
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolar fuel photoanodes prepared by inkjet printing of copper vanadates†
P. F.
Newhouse
a,
D. A.
Boyd
ab,
A.
Shinde
a,
D.
Guevarra
a,
L.
Zhou
a,
E.
Soedarmadji
a,
G.
Li
 c,
J. B.
Neaton
cd and
J. M.
Gregoire
*a
c,
J. B.
Neaton
cd and
J. M.
Gregoire
*a
aJoint Center for Artificial Photosynthesis, California Institute of Technology, Pasadena, CA 91125, USA. E-mail: gregoire@caltech.edu
bDivision of Physics, Mathematics & Astronomy, California Institute of Technology, Pasadena, CA 91125, USA
cJoint Center for Artificial Photosynthesis and Molecular Foundry, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
dDepartment of Physics, University of California and Kavli Energy NanoSciences Institute, Berkeley, CA 94720, USA
First published on 11th April 2016
Abstract
Widespread deployment of solar fuel generators requires the development of efficient and scalable functional materials, especially for photoelectrocatalysis of the oxygen evolution reaction. Metal oxides comprise the most promising class of photoanode materials, but no known material meets the demanding photoelectrochemical requirements. Copper vanadates have recently been identified as a promising class of photoanode materials with several phases exhibiting an indirect band gap near 2 eV and stable photoelectrocatalysis of the oxygen evolution reaction in a pH 9.2 electrolyte. By employing combinatorial inkjet printing of metal precursors and applying both calcination and rapid thermal processing, we characterize the phase behaviour of the entire CuO–V2O5 composition space for different thermal treatments via automated analysis of approximately 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Raman spectra acquired using a novel Raman imaging technique. These results enable the establishment of structure–property relationships for optical absorption and photoelectrochemical properties, revealing that highly active photoelectrocatalysts containing α-Cu2V2O7 or α-CuV2O6 can be prepared using scalable solution processing techniques. An additional discovery results from the formation of an off-stoichiometric β-Cu2V2O7 material that exhibits high photoelectroactivity in the presence of a ferri/ferrocyanide redox couple with excellent stability in a pH 13 electrolyte, demonstrating that copper vanadates may be viable photoanodes in strong alkaline electrolytes.
000 Raman spectra acquired using a novel Raman imaging technique. These results enable the establishment of structure–property relationships for optical absorption and photoelectrochemical properties, revealing that highly active photoelectrocatalysts containing α-Cu2V2O7 or α-CuV2O6 can be prepared using scalable solution processing techniques. An additional discovery results from the formation of an off-stoichiometric β-Cu2V2O7 material that exhibits high photoelectroactivity in the presence of a ferri/ferrocyanide redox couple with excellent stability in a pH 13 electrolyte, demonstrating that copper vanadates may be viable photoanodes in strong alkaline electrolytes.
Introduction
The development of efficient, stable photoanodes is a formidable materials challenge that is critical for establishing a solar fuel technology. Photoelectrochemical conversion of H2O and CO2 into either H2 or carbon-containing fuel is a promising renewable energy technology that can be efficiently powered by solar radiation using a tandem-light-absorber architecture.1–3 The photoanode component of a tandem device for either H2 or carbon-containing fuel must provide solar-driven electrocatalysis of the oxygen evolution reaction (OER). Metal oxides are of primary interest for photoanodes due to their stability against oxidation,4 presenting the materials science challenge of identifying metal oxide semiconductors that exhibit this stability along with the requisite band energetics, in particular a band gap energy below approximately 2.2 eV with a valence band position that enables efficient OER photoelectrocatalysis.1–3,5 The challenges of developing a suitable metal oxide photoanode are well documented,4,6 and substantial progress has been made recently with the identification of manganese and copper vanadates as low-band gap semiconductors with desirable band energetics and promising aqueous stability.7,8 In the present work, we demonstrate that high performance copper vanadate photoanodes can be synthesized using an affordable, scalable inkjet printing technique.Monoclinic BiVO4 is a widely studied photoanode material with an undesirably large direct band gap of approximately 2.4 eV9–11) and suffers from poor stability under operational conditions.9 More recent developments include the identification of a divalent Mn vanadate, β-Mn2V2O7, which is a semiconductor with a 1.8 eV direct band gap and excellent band alignment to the OER equilibrium potential.7 A suite of divalent Cu vanadates have also been reported as photoanodes, and indeed the CuO–V2O5 system is rapidly emerging as the most prominent composition system for OER photoelectrocatalysts. Seabold and Neale12 reported OER photoelectrocatalysis with β-Cu3V2O8 (pseudolyonsite) in a pH 9.2 electrolyte. Zhou et al.8 reported OER photoelectrocatalysis with Cu11V6O26 (fingerite), γ-Cu3V2O8 (McBirneyite), α-Cu2V2O7 (blossite), and β-Cu2V2O7 (ziesite) in pH 7, pH 9.2 and pH 13 electrolytes. Guo et al.13 reported OER photoelectrocatalysis with β-Cu2V2O7 (ziesite) and α-CuV2O6 in a pH 9.2 electrolyte. The experimental8,12,13 and computational8 assessments of the optical properties of these phases indicate that they are all indirect band gap semiconductors with band gap energy near or below 2 eV, and the photoelectrochemical activity of these 6 photoelectrocatalysts is particularly promising in a borate-buffered pH 9.2 electrolyte.
Out of the 6 copper vanadate phases, 5 are synthetic analogues of naturally occurring minerals, as noted above by the mineral names in parentheses. The phase behaviour of the CuO–V2O5 system has deep roots in the mineralogy field14–18 and has been studied over several decades.15–25 In the present work, we further explore this composition system using combinatorial materials science techniques, which have been successfully applied to the exploration of materials in complex, multi-component composition spaces.26–31
Combinatorial and high throughput techniques have enabled the discovery and investigation of electrocatalytic and photoelectrocatalytic materials for applications in fuel cells and solar fuel generators.30–45 Recent combinatorial efforts for the discovery of light absorbers and photocatalysts for solar fuel reactions include sputter-deposited photocathodes46 and photoanodes,47 including the copper vanadate study noted above.8 While physical vapour deposition offers excellent control of material properties and quality, materials synthesis techniques based on solution processing are desirable for scalable and affordable manufacturing, with inkjet printing being a particularly attractive technology for the synthesis of functional metal oxides.48,49 The use of inkjet-deposited composition libraries for the discovery of solar fuel materials was pioneered by Parkinson et al.,31,50 and has been effective in the identification of OER electrocatalysts51–53 and their integration onto light absorbers.54 In the reports on copper vanadate photoanodes, β-Cu3V2O8 has been synthesized by a multi-step wet chemical method involving the synthesis of Cu3V2O7(OH)2·2H2O nanoparticles,12 and both β-Cu2V2O7 and α-CuV2O6 were synthesized via drop casting and calcination of Cu(NO3)2 and VCl3 metal precursor solutions,13 indicating that copper vanadates may be a class of functional metal oxides that are particularly amenable to synthesis by solution processing techniques.
In the present work, the complete CuO–V2O5 composition system is studied as a discrete composition library with 4% composition intervals using combinatorial inkjet printing. The calcination of elemental precursors yields phase behaviour that is markedly different from traditional synthesis techniques and investigation of this phase behaviour is enabled through a novel Raman imaging technique. By comparing composition trends in photoelectrochemical activity with the observed crystalline phase map, the photoelectrochemical properties of inkjet-printed β-Cu2V2O7 (ziesite), α-Cu2V2O7 (blossite), and α-CuV2O6 are revealed. Additional application of rapid thermal processing (RTP) produces increased optical absorption and greatly enhanced the photoelectrochemical activity of β-Cu2V2O7, demonstrating that this phase is a viable photoanode in strong alkaline electrolytes. Our demonstration of excellent photoelectrochemical activity and stability of materials synthesized via inkjet-printing of metal precursor solutions further bolsters the potential of copper vanadates to enable an efficient, scalable, solar fuel technology.
Experimental
Inkjet printing and thermal processing
The (Cu–V)–O pseudo-binary composition libraries were deposited onto 100 × 150 × 2.2 mm FTO-coated soda lime glass (15 ohm sq−1, Hartford Glass) using a Microfab Technologies JetLab4 inkjet printer. The precursor inks were 0.13 M aqueous solutions of Cu (Cu(NO3)2·3H2O, Sigma, 99.9%) and V (VOSO4·3H2O, Sigma, 99.9%) dissolved in Millipore 18 Mohm water containing 10 and 20 vol% diethylene glycol, respectively. Thermal gravimetric analysis (TA Instruments Q600) was used to determine the formula weight (hydrate content) of the V precursor to enable accurate preparation of the precursor ink.The Cu1−xVxOz pseudo-binary space was synthesized with 4 at% composition intervals by depositing 26 individual composition samples with the corresponding ratio of the precursor inks. To print each discrete composition sample over a substrate area of approximately 1 mm × 1 mm, a total of 300 drops were dispensed, corresponding to a total metal loading of approximately 4 nmol per sample. The as-printed libraries were then dried in two steps (60 min at 37 °C, then 30 min at 65 °C). The precipitation of salts during the drying processes resulted in non-uniform salt thickness over the small area and small variations in the nominal 1 mm2 substrate area covered by the samples. The metal precursors were converted to metal oxides through calcination at 535 °C for 90 min in a box oven in air with ramp up and ramp down rates of approximately 4.9 and 10.3 °C min−1, respectively. The libraries processed in this manner are hereafter referred to in the text as “as-calcined”. Given the average density of the observed oxide compounds, the nominal average thickness of the samples is approximately 100 nm.
To enable a variety of photoelectrochemical experiments, duplicate library plates were prepared by this method and select libraries were further processed using rapid thermal processing (RTP, Modular Process Technology Corp. RTP-600xp). Only the top lamps of the furnace were used and the RTP was performed in ambient air. The library plate was placed on a 2 mm-thick graphite support and strong radiative heating was used to ramp the temperature to 671 °C in 38 s, as measured by a thermocouple underneath the graphite support. A lower lamp power was used to lower the temperature to 636 °C over 14 s and then the plate was allowed to free-cool at a rate of approximately 13 °C s−1. Given the instrument configuration and thermocouple placement, the temperature profile experienced by the printed samples likely differs from that measured by the thermocouple. As noted below, this extra thermal processing alters material properties, indicating that the maximum temperature of the samples exceeded the 535 °C temperature of the box oven calcination and may indeed be near or appreciably greater than the intended temperature of 636 °C. Samples with additional RTP processing are labelled as “post-RTP”.
Sample imaging
After thermal processing, each library plate was imaged using an Epson V600 photoscanner, providing sample images to assist in quality control and in interpretation of Raman and optical characterization data. Images of the duplicate composition library samples can be found in the ESI,† with select sample images included in the following figures.SEM experiments were performed with a FEI Nova NanoSEM 450. The (Cu, V) composition of the samples was measured with an EDS detector (Oxford Instruments X-Max 80 mm2) using a 10 kV electron beam and 7 mm working distance. Secondary electron imaging was used to observe the sample microstructure.
Raman imaging
A 26-sample library from both an as-calcined and a post-RTP library plate was characterized using a novel Raman imaging technique enabled by a Renishaw inVia Reflex Micro Raman spectrometer with Wire 4.1 control and analysis software. The laser wavelength was 532 nm and the grating was 2400 lines mm−1 (visible). Spectra were taken over the range of 78 to 1339.9 cm−1 using a 50× objective (numerical aperture 0.75). Raman imaging was performed using the Renishaw Streamline™ mapping system in which the laser beam spot was expanded into a line of approximate size 26 × 2 μm using a cylindrical lens and scanned over the desired area. The streamline maps incorporated the surface map capability of Wire 4.1 whereby a three dimensional topographical map of the area of interest was created beforehand and used to dynamically maintain focus during acquisition. Raman spectra were acquired at 50 μm spatial resolution and a 2 s exposure time.The Renishaw Empty Modelling data analysis tool was used to derive basis spectra from the Raman imaging dataset, and these basis spectra were identified and matched to reference patterns from the literature and the RRUFF database (http://rruff.info/). The resulting 6 reference patterns and a representative spectrum of the FTO/glass substrate were then used in a Direct Classical Least Squares (DCLS) fitting to determine the intensity of each basis pattern in each measured Raman spectrum. The Renishaw data visualization software was then utilized to generate false colour maps in which spectra exhibiting a sufficient intensity of a given reference pattern are coloured accordingly, as detailed below.
To resolve conflicting literature reports on the association of Raman patterns with specific copper vanadate polytypes, the vibrational modes of several copper vanadate phases were calculated using VASP55 and the script vasp_raman.py.56 Further details on these calculations and their implications for phase identification are provided in the ESI.†
UV-vis spectroscopy
Ultraviolet-visible (UV-vis) absorption spectroscopy was performed using a custom scanning spectroscopy instrument described in detail previously.57 Briefly, the dual integrating sphere system measured both the fractional transmittance (T) and reflectance (R) of each sample using illumination from a 200 W (Hg)Xe broadband source (Newport/Oriel Apex) and Spectral Products, Inc. model SM303 spectrometers. These signals were used to calculate the spectral absorption coefficient (α) up to a factor of film thickness (τ): α × τ = −ln[T × (1 − R)−1].Photoelectrochemistry
High throughput photoelectrochemical (PEC) measurements were used to measure the photocurrent from each sample under a variety of conditions. Typically 10 copies of the 26-sample composition library were deposited onto a single plate to enable a variety of experimental testing conditions. While the samples used for SEM and Raman characterization were ordered systematically with respect to composition, samples prepared for PEC measurements were printed with a randomized order so that any spatial or temporal artefacts in the PEC measurements along the line of samples were not convoluted with composition. For each type of PEC measurement, 3 copies of the 26-sample library were measured, each with a unique randomization. Manual inspection of the data revealed occasional outliers with poor PEC performance, which as noted in the ESI† was due to defective sample synthesis. To apply consistent data processing to each PEC measurement and each composition sample, the 3 values of the photocurrent from a PEC measurement of a given library composition were processed as follows: the lowest value was disregarded and the remaining 2 values were averaged.The PEC experiments to measure OER photoelectrochemical activity were performed using our previously reported scanning droplet cell (SDC) instrumentation with a Gamry G 300 potentiostat controlled by custom automation software.58 The SDC enabled the formation of a 3-electrode cell for each sample, including a Pt wire counter electrode and a custom Ag/AgCl reference electrode that was calibrated using commercial electrodes. Working electrode contact to the Cu1−xVxOz film was made by adhering Cu tape to the FTO conducting layer along the perimeter of the library plate. A 0.4 mm-diameter optical fiber (Ocean Optics P400-1-UV-VIS) was placed in the central axis of the SDC and terminated approximately 1 mm from the sample, providing a circular illumination footprint with 1.5 mm diameter that encircled the measurement sample. The SDC was operated as a flow cell in which an aqueous electrolyte flowed at approximately 10 μL s−1, flushing the active drop volume approximately 10 times per second. It is worth noting that time-resolved measurements under this rapid flow create an accelerated assessment of material stability because the concentration of dissolved metal species in the analyte cannot increase over time.
Experiments were performed with one of the following aqueous electrolytes, which were continuously bubbled with 1 atm O2 for at least 30 minutes prior to measurements and for the entire duration of the measurements: 0.1 M sodium hydroxide with 0.5 M sodium sulfate as a supporting electrolyte (pH 13), 0.1 M boric acid with approximately 0.05 M potassium hydroxide as both the supporting electrolyte and pH buffer (pH adjusted to 9.2, referred to as “pH 9”), and 0.5 M each of potassium phosphate dibasic and potassium phosphate monobasic as both the supporting electrolyte and pH buffer (pH 7). In each of these electrolytes, the anodic photocurrent was measured with an applied bias of 1.23 V vs. RHE, the Nernstian potential of the OER, and the photocurrent measurements in these 3 electrolytes are referred to as OER-13, OER-9, and OER-7, respectively.
To identify photoactive materials which may suffer from poor OER catalysis or insufficiently energetic holes, additional measurements were performed in the presence of a [Fe(CN)6]3−/4− redox couple by dissolving 50 mM each of potassium ferricyanide and potassium ferrocyanide in the respective aqueous electrolyte. It is worth noting that these salts may chemically interact with the electrode surface, and we assert that such interactions can only create a false negative result in the photocurrent measurement since any surface contamination is unlikely to enhance the photoresponse of the composition library samples. Measurements performed with this redox couple are referred to collectively as FCN and as FCN-13, FCN-9, and FCN-7 for pH 13, pH 9.2, and pH 7 electrolytes, respectively. These measurements were performed in a different configuration of the SDC in which separate Pt wires were used as reference and counter electrodes. Due to the high optical absorption of the redox couple, these experiments were performed by applying illumination from the back-side of the library plate with an illumination footprint that is approximately 1.8 mm in diameter, covering the nominal 1 mm × 1 mm sample area.
While a standard illumination spectrum, such as AM 1.5, would provide the most technologically pertinent characterization, the materials discovery experiments were performed using an alternate illumination scheme. To provide sample illumination energy for any light absorber of interest, experiments were performed with a 385 nm light emitting diode (LED, Thor Labs, M385F1, 0.7 A current limit) unless otherwise noted. The illumination power was measured by placing the operational SDC on a quartz plate and collecting the transmitted light with an optical meter (Newport Corporation, 1918-R) configured with a silicon-UV enhanced photodetector (Newport Corporation, 818-UV). The illumination power was 3.0 mW for the OER-13 and OER-9 measurements and 2.8 mW for the OER-7 measurement. For all measurements done with the [Fe(CN)6]3−/4− redox couple and backside illumination, the illumination power was 3.7 mW.
The short circuit photocurrent density J was calculated from toggled-illumination chronoamperometry (CA) measurements and the nominal 1 mm2 sample area since the illumination footprint enclosed the sample. Since the metal oxide material is typically within the 1 mm2 intended sample area and does not conformally coat the substrate, each J value is an underestimate of the geometric current density. For each sample, the SDC was positioned above the sample and the OER short circuit potential (1.23 V vs. RHE) was applied; data were not acquired for the initial 1 s of polarization to avoid any capacitive transient. The illumination was then toggled at 1 Hz (approximate illumination duration of 0.4 s for OER measurements and 0.5 s for FCN measurements). Each photocurrent measurement proceeded for 44 s and the photocurrent densities after 4 s (J4) and 44 s (J44) of illumination toggling were calculated. To avoid the initial transients upon toggling illumination in the photocurrent calculations, and to account for any non-zero dark current, the average current from a portion of the illuminated period was subtracted by the average current from a portion of the previous dark period, as illustrated below. Additional cyclic voltammetry (CV, 0.01 V s−1, illumination toggling with 2 s on and 1 s off) and 30 minute CA (at 1.23 V vs. RHE, illumination toggling with 10 s on and 2.5 s off) measurements were performed using the OER-9 electrolyte on representative samples.
Select compositions from initial experiments were chosen for illumination with additional wavelengths to measure the external quantum efficiency (EQE) as a function of incident photon energy, which is also referred to as the incident photon-to-current efficiency (IPCE). CA measurements at the OER short circuit potential were performed in either pH 13 or pH 7 electrolyte with four different LED illumination sources (Thor Labs M-series): 385 nm (3.22 eV), 455 nm (2.72 eV), 530 nm (2.34 eV), and 617 nm (2.01 eV). EQE was calculated as
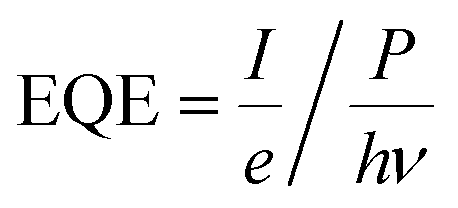
Results and discussion
Chemical and structural characterization
EDS measurements of the as-calcined library samples indicated that the Cu:V stoichiometry is within the measurement uncertainty of the intended library compositions. We refer to the samples by the intended V concentration of the library samples, x in Cu1−xVxOz, which varies from 0 to 1 in intervals of 0.04. While the oxygen stoichiometry (z) was not measured, we expect that it is approximately 1 + 1.5x.
SEM imaging revealed that each library sample is a discontinuous thin film and the morphology is composition-dependent. Representative images are shown in Fig. 1 where the Cu-rich sample (x = 0.16) appears to be a densely packed agglomeration of particles with characteristic feature sizes in the 50–200 nm range. The V-rich sample (x = 0.92) contains rod-shaped features that are typical of V2O5-containing materials. The intermediate compositions all exhibited features similar to those seen in the images for the x = 0.32 and x = 0.72 samples. These samples are composed of discontinuous mounds of materials with the characteristic diameter between 50 and 700 nm. Upon RTP processing, the morphology is mostly unchanged, with the image of the x = 0.72 sample appearing very similar to the as-calcined version and the x = 0.32 sample containing the same morphology with some additional unidentified particles that are likely contamination.
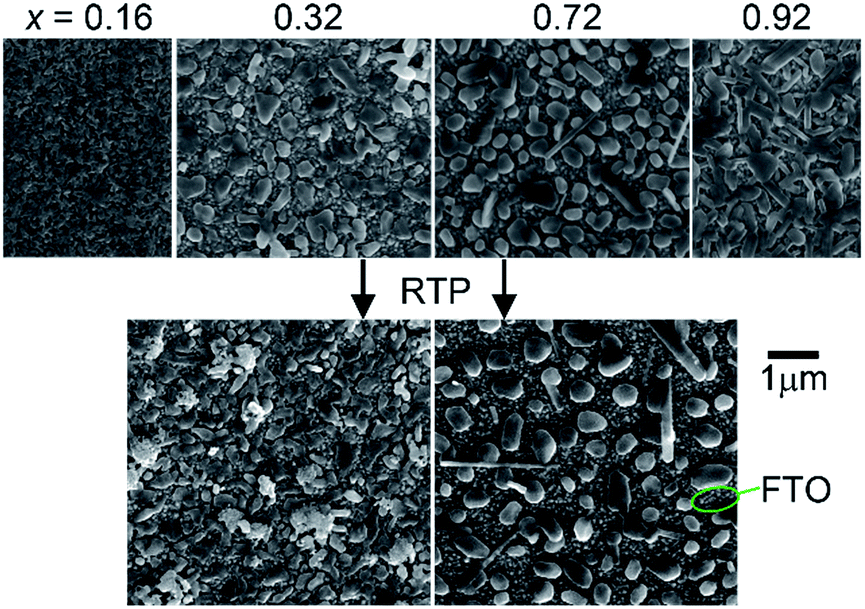 | ||
| Fig. 1 SEM images of the representative as-calcined (top) and post-RTP (bottom) composition samples. Each sample is discontinuous and a bare FTO region is noted. | ||
Despite substantial effort to perform X-ray diffraction (XRD)-based structural characterization of the as-calcined and post-RTP libraries, we were unable to obtain an interpretable scattering signal from the Cu1−xVxOz samples, particularly due to the large signal from the underlying FTO thin film. While a grazing incidence XRD technique would provide a relatively larger signal from the Cu1−xVxOz thin films, we obtained this surface sensitivity through a novel application of Raman microscopy. By employing a 50× objective in the Raman microscope, the depth of focus was limited to approximately 2 μm such that the metal oxide sample constituted a substantial fraction of the measurement volume. Since the concomitantly small sampling area provided by the high magnification lens can only characterize a small portion of a 1 mm2 sample in a single-point Raman measurement, the StreamLine Raman imaging capability was employed to rapidly acquire 47360 Raman spectra with 50 μm steps to cover the 2 × 59.2 mm substrate area containing a 26-sample Cu1−xVxOz library.
Through manual comparison with the literature and database patterns, we identified 6 patterns corresponding to Cu1−xVxOz phases,‡ which are shown in Fig. 2. The following patterns were extracted from published figures or the RRUFF database: CuO,59 Cu5V2O10,60 β-Cu2V2O7,61 α-Cu2V2O7,60 α-CuV2O6,62 and V2O5.63 Raman imaging was performed on both an as-calcined and a post-RTP library, and the same set of phases was identified in both libraries. To illustrate the excellent match between the Raman spectra and reference patterns, a single representative Raman spectrum (1 of the 47360) of each of the 6 phases is shown in Fig. 2 for both the as-calcined and post-RTP datasets. A representative signal from the FTO/glass substrate is also shown.
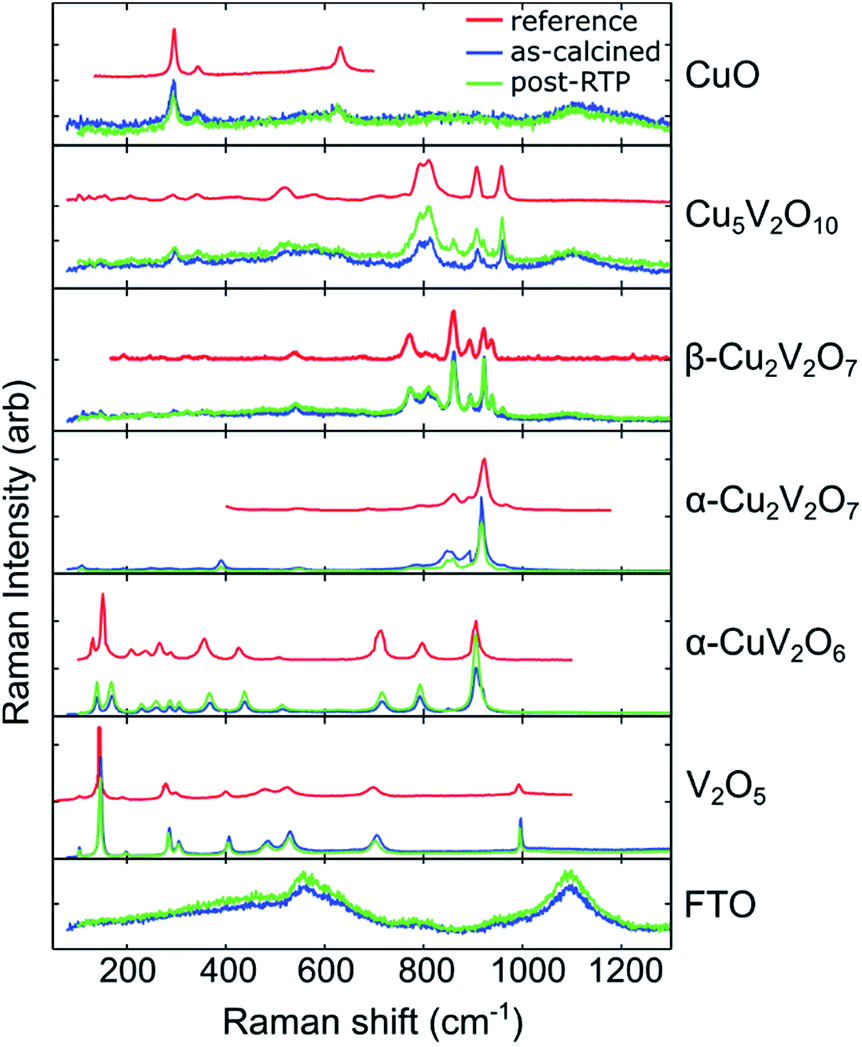 | ||
| Fig. 2 Raman spectra identifying the presence of 6 phases in the composition libraries. Reference patterns of each phase are shown in red along with a representative Raman measurement from the as-calcined library (blue) and post-RTP library (green). An example pattern from the bare substrate is shown in the bottom panel. | ||
To visualize the intensity of each of these reference patterns in the composition library, a colour was assigned to each phase and the results of DCLS fitting routing were used to produce a threshold image for each phase. Raman spectra with sufficient intensity of a given reference pattern were coloured accordingly, producing a series of 6 false colour images that indicate the spatial distribution of each of the 6 phases. These phase distribution maps and the analogous map of the substrate spectrum are shown in Fig. 3 for the post-RTP library. The results of the as-calcined library are similar and are shown in the ESI.†
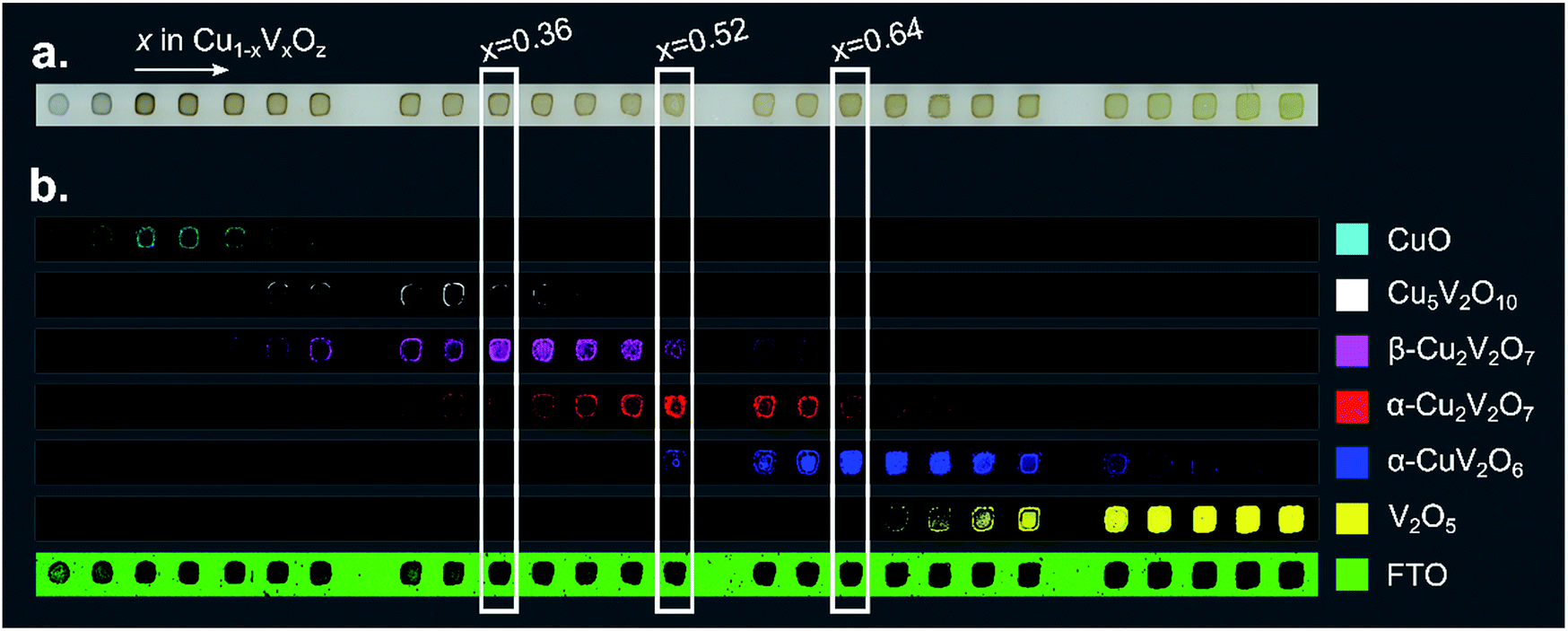 | ||
| Fig. 3 (a) An image of a 26-sample post-RTP composition library. (b) Each of the 7 false color images is 40 × 1184 pixels where each pixel corresponds to a Raman measurement and the pixel is colored according to the phase-color legend if the respective reference pattern from Fig. 2 was identified in the Raman measurement. These images provide a map of the phase distribution, which can be directly compared to the image in part (a). Representative composition samples of β-Cu2V2O7, α-Cu2V2O7, and α-CuV2O6 are denoted with white rectangles. | ||
The basis spectrum corresponding to the FTO/glass substrate (green) is only prominent where no Cu1−xVxOz sample was printed, and each of the 6 basis spectra corresponding to Cu1−xVxOz phases is observed over a range of composition samples.
An important observation from the Raman imaging results is that each composition sample exhibits non-uniformity, most notably in that the edge of the sample exhibits stronger Raman intensity than the centre, which is commensurate with visual inspection of the sample images (Fig. 3a). The dark outline of each sample corresponds to enhanced precipitation of the dissolved salts near the sample perimeter, a phenomenon commonly referred to as the coffee-ring effect. After thermal processing, the metal oxide loading is higher along the sample perimeter, leading to increased optical density and a larger Raman signal.
The intensity of the Raman signal generally increases with x (V concentration), and in particular the V2O5 phase is identified throughout the x = 1 sample area but the CuO phase is identified in a subset of the x = 0 sample area, primarily due to the weak Raman scattering of CuO. The most Cu-rich copper vanadate observed is Cu5V2O10, the synthetic analogue of the Stoiberite mineral phase. The other 3 copper vanadate phases have been reported as OER photoelectrocatalysts and are the primary focus of the present work.
At a calcination temperature of 535 °C, the expected phases at x = 0.4 and x = 0.5 are γ-Cu3V2O8 and β-Cu2V2O7, respectively.19,64 The observed phase behaviour in these composition regions is notably different, with α-Cu2V2O7 (an equilibrium phase above 605 °C19,64) obtained near x = 0.5 instead of the low-temperature polymorph, yet β-Cu2V2O7 is observed in the library at Cu-rich compositions near the stoichiometry of γ-Cu3V2O8 and β-Cu3V2O8. In the ESI,† we provide detailed inspection of the Raman patterns and support the identification of β-Cu2V2O7 through comparison with calculated Raman patterns. While it is possible that a small amount of Cu3V2O8 phases may be present in the x = 0.36 composition region, the primary contribution to the Raman signal is from the β-Cu2V2O7 structure, and we refer to the off-stoichiometric β-Cu2V2O7 at Cu-rich compositions as β-Cu2V2O7*. We note the distinct possibility that the Cu-rich samples exhibiting this phase contain stoichiometric β-Cu2V2O7 with the excess Cu forming a material that is not detectable in the Raman measurements. The detailed structure of these complex metal oxides, the possibility of forming an off-stoichiometric β-Cu2V2O7 structure, and the role of kinetic processes during calcination of mixed elemental precursors are important research areas for further development of these photoanode materials.
The results of Fig. 3 suggest that only the end-member Cu1−xVxOz samples (x = 0 and x = 1) are phase-pure. While the reference patterns contain overlapping features that can lead to false detection of a given phase in a Raman spectrum in the DCLS analysis, we expect that most of the composition samples do contain multiple phases. Still, representative samples can be chosen for each phase where the Raman intensity of the corresponding basis pattern is largest. In particular, representative samples of β-Cu2V2O7*, α-Cu2V2O7, and α-CuV2O6 are marked in Fig. 3 at compositions x = 0.36, 0.52, and 0.64, respectively. The phase and composition labels from these samples are noted in the following figures to indicate structure–property relationships.
Optical absorption
Transmission and reflection spectra were measured for each composition sample in the as-calcined and post-RTP libraries, providing composition maps of the spectral absorption as summarized in Fig. 4. The UV-vis absorption spectra are represented as the absorption coefficient up to a factor of the film thickness (α × τ) since the film thickness was not explicitly measured and is not well defined given the discontinuous morphology (see Fig. 1). Using the nominal thickness of 100 nm, α is of the order 105 cm−1 at 3 eV for each of the representative composition samples, in agreement with recently reported absorption of copper vanadates.8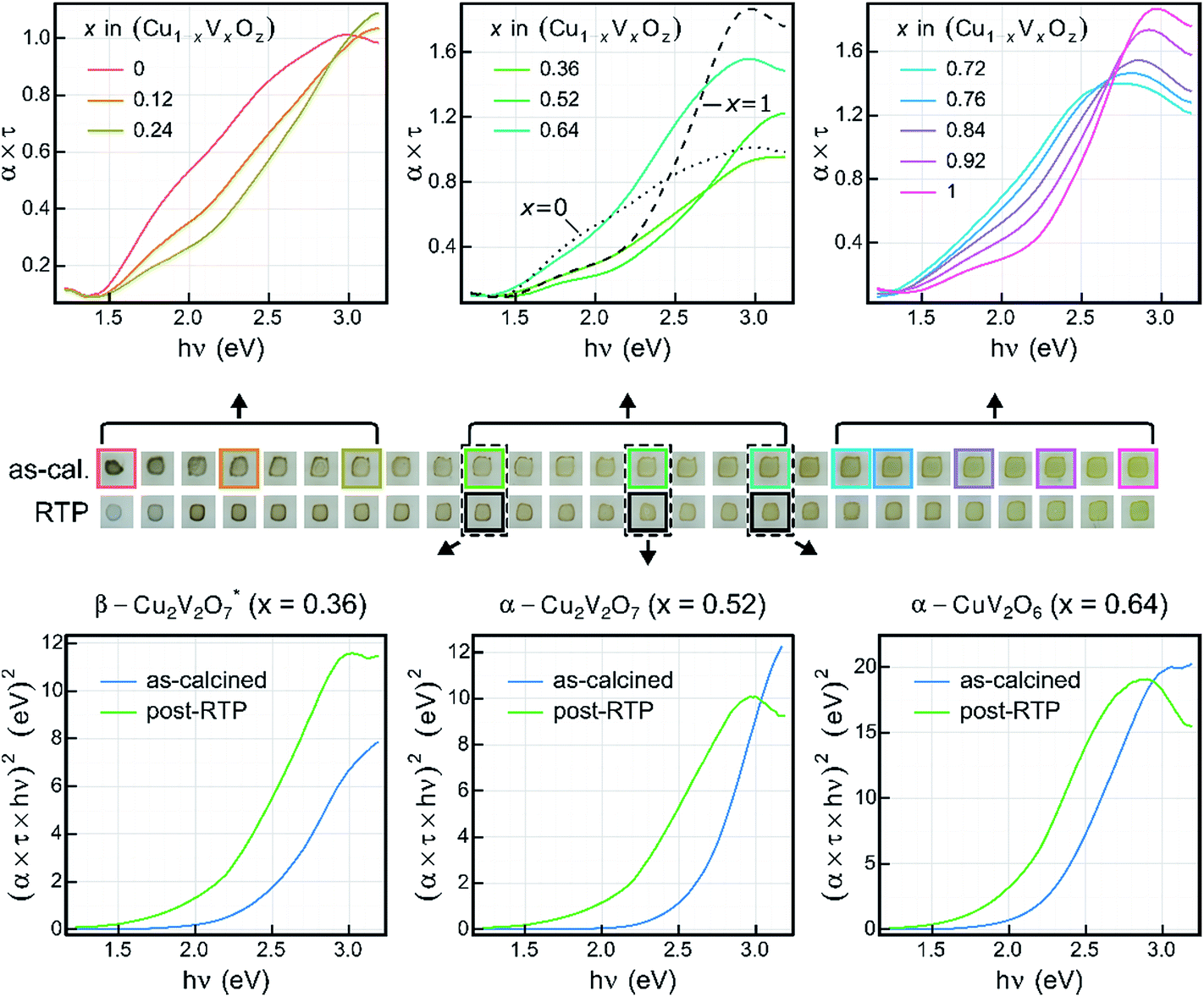 | ||
| Fig. 4 Optical absorption of Cu1−xVxOz samples from UV-vis spectroscopy. The sample images (centre row) show the visual appearance of the samples and provide a guide to the optical spectra figures. The absorption spectra of the select as-calcined samples (top) are shown in a series of the panels. For the 3 representative samples of copper vanadate phases, the direct-allowed Tauc transformation (bottom, blue) of the absorption spectra is shown. As evident in the sample images, many post-RTP samples exhibited higher absorption, as indicated by the Tauc spectra of the representative samples (bottom, green). | ||
To visualize the composition trends in spectral absorption, representative Cu-rich, intermediate, and V-rich samples are shown in a series of 3 panels. Starting from the CuO sample (x = 0), addition of V results in blue-shifting of the absorption. Starting from the V2O5 sample (x = 1), addition of Cu results in red-shifting of the absorption in the visible and near-visible region with lowered absorption in the UV region. These monotonic trends with respect to composition do not continue in the x = 0.28 to 0.68 composition region. As shown in Fig. 3, the phase behaviour changes rapidly in this composition region and the spectra of the representative composition samples for β-Cu2V2O7*, α-Cu2V2O7, and α-CuV2O6 are shown in the top-centre panel, with α-CuV2O6 exhibiting the highest absorption. The direct-allowed Tauc spectra of these 3 samples are shown in the bottom panels of Fig. 4 (blue traces) and indicate that the direct-allowed optical transition in these samples is obscured by substantial absorption at lower photon energies. This behaviour is expected given that these phases are indirect-gap semiconductors, although the indirect-allowed Tauc spectra (see ESI†) do not consistently demonstrate the presence of a near-2 eV band gap.
The direct-allowed Tauc plots of these 3 compositions also include the analogous data of samples from the post-RTP library (green traces). These are not the same samples but a library that was prepared as a duplicate of the as-calcined library with additional RTP thermal processing. The post-RTP samples appear less absorbing than the as-calcined samples in the extreme Cu-rich portion of the library, but for the representative composition samples of β-Cu2V2O7*, α-Cu2V2O7, and α-CuV2O6, the post-RTP samples appear slightly darker, which is evident in the red-shifting of the Tauc spectra. Since the Raman-determined phase behaviour of the 2 libraries is indistinguishable, the considerable difference in optical absorption is somewhat surprising and is similar to that observed in previous studies where highly absorbing, oxygen-deficient metal oxides were prepared with the same crystal structure as that of more transparent, stoichiometric oxides.65,66 The direct-allowed Tauc plots of the post-RTP samples have a more readily identifiable linear region that would suggest a direct band gap near 2 eV for each of the 3 phases, motivating further investigation of defect-mediated alterations to the electronic structure of copper vanadates. For present purposes, we note that the optical absorption of these copper vanadates is sensitive to thermal processing and that the substantial absorption in the 2 to 3 eV energy range confirms the potential of these materials as solar photoanodes.
Photoelectrochemistry
Photocurrent measurements were conducted using two different SDC configurations, schematically depicted in Fig. 5, which also shows representative data for an OER and FCN experiment. The figure also highlights data points used in the photocurrent density calculation. The OER experiments were performed with an applied bias of 1.23 V vs. RHE, where the absence of anodic current in the dark provides an indication that the material does not electrochemically corrode in the given electrolyte. As evident in Fig. 5, the dark current is near zero for these 2 representative experiments, and indeed no appreciable dark current was observed for any of the composition samples in any of the electrolytes.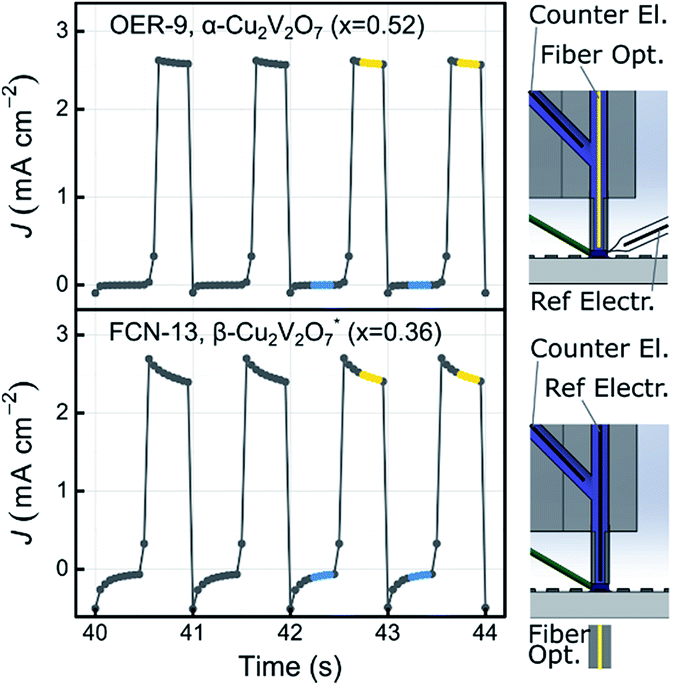 | ||
| Fig. 5 SDC measurements of the photocurrent for the OER-9 measurement at 1.23 V vs. RHE (top) and the FCN-13 measurement at 0 V vs. [Fe(CN)6]3−/4− (bottom). The dark current is near zero and for photocurrent density calculations J44 is measured as the average of the points highlighted in blue. The illuminated current is measured as the average of the points highlighted in yellow. The configuration of the SDC is illustrated on the right hand side for each measurement using a cross-section view in which a single sample is contacted with an electrolyte drop (dark blue) that is continually flushed through suction tubes (green). The electrolyte within the body of the cell is shown as light blue and the fiber optic for top-side illumination (OER measurements) and bottom-side illumination (FCN measurements) is shown in yellow. | ||
For the purpose of high throughput screening, the photocurrent was measured under intense UV illumination, which provides large photocurrent signals in active photoanode samples and also provides an accelerated assessment of photoelectrochemical stability. The combination of intense UV illumination, 1 Hz illumination cycling, and a rapidly flowing electrolyte induces rapid degradation in the photocurrent performance of unstable materials. For example, a material whose photo-response is purely due to corrosion with an illuminated current density of 2 mA cm−2 will pass approximately 4.5 nmol of 1e charge during the 44 s toggled-illumination experiments, which would correspond to complete corrosion of the metal oxide samples (approximately 4 nmol metal per sample) via a 1e corrosion process. It is worth noting that corrosion of even a small fraction of a sample can be deleterious to its photoelectrochemical properties. Consequently, we used the performance metric J44 to identify photoanode materials that exhibit appreciable short-term stability. To identify materials that are photoactive but lack stability on this time scale, the metric J4 is also used.
Fig. 6a shows both J44 and J4 for the 26 as-calcined library compositions in the 3 different OER electrolytes. The results clearly reveal the strong photo-responses of the copper vanadates, particularly in comparison to the binary oxides CuO and V2O5. In the OER-13 electrolyte, all samples containing either, α-Cu2V2O7 or α-CuV2O6 exhibit anodic photocurrent density, with the representative sample of the α-CuV2O6 phase exhibiting the highest J4 value. While some photocurrent density is still evident after 44 s of operation, the J44 values are more than 10 times lower than the J4 values, indicating that these photoanode materials are not stable in this strong alkaline environment, which has been shown for PVD-synthesized α-Cu2V2O7 and is hereby demonstrated for α-CuV2O6.8 The composition trends in the OER-7 electrolyte are quite similar, with lower J4 values and a smaller level of degradation over 44 s, which is commensurate with both lower OER kinetics at neutral pH and slower photo-corrosion processes.
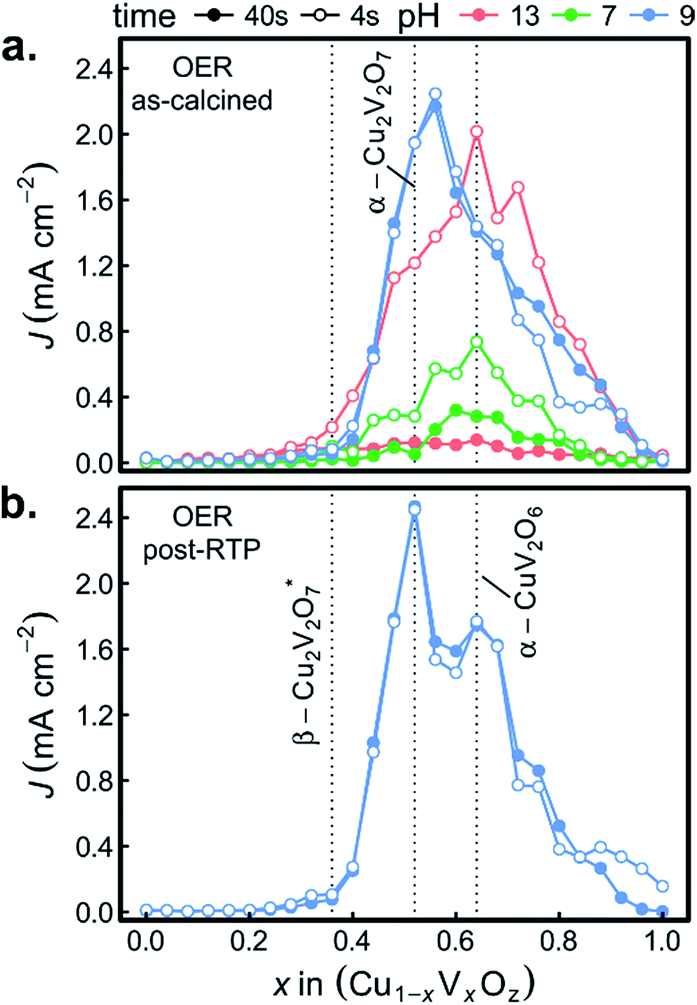 | ||
| Fig. 6 The photocurrent from OER measurements (1.23 V vs. RHE) is shown for 3 different electrolytes with the as-calcined library (a) and for the pH 9 electrolyte with the post-RTP library (b). For each experiment, the photocurrent after 4 s and 44 s of illumination is shown to assess initial transients in the photo-response of each composition sample. Representative composition samples of β-Cu2V2O7, α-Cu2V2O7, and α-CuV2O6 are indicated by vertical dashed lines. | ||
Substantially different behaviour is observed in the OER-9 electrolyte where the photocurrent density of α-Cu2V2O7 exceeds that of α-CuV2O6, and at both of these representative compositions, the photocurrent density does not degrade over the 44 s measurement. The stable OER photoelectrocatalysis in this electrolyte is in agreement with previous reports,8,13 and these measurements were repeated on a post-RTP library, as shown in Fig. 6b. The J4 and J44 signals are nearly identical and exhibit clear local maxima at the representative compositions for α-Cu2V2O7 and α-CuV2O6. In both the as-calcined and post-RTP libraries, samples containing an appreciable amount of either of these phases exhibit OER-9 photoactivity. The crystallinity is likely improved in the post-RTP library, providing a more pronounced relationship between the structure and photocurrent. The results indicate that only marginal improvements in photoelectrochemical performance are attained through RTP processing of these phases and that α-Cu2V2O7 is slightly more photoactive than α-CuV2O6 under these conditions.
To confirm the activity and stability of these 2 photoelectrocatalyst phases, the representative samples from the RTP library (x = 0.52 and 0.64 in Fig. 6b) were further characterized with both CV and 30 minute CA measurements in the OER-9 electrolyte. The results are shown in Fig. 7 and reveal several important properties of these photoelectrocatalysts: α-Cu2V2O7 exhibits a higher photocurrent than α-CuV2O6 over the full potential range; the illuminated open circuit is between 0.8 and 0.9 V vs. RHE for both photoelectrocatalysts; the consistent dark current near 0 demonstrates excellent electrochemical stability; both photoelectrocatalysts suffer some degradation over the first several of the 144 illumination cycles; from 10 to 30 minutes of illumination cycling, the photoelectrocatalysts exhibit near-perfect stability. The CA measurements were performed at 1.23 V vs. RHE and the initial values appear to be slightly smaller than the corresponding values from Fig. 6b, which may be due in part to the lower illumination toggling frequency, although it is worth noting that after the measurements of Fig. 6b the electrolyte-wetted samples were left to dry and then stored in air for 4 months before the CV and 30 minute CA experiments were performed, with the only sample pre-treatment being approximately 10 s of exposure to the electrolyte before data acquisition. The use of intense UV illumination provides an accelerated measurement of stability, and the excellent stability after exposure to both ambient conditions and prolonged illumination toggling highlights the potential of these photoelectrocatalysts as durable solar fuel photoanodes.
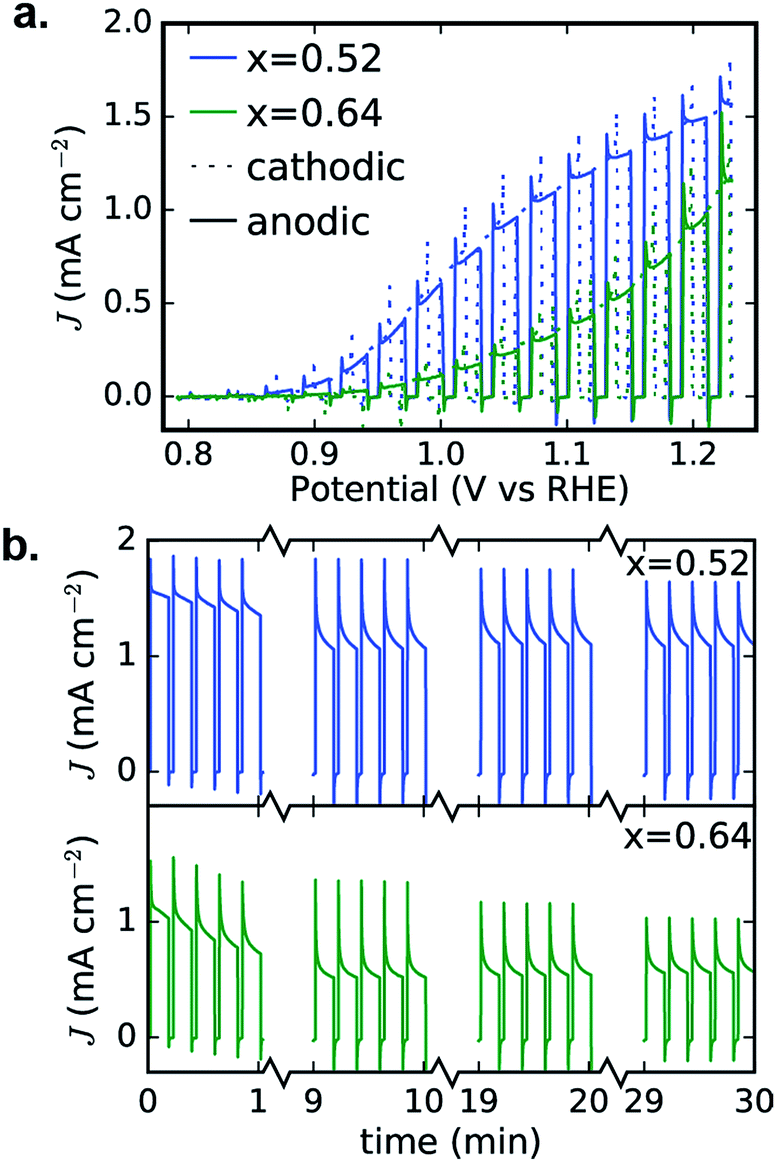 | ||
| Fig. 7 Detailed PEC characterization of α-Cu2V2O7 (x = 0.52) and α-CuV2O6 (x = 0.64) from Fig. 6b. The 0.01 V s−1 CVs (a) and 30 minute CA (b) measurements exhibit excellent performance in the OER-9 electrolyte. The cathodic and anodic sweeps of each CV are nearly identical, demonstrating stability during this voltage cycle, and the 30 minute toggled illumination data are shown as 4 sets of 5 illumination cycles from different points in the 30 minute measurement, demonstrating that both photoelectrocatalysts suffer some photocurrent loss during the first few minutes and then remain quite stable during the remainder of the experiment. | ||
The combinatorial PEC measurements were also performed on the as-calcined samples in the presence of the [Fe(CN)6]3−/4− redox couple, as shown in Fig. 8a. The use of a bare Pt reference electrode and 0 V bias correspond to well-poised solution potentials of 1.13, 0.90, and 0.77 V vs. RHE at pH 13, pH 9.2, and pH 7, respectively. The results in the FCN-13 electrolyte are of particular interest due to the similarity of the redox couple potential to the Nernstian potential for the OER, as previously discussed.7,67 For the samples containing α-Cu2V2O7 or α-CuV2O6, the composition trends in J4 and J44 are quite similar to those of Fig. 6a.
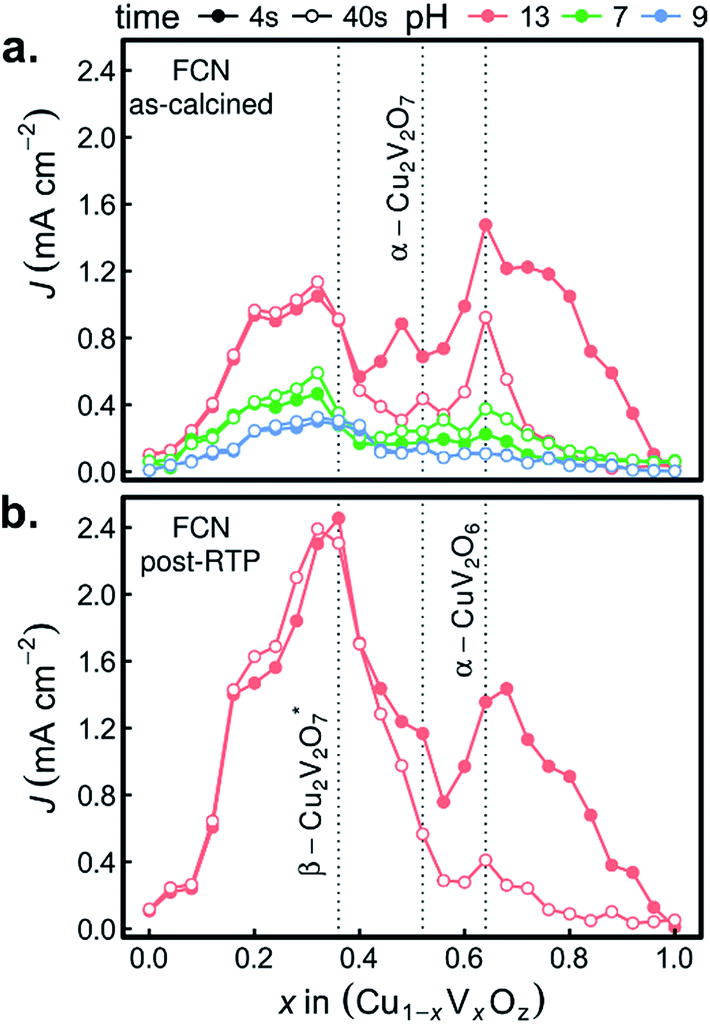 | ||
| Fig. 8 The photocurrent from FCN measurements (0 V vs. [Fe(CN)6]3−/4−) is shown for 3 different electrolytes with the as-calcined library (a) and for the pH 13 electrolyte with the post-RTP library (b). For each experiment, the photocurrent after 4 s and 44 s of illumination is shown to assess initial transients in the photo-response of each composition sample. Representative composition samples of β-Cu2V2O7, α-Cu2V2O7, and α-CuV2O6 are indicated by vertical dashed lines. | ||
The remarkable result is the emergence of the Cu-rich samples, and in particular the representative composition for the β-Cu2V2O7* phase, as the top PEC performer in each of the FCN electrolytes. In addition, the high photocurrent density is retained over the 44 s measurement, even in the pH 13 electrolyte. The observation of lower but finite photocurrent density in the lower pH electrolytes leads to several conjectures concerning the properties of the β-Cu2V2O7* phase. The anodic photocurrent at 0.77 V vs. RHE for FCN-7 indicates that the flat band potential of the semiconductor lies below that value, which is commensurate with results for other copper vanadates in Fig. 7a and in previous reports using both experimental and theoretical methods.8,12,13 In the FCN-13 electrolyte, the solution potential of 1.13 V vs. RHE corresponds to a larger difference from the flat band potential and hence the propensity for higher band banding and increased charge extraction from the photoanode, which is supported by the observation of higher photocurrent density. Since the oxidation of the redox couple at the photoanode surface is reversible and there is an appreciable area fraction of exposed FTO (see Fig. 1), which is not photoactive, these FCN photocurrent measurements have an appreciable current shunt which, as shown in mass transport modelling,67 can lead to this measurement substantially underrepresenting the true photo-activity of the material. The lack of photocurrent density in the OER measurements despite the excellent photocurrent in FCN measurements suggests that the β-Cu2V2O7* materials are limited by catalysis of the OER, which could be due to a valence band energy that is near the OER Nernstian potential. In this case, a highly efficient OER photoanode may be created through coupling of this light absorber with a suitable electrocatalyst. It is worth noting that this may be an off-stoichiometric compound with trap states that create a pseudo-valence band near the OER potential with a deeper valence band from which the majority of photo-excitations occur. To differentiate these phenomena and ascertain the capacity of β-Cu2V2O7* to enable efficient OER photoelectrocatalysis, the electronic structure and defect chemistry of the off-stoichiometric compound must be further explored.
The FCN-13 measurement was repeated on a post-RTP library, leading to a 2-fold enhancement in photocurrent in the β-Cu2V2O7* composition region for both J4 and J44, as shown in Fig. 8b. As noted above, the post-RTP material is more absorbing over the entire UV-vis range, which may contribute to the enhanced photo-activity. We also note that the RTP processing may create oxygen deficiencies, which in wider-band-gap metal oxides have been shown to improve photoanode performance due to a variety of factors including enhancement of charge transport.65,66
We recently reported that no copper vanadate phases are thermodynamically stable at any pH and potential but that self-passivation in the OER-9 electrolyte can provide long-term operational stability.68 The results of Fig. 7b indicate that indeed these inkjet-printed materials self-passivate and remain photoactive in the OER-9 electrolyte. The stability of β-Cu2V2O7* in the FCN-13 electrolyte suggests that this phase may also self-passivate in the pH 13 electrolyte, and given that the passivation mechanisms involve the formation of a Cu-rich surface layer,68 the Cu-rich off-stoichiometry of the inkjet-printed β-Cu2V2O7* may be critical for enabling pH 13 operation.
The UV-illumination experiments in Fig. 6–8 provide a wealth of information on the photoelectrochemical properties of these copper vanadates, and the most promising combinations of sample compositions and electrolytes were further investigated at lower photon energy. By repeating the 44 s chopped-illumination experiments using a series of 4 LEDs, the spectral EQE values of the representative α-Cu2V2O7 composition in the OER-9 experiment and the representative β-Cu2V2O7* composition in the FCN-13 experiment were measured, as shown in Fig. 9a (both post-RTP samples). By additionally scaling by the fraction of light absorbed by the materials at the respective photon energies, which is notably an ex situ measurement, the IQE was also determined, as shown in Fig. 9b. The EQE and IQE of β-Cu2V2O7* have not been reported and are among the best for metal oxide photoanodes in the pH 13 electrolyte. The EQE spectrum of α-Cu2V2O7 has a similar dependence on photon energy as that previously reported with a 1 μm-thick film in the same electrolyte at 1.23 V vs. RHE,13 with our material exhibiting approximately twice the EQE efficiency. Given the approximately 10-fold lower thickness of our inkjet-printed film, the IQE is likely much higher, and additional improvements may result by optimizing the thickness of each copper vanadate photoanode. 10% efficiency at 3.2 eV with finite efficiency down to 2 eV, combined with the noted underestimation of the efficiency due to the current shunt, demonstrates the potential of this material to enable efficient photoelectrocatalysis of the OER using solar radiation.
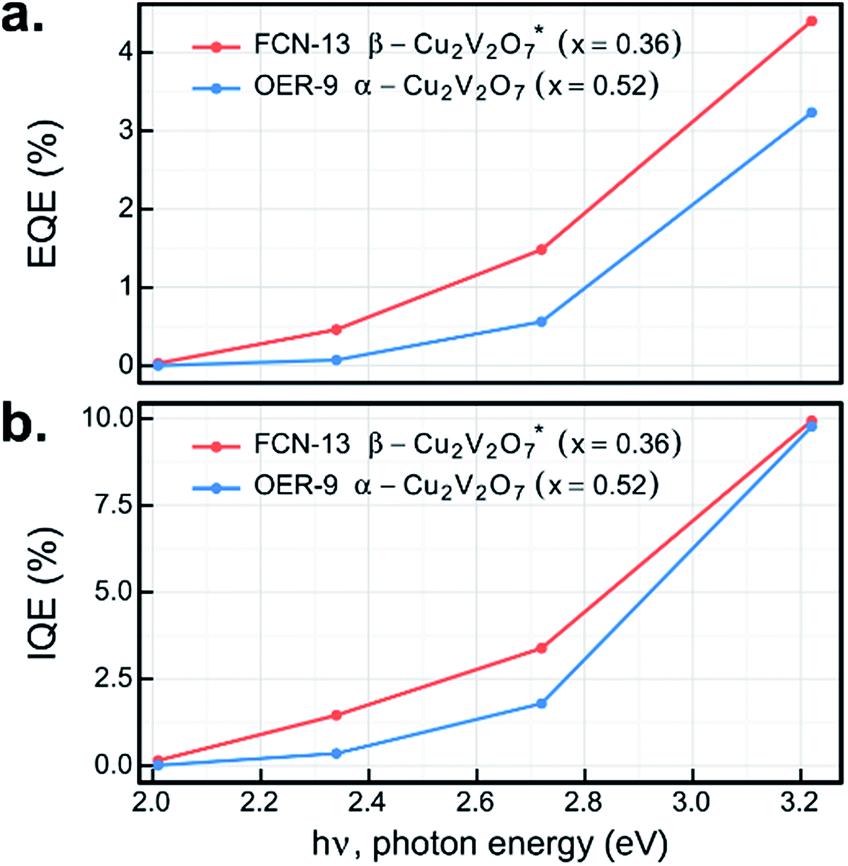 | ||
| Fig. 9 The external (a) and internal (b) quantum efficiencies at 4 photon energies are shown using the J44 SDC measurements for select post-RTP samples. The representative β-Cu2V2O7 sample is shown in the FCN-13 electrolyte and the representative α-Cu2V2O7 sample is shown in the OER-9 electrolyte. The vanishing QE and IQE values near 2 eV are in agreement with previous reports of an indirect band gap at this energy. | ||
Conclusions
The copper vanadates Cu5V2O10, β-Cu2V2O7, α-Cu2V2O7, and α-CuV2O6 were synthesized via inkjet printing of precursor salts, a scalable solution processing method for synthesis of functional metal oxides. The development of a novel Raman imaging technique with associated data processing and visualization enabled phase mapping of the composition library, leading to the establishment of structure–property relationships with respect to optical absorption (using UV-vis spectroscopy) and photoelectrocatalysis (using high throughput electrochemistry). By evaluating the photoelectroactivity of the 26-sample composition library in 3 different electrolytes and both with and without the addition of a facile redox couple, β-Cu2V2O7, α-Cu2V2O7, and α-CuV2O6 were identified as promising photoanodes for solar fuel applications. Both α-Cu2V2O7 and α-CuV2O6 are highly stable and active for photoelectrocatalysis of the OER in a borate-buffered pH 9.2 electrolyte. While the β-Cu2V2O7 phase obtained at Cu-rich compositions does not show direct OER activity, it is highly photoactive in the presence of [Fe(CN)6]3−/4− and is the only photoelectrocatalyst to exhibit excellent stability in the pH 13 electrolyte. The activity of this phase is greatly enhanced by rapid thermal processing, creating numerous opportunities for enabling efficient solar fuel generation using metal oxide photoanodes.Acknowledgements
This material is based upon work performed by the Joint Center for Artificial Photosynthesis, a DOE Energy Innovation Hub, supported through the Office of Science of the U.S. Department of Energy under Award Number DE-SC0004993. The authors thank Natalie Becerra-Stasiewicz for assistance with acquisition of the SEM images, Santosh Suram for assistance with UV-vis data, Joel Haber for helpful discussion and Florian Brown-Altvater for assistance with the Raman calculations.Notes and references
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- H.-J. Lewerenz and L. Peter, Photoelectrochemical Water Splitting, The Royal Society of Chemistry, 2013 Search PubMed.
- S. Hu, C. Xiang, S. Haussener, A. D. Berger and N. S. Lewis, Energy Environ. Sci., 2013, 6, 2984 CAS.
- F. E. Osterloh, Chem. Mater., 2008, 20, 35–54 CrossRef CAS.
- S. Haussener, C. Xiang, J. M. Spurgeon, S. Ardo, N. S. Lewis and A. Z. Weber, Energy Environ. Sci., 2012, 5, 9922–9935 CAS.
- A. A. Ismail and D. W. Bahnemann, Sol. Energy Mater. Sol. Cells, 2014, 128, 85–101 CrossRef CAS.
- Q. Yan, G. Li, P. F. Newhouse, J. Yu, K. A. Persson, J. M. Gregoire and J. B. Neaton, Adv. Energy Mater., 2015, 5, 1401840 Search PubMed.
- L. Zhou, Q. Yan, A. Shinde, D. Guevarra, P. F. Newhouse, N. Becerra-Stasiewicz, S. M. Chatman, J. A. Haber, J. B. Neaton and J. M. Gregoire, Adv. Energy Mater., 2015, 5, 1500968 Search PubMed.
- Y. Park, K. J. McDonald and K.-S. Choi, Chem. Soc. Rev., 2013, 42, 2321–2337 RSC.
- Z.-F. Huang, L. Pan, J.-J. Zou, X. Zhang and L. Wang, Nanoscale, 2014, 6, 14044–14063 RSC.
- J. K. Cooper, S. Gul, F. M. Toma, L. Chen, Y.-S. Liu, J. Guo, J. W. Ager, J. Yano and I. D. Sharp, J. Phys. Chem. C, 2015, 119, 2969–2974 CAS.
- J. A. Seabold and N. R. Neale, Chem. Mater., 2015, 1005–1013, DOI:10.1021/cm504327f.
- W. Guo, W. D. Chemelewski, O. Mabayoje, P. Xiao, Y. Zhang and C. B. Mullins, J. Phys. Chem. C, 2015, 119, 27220–27227 CAS.
- P. D. Robinson, J. M. Hughes and M. L. Malinconico, Am. Mineral., 1987, 72, 397–400 CAS.
- J. M. Hughes, B. S. Christian, L. W. Finger and L. L. Malinconico, J. Volcanol. Geotherm. Res., 1987, 33, 183–190 CrossRef CAS.
- J. M. Hughes, Am. Mineral., 1985, 70, 193–196 CAS.
- M. E. Zelenski, N. V. Zubkova, I. V. Pekov, M. M. Boldyreva, D. Yu. Pushcharovsky and A. N. Nekrasov, Eur. J. Mineral., 2011, 23, 475–481 CrossRef CAS.
- L. W. Finger, Am. Mineral., 1985, 70, 197–199 CAS.
- B. V. Slobodin and R. F. Samigullina, Inorg. Mater., 2010, 46, 196–200 CrossRef CAS.
- G. M. Clark and R. Garlick, J. Inorg. Nucl. Chem., 1978, 40, 1347–1349 CrossRef CAS.
- R. D. Shannon and C. Calvo, Can. J. Chem., 1972, 50, 3944 CrossRef CAS.
- P. J. Coing-Boyat, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 1546–1548 CrossRef.
- N. S. Rao and O. G. Palanna, Bull. Mater. Sci., 1993, 16, 261–266 CrossRef CAS.
- M. E. Zelenski, N. V. Zubkova, I. V. Pekov, M. M. Boldyreva, D. Yu. Pushcharovsky and A. N. Nekrasov, Eur. J. Mineral., 2011, 23, 475–481 CrossRef CAS.
- J. M. Hughes, B. S. Christian, L. W. Finger and L. L. Malinconico, J. Volcanol. Geotherm. Res., 1987, 33, 183–190 CrossRef CAS.
- M. L. Green, I. Takeuchi and J. R. Hattrick-Simpers, J. Appl. Phys., 2013, 113, 53 CrossRef.
- H. Koinuma and I. Takeuchi, Nat. Mater., 2004, 3, 429–438 CrossRef CAS PubMed.
- J. C. Zhao, Prog. Mater. Sci., 2006, 51, 557–631 CrossRef.
- S. S. Mao, Appl. Phys. A: Mater. Sci. Process., 2011, 105, 283–288 CrossRef CAS.
- T. H. Muster, A. Trinchi, T. A. Markley, D. Lau, P. Martin, A. Bradbury, A. Bendavid and S. Dligatch, Electrochim. Acta, 2011, 56, 9679–9699 CrossRef CAS.
- M. Woodhouse and B. A. Parkinson, Chem. Soc. Rev., 2009, 38, 197–210 RSC.
- J. E. Katz, T. R. Gingrich, E. A. Santori and N. S. Lewis, Energy Environ. Sci., 2009, 2, 103–112 CAS.
- S. P. Berglund, H. C. Lee, P. D. Nunez, A. J. Bard and C. B. Mullins, Phys. Chem. Chem. Phys., 2013, 15, 4554–4565 RSC.
- J. H. He and B. A. Parkinson, ACS Comb. Sci., 2011, 13, 399–404 CrossRef CAS PubMed.
- J. S. Jang, J. Lee, H. Ye, F. R. F. Fan and A. J. Bard, J. Phys. Chem. C, 2009, 113, 6719–6724 CAS.
- J. S. Jang, K. Y. Yoon, X. Y. Xiao, F. R. F. Fan and A. J. Bard, Chem. Mater., 2009, 21, 4803–4810 CrossRef CAS.
- T. F. Jaramillo, S. H. Baeck, A. Kleiman-Shwarsctein, K. S. Choi, G. D. Stucky and E. W. McFarland, J. Comb. Chem., 2005, 7, 264–271 CrossRef CAS PubMed.
- T. F. Jaramillo, S. H. Baeck, A. Kleiman-Shwarsctein and E. W. McFarland, Macromol. Rapid Commun., 2004, 25, 297–301 CrossRef CAS.
- C. P. Jiang, R. L. Wang and B. A. Parkinson, ACS Comb. Sci., 2013, 15, 639–645 CrossRef CAS PubMed.
- J. W. Lee, H. C. Ye, S. L. Pan and A. J. Bard, Anal. Chem., 2008, 80, 7445–7450 CrossRef CAS PubMed.
- G. J. Liu and A. J. Bard, J. Phys. Chem. C, 2010, 114, 17509–17513 CAS.
- M. Woodhouse, G. S. Herman and B. A. Parkinson, Chem. Mater., 2005, 17, 4318–4324 CrossRef CAS.
- M. Woodhouse and B. A. Parkinson, Chem. Mater., 2008, 20, 2495–2502 CrossRef CAS.
- H. Ye, J. Lee, J. S. Jang and A. J. Bard, J. Phys. Chem. C, 2010, 114, 13322–13328 CAS.
- J. M. Gregoire, M. E. Tague, S. Cahen, S. Khan, H. C. D. Abruña, F. J. DiSalvo and R. B. van Dover, Chem. Mater., 2009, 22, 1080–1087 CrossRef.
- H. S. Stein, R. Gutkowski, A. Siegel, W. Schuhmann and A. Ludwig, J. Mater. Chem. A, 2016, 4, 3148–3152 CAS.
- R. Meyer, K. Sliozberg, C. Khare, W. Schuhmann and A. Ludwig, ChemSusChem, 2015, 8, 1279–1285 CrossRef CAS PubMed.
- A. Lesch, F. Cortés-Salazar, V. C. Bassetto, V. Amstutz and H. H. Girault, CHIMIA International Journal for Chemistry, 2015, 69, 284–289 CrossRef CAS PubMed.
- X. Liu, T.-J. Tarn, F. Huang and J. Fan, Particuology, 2015, 19, 1–13 CrossRef CAS.
- M. Woodhouse and B. A. Parkinson, Chem. Mater., 2008, 20, 2495–2502 CrossRef CAS.
- J. A. Haber, C. Xiang, D. Guevarra, S. Jung, J. Jin and J. M. Gregoire, ChemElectroChem, 2014, 1, 524–528 CrossRef.
- J. A. Haber, D. Guevarra, S. Jung, J. Jin and J. M. Gregoire, ChemElectroChem, 2014, 1, 1613–1617 CrossRef CAS.
- J. A. Haber, Y. Cai, S. Jung, C. Xiang, S. Mitrovic, J. Jin, A. T. Bell and J. M. Gregoire, Energy Environ. Sci., 2014, 7, 682–688 CAS.
- D. Guevarra, A. Shinde, S. K. Suram, I. D. Sharp, F. M. Toma, J. A. Haber and J. M. Gregoire, Energy Environ. Sci., 2016, 9, 565–580 CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- A. Fonari and S. Stauffer, in vasp_raman.py, https://github.com/raman-sc/VASP/, 2013 Search PubMed.
- S. Mitrovic, E. W. Cornell, M. R. Marcin, R. J. R. Jones, P. F. Newhouse, S. K. Suram, J. Jin and J. M. Gregoire, Rev. Sci. Instrum., 2015, 86, 013904 CrossRef PubMed.
- J. M. Gregoire, C. Xiang, X. Liu, M. Marcin and J. Jin, Rev. Sci. Instrum., 2013, 84, 024102 CrossRef PubMed.
- L. Debbichi, M. C. Marco de Lucas, J. F. Pierson and P. Krüger, J. Phys. Chem. C, 2012, 116, 10232–10237 CAS.
- T. Kawada, S. Hinokuma and M. Machida, Catal. Today, 2015, 242, 268–273 CrossRef CAS.
- B. Lafuente, R. T. Downs, H. Yang and N. Stone, in Highlights in Mineralogical Crystallography, ed. T. Armbruster, R. M. Danisi and W. De Gruyter, Berlin, Germany, 2015, pp. 1–30 Search PubMed.
- Y. Wei, C. W. Ryu, G. Chen and K. B. Kim, Electrochem. Solid-State Lett., 2006, 9, A487–A489 CrossRef CAS.
- X. J. Wang, H. D. Li, Y. J. Fei, X. Wang, Y. Y. Xiong, Y. X. Nie and K. A. Feng, Appl. Surf. Sci., 2001, 177, 8–14 CrossRef CAS.
- B. V. Slobodin, L. L. Surat and R. F. Samigullina, Russ. J. Inorg. Chem., 2009, 54, 857–863 CAS.
- G. Wang, Y. Ling and Y. Li, Nanoscale, 2012, 4, 6682–6691 RSC.
- G. Wang, Y. Yang, Y. Ling, H. Wang, X. Lu, Y.-C. Pu, J. Z. Zhang, Y. Tong and Y. Li, J. Mater. Chem. A, 2016, 4, 2849–2855 CAS.
- C. Xiang, J. Haber, M. Marcin, S. Mitrovic, J. Jin and J. M. Gregoire, ACS Comb. Sci., 2014, 16, 120–127 CrossRef CAS PubMed.
- L. Zhou, Q. Yan, J. Yu, R. J. R. Jones, N. Becerra-Stasiewicz, S. K. Suram, A. Shinde, D. Guevarra, J. B. Neaton, K. A. Persson and J. M. Gregoire, Phys. Chem. Chem. Phys., 2016, 18, 9349–9352 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta01252c |
| ‡ For details on the structures of the copper vanadate phase, we refer the reader to http://materialsproject.org where detailed information is available at the following material IDs: mp-559440 (Cu5V2O10), mp-559660 (β-Cu2V2O7), mp-505508 (α-Cu2V2O7), and mp-741706 (α-CuV2O6). |
| This journal is © The Royal Society of Chemistry 2016 |