
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComplementary HPLC-ICP-MS and synchrotron X-ray absorption spectroscopy for speciation analysis of chromium in tobacco samples
Susana
Cuello
a,
John
Entwisle
a,
Jocelyn
Benning
b,
Chuan
Liu
b,
Steven
Coburn
b,
Kevin G.
McAdam
b,
Julian
Braybrook
a and
Heidi
Goenaga-Infante
*a
aLGC Limited, Queens Road, Teddington, Middlesex TW11 0LY, UK. E-mail: Heidi.Goenaga-Infante@lgcgroup.com
bGroup R&D Centre, British American Tobacco, Regents Park Road, Southampton, SO15 8TL, UK
First published on 9th December 2015
Abstract
Speciation data for chromium in tobacco products, as obtained by complementary HPLC-ICP-MS and synchrotron-based X-ray Absorption Near-Edge Structure spectroscopy (XANES), are presented for the first time. Non-denaturing extraction conditions were investigated to avoid Cr species redox inter-conversion before analysis of extracts using HPLC-ICP-MS. Methodology based on HPLC-ICP-MS, which is compatible with the extraction conditions, was developed for separation and detection of inorganic Cr species such as Cr(III) and Cr(VI) in aqueous standard solutions. The instrumental limits of detection (3σ criterion) obtained for Cr(III) and Cr(VI) were 0.12 and 0.08 ng g−1 Cr, respectively. The total Cr extracted from 3R4F cut tobacco with water was around 10% of the total Cr in the solid (1949 ± 171 ng g−1 of Cr on a dry weight basis), with 75% of the aqueous Cr associated with species of molecular mass > 3 kDa. Cr(III) was the main identified species in the tobacco extracts using HPLC-ICP-MS, whilst Cr(VI) could not be detected. In situ XANES analysis revealed that the cut tobacco from 3R4F reference cigarettes contained only Cr(III). Following leaching with water, leaching with sodium dodecylsulfate (SDS) on the solid residue led to extraction of a further 10% of the Cr contained in the solid tobacco. The total Cr data obtained by ICP-MS for HNO3 and HNO3/HF acid digests of 3R4F cut tobacco suggested that additional 12% of the total Cr in the solid appears to be associated with silicates, which are known to occur naturally in tobacco products. Although Cr species could not be detected in water leachates from 3R4F smoke condensates using the HPLC-ICP-MS method developed here, XANES measurements identified Cr(III) as the main Cr species present in cigarette smoke condensate, with no detectable Cr(VI). HPLC-ICP-MS data obtained for smoke condensates from cigarettes spiked with Cr(III) before smoke collection revealed that Cr(III) is the main Cr species in present the water soluble fraction of the condensate. Spiking experiments demonstrated that Cr(VI) was highly unstable in trapped smoke condensate. In this work no evidence was observed for the presence of Cr(VI) in mainstream smoke generated from 3R4F cigarettes.
Introduction
Tobacco plants absorb heavy metals from the environment and accumulate them in the leaves while growing.1 Tobacco is also exposed to metallic sources during leaf cutting and cigarette production. When cigarettes are smoked, metals in the tobacco are found to transfer into mainstream smoke.2 Cigarette smoke is, therefore, a source of heavy metals intake for smokers, via the respiratory tract.Traditionally, tobacco and cigarette smoke are mainly analysed for seven trace elements (As, Cd, Cr, Hg, Ni, Pb and Se) using various atomic absorption or mass spectroscopic techniques to quantify their total levels in these matrices. However, these techniques provide no information on the chemical identity and relative amounts of the different species potentially present for each metal. Speciation studies are important due to the fact that the potential hazard of a metal (e.g. its mobility, reactivity and toxicity) varies with the chemical form and may change with time and environmental conditions. For example, reactions within the growing plant and subsequent curing may modify metal species, while the redox conditions of combustion can lead to major changes in speciation. Such factors may markedly alter the toxicity risk associated with a given metal, depending on the way that tobacco is consumed. Therefore, extending the existing knowledge to the area of metal speciation could potentially improve current understanding of the drivers of cigarette smoke toxicity.
Whilst a large amount of data has been made available for total metal concentrations in tobacco samples, the published speciation data is still very limited and focuses mainly on arsenic speciation.3–5 Main challenges include the very low levels of relevant elements in tobacco and smoke samples, the complexity of the matrix where they exist, and also potential changes in speciation as a result of sample preparation, ageing and environmental interactions (particularly for condensed cigarette smoke or “tar”).
Tobacco and cigarette smoke have been shown to contain chromium.5 Chromium toxicity is very much dependent on its oxidation state. Chromium can exist in several oxidation states. However, only elemental Cr(0), trivalent Cr(III) and hexavalent Cr(VI) are common in the natural environment. Cr(VI) compounds are capable of inducing DNA damage and have potential cell transforming effects.6 In contrast, Cr(III) is physiologically essential in sugar and lipid metabolism.7 The IARC has classified Cr(VI) as a Group 1 carcinogen (carcinogenic to humans), while Cr(0) and Cr(III) belong to Group 3 carcinogens (unclassifiable as to carcinogenicity in humans).8 Fowles and Dybing9 conducted a quantitative risk assessment of the contribution of individual cigarette smoke constituents towards the disease incidence associated with smoking. In their calculations they assumed that all Cr in cigarette smoke was present as Cr(VI), and on this basis, they calculated that Cr(VI) was the ninth most important compound for cancer risks and the fifth most important for respiratory effects of the more than a hundred and fifty compounds examined. Despite the concerns that the presence of toxic Cr species in tobacco smoke may pose health risks to smokers, to the author's knowledge, no quantitative Cr speciation data obtained with well characterised methods has been reported so far for cut tobacco or cigarette smoke.
One major challenge associated with the accurate quantification of inorganic Cr species is that the oxidation state of Cr can readily change, depending on pH and electrochemical potential of the exposing environment. In order to achieve maximised extraction efficiency of Cr(III) and Cr(VI) species from solids, previous studies have used alkaline hydrolysis but such methodology has only produced reliable data when used in combination with isotope dilution mass spectrometry calibration in order to minimise the effect of species redox inter-conversion on the accuracy of the Cr species concentration.10,11
For the determination of Cr(III) and Cr(VI) species in a range of sample extracts, use of HPLC-ICP-MS has proven successful. ICP-MS offers one of the most sensitive and robust detectors with pronounced advantages of elemental specificity, wide linear dynamic range and extremely low detection limits.12 The use of dynamic reaction cell (DRC) with ICP-MS greatly reduces the spectroscopic interferences of 40Ar12C+ and 35Cl16O1H+, and thus improves the sensitivity of chromium analysis further.13 The chromatographic methods for chromium speciation analysis may involve either ion exchange chromatography,14,15 with Cr(III) often eluting in the dead volume, or ion-pair reversed-phase chromatography (RPIPC),16 which has been shown to offer higher degree of flexibility, in terms of mobile phase composition to achieve the desired chromatographic selectivity.
Alternatively, X-ray absorption spectroscopy (XAS) is a technique that is able to obtain elemental information on the local atomic structure and the electronic states with minimum sample interference and no need for species extraction.17 A XAS spectrum usually contains three characteristic energy regions, the pre-edge region indicating the presence of an element, the X-ray Absorption Near Edge Structure (XANES) which gives information on the oxidation states of the element, and the Extended X-ray Absorption Fine Structure (EXAFS) which gives information pertaining to the chemical coordination environment of the element.18 Previous work on As speciation using XANES analysis reported a mixture of As(V) and As(III) in 3R4F mainstream cigarette smoke condensate.3
In this work, a systematic approach to the determination of low ng g−1 of Cr species in tobacco and mainstream smoke based on the use of HPLC-ICP-MS with highly compatible extraction and chromatographic separation conditions is presented for the first time. The use of solvents containing tetrapropylammoniumbromide (TPABr) and EDTA for both, species separation and extraction under mild conditions, was investigated to minimise species redox inter-conversion. To verify this, recovery experiments on samples and extracts spiked with Cr species were undertaken. A homogenate of cut tobacco from 3R4F Kentucky reference cigarettes (a replacement of 2R4F cigarettes) was used as a model sample for Cr speciation in tobacco. These research cigarettes, which are typical, blended cigarettes containing all major types of tobacco varieties, have been designed as consistent and uniform test items for research and were recently studied for As speciation.3,4 Following Cr extraction from 3R4F cut tobacco with aqueous TPABr/EDTA, extraction with driselase or SDS or proteolytic enzymes were investigated for further extraction of Cr from the solid residue. Alternatively, procedures based on alkaline hydrolysis of the tobacco homogenate and HNO3/HF versus HNO3 microwave digestion with ICP-MS analysis were also investigated to get a deeper insight into the fractionation of Cr in the solid sample. The developed methodology was then applied to study the speciation of Cr in aqueous TPABr/EDTA extracts of tobacco smoke generated from 3R4F Kentucky reference cigarettes, from cigarettes spiked with Cr(III) before smoking, and smoke condensates spiked with Cr(III) and Cr(IV) after smoke trapping. Finally, the speciation of Cr in cut tobacco and mainstream smoke from 2R4F cigarettes (equivalent to 3R4F in terms of leaf composition and smoke chemistry19) was investigated using XANES. The feasibility of this technique to examine the oxidation states of species present in tobacco, cigarette ash, and mainstream smoke condensate of 2R4F cigarettes was investigated.
Experimental
Reagents, standards and samples
Ethylenediaminetetraacetic acid (EDTA, 99.995%), tetrapropyl ammonium bromide (TPABR, >99%), tetrapropylammonium hydroxide (TPAOH), ammonium hydroxide solution (28–30% m/v) and chromium(III) chloride hexahydrate (98%) were obtained from Sigma-Aldrich (Poole, Dorset, UK). Potassium dichromate certified reference material (NIST SRM 136f) were supplied by LGC Standards (Teddington, Middlesex, UK). Super-purity nitric acid, hydrogen peroxide and hydrofluoric acid were all purchased from Romil (Cambridge, UK). De-ionized water (18.2 MΩ cm−1) was obtained from an Elga water purification unit (Marlow, UK).For quantification of the total chromium content, working standard solutions were prepared by dilution of the 1000 mg L−1 of Cr(III) stock solution with 2% (v/v) nitric acid aqueous solution containing Ge as an internal standard. The NIST 1575 (pine needles) standard reference material was used for quality control of total chromium determination by microwave digestion and ICP-MS.
The research reference cigarettes 2R4F and 3R4F used in this study were provided from Kentucky Tobacco Research & Development Center (Kentucky, Louisville, USA). 2R4F cigarettes were firstly produced in 2003 to substitute the 1R4F cigarettes. In 2008, due to diminishing stock of 2R4F, a replacement 3R4F was made to match 2R4F as much as possible in leaf blend and smoke composition.19 The research described in this work occurred during this stock transition; hence results from both cigarettes were obtained and reported here.
For the HPLC-ICP-MS studies 3R4F cigarettes were used due to their availability. They were kept in a plastic bag and stored in the dark at 4 °C before homogenization and sample preparation.
For the synchrotron studies, 2R4F cigarettes were used following storage in a conditioned room (temperature 22 ± 2 °C and relative humidity 60 ± 5%) for at least 48 hours prior to smoking.
The cigarettes were smoked under ISO 3308 puffing parameters (35 mL puff volume, 2 s puff duration and once every 60 seconds).
Instrumentation
HPLC-Q-ICP-MS measurements were performed using an Agilent 1100 HPLC with quaternary gradient pump, thermostatted autosampler and thermostatted column compartment (Agilent Technologies, Palo Alto, CA, USA). The chromatographic species separation was carried out isocratically using a Agilent PLRP-S 100 Å polymeric reversed phase column made of PEEK (150 mm × 4.6 mm, 3 μm) (Agilent Technologies UK Ltd, Wokingham, Berkshire, UK) at a flow rate of 0.8 mL min−1. The column was kept at 40 °C and the volume of sample injected in the chromatographic system was 50 μL. The outlet of column was connected directly via PEEK tubing to an Agilent 7700 collision cell ICP-MS (Agilent Technologies, Palo Alto, CA, USA) equipped with micromist nebuliser and cooled Scott spray chamber. The ICP-MS was operated in He mode to remove the interferences. Table 1 summarizes the ICP-MS and HPLC conditions.| ICP-MS settings | |
|---|---|
| 7700 ICP-MS | |
| RF power | 1500 W |
| Carrier Ar flow rate | 1.09 L min−1 |
| Sample/skimmer cones | Ni/Ni |
| Spray chamber temperature | 2 °C |
| C/RC gas (He) flow rate | 4.5 mL min−1 |
| Data acquisition | |
| Points per spectral peak | 1 |
| Isotopes monitored | 52Cr, 53Cr |
| Integration time per mass | 500 ms |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|
| HPLC conditions | |
| Analytical column | Agilent PLRP-S 100 Å in PEEK column (150 mm × 4.6 mm, 3 μm) |
| Injection volume | 50 μL |
| Mobile phase | 0.18 mM TPABr/1 mM EDTA at pH 8 |
| Flow rate | 0.8 mL min−1 |
| Temperature | 40 °C |
Microwave digestions for the quantification of the total chromium in the samples were performed in an Ethos EZ microwave system (Milestone, Sorisole, Italy).
Fisherbrand (Fisher Scientific, New Hampshire, USA) ultrasonic bath with temperature control was used for sonication assisted extraction.
The oxidation states of Cr in powdered tobacco, cigarette ash and mainstream smoke condensate were measured at a UK Synchrotron Station (USS) and also at Hamburger Synchrotronstahlungslabor (HASSYLAB) facility of the Deutsches Elektronen Synchrotron (DESY).
Procedures
Preparation of cut tobacco homogenate for HPLC-ICP-MS speciation studies
To avoid possible metal contamination, cut tobacco from the cigarettes were manually ground with an agate mortar and a pestle (previously decontaminated with 45 min sonication in 10% HNO3, thoroughly rinsed with deionised water and air dried). The ground tobacco was stored in amber vials at −80 °C.Preparation of tobacco for XANES speciation studies
The cut tobacco powder was prepared by grinding 2R4F tobacco with a titanium coated grinder (to avoid contamination by the metals of interest). The ground cut tobacco powder was then made into a pellet for X-ray detection using a laboratory pellet press (15T-Presse, Maassen GmbH, Reutlingen, Germany). Plastic tapes were used to cover the metal surfaces of the press which would be in contact with tobacco powder.Preparation of cigarettes with enhanced Cr levels
In addition to speciation studies on reference cigarettes and their tobaccos, experiments were also conducted on cigarettes which were injected with Cr(III) solutions to artificially enhance their levels of Cr. Briefly, 3R4F cigarettes were injected with 25 μL Cr(III) solutions at two different levels (20- and 40-fold the water soluble chromium concentration in cigarettes). The expected total concentrations for the low and the high spike levels were 5.80 and 9.80 μg g−1 of chromium, respectively.Collection of mainstream smoke condensates for HPLC-ICP-MS speciation studies
Mainstream smoke condensates from non-spiked and Cr-spiked 3R4F cigarettes were generated at British American Tobacco's research laboratory (GR&D, Southampton, UK) following a standard ISO machine-smoking procedure reported elsewhere.3 Briefly, ten conditioned cigarettes were smoked for each of the sample tested, using a 20-port Borgwaldt LM20 linear smoking machine (Borgwaldt KC GmbH, Hamburg, Germany). The mainstream smoke passed into a metal-free smoke trap assembly (impingers), which is a trap containing approximately 6 cm depth of soda lime glass balls and a small wad of quartz wool. During the smoking process, the smoking traps were held in polystyrene boxes and surrounded by dry ice to promote condensation and reduce volatility. The smoke condensates were transported in the impingers to LGC in dry ice and stored at −80 °C immediately after delivery.Collection of mainstream smoke condensates for XANES oxidation state studies
In order to examine the oxidation state of the metals in smoke under conditions relevant to human smoking, it is necessary to preserve any possible redox character of the trapped mainstream smoke. The particulate matter collected from cigarette mainstream smoke has been shown to have complex redox behaviour.20 In order to minimise any metal redox reactions occurring in the trapped mainstream smoke during or after smoking, a piece of specially designed glassware with a dry ice cooling chamber was made (Fig. 1). Cigarettes were smoked under ISO puffing conditions using a single-port smoking machine (Borgwaldt KC GmbH, Hamburg, Germany). The smoke was trapped using either a CFP cut-down to 25 mm, or a piece of metal-free Kapton tape (a metal-free polyimide film). The dead volume between the exit of the cigarette filter and the CFP or Kapton tape is approximately 1 mL. At a volumetric flow rate in a puff of 17.5 mL s−1, this ensures that the smoke ageing time is minimised to milliseconds. Samples were stored at solid-CO2 temperatures prior to analysis. For the extended analysis times necessary for smoke condensate analysis samples were stored at liquid nitrogen temperatures to minimise sample ageing.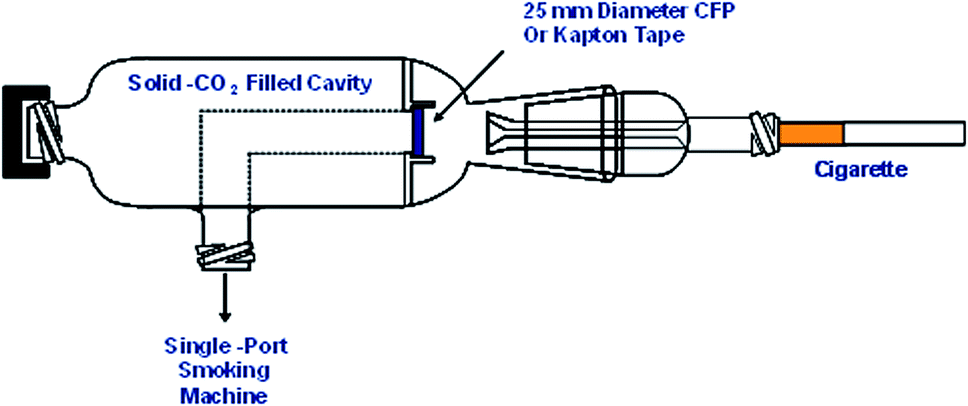 | ||
| Fig. 1 A schematic of the smoking glassware with a dry ice chamber. The cigarette is fitted into the glass tube on the right and a plastic tube is used to connect to a single-port smoking machine. During the experiment, the glass tube was inserted further so that the smoke from the cigarettes sticks to the CFP or a Kapton tape directly. The cap for the dry ice chamber is not kept tight during smoking in order to prevent any increase in pressure in the chamber. | ||
Generation of ash samples for XANES speciation studies
Cigarette ash was collected by smoking 2R4F cigarettes with a 20-port rotary smoking machine under the ISO smoking regime. It was collected and made into pellets for X-ray detection in the same way as the tobacco powder. Metal contamination was minimised during the smoking and ash collection by using clean non-metallic tools for sample handling.Extraction of Cr from cut tobacco using non-denaturing conditions
0.4 g of the cut tobacco was weighed into 15 mL Falcon tubes and 4 g of extract solution (2.8 mM TPABr/4 mM EDTA pH 8.5) was added to the sample. To ensure the stability of Cr(VI), samples were titrated with ammonia solution to get a pH 8. Tubes were placed in ultrasonication bath for 2 hours at 25 °C. To collect the supernatant, samples were centrifuged at 3000 rpm for 30 min. An aliquot of the sample was diluted 1:3 with the extract solution. The sample was heated for 30 min at 70 °C to make sure that all Cr(III) was complexed with the EDTA to stabilize the Cr(III). Finally, the sample was injected onto the HPLC column.
Spiking experiments were performed to investigate the possible occurrence of species interconversion and losses during sample preparation. The same procedure, as described above was followed but samples were spiked with 1 ng g−1 of Cr(III) or Cr(VI). Spike recoveries were calculated by HPLC-ICP-MS analysis of the extracts using Cr(III) and Cr(VI) standard solutions and external calibration.
Chromium fractionation by sequential extraction of cut tobacco
Following Cr extraction from 3R4F cut tobacco with aqueous TPABr/EDTA, extraction with driselase or SDS or proteolytic enzymes were investigated for further extraction of Cr from the solid residue. To achieve this, the sequential extraction procedure developed by Taebunpakul et al. for As in tobacco was adopted.4Ultrafiltration of aqueous extracts from cut tobacco
Aqueous buffered extracts of cut tobacco, prepared as described above, were submitted to ultrafiltration using 3 kDa cellulose acetate centrifugal filter devices (Millipore). Three ultrafiltration replicates and corresponding blanks were prepared. The total Cr contents of the filter retentate and filtrate were determined by ICP-MS using the procedure described below.Extraction of water-soluble chromium from smoke condensates
15 g of the extract solution (2.8 mM TPABr + 4 mM EDTA, pH 8.5) was added to the impingers and placed, bottom up, in an ultrasonic bath for 30 min at 25 °C and 5 min sonication. Water extracts were collected from the impingers and heated at 70 °C during 30 min. In the case of the smoke condensates, several sets of spiking experiments were performed due to the species interconversion. These experiments are explained further in the Results and discussion section.Moisture content determination
The moisture content of the cut tobacco homogenate was determined in three sub-samples of the material. Briefly, 0.5 g of sample was heated at 100 °C in an oven for 3 hours, cooled to ambient temperature in a desiccator and then weighed. This procedure was repeated with 1 h heating cycles until constant weight (difference of <0.001 g between to measurements) was reached.Determination of total chromium in cut tobacco
About 0.48 g of homogenised cut tobacco was weighed accurately into PTFE microwave digestion vessels followed by the addition of 6 mL of HNO3:H2O2:HF (50:48:2, v/v/v). Addition of HF was needed to dissolve any silica, which occurs naturally in tobacco products.21 After mineralization, the digests were diluted to 18 g with de-ionized water. Quantification was performed by standard addition and adding 10 ng g−1 of Ge as internal standard. The isotopes 52Cr and 53Cr were monitored by ICPMS using He as a collision cell gas to remove the potential polyatomic interferences on the detection of m/z 52 (e.g.40Ar12C+). Analysis of the NIST SRM 1575 (pine needles) with Cr certified values of 2.6 ± 0.2 μg g−1 was used to evaluate the accuracy of the procedure. For comparison, another set of samples were submitted to the same microwave digestion protocol but only a mixture of HNO3:H2O2 (50:50, v/v) in order to estimate the fraction of Cr that may be associated of silicates as the Cr difference between the two digestion procedures.
For alkaline hydrolysis, the cut tobacco sample (0.25 g) was weighed into a 15 mL Falcon tube. This was followed immediately by the addition of 8 g of extract solution (94 mM EDTA in 1.0 M TPAOH). The Falcon tube was placed in an ultrasonic bath to ensure that the sample was dispersed in the liquid, then the samples were heated using a Modblock set at 95 °C for 5 hours. After 5 hours, an aliquot of 4 g was digested with HNO3:H2O2 (50:50, v/v). After acid digestion, the samples were diluted up to 12.5 g with ultrapure deionised water.
Determination of total chromium in smoke condensates
25 g of a mixture containing 10% HNO3:H2O2 (96:4, v/v) with 10 ng g−1 Ge was added to the impingers and immediately sonicated in an ultrasonic bath for 15 min. After sonication, the liquid in the impingers was decanted through the quartz wool into a 50 mL polypropylene centrifuge. A second extraction was performed following the same procedure. The total Cr content of the pooled digests was determined by ICP-MS using the procedure described above.
Chromium speciation by HPLC-ICP-MS
Polymeric phase PLRP-S (polydivinylbenzene) packed in a PEEK column was selected for the separation of inorganic Cr species. The chromatographic species separation was carried out isocratically using a 1100 series HPLC (Agilent Technologies UK Ltd. Wokingham, Berkshire, UK) with a mobile phase containing 0.18 mM TPABr and 1 mM EDTA at pH 8. Ion pair/chelation reversed phase chromatography was performed using a PLRP-S 100 Å in PEEK column (150 mm × 4.6 mm, 3 μm) (Agilent Technologies UK Ltd, Wokingham, Berkshire, UK). The column was kept at 40 °C and injection volume of 50 μL was used with mobile phase flow rate of 0.8 mL min−1. The column eluent was introduced directly on to a 7700 ICP-MS (Agilent Technologies UK Ltd, Wokingham, Berkshire, UK) via a microflow quartz concentric nebulizer coupled to a 2 °C cooled Scott type double pass spray chamber. The ICP-MS was operated in He mode (4.5 mL min−1) to remove the potential polyatomic interferences on the detection of m/z 52 (e.g.40Ar12C+).Throughout, precisions are standard deviations of three independent replicates (n = 3).
Synchrotron X-ray absorption spectroscopy analysis
Chromium spectra were collected on two synchrotron beamlines, initially at the UK (Daresbury) facility which was shortly shut down during the period of this work and at a later date in Germany (HASYLAB). The Daresbury facility comprises a 3 GeV electron storage ring, injected from a 100 MeV linear accelerator through a full energy booster synchrotron. The beamline delivers monochromatic X-rays in the energy range of 2 to 20 keV using a pair of Si(111) crystals. The beam size for the sample analysis was approx 0.5 mm by 0.5 mm and 0.5 eV steps around the edge was used. At HASYLAB, X-rays were produced using a blending magnet on the DORIS III storage ring. Monochromatic X-rays were generated using a double-crystal monochromator with Si(111) crystal and focused using nickel coated mirrors. The monochromator energy was calibrated using a Cr metal foil. Fluorescence spectra were measured using a 7 pixel lithium-drifted silicon fluorescence detector. The smoke condensate samples were measured at liquid nitrogen temperature in a cryostat at the A1 beamline. Further details about the beamline can be found at http://hasyweb.desy.de/science/annual_reports/2007_report/part1/contrib/25/22216.pdf.Spectra were collected in transmission and fluorescence modes for standards and samples respectively. One of the mainstream smoke samples collected was kept at −140 °C using a Linkam (Linkam Scientific Instruments) temperature stage during the data acquisition. Some spectra were collected at room temperature. The energy scale was calibrated with Cr metal foil. Data reduction and XANES analyses were carried out using Athena XAS processing software.22
Results and discussion
Determination of total chromium in cut tobacco and smoke condensates
The total Cr content of 3R4F, as found by microwave acid digestion (using the acid mixture HNO3:H2O2:HF) was 1949 ± 171 ng g−1 of Cr (on a dry weight basis and precision as SD, n = 3). This result agrees well with the total content of Cr found for 2R4F, which was 1736 ± 233 ng g−1 of Cr (precision as SD, n = 3). When using the acidic mixture without HF, the total Cr content of 3R4F tobacco was found to be 1715 ± 85 ng g−1 (precision as SD, n = 3), suggesting that approximately 12% of the total Cr in the solid seems to be present in the silicate-containing fraction. The results reported above are in good agreement with the values reported by Fresquez et al.23 for total Cr in 3R4F and 2R4F cigarettes (1300 ± 300 ng g−1 of Cr). On the other hand, the total Cr levels in 2R4F cigarettes found by Counts et al.24 (980 ± 60 ng g−1) is significantly lower than the amount of chromium reported in this paper (1949 ± 171 ng g−1). This could be due to the fact that the microwave digestion was performed without the addition of HF,24 which has been proven essential to release the Cr from the silicate-containing fraction.
For quality control purposes, the NIST SRM 1575 (pine needles) was analysed under the optimal conditions described above. An average recovery of 99 ± 3% (RSD, n = 3) of the certified value was obtained using the HF digestion procedure.
The smoke condensates used in this study were generated from injected cigarettes with Cr(III). This was required since Cr(III) could not be detected well above the LOQ (0.41 ng g−1 Cr, based on a signal-to-noise ratio of 10:1) in aqueous extracts of smoke condensates of unspiked cigarettes. More than 99% of Cr in the solution used for injection of cigarettes remained as Cr(III), for at least 48 h after preparation. The total chromium in the injected cigarettes represented approximately 95% of the expected Cr values (5520 ± 323 ng g−1 and 9246 ± 198 ng g−1 for the low and high spike level of Cr, respectively). Only 26 ± 7 ng (or 4.3 ± 1.2 ng g−1) of Cr was found in smoke for both levels of injected Cr(III), suggesting that less than 1% of the chromium present was transferred from the cigarette to the smoke. This agrees well with the transfer rate reported in the literature (0.4–1.8%), which is relatively low due to low volatility of the chromium compounds.25
Separation and detection of Cr species by HPLC-ICP-MS
Methodology for the separation of Cr(III) and Cr(VI) using reversed phase ion pairing HPLC26,27 (RPIPHPLC) with ICP-MS detection was developed with the purpose to achieve sufficient retention and chromatographic selectivity for both species in the complex matrix, to avoid species redox inter-conversion during the separation step and to minimise introduction of organics into the ICP-MS. The latter is due to the fact that significant quantities of organic modifiers in the mobile phase can significantly affect the limit of detection of Cr species due to generation of 40Ar12C+, interfering the detection of m/z 52.The column selected was a PLRP-S (polydivinylbenzene) column made of PEEK instead of stainless steel to avoid chromium bleeding from the column. PLRP-S columns contain rigid macro porous spherical particles of polystyrene and divinylbenzene and therefore, have the advantage that they are physically and chemically stable across the complete pH range. Moreover the thermal stability of PLRP-S media is superior to that of silica-based material.
Regarding the selection of ion-pair reagent, tetrabutylammonium has previously been used in the mobile phase for ion-pair formation and retention of Cr(VI) in combination with methanol or acetonitrile.25,26,28,29 However, this approach results in a high content of organics reaching the ICP. To minimize this, tetrapropyl ammonium bromide (TPABr)30,31 was used in this work. This enabled sufficient chromatographic selectivity to be achieved for the inorganic Cr species without the need for organic modifiers such as methanol or acetonitrile.
The pH of the mobile phase is another factor that critically affects Cr species stability, retention and separation. At pH > 8 Cr(VI) exists mainly as chromate (CrO42−), which can react with the positive charge of the cationic ion-pairing reagent (TPABr), thus interacting with the stationary phase to enable retention of CrO42−. On the other hand, the Cr(III) species can precipitate as Cr(OH)3 at basic pHs.32 In this work, in order to solubilise, stabilise and retain the Cr(III) species at basic pHs, its complexation with EDTA was achieved by heating for 30 min at 70 °C prior to injection of the Cr extract onto the column. The Cr(III)–EDTA complex is negatively charged33 and it can interact with the TPABr. By adding EDTA to the mobile phase and maintaining the column heated at 40 °C, this work ensured stability of the Cr(III)–EDTA complex.
Fig. 2 shows the chromatogram obtained for a Cr standard mixture containing Cr(VI) and Cr(III), each at a concentration of 10 ng g−1 Cr. As shown, baseline separation of both species can be achieved in less than 20 minutes using a mobile phase containing 0.18 mM TPABr/1 mM EDTA at pH 8 and a flow rate of 0.8 mL min−1. Using these optimal conditions, chlorine-based interferences (35Cl16O1H+ and 37Cl16O+) on Cr(III) and Cr(VI) coming from the chlorine present in the Cr(III) standard, were overcome since they were detected in the chromatographic void volume. Evidence for this was found by injecting 100 mg kg−1 sodium chloride onto the column and consequently, monitoring m/z 53 by HPLC-ICP-MS. The instrumental limits of detection (3σ criterion) obtained for Cr(III) and Cr(VI) using the HPLC-ICP-MS methodology developed here were 0.12 and 0.08 ng g−1 Cr, respectively.
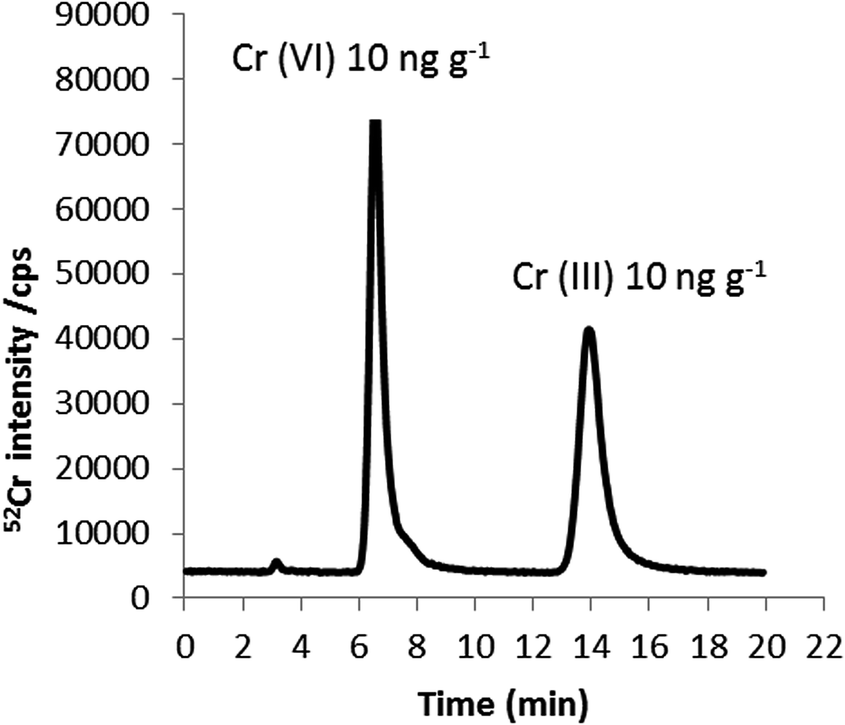 | ||
| Fig. 2 HPLC-ICP-MS chromatogram of Cr(VI) and Cr(III) in an aqueous standard solution (10 ng g−1 of each Cr species). 0.18 mM tetrapropylammonium bromide (TPABr), 1 mM EDTA (pH 8) as mobile phase at a flow rate of 0.8 mL min−1. | ||
Extraction of Cr species from cut tobacco
Both 2.8 mM tetrapropylammonium bromide (TPABr) containing 4 mM EDTA (pH 8.5), which is similar to the HPLC mobile phase, and 1 M ammonium acetate (pH 7.1) were investigated as solvents for chromium extraction from cut tobacco. The extractant choice was based on their neutral/basic pHs as Cr(VI) is likely to be transformed to Cr(III) at acidic pHs.The total Cr extracted using both solvents (0.4 g sample in 4 mL extractant) and a sonication time of 2 h was found to be 10 ± 2% of the total Cr in the homogenate of 3R4F cut tobacco (1949 ± 171 ng g−1 Cr). Increasing sonication time did not further improve the Cr extraction efficiency using these highly aqueous solvents.
Alkaline hydrolysis, using TPAOH, was investigated in order to increase the Cr extraction yield because it has been proven to help dissolve insoluble Cr(VI) compounds.34 EDTA was added to prevent the precipitation and/or oxidation of Cr(III) due to the high pH.31 Using this method, approximately 14 ± 2% of the total Cr was extracted. This did not represent a significant improvement in extraction efficiency in comparison to that of aqueous extractants (10 ± 2% of the total Cr). Moreover, the use of harsh alkaline conditions is known to lead to species inter-conversion, requiring the need of tedious and expensive calibration procedures such as isotope dilution mass spectrometry to achieve accurate speciation data.
Finally, a multi-step sequential extraction procedure using aqueous solvents followed by enzymes and surfactants was studied and the results have been summarised in Table 2. This has been applied successfully to the extraction of arsenic species from cut tobacco4 and therefore, previously reported methodology was adopted here. Initially water extraction removed the water soluble chromium compounds. The total Cr results suggested that only the extraction with SDS (to solubilise water-insoluble proteins) leached a further 10% from the total Cr in the solid matrix. The use of either Driselase (to remove cell-wall components) or proteolytic enzymes (to hydrolyse proteins remaining in the residual solid) did not help to increase the extraction efficiency of Cr from the cut tobacco matrix.
| Sample treatment | % of Cr extracted vs. total |
|---|---|
| MW digestion (HNO3:H2O2:HF) |
100 ± 2 |
| MW digestion (HNO3:H2O2) |
88 ± 1 |
| Extraction using non-denaturing conditions | 10 ± 2 |
| Sequential extraction (water + driselase + SDS + proteolytic enzymes) | 20 ± 3 |
| Alkaline hydrolysis | 14 ± 2 |
In total, a maximum of 32% of the total Cr in the solid cut tobacco could be extracted as found present in the water soluble fraction, the water-insoluble protein fraction (SDS fraction) and the silicate fraction (HF digestion). The rest of the Cr is likely to be associated with Cr(III) species (e.g. Cr2O3),35 which can only be dissolved using strong acid conditions (e.g. by microwave acid digestion). This is examined through the XANES experiments reported below.
For further work, leaching with 2.8 mM tetrapropylammonium bromide (TPABr) containing 4 mM EDTA (pH 8.5) was chosen as a trade-off between the extraction yield, chromium species preservation and compatibility with the chromatographic system for further speciation studies in cut tobacco and smoke condensates. The choice is also driven by the need to determine whether the easily leachable Cr is present as inorganic Cr(III) or Cr(VI), due to the relative high toxicity of the latter.
Chromium speciation analysis of cut tobacco by HPLC-ICP-MS and XANES
Detection and quantification of Cr species in the aqueous leachable fraction (non-denaturing conditions) of 3R4F cut tobacco was performed using 2.8 mM tetrapropylammonium bromide (TPABr) and 4 mM EDTA (pH 8.5) and the conditions described above.The chromatogram obtained using the HPLC-ICP-MS conditions summarised in Table 1 is shown in Fig. 3. As can be seen, Cr(III) appears to be the main Cr species detected at 14 minutes. Species identification is on the basis of retention time matching with a Cr(III) standard. Quantification of the chromium species present in the cut tobacco was performed by external calibration using HPLC-ICP-MS with Cr(III) standards. The amount of Cr associated with Cr(III) (9.1 ± 0.5 ng g−1) was found to comprise around 5% of the total chromium in the aqueous leachate (195 ± 36 ng g−1). Spike experiments with Cr(VI) and Cr(III) achieved recoveries of 107 ± 7% and 90 ± 6%, respectively, indicating that there were no significant losses during the sample preparation or in the chromatographic system and that species inter-conversions were kept to a minimum or avoided completely.
 | ||
| Fig. 3 HPLC-ICP-MS chromatogram of Cr species in cut tobacco extracted with 2.8 mM TPABr and 4 mM EDTA (pH 8.5). 0.18 mM, 1 mM EDTA as mobile phase (pH 8) at a flow rate of 0.8 mL min−1. | ||
The chromatographic profile shown in Fig. 3 alone does not seem to support the fact that only 5% of the total Cr in the aqueous extract corresponds to leachable Cr(III), as discussed above. The figure shows only two chromatographic peaks corresponding to unretained or positively charged Cr species (void) and inorganic Cr(III), which seem to comprise approximately 40% and 45%, respectively, of the total Cr in the extract. This is calculated on the basis of peak area percentage distribution and considering that most injected Cr is eluting from the column. This discrepancy may suggest that not all the Cr present in the aqueous extract is detected using the HPLC-ICP-MS methodology developed here for leachable inorganic Cr(III) and Cr(VI) fractionation.
In order to understand the discrepancy mentioned above, further investigation on the fractionation of Cr in the aqueous leachate was performed by offline ultrafiltration experiments, as described in Procedures. Interestingly, the results revealed that 75 ± 8% (RSD, n = 3) of the total Cr in the aqueous extract of cut tobacco seems to be associated with species of Mw > 3 kDa, which cannot be easily eluted from the reversed phase column under the selected chromatographic conditions (e.g. low content of organic solvents). Again, it is important to note that such conditions were selected for the purpose of speciation of inorganic Cr species with focus on leachable Cr(III) and Cr(VI). The remaining Cr (Mw < 3 kDa) represents only 25 ± 8% of the total Cr in the leachate with Cr(III) being only a fraction of it. This is more in accordance with the quantitative data reported for Cr(III) above, as obtained by using external calibration with HPLC-ICP-MS, leaving only 25 ± 8% (RSD, n = 3) of the total Cr present in a fraction with Mw < 3 kDa.
Further insight into the Cr content of tobacco was provided by experiments using XANES. This approach avoids the complexities of Cr species extraction, from solid tobacco, as the technique examines the solid tobacco sample without complex pre-treatment. The XANES spectra of Cr standards are shown in Fig. 4, the absorption edges being used as fingerprints to identify the presence of the metal, the valence state and the symmetry round the absorbing atoms. The Cr K-edge measured at 5989 eV was used for calibration. The Cr(II) edge position was identified at 5992.6 eV by the Cr acetate standard. Trivalent and hexavalent oxidation states were measured on Cr2O3 and CrO3 at the edge position of 5997.4 and 6004.7 eV, respectively.
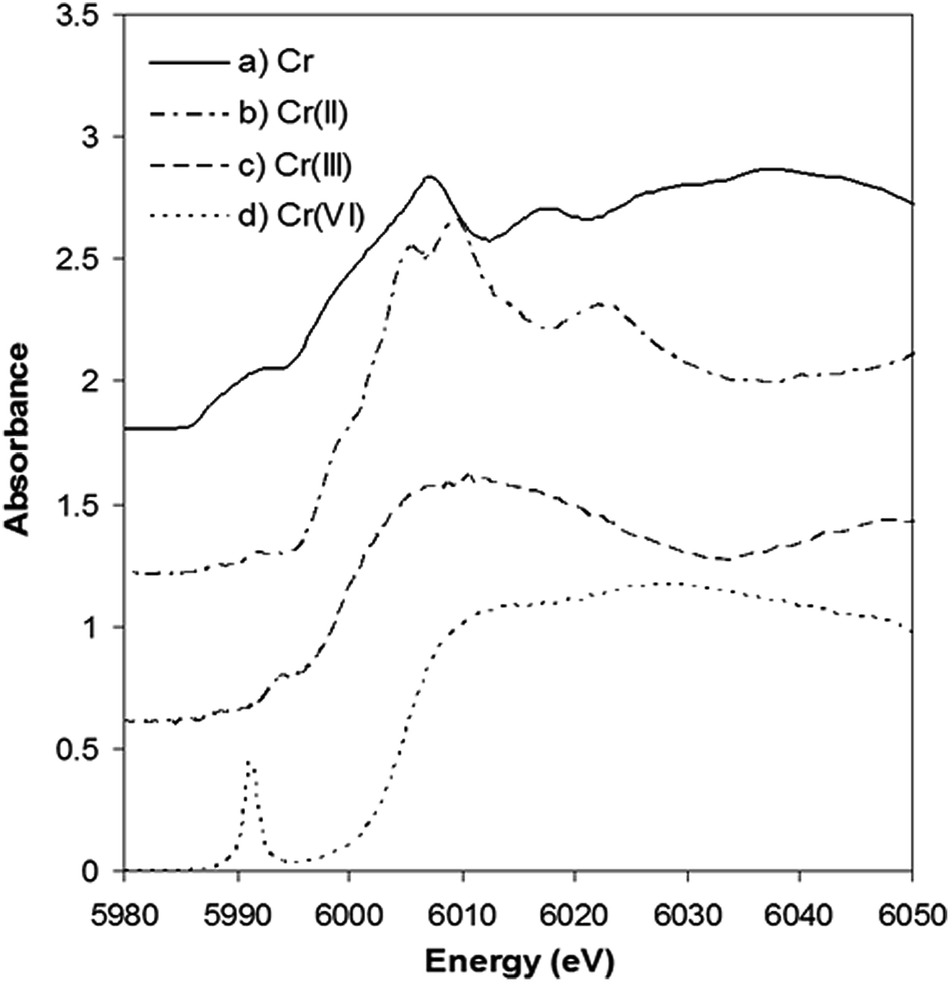 | ||
| Fig. 4 Normalized Cr K-edge XANES spectra of (a) Cr(0) metal, (b) Cr(II) acetate, (c) Cr2O3, and (d) CrO3. These spectra have been vertically shifted for clarity | ||
It is clear that the absorption edge shifts to higher energy values with increasing valence for chromium. It can also be seen that the pre-edge and edge-step features vary considerably from one spectrum to another. A sharp pre-edge peak at 5991 eV is only present in the CrO3 spectrum corresponding to the existence of Cr(VI).
The XANES spectra of chromium in the tobacco powder is shown in Fig. 5(a). The spectrum shows a clear edge signal, confirming the presence of chromium in the sample. The lack of the pre-edge peak associated with Cr(VI) and the edge position of the tobacco sample (∼5996 ± 2 eV) suggests that very little Cr(VI) is present, and indicates that the most likely Cr species present in the sample is Cr(III), which agrees with the results obtained by HPLC-ICP-MS and with the results of Zayed et al.,36 who showed that the chromium absorbed by the roots of plants is converted to Cr(III) and then translocated to the leaves.
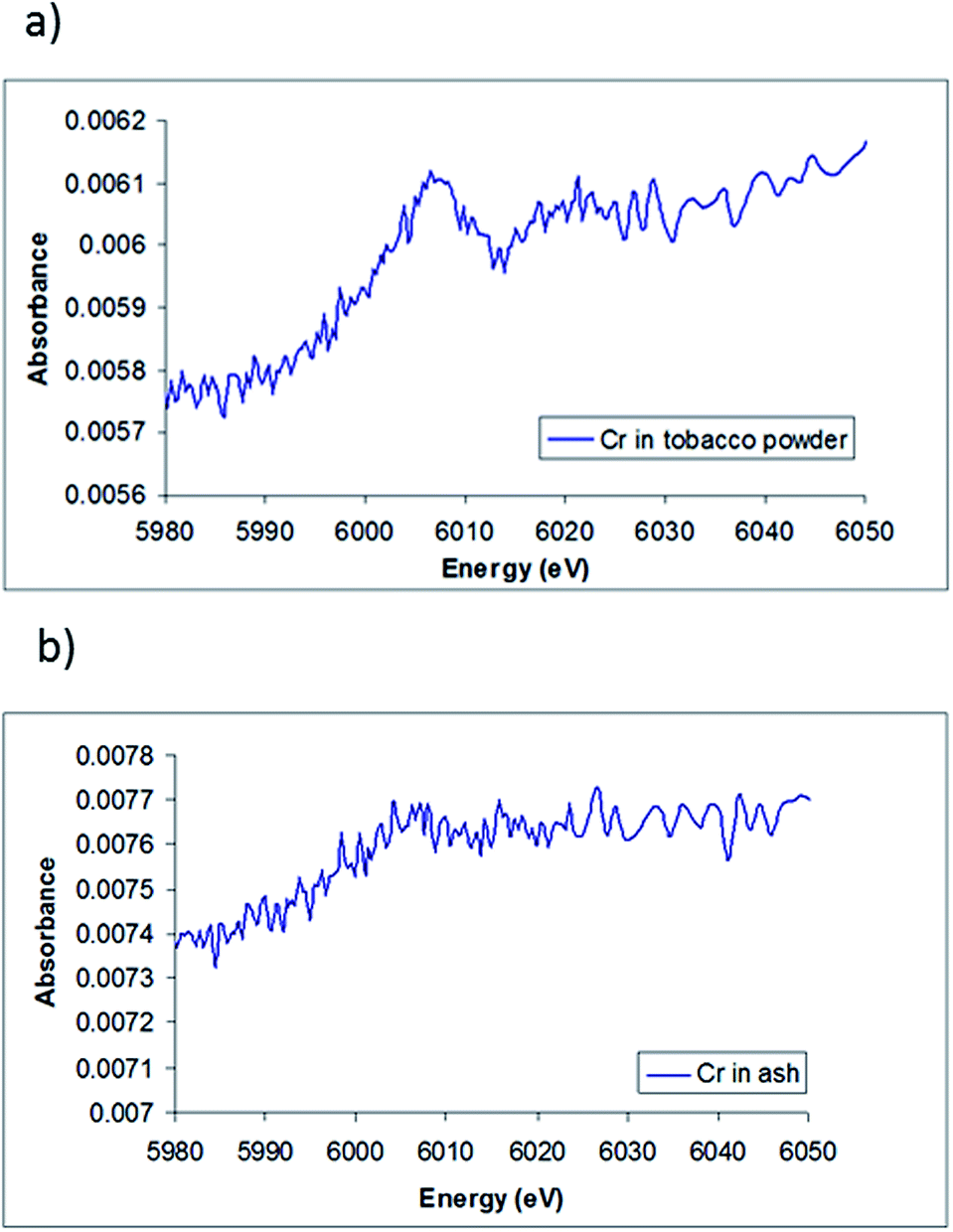 | ||
| Fig. 5 The XANES spectra of Cr (a) in tobacco powder samples, (b) in cigarette ash samples. | ||
Compared to the chromium in tobacco powder spectra, Fig. 5(b) shows the XANES spectra of chromium in the cigarette ash sample. It appears that the spectrum has a rather weak edge signal, making it difficult to accurately determine an oxidation state. However, the edge position of the ash sample combined with the lack of the pre-peak features for Cr(VI) also suggests the oxidation state of chromium in tobacco ash is more likely to be Cr(III).
Chromium speciation analysis of mainstream smoke condensates by HPLC-ICP-MS and XANES
In order to investigate the fractionation of chromium into Cr(VI) and Cr(III) in mainstream smoke condensates, speciation analysis of condensate water extracts, as obtained by using the same extraction procedure employed for 3R4F cut tobacco, was undertaken using RPIPHPLC-ICP-MS.Fig. 6 shows chromatograms of the smoke condensates extracts from cigarettes spiked at different levels. The main chromium species detected was Cr(III). For both spike levels, the amount of Cr(III) was very similar, being 4 ± 1 ng (or 0.7 ± 0.2 ng g−1) of Cr(III) for the low spike level and 4.4 ± 0.3 ng (or 0.73 ± 0.05 ng g−1) of Cr(III) for the high spike level. These values represent approximately 16% of the total Cr in the solid smoke condensate. It is interesting to note that Cr(III) concentration found in the smoke condensate extracts by HPLC-ICPMS seems to be independent on the level of chromium fortification. This result is supported by a good agreement found between the total Cr levels of the smoke extracts for both spike levels. Either saturation of the smoke with Cr at the lowest Cr spike level and/or the lack of detectability of the Cr transferred from tobacco to smoke [due to the very small (only 2-fold) difference between spike levels combined with the poor Cr transference efficiency] are likely to be responsible for the observed results. Having said that, in the absence of data for spike levels below 5520 ng g−1 and well above 9246 ng g−1 of Cr, it is very difficult to draw a definitive explanation for this observation.
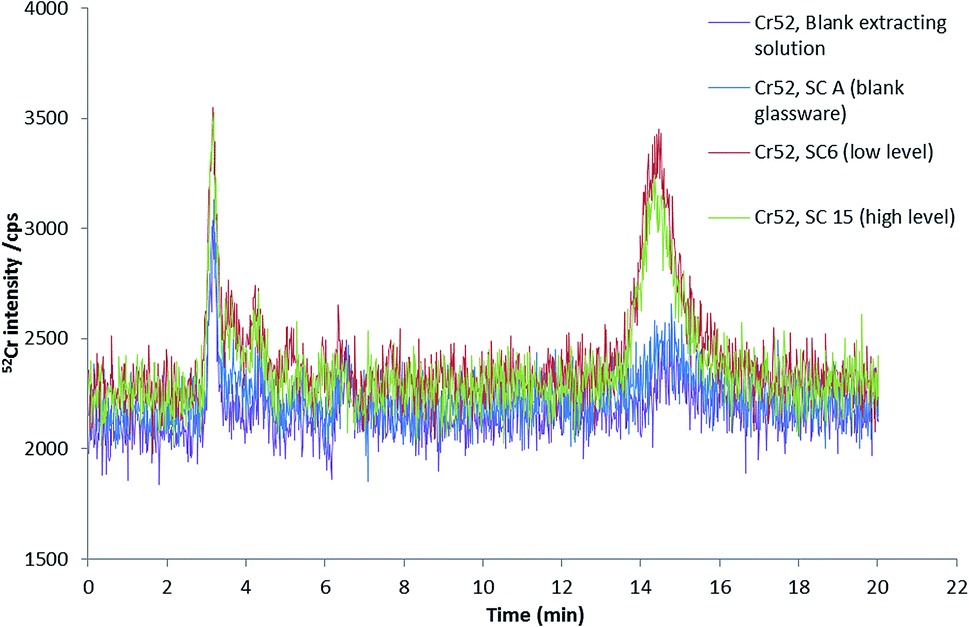 | ||
| Fig. 6 HPLC-ICP-MS chromatogram of Cr species in mainstream smoke from cigarette spiked two different spike levels (5.80 and 9.80 μg g−1 of chromium, respectively) using 2.8 mM TPABr and 4 mM EDTA (pH 8.5) as extractant. 0.18 mM, 1 mM EDTA (pH 8) as mobile phase at a flow rate of 0.8 mL min−1. | ||
To investigate whether or not Cr(VI) is present in the large fraction of the smoke condensate (84%) that was not amenable to HPLC-ICP-MS speciation analysis, XANES was employed. For the XANES experiments, the mainstream smoke samples collected using two separate trapping systems, using a Cambridge filter pad (CFP) and, as an alternative trapping system, using impaction onto Kapton tape. XANES spectra were captured for blank CFPs, mainstream smoke condensate trapped on CFPs, and smoke impacted on Kapton tape. Fig. 7(a) shows the summed absorbance spectra taken from both the CFP trapped mainstream smoke samples and blank CFP. In both spectra, a significant edge step due to Cr(III) was observed. Clearly the presence of Cr(III) intrinsic to the CFP influences the spectrum observed with the CFP/smoke sample, however it is notable that there is no evidence for any Cr(VI) in the smoke/CFP spectrum. In Fig. 7(b), when the smoke condensate was collected on a Cr-free Kapton tape, a clear presence of Cr(III) edge was also noted. Fig. 8 compares normalised spectra for Cr(III) and Cr(VI) standards to the smoke/Kapton signal, and clearly demonstrates that Cr(III) is the most significant chromium species in mainstream smoke condensate. The presence of two small pre-edge peaks in the smoke sample coincide with the two pre-edge peaks seen with the Cr(III) standard. One of the pre-edge peaks in the smoke condensate analysis experiment appeared to be enhanced over that of the Cr(III) sample. The position of the enhanced sample occurs at a different (higher) energy than the Cr(VI) pre-edge peak and therefore is unlikely to be caused by trace levels of Cr(VI) in the smoke condensate sample. The increased signal at this energy may reflect differences in the chemical environment of Cr in the standards and in smoke condensate.
 | ||
| Fig. 7 Comparison between the absorbance spectra of the mainstream smoke condensate samples and the blank Cambridge filter pad (CFP) substrates (a); and smoke condensate collected on a Kapton substrate (b). | ||
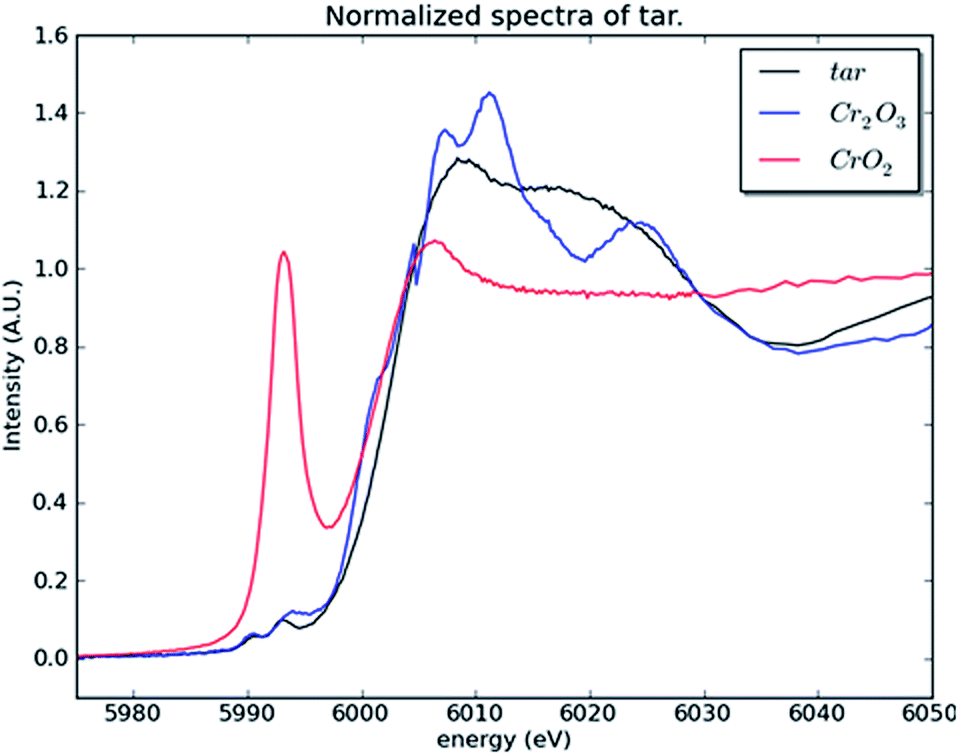 | ||
| Fig. 8 Normalised absorption spectra of the mainstream smoke condensate (tar) sample collected on a Kapton substrate and the two Cr standards. | ||
To further examine the possible presence of trace quantities of Cr(VI) in cigarette smoke, experiments were conducted in which smoke condensates were spiked separately with Cr(III) and Cr(VI) prior to quantitative speciation by HPLC-ICP-MS. With Cr(III)-spiked smoke samples, the Cr(III) the recovery obtained was approximately 100%, suggesting that Cr(III) is stable in smoke condensate during sample treatment. On the other hand, when samples were spiked with Cr(VI), 96% of the added chromium was recovered as Cr(III) at a time > 1 h (sample treatment time). This is represented in Fig. 9. The acidic nature of the smoke condensate matrix37,38 and the contact of the added species with the matrix seem to be responsible for this conversion; Cr(VI) is not stable at acidic pHs. To further investigate whether species conversion is due to a matrix effect rather than the influence of sample preparation conditions and/or the combination of both, two sets of experiments were performed. Firstly, smoke condensates were spiked with Cr(VI) after sample treatment and prior to analysis. In this case, Cr(VI) was reduced over time to Cr(III); 80% and 100% conversion was observed after 8 and 24 h of Cr(VI) addition, respectively (data not shown). Secondly, standards of Cr(VI) in the extract solvent, without the presence of the matrix, were submitted to the entire sample preparation process described above before analysis. No evidence of conversion into Cr(III) was detected.
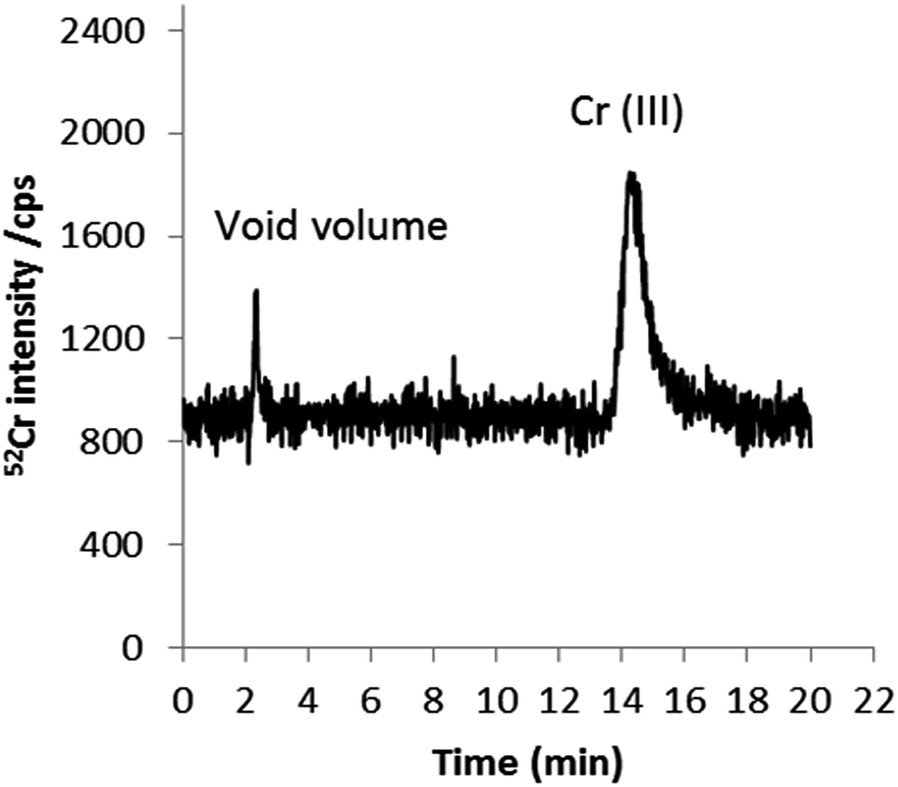 | ||
| Fig. 9 HPLC-ICP-MS chromatogram of Cr species in mainstream smoke spiked with 1 ng g−1 Cr(VI) (added to the extractant). 0.18 mM, 1 mM EDTA (pH 8) as mobile phase at a flow rate of 0.8 mL min−1. | ||
The results reported above suggest that Cr(VI) is not stable in cigarette smoke condensate. This finding does not agree with that reported by Sógor et al. where 0.8–1.2% of the original chromium content of the cigarettes is reported to be present in smoke as the toxic Cr(VI) form.35 However, quality control of the proposed methodology, e.g. by spiking experiments to prove the lack of Cr(III) and Cr(VI) redox interconversion was not undertaken in this previous work.35
Support for the instability of Cr(VI) in cigarette smoke condensate is provided by previous reports of the reducing nature and pH of the cigarette smoke. Schmeltz et al.39 measured the redox potential of cigarette smoke on a puff-by-puff basis. It was found that the reducing activity of the smoke increased as the cigarette was puffed.
The electrochemical potential of the smoke was found to be between 0.24 V (the first puff) to 0.17 V (last puff) vs. calomel reference electrode (SCE). Values for the pH of mainstream smoke condensate from US blended cigarettes can be measured at pH 5.5–6.5 depending upon blend composition and the measurement technique.29,40 According to Pourbaix's diagram for chromium,41 Cr(III) is the most stable form in this pH–potential region for the mainstream smoke, since Cr(III) → Cr(VI) conversion will not take place at a potential of below 0.51 V vs. SCE, and Cr(III) → Cr(II) is not possible at a potential more than −0.82 V vs. SCE. Smoke pH is generally between 5 to 6.5 for contemporary commercial cigarettes of either US blended or Virginia type, hence the findings of this study is likely to be relevant to the majority of commercial cigarettes, despite the fact that our measurements were only performed on reference cigarettes. However, our speciation findings on the Cr content of tobacco may be somewhat less general, as some non-cigarette tobaccos have more alkaline pHs (e.g., pH 8–9) such as many smokeless tobaccos.42 Further studies are required to investigate Cr speciation in these tobaccos.
Conclusion
This work demonstrates that the main Cr species found either in 3R4F cut tobacco or smoke condensate from Cr(III)-spiked tobacco corresponds to Cr(III). This was achieved for the first time by using HPLC-ICP-MS as a complementary technique to XANES. The quantitative speciation data obtained also suggests that approximately 12% of the chromium in 3R4F tobacco is associated within the silica-containing fraction, 10% is water soluble (75% of which is associated with species of molecular mass > 3 kDa), and a further 10% is likely associated with water-insoluble proteins. The chemical environment of the remaining 68% of the Cr, other than it's oxidation state of +3, in tobacco remains unclear. Results from specie-spiking experiments, either on smoke condensates before species extraction or on water leachates of smoke condensates, pointed to the likely conversion of Cr(VI) to Cr(III) due to the acidic and reductive nature of the smoke condensates, thus negating the possibility of finding Cr(VI) in tobacco smoke.The results of total Cr measurements obtained for Cr-spiked cut tobacco and the corresponding smoke condensate using ICP-MS after microwave acid digestion suggest that less than approximately 1% of the total Cr is transferred from the leaf to the smoke. Therefore, for Cr speciation in smoke from unspiked 3R4F tobacco to be undertaken, the feasibility of a different instrumental set up (e.g. use of ammonia reaction gas in single or triple quadrupole ICP-MS, sector field ICP-MS and/or metal-free HPLC) offering improved sample-to-blank signal ratio and Cr limit of detection has to be investigated.
Acknowledgements
This work was funded by British American tobacco (BAT). The authors thank the assistance of Julien Heroult with the initial method development for total Cr and Cr species in cut tobacco and the determination of moisture content and Jin Hu and Adam Webb for conducting the XANES experiments.References
- C. D. Tsadilas, N. A. Karaivazoglou, N. C. Tsotsolis, S. Stamatiadis and V. Samaras, Environ. Pollut., 2005, 134, 239–246 CrossRef CAS PubMed.
- C. D. Tsadilas, J. Plant Nutr., 2000, 23, 1167–1178 CrossRef CAS.
- C. Liu, J. Hu and K. G. McAdam, Spectrochim. Acta, Part B, 2009, 64, 1294–1301 CrossRef.
- S. Taebunpakul, C. Liu, C. Wright, K. McAdam, J. Heroult, J. Braybrook and H. Goenaga-Infante, J. Anal. At. Spectrom., 2011, 26, 1633–1640 RSC.
- R. C. J. Campbell, W. E. Stephens and A. A. Meharg, Tob. Induced Dis., 2014, 12, 1–8 CrossRef PubMed.
- IARC Working Group, in IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans, International Agency for Research on Cancer, World Health Organization, 1986, 38, 115–116 Search PubMed.
- M. Chiba and R. Masironi, Bull. World Health Organ., 1992, 70, 269–278 CAS.
- IARC Working Group, in IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans, International Agency for Research on Cancer, World Health Organization, 1990, 49, 213–214 Search PubMed.
- J. Fowles and E. Dybing, Tobacco Control, 2003, 12, 424–430 CrossRef CAS.
- L. Yang, E. Ciceri, Z. Mester and R. E. Sturgeon, Anal. Bioanal. Chem., 2006, 386, 1673–1680 CrossRef CAS PubMed.
- M. E. Soares, E. Vieira and M. de Lourdes Bastos, J. Agric. Food Chem., 2010, 58, 1366–1370 CrossRef CAS PubMed.
- K. Wrobel and J. A. Caruso, in The encyclopedia of mass spectrometry, volume 5: elemental and isotope ratio mass spectrometry, ed. M. L. Gross and R. M. Caprioli, Elsevier, Oxford, 2010 Search PubMed.
- G. Eiden, C. J. Barinaga and D. W. Koppenaal, in The encyclopedia of mass spectrometry, volume 5: elemental and isotope ratio mass spectrometry, ed. M. L. Gross and R. M. Caprioli, Elsevier, Oxford, 2010 Search PubMed.
- I. K. Dimitrakopoulos, H. D. Tserepa and N. S. Thomaidis, Anal. Lett., 2012, 45, 570–580 CrossRef CAS.
- Z. L. Chen, M. Megharaj and R. Naidu, Talanta, 2007, 73, 948–952 CrossRef CAS PubMed.
- R. E. Wolf, J. M. Morrison and M. B. Goldhabe, J. Anal. At. Spectrom., 2007, 22, 1051–1060 RSC.
- N. Unceta, F. Seby, J. Malherbe and O. Donard, Anal. Bioanal. Chem., 2010, 397, 1097–1111 CrossRef CAS.
- P. M. Bertsch and D. B. Hunter, Chem. Rev., 2001, 101, 1809–1842 CrossRef CAS PubMed.
- E. Roemer, H. Schramke, H. Weiler, A. Buettner, S. Kausche, S. Weber, A. Berges, M. Stueber, M. Muench, E. Trelles-Sticken, J. Pype, K. Kohlgrueber, H. Voelkel and S. Wittke, Beitr. Tabakforsch. Int., 2012, 25, 316–335 CAS.
- M. Borgerding and H. Klus, Exp. Toxicol. Pathol., 2005, 57, 43–73 CrossRef CAS PubMed.
- A. Rodgman and T. A. Perfetti, in The Chemical Components of Tobacco and Tobacco Smoke, ed. A. Rodgman and T. A. Perfetti, Taylor and Francis Group, CRC Press, Boca Raton, FL, 2009 Search PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- M. R. Fresquez, R. S. Pappas and C. H. Watson, J. Anal. Toxicol., 2013, 37, 298–304 CrossRef CAS PubMed.
- M. E. Counts, F. S. Hsu and F. J. Tewes, Regul. Toxicol. Pharmacol., 2006, 46, 225–242 CrossRef CAS PubMed.
- A. Rodgman and T. A. Perfetti, The Chemical Components of Tobacco and Tobacco Smoke, CRC Press, 2nd edn, 2013 Search PubMed.
- J. Lintschinger, K. Kalcher, W. Gössler, G. Kölbl and M. Novic, J. Anal. Chem., 1995, 351, 604–609 CAS.
- R. E. Wolf, J. M. Morrison and M. B. Goldhaber, J. Anal. At. Spectrom., 2007, 22, 1051–1060 RSC.
- Y.-L. Chang and S.-J. Jiang, J. Anal. At. Spectrom., 2001, 16, 858–862 RSC.
- J.-F. Jen, G.-L. Ou-Yang, C.-S. Chen and S. M. Yang, Analyst, 1993, 118, 1281–1284 RSC.
- J. Entwisle and H. Goenaga-Infante, LGC/R/2013/271 March 2013, https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/318687/Chromium_Speciation_GC_Report.pdf, accessed October 2015.
- J. Entwisle and H. Goenaga-Infante, LGC/R/2013/312 October 2013, https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/318505/GC_Chromium_Speciation_Quantitative_Report.pdf, accessed October 2015.
- L. Xing and D. Beauchemin, J. Anal. At. Spectrom., 2010, 25, 1046–1055 RSC.
- M. L. Marina, P. Andres and J. C. Díez-Masa, Chromatographia, 1993, 35, 621–626 CAS.
- A. Katz, Environ. Health Perspect., 1991, 92, 13–16 CrossRef PubMed.
- C. Sógor, A. Gáspar and J. Posta, Microchem. J., 1998, 58, 251–255 CrossRef.
- A. Zayed, C. M. Lytle, J. H. Qian and N. Terry, Planta, 1998, 206, 293–299 CrossRef CAS.
- R. R. Baker, Smoke Chemistry, in Tobacco Production, Chemistry and Technology, ed. Davis DL and Nielsen MT, Blackwell Publishing Ltd, 1999, pp. 398–439 Search PubMed.
- J. I. Seeman, Chem. Res. Toxicol., 2007, 20, 326–343 CrossRef CAS PubMed.
- I. Schmeltz, J. Tosk, G. Jacobs and D. Hoffmann, Anal. Chem., 1977, 49, 1924–1929 CrossRef CAS PubMed.
- R. R. Baker, Combust. Flame, 1977, 30, 21–32 CrossRef CAS.
- M. Pourbaix, Atlas of Electrochemical Equilibria in Aqueous Solutions, National Association of Corrosion Engineers, 2nd edn, 1974 Search PubMed.
- A. Rodgman, Contrib. Tob. Res., 2000, 19, 117–235 CAS.
| This journal is © The Royal Society of Chemistry 2016 |