
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Green chain-shattering polymers based on a self-immolative azobenzene motif†
Hatice
Mutlu
 ab and
Christopher
Barner-Kowollik
*ab
ab and
Christopher
Barner-Kowollik
*ab
aPreparative Macromolecular Chemistry, Institut für Technische Chemie und Polymerchemie, Karlsruhe Institute of Technology (KIT), Engesserstr. 18, 76131 Karlsruhe, Germany. E-mail: christopher.barner-kowollik@kit.edu
bSoft Matter Synthesis Laboratory, Institut für Biologische Grenzflächen (IBG), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 22nd February 2016
Abstract
A chain-shattering polymer system consisting of nontoxic, renewable resource-based monomers via acyclic diene metathesis (ADMET) chemistry is introduced. Amphiphilic triblock copolymers with apparent molecular weights in the range from 10 to 23 kDa are synthesized using a monofunctional polyethylene glycol monoacrylate, which acts as a selective chain-transfer agent during the polymerization process. Most importantly, the functional polymers possess repetitive midchain azobenzene moieties imparting them with self-immolative properties. By virtue of the enzyme degradable azobenzene chain elements, the amphiphilic macromolecules can be efficiently degraded via a self-immolative reaction into small fragments. The construction of the macromolecules along with their degradation is evidenced by nuclear magnetic resonance spectroscopy, electrospray ionization mass spectrometry and size exclusion chromatography. In addition, the triggered degradation leads to a strong reduction in the UV absorptivity of the polymeric material.
Introduction
The current drive towards sustainable processes and environmental responsibility in macromolecular science requires alternative polymers with improved degradability based on renewable source-derived materials.1 The use of biomass, for instance castor oil or many other plant oils, has been recognized as one of the most promising routes, especially as a substitute source of chemicals which prospectively may replace the known diminishing petrochemicals.2 Importantly, degradation is not dependent on the origin of the building blocks but only on the chemical linkages present in the polymer.3 Nevertheless, one approach to obtain a biodegradable material in a sustainable fashion is to synthesize polymers constituted of a polymer backbone based on renewable source-derived components in which one or more degradable units (hydrodegradable, photodegradable or biodegradable) are embedded.4 However, the variety of degradation units in such materials remains limited. Thus, increasing the diversity of degradable polymers – including those derived from renewable resources – might allow for better matching soft matter materials to specific applications. The azobenzene functionality is unique as it is capable of responding to changes in environmental conditions5 as well as being cleavable under an enzymatic impact (in vivo) via a two-electron reduction process.6,7 Despite the fact that azo containing compounds display a high structural diversity, the degradation is induced by specific enzymes, among which are azoreductases and glycosidases.6 While the azo bond reduction has been described in both anaerobic and aerobic bacteria, anaerobic conditions are prevalent in the human body.8,9 The operation of these enzymes has common mechanistic features as they all catalyse redox reactions and moreover exhibit relatively wide substrate specificities.6 More specifically, azoreductases – present and active both in the gut microflora and mammalian tissues (such as the skin9 and the liver)10 – are redox cofactor11 (i.e. flavin mononucleotide (FMN) or flavin adenine dinucleotide (FAD)) dependent enzymes that require the addition of a cofactor such as nicotinamide adenine dinucleotide phosphate (NAD(P)H) as electron donors for the reduction of the azo linkage. Thus, the reductive cleavage of the azobenzene bond in the presence of azoreductase results in a self-immolative aniline derivative, which degrades entirely by intramolecular 1,6-elimination reactions via iminoquinone methide intermediates to 4-aminobenzyl alcohol, a by-product with low toxicity.12,13 The driving force is – in addition to the increase in entropy – the irreversible formation of CO2. Substantial efforts have been devoted to designing polymers incorporating azo moieties into the main chain including poly(ether ester),14 polyurethanes,15 and polyamides,16 however these are rarely discussed with regard to the structural features of the azo compound. The azo unit can accelerate the rate of reduction and in turn the degradation of the polymer in a self-immolative manner. Besides the typical self-immolative polymers end-capped with variable trigger responsive functional groups,17 there are rare examples of self-immolative polymeric systems which possess multiple triggers linked either as side chain motifs or directly within the backbone, termed chain-shattering polymers.Chain-shattering polymers spontaneously disintegrate along the main chain into small components only after activation by a specific stimulus. The systems whose multiple triggers are connected as pendant groups are generally synthesized using classical step-growth copolymerization. Typically, activated bifunctional self-immolative linkers capped by a trigger are reacted in a step-growth manner with bifunctional comonomers to yield polymers which degrade either through a consecutive quinone methide rearrangement (via 1,4-/1,6-elimination) or intramolecular cyclization upon a specific stimulus.18 Alternatively, the toolbox for the synthesis of chain-shattering polymers was expanded by employing chain-growth ring-opening polymerization (ROP)19 and olefin metathesis chemistry20 (specifically acyclic diene metathesis). However, none of the systems described above were targeted to develop a chain-shattering polymer whose enzyme-labile self-immolative triggers are directly incorporated as repetitive chain motifs within the polymer backbone. Thus, with the aim to critically diversify the available chain-shattering polymer toolbox and enlarge the variety of self-immolative monomers, we describe the development of the strategy presented in Fig. 1.
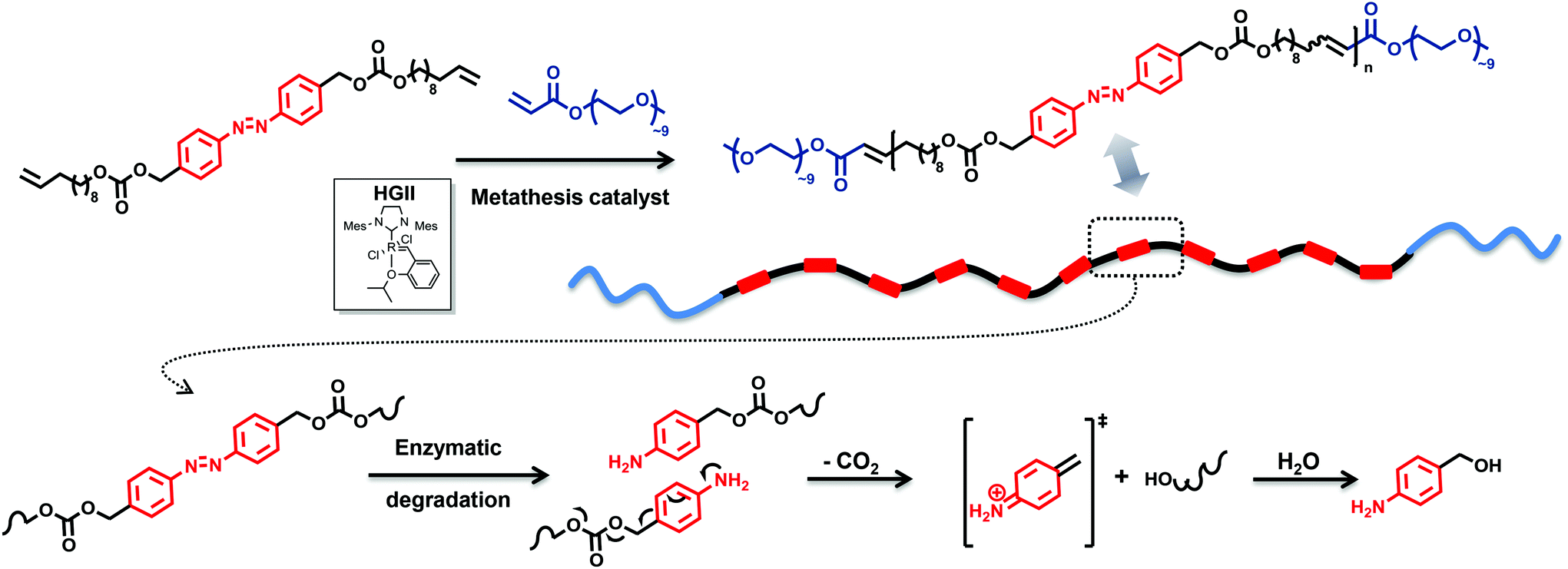 | ||
| Fig. 1 Enzyme-responsive chain-shattering amphiphilic ADMET triblock copolymer containing multiple self-immolative degradable azobenzene moieties as a repetitive constraint. | ||
Results and discussion
The construction of polymers conjugated with triggers in the backbone from a building block with two terminal alkenes and an azo moiety serving as an enzyme-responsive group will result in elimination reactions at each repeating unit after the application of a trigger, here an enzyme. Thus, a depolymerisation reaction will occur by means of a chain-shattering mechanism, thereby accelerating the depolymerisation. The challenge and innovation inherent to the current approach is the design of an enzyme-responsive polymer segment. Inspired by the synthetic possibilities to introduce repetitive constraining elements via acyclic diene metathesis (ADMET) polymerization,21 a precision polyolefin containing a degradable azobenzene moiety as a repetitive constraint (Fig. 1) is introduced. Indeed, Wagener has explored the use of this polymerization technique to elegantly synthesize precision polyolefins containing diverse functional groups.22 Moreover, Meier and colleagues used the ADMET polymerization of renewable bio-based α,ω-dienes incorporating hydrodegradable linkages (such as anhydride, esters)23 to yield (bio)degradable polymers. Undoubtedly, fatty acids and their derivatives are suitable components for the preparation of (bio)degradable polymers since they are hydrophobic entities that naturally occur in the body, and their application as building blocks for such polymers introduces additional pliability.24 Thus, 10-undecene-1-ol, a subsidiary product of castor oil,2 was used as a renewable-based alcohol derivative for the synthesis of the self-immolative α,ω-diene monomer M1 (Fig. 2). The synthesis of a suitable monomer for ADMET polymerization was achieved by placing a carbonate moiety as a leaving group at the benzylic position of the azobenzene functionality in the hydroxyl alkene-based M1 (for preparative details refer to the ESI†). Our efforts for ensuring the environmentally benign nature of M1 were not limited to biomass as a source. We employed glucose as a ‘green’ reducing agent for the synthesis of the azo derivative, 4,4′-bis(hydroxymethyl)-azobenzene (Fig. 2) starting from 4-nitrobenzyl alcohol. Moreover, the aforementioned nitro benzyl derivative was obtained via the oxone mediated oxidation of 4-aminobenzyl alcohol.25 In addition, 1,1-carbonyl diimidazole (CDI)26 was used as a safe and non-toxic reagent to couple the azo diol moiety with 10-undecen-1-ol yielding the self-immolative α,ω-diene monomer M1 (Fig. 2).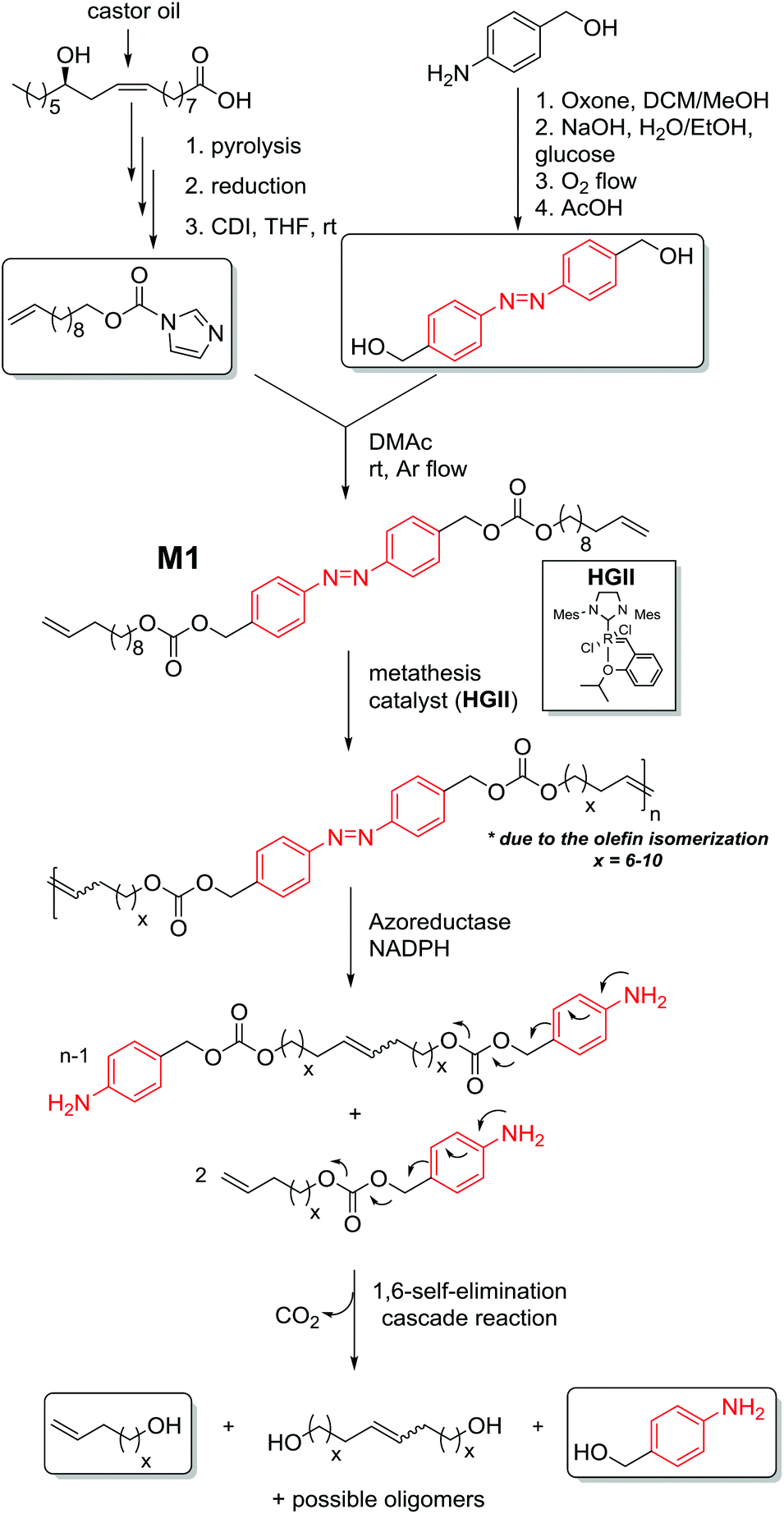 | ||
| Fig. 2 Synthesis of the monomer, M1, possessing a self-immolative character and the respective ADMET polymer P1. The plausible cleavage of azobenzene linkages in P1 upon enzymatic action and subsequent 1,6-self elimination to smaller molecular weight species. | ||
The synthesized azo derivative, α,ω-diene monomer M1, enabled the introduction of a degradable constraint every 20 carbons in the polymer obtained by ADMET chemistry. Polymer P1, with an apparent molar mass of 25 kDa and with a dispersity of 2.0 (as determined by size exclusion chromatography (SEC) relative to polystyrene standards), was generated in the presence of 1 mol% second-generation Hoveyda-Grubbs (HGII) catalyst per double bond (i.e. 2 mol% HGII catalyst/molecule) under reduced pressure (compare Fig. 2 and Table S1 in the ESI†). HGII was the choice of catalyst due to its outstanding tolerance and activity toward coordinative and non-coordinative heteroatom containing dienes.27
To remove the quenched metathesis catalyst from the crude polymer reaction mixture, the mixture was eluted with chloroform and passed through a short plug of silica/basic alumina. In comparison with the 1H NMR of the corresponding monomer M1 (Fig. 3a), a clean and complete transformation of the α,ω-diene to the unsaturated polymer P1 was observed (Fig. 3b). 1H NMR spectroscopic analysis of the polymer revealed resonances characteristic of the newly formed internal double bond at δH 5.35 ppm. Furthermore, it is worth highlighting that along 1H NMR, 13C NMR analysis also demonstrated that the azo functionality remained intact, indicating the compatibility of ADMET chemistry with the azobenzene unit (compare Fig. S2 in the ESI†).
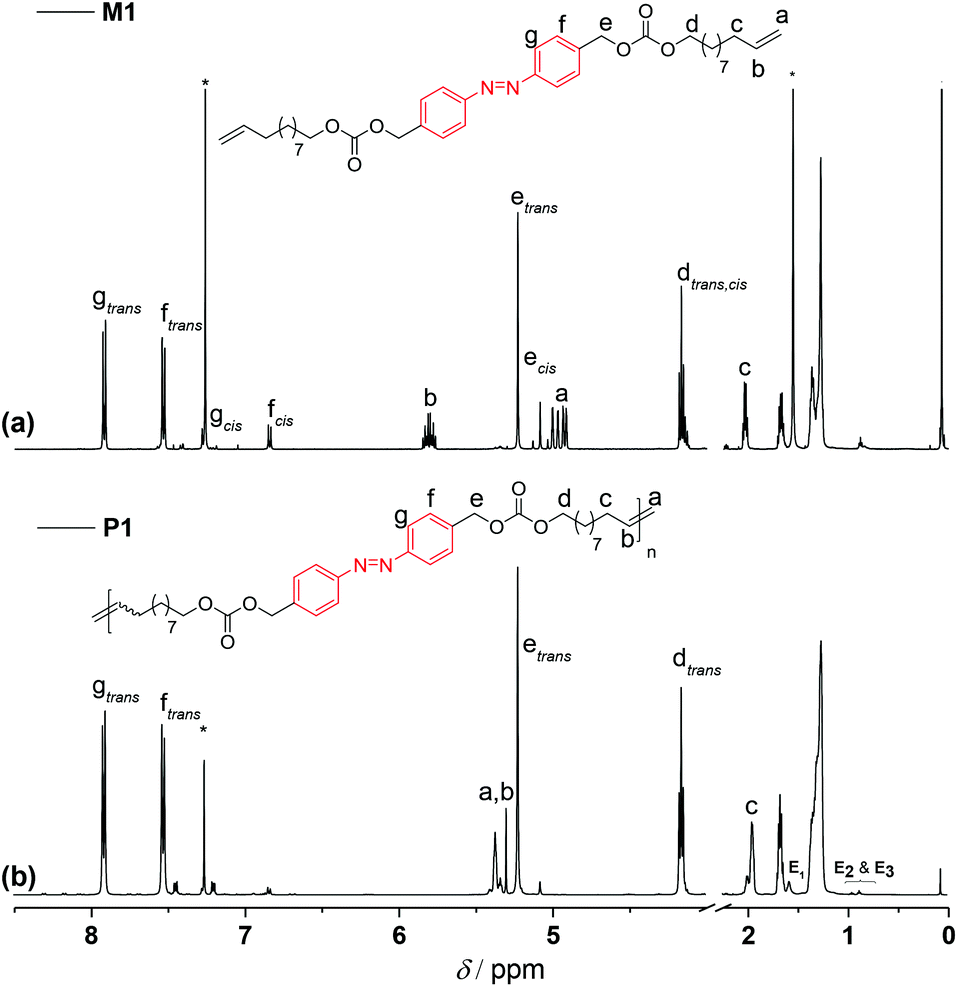 | ||
| Fig. 3 1H NMR (500 MHz, CDCl3) spectra of (a) the azo derivative α,ω-diene monomer M1 and (b) of a representative homopolymer P1 (shown in Table S1 in the ESI†). The end groups associated with the olefin isomerization in the homopolymer P1 are indicated as E1, E2, and E3, respectively. | ||
To determine the distribution and the degree of polymerization (DPn) of the polymer, P1 was subjected to matrix assisted laser desorption ionization time-of-flight mass spectrometry. However, due to the broad dispersity of P1, initial attempts to obtain mass spectra representative of the complete distribution of the polymer failed. On the other hand, the undesired isomerization side reaction28 which usually occurs during ADMET polymerization facilitated to define the DPn by 1H NMR via end-group resonance integration, yielding a DPn of 42 for P1 (Mn of 25.5 kDa, refer to the NMR discussion part in the ESI†). The end groups associated with the olefin isomerization are indicated as E1, E2, and E3, respectively (refer to the 1H NMR assignments in Fig. 3). Commonly, additives such as benzoquinone29 are used to suppress the isomerization, however any attempts to employ benzoquinone during the ADMET polymerization reactions failed. It has been reported that the azobenzene group does not undergo any 1,4-cycloaddition with benzoquinone,30 yet the polymers obtained from the polymerization of M1 in the presence of 1,4-benzoquinone were not completely soluble in common organic solvents, suggesting that some limited crosslinking reactions might have taken place.
In order to evaluate the efficiency of the anticipated elimination reactions in the new polymer system, the reductive cleavage of the azo functionality within M1 itself was assessed by NMR spectroscopic methods. Na2S2O4, a substitute that mimics the azoreductase enzyme, was employed as the reducing agent.31 Upon treatment with 1 equivalent of the reducing agent in phosphate-buffered saline (PBS, pH 7.4),32 the reaction was incubated at 37 °C for several hours. 1H NMR analysis of the final reaction mixture showed far different results than expected, i.e. no degradation. The limited solubility of the hydrophobic M1 in PBS is identified as the main reason for the incomplete reduction.8 However, this attempt also clearly evidenced the hydrolytic stability of M1. To facilitate a complete reduction, a mixture of dichloromethane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol:water (3:20:20), which has proven to be a useful solvent for the conversion of azobenzenes into N,N′-diarylhydrazines,33 was employed to drive the reduction to the corresponding aniline derivative. By using 5.5 equivalents of Na2S2O4, a loss of the characteristic strong yellow colour of M1 is detected within 40 min, and the reduction was confirmed by UV-Vis and 1H NMR (Fig. 4 and S3 in the main text and the ESI,† respectively). The UV–visible absorption spectra of M1 were characterized by a band at 330 nm, the intensity of which decreased gradually with time during the degradation process (Fig. 4). For the complete reduction, the necessary reaction time was 5 h in total.
:methanol:water (3:20:20), which has proven to be a useful solvent for the conversion of azobenzenes into N,N′-diarylhydrazines,33 was employed to drive the reduction to the corresponding aniline derivative. By using 5.5 equivalents of Na2S2O4, a loss of the characteristic strong yellow colour of M1 is detected within 40 min, and the reduction was confirmed by UV-Vis and 1H NMR (Fig. 4 and S3 in the main text and the ESI,† respectively). The UV–visible absorption spectra of M1 were characterized by a band at 330 nm, the intensity of which decreased gradually with time during the degradation process (Fig. 4). For the complete reduction, the necessary reaction time was 5 h in total.
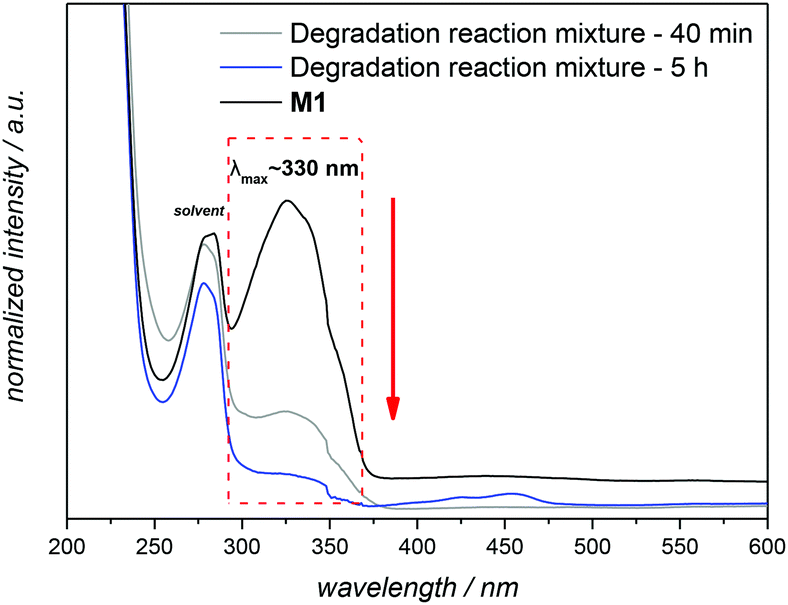 | ||
| Fig. 4 UV-visible spectra illustrating the degradation of M1 in the presence of 5.5 equivalents of Na2S2O4 in a mixture of dichloromethane:methanol:water at reflux temperature (for details see section A.8 in the ESI†). Spectra recorded in THF at 25 °C. | ||
Once the reduction of M1 had been established, the subsequent step was to perform reduction studies on the polymer itself. While Na2S2O4 is a specific, bio-compatible34 and effective chemical stimulus that may be useful in some applications, it was also of interest to demonstrate that the reduction of the specified system is possible under biologically relevant conditions, as this would significantly expand the utility of the polymer. Thus, the following experiments were conducted with the enzyme azoreductase (commercially known as DT-diaphorase) in the presence of the coenzyme NADPH.
The initial degradation studies were performed in the same manner as those previously reported by Khan et al., by suspending the ADMET polymer P1 in PBS, and subsequently exposing it to a mixture of the enzyme azoreductase and the coenzyme NADPH, followed by incubation at 37 °C for a period of 12 h. As noted, a crucial point in the reduction reactions is to avoid oxygen due to the fact that dioxygen can serve as an alternative electron acceptor, thus inhibiting the azoreductase activity.35 Accordingly, the reduction was performed under anaerobic conditions. To remove the cofactor and its by-products, the solution was dialyzed against water. Unexpectedly, 1H NMR and SEC analysis of P1′ (Fig. S4 and S5 in the ESI,† respectively) did not indicate any degradation, moreover no specific colour change was observed during the reaction. On the basis of the 1H NMR analysis, the trans–cis isomerisation of the azo bond was detected as the only significant change (compare Fig. S4 in the ESI†). Indeed, this transformation can be of particular interest for future studies in the context of the reduction-sensitive moiety due to the fact that cis azobenzene has an increased rate of reduction relative to the trans isomer.36 Nevertheless, having in mind that the specified enzyme can show ‘degressive’ prolonged stability under the aforementioned conditions, the influence of the reaction time was subsequently investigated. However, even after monitoring the degradation reaction over a period of 11 days (P1″), there was only a minimal decrease in the molecular weight of the polymer (due to a possibly competing hydrolytic mechanism) as observed in the SEC chromatogram of the isolated product (Fig. S6 in the ESI†).
The ineffective degradation of P1 clearly indicates that to achieve the desired degradability, the polymer structure needs to be adjusted. Indeed, an advantage of the ADMET polymerization approach is the possibility to use olefins other than the terminal ones not only to introduce defined end groups but also as a way to obtain ABA triblock copolymers with finely selected solubility properties. Thus, a poly(ethylene glycol) (PEG) moiety possessing a methyl ether acrylate functionality, considered to be inert and possessing a low level of toxicity,37 can provide the desired water solubility. By incorporation of water soluble blocks in the polymer system, one can avoid the potential steric inhibition of the azoreductase. Thus, an amphiphilic copolymer was prepared under similar conditions to those for the ADMET homopolymer, but with the addition of a selective and irreversible chain-stopper (CS), a monofunctional PEG methyl ether acrylate (Mn = 480 Da) (compare Fig. 1, 2 and S7, respectively in the main text and the ESI†).38 The addition of variable amounts of CS (2, 5, 10 and 20 mol%, respectively) to the polymerization mixture led to efficient control of the molecular weight of the resulting polymers P2–P5 (see Fig. 5 as well as Table S1, entries 2–5 in the ESI†), thus resulting in defined amphiphilic ABA triblock copolymer architectures. The Mn values determined for the copolymers by SEC are slightly different compared with the theoretical ones (Table S1 in the ESI†), since SEC molecular weights were determined relative to the polystyrene (PS) standards. The ABA triblock copolymers were tested for their degradation behaviour. The triblock P4, synthesized with 10 mol% CS in the presence of 1 mol% second-generation Hoveyda-Grubbs (HGII) catalyst per double bond (i.e. 2 mol% HGII catalyst/molecule) under reduced pressure, was subjected to the same degradation conditions as the homopolymer P1 for reaction times of 12, 24 and 36 h, respectively. The degradation mixture of the polymer incubated for 36 h indicated a similar degree of degradation to that after 24 h (compare Fig. S8 in the ESI†). We suggest that changes in the pH (from the initial pH value of 7.4 to 6.5) during the degradation due to increased CO2 levels in the sealed vials likely affected the degree of degradation. Indeed, CO2 is acidic in nature, thus a high CO2 saturation will lead to low pH and consequently reduce the activity of azoreductase, which is mainly active in the pH range of 5.5 to 8.3 (optimal reduction conditions being structure specific).39
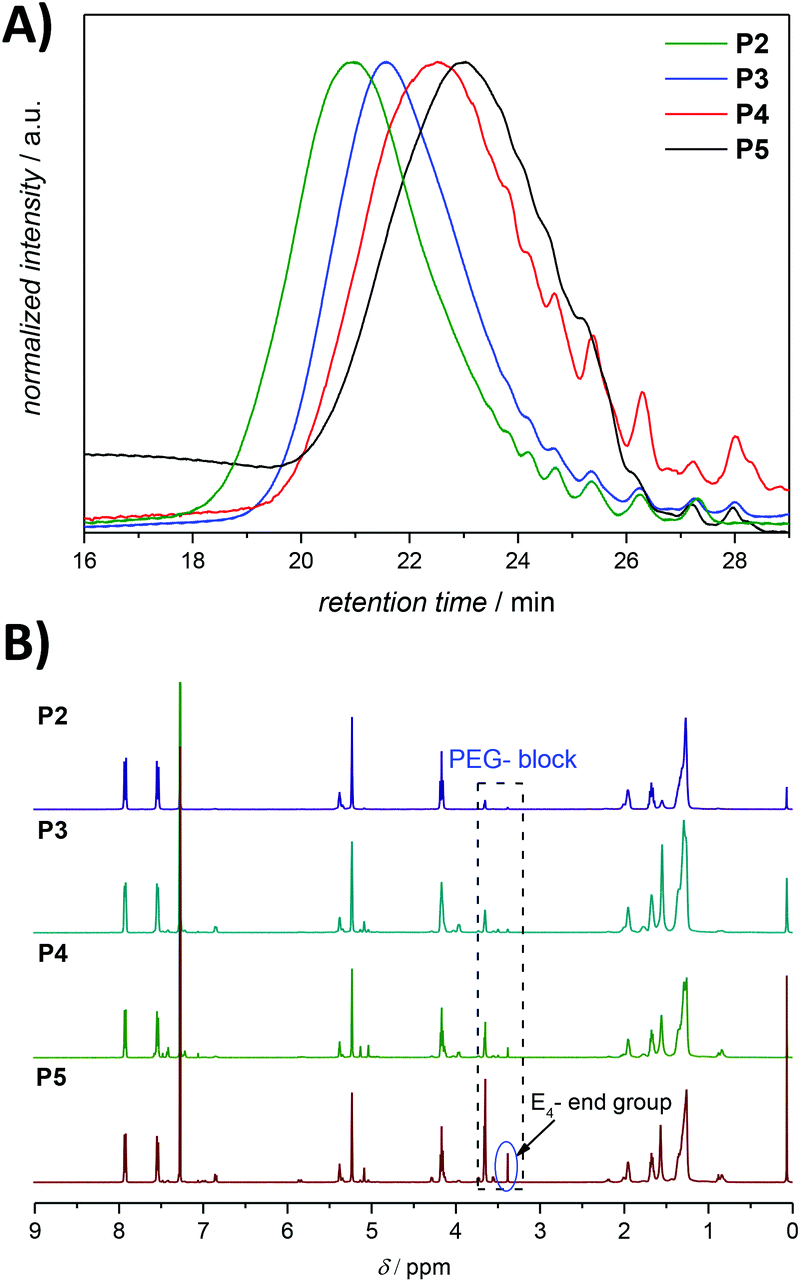 | ||
| Fig. 5 (A) SEC chromatograms for ADMET amphiphilic triblock copolymers P2–P5 presented in Table S1 in the ESI.† The SEC traces were measured in THF at 30 °C. (B) 1H NMR (500 MHz, CDCl3) spectra of the amphiphilic triblock copolymers P2–P5 presented in Table S1,† respectively. Refer to Fig. 3 for the assignment of key resonances associated within the aromatic region. The major end group associated with the olefin isomerization in the copolymers P2–P5 is indicated as E4. | ||
UV-vis spectroscopy analysis established a broad adsorption band at 330 nm for the amphiphilic triblock copolymer P4, which is typical of the azobenzene chromophore having a dihydroxy substitution pattern at the para-position.40 The examination of the purified degradation mixture after 24 h treatment, P4″ by UV-vis spectroscopy confirmed that the azobenzene linkages were cleaved, as no absorption signal could be observed at 330 nm after the enzymatic treatment of polymer P4 (refer to Fig. 6B).
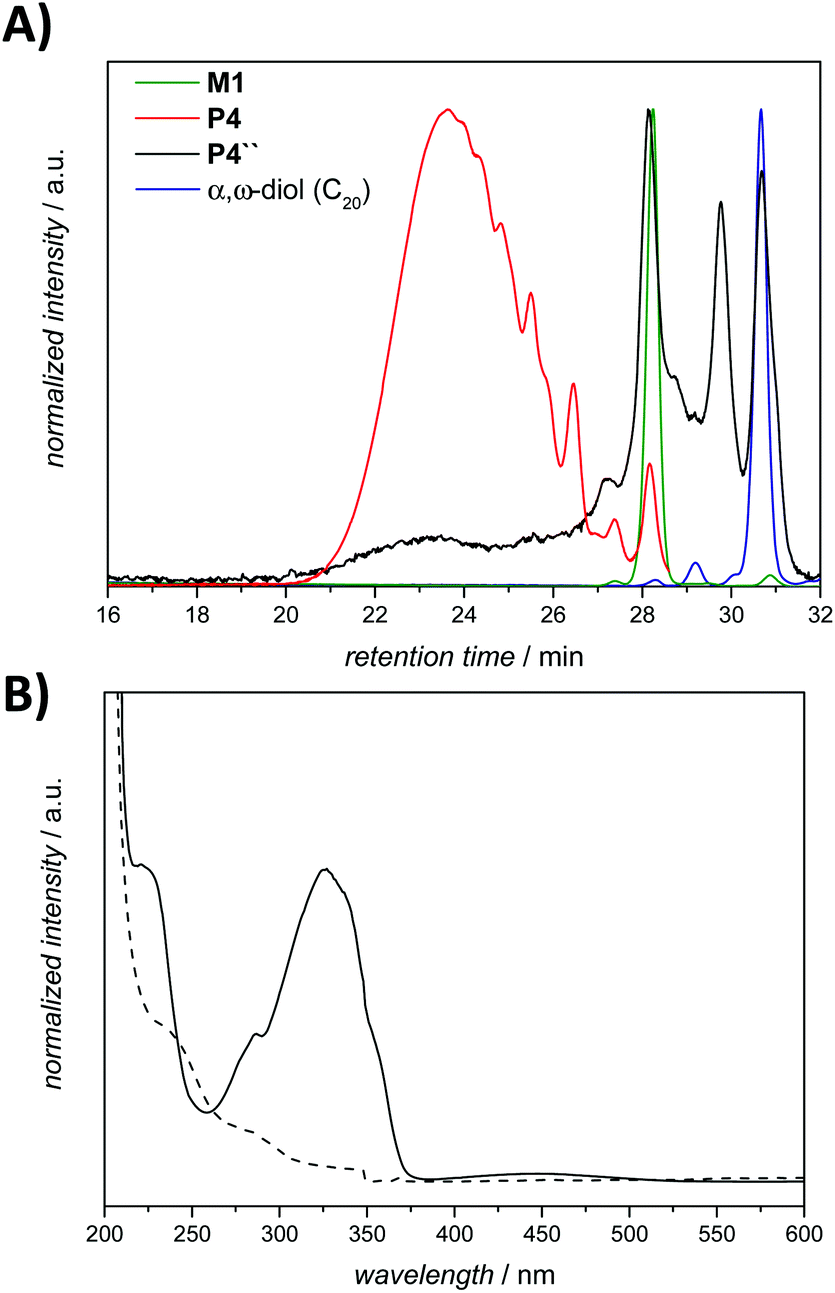 | ||
| Fig. 6 (A) SEC chromatograms obtained in THF for monomer M1 (green line), polymer P4 (red line, due to an artefact of the measurement, the elugram finished at 28.6 min), the resulting mixture after treatment with the enzyme for 24 h – P4″ (black line), and the model degradation product α,ω-diol (blue line), respectively. (B) UV-Vis spectra of the polymer P4 (solid line) and the reaction mixture P4″ after the enzymatic reaction and purified (dashed line). The sample concentration is 0.4 mg mL−1 in CH2Cl2. Solvent cut-off wavelengths L0: 245 and L1: 230 nm, respectively (λmax: 253 nm for CH2Cl2). | ||
In addition, in the 1H NMR spectra, resonances corresponding to the expected depolymerization products are observed to increase in intensity, while the resonances in the aromatic region associated with the polymer decreased (Fig. S9 in the ESI†). Although SEC, UV-Vis and 1H NMR spectroscopy evidence the successful degradation of the amphiphilic triblock copolymer, these data alone do not provide insight into the exact composition of the small molecule degradation fragments. Therefore, additional high resolution ESI-MS allowed assigning the degradation products, evidencing all possible fragments which can arise (Fig. 7). Analysis of a solution of P4″ in CH2Cl2:MeOH (3:1, v/v) by using a Q-Exactive Orbitrap mass spectrometer indicated that F1–F4 (shown in Fig. 7) were the major degradation products, suggesting that the desired degradation occurred at the majority of the self-immolative repeating units. Surprisingly, both the α,ω-diol (F4, m/zcalc = 335.29, [M + Na]+) and the unstable species F2 (m/zcalc = 633.38, [M + Na]+), before its 1,6-elimination to form F4, were detected. Moreover, we were able to detect the fragment F1 (m/zcalc = 1253.77, [M + Na]+) before its self-immolation to F3 (m/zcalc = 647.40, [M + Na]+), which is the hydrophilic block of the polymer P4. Therefore, the mass difference between the parent ion m/z 1253.7646 and the product ion m/z 647.3977 can be tentatively assigned to the repeating unit of the hydrophobic part of the triblock copolymer P4 with a nominal mass of 606.3669 Da. The fragments F1 and F3 show the typical PEG pattern with mass distributions of the adjacent peaks separated by 44.08 Da, corresponding to the mass of the repeat unit, ethylene oxide.41 In addition, the expected compound with m/z 123.07 (4-aminobenzyl alcohol) was not detected. However, as demonstrated by using a standard of 4-aminobenzyl alcohol, this compound is not stable under the experimental conditions (MS), and several coupling reactions could have taken place.
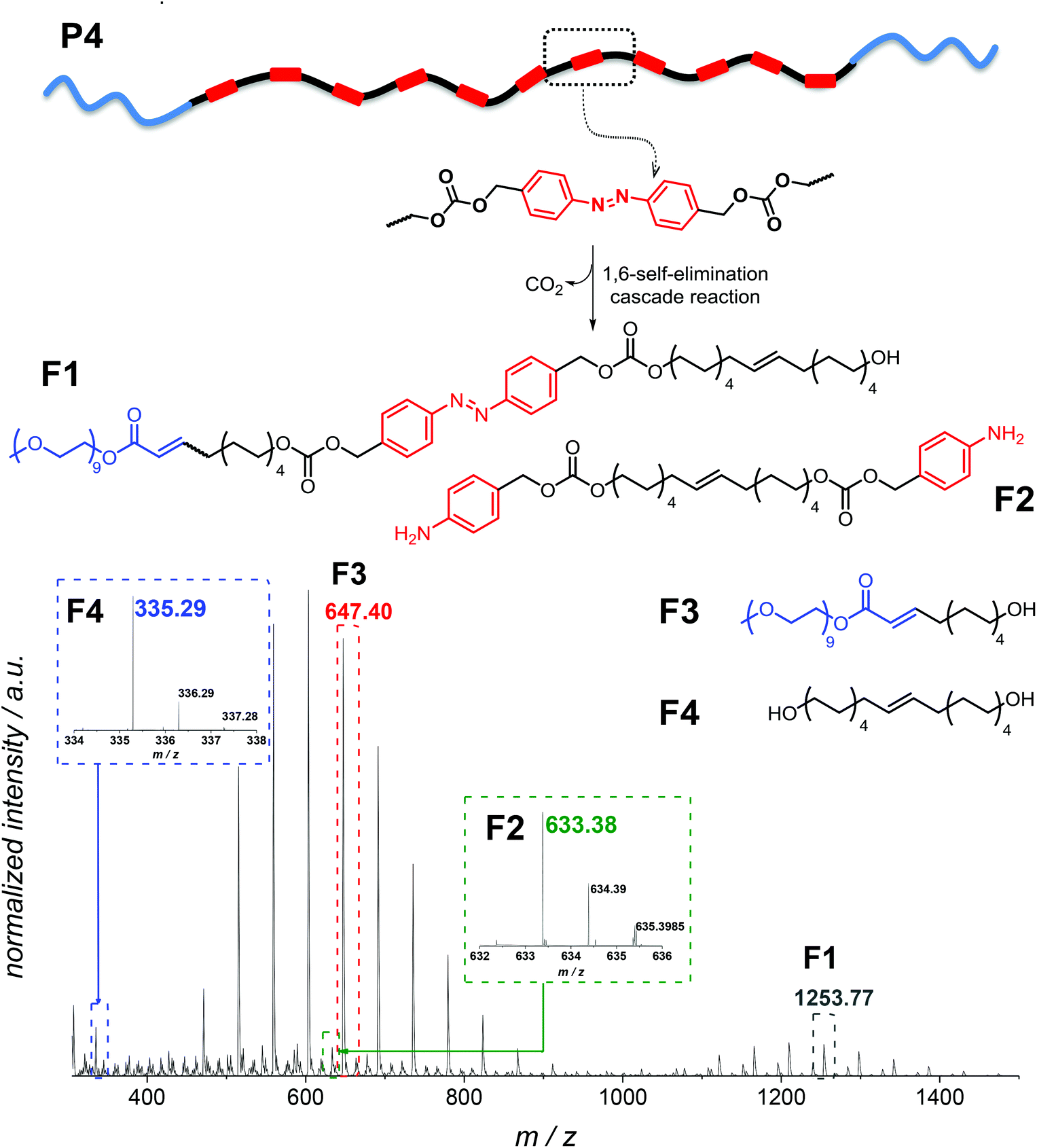 | ||
| Fig. 7 High resolution ESI-MS analysis of the degradation fragments of P4 after enzymatic treatment for 24 h (P4″). | ||
To confirm that the observed degradation products are associated with the proposed 1,6-elimination reaction, control reactions were performed with polymer P4 in water without any reducing agent for a period of 24 h (compare the SEC traces in Fig. S10 in the ESI†). Although the control reaction indicates a slight shift compared to the initial polymer P4, it is clear that the proposed reductive/self-immolative mechanism is dominant in comparison with a possibly competing hydrolytic mechanism, which is largely inoperational unless the exposure time is very long (compare Fig. S6 in the ESI†).
Conclusions
In summary, we introduce a strategy for the synthesis of renewable resource-based amphiphilic ADMET polymers which possess self-immolative degradable enzyme-triggered azobenzene units, thus establishing a new class of chain-shattering polymers. Upon treatment with an enzyme and a coenzyme, the azo units were transformed into amine groups, which triggered the sequential self-immolative process to degrade the polymer main chain. The in situ formed quinone methide intermediates were quenched by water molecules to form stable and safe hydroxymethylphenol derivatives. The degradation of the introduced system contrasts with conventional self-immolative polymers, which degrade from one chain end to the other. The chemistry of the system is unique since the polymer degrades from the trigger cleavage side to its tail. In addition, the enzyme employed for the degradation, DT-diaphorase, does not form any free-radical or reactive oxygen intermediates during the degradation process. The developed systems thus open the prospect to function as delivery vehicles in cancer cells/tissues in the human body which show higher turnovers of NADPH than in normal cells/tissues.42 The introduced system is a prime example of a stimuli responsive biodegradable polymer and future studies will investigate the applicability of the new polymeric material for single chain nanoparticle synthesis43 for applications in imaging and delivery.Acknowledgements
C. B.-K. acknowledges support for the current project in the context of the Biointerfaces in Technology and Medicine program (BIFTM) of the Helmholtz association as well as the Karlsruhe Institute of Technology (KIT). Additional funding from the German Research Council (Large Equipment Scheme) is acknowledged. The authors thank Prof. Michael A. R. Meier (KIT) for GC-MS access and Dr Ana Beloqui Elizazu (KIT) for the fruitful discussion on the enzymatic degradation experiments.Notes and references
- K. Yao and C. Tang, Macromolecules, 2013, 46, 1689 CrossRef CAS.
- H. Mutlu and M. A. R. Meier, Eur. J. Lipid Sci. Technol., 2010, 112, 10 CrossRef CAS.
- K. E. Uhrich, S. M. Cannizaro, R. S. Langer and K. M. Shakesheff, Chem. Rev., 1999, 99, 3181 CrossRef CAS PubMed.
- M. Mitrus, A. Wojtowicz and L. Moscicki, in Thermoplastic Starch, Wiley-VCH Verlag GmbH & Co. KGaA, 2010, pp. 1–33 Search PubMed; R. R. Karlsson and A.-C. Albertsson, Polym. Eng. Sci., 1998, 38, 1251 Search PubMed.
- T. Shang, K. A. Smith and T. A. Hatton, Langmuir, 2003, 19, 10764 CrossRef CAS; L. Ding, M. Xu, J. Wang, Y. Liao and J. Qiu, Polymer, 2014, 55, 1681 CrossRef.
- For detailed mechanistic information on such enzymatic degradation: J. Chacko and K. Subramaniam, Int. J. Environ. Sci., 2011, 1, 1250 Search PubMed; K. Golka, S. Kopps and Z. W. Myslak, Toxicol. Lett., 2004, 151, 203 CrossRef CAS PubMed.
- J. P. Brown, G. V. McGarraugh, T. M. Parkinson, R. E. Wingard and A. B. Onderdonk, J. Med. Chem., 1983, 26, 1300 CrossRef CAS PubMed; M. Saffran, G. S. Kumar, C. Savariar, J. C. Burnham, F. Williams and D. C. Neckers, Science, 1986, 233, 1081 Search PubMed; J. Kopeček, P. Kopečková, H. Brondsted, R. Rathi, B. Rihova, P.-Y. Yeh and K. Ikesue, J. Controlled Release, 1992, 19, 121 CrossRef; G. Van den Mooter, C. Samyn and R. Kinget, Int. J. Pharm., 1992, 87, 37 CrossRef; S.-Q. Gao, Z.-R. Lu, B. Petri, P. Kopečková and J. Kopeček, J. Controlled Release, 2006, 110, 323 CrossRef PubMed; J. Rao and A. Khan, J. Am. Chem. Soc., 2013, 135, 14056 CrossRef PubMed; J. Rao, C. Hottinger and A. Khan, J. Am. Chem. Soc., 2014, 136, 5872 CrossRef PubMed; A. D. Wong, T. M. Güngör and E. R. Gillies, ACS Macro Lett., 2014, 3, 1191 CrossRef; J. Rao and A. Khan, Polym. Chem., 2015, 6, 686 RSC.
- A. Stolz, Appl. Microbiol. Biotechnol., 2001, 56, 69 CrossRef CAS PubMed.
- R. L. Stingley, W. Zou, T. M. Heinze, H. Chen and C. E. Cerniglia, J. Med. Microbiol., 2010, 59, 108 CrossRef CAS PubMed.
- C. N. Martin and J. C. Kennelly, Carcinogenesis, 1981, 2, 307 CrossRef CAS PubMed.
- S. Deller, P. Macheroux and S. Sollner, Cell. Mol. Life Sci., 2008, 65, 141 CrossRef CAS PubMed.
- The resulting amines are then generally excreted from the body; the less soluble amines, which may be absorbed through the intestinal lining, are processed in the liver before reaching the large intestine.
- P. H. Houba, E. Bouven, C. A. Erkelens, R. G. Leenders, J. W. Scheeren, H. M. Pinedo and H. J. Haisma, Br. J. Cancer, 1998, 78, 1600 CrossRef CAS PubMed; S. H. Lee, E. Moroz, B. Castagner and J.-C. Leroux, J. Am. Chem. Soc., 2014, 136, 12868 CrossRef PubMed.
- W. Kalala, R. Kinget, G. Van den Mooter and C. Samyn, Int. J. Pharm., 1996, 139, 187 CrossRef CAS.
- Y. Kimura, Y. Makita, T. Kumagai, H. Yamane, T. Kitao, H. Sasatani and S.-I. Kim, Polymer, 1992, 33, 5294 CrossRef CAS; T. Yamaoka, Y. Makita, H. Sasatani, S.-I. Kim and Y. Kimura, J. Controlled Release, 2000, 66, 187 CrossRef PubMed.
- E. Schacht, A. Gevaert, E. R. Kenawy, K. Molly, W. Verstraete, P. Adriaensens, R. Carleer and J. Gelan, J. Controlled Release, 1996, 39, 327 CrossRef CAS.
- A. Sagi, R. Weinstain, N. Karton and D. Shabat, J. Am. Chem. Soc., 2008, 130, 5434 CrossRef CAS PubMed; W. Wang and C. Alexander, Angew. Chem., Int. Ed., 2008, 120, 7921 CrossRef; C. A. Blencowe, A. T. Russell, F. Greco, W. Hayes and D. W. Thornthwaite, Polym. Chem., 2011, 2, 773 RSC; S. T. Phillips and A. M. DiLauro, ACS Macro Lett., 2014, 3, 298 CrossRef and references therein; G. I. Peterson, M. B. Larsen and A. J. Boydston, Macromolecules, 2012, 45, 7317 CrossRef; S. Gnaim and D. Shabat, Acc. Chem. Res., 2014, 47, 2970 CrossRef PubMed; J. A. Kaitz, O. P. Lee and J. S. Moore, MRS Commun., 2015, 5, 191 CrossRef; A. Alouane, R. Labruère, T. Le Saux, F. Schmidt and L. Jullien, Angew. Chem., Int. Ed., 2015, 127, 7600 CrossRef; M. E. Roth, O. Green, S. Gnaim and D. Shabat, Chem. Rev., 2016, 116, 1309 CrossRef PubMed.
- N. Fomina, C. McFearin, M. Sermsakdi, O. Edigin and A. Almutairi, J. Am. Chem. Soc., 2010, 132, 9540 CrossRef CAS PubMed; C. de Gracia Lux, S. Joshi-Barr, T. Nguyen, E. Mahmoud, E. Schopf, N. Fomina and A. Almutairi, J. Am. Chem. Soc., 2012, 134, 15758 CrossRef PubMed; C. de Gracia Lux, C. L. McFearin, S. Joshi-Barr, J. Sankaranarayanan, N. Fomina and A. Almutairi, ACS Macro Lett., 2012, 1, 922 CrossRef PubMed; Y. Zhang, Q. Yin, L. Yin, L. Ma, L. Tang and J. Cheng, Angew. Chem., Int. Ed., 2013, 125, 6563 CrossRef; Y. Zhang, L. Ma, X. Deng and J. Cheng, Polym. Chem., 2013, 4, 224 RSC; C. de Gracia Lux, J. Olejniczak, N. Fomina, M. L. Viger and A. Almutairi, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3783 CrossRef; B. Fan, J. F. Trant, A. D. Wong and E. R. Gillies, J. Am. Chem. Soc., 2014, 136, 10116 CrossRef PubMed.
- C. de Gracia Lux and A. Almutairi, ACS Macro Lett., 2013, 2, 432 CrossRef CAS PubMed; J. Olejniczak, M. Chan and A. Almutairi, Macromolecules, 2015, 48, 3166 CrossRef; F.-Y. Qiu, C.-C. Song, M. Zhang, F.-S. Du and Z.-C. Li, ACS Macro Lett., 2015, 4, 1220 CrossRef.
- A. Lv, Z.-L. Li, F.-S. Du and Z.-C. Li, Macromolecules, 2014, 47, 7707 CrossRef CAS.
- H. Mutlu, L. M. de Espinosa and M. A. R. Meier, Chem. Soc. Rev., 2011, 40, 1404 RSC.
- S. E. Lehman Jr. and K. B. Wagener, in Handbook of Metathesis, Wiley-VCH, Weinheim, Germany, 2003, pp. 283–353 and references therein CrossRef CAS; T. W. Baughman and K. B. Wagener, in Advances in Polymer Science, Springer, Heidelberg, Germany, 2005, pp. 1–42 CrossRef CAS; E. B. Berda and K. B. Wagener, in Polymer Science: A Comprehensive Reference, Elsevier BV, Amsterdam, Netherlands, 2012, pp. 195–216 CrossRef CAS; E. B. Berda and K. B. Wagener, in Synthesis of Polymers; New Structures and Methods, Wiley-VCH, Weinhein, Germany, 2012, pp. 587–600 CrossRef CAS; P. Atallah, K. B. Wagener and M. D. Schulz, Macromolecules, 2013, 46, 4735 CrossRef CAS; C. Simocko, P. Atallah and K. B. Wagener, Curr. Org. Chem., 2013, 17, 2749 CrossRef; N. F. Sauty, L. C. da Silva, M. D. Schulz, C. S. Few and K. B. Wagener, Appl. Petrochem. Res., 2014, 4, 225 CrossRef.
- O. Türünc and M. A. R. Meier, Green Chem., 2011, 13, 314 RSC; P. A. Fokou and M. A. R. Meier, J. Am. Chem. Soc., 2009, 131, 1664 CrossRef CAS PubMed.
- M. Sokolsky-Papkov, A. Shikanov, N. Kumar, B. Vaisman and A. J. Domb, Bull. Isr. Chem. Soc., 2008, 23, 12 Search PubMed.
- P. S. Mukherjee, N. Das, Y. K. Kryschenko, A. M. Arif and P. J. Stang, J. Am. Chem. Soc., 2004, 126, 2464 CrossRef CAS PubMed.
- S. P. Rannard and N. J. Davis, Org. Lett., 1999, 1, 933 CrossRef CAS.
- T. Lebarbé, A. S. More, P. S. Sane, E. Grau, C. Alfos and H. Cramail, Macromol. Rapid Commun., 2014, 35, 479 CrossRef CAS PubMed; K. Terada, E. B. Berda, K. B. Wagener, F. Sanda and T. Masuda, Macromolecules, 2008, 41, 6041 CrossRef; F. N. Führer and H. Schlaad, Macromol. Chem. Phys., 2014, 215, 2268 CrossRef; A. Tüzün, G. Lligadas, J. C. Ronda, M. Galia and V. Cadiz, Eur. Polym. J., 2015, 67, 503 CrossRef.
- B. Schmidt, Eur. J. Org. Chem., 2004, 1865 CrossRef CAS.
- S. H. Hong, D. P. Sanders, C. W. Lee and R. H. Grubbs, J. Am. Chem. Soc., 2005, 127, 17160 CrossRef CAS PubMed.
- J. M. Fontan, Ph.D. thesis, University of Madrid, 1948.
- X. Li, J. Li, Y. Gao, Y. Kuang, J. Shi and B. Xu, J. Am. Chem. Soc., 2010, 132, 17707 CrossRef CAS PubMed; G. Leriche, G. Budin, L. Brino and A. Wagner, Eur. J. Org. Chem., 2010, 2010, 4360 Search PubMed; Y. Y. Yang, M. Grammel, A. S. Raghavan, G. Charron and H. C. Hang, Chem. Biol., 2010, 17, 1212 CrossRef PubMed; J. B. Denny and G. Blobel, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 5286 CrossRef; J. M. Bergen, E. J. Kwon, T. W. Shen and S. H. Pun, Bioconjugate Chem., 2008, 19, 377 CrossRef PubMed; O. A. Kucherak, S. Oncul, Z. Darwich, D. A. Yushchenko, Y. Arntz, P. Didier, Y. Mely and A. S. Klymchenko, J. Am. Chem. Soc., 2010, 132, 4907 CrossRef PubMed.
- M. W. Jones, G. Mantovani, C. A. Blindauer, S. M. Ryan, X. Wang, D. J. Brayden and D. M. Haddleton, J. Am. Chem. Soc., 2012, 134, 7406 CrossRef CAS PubMed.
- L. K. Sydnes, S. Elmi, P. Heggen, B. Holmelid and D. Malthe-Sørensen, Synlett, 2007, 1695 CrossRef CAS.
- G. Leriche, G. Budin, Z. Darwich, D. Weltin, Y. Mely, A. S. Klymchenko and A. Wagner, Chem. Commun., 2012, 48, 3224 RSC.
- F. Rafii, W. Franklin and C. E. Cerniglia, Appl. Environ. Microbiol., 1990, 56, 2146 CAS.
- C. Boulègue, M. Löweneck, C. Renner and L. Moroder, ChemBioChem, 2007, 8, 591 CrossRef PubMed.
- V. S. Chadwich, S. F. Phillips and A. F. Hoffman, Gastroenterology, 1977, 73, 241 Search PubMed; C. Fruijtier-P€olloth, Toxicology, 2005, 214, 1 CrossRef CAS PubMed; R. P. Gullapalli and C. L. Mazzitelli, Int. J. Pharm., 2015, 496, 219 CrossRef PubMed.
- A. Rybak and M. A. R. Meier, ChemSusChem, 2008, 1, 542 CrossRef CAS PubMed.
- H. E. Johansson, M. K. Johansson, A. C. Wong, E. S. Armstrong, E. J. Peterson, R. E. Grant, M. A. Roy, M. V. Reddington and R. M. Cook, Appl. Environ. Microbiol., 2011, 77, 4223 CrossRef CAS PubMed.
- L. Wu, Y. He and X. Tang, Bioconjugate Chem., 2015, 26, 1070 CrossRef CAS PubMed.
- R. Chen and L. Li, J. Am. Soc. Mass Spectrom., 2001, 12, 832 CrossRef CAS PubMed.
- S. K. Jonas, C. Benedetto, A. Flatman, L. Micheletti, C. Riley, P. A. Riley, D. Spargo, M. Zonca and T. F. Slater, Br. J. Cancer, 1992, 66, 185 CrossRef CAS PubMed.
- J. Willenbacher, K. N. R. Wuest, J. O. Mueller, M. Kaupp, H.-A. Wagenknecht and C. Barner-Kowollik, ACS Macro Lett., 2014, 3, 574 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization details. See DOI: 10.1039/c5py01937k |
| This journal is © The Royal Society of Chemistry 2016 |