura, Y. Kazue, N. Tachikawa, K. Dokko and M. Watanabe, Chem. Lett., 2010, 39, 753–755 CrossRef CAS.
- T. Murphy, S. K. Callear, N. Yepuri, K. Shimizu, M. Watanabe, J. N. C. Lopes, T. Darwish, G. G. Warr and R. Atkin, Phys. Chem. Chem. Phys., 2016, 18, 17224–17236 RSC.
- K. Shimizu, A. A. Freitas, R. Atkin, G. G. Warr, P. A. FitzGerald, H. Doi, S. Saito, K. Ueno, Y. Umebayashi and M. Watanabe, Phys. Chem. Chem. Phys., 2015, 17, 22321–22335 RSC.
- B. McLean, H. Li, R. Stefanovic, R. J. Wood, G. B. Webber, K. Ueno, M. Watanabe, G. G. Warr, A. Page and R. Atkin, Phys. Chem. Chem. Phys., 2015, 17, 325–333 RSC.
- H. Li, F. Endres and R. Atkin, Phys. Chem. Chem. Phys., 2013, 15, 14624–14633 RSC.
- F. Endres, N. Borisenko, S. Z. El Abedin, R. Hayes and R. Atkin, Faraday Discuss., 2012, 154, 221–233 RSC.
- Y. Yokota, T. Harada and K.-i. Fukui, Chem. Commun., 2010, 46, 8627–8629 RSC.
- Y. Z. Su, Y. C. Fu, J. W. Yan, Z. B. Chen and B. W. Mao, Angew. Chem., 2009, 121, 5250–5253 CrossRef.
- D. Wakeham, R. Hayes, G. G. Warr and R. Atkin, J. Phys. Chem. B, 2009, 113, 5961–5966 CrossRef CAS PubMed.
- R. Hayes, S. Z. El Abedin and R. Atkin, J. Phys. Chem. B, 2009, 113, 7049–7052 CrossRef CAS PubMed.
- R. Atkin and G. G. Warr, J. Phys. Chem. C, 2007, 111, 5162–5168 CAS.
- A. Elbourne, S. McDonald, K. Voïchovsky, F. Endres, G. G. Warr and R. Atkin, ACS Nano, 2015, 9, 7608–7620 CrossRef CAS PubMed.
- H. Li, R. J. Wood, F. Endres and R. Atkin, J. Phys.: Condens. Matter, 2014, 26, 284115 CrossRef PubMed.
- H. Li, R. J. Wood, M. W. Rutland and R. Atkin, Chem. Commun., 2014, 50, 4368–4370 RSC.
- J. Sweeney, F. Hausen, R. Hayes, G. B. Webber, F. Endres, M. W. Rutland, R. Bennewitz and R. Atkin, Phys. Rev. Lett., 2012, 109, 155502 CrossRef PubMed.
- A. Elbourne, K. Voïtchovsky, G. G. Warr and R. Atkin, Chem. Sci., 2015, 6, 527–536 RSC.
- J. M. Black, D. Walters, A. Labuda, G. Feng, P. C. Hillesheim, S. Dai, P. T. Cummings, S. V. Kalinin, R. Proksch and N. Balke, Nano Lett., 2013, 13, 5954–5960 CrossRef CAS PubMed.
- X. Zhang, Y.-X. Zhong, J.-W. Yan, Y.-Z. Su, M. Zhang and B.-W. Mao, Chem. Commun., 2011, 48, 582–584 RSC.
- C. Lee, Q. Li, W. Kalb, X.-Z. Liu, H. Berger, R. W. Carpick and J. Hone, Science, 2010, 328, 76–80 CrossRef CAS PubMed.
- A. Socoliuc, R. Bennewitz, E. Gnecco and E. Meyer, Phys. Rev. Lett., 2004, 92, 134301 CrossRef CAS PubMed.
- H. Moon, R. Tatara, T. Mandai, K. Ueno, K. Yoshida, N. Tachikawa, T. Yasuda, K. Dokko and M. Watanabe, J. Phys. Chem. C, 2014, 118, 20246–20256 CAS.
- H. Li, R. Atkin and A. Page, Phys. Chem. Chem. Phys., 2015, 17, 16047–16052 RSC.
- A. Elbourne, J. Sweeney, G. B. Webber, E. J. Wanless, G. G. Warr, M. W. Rutland and R. Atkin, Chem. Commun., 2013, 49, 6797–6799 RSC.
- J. Sweeney, G. B. Webber, M. W. Rutland and R. Atkin, Phys. Chem. Chem. Phys., 2014, 16, 16651–16658 RSC.
- A. I. Frolov, K. Kirchner, T. Kirchner and M. V. Fedorov, Faraday Discuss., 2012, 154, 235–247 RSC ; discussion 313–233, 465–271.
- T. Carstens, R. Gustus, O. Hoefft, N. Borisenko, F. Endres, H. Li, R. J. Wood, A. J. Page and R. Atkin, J. Phys. Chem. C, 2014, 118, 10833–10843 CAS.
- R. Atkin, S. Z. El Abedin, R. Hayes, L. H. S. Gasparotto, N. Borisenko and F. Endres, J. Phys. Chem. C, 2009, 113, 13266–13272 CAS.
- T. Carstens, R. Hayes, S. Z. E. Abedin, B. Corr, G. B. Webber, N. Borisenko, R. Atkin and F. Endres, Electrochim. Acta, 2012, 82, 48–59 CrossRef CAS.
Footnote |
| † Current address School of Molecular Sciences, The University of Western Australia, WA 6009, Australia |
| This journal is © The Royal Society of Chemistry 2017 |