DOI:
10.1039/C5RA25716F
(Paper)
RSC Adv., 2016,
6, 8829-8837
Sulfonated carbon as a new, reusable heterogeneous catalyst for one-pot synthesis of acetone soluble cellulose acetate†
Received
2nd December 2015
, Accepted 12th January 2016
First published on 15th January 2016
Abstract
A sulfonated carbon (SO3H/PhSO3H-carbon) catalyzed novel process was developed for the solvent free synthesis of acetone soluble-cellulose acetate (CA) via acetylation of cellulose with acetic anhydride. The SO3H/PhSO3H functionalized carbons easily outperformed the traditional solid acids (zeolites, heteropoly acids, Amberlyst-15 etc.) producing acetylated products with DS values between 1.6 and 2.94, in high yield (48–77% isolated yield) and under solvent free conditions, in a one-pot process. Further, it was possible to produce the commercially desired, soluble CA (DS values 2–2.7) in excellent yields (∼70%) from microcrystalline cellulose under optimized reaction conditions over the highly active mesoporous sulfonated catalyst (AC500S). A catalyst-to-cellulose (w/w) ratio of 1, acetic anhydride-to-AGU (anhydroglucose unit) mole ratio of 4.5 and reaction time of 12 h was applied. Additionally, the sulfonated catalyst could be easily recovered by centrifugal separation of the reaction mixture (diluted with acetone) and subsequently applied in the next reaction cycle with no significant reduction in yield and DS of CA over multiple reaction cycles.
1. Introduction
Cellulose acetate (CA) is one of the most commercially important cellulose derivatives with wide range of application in coatings, films, membrane separation, textile, pharmaceutical and cigarette industries. The most commonly used and industrially important CAs are the acetone-soluble diacetates (CDA) with an average degree of substitution (DS value) in the range of 2.2–2.7.1,2 Meanwhile, the less soluble cellulose triacetates (CTA), with DS values of 2.8 and above have not found a great number of commercial applications. Industrial production of CAs has been well recognized for over 100 years and has been traditionally carried out by reacting cellulose with an excess of acetic anhydride in the presence of strong mineral acids such as sulfuric or perchloric acid as the catalyst. Although mineral acids show good catalytic activity but their use also unnecessarily complicates the entire production process as product separation, product purification and generation of neutralization wastes become unavoidable which ultimately contribute to higher production costs. Moreover, due to the nature of the mineral acid catalyzed reaction it is also impossible to synthesize the partially substituted cellulose acetates directly and therefore the commercially sought-after acetone-soluble cellulose diacetates (CDA) are obtained by hydrolyzing fully substituted CTA in a multi-step process.1–3 Hence, development of a “green” approach based on recyclable strong solid acids for the one-pot synthesis of acetone-soluble CA is of great industrial importance.
Recently, acetone-soluble CA has been obtained by employing acidic ionic liquids (ILs) as catalysts.4–7 However, the process has drawbacks in terms of industrial implementation because of the often expensive nature of ILs, limited solubility of cellulose in ILs and the difficulties associated with IL recycling as well as product separation, even if they are insoluble and applied in a heterogeneous manner.7 On the other hand, despite the apparent processing advantages, the efficiency of reported solid acid catalysts in cellulose acetylation is significantly lower than that of the mineral acids and the only successful application of solid catalysts reported for cellulose acetylation include the extremely strong Brønsted acidic solid acids such as sulfated zirconia, heteropolyacids (H3PW12O40·6H2O) and Amberlyst-15.8–10 A common drawback of all these processes is, however, the low CA yield, separation of CA, use of solvents, requirement of large catalyst amounts (loading) and catalyst reusability. Overall, literature suggest the important role of strong Brønsted acidic sites (SO42−/–SO3H, having H0 less than or comparable to conc. H2SO4) in the reaction.7,8 Besides, one of the biggest difficulty with regard to cellulose conversion by chemical reactions (acetylation/hydrolysis etc.) as compared to the traditional liquid phase reactions originate from its chemical structure. Cellulose has a well packed crystalline structure resulting from the presence of strong inter and intra hydrogen bonds which make is insoluble in most of the known organic solvents and also reduce accessibility of the reactive hydroxyl groups of cellulose for chemical transformtions.2,7b,12,13
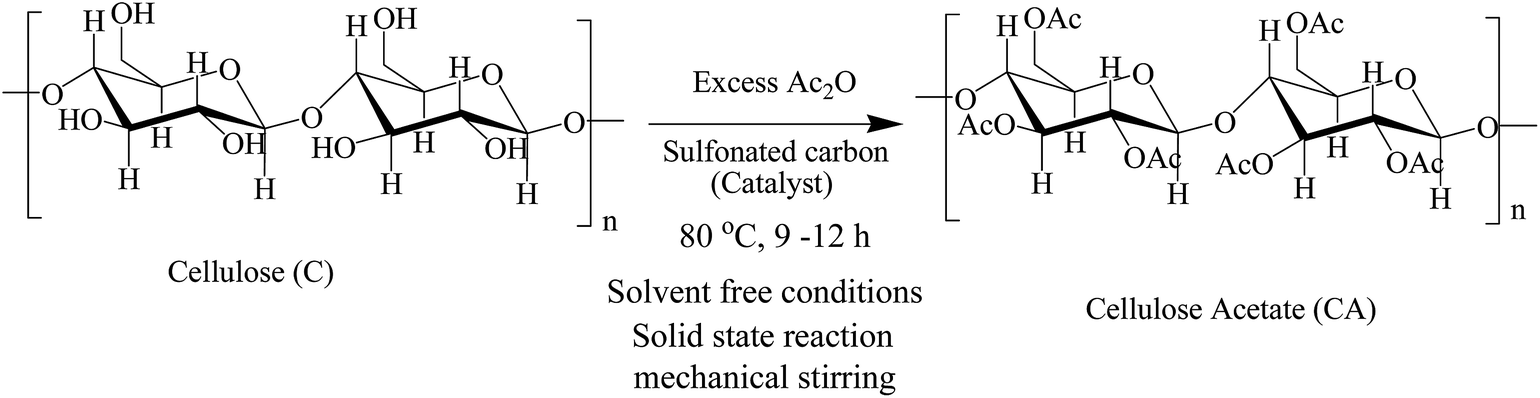
The –SO3H functionalized carbon based materials (sulfonated carbons) are relatively new addition to the family of solid protonic acids and have been successfully applied as heterogeneous catalysts in various liquid phase reactions (esterification, hydrolysis, acetylation etc.).11,12 These sulfonated materials possess all the features of an ideal solid strong protonic acid: H0 = −11, tailorable textural properties and high thermal, chemical and mechanical stability which render them an excellent substitute for liquid acid catalysts in acid catalyzed reactions.11–14 Although such materials have been successfully applied as catalyst for upon saccharification of cellulose and cellulosic materials, till date there are no earlier reports on the application of sulfonated carbons as a catalyst for CA synthesis. Herein, we describe a new process for the solventless synthesis of CA from microcrystalline cellulose over –SO3H/PhSO3H functionalized carbon based solid acids or sulfonated carbons (Scheme 1).12,13
 |
| | Scheme 1 Esterification of cellulose with acetic anhydride to cellulose acetate. | |
The present study was motivated by the recently demonstrated excellent catalytic activity of sulfonated carbons in glycerol acetylation which prompted us to further investigate the potential of sulfonated carbon materials as catalyst in the solid phase acetylation of cellulose with acetic anhydride.14 To the best of our knowledge, this is also the 1st study investigating the catalytic potential of these novel materials in solvent free acetylation of cellulose to cellulose acetates under heterogeneous conditions (Scheme 1). Some zeolites and un-modified active carbon were also investigated for the sake of comparison.
2. Experimental
2.1. Materials
Cellulose (microcrystalline, Sigma), starch (99.9%, Sigma), ortho-phosphoric acid (88%, Merck), sulfanilic acid (99%, Merck), H3PO2 (aq. 30–32%, SRL), NaNO2 (98%, Merck), NaOH (99%, Merck), H2SO4 (98%, Sigma), HCl (35%, Sigma), acetone (99.5%, Sigma), ethanol (99.9%, Sigma),CH2Cl2 (99.9%, Sigma), DMSO (99.9%, Sigma), DMSO-d6 (99.9%, Merck), oleic acid (90%, Sigma) and acetic anhydride (99.9%, Sigma) were purchased from commercial sources and used as received.
2.2. Catalyst preparation
2.2.1. Sulfonated carbons. To obtain the –SO3H acid functionalized mesoporous carbons, Pongamia glabra cake derived mesoporous active carbon (obtained by phosphoric acid activation at 500 °C) and commercial mesoporous active carbon (Sigma) was subjected to sulfonation with 4-benzenediazonium sulfonate (4-BDS) according to procedures reported in literature.13,14 Here, we opted for 4-BDS instead of H2SO4 (conc. or fuming) as a sulfonating agent as active carbons have a aromatized carbon structure with large number of graphitic sp2 sites and the former reagent is reported to be more efficient in sulfonating sp2 carbons, whereas H2SO4 is more effective with non-graphitic (sp3) carbons. Besides, the use of mild sulfonation conditions and higher stability of –PhSO3H/–SO3H sites make this process more attractive for our purpose (also we were interested in introducing –PhSO3H/–SO3H only but not –OH and –COOH which are also generated with stronger/oxidizing agents such as H2SO4).13 The non-porous sulfonated carbon was also obtained from Pongamia glabra cake according to the one-step hydrothermal method.14 The detailed synthesis procedures and characterization of each of these materials can be found in our previous communication.14The –SO3H functionalized magnetic Fe@C composite catalyst was prepared using potato starch (SRL) and Fe salts (Sigma) as raw materials in a three-step process. First, the magnetite (Fe3O4) nanoparticles (NP) were prepared by a co-precipitation method using 0.4 N HCl solution containing FeCl3 and FeCl2 (the molar ration of Fe2+/Fe3+ was 0.5).15 In brief, 25 ml of the (Fe2+/Fe3+) solution was added drop-wise to 250 ml of 1.5 N NaOH solution under vigorous stirring where upon Fe3O4 nanoparticles (NPs) were formed. To stabilize the NPs, 100 μl of oleic acid (Sigma) was added. Finally, impurity-free, oleic acid-stabilized magnetite NPs were obtained by repeated centrifugation and washing with deionized water. In the next step, carbon coated magnetite NP composite was prepared by hydrothermal treatment of 5 g of the obtained NPs in 100 ml deionised water containing 5 g starch (SRL) in a 200 ml Teflon-lined autoclave at 180 °C for 24 h. Finally, the carbon coated magnetite NPs were activated at 500 °C under constant N2 flow (75 ml min−1) for sp2 carbon enrichment and subsequently sulfonated with freshly prepared 4-BDS to obtain the sulfonated Fe3O4@C composite catalyst (FeCS).14,16
2.2.2. Acidic zeolites. For comparison, commercial zeolites H-ZSM-5 {SiO2/Al2O3 mole ratio 23, specific surface area 443 m2 g−1} and H–Y {SiO2/Al2O3 mole ratio 12, specific surface area 884 m2 g−1} obtained from Zeolyst International were used. Prior to use zeolites were calcined at 450 °C in a muffle oven under stagnant atmospheric air to convert them from NH4+ to H+ forms.14 Some zeolites were also similarly sulfonated with 4-BDS for the introduction of –SO3H groups.17
2.3. Catalytic reaction procedure for cellulose acetylation
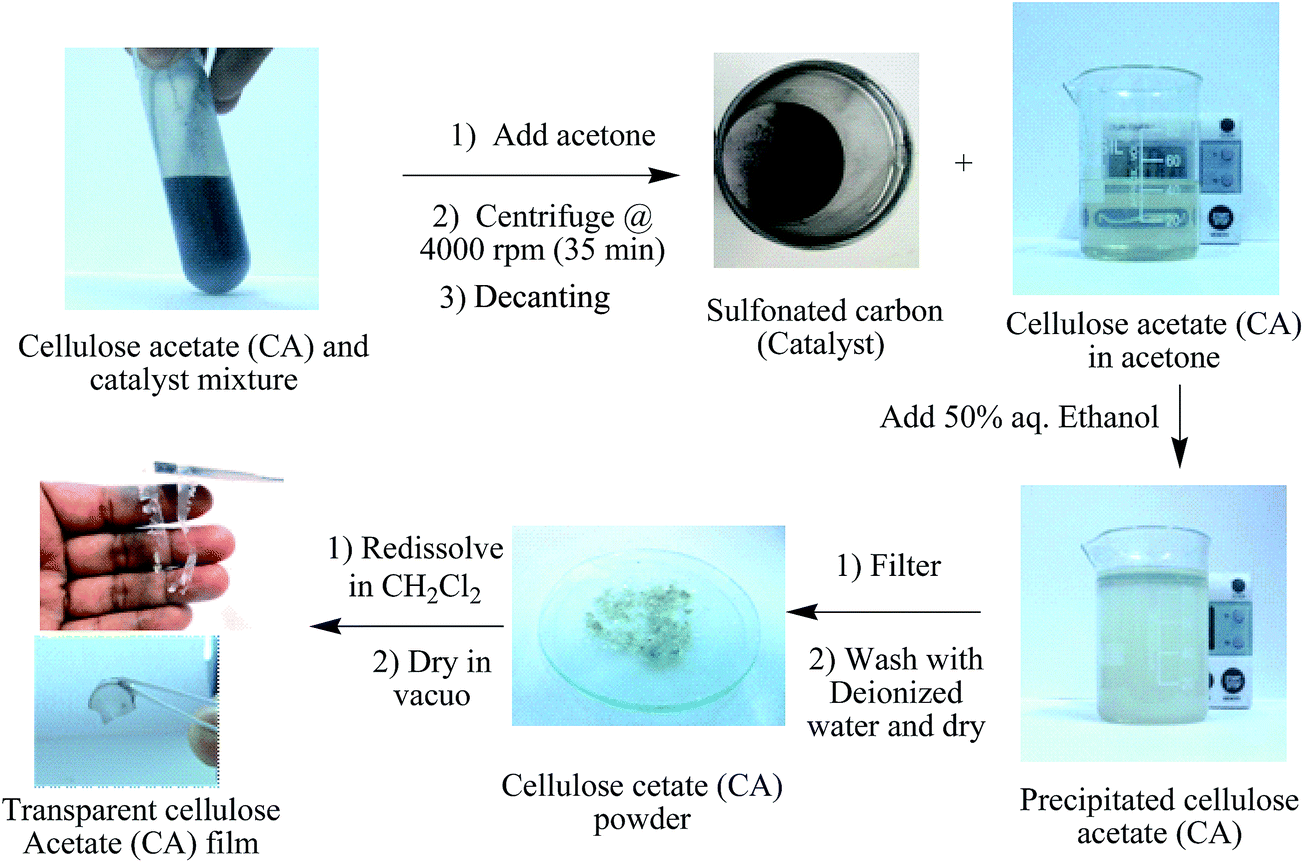
Reactions were performed in a 50 ml 2 necked ground flask equipped with mechanical stirring and a reflux condenser in a temperature controlled oil bath. In a typical experiment, 1 g (∼6.1 mmol AGU) microcrystalline cellulose (Sigma, crystallinity index ∼82%, degree of polymerization 789, <70 μm and vacuum dried at 70 °C for 24 h), 2.8–5.7 g acetic anhydride and 0.25–1 g catalyst (7–70 μm) was heated at 80 °C with constant mechanical stirring (1000 rpm) for 12–24 h. After completion of the reaction, the resultant viscous liquid (containing product and catalyst) was allowed to cool to room temperature, diluted with 40 ml acetone or DMSO and centrifuged at 4000 rpm for 35 min to separate the product and solid catalyst particles. The obtained transparent liquid was poured into 50 ml of 50% (v/v) aqueous ethanol and stirred for 30 min to precipitate CA. Finally, the acetylated product (CA) was filtered, thoroughly washed with deionized water in order to remove excess acetic anhydride and acetic acid (formed during the reaction), vacuum dried at 70 °C and weighed (Scheme 2). Here, the catalytic performances of the investigated material were expressed as a function of isolated product yield (after separation, washing and drying steps) and degree of substitution (DS) value. The isolated product yields were calculated based on the complete substitution of cellulose using the equations suggested by Fan et al., 2014.10| |
 | (1) |
where, mCA, mC represent the mass of acetylated product, cellulose and 162 and 291 represent the mass of anhydroglucose unit (AGU) of cellulose and cellulose triacetate, respectively.
 |
| | Scheme 2 Separation off CA from reaction mixture and preparation of CA film. | |
2.4. Analytical section
The elemental composition (bulk) of the carbonized materials and the carbon sources were determined by organic elemental analysis on a Thermo Scientific FLASH 2000 apparatus. The powder X-ray powder diffraction (XRD) patterns of carbon samples were recorded on a Rigaku miniflex diffractometer (Cu-Kα radiation, λ = 1.5406 Å) in 2θ range 10–70° at a scanning rate of 4 °C min−1. FT-IR spectra were recorded in KBr pellets on a Nicolet (Impact 410) FT-IR spectrophotometer. Transmission electron micrographs (TEM) were recorded on a Jeol JEM-2100 electron microscope operating at 200 kV. The resolution was around 0.4 nm. Samples were suspended in ethanol and deposited straight away on a copper grid prior to analysis. The specific surface area, pore size and pore volume of the carbon materials were determined by means of N2 physisorption at liquid nitrogen temperatures on a Carlo Erba Sorptomatic 1990 instrument. The samples were pre-treated at 150 °C while degassing (∼0.1 Pa). The thermal stability of the catalytic materials were investigated by thermo gravimetric analysis (TGA 6000, PerkinElmer) from room-temperature to 500 °C at a ramping rate of 10 °C min−1 under N2 flow (UHP grade). The surface acidities of the carbon materials were measured by temperature-programmed adsorption–desorption of ammonia on an AutoChem 2910, Micromeritics instrument. TPD was carried out from 100 to 500 °C at a heating rate of 10 °C min−1 with He flow rate of 35 ml min−1. After each TPD measurements, the amount of ammonia adsorbed was determined form the calibration curve obtained from varying volumes of ammonia in He. The –SO3H densities of sulfonated carbons were estimated from elemental analysis assuming all sulfur presented in the carbon samples are due to –SO3H/–PhSO3H groups.13,14
The obtained products (CA) were analyzed by FT-IR ATR (Agilent Cary 670 spectrometer), thermogravimetric analysis (TGA 6000, PerkinElmer) under N2 flow (UHP garde) and 1H-NMR spectroscopy (Jeol JNM-ECS400 NMR spectrometer operating at 25.5 °C using DMSO-d6 as a solvent and TMS as an internal standard). The DS of the product was determined from 1H-NMR data using the equation given below.6,7
| |
 | (2) |
where,
Aacetate (1.5–2.3 ppm) is the area of the methyl proton signals and
AAGU (3.5–5.8 ppm) is the area of the proton signals of the cellulose AGU unit (the DS analyses were performed as triplicates). In order to conduct reusability tests, the catalyst separated by centrifugation was thoroughly washed with acetone, deionised water and dried in a vacuum oven at 80 °C (
Scheme 2). Finally, the catalytic tests were repeated with recycled catalyst and maintaining similar reaction conditions as during the first run.
3. Results and discussion
3.1. Catalyst characterization
The textural and acidic properties of the investigated catalytic materials are summarized in Table 1. In terms of pore structure, among the SO3H/PhSO3H-carbons, the sulfonated Pongamia active carbon (AC500S) and commercial active carbon (CACS) were mesoporous; while the hydrothermally sulfonated catalyst (ACSHT) and sulfonated Fe@C nanocomposite (FeCS) catalysts were non-porous. Overall, the textural and acidic properties of these materials were similar to the sulfonated carbon catalysts reported in our previous papers (Table S1, ESI†).13,14 Further details on AC500, AC500S and ACSHT can be found in Konwar et al., 2015.14 As well-known, contrary to the carbons, the zeolites H–Y and H-ZSM-5 are microporous in nature and presented large specific surface area upto 884 and 443 m2 gcat−1, respectively. In terms of total surface acid site density, ACSHT contained the highest amount of surface acidic and –SO3H sites (mmol gcat−1), followed by AC500S, CACS, FeCS, H-ZSM-5, non-sulfonated active carbons (AC500, CAC) and H–Y respectively (Table 1). Further, in terms of strength of acidic sites while the sulfonic acid (–SO3H/–PhSO3H) functionalized materials are known to be comparable to 100% H2SO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11,13 zeolites are reported comparatively less acidic (H0 comparable to 80% H2SO4) on the Hammett scale (Table 1).18,19 In contrast, the non-sulfonated carbons (or the parent active carbon: CAC and AC500) with –COOH and –OH groups are the least acidic in terms of acid site strength among all the investigated catalysts.
11,13 zeolites are reported comparatively less acidic (H0 comparable to 80% H2SO4) on the Hammett scale (Table 1).18,19 In contrast, the non-sulfonated carbons (or the parent active carbon: CAC and AC500) with –COOH and –OH groups are the least acidic in terms of acid site strength among all the investigated catalysts.
Table 1 Properties of catalytic materials used in this worke
| Catalyst |
Acidityb (mmol gcat−1) |
–SO3Hc (mmol gcat−1) |
Surface area (m2 gcat−1) |
Pore diameter (nm) |
Pore volume (cm3 gcat−1) |
| Prepared by treating H–Y zeolite with 4-benzenediazoniumsulfonate. Measured by pyridine adsorption desorption in FT-IR for zeolites and NH3-TPD for carbons. Based on CHNS analysis. Compiled from ref. 14. n.d = not determined. |
| H-ZSM-5 |
1.142 |
— |
443 |
0.62 & 0.63d |
n.d |
| H–Y |
0.825 |
— |
884 |
0.81d |
n.d |
| H–Y–Sa |
n.d |
0.12 |
n.d |
n.dd |
n.d |
| CAC |
1.1 |
— |
201 |
5.5 |
0.14 |
| AC500 |
1.17d |
— |
820d |
4.7 |
0.66 |
| CACS |
5.12d |
0.53 |
119 |
4.01 |
0.07 |
| AC500S |
6.07d |
0.82 |
483d |
4.8 |
0.46 |
| ACSHT |
6.84d |
1.13d |
<1d |
Non-porous |
— |
| FeCS |
1.34 |
0.3 |
8 |
Non-porous |
— |
The structural features of the newly reported magnetic composite catalyst (FeCS) were investigated by XRD and TEM techniques. The formation of well-defined magnetite (Fe3O4) nanoparticles was confirmed by XRD (2θ at 30.3°, 35.7°, 54.8°, 57.22° and 62.9°, ESI†) and SEDA patterns (Fig. 1),15,20 while TEM images clearly show the successful encapsulation of the Fe3O4 nanoparticles of size 2–20 nm by aromatic carbon sheets formed by hydrothermal carbonisation of starch (Fig. 1). Comparison, of the TEM pictures of FeCS and AC500S also confirm the non-porous structure of the former while the later exhibited several pores with diameters ≥10 nm.
 |
| | Fig. 1 TEM images of (a) FeCS (20 nm), (b) AC500S (20 nm), (c) FeCS (100 nm) and (d) SEDA diffraction patterns of FeCS. | |
3.2. Catalytic activity in cellulose acetylation
3.2.1. Influence catalyst material used. Table 2 summarizes the results of the preliminary catalytic tests conducted over different solid acids at 80 °C. The data in Table 2 clearly show that with respect to isolated product yield microporous zeolites and non-sulfonated ACs (materials with Hammett acidity greater/weaker than 100% or conc. H2SO4) to be inactive in cellulose acetylation; whereas, the strongly Brønsted acidic sulfonated carbons (H0 comparable to 100% H2SO4) were catalytically active and produced cellulose acetate in yields approaching as high as 77% (Table 2). In fact, catalytic performance of the best sulfonated catalyst (AC500S, a large pore catalyst with a sulfonic group density of 0.82 mmol gcat−1) was en par to that of mineral acids (complete acetylation, DS of 2.94).1–3 Also among the sulfonated materials, the acetylation activity (i.e. yield and extent of –OH substitution among acetylated products) varied directly as a function of material sulfonic acid (–SO3H/–PhSO3H) density and pore sizes. The order of activity being: AC500S > CACS > ACSHT > FeCS, a trend consistent with the collective effects of the two aforesaid parameters. On the contrary, in the absence of a strong Brønsted acid catalyst having strength comparable to 100% H2SO4 (blank reaction, with microporous zeolites and non-sulfonated active carbons), the extent of cellulose acetylation was insignificant (indicated by the very low DS of esterified products 0.1–0.5) and it was practically impossible to separate these partially substituted, insoluble products from solid catalyst particles. So, CA yield were considered to be essentially ∼0% (Table 2), in such reactions. Thus, our preliminary results indicate that for the solid acids to be catalytically active in cellulose acetylation (Scheme 1) the presence of large pores (mesoporosity) (Fig. S1, ESI†) and a high concentration of sulfonic acid (–SO3H/–PhSO3H) sites (or sites with comparable acidic strength) are essential. Accordingly, in the current work the sulfonated catalyst with the largest pore volume (AC500S) exhibited highest activity while those with lower concentration of sulfonic acidic sites and/or lower porosity were considerably less active (Table 2). Particularly, catalyst porosity had a distinct effect on acetylation activity and herein, as the large pore sulfonated catalysts (AC500S and CACS, ESI†) offered better accessibility for the bulky substrate molecules (cellulose, critical diameter ∼10 nm) to the active strong acid sites, superior activity resulted (i.e. high CA yield and DS value were obtained) (Table 2).21 Presumably, this could be the most important factor contributing to the poor acetylation activity of the zeolites as the strong (active) acid sites of such materials are located within the small micropores and it would be virtually impossible for the large polymeric cellulose molecules to enter such pores (<2 nm).13 Although, the average pore size of both AC500S and CACS (Table 1) were less than cellulose critical diameter they still contained abundant pores with sizes greater 10 nm and accordingly exhibited high activity in acetylation of cellulose (Fig. S1(b), ESI†). In contrast, the unexpectedly high activity of the non-porous sulfonated catalyst, ACSHT originates from its high concentration of –SO3H groups which eventually contribute to surface reactions.
Table 2 Overview of catalytic performance of solid acids in cellulose acetylation
| Catalyst |
Reaction time (h) |
Ac2O/AGU (mole ratio) |
Catalyst (g) |
Yielda (%) |
DS |
| Isolated product yield after separation, washing and vacuum drying at 70 °C, conditions: in all experiments the amount of cellulose was 1 g (∼6.1 mmol AGU), stirring rate was fixed at 1000 rpm in all the experiments. The crystallinity index and degree of polymerization (DP) of cellulose substrate (microcrystalline, Sigma) were ∼82% and 789 respectively (values adapted from ref. 7(b) and 13(a)). |
| Blank |
24 |
9 |
— |
0 |
— |
| H–Y |
24 |
9 |
1 |
0 |
— |
| H–Y–S |
24 |
9 |
1 |
0 |
— |
| H-ZSM-5 |
24 |
9 |
1 |
0 |
— |
| CAC |
24 |
9 |
1 |
0 |
— |
| AC500 |
24 |
9 |
1 |
0 |
— |
| CACS |
12 |
9 |
1 |
50 |
2.1 |
| AC500S |
12 |
9 |
1 |
77 |
2.94 |
| ACSHT |
24 |
9 |
1 |
54 |
2.3 |
| FeCS |
24 |
9 |
1 |
20 |
1.2 |
Visually, the success of cellulose acetylation (with the sulfonated materials) could be easily recognized from the gradual transformation of the reaction system from an insoluble solid–liquid mixture to highly viscous black liquid which upon dilution with acetone (or DMSO) and separation of the catalyst particles gave a transparent solution of soluble CA (Scheme 2). Overall, the results of our catalytic tests indicate that the strong Brønsted acid sites (–PhSO3H/–SO3H) present in sulfonated carbons are responsible for the catalytic action as the non-sulfonated carbons were inactive in cellulose acetylation, an observation also consistent with findings of Zhang et al., 2013.7 It is most likely that the weakly acidic –COOH and –OH groups of non-sulfonated carbons failed to activate acetic anhydride molecule.14 While, the inability of zeolites to acetylate cellulose could be accredited to their narrow pore structure (microporosity) preventing interaction between the active acid sites and the cellulose molecules;21 the same zeolites have been successfully demonstrated to acetylate smaller substrates molecules such as glycerol, 1,2-diacetin and 1,3-diacetin having critical diameter of 0.646 nm, 0.78 nm and 0.943 nm, respectively under comparable reaction conditions.14 In a related study, Zhang et al., 2013 also obtained comparable results for [Hmim]HSO4 (a –SO3H containing IL), further the authors also proposed that the Brønsted acidic HSO4 group of IL activate the carbonyl group of acetic anhydride, thus making it more reactive for acetylation.7 Here also, we believe that catalytic activity of the sulfonated materials could be accredited to the operation of a similar reaction mechanism whereupon the –SO3H/PhSO3H groups present in these materials activate the carbonyl carbon in an analogous manner.
3.2.2. Effect of reaction parameters. For optimization of process conditions further investigation were made upon the best sulfonated catalyst (AC500S) as a reference. The results showed that in addition to the catalyst properties, CA yield and quality (DS) were also affected by duration of reaction, the amount of catalyst used and molar ratio of acetic anhydride-to-AGU (Fig. 2 and 3). Both the DS value and yield of CA were observed to increase as a function of reaction time, catalyst loading and acetic anhydride amount, reaching a maximum (DS value of 2.94 and yield of 77%) in 12 h (reaction time) with 1 g catalyst and Ac2O-to-AGU molar ratio of 9:1, respectively (Table 2 and Fig. 2). In contrast, reaction temperature was found have no effect on DS or CA yield (investigated at 80 °C and 100 °C). Accordingly, with AC500S, partially substituted and acetone soluble CA with a DS value of 2–2.7 and yield close to ∼70% could be obtained in 9–12 h under mild reaction conditions (Fig. 2). Also, from the comparison of the individual –CH3 signals (C2, C3, and C6) of AGU, DS of individual –OH groups could be calculated and which showed the order of reactivity of the of –OH groups of AGU to be C6–OH > C2–OH > C3–OH similar to the results obtained for ionic liquid catalysts [Amim]Cl and [Hmim]HSO4, respectively (Fig. 3).5,7 Overall, over AC500S, the optimized conditions for producing the commercially desirable CDA (DS 2.7) was found to be 80 °C, Ac2O-to-AGU molar ratio of 4.5 and 12 h reaction time using catalyst to cellulose (w/w) ratio of 1. Also, in this work, the use of AGU-to-Ac2O molar ratio less than 4.5 always resulted in incomplete/partial cellulose conversion (data not shown) for all the sulfonated catalysts as a significant amount of Ac2O was also lost to the reaction with free moisture and –OH groups present on catalyst surface.14 In fact, the inferior catalytic activity of ACSHT could most likely be linked with the loss of Ac2O to such side reactions as it was only partially carbonized and possessed a surface which was highly functionalized with –OH groups (FT-IR and TGA, ESI†). A similar, trend was also observed upon acetylation of glycerol with acetic anhydride over sulfonated carbons, always requiring greater than stoichiometric (i.e. >3:1) molar ratio of anhydride to glycerol to reach 100% triacetin (triester) selectivity.14 Thus, for reactions involving anhydrides as one of the substrates sulfonated carbons with a lower density of –OH and –COOH groups are ideal/suitable catalysts (i.e. those obtained by 4-BDS treatment).
 |
| | Fig. 2 (a) Yield and DS value as a function of catalyst (AC500S) loading at fixed acetic anhydride -to-AGU mole ratio of 9 at 80 °C and (b) influence of reaction time on yield and DS of CA at fixed catalyst/cellulose ratio (w/w) of 1 (Hollow symbols represent Ac2O-to-AGU mole ratio = 9 and solid symbols represent Ac2O-to-AGU mole ratio = 4.5, 80 °C). | |
 |
| | Fig. 3 Effect of reaction duration and acetic anhydride-to-AGU molar ratio (4.5 and 9) on DS of acetylated product. Reaction conditions: catalyst (AC500S)/cellulose ratio (w/w) = 1, reaction temperature = 80 °C, stirring rate = 1000 rpm. | |
3.2.3. Influence of reuse. In order to investigate the operational stability of sulfonated carbons during the reaction, reusability tests were also conducted with the optimum catalyst AC500S under standardized reaction conditions. The results in Fig. 4 showed that the sulfonated materials demonstrated excellent operational stability during acetylation reactions showing no significant loss of activity (constant DS and CA yield) during three successive reaction cycles. These results are in fact in good agreement with our previous results whereupon the high stability of –PhSO3H groups of the sulfonated catalysts obtained by 4-BDS treatment was also demonstrated in liquid phase reactions like esterification, transesterification and acetylation.13,14 The presence of –SO3H/–PhSO3H groups in the spent catalytic materials were confirmed by FT-IR analysis which clearly showed the presence of characteristic bands of –SO3H and S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (ESI†) (SO3− stretching at 1011 and 1085 cm−1 and O–S–O stretching in SO3H at 1176 and 1280 cm−1) and the corresponding density of –SO3H functions determined by elemental analysis to be roughly same as the fresh catalyst (0.8 mmol gcat−1).
O (ESI†) (SO3− stretching at 1011 and 1085 cm−1 and O–S–O stretching in SO3H at 1176 and 1280 cm−1) and the corresponding density of –SO3H functions determined by elemental analysis to be roughly same as the fresh catalyst (0.8 mmol gcat−1).
 |
| | Fig. 4 (a) Influence of reuse on the yield (isolated) and DS of the acetylated product and (b) corresponding 1H NMR of cellulose acetate (CA) obtained with sulfonated carbon catalyst (AC500S). Reaction conditions: catalyst (AC500S)/cellulose ratio (w/w) = 1, acetic anhydride-to-AGU molar ratio = 9, reaction temperature = 80 °C, stirring rate = 1000 rpm, reaction duration = 12 h. | |
3.2.4. Comparison with other solid acids. When compared to the heterogeneous catalysts reported (best results) in earlier studies (SO42−/ZrO2, H3PW12O40·6H2O and Amberlyst-15), the yield and DS obtained over sulfonated carbons were clearly higher. In addition our process also present several advantages: the main improvements are (a) the straight-forward recycling and reuse of the catalyst (Fig. 4) and (b) the possibility to directly convert microcrystalline cellulose under mild, solventless conditions without any prior treatments like ball milling (Table 3). Further, compared to the traditional solid catalysts, sulfonated carbons are reasonably cheap, easier to synthesize and easy to separate from post reaction mixtures (due to the distinct appearance as a fine black powder, Scheme 2).11–14 Overall, catalytic behavior of sulfonated carbons are analogous to liquid H2SO4 and [Hmim]HSO4 and consistent with the role of –SO3H groups in cellulose conversion.7,12,13 However, in contrast to the liquid mineral acid catalysts which offer little control over DS (they produce triacetate exclusively), upon use of sulfonated carbon catalysts it was possible to control the process and selectively produce CA with different DS values (Table 2, Fig. 2 and 3). Besides, in comparison to most of the solid catalysts reported in open literature, our process also worked with a lower catalyst dosage. On the downside, the current process suffered from a minor drawback as the partly esterified/unreacted cellulose (DS < 1.5, insoluble) and catalyst particles could not be easily separated; thus, the process could not be applied to produce insoluble CA with low DS values (0.5–1.5). Nevertheless, we were able to partially address this issue with the magnetically separable sulfonated carbon composite catalyst (FeCS); however, the same catalyst was considerably less active due to its low –SO3H density nonporous structure and could not be applied to produce the commercially desired soluble CA (Tables 1 and 2).
Table 3 Catalytic performance of sulfonated carbons in comparison to solid acids reported in previous worksa
| Catalyst |
Time (h) |
T (°C) |
Solvent |
Cellulose (g) |
Ac2O (g) |
AcOH (g) |
Catalyst (g) |
Yield (%) |
DS |
Ref. |
| The yield of cellulose acetate was calculated based on the complete substitution of cellulose except in the experiment marked*. |
| [Hmim]HSO4 |
12 |
100 |
— |
3.24 |
20.4 |
0 |
1.35 |
149.7* |
2.41 |
7 |
| SO42−/ZrO2 |
7.5 |
RT |
@Ball-milled |
10 |
15 ml |
0 |
0.553 |
75.6 |
1.8 |
8 |
| H3PW12O40·6H2O |
6 |
45 |
CH2Cl2 |
2 |
5 |
0.5 |
6.0 |
20.6 |
2.2 |
9 |
| Amberlyst-15 |
10 |
45 |
CH2Cl2 |
2 |
8.8 |
0.55 |
1.5 |
54.1 |
2.38 |
10 |
| CACS |
12 |
80 |
— |
1 |
5.7 |
0 |
0.25 |
50 |
2.1 |
This work |
| AC500S |
12 |
80 |
— |
1 |
5.7 |
0 |
0.5 |
69 |
2.67 |
| AC500S |
12 |
80 |
— |
1 |
2.8 |
0 |
1 |
70 |
2.7 |
| ACSHT |
24 |
80 |
— |
1 |
5.7 |
0 |
1 |
50 |
2.3 |
| FeCS |
24 |
80 |
— |
1 |
5.7 |
0 |
1 |
18 |
1.2 |
3.3. Characterization of cellulose acetate
From the practical (industrial) point of view, the most important property of CA is its solubility in organic solvents. In terms of solubility, we observed a significant variation among the obtained CA products, which was consistent with the effect of DS on CA properties and at par the trends reported by other researchers.1–3,5,9 Here, CA with DS values between 2 and 2.7 were soluble in acetone, CH2Cl2 and DMSO; while the products with DS > 2.8 were found to be soluble only in CH2Cl2 and DMSO. On the contrary, the partially acetylated products (DS 0.5–1.2) were insoluble in acetone and also difficult to solubilize in DMSO.
The 1H NMR of a representative CA samples illustrating the characteristic –CH3 signals of acetate and cellulose AGU unit –CH at 1.8–2.1 ppm and 3.5–5.0 ppm, respectively, is shown in Fig. 4(b). FT-IR patterns of CA also show the characteristic CO acetate peak at 1750 cm−1 and a correspondingly decreased –OH stretch signal near 3400 cm−1 (Fig. 5).8,22 The thermal properties of non-derivatized (native) cellulose and soluble cellulose acetate (with a DS value of 2.7) were also characterized by thermogravimetric measurements under N2 atmosphere (heating rate of 10 °C min−1), the results of which are shown in Fig. 6. It can be noticed from the TGA and DTA plots that only one major weight loss event occurred in the two samples in the temperature range of 300–400 °C. Correspondingly, from the DTA plots the maximum decomposition temperatures of cellulose and cellulose acetate were observed to be at 345 °C and 364 °C, respectively (Fig. 6(b)). This, indicated that both the onset temperature and the temperature at maximum decomposition rate of CA were higher than those observed for pure cellulose; an observation that is also in accordance with the trends reported in literature.5,9,10,22
 |
| | Fig. 5 FT-IR spectra of cellulose acetate (CA) obtained with sulfonated carbon catalyst (AC500S). | |
 |
| | Fig. 6 (a) TGA and (b) DTA patterns of a representative cellulose acetate (CA) sample obtained with sulfonated carbon catalyst (AC500S). | |
4. Conclusions
In summary, we have demonstrated that it is possible to catalyze the acetylation of cellulose with acetic anhydride over –SO3H functionalized carbon materials under mild (80 °C, acetic anhydride-to-AGU mole ratio of 4.5–9), solventless conditions. The results of our catalytic test cycles indicated that cellulose acetates with various DS values ranging between 1.2 and 2.94 could be obtained in excellent yields (≥70%) by employing sulfonated carbons as catalysts. Most importantly, it was possible to control yield and DS by simple adjustment of reaction conditions (duration, Ac2O-to-AGU mole ratio) and catalyst type, and selectively obtain the soluble diacetates directly in a one-pot process. These results corroborate the suitability of sulfonated carbons as a green substitute to liquid acid catalysts for production of commercially important cellulose esters.
Acknowledgements
This work is part of the activities at the Åbo Akademi University Johan Gadolin Process Chemistry Centre within the Centre of Excellence Programme appointed by ÅAU, Finland. Centre for International Mobility – CIMO (Finland) is acknowledged for providing visiting doctoral fellowship to Lakhya Jyoti Konwar at ÅAU-PCC. In Sweden the Bio4Energy programme, Wallenberg Wood Science Center and Kempe Foundations are acknowledged. Department of Energy, Tezpur University is gratefully acknowledged for donating the de-oiled cake.
References
- S. Fischer, K. Thümmler, B. Volkert, K. Hettrich, I. Schmidt and K. Fischer, Macromol. Symp., 2008, 262, 89–96 CrossRef CAS.
- T. Heinze and T. Liebert, Prog. Polym. Sci., 2001, 26, 1689–1762 CrossRef CAS.
- A. Hummel, Macromol. Symp., 2004, 208, 61–79 CrossRef CAS.
- J. Wu, J. Zhang, H. Zhang, J. He, Q. Ren and M. Guo, Biomacromolecules, 2004, 5, 266–268 CrossRef CAS PubMed.
- Y. Cao, J. Wu, T. Meng, J. Zhang, J. He, H. Li and Y. Zhang, Carbohydr. Polym., 2007, 69, 665–672 CrossRef CAS.
- X. Sun, C. Lu, W. Zhang, D. Tian and X. Zhang, Carbohydr. Polym., 2013, 98, 405–411 CrossRef CAS PubMed.
-
(a) X. Zhang, W. Zhang, D. Tian, Z. Zhou and C. Lu, RSC Adv., 2013, 3, 7722–7725 RSC;
(b) D. G. Raut, O. Sundman, W. Su, P. Virtanen, Y. Sugano, K. Kordas and J.-P. Mikkola, Carbohydr. Polym., 2015, 130, 18–25 CrossRef CAS PubMed.
- L. Yan, W. Li, Z. Qi and S. Liu, J. Polym. Res., 2006, 13, 375–378 CrossRef CAS.
- G. Fan, M. Wang, C. Liao, T. Fang, J. Li and R. Zhou, Carbohydr. Polym., 2013, 94, 71–76 CrossRef CAS PubMed.
- G. Fan, C. Liao, T. Fang, S. Luo and G. Song, Carbohydr. Polym., 2014, 112, 203–209 CrossRef CAS PubMed.
- K. Nakajima and M. Hara, ACS Catal., 2012, 2(7), 1296–1304 CrossRef CAS.
- M. Hara, Energy Environ. Sci., 2010, 3, 601–607 CAS.
-
(a) L. J. Konwar, P. Mäki-Arvela, E. Salminen, N. Kumar, A. J. Thakur, J.-P. Mikkola and D. Deka, Appl. Catal., B, 2015, 176–177, 20–35 CrossRef CAS;
(b) L. J. Konwar, D. Das, A. J. Thakur, E. Salminen, P. Mäki-Arvela, N. Kumar, J.-P. Mikkola and D. Deka, J. Mol. Catal. A: Chem., 2014, 388–389, 167–176 CrossRef CAS.
- L. J. Konwar, P. Mäki-Arvela, P. Begum, N. Kumar, A. J. Thakur, J.-P. Mikkola, R. C. Deka and D. Deka, J. Catal., 2015, 329, 237–247 CrossRef CAS.
- S. Zhang, H. Niu, Z. Hu, Y. Cai and Y. Shi, J. Chromatogr. A, 2010, 1217, 4757–4764 CrossRef CAS PubMed.
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8(6), 1679–1682 CrossRef CAS PubMed.
- M. P. Stewart, F. Maya, D. V. Kosynkin, S. M. Dirk, J. J. Stapleton, C. L. McGuiness, D. L. Allara and J. M. Tour, J. Am. Chem. Soc., 2004, 126(1), 370–378 CrossRef CAS PubMed.
- T. Yamanaka and K. Tanabe, J. Phys. Chem., 1975, 79(22), 2409–2411 CrossRef CAS.
-
(a) H. A. Benesi, J. Am. Chem. Soc., 1956, 78(21), 5490–5494 CrossRef CAS;
(b) P. A. Wright, Microporous Solid Acid Catalysts and their Applications, Microporous Framework Solids, 2007, 312–371 Search PubMed.
- W. Liu, K. Tian, H. Jiang and H. Yu, Sci. Rep., 2013, 3, 2419, DOI:10.1038/srep02419.
-
(a) J. Jaea, G. A. Tompsetta, A. J. Fosterb, K. D. Hammonda, S. M. Auerbacha, R. F. Lobob and G. W. Huber, J. Catal., 2011, 279, 257–268 CrossRef;
(b) D. Topgaard and O. Soederman, Langmuir, 2001, 17, 2694 CrossRef CAS.
-
(a) J. Li, L.-P. Zhang, F. Peng, J. Bian, T.-Q. Yuan, F. Xu and R.-C. Sun, Molecules, 2009, 14, 3551–3566 CrossRef CAS PubMed;
(b) A. El Nemr, S. Ragab, A. El Sikaily and A. Khaled, Carbohydr. Polym., 2015, 130, 41–48 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra25716f |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.