
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An aromatic/aliphatic polyester prepared via ring-opening polymerisation and its remarkably selective and cyclable depolymerisation to monomer†
Jarret P.
MacDonald
and
Michael P.
Shaver
*
EaStCHEM, School of Chemistry, University of Edinburgh, Joseph Black Building, EH9 3JF, David Brewster Road, Edinburgh, UK. E-mail: michael.shaver@ed.ac.uk
First published on 12th November 2015
Abstract
The ring-opening polymerisation of 2,3-dihydro-5H-1,4-benzodioxepin-5-one (2,3-DHB) with aluminium salen or organocatalysts gives polyester homopolymers and copolymers with L-lactide or rac-β-butyrolactone that contain both aromatic and aliphatic linkages, the first polymers with an aromatic ring in the backbone prepared by this key method. The same Al salen catalyst catalyses a remarkably selective depolymerisation to monomer under modified reaction conditions. The process may be cycled to repeatedly recycle polymer to monomer and maintain the polymer's low dispersity.
Introduction
The exceptional growth in both fundamental research and industrial importance of (bio)degradable polymers stems from the controlled ring-opening polymerisation (ROP) of ε-caprolactone (ε-CL), lactide (LA) and β-butyrolactone (β-BL).1 Challenges remain as the resultant polyesters (i.e. poly(lactic acid), PLA) often have non-ideal thermal properties, slow hydrolytic degradation and industrial enzymatic composting to degrade to lactic acid rather than the original cyclic ester. Many elegant strategies exist to both expand scope and tune polymer properties including control of microstructure,2–5 macrostructure6–11 and composition.12–22 This includes mimicry of petroleum-derived polymers such as the ROP of macrolactones that introduce long polyolefin-like segments into polyester repeat units to prepare polyethylene-like structures23–29 including high molecular weight polymacrolactones prepared using aluminium salen catalysts.30,31Pioneering work extended mimicry in monomer design to the incorporation of aromatic substituents32,33 when Baker polymerised phenyllactide34 and mandelide35 to introduce pendant phenyl rings and produce polymers akin to polystyrene. Poly(mandelic acid) was later prepared via the ROP of 5-phenyl-1,3-dioxolane-2,4-dione with concomitant loss of CO2,36 which was extended to high molecular weight stereoregular polymers using organocatalysts.37
Introduction of aromatic functionalities within the polymer backbone rather than pendant to the chain is more exceptional. While a copolymerisation of styrene oxide with phthalic anhydride has been published,38 there have been no reports of polymers prepared via cyclic ester ROP that incorporate phenyl moieties into the polymer backbone. This is surprising as many aromatic polyesters such as poly(ethylene terephthalate) (PET), are important commodity plastics. This work has been extended to include other epoxides,39 as well as replacing phthalic anhydride with an ester, dihydrocoumarin.40
The synthesis of PET and similar aromatic/aliphatic polyesters has been achieved through ROP.41 This method involved first synthesising cyclic oligomers from monomers, which are then used in ROP. While this allowed for synthesis of a wide range of alkylene phthalate polymers, reactions required high temperatures and resulted in broad dispersities. Recent advances in this field have investigated catalyst choice, broadening monomer scope and copolymerisation.42–55 Despite such advances, the polymerisations were typically uncontrolled and required synthesis of oligomers with varying size before polymerisation.
We thus targeted the production of polyesters that contained an aromatic and aliphatic linkage in the polymer backbone that could be readily synthesised from a well-defined monomer, identifying the benzodioxepinones as a class of aromatic cyclic esters that may serve as monomers for ROP. In particular, 2,3-dihydro-5H-1,4-benzodioxepin-5-one (2,3-DHB) is commercially available and facile to synthesise.56–58 Additionally, a highly enantioselective synthesis of substituted 2,3-DHBs suggested a potentially broad monomer scope.59,60 We hypothesised that 2,3-DHB would undergo ROP (Scheme 1) in a manner similar to other seven-membered cyclic esters (i.e. ε-CL) whose ROP reactions are readily controlled to high conversion by judicious choice of catalyst.61–67
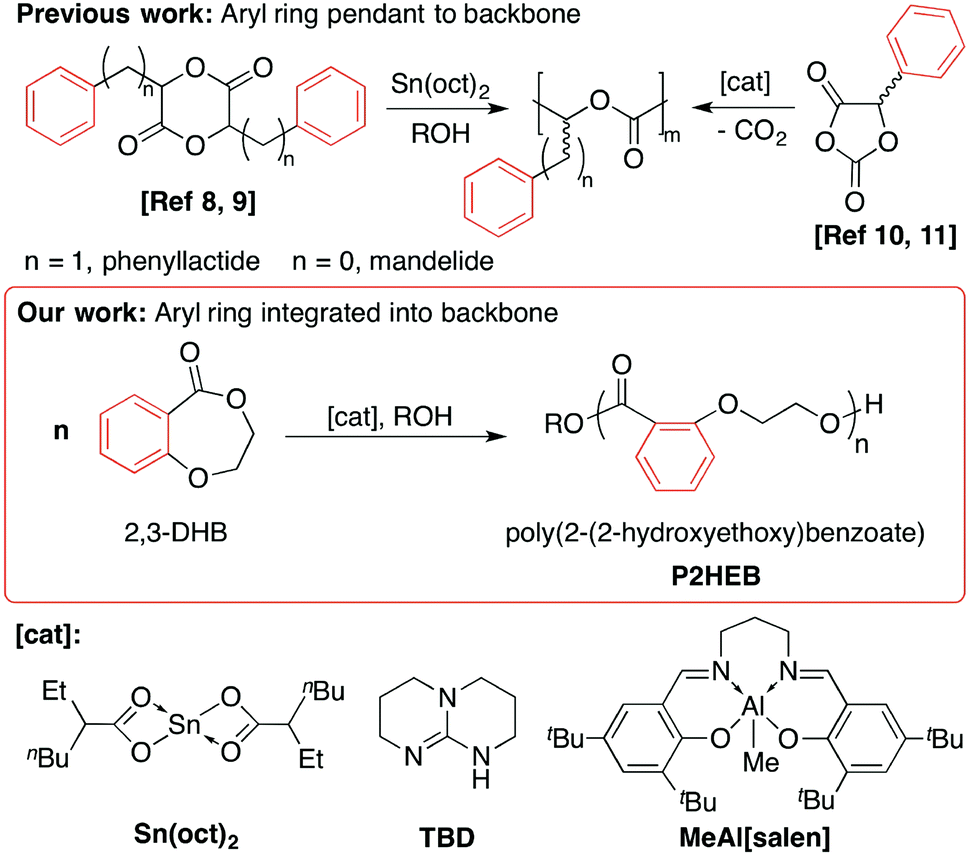 | ||
| Scheme 1 Polymerisation of phenyllactide, mandelide, 5-phenyl-dioxolanedione and, in this work, benzodioxipinone monomers to prepare polyesters with pendant (poly(phenyllactic acid), poly(mandelic acid)) and backbone-incorporated (poly(2–2-hydroxyethoxybenzoate) phenyl substituents using organo- and metal-based cataylsts. | ||
Results and discussion
Homopolymerisation of 2,3-DHB
Polymerisation of 2,3-DHB using Sn(oct)2 gave no or low monomer conversion to poly(2-(2-hydroxyethoxy)benzoate), P2HEB, at 70–120 °C (Table S1†). Longer reaction times did not significantly increase conversion but did promote a loss of control. Higher polymer dispersities (Đ = 1.44) suggested transesterification dominated after polymer–monomer equilibrium was established. The relatively low solubility of P2HEB in THF means that dn/dc values would be inaccurate. We thus report molecular weights calculated by relative integration of benzylic end-group resonances to polymer resonances.Aluminium salen complexes are excellent catalysts for ROP and exhibit a lower tendency towards transesterification than tin catalysts.68,69 Switching to an aluminium salen complex, MeAl[salen], gave much improved 2,3-DHB polymerisation (Fig. S2, Table S2†) after optimisation. Bulk and solution polymerisations conducted at 120 °C were uncontrolled (Đ > 1.6) and reached a maximum conversion of 66%. Furthermore, solution polymerisations required a high initial 2,3-DHB concentration ([2,3-DHB]0); no productive polymerisation was observed under dilute conditions. Interestingly, decreasing the polymerisation temperature to 70 °C solved these challenges. Neat polymerisation at 70 °C for one hour yielded P2HEB with 64% conversion, low Đ (1.13) and predictable molecular weights. Performing the reaction in toluene for one hour resulted in a modest increase in conversion (75%) with only a slight increase in Đ (1.16). Extending the polymerisation time to three hours under identical conditions yielded no significant change in polymer characteristics. Decreasing the temperature minimised transesterification side reactions. Building on this promising result, the Al-mediated ROP was explored (Table 1). Polymerisations were exceptionally well controlled while reaching higher conversion when T ≤ 60 °C, even permitting room temperature ROP. Higher molecular weight P2HEB was also synthesised by increasing [2,3-DHB]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[Al]0:[BnOH]0 to 200:1:1 and 500:1:1 without sacrificing polymerisation control.
:[Al]0:[BnOH]0 to 200:1:1 and 500:1:1 without sacrificing polymerisation control.
| T (°C) | Time (h) | [M]0/[Al]0 | Conv.b (%) |
M
n,thc |
M
nb |
Đ |
|---|---|---|---|---|---|---|
|
a 2.3-DHB polymerisation conducted in toluene (1:1 m/m).
b Determined by 1H NMR spectroscopy.
c
M
n,th = ([2,3-DHB]0/[BnOH]0) × % conversion × MW2,3-DHB.
d Determined by gel permeation chromatography (details in ESI).
|
||||||
| 60 | 4 | 100 | 92 | 15270 |
13000 |
1.08 |
| 60 | 6 | 100 | 92 | 15270 |
13500 |
1.09 |
| 50 | 6 | 100 | 91 | 15060 |
15220 |
1.07 |
| 50 | 24 | 200 | 88 | 28870 |
25210 |
1.10 |
| 50 | 24 | 500 | 78 | 63740 |
52050 |
1.11 |
| 22 | 6 | 50 | 80 | 6690 | 6790 | 1.13 |
| 22 | 6 | 200 | 46 | 15350 |
14210 |
1.12 |
| 22 | 24 | 200 | 85 | 27910 |
27010 |
1.11 |
Monomer equilibrium and P2HEB depolymerisation
The odd observation of higher conversions at lower temperatures can be explained by the monomer-polymer equilibrium. That is, the relative rate of transesterification leading to depolymerisation (kd) compared to rate of productive transesterification polymerisation (kp) increases when T ≥ 70 °C, shifting the equilibrium towards higher [2,3-DHB], the importance of which was noted in a recent paper on the ROP of morpholinones.70 In our study, we verified this equilibrium by performing a variable temperature NMR scale polymerisation (see ESI†). Polymerisation of 50 eq. of 2,3-DHB at room temperature gave an NMR conversion of 88%. Subsequent heating of the sample for 10 hours at 90 °C resulted in a decrease in conversion (70%) with an increase in monomer signals, indicating depolymerisation had occurred with no apparent degradation.As mentioned previously, performing polymerisations under dilute conditions resulted in no conversion; successful polymerisations mediated by MeAl[salen] were conducted at [2,3-DHB]0 of 4–5 M. We noted that it was possible to polymerise at lower concentrations using organocatalysts (Table S3†). While basic or acidic organocatalysts 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and diphenyl phosphate (DPP) respectively, did not yield any polymer, the bifunctional 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) catalysed a very well controlled polymerisation at relatively rapid rates (Table S4†) even under much lower concentrations. Polymerisations at 2.4 M reached 87% after just 30 minutes. Decreasing [2,3-DHB]0 further to 1.3 and 0.5 M decreased polymer conversion to 80 and 51%, respectively as the polymer–monomer equilibrium favoured depolymerisation at lower concentrations.
Beyond the novelty of the first aromatic backbone components accessed through cyclic ester monomer ROP, these two observations led us to the most interesting feature of this polymerisation system: a clean, reversible and cyclable depolymerisation. While depoylemerisation of aliphatic polyesters is observed in other systems, it is not typically selective.65,71,72 As a result, oligomers are typically produced. The synthesis of lactide from PLA can be achieved through ring-closing depolymerisation (Scheme 2), though a high degree of depolymerisation is driven by the removal of monomer by distillation.73 For the MeAl[salen] catalyst we observed both a temperature dependent and concentration dependent monomer/polymer equilibrium (vide supra). When we exploit only concentration, depolymerisation occurs selectively to monomer with no oligomers observed. To demonstrate this, P2HEB homopolymer was synthesised and purified to remove any residual monomer. P2HEB was then added to MeAl[salen] in toluene at 60 °C for six hours. Two methods were used to establish the equilibrium monomer concentration of P2HEB. First, this was done by polymerisation of 2,3-DHB (Table S9†). In a glovebox, 2,3-DHB polymerisation was set up as described previously at three different [2,3-DHB]0. The polymerisations were continued for six hours at 60 °C. After six hours, 1H NMR spectroscopy was used to calculate relative [2,3-DHB]:[P2HEB] and consequently [2,3-DHB]eq. Longer times did not change conversion. It was also calculated by depolymerisation (Table S10†). Solutions of MeAl[salen] (1.4 mol%) and three different [P2HEB] in C6D6 were heated to 60 °C for 12 hours and analysed by 1H NMR spectroscopy. Using relative integration of [P2HEB]:[2,3-DHB] and initial concentration, [2,3-DHB]eq was calculated. By both methods, [P2HEB]eq was 0.36 M at 60 °C in toluene.
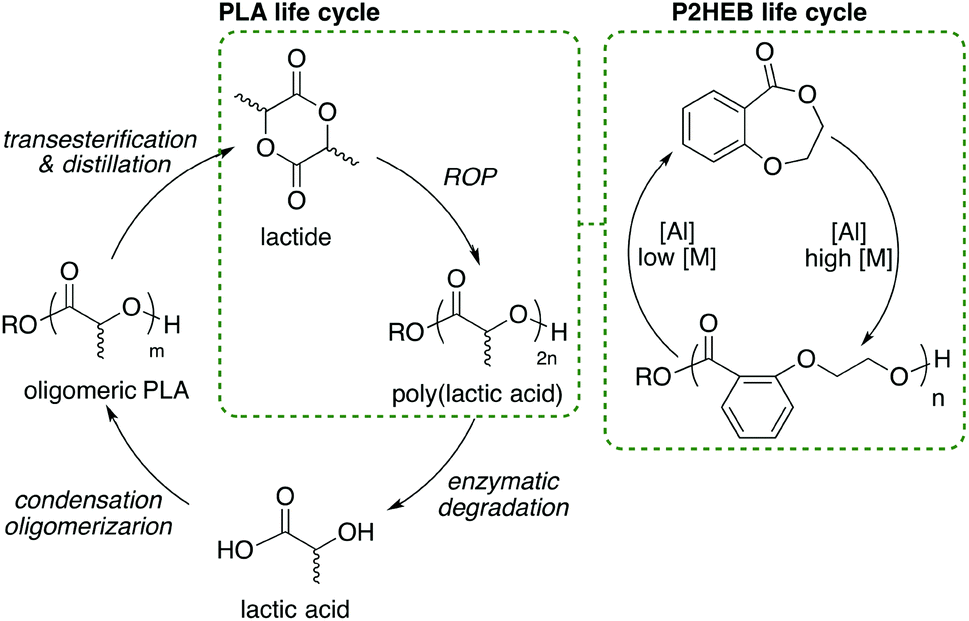 | ||
| Scheme 2 Idealized life cycles of poly(lactic acid) and poly(2-(2-hydroxyethoxy)benzoate) moving from four energy intensive steps in PLA recycling to simple ring-opening polymerization and ring closing depolymerization steps for P2HEB. | ||
We established a 0.17 M [P2HEB]0 and selectively converted to 2,3-DHB (>90%) in <12 hours (Fig. S2, Table S6†). Importantly, this behaviour is unique to this new polymer and does not occur with poly(lactic acid)s under similar conditions with this catalyst system. The MeAl[salen] catalyst is also essential as subjecting P2HEB to similar conditions in the presence of TBD did not yield significant 2,3-DHB (≤5%) after 12 hours at 60 °C, instead leading to transesterified polymer products.
We envisage this control being beneficial in the life cycle of the polymer as monomer can be recovered directly, bypassing hydroxyacids as intermediates in energy-intensive monomer synthesis. In the case of Al, the same catalyst can be used for polymerisation and depolymerisation (Scheme 2).
To exemplify reversibility, a one-pot reaction was run with an initial monomer concentration of 4.1 M. After 6 h at 60 °C, conversion to P2HEB was 82% (entry 1, Table 2). Addition of toluene gave an apparent [2,3-DHB]0 of 0.2 M and resulted in a depolymerisation with 2,3-DHB:P2HEB = 94:6 (entry 2, Table 2). Reconcentration of the reaction in vacuo to give an apparent [2,3-DHB]0 of 4.1 M resulted in 84% conversion to P2HEB after heating to 60 °C for 6 h. The polymerisation is nearly fully reversible and that the depolymerisation yields monomer with a surprisingly high degree of purity. Importantly, no degradation products are produced, allowing for clean repolymerisation with exceptional levels of control over dispersity and molecular weight.
| Entry | Concentrationb (M) | 2,3-DHBc (%) | P2HEBc (%) |
M
n,thd |
M
ne |
Đ |
|---|---|---|---|---|---|---|
|
a One-pot polymerization–depolymerization of 2,3-DHB/P2HEB by varying concentration with [2,3-DHB]0:[Al]0:[BnOH]0 = 100:1:1.
b Concentration = [2,3-DHB]0/volume toluene.
c Determined by gel permeation chromatography.
d
M
n,th = ([2,3-DHB]0/[BnOH]0) × % conversion × MW2,3-DHB.
e Determined by 1H NMR spectroscopy.
|
||||||
| 1 | 4.1 | 18 | 82 | 13600 |
12200 |
1.08 |
| 2 | 0.2 | 94 | 06 | 1100 | 1090 | 1.09 |
| 3 | 4.1 | 16 | 84 | 13850 |
13720 |
1.07 |
Excellent retention of control upon repolymerisation is also exemplified by gel permeation chromatography (GPC) of the crude samples (Fig. 1). The trace corresponding to entry 2 shows that polymer has been converted near quantitatively to monomer (94%).
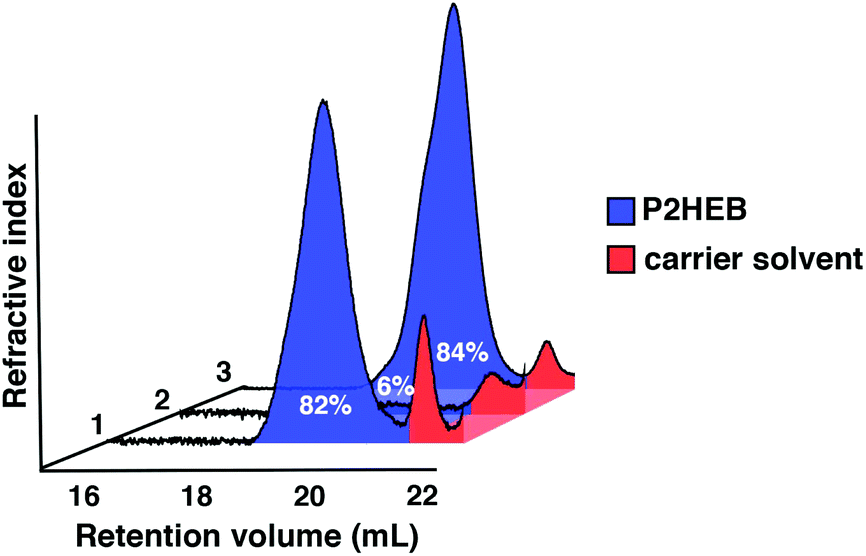 | ||
| Fig. 1 GPC traces of crude samples from entries 1, 2 and 3. | ||
Copolymers with P2HEB
Finally, we wanted to extend the utility of this new monomer and the livingness of the ROP by incorporating 2,3-DHB into copolymers. AB diblock and ABA triblock copolymers are readily prepared by sequential monomer addition. Polymerisation of 2,3-DHB was followed by addition of L-LA and an increase in reaction temperature to incorporate the more inert lactide monomer. The copolymerisation reactions were high yielding and well controlled (Tables S5 and S6†). 1H NMR spectroscopy of the P(L-LA) methine region suggested that the copolymers had undergone some scrambling (Fig. S1†) indicating the AB copolymers were likely gradient copolymers instead of true block copolymers. AB block copolymers were prepared using a monofunctional alcohol initiator (BnOH) while ABA block copolymers were synthesised using a propanediol core to build the central P2HEB mid-block followed by growth of the two PLA A blocks. Sequential addition of 2,3-DHB to growing poly(3-hydroxybutyrate), P3HB, resulted in the AB block copolymer P(3HB-b-2HEB). Scrambling was unlikely in this copolymer as the P2HEB block is grown onto the P3HB block.The scrambling of P2HEB/PLA in copolymers made depolymerisation of P2HEB/PLA copolymers difficult as depolymerisation was halted once a lactic acid unit was encountered. However, P(3HB-b-2HEB) copolymers readily depolymerised the P2HEB block (>90%) leaving only P3HB chains (Fig. 2). This exemplified the utility of a depolymerisable monomer within a larger macromolecular structure. The thermal properties of the polymers were also studied (Table S7†). Pure P2HEB stability was tuned by the copolymer composition where the onset of decomposition (Td,onset) was increased from 219 °C to 279 °C in changing from homo to ABA copolymer, with 95% sample decomposition at 262 °C and 319 °C respectively (Fig. S4†). Other thermal properties were unexceptional (Table S7†) and the tuning of these in homo and copolymers is a current target of our research group.
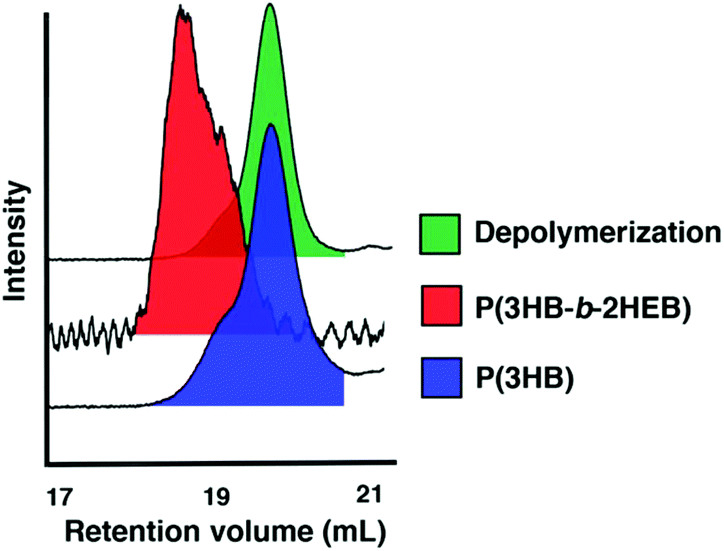 | ||
| Fig. 2 GPC traces of P(3HB) and P(2HEB) copolymers and depolymerisation. | ||
Conclusions
In conclusion, the polymerisation of a novel aromatic/aliphatic monomer, 2,3-DHB, was achieved using an aluminium based catalyst or an organocatalyst allowed for the synthesis of very well controlled aromatic/aliphatic polyester from a well defined monomer. Copolymers with lactide and β-butyrolactone were synthesised with similarly high levels of control. Importantly, when the MeAl[salen] catalyst is used the polymers are easily ring-closed back to monomer at lower concentrations, providing a clean and selective route to recycle the polymer back to monomer. The concentration of the reaction could be manipulated in situ to achieve a fully reversible polymerisation and repolymerisation without a loss of control. We continue to work beyond the scope of this report, exploring benzodioxepinone monomers as components to tune thermal properties in copolymers, build better polymer degradation strategies and expand the monomer scope to substituted DHBs.Experimental
General considerations
All experiments involving moisture- and air-sensitive compounds were performed under a nitrogen atmosphere using an MBraun LABmaster sp glovebox system or a Vigor glovebox equipped with a −35 °C freezer and [H2O] and [O2] analysers or using standard Schlenk techniques. Gel permeation chromatography (GPC) was used to determine polymer dispersities and was carried out in THF at a flow rate of 1 mL min−1 on a Malvern Instruments Viscotek 270 GPC Max triple detection system with 2× mixed bed styrene/DVB columns (300 × 7.5 mm). GPC analysis was performed using OmniSEC 5.0 software. Polymer molecular weights were calculated by 1H NMR spectroscopy by relative integration of benzylic end-group resonances to polymer resonances. 1H and 13C NMR spectra were recorded at 298 K with Bruker Avance spectrometers (400 or 500 MHz) in CDCl3 or C6D6. TGA samples were heated at 10 °C min−1 to 150 °C and held for 10 minutes to remove residual solvent and cooled to room temperature at 10 °C min−1 until decomposition was observed.Materials
MeAl[salen] was synthesised via modified literature procedures.69,74 Benzyl alcohol was dried by refluxing over calcium hydride for 24 hours, distilled under inert atmosphere and degassed by three freeze–pump–thaw cycles prior to use. 1,3-Propanediol was distilled under vacuum and stored under inert atmosphere prior to use. Toluene was obtained from an Innovative Technologies solvent purification system, consisting of columns of alumina and copper catalyst and was degassed by three freeze–pump–thaw cycles prior to use. C6D6 was refluxed over potassium for 72 hours, distilled under inert atmosphere and degassed by three freeze–pump–thaw cycles prior to use. L-Lactide was purified by three vacuum sublimations and dried under reduced pressure for 18 hours prior to use. 2,3-Dihydro-5H-1,4-benzodioxepin-5-one (2,3-DHB) was purchased from AEchem Scientific Corporation and recrystallised three times from EtOAC:hexanes (50:50) followed by drying under vacuum at 60 °C for 18 hours prior to use.
Representative homopolymerization of 2,3-DHB
In a glovebox, 2,3-DHB (116 mg, 0.71 mmol), MeAl[salen] (3.8 mg, 0.01 mmol), BnOH (0.7 μL, 0.01 mmol) and toluene (150 mg) were added to an ampoule. The ampoule was sealed, removed from the glovebox and placed in a preheated oil bath at 60 °C for six hours. After six hours, 0.5 mL of a 10% MeOH in CHCl2 solution was added to the ampoule to quench polymerisation. The solution was then added dropwise to cold MeOH to leave a white solid. Precipitation was repeated twice to remove residual 2,3-DHB.1H NMR (500 MHz, CDCl3): δ 7.72 (m, 1H, ArH), 7.36 (m, 1H ArH), 6.94–6.88 (m, 2H, ArH), 4.55 (t, J = 5.0 Hz, 2H, C(O)OCH2CH2O), 4.25 (t, J = 5.0 Hz, 2H, C(O)OCH2CH2O). 13C NMR (126 MHz, CDCl3) δ 166.00 (C(O)OR), 158.31, 133.70, 131.90, 121.00, 120.85, 114.40 (Ar), 67.40 (C(O)OCH2CH2O), 63.05 (C(O)OCH2CH2O).
Representative AB block copolymerization of 2,3-DHB/L-lactide
In a glovebox, 2,3-DHB (164 mg, 1.00 mmol), MeAl[salen] (5.4 mg, 0.01 mmol), 1,3-propanediol (1.0 μL, 0.01 mmol) and toluene (265 mg) were added to an ampoule. The ampoule was sealed, removed from the glovebox and placed in a preheated oil bath at 60 °C for six hours. After six hours, 0.5 mL of a 10% MeOH in CHCl2 solution was added to the ampoule to quench polymerisation. The solution was then added dropwise to cold MeOH to leave a white solid. Precipitation was repeated twice to remove residual 2,3-DHB. The isolated polymer was then dried under vacuum for 18 hours. To P2HEB (0.005 mmol) was added L-lactide (36 mg, 0.25 mmol) in toluene (0.3 mL) at 70 °C. The reaction was stirred for three hours followed by addition on 0.5 mL of a 10% MeOH in CHCl2 solution was added to the ampoule to quench polymerisation. The solution was then added dropwise to cold MeOH to leave the desired copolymer.Representative ABA block copolymerization of 2,3-DHB/L-lactide
In a glovebox, 2,3-DHB (323 mg, 2.00 mmol), MeAl[salen] (10.8 mg, 0.02 mmol), 1,3-propanediol (0.7 mg, 0.01 mmol) and toluene (265 mg) were added to an ampoule. The ampoule was sealed, removed from the glovebox and placed in a preheated oil bath at 60 °C for six hours. After six hours, 0.5 mL of a 10% MeOH in CHCl2 solution was added to the ampoule to quench polymerisation. The solution was then added dropwise to cold MeOH to leave a white solid. Precipitation was repeated twice to remove residual 2,3-DHB. The isolated polymer was then dried under vacuum for 18 h. To P2HEB (0.01 mmol) was added L-lactide (72 mg, 0.5 mmol) in toluene (0.3 mL) at 70 °C. The reaction was stirred for three hours followed by addition on 0.5 mL of a 10% MeOH in CHCl2 solution was added to the ampoule to quench polymerization. The solution was then added dropwise to cold MeOH to give the desired copolymer.Representative AB block copolymerization of 2,3-DHB/rac-β-butyrolactone
In a glovebox, rac-β-butyrolactone (100 mg, 1.31 mmol), MeAl[salen] (14.4 mg, 0.01 mmol), BnOH (2.7 μL, 0.01 mmol) and toluene (250 mg) were added to an ampoule. The ampoule was sealed, removed from the glovebox and placed in a preheated oil bath at 85 °C for two hours. After two hours, the ampoule was degassed three times and returned to a glovebox, where 2,3-DHB (215 mg, 1.31 mmol) was added. The ampoule was then sealed, removed from the glovebox and placed in preheated oil bath at 60 °C for two hours, followed by addition of 0.5 mL of a 10% MeOH in CH2Cl2 to the ampoule to quench polymerisation. The solution was then added dropwise to cold MeOH and filtered to give the desired copolymer.Acknowledgements
The authors would like to thank the University of Edinburgh for a Chancellor's Fellowship and the Marie-Curie Actions Programme (Grant FP7-PEOPLE-2013-CIG-618372).References
- C. K. Williams and M. A. Hillmyer, Polym. Rev., 2008, 48, 1–10 CrossRef CAS.
- N. Spassky, M. Wisniewski, C. Pluta and A. Le Borgne, Macromol. Chem. Phys., 1996, 197, 2627–2637 CrossRef CAS.
- T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 1999, 121, 4072–4073 CrossRef CAS.
- T. M. Ovitt and G. W. Coates, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4686–4692 CrossRef CAS.
- T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1316–1326 CrossRef CAS PubMed.
- M. J. Stanford and A. P. Dove, Macromolecules, 2008, 42, 141–147 CrossRef.
- M. J. Stanford, R. L. Pflughaupt and A. P. Dove, Macromolecules, 2010, 43, 6538–6541 CrossRef CAS.
- D. J. A. Cameron and M. P. Shaver, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1477–1484 CrossRef CAS.
- M. P. Shaver and D. J. A. Cameron, Biomacromolecules, 2010, 11, 3673–3679 CrossRef CAS PubMed.
- G. K. Tennekone, B. D. Wagner and M. P. Shaver, Green Mater., 2013, 2, 31–42 CrossRef.
- W. Zhao, Y. Wang, X. Liu, X. Chen, D. Cui and E. Y. X. Chen, Chem. Commun., 2012, 48, 6375–6377 RSC.
- J. P. MacDonald, M. P. Parker, B. W. Greenland, D. Hermida-Merino, I. W. Hamley and M. P. Shaver, Polym. Chem., 2015, 6, 1445–1453 RSC.
- C.-M. Dong and G. Liu, Polym. Chem., 2013, 4, 46–52 RSC.
- S. Doppalapudi, A. Jain, W. Khan and A. J. Domb, Polym. Adv. Technol., 2014, 25, 427–435 CrossRef CAS.
- H. Abe, Y. Doi, Y. Hori and T. Hagiwara, Polymer, 1998, 39, 59–67 CrossRef CAS.
- Y. Hori, Y. Takahashi, A. Yamaguchi and T. Nishishita, Macromolecules, 1993, 26, 4388–4390 CrossRef CAS.
- M. Basko, A. Duda, S. Kazmierski and P. Kubisa, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4873–4884 CrossRef CAS.
- J.-H. Kim and J. H. Lee, Polym. J., 2002, 34, 203–208 CrossRef CAS.
- D. C. Aluthge, C. Xu, N. Othman, N. Noroozi, S. G. Hatzikiriakos and P. Mehrkhodavandi, Macromolecules, 2013, 46, 3965–3974 CrossRef CAS.
- E. M. Frick and M. A. Hillmyer, Macromol. Rapid Commun., 2000, 21, 1317–1322 CrossRef.
- E. M. Frick, A. S. Zalusky and M. A. Hillmyer, Biomacromolecules, 2003, 4, 216–223 CrossRef CAS PubMed.
- N. Kasyapi and A. K. Bhowmick, RSC Adv., 2014, 4, 27439–27451 RSC.
- B. Lebedev and A. Yevstropov, Makromol. Chem., 1984, 185, 1235–1253 CrossRef CAS.
- M. Letizia Focarete, M. Scandola, A. Kumar and R. A. Gross, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 1721–1729 CrossRef.
- S. Strandman, J. E. Gautrot and X. X. Zhu, Polym. Chem., 2011, 2, 791–799 RSC.
- A. Duda, A. Kowalski, S. Penczek, H. Uyama and S. Kobayashi, Macromolecules, 2002, 35, 4266–4270 CrossRef CAS.
- Y. Wang and M. Kunioka, Macromol. Symp., 2005, 224, 193–206 CrossRef CAS.
- L. van der Mee, F. Helmich, R. de Brujin, J. A. J. M. Vekemansm, A. R. A. Palmans and E. W. Meijer, Macromolecules, 2006, 39, 5021–5027 CrossRef CAS.
- S. Namekawa, S. Suda, H. Uyama and S. Kobayashi, Int. J. Biol. Macromol., 1999, 25, 145–151 CrossRef CAS PubMed.
- I. van der Meulen, E. Gubbels, S. Huijser, R. Sablong, C. E. Koning, A. Heise and R. Duchateau, Macromolecules, 2011, 44, 4301–4305 CrossRef CAS.
- M. P. F. Pepels, M. Bouyahyi, A. Heise and R. Duchateau, Macromolecules, 2013, 46, 4324–4334 CrossRef CAS.
- H. Fukuzaki, M. Yoshida, M. Asano, M. Kumakura, K. Imasaka, T. Nagai, T. Mashimo, H. Yuasa, K. Imai and H. Yamanaka, Eur. Polym. J., 1990, 26, 1273–1277 CrossRef CAS.
- I. Tabushi, H. Yamada, H. Matsuzaki and J. Furukawa, J. Polym. Sci., Polym. Lett. Ed., 1975, 13, 447–450 CrossRef CAS.
- T. L. Simmons and G. L. Baker, Biomacromolecules, 2001, 2, 658–663 CrossRef CAS PubMed.
- T. Liu, T. L. Simmons, D. A. Bohnsack, M. E. Mackay, M. R. Smith and G. L. Baker, Macromolecules, 2007, 40, 6040–6047 CrossRef CAS.
- I. J. Smith and B. J. Tighe, Makromol. Chem., 1981, 182, 313–324 CrossRef CAS.
- A. Buchard, D. R. Carbery, M. G. Davidson, P. K. Ivanova, B. J. Jeffery, G. I. Kociok-Köhn and J. P. Lowe, Angew. Chem., Int. Ed., 2014, 53, 13858–13861 CrossRef CAS PubMed.
- E. Hosseini Nejad, A. Paoniasari, C. E. Koning and R. Duchateau, Polym. Chem., 2012, 3, 1308–1313 RSC.
- P. K. Saini, C. Romain, Y. Zhu and C. K. Williams, Polym. Chem., 2014, 5, 6068–6075 RSC.
- N. J. Van Zee and G. W. Coates, Chem. Commun., 2014, 50, 6322–6325 RSC.
- D. J. Brunelle, J. E. Bradt, J. Serth-Guzzo, T. Takekoshi, T. L. Evans, E. J. Pearce and P. R. Wilson, Macromolecules, 1998, 31, 4782–4790 CrossRef CAS PubMed.
- H. Chen, C. Huang, W. Yu and C. Zhou, Polymer, 2013, 54, 1603–1611 CrossRef CAS.
- H. Chen, W. Yu and C. Zhou, Polym. Eng. Sci., 2012, 52, 91–101 CAS.
- P. Fabbri, E. Bassoli, S. B. Bon and L. Valentini, Polymer, 2012, 53, 897–902 CrossRef CAS.
- G. Giammanco, A. Martinez de Ilarduya, A. Alla and S. Munoz-Guerra, Biomacromolecules, 2010, 11, 2512–2520 CrossRef CAS PubMed.
- N. Gonzalez-Vidal, A. Martinez de Ilarduya and S. Munoz-Guerra, Eur. Polym. J., 2010, 46, 792–803 CrossRef CAS.
- P. Hodge, React. Funct. Polym., 2014, 80, 21–32 CrossRef CAS.
- S. D. Kamau, P. Hodge, R. T. Williams, P. Stagnaro and L. Conzatti, J. Comb. Chem., 2008, 10, 644–654 CrossRef CAS PubMed.
- C. Labruyere, O. Talon, N. Berezina, E. Khousakoun and C. Jerome, RSC Adv., 2014, 4, 38643–38648 RSC.
- Z. A. Mohd. Ishak, K. G. Gatos and J. Karger-Kocsis, Polym. Eng. Sci., 2006, 46, 743–750 CAS.
- E. J. Shin, A. E. Jones and R. M. Waymouth, Macromolecules, 2012, 45, 595–598 CrossRef CAS.
- A. R. Tripathy, W. J. MacKnight and S. N. Kukureka, Macromolecules, 2004, 37, 6793–6800 CrossRef CAS.
- Q. Xu, J. Chen, W. Huang, T. Qu, X. Li, Y. Li, X. Yang and Y. Tu, Macromolecules, 2013, 46, 7274–7281 CrossRef CAS.
- J. H. Youk, A. Boulares, R. P. Kambour and W. J. MacKnight, Macromolecules, 2000, 33, 3600–3605 CrossRef CAS.
- J. Zhang, Z. Wang, B. Wang, Q. Gou, J. Zhang, J. Zhou, Y. Li, P. Chen and Q. Gu, Polym. Chem., 2013, 4, 1648–1656 RSC.
- C. A. Rose and K. Zeitler, Org. Lett., 2010, 12, 4552–4555 CrossRef CAS PubMed.
- L. Liu, S. Xu and H. Zhou, Tetrahedron, 2013, 69, 8386–8391 CrossRef CAS.
- M. T. Berry, D. Castrejon and J. E. Hein, Org. Lett., 2014, 16, 3676–3679 CrossRef CAS PubMed.
- Z. Shen, H. A. Khan and V. M. Dong, J. Am. Chem. Soc., 2008, 130, 2916–2917 CrossRef CAS PubMed.
- Z. Shen, P. K. Dornan, H. A. Khan, T. K. Woo and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 1077–1091 CrossRef CAS PubMed.
- K. Odelius and A.-C. Albertsson, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1249–1264 CrossRef CAS.
- M. Hakkarainen, G. Adamus, A. Höglund, M. Kowalczuk and A.-C. Albertsson, Macromolecules, 2008, 41, 3547–3554 CrossRef CAS.
- T. Mathisen, K. Masus and A.-C. Albertsson, Macromolecules, 1989, 22, 3842–3846 CrossRef CAS.
- A.-C. Albertsson and R. Palmgren, J. Macromol. Sci., Pure Appl. Chem., 1993, 30, 919–931 CrossRef.
- K. Stridsberg and A.-C. Albertsson, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 3407–3417 CrossRef CAS.
- A. Löfgren and A.-C. Albertsson, Polymer, 1995, 36, 3753–3759 CrossRef.
- A. Loefgren, A.-C. Albertsson, P. Dubois, R. Jerome and P. Teyssie, Macromolecules, 1994, 27, 5556–5562 CrossRef CAS.
- J. P. MacDonald and M. P. Shaver, in Green Polymer Chemistry: Biobased Materials and Biocatalysis, American Chemical Society, 2015, ch. 10, vol. 1192, pp. 147–167 Search PubMed.
- P. Hormnirun, E. L. Marshall, V. C. Gibson, R. I. Pugh and A. J. P. White, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15343–15348 CrossRef CAS PubMed.
- T. R. Blake and R. M. Waymouth, J. Am. Chem. Soc., 2014, 136, 9252–9255 CrossRef CAS PubMed.
- M. Nelißen, H. Keul and H. Höcker, Macromol. Chem. Phys., 1995, 196, 1645–1661 CrossRef.
- F. Nederberg, E. F. Connor, T. Glausser and J. L. Hedrick, Chem. Commun., 2001, 2066–2067 RSC.
- D. K. Yoo, D. Kim and D. S. Lee, Macromol. Res., 2006, 14, 510–516 CrossRef CAS.
- D. A. Atwood, M. S. Hill, J. A. Jegier and D. Rutherford, Organometallics, 1997, 16, 2659–2664 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR characterisation, polymerisation data. See DOI: 10.1039/c5py01606a |
| This journal is © The Royal Society of Chemistry 2016 |