
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural effects of dibromocarbazoles on direct arylation polycondensation with 3,4-ethylenedioxythiophene†
Wei
Li
and
Tsuyoshi
Michinobu
*
Department of Organic and Polymeric Materials, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8552, Japan. E-mail: michinobu.t.aa@m.titech.ac.jp
First published on 11th April 2016
Abstract
The direct arylation reaction of a C–H bond with aromatic halides has been employed as a novel environmentally-friendly polymerization method for producing conjugated polymers. In this article, 3,4-ethylenedioxythiophene (EDOT) was copolymerized with a series of dibromocarbazoles (Cbzs) by the phosphine-free and microwave-assisted direct arylation polycondensation. The microwave heating dramatically shortened the reaction time compared to conventional heating. Under optimal conditions, a solution processable linear polymer, 3,6-Cbz-EDOT, with the number-average molecular weight (Mn) of 5600 and polydispersity index (Mw/Mn) of 1.9 was successfully synthesized. In contrast, the other dibromocarbazole monomers with different substitution patterns, i.e., 2,7-Cbz and 1,8-Cbz, did not afford the corresponding soluble linear polymers due to the occurrence of undesired crosslinking and the poor reactivity caused by steric hindrance or unsuitable catalysts, respectively. The electrochromic properties of 3,6-Cbz-EDOT were investigated. The thin polymer film displayed two-step color changes with the increasing potentials. The first color change was highly reversible with the switching times of the coloring and bleaching processes being 3.2 and 2.0 s, respectively.
Introduction
Carbazole-based conjugated polymers have attracted much attention due to their excellent optical, electrochemical, and conducting properties, which can be controlled by the connectivity patterns of the carbazole unit and comonomer structures.1–8 The initially developed conjugated carbazole polymers were poly(3,6-carbazole) derivatives because the 3,6-positions of the carbazole showed the highest reactivity for oxidative polymerization as well as towards various electrophiles.9 In 2001, Leclerc et al. and Müllen et al. independently reported the efficient synthesis of the 2,7-dihalogeno-carbazole derivatives.10,11 Their pioneering studies opened the door to a new class of poly(2,7-carbazole) derivatives, which served as a chemically stable alternative of blue-emitting polyfluorene derivatives.12 In addition to the emissive properties, excellent p-type semiconducting properties were demonstrated in bulk-heterojunction organic solar cells, thin film transistors, thermoelectric and electrical memory devices when donor–acceptor alternating poly(2,7-carbazole)s were designed.13–19 In contrast, our group has successfully developed poly(1,8-carbazole) derivatives by the stepwise functionalization of the monomeric carbazole unit.20,21 The poly(1,8-carbazole)s displayed the combined properties of the poly(3,6-carbazole)s in terms of the connectivity pattern and poly(2,7-carbazole)s in terms of the effective conjugation length evaluated on the basis of the longest absorption maxima (λmax).22–24The conventional synthesis of the aforementioned conjugated carbazole polymers was based on the Pd-catalyzed polycondensation, such as the Suzuki, Stille, and Sonogashira polycondensations. However, in order to construct an environmentally benign and cost-effective synthetic route of conjugated polymers, the direct arylation protocol, which activates the C–H bonds of the unprepared arenes or heteroarenes to directly react with aryl halides, was recently introduced.25–27 In contrast to the necessity of the multistep tedious preparation of bifunctional boronic acids (for Suzuki coupling) and tin compounds (for Stille coupling), the direct arylation protocol can reduce the synthetic steps and, more importantly, does not produce a stoichiometric amount of toxic byproducts. Therefore, the number of publications using this protocol is currently increasing. Some of the most frequently used C–H activated reactants or bifunctional monomers are the thiophene-based substances. For example, 3,4-dimethylthiophene and 3,4-ethylenedioxythiophene (EDOT) possess activated C–H bonds only at the 2- and 5-positions of the thiophene ring, and accordingly, they can serve as a bifunctional monomer for the direct arylation polycondensation.28–32 Kanbara and his coworkers recently succeeded in optimizing the polycondensation of such monomers using Pd(OAc)2 but without any phosphine ligands.33 Furthermore, they demonstrated the effectiveness of microwave heating in order to increase the monomer reactivities and obtain high molecular weight polymers.34 It is indeed widely recognized that the microwave-assisted direct arylation polycondensation is an efficient process for synthesizing optoelectronic organic polymers.35–37
We now describe the structural effects of the dibromocarbazole monomers on the microwave-assisted direct arylation polycondensation with EDOT. We show that 3,6-dibromocarbazole is the most suitable for producing linear soluble polymers as compared to the 2,7- and 1,8-dibromocarbazole counter structures. We also demonstrate the reversible electrochromic properties of the resulting polymer based on 3,6-carbazole and EDOT. Overall, the results reported here indicate the important aspects of the monomer structures and polymer properties regarding the emerging polymerization technique of direct arylation polycondensation.
Experimental
Materials
3,6-Dibromo-9-(2-ethylhexyl)carbazole 1,38 2,7-dibromo-9-(2-ethylhexyl)carbazole 2,11 and 3,6-di-tert-butylcarbazole20 were prepared according to the reported literature procedure. Commercially available solvents and reagents were used without further purification unless stated otherwise.General methods
1H NMR and 13C NMR spectra were measured on a JEOL model AL300 spectrometer at 20 °C. Chemical shifts are reported in ppm downfield from SiMe4, using the solvent's residual signal as an internal reference. Fourier transform infrared (FT-IR) spectra were recorded on a JASCO FT/IR-4100 spectrometer in the range from 4000 to 600 cm−1. MALDI-TOF mass spectra were measured on a Shimadzu/Kratos AXIMA-CFR mass spectrometer equipped with nitrogen laser (λ = 337 nm) and pulsed ion extraction, which was operated in a linear-positive ion mode at an accelerating potential of 20 kV. Dichloromethane solutions containing 1 g L−1 of a sample, 10 g L−1 of dithranol, and 1 g L−1 of sodium trifluoroacetate were mixed at the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, and 1 mL aliquot of this mixture was deposited onto a sample target plate. Gel permeation chromatography (GPC) was measured on a JASCO GULLIVER 1500 equipped with a pump (PU-2080 Plus), an absorbance detector (RI-2031 Plus), and two Shodex GPC KF-803 columns (8.0 mm I.D. × 300 mm L) based on a conventional calibration curve using polystyrene standards. Tetrahydrofuran (40 °C) was used as a carrier solvent at the flow rate of 1.0 mL min−1. UV-vis spectra were measured on a JASCO V-670 spectrophotometer. Fluorescence spectra were measured on a JASCO FP6500 spectrophotometer. Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were carried out on a Rigaku Thermoplus TG8120 and DSC8230, respectively, under flowing nitrogen at the scan rate of 10 °C min−1. Electrochemistry measurements were carried out on a BAS electrochemical analyzer model 612C at 25 °C in dehydrated CH3CN containing 0.1 M (nC4H9)4NPF6 in the three electrode cell. The working, reference, and auxiliary electrodes were an indium–tin–oxide (ITO) electrode, Ag/Ag+/CH3CN/(nC4H9)4NPF6, and a Pt wire, respectively. Ferrocene/ferrocinium (Fc/Fc+) couple was 0.13 V (vs. Ag/Ag+). All potentials are referenced to the Ag/Ag+ couple unless otherwise stated. Polymer thin films were prepared on an ITO electrode (10 Ω, about 0.8 × 2.5 cm2) by spray-coating of polymer solutions (5.0 g L−1 in CH2Cl2). Absorption spectra of spectroelectrochemistry experiments were measured on an Agilent 8453 UV-vis spectrophotometer.
:1:1, and 1 mL aliquot of this mixture was deposited onto a sample target plate. Gel permeation chromatography (GPC) was measured on a JASCO GULLIVER 1500 equipped with a pump (PU-2080 Plus), an absorbance detector (RI-2031 Plus), and two Shodex GPC KF-803 columns (8.0 mm I.D. × 300 mm L) based on a conventional calibration curve using polystyrene standards. Tetrahydrofuran (40 °C) was used as a carrier solvent at the flow rate of 1.0 mL min−1. UV-vis spectra were measured on a JASCO V-670 spectrophotometer. Fluorescence spectra were measured on a JASCO FP6500 spectrophotometer. Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were carried out on a Rigaku Thermoplus TG8120 and DSC8230, respectively, under flowing nitrogen at the scan rate of 10 °C min−1. Electrochemistry measurements were carried out on a BAS electrochemical analyzer model 612C at 25 °C in dehydrated CH3CN containing 0.1 M (nC4H9)4NPF6 in the three electrode cell. The working, reference, and auxiliary electrodes were an indium–tin–oxide (ITO) electrode, Ag/Ag+/CH3CN/(nC4H9)4NPF6, and a Pt wire, respectively. Ferrocene/ferrocinium (Fc/Fc+) couple was 0.13 V (vs. Ag/Ag+). All potentials are referenced to the Ag/Ag+ couple unless otherwise stated. Polymer thin films were prepared on an ITO electrode (10 Ω, about 0.8 × 2.5 cm2) by spray-coating of polymer solutions (5.0 g L−1 in CH2Cl2). Absorption spectra of spectroelectrochemistry experiments were measured on an Agilent 8453 UV-vis spectrophotometer.
General procedure for direct arylation polycondensation
Dibromocarbazole monomer (0.5 mmol) and 3,4-ethylenedioxythiophene (EDOT, 0.5 mmol) were placed on a reaction tube. The tube was transferred into a glove box under argon atmosphere. Then, 1.0 mol% of Pd(OAc)2, 1.5 equiv. of potassium pivalate (KOPiv), and dry N,N-dimethylacetamide (DMAc, 2.5 mL) were added. The tube was sealed, removed from the glove box, and subjected to the microwave synthesis. After the reaction was completed, it was cooled to room temperature and the resulting product was washed with water, methanol, and hexane. The solid product was dissolved into chloroform and reprecipitated into methanol.3,6-Cbz-EDOT: 87% yield. 1H NMR (300 MHz, CDCL3): δ 8.47 (s, 2H), 7.91 (s, 2H), 7.26 (s, 2H), 4.39 (s, 4H), 4.10 (d, J = 4.2 Hz, 2H), 2.00 (s, 1H), 1.26 (d, J = 4.5 Hz, 8H), 0.85 ppm (s, 6H). IR (neat): ν = 2923, 2857, 1596, 1514, 1432, 1359, 1292, 1213, 1149, 1090, 1034, 915, 865, 802, 734, 632 cm−1.
Results and discussion
Polymer synthesis
Three symmetric dibromocarbazole monomers with different substitution patterns, 1–3, were prepared (ESI†). A branched ethylhexyl group was introduced into the 9-position of the carbazole in order to increase the solubility of the resulting polymers. The dibromocarbazole monomers were copolymerized with 3,4-ethylenedioxythiophene (EDOT) under direct arylation conditions using Pd(OAc)2 as a catalyst and potassium pivalate (KOPiv) as a base in N,N-dimethylacetamide (DMAc) (Scheme 1). First, the reactivity of 3,6-dibromocarbazole 1 was investigated (Table 1). The direct arylation polycondensation of 1 and EDOT at 100 °C for 6 h produced the corresponding polymer, 3,6-Cbz-EDOT, with the number-average molecular weight (Mn) of 3000 and polydispersity index (Mw/Mn) of 1.6 in 72% yield (run 6 in Table 1). It should be noted that the employed conditions were comparable to or even better than other reported conditions with different catalyst/ligand combinations, such as Pd(OAc)2/PCy3·HBF439 and Pd2(dba)3/P(o-MePh)340 (Table S1†). In order to increase the molecular weights, microwave synthesis was adopted due to its high efficiency, short reaction time, and ready accessibility. Based on the temperature dependence for the constant polymerization time of 30 min (runs 1, 2, 4, and 7 in Table 1), it was found that 80 °C was the most suitable temperature for producing the highest Mn of 5600. In order to further increase the Mn, the polymerization time was increased to 60 min (run 3 in Table 1). However, this change resulted in a decrease in the Mn to 2500. Similar to this result, there was an optimum polymerization time at 100 °C. A prolonged polymerization time from 30 to 60 min led to a slight increase in the Mn from 4100 to 5000 (runs 4 and 5 in Table 1). All the polymerization conditions produced the desired 3,6-Cbz-EDOT with a reasonable polydispersity in moderate yields except for the polymerization at 60 °C for 30 min (run 1 in Table 1).
 | ||
| Scheme 1 Polycondensation of EDOT with various dibromocarbazole comonomers. | ||
| Run | Temp. (°C) | Time (min) |
M
nb (g mol−1) |
M
w/Mnb |
Yield (%) |
|---|---|---|---|---|---|
| a Performed in the presence of 1.0 wt% Pd(OAc)2 and 1.5 equiv. KOPiv in DMAc under microwave irradiation. b Determined by GPC (THF eluent, calibrated by polystyrene standards). c Regular heating. | |||||
| 1 | 60 | 30 | 600 | 2.4 | 14 |
| 2 | 80 | 30 | 5600 | 1.9 | 87 |
| 3 | 80 | 60 | 2500 | 1.6 | 56 |
| 4 | 100 | 30 | 4100 | 1.7 | 68 |
| 5 | 100 | 60 | 5000 | 2.1 | 75 |
| 6c | 100 | 360 | 3000 | 1.6 | 72 |
| 7 | 120 | 30 | 1700 | 1.4 | 45 |
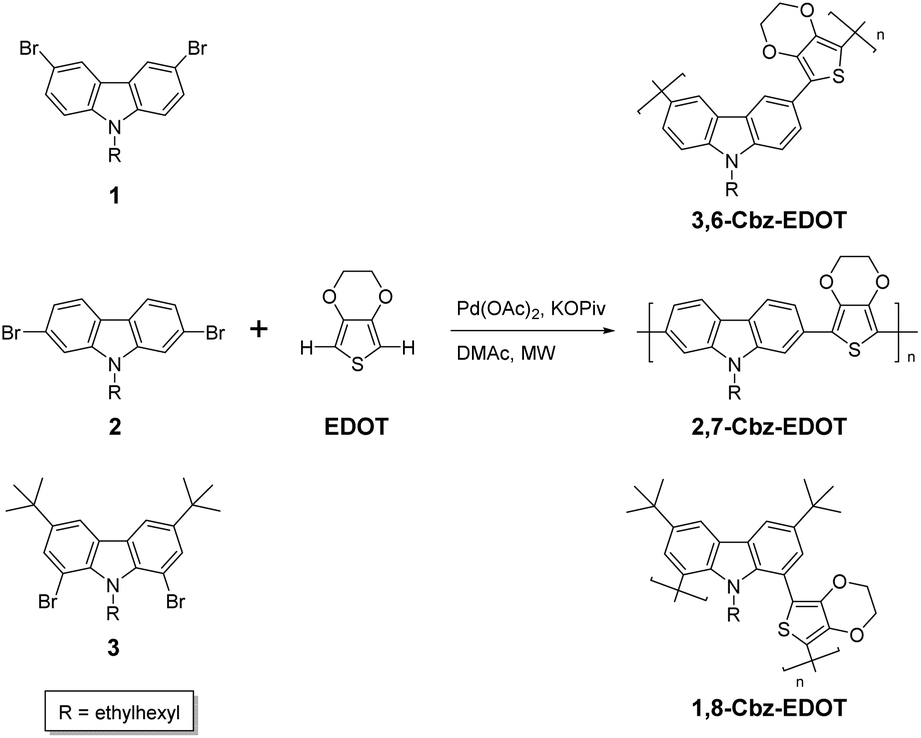
The chemical structure of 3,6-Cbz-EDOT was confirmed by its 1H NMR and IR spectra. In the 1H NMR spectrum, the aromatic peaks were ascribed to the 3,6-carbazole unit. In addition, the methylene group directly attached to the nitrogen atom of the carbazole and the ethylene groups of EDOT were detected at 4.39 and 4.21 ppm, respectively (Fig. 1). In the IR spectrum, the peak at 890 cm−1 attributed to the C–H bending of EDOT disappeared, suggesting the polymer formation. The thermal properties of the 3,6-Cbz-EDOT were measured by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). The TGA showed that the 5% weight loss temperature (Td5%) of 3,6-Cbz-EDOT was 386 °C, indicating a high thermal stability (Fig. S7†). The DSC curve did not reveal clear melting and crystallization peaks, but it exhibited a weak endothermic transition at 148 °C upon heating and an exothermic one at 111 °C upon cooling, attributable to a glass transition (Fig. S8†).
 | ||
| Fig. 1 1H NMR of 3,6-Cbz-EDOT in CDCl3. | ||
With the success of 3,6-Cbz-EDOT in mind, other dibromocarbazole monomers with different substitution patterns, namely the 2,7-dibromocarbazole and 1,8-dibromocarbazole derivatives, 2 and 3, were also subjected to the microwave-assisted direct arylation polycondensation with EDOT under the same conditions (Scheme 1). Surprisingly, the polymerization of 2 and EDOT produced insoluble gels even when the lower temperature of 60 °C and shorter time of 30 min were adopted. This result suggested that the C–H activation most likely occurred at the 3,6-positions of 2 in addition to the planned direct arylation coupling at the 2,7-positions, resulting in the cross-linking reactions. Also, this was not caused by the solubility problem of the resulting polymer, because another 2,7-dibromocarbazole monomer 4 with a longer branched alkyl chain (2-hexyldecyl group) gave the same result when polymerized under the microwave-assisted direct arylation conditions. It should be noted that the 3,6-positions of the carbazole derivatives are active C–H units in the direct arylation coupling.41 Accordingly, the gels composed of 2,7-Cbz-EDOT were not further characterized due to the insolubility in common organic solvents, such as chloroform, dichloromethane, THF, and DMSO. In contrast, the direct arylation polycondensation of 3 and EDOT did not proceed very well under the employed conditions. This might be due to the steric hindrance of the 1,8-positions mainly caused by the bulky ethylhexyl group at the adjacent 9-position or unsuitable catalysts for this substance.42 In addition, the 3,6-positions of 3 were perfectly blocked by the tert-butyl group, thereby preventing self-polycondensation.
Optical and electrochemical properties
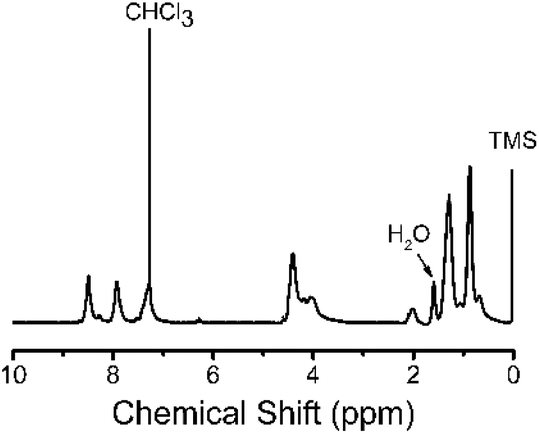
The UV-vis absorption and fluorescence spectra of 3,6-Cbz-EDOT were measured in CH2Cl2 and in a thin film (spin-coated on an ITO glass). We recently reported that the effective conjugation length of the 3,6-Cbz units saturates at the pentamer.43 Hence, it was postulated that the 3,6-Cbz-EDOT with the Mn of 5600 (14 repeat units) sufficiently shows the polymer properties. The absorption maximum (λmax) in CH2Cl2 was 354 nm, while the λmax of the thin film bathochromically shifted to 374 nm (Fig. 2). This red shift was due to the strong intermolecular interaction and the closer packing of the polymer backbones in the thin film. The fluorescence spectra of 3,6-Cbz-EDOT exhibited two emission bands at 438 and 469 nm in CH2Cl2 and 486 and 514 nm in the thin film. The red shift in the fluorescence spectra was more significant than that in the absorption spectra. Thus, the difference in the emission colors between the blue in solutions and green in films could be visually distinguished (Fig. S9†).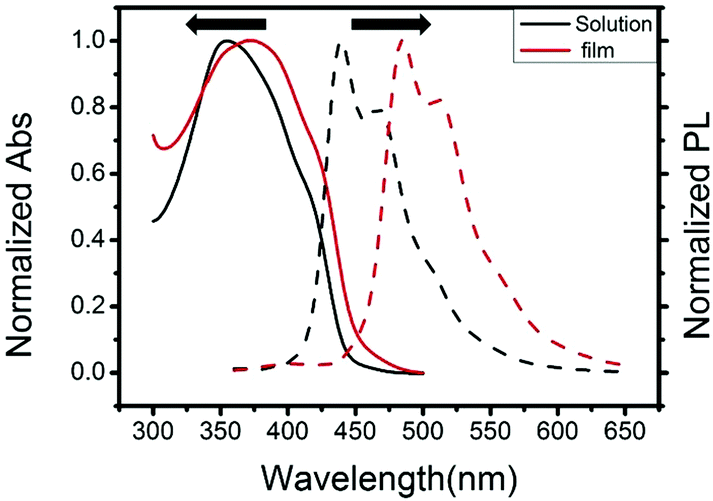 | ||
| Fig. 2 Normalized UV-vis absorption and fluorescence spectra of 3,6-Cbz-EDOT in CH2Cl2 measured at 10−6 M repeat unit−1 and in thin films. | ||
The cyclic voltammetry (CV) of 3,6-Cbz-EDOT was measured in CH3CN with 0.1 M (nC4H9)4NPF6 as the supporting electrolyte at 20 °C (Fig. 3). The film showed a reversible multistep oxidation due to the highly electron-rich carbazole and EDOT units as well as the electroactive connectivity position of the 3,6-carbazoles.44–47 In order to determine the exact oxidation potentials, differential pulse voltammetry (DPV) was measured using the same setup. The DPV revealed three oxidation peaks at 0.52, 0.80, and 1.42 V (vs. Ag/Ag+). This result suggests the stepwise generation of three different charge states in the oligomeric repeat units, namely a cation radical, bication, and trication radical as illustrated in Scheme 2. Similar to the emeraldine base of polyaniline, the positive charges are formally located at the carbazole nitrogens up to the second oxidation steps. However, the third oxidation occurs at the neutral EDOT moieties.
 | ||
| Fig. 3 Cyclic voltammetry (solid line) and differential pulse voltammetry (dotted line) of 3,6-Cbz-EDOT films on an ITO electrode, measured in CH3CN with 0.1 M (nC4H9)4NPF6 at the scan rate of 0.1 V s−1 under flowing nitrogen. | ||
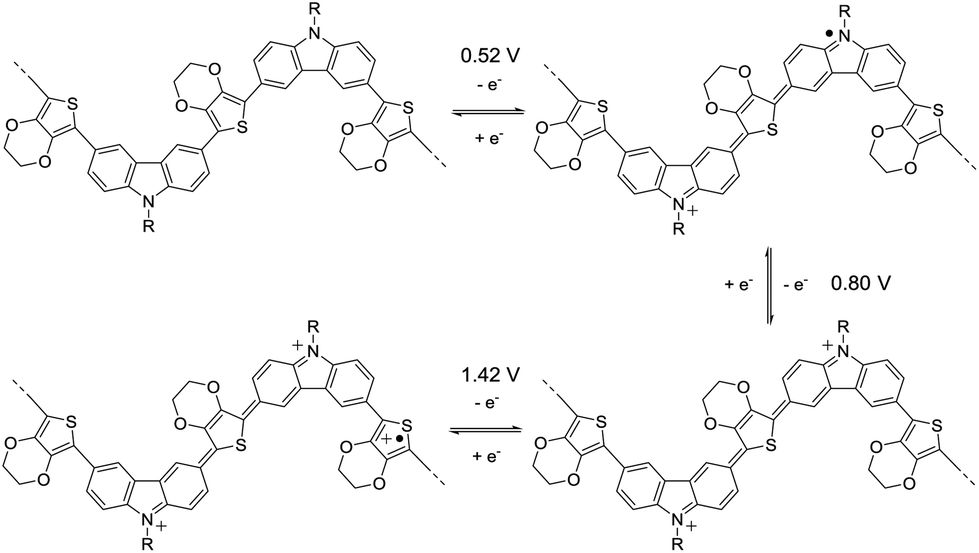 | ||
| Scheme 2 Schematic illustration of the stepwise oxidation of 3,6-Cbz-EDOT. | ||
Electrochromism
Based on the optical and electrochemical studies, the electrochromic properties of the 3,6-Cbz-EDOT thin film were investigated. Spectroelectrochemistry was used to monitor the spectral changes in the electrochromic films. The spray-coated thin films on an ITO-coated glass plate together with the reference and counter electrodes were placed in a 1-cm cuvette. The UV–vis-near infrared spectra were measured in CH3CN containing 0.1 M (nC4H9)4NPF6 at 20 °C. The polymer films displayed three distinct colors in response to the application of different potentials. When potentials more positive than 0.50 V were applied to the film, the original absorption band at 356 nm started to decrease and a new absorption band at 519 nm appeared (Fig. 4). This spectral change continued up to the potential application of 0.80 V. As a result of this electrochromism, the pale yellow color of the neutral polymer film changed to red. However, during the course of the continuous spectral changes up to 0.80 V, there were two isosbestic points at 443 nm (0.00–0.50 V) and 431 nm (0.50–0.80 V) (Fig. S10†). The first isosbestic point corresponds to the generation of cation radicals from the neutral state, and the formed cation radical species smoothly changed into the closed-shell structure of bication during the spectral changes with the second isosbestic point. Accordingly, the stable red color of the film is attributed to the bication. A further continuous increase in the applied potential from 0.80 to 1.40 V resulted in a well-defined low-energy band at 802 nm ascribed to the trication radical state. Thus, the film color eventually turned dark blue. It was thought that this state is the full oxidation form of the polymer main chain of which the chemical stability is slightly beyond the limit (vide infra).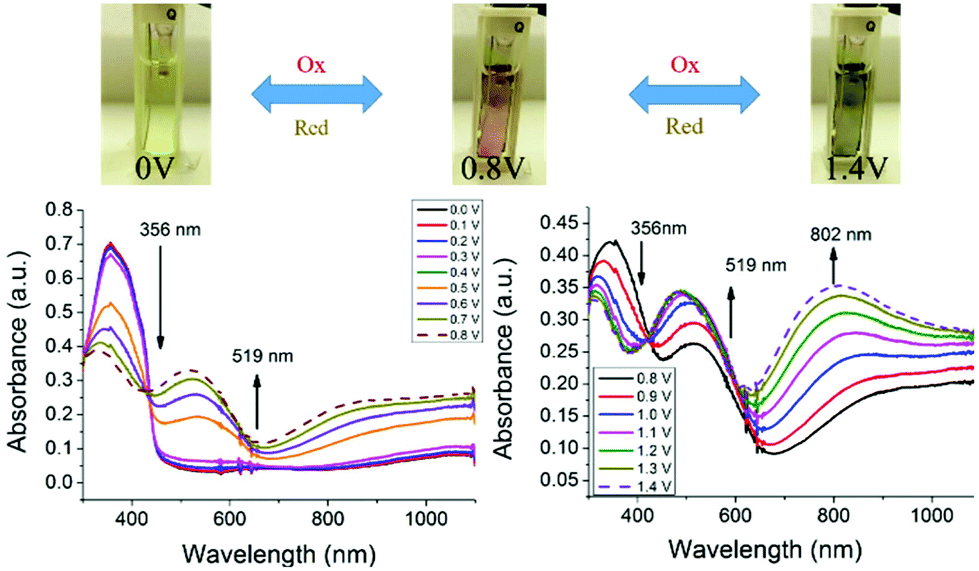 | ||
| Fig. 4 Spectroelectrochemistry of 3,6-Cbz-EDOT films on an ITO electrode measured in CH3CN with 0.1 M (nC4H9)4NPF6. | ||
Reversible color production by controlled potential applications is essential for the fabrication of practical electrochromic and electrofluorochromic devices.48–53 To gain a detailed insight into the film stability and coloring mechanism, the response time and switching processes were investigated. The original absorbance at 356 nm and the bication absorbance at 519 nm were monitored as a function of time (Fig. 5a). The pulse time was determined to be 20 s. The UV–vis-near infrared spectral changes in the oxidation process from the neutral to carbazole bication were fully reversible. The switching time was evaluated as 90% of the full switch, because it was difficult to visually perceive any further color changes beyond this point. The results revealed that the switching times of the coloring and bleaching process for the polymer film were 3.2 and 2.0 s, respectively (Fig. 5b). The polymer film quickly switched between the neutral yellow and oxidized red states. Unfortunately, the second electrochromic color change of the 3,6-Cbz-EDOT film from red to dark blue was not fully reversible upon the repeated pulse applications. This was probably due to the unstable trication radical state of the fully oxidized polymer backbone (vide supra).
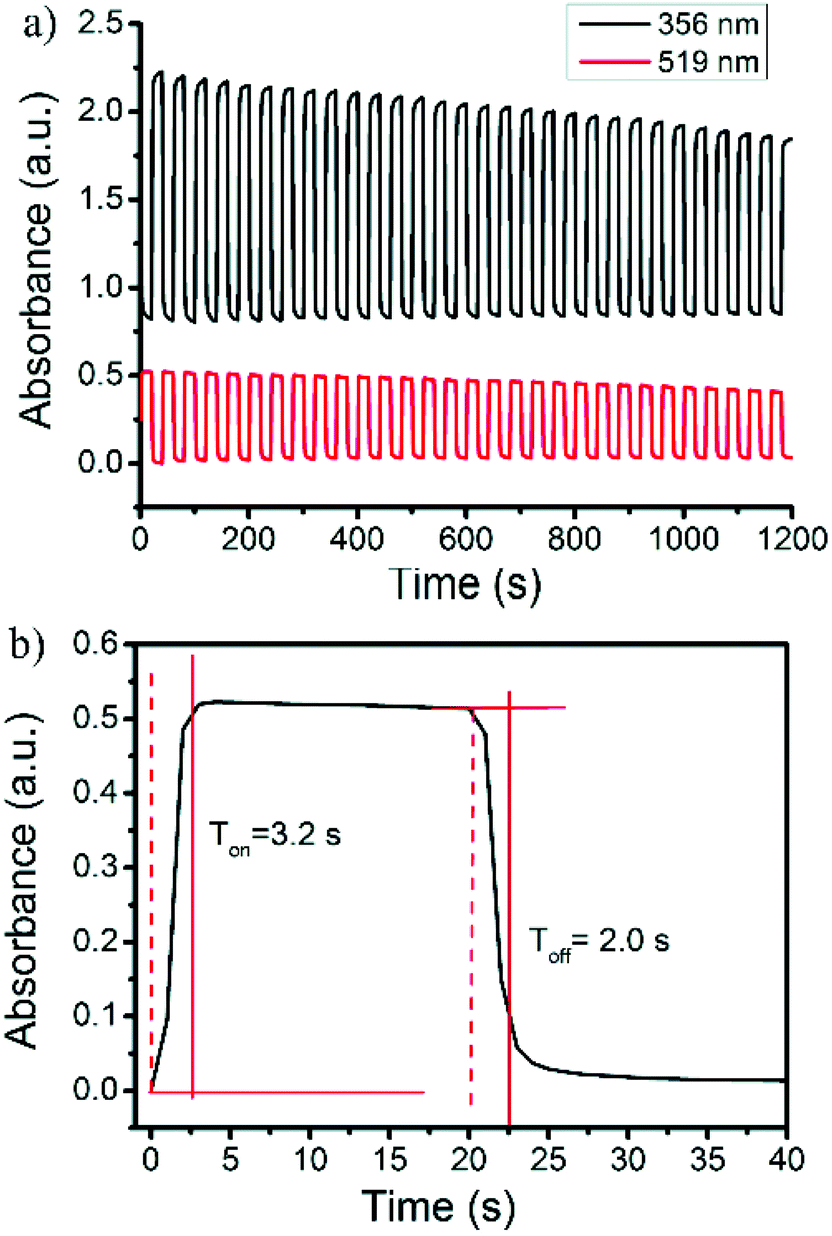 | ||
| Fig. 5 (a) Repeated absorbance changes at 356 nm and 519 nm and (b) enlarged change at 356 nm upon electrochromic switching between 0 and 0.8 V of the 3,6-Cbz-EDOT film on the ITO-coated glass plate. | ||
The proportionality factor that relates the optical absorbance change of an electrochrome at a given wavelength (ΔA) to the density of the injected/ejected electrochemical charge necessary to induce a full switch (Qd) is called the coloration efficiency (CE). CE values are inherent to the electrochromic material under characterization, some being intensely colored by nature in at least one redox state which others display fainter tones over their doping/dedoping cycles regardless of the deposited film thicknesses. The quantitative determination of the CE was estimated using the following equation:
| CE = ΔA/Qd | (1) |
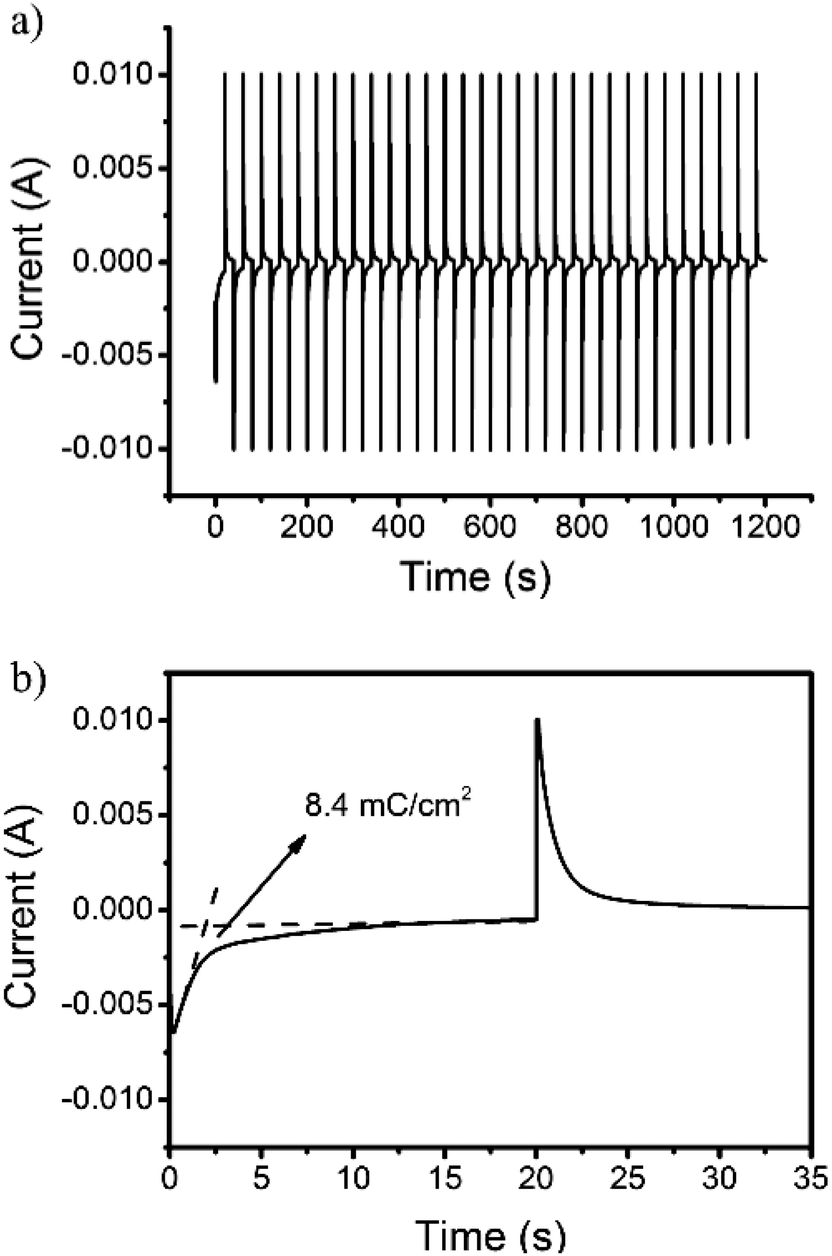 | ||
| Fig. 6 Current consumption upon (a) repeated and (b) enlarged electrochromic switching between 0 and 0.8 V of the 3,6-Cbz-EDOT on the ITO-coated glass plate. | ||
Conclusions
In summary, the microwave-assisted direct arylation polymerization of dibromocarbazole derivatives and EDOT was investigated. A short polymerization time was the prominent feature of the microwave heating. Only the 3,6-dibromocarbazole derivative afforded the corresponding soluble polymer, 3,6-Cbz-EDOT, whereas the other counter dibromocarbazole monomers produced insoluble gels (2,7-dibromocarbazole) and oligomers (1,8-dibromocarbazole). The electrochromic application of 3,6-Cbz-EDOT was investigated based on the electroactive structure of the 3,6-carbazole-based polymers. The 3,6-Cbz-EDOT films showed two-step color changes from light-yellow to red and finally to dark blue under a continuous potential increase. The detailed analysis of the electrochromic behavior revealed that the first step was fully reversible with the fast response time of 2.0 s and the coloration efficiency of 61.9 cm2 C−1. This is the first report that demonstrates the advantageous features of the 3,6-dibromocarbazole monomer over the other symmetric dibromocarbazole monomers in the microwave-assisted direct arylation polymerization. In addition, it was clearly shown that the resulting 3,6-Cbz-EDOT is a very promising electrochromic polymer.Acknowledgements
This work was supported by JSPS KAKENHI Grant Number 26620173, Kurata Memorial Hitachi Science and Technology Foundation, Tokuyama Science Foundation, TOBE MAKI Scholarship Foundation, Maekawa Houonkai Foundation, and the Support for Tokyotech Advanced Researchers (STAR).Notes and references
- J. V. Grazulevicius, P. Strohriegl, J. Pielichowski and K. Pielichowski, Prog. Polym. Sci., 2003, 28, 1297 CrossRef CAS.
- P. Sigwalt, G. Wegner, J.-F. Morin, M. Leclerc, D. Ades and A. Siove, Macromol. Rapid Commun., 2005, 26, 761 CrossRef CAS.
- N. Blouin and M. Leclerc, Acc. Chem. Res., 2008, 41, 1110 CrossRef CAS PubMed.
- P.-L. T. Boudreault, N. Blouin and M. Leclerc, Adv. Polym. Sci., 2008, 212, 99 CrossRef CAS.
- S. Beaupre, P.-L. T. Boudreault and M. Leclerc, Adv. Mater., 2010, 22, E6 CrossRef CAS PubMed.
- P.-L. T. Boudreault, S. Beaupre and M. Leclerc, Polym. Chem., 2010, 1, 127 RSC.
- J. Li and A. C. Grimsdale, Chem. Soc. Rev., 2010, 39, 2399 RSC.
- F. Dumur, Org. Electron., 2015, 25, 345 CrossRef CAS.
- X. T. Tao, Y. D. Zhang, T. Wada, H. Sasabe, H. Suzuki, T. Watanabe and S. Miyata, Adv. Mater., 1998, 10, 226 CrossRef CAS.
- J.-F. Morin and M. Leclerc, Macromolecules, 2001, 34, 4680 CrossRef CAS.
- F. Dierschke, A. C. Grimsdale and K. Müllen, Synthesis, 2003, 2470 CAS.
- T. Mori, T. Shinnai and M. Kijima, Polym. Chem., 2011, 2, 2830 RSC.
- N. Blouin, A. Michaud and M. Leclerc, Adv. Mater., 2007, 19, 2295 CrossRef CAS.
- N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. Neagu-Plesu, M. Belletete, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2008, 130, 732 CrossRef CAS PubMed.
- R. B. Aich, N. Blouin, A. Bouchard and M. Leclerc, Chem. Mater., 2009, 21, 751 CrossRef CAS.
- S. Beaupre, A.-C. Breton, J. Dumas and M. Leclerc, Chem. Mater., 2009, 21, 1504 CrossRef CAS.
- Y. Zou, D. Gendron, R. Badrou-Aich, A. Najari, Y. Tao and M. Leclerc, Macromolecules, 2009, 42, 2891 CrossRef CAS.
- S. G. Hahm, T. J. Lee, D. M. Kim, W. Kwon, Y.-G. Ko, T. Michinobu and M. Ree, J. Phys. Chem. C, 2011, 115, 21954 CAS.
- T. Umeyama, Y. Watanabe, E. Douvogianni and H. Imahori, J. Phys. Chem. C, 2013, 117, 21148 CAS.
- T. Michinobu, H. Osako and K. Shigehara, Macromol. Rapid Commun., 2008, 29, 111 CrossRef CAS.
- T. Michinobu, H. Osako and K. Shigehara, Macromolecules, 2009, 42, 8172 CrossRef CAS.
- S. Habuchi, H. Fujita, T. Michinobu and M. Vacha, J. Phys. Chem. B, 2011, 115, 14404 CrossRef CAS PubMed.
- T. Michinobu, H. Osako, C. Seo, K. Murata, T. Mori and K. Shigehara, Polymer, 2011, 52, 5756 CrossRef CAS.
- H. Fujita and T. Michinobu, Macromol. Chem. Phys., 2012, 213, 447 CrossRef CAS.
- A. Facchetti, L. Vaccaro and A. Marrocchi, Angew. Chem., Int. Ed., 2012, 51, 3520 CrossRef CAS PubMed.
- L. G. Mercier and M. Leclerc, Acc. Chem. Res., 2013, 46, 1597 CrossRef CAS PubMed.
- S. Kowalski, S. Allard, K. Zilberberg, T. Riedl and U. Scherf, Prog. Polym. Sci., 2013, 38, 1805 CrossRef CAS.
- H. Zhao, C.-Y. Liu, S.-C. Luo, B. Zhu, T.-H. Wang, H.-F. Hsu and H.-H. Yu, Macromolecules, 2012, 45, 7783 CrossRef CAS.
- K. Yamazaki, J. Kuwabara and T. Kanbara, Macromol. Rapid Commun., 2013, 34, 69 CrossRef CAS PubMed.
- J. Kuwabara, T. Yasuda, S. J. Choi, W. Lu, K. Yamazaki, S. Kagaya, L. Han and T. Kanbara, Adv. Funct. Mater., 2014, 24, 3226 CrossRef CAS.
- Y. Nohara, J. Kuwabara, T. Yasuda, L. Han and T. Kanbara, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1401 CrossRef CAS.
- F. Grenier, B. R. Aich, Y.-Y. Lai, M. Guerette, A. B. Holmes, Y. Tao, W. W. H. Wong and M. Leclerc, Chem. Mater., 2015, 27, 2137 CrossRef CAS.
- Y. Fujinami, J. Kuwabara, W. Lu, H. Hayashi and T. Kanbara, ACS Macro Lett., 2012, 1, 67 CrossRef CAS.
- S. J. Choi, J. Kuwabara and T. Kanbara, ACS Sustainable Chem. Eng., 2013, 1, 878 CrossRef CAS.
- T. N. Glasnov and C. O. Kapper, Macromol. Rapid Commun., 2007, 28, 395 CrossRef CAS.
- Y. Yuan, T. Michinobu, M. Ashizawa and T. Mori, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1013 CrossRef CAS.
- M. B. Gawande, S. N. Shelke, R. Zboril and R. S. Varma, Acc. Chem. Res., 2014, 47, 1338 CrossRef CAS PubMed.
- S.-H. Jung, H. K. Kim, S.-H. Kim, Y. H. Kim, S. C. Jeoung and D. Kim, Macromolecules, 2000, 33, 9277 CrossRef CAS.
- J. Kuwabara, K. Yamazaki, T. Yamagata, W. Tsuchida and T. Kanbara, Polym. Chem., 2015, 6, 891 RSC.
- M. Wakioka, N. Ichihara, Y. Kitano and F. Ozawa, Macromolecules, 2014, 47, 626 CrossRef CAS.
- J. Kuwabara, Y. Nohara, S. J. Choi, Y. Fujinami, W. Lu, K. Yoshimura, J. Oguma, K. Suenobu and T. Kanbara, Polym. Chem., 2013, 4, 947 RSC.
- J.-H. Chu, C.-C. Wu, D.-H. Chang, Y.-M. Lee and M.-J. Wu, Organometallics, 2013, 32, 272 CrossRef CAS.
- H. Fujita and T. Michinobu, Heterocycles, 2014, 89, 2346 CrossRef CAS.
- G. A. Sotzing, J. L. Reddinger, A. R. Katritzky, J. Soloducho, R. Musgrave, J. R. Reynolds and P. J. Steel, Chem. Mater., 1997, 9, 1578 CrossRef CAS.
- D. Witker and J. R. Reynolds, Macromolecules, 2005, 38, 7636 CrossRef CAS.
- P. Taranekar, T. Fulghum, A. Baba, D. Patton and R. Advincula, Langmuir, 2007, 23, 908 CrossRef CAS PubMed.
- R. Ponnapati, M. J. Felipe, V. Muthalagu, K. Puno, B. Wolff and R. Advincula, ACS Appl. Mater. Interfaces, 2012, 4, 1211 CAS.
- N. Kobayashi, K. Teshima and R. Hirohashi, J. Mater. Chem., 1998, 8, 497 RSC.
- P. M. Beaujuge and J. R. Reynolds, Chem. Rev., 2010, 110, 268 CrossRef CAS PubMed.
- P. M. Beaujuge, C. M. Amb and J. R. Reynolds, Acc. Chem. Res., 2010, 43, 1396 CrossRef CAS PubMed.
- H.-J. Yen and G.-S. Liou, Polym. Chem., 2012, 3, 255 RSC.
- H.-J. Yen and G.-S. Liou, Chem. Commun., 2013, 49, 9797 RSC.
- J.-H. Wu and G.-S. Liou, Adv. Funct. Mater., 2014, 24, 6422 CrossRef CAS.
- D. Chao, S. Wang, B. T. Tuten, J. P. Cole and E. B. Berda, Macromolecules, 2015, 48, 5054 CrossRef CAS.
- X. Ma, H. Niu, H. Wen, S. Wang, Y. Lian, X. Jiang, C. Wang, X. Bai and W. Wang, J. Mater. Chem. C, 2015, 3, 3482 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Monomer synthesis, NMR characterization, and thermal analyses. See DOI: 10.1039/c6py00381h |
| This journal is © The Royal Society of Chemistry 2016 |