
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAluminium salabza complexes for fixation of CO2 to organic carbonates†
L.
Cuesta-Aluja
*,
J.
Castilla
and
A. M.
Masdeu-Bultó
*
Department of Physical and Inorganic Chemistry, University Rovira i Virgili, Marcel·lí Domingo, s/n. 43007 Tarragona, Spain. E-mail: annamaria.masdeu@urv.cat
First published on 27th April 2016
Abstract
A highly stable and easy to synthesize aluminium complex bearing a flexible N2O2-donor salabza ligand (N,N′-bis(salicylene)-2-aminobenzylamine) in combination with tetrabutylammonium bromide forms an active binary catalytic system for the cycloaddition of CO2 to epoxides (TOFs 120–3434 h−1) under mild conditions (10 bar, 80 °C) and low catalyst loadings (0.05–0.2 mol%). Kinetic experiments have shown that the cycloaddition of CO2 to styrene oxide catalyzed by 1/TBAB is first order in 1, TBAB, CO2 and epoxide. A reaction mechanism is proposed based on these observations. Fe(III) and Co(III) related complexes are less active catalysts for this reaction.
Introduction
The metal-catalysed coupling of CO2 and epoxides has become one of most studied topics in CO2 activation, as it is an atom-efficient reaction to selectively obtain highly value-added cyclic carbonates or polycarbonates from inexpensive and readily available starting materials (Scheme 1). Cyclic carbonates are valuable synthetic targets that are widely used as raw materials for the synthesis of small molecules1,2 and polymers.3,4 They are also used as electrolytes in lithium-ion secondary batteries5 and have applications in the chemical industry as excellent polar aprotic solvents.6–8 Polycarbonates are high performance and eco-efficient materials with high transparency, durability, safety, heat and shatter resistance, good electrical insulation, strength, lightness and biodegradability with application in many areas of industry.9 The increasing demand of these compounds implies the development of new, commercially viable, catalysts and processes which operate under mild reaction conditions.10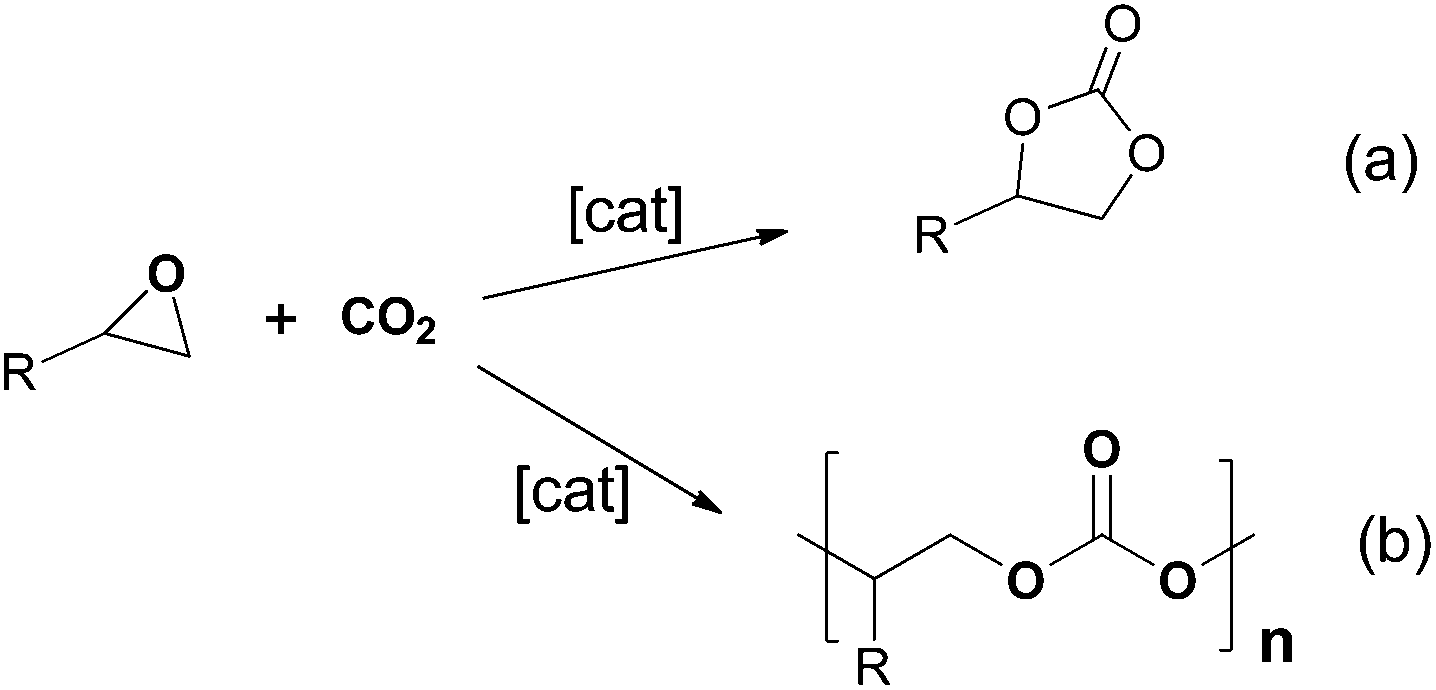 | ||
| Scheme 1 (a) Cycloaddition and (b) copolymerization of CO2 and epoxides. | ||
Catalysts for the coupling of CO2 and epoxides include halide, quaternary alkyl ammonium or phosphonium salts, as well as ionic liquids and metal complexes.11,12 Halide, quaternary salts and ionic liquids are known to produce the most stable thermodynamic product, the cyclic carbonate.11 This can be explained by the fact that in the absence of a Lewis acid the reaction requires higher temperatures.13 On the other hand, metal complexes can catalyse the formation of polymer and/or cyclic carbonate products depending on the co-catalyst, substrate and reaction conditions used. In this context, N-heterocyclic amines, phosphines or anions derived from PPN+ (PPN+ = [Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NPPh3]+) and ammonium salts,14 which act as nucleophiles, have been employed as co-catalysts.15 In most cases, such binary catalytic systems, Lewis acid/nucleophile, lead to an enhanced activity under milder reaction conditions.16–18
NPPh3]+) and ammonium salts,14 which act as nucleophiles, have been employed as co-catalysts.15 In most cases, such binary catalytic systems, Lewis acid/nucleophile, lead to an enhanced activity under milder reaction conditions.16–18
Recently, there has been an increased interest in developing new catalytic systems based on earth-abundant and widely distributed metals such as aluminium and iron. Early studies by Inoue and co-workers showed the possibility of CO2 activation using Al(III) porphyrin complexes in the presence of imidazole, quaternary ammonium or phosphonium salts.19 At room temperature and atmospheric pressure of CO2 tetraphenylporphyrinatoaluminium(III) methoxy derivative [(TPP)AlOMe] in the presence of 1-methylimidazole catalysed the formation of propylene carbonate from CO2 and propylene oxide. The formation of polycarbonates from CO2 and epoxides was also achieved using [(TPP)AlCl]/R4NBr or R4PBr under CO2 pressure.20
A recent outstanding example of highly active catalysts under mild reaction conditions is the hexachlorinated aluminum(III)–aminetriphenolate A (Chart 1) combined with tetrabutylammonium iodide. This catalytic system gave an initial TOF of 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 in the cycloaddition of CO2 to 1,2-epoxyhexane at 90 °C and 10 bar of initial CO2 pressure at very low catalyst loading. However, the molar ratio of catalyst:co-catalyst was high (100).21
000 h−1 in the cycloaddition of CO2 to 1,2-epoxyhexane at 90 °C and 10 bar of initial CO2 pressure at very low catalyst loading. However, the molar ratio of catalyst:co-catalyst was high (100).21
 | ||
| Chart 1 | ||
Some of the most widely employed catalysts in the literature for these reactions are metal–salen type complexes (salen = N,N′-ethylenebis(salicylimine)) mainly as a result of the pioneering work of Darensbourg with chromium(salen) based catalysts.22 These catalysts are easily prepared, with the possibility of performing large-scale synthesis. Moreover, they are highly stable and robust. Their reactivity properties can be easily fine-tuned by changing the diimine skeleton or the substituents in the phenolate moiety. Dimeric Al(III) salen complex [Al(salen)2(μ-O)] (B, Chart 1) reported by Meléndez et al.23 under solvent-free conditions and using tetrabutylammonium bromide (TBAB) as a co-catalyst displayed a unique activity under atmospheric pressure of CO2 and at 25 °C.24 Mononuclear analogous Al(III) salen based catalytic systems require higher pressure and temperature. Thus, Lu et al. have reported an Al(salen)Cl/TBAB catalytic system for the synthesis of ethylene carbonate, which resulted in a rapid formation of the carbonate under supercritical carbon dioxide conditions,25 however, the rate of conversion was reduced to half when the reaction was carried out at less than 40 bar pressure. Bifunctional aluminum(salen) complexes with appended pyridinium salt substituents,26 imidazolium-based ionic liquid moiety27 or N-methylhomopiperazine moieties28 in the substituents of the phenolate moieties were efficient catalysts for propylene carbonate formation (TOF up to 297 h−1). However, the introduction of a second functionality in the skeleton of the catalyst required additional synthesis steps.
Aluminum(salen) complexes together with a series of anionic and neutral co-catalysts were active for the copolymerization of CO2 and CHO. Darensbourg and co-workers showed that a more electron-withdrawing salen framework was necessary to produce significant quantities of copolymers with a high CO2 content in the absence of a cyclic carbonate by-product. Nevertheless, the TOFs obtained, ranged from 5.2 to 35.4 h−1, while chromium(salen) systems under similar conditions provided TOFs as high as 1150 h−1.29
To sum up, aluminum complexes are indeed efficient for the activation of CO2, however, they provide low activities and selectivity towards the desired product. Taking this into account we were interested in N,N′-bis(salicylene)-2-aminobenzylamine(salabza) derivatives as ligands for metal complexes. These complexes would have a more flexible structure than salen derivatives and an open active site, which may provide higher reactivity maintaining the high stability of the tetradentate coordination (Scheme 2). For this reason, we decided to prepare Al(III) complexes with the tetradentate-N2O2 ligand N,N′-bis(3,5-di-tert-butylsalicylene)-2-aminobenzyl-amine derivative (H2L, Scheme 2), which is easily synthesised in one step.30 Iron(III), chromium(III) and cobalt(III) complexes were also prepared. The activity of these metal-salabza complexes in the coupling of CO2 and epoxides is presented. The synthesis of other Cu, Ti, Mn, Co, Zn and Al complexes with similar salabza-type ligands have already been reported.31–34 A Co(III) complex with a 2,2-dimethylpropyldiamine related skeleton was reported to produce low conversion in the copolymerisation of CO2 with cyclohexene oxide.35
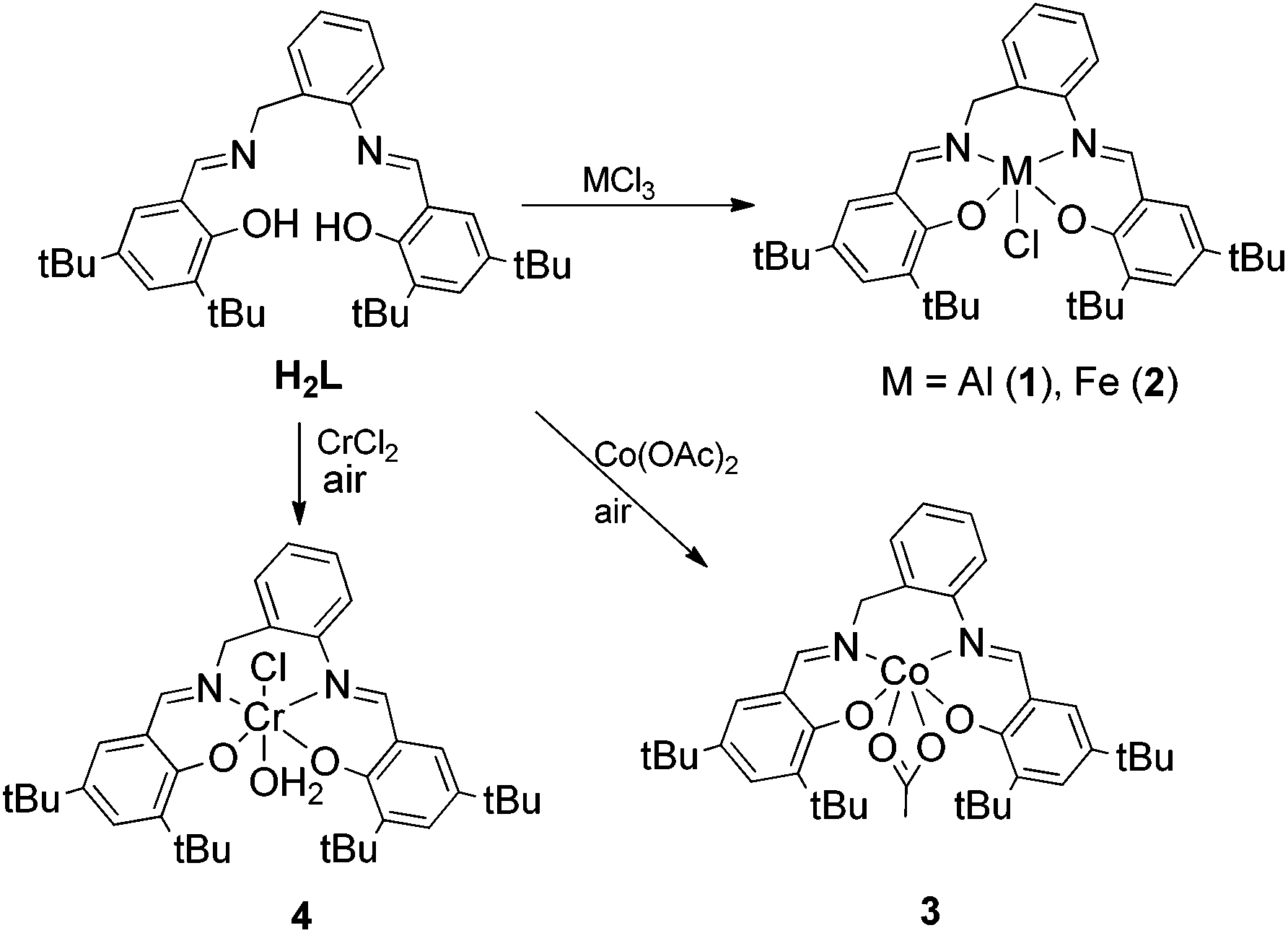 | ||
| Scheme 2 Syntheses of complexes 1–4. | ||
Results and discussion
Syntheses of complexes
Ligand H2L was prepared following the general procedure described by Lin and co-workers by the reaction of 3,5-di-tert-butyl salicylaldehyde and 2-aminobenzylamine in ethanol.30 Treatment of H2L with different metal halide precursors MCl3 (M = Al(III), Fe(III)) afforded the corresponding metal(III) complexes 1 and 2 in good to moderate yield (Scheme 2). Cr(III) and Co(III) complexes were prepared in moderate yields from M(II) salts (CrCl2 and Co(OAc)2·H2O) by addition of H2L and subsequent oxidation with air (Scheme 2). Compounds 1–4 were isolated as stable solids except in the case of 4, which decomposed under air at room temperature. They were characterized by mass spectrometry, elemental analysis, 1H and 13C NMR, IR, UV-visible spectroscopy, magnetic susceptibility (2–4) and X-ray diffraction analysis (2–4).According to mass spectra (1–4) and X-ray diffraction data (2–4), they all formed monometallic species and no evidence of bimetallic species was found. Probably, the tert-butyl substituents together with the more hindered six membered diamine ring prevented the formation of dinuclear species.
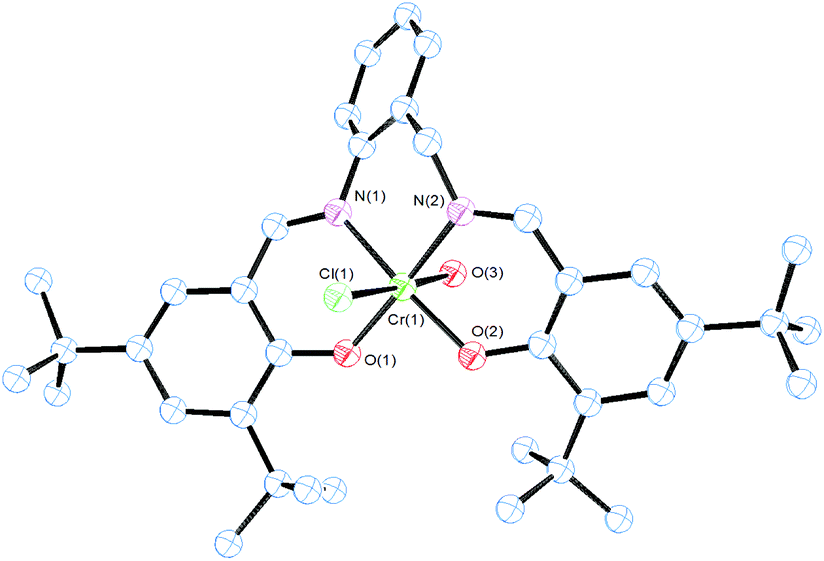
The molecular structures of 2–4 were obtained by single-crystal X-ray diffraction analysis (Fig. 1–3). Purple crystals of 2 were obtained by slow evaporation of a diluted solution of the complex in diethyl ether/hexane. 2 presented a 5-coordinated environment (Fig. 1) with the geometry around the iron center intermediate between trigonal bipyramidal and square-pyramidal.36 This geometry differed from those reported for [Fe(salenR)Cl]·C6H6,37 [Fe(salphen)Cl],38 and other iron salen-type complexes,39 which had a perfect square-pyramidal geometry. The length of the diimine ligand could be considered to influence the iron environment producing this change in the geometry. The Fe–N(imine) (2.139(2) and 2.0858(19) Å), Fe–O(phenolate) (1.8659(16) and 1.8995(16) Å) and Fe(Cl) (2.2466(7) Å) bond distances are in agreement with the reported data from other salen-type iron complexes.37,38,40 The Fe–N(imine) distance has been suggested as an indicator of the spin state for Fe(III) in salen iron complexes, with a distance of 2.00–2.10 Å for the high-spin state, and 1.93–1.96 Å for the low-spin state.40 The mean bond distance in the structure reported here suggests that the metal ion in 2 is in the high-spin state, which is consistent with the results of room temperature magnetic susceptibility (μeff = 5.90μB).40
 | ||
| Fig. 1 ORTEP drawing of complex 2. All hydrogen atoms and solvent molecules are omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°): Fe1–O1 1.8659(16), Fe1–O2 1.8995(16), Fe1–N1 2.139(2), Fe1–N2 2.0858(19), Fe1–Cl1 2.2466(7), O1–Fe1–O2 91.99(7), O1–Fe1–N2 126.35(8), O2–Fe1–N2 86.03(7), O1–Fe1–N1 85.73(7), O2–Fe1–N1 167.84(7), N2–Fe1–N1 85.72(7), O1–Fe1–Cl1 116.36(6), O2–Fe1–Cl1 100.42(6), N2–Fe1–Cl1 116.70(6), N1–Fe1–Cl1 91.27(6). | ||
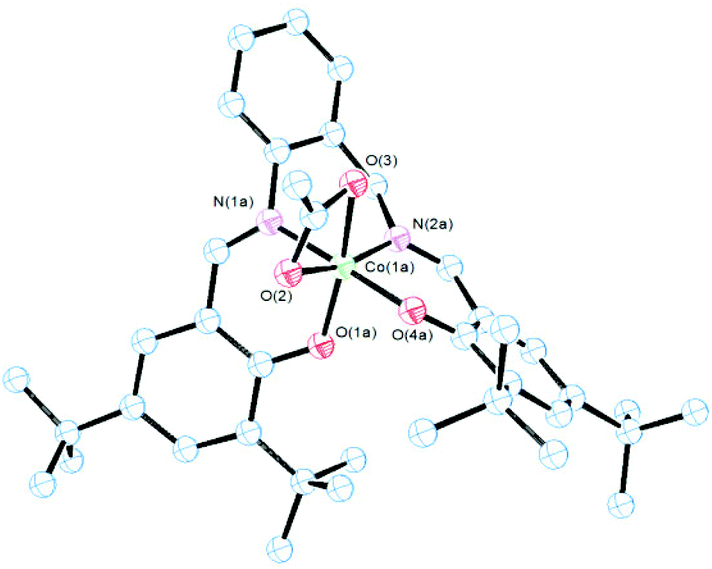 | ||
| Fig. 2 ORTEP drawing of complex 3. All hydrogen atoms and solvent molecules are omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°): Co(1a)–O(1a) 1.866(8), Co(1a)–O(4a) 1.867(6), Co(1a)–N(1a) 1.894(7), Co(1a)–N(2a) 1.898(6), Co(1a)–O(2) 1.975(5), Co(1a)–O(3) 2.053(5), O(1a)–Co(1a)–O(3) 166.5(4), O(4a)–Co(1a)–N(2a) 94.2(3), N(2a)–Co(1a)–N(1a) 90.8(5), O(1a)–Co(1a)–O(2) 101.7(3), O(2)–Co(1a)–O(4a) 85.9(3) and O(2)–Co(1a)–O(3) 64.8(18). | ||
 | ||
| Fig. 3 ORTEP drawing of complex 4. All hydrogen atoms and solvent molecules are omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°):Cr1–O1 1.916(5), Cr1–O2 1.923(5), Cr1–N2 2.012(6), Cr1–N1 2.059(6), Cr1–Cl1 2.299(3), Cr1–O3 2.052(5), O(1)–Cr(1)–O(2) 91.8(2)°, O(1)–Cr(1)–N(1) 88.3(2)°, O(2)–Cr(1)–N(2) 90.2(2)°, N(1)–Cr(1)–N(2) 89.8(3)°. | ||
Brown crystals of 3 were obtained by slow evaporation of a diluted solution of the complex in a mixture of diethyl ether/hexane. As shown in Fig. 2 the molecular structure of 3 was also monomeric with a six-coordinated central cobalt atom. In this case L2− acted also as a tetradentated anionic ligand and one acetate ion as a bidentate chelate. Evidence of the chelate acetate was also obtained from IR spectra41,42 by the presence of one strong band at 1525 cm−1 and another presumably in the overcrowded ν(CC) range of 1478–1409 cm−1 (νa(COO) and νs(COO) respectively) with a Δν value between 116–47 cm−1.42 The geometry around the cobalt atom (Fig. 2) was a distorted octahedral similar to analogous Co(III) salen complexes in the literature.35,43 Bond distances Co–N (imino), Co–O (phenolate) and Co–O (acetate) (Fig. 2) are in the average range as the corresponding values in similar octahedral Co(III) systems.43
Complex 4 adopted also an octahedral geometry around the chromium metal centre where the anionic ligand L2− was coordinated in the equatorial plane in a tetradentate fashion through the imine and phenolate O atoms (Fig. 3). One chloride anion and a water molecule, coming from wet air in the oxidation step, were located at the axial positions. The equatorial donor atoms presented a nearly planar geometry (Fig. 3). The chloride anion and the water molecule were located with an almost linear disposition (O(3)–Cr(1)–Cl(1) bond angle of 177.13(18)°). The Cr–N (imine), Cr–O (phenolate) as well as Cr–Cl bond lengths observed were in concordance with the chromium(III) salen-type complexes reported in the literature.44,45
Cycloaddition of CO2 to monosubstituted epoxides
Initially, complexes 1–4 were tested as catalysts, in conjunction with TBAB, for the coupling reaction of CO2 and 1,2-epoxyhexane as a benchmark substrate. The complexes were soluble in the net substrate; therefore no additional solvent was required. The initial reaction conditions were 10 bar CO2, 45 °C using 0.2 mol% complex and 0.2 mol% TBAB over 18 h. Catalysts 2–4/TBAB catalysed the coupling of CO2 and 1,2-epoxyhexane to produce the cyclic carbonate selectively (Table 1). In the case of catalyst 4/TBAB we obtained nonreproducible results probably due to decomposition. Under these conditions, the best result was achieved with the 1/TBAB catalytic system with almost 50% of conversion and total selectivity towards cyclic carbonate (entries 1–3, Table 1). The higher Lewis acidity of the Al(III) centre compared to Fe(III) and Co(III) may explain the higher conversion obtained with 1/TBAB although other factors related with the stability of the intermediates should also be taken into account. With this catalytic system, an increase of the catalyst/co-catalyst ratio to 1/5 (1.0 mol% co-catalyst loading) produced an enhancement in the catalytic activity up to 100% conversion (entry 4, Table 1). However, in the absence of a co-catalyst almost no cyclic carbonate was formed even when running the reaction at 80 °C (entry 5, Table 1). It is important to note that TBAB alone showed very low conversion under the employed catalytic conditions (entry 6, Table 1). This synergistic effect between the aluminium complex 1 and TBAB is in concordance with the analogous behaviour observed with other catalytic systems for the same reaction.21,46| Entry | Cat. | Co-cat. | T (°C) | Cat./TBABb (mol%) | Conv.c,d (%) | Ye (%) |
|---|---|---|---|---|---|---|
| a Reaction conditions: time = 18 h, PCO2 = 10 bar, 1,2-epoxyhexane: 33.15 mmol (4 mL), reactions were run in duplicate. b mol% with respect to the substrate. c Measured by 1H NMR. d Selectivity for the cyclic carbonate product > 99%. e Yield of carbonate product determined by 1H NMR using mesitylene as the internal standard. | ||||||
| 1 | 1 | TBAB | 45 | 0.2/0.2 | 49 | n.d. |
| 2 | 2 | TBAB | 45 | 0.2/0.2 | 17 | 16 |
| 3 | 3 | TBAB | 45 | 0.2/0.2 | 31 | 31 |
| 4 | 1 | TBAB | 45 | 0.2/1.0 | 100 | 73 |
| 5 | 1 | — | 80 | 0.2/— | 1 | 1 |
| 6 | — | TBAB | 80 | —/0.2 | 11 | 8 |
The effect of pressure (10, 30 and 50 bar) and temperature (45, 60, 80 °C) was also evaluated at 18 h of reaction time with 1/TBAB (catalyst–co-catalyst = 1) (Fig. 4). As expected the temperature has a beneficial effect in the conversion even when the CO2 pressure increases up to 50 bar. On the other hand, raising the pressure produced an increase of conversion when working at 45 and 60 °C. When the reaction was run at 80 °C, no positive effect was observed by increasing the pressure from 10 to 50 bar.
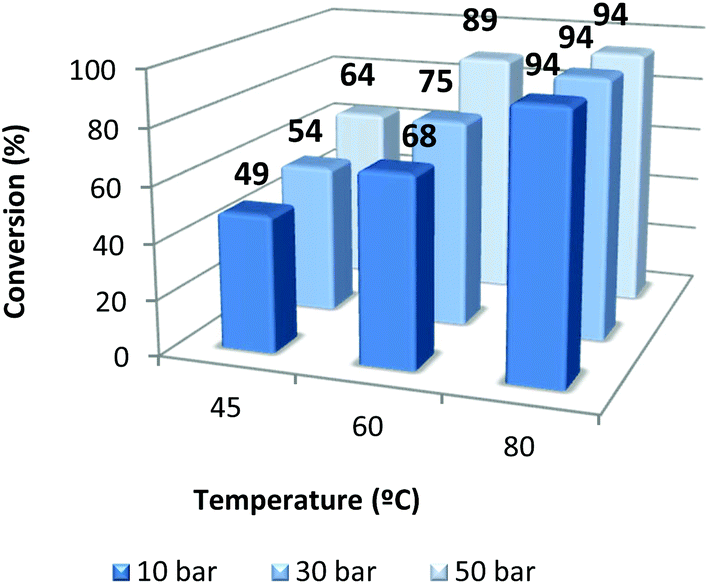 | ||
| Fig. 4 Effect of CO2 pressure and temperature on the coupling of 1,2-epoxyhexane and CO2 using the 1/TBAB catalyst system. Reaction conditions: catalyst 0.2 mol%, TBAB 0.2 mol%, 18 h. Reactions were run in duplicate. | ||
At the optimized temperature (80 °C), pressure (10 bar) and catalyst/co-catalyst ratio of 5 we proceeded to optimize the catalyst loading and reaction time to obtain the initial maximum TOF at low conversion. With reduced amounts of catalysts and co-catalysts and also decreasing the reaction time to 1 h it was found that significant conversion could still be achieved (up to 53%, initial TOF up to 531 h−1, entry 1, Table 2).
a
| Entry | 1/TBABb (mol%) | T (°C) | P (bar) | t (h) | Conv.c,d (%) | TOFe (h−1) | Yf (%) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: 1,2-epoxyhexane: 24.86 mmol (3 ml), reactions were run in duplicate. b mol% with respect to the substrate. c Measured by 1H NMR. d Selectivity for the cyclic carbonate product >99%. e Averaged TOF (mol substrate converted) (mol catalyst)−1 h−1. f Yield of carbonate product determined by 1H NMR using mesitylene as the internal standard. | |||||||
| 1 | 0.1/0.5 | 80 | 10 | 1 | 53 | 531 | 50 |
| 2 | 0.1/0.5 | 45 | 10 | 1 | 9 | 90 | 9 |
| 3 | 0.1/0.5 | 80 | 1 | 1 | 27 | 267 | 21 |
| 4 | 0.05/0.25 | 80 | 10 | 0.5 | 20 | 800 | 19 |
To assess if the catalytic activity was maintained under milder reaction conditions the same reaction was performed at 45 °C (entry 2, Table 2) and under an atmospheric pressure of CO2 (entry 3, Table 2). In both cases a decrease in the catalytic activity was observed (TOF 90 and 267 h−1 respectively), but the initial TOF at atmospheric pressure was still encouraging. A maximum TOF of 800 h−1 was obtained at a catalyst loading of 0.05 mol% in 0.5 h of reaction time (entry 4, Table 2). This value is slightly lower than the one reported using the hexachloro Al(III)(amine triphenolate)/TBAI catalytic system at a similar catalyst/cocatalyst ratio (0.05/0.25 mol%) at 90 °C and 10 bar CO2 initial pressure (TOF 900 h−1).21 However, it is higher than the one obtained with the dimeric aluminium(salen) ([Al(salen)2(μ-O)]/TBAI, (B, Chart 1) under the same conditions (0.05/0.25 mol%) at 90 °C and 10 bar CO2 initial pressure).21 Compared to these two catalytic systems, the advantages of 1/TBAB are that it does not require the introduction of chlorinated groups in the complex skeleton and can be used at a lower co-catalyst/catalyst ratio. Nevertheless, hexachloro Al(III) (amine triphenolate)/TBAI can be used at a much lower complex concentration21 and [Al(salen)2(μ-O)]/TBAI is active at 1 bar CO2 and room temperature.23
The catalytic activity of 1/TBAB was studied in the cycloaddition of CO2 to a variety of terminal and functionalized epoxides providing the corresponding cyclic carbonates (Fig. 5). All of these selected substrates were converted into the corresponding carbonates with total selectivity in the cyclic product at a turn over frequency between 120 and 3434 h−1 (Fig. 5). This information indicated the high tolerance of the catalytic system especially with alkyl halide functionalities such as 1-chloro-2,3-epoxypropane, which presented the highest catalytic activity. It is remarkable that the catalyst system 1/TBAB was also active for internal hindered substrates, such as methyl epoxyoleate derived from a natural product, although higher concentration of the catalyst was required (Fig. 5). Leitner and co-workers found that TBAB alone (2 mol%) gave very low conversion of 11% with a cis/trans ratio of 71/29 at 100 °C, 125 bar over 6 h.47
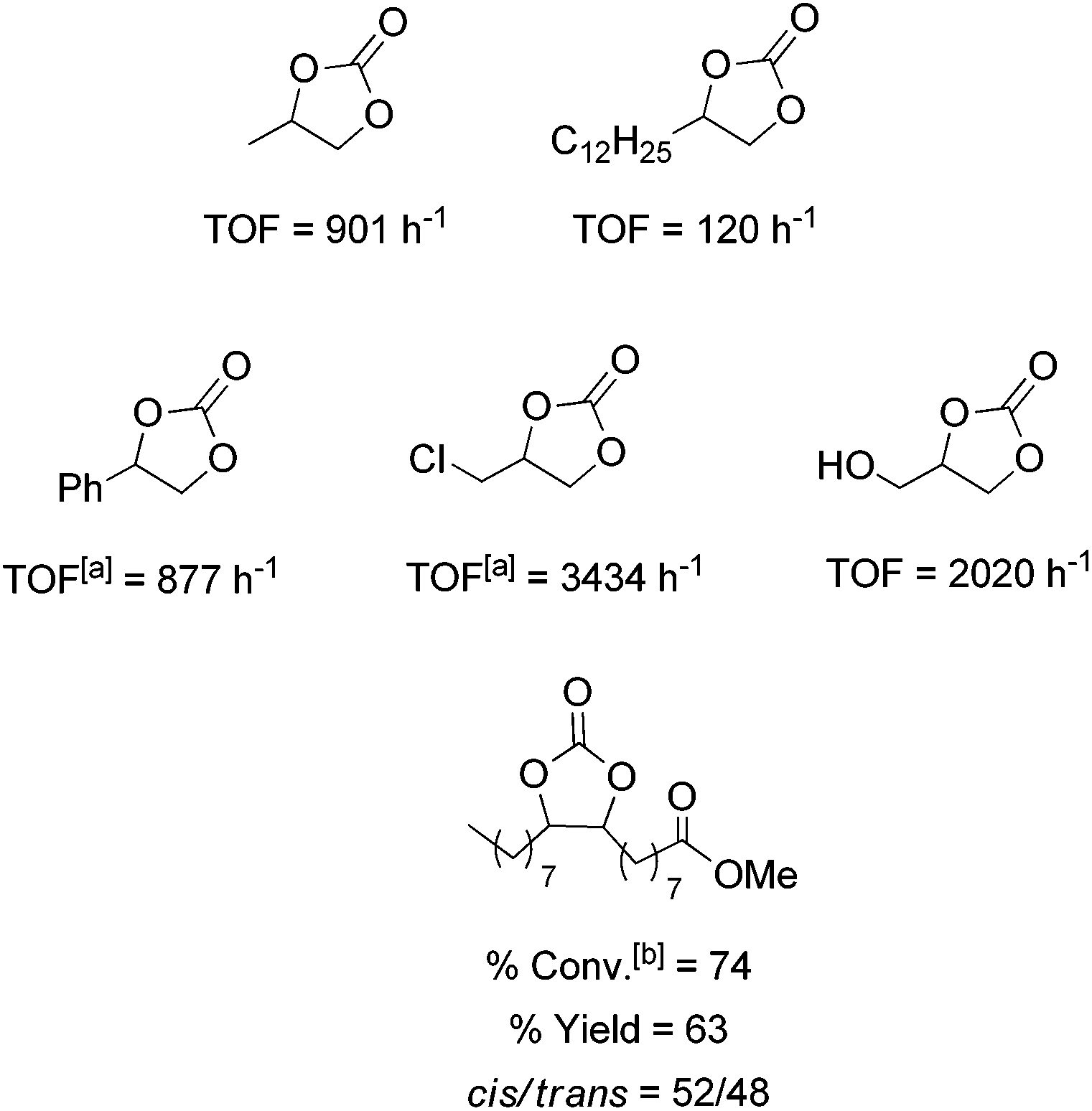 | ||
| Fig. 5 Cycloaddition of different epoxides to CO2 with the catalytic system 1/TBAB. Reaction conditions: T = 80 °C, time = 0.5 h, PCO2 = 10 bar, substrate: 3 ml; 1: 0.05 mol%, TBAB: 0.25 mol%. a1: 0.03 mol%; TBAB: 0.15 mol; b1: 2 mol%, TBAB: 2 mol%; T = 100 °C; PCO2 = 100 bar. Reactions were run in duplicate. | ||
Polymerization of CO2 with cyclohexene oxide
The reaction of CO2 and cyclohexene oxide (CHO) was then investigated using catalyst 1 and changing the co-catalyst to a most effective Lewis base co-catalyst for this reaction such as (bis(triphenylphosphine)iminium chloride (PPNCl)).29 The copolymerization reaction was carried out initially at 80 °C and 50 bar over 18 h with 1 complex (0.2 mol%) in the presence of an equimolar amount of PPNCl without the addition of solvent (entry 1, Table 3). The 1H NMR of the crude of the reaction showed a peak at δ 4.6 ppm characteristic of poly(cyclohexene carbonate) (PCHC)22 together with signals at δ 4.5 and δ 4.0 ppm attributed to the cis- and trans-cyclohexene carbonate (CHC)48,49 respectively (ratio cis/trans 75/25, ratio PCHC/CHC 71/29). The formation of mixtures of polycarbonate and cyclic carbonate in the reaction of CHO and CO2 is consistent with the fact that the activation energy for the formation of PCHC is lower compared to other substrates; nevertheless, the cyclic product is still the most stable thermodynamically.50 Indeed, Darensbourg and co-workers observed almost total selectivity towards the cyclic carbonate by-product using PPNCl with similar aluminum–salen complexes.29 The polymer was isolated by extraction of the cyclic product with hexane obtaining a polycarbonate with a high degree of incorporation of CO2 (92%) measured by 1H NMR spectroscopy. The number-average molecular weight (Mn) of the alternating copolymer and polydispersity (Mw/Mn), estimated by gel permeation chromatography (GPC), was 1700 and 1.3, respectively (entry 1, Table 3). Polycarbonate oligomers with diol functionalities are interesting materials for the synthesis of polyurethanes.51| Entry | T (°C) | P (bar) | Time (h) | Conv.b (%) | Yc (%), (Mw·103, Mw/Mn)d | % CO2 contentb | PCHC/CHCe (%) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: cyclohexene oxide: 29.70 mmol (3 ml); 1: 0.2 mol% with respect to the substrate: PPNCl: 0.2 mol% with respect to the substrate, reactions were run in duplicate. b Measured by 1H NMR. c Yield of PCHC determined by 1H NMR using mesitylene as the internal standard. d Determined by GPC using polystyrene as the standard. e Selectivity determined by 1H NMR. | |||||||
| 1 | 80 | 50 | 18 | 66 | 41 (1700, 1.3) | 92 | 71/29 |
| 2 | 45 | 50 | 24 | 63 | 50 (2100, 1.2) | 96 | 82/18 |
| 3 | 25 | 50 | 90 | 58 | 45 (2900, 1.3) | 92 | 84/16 |
| 4 | 45 | 10 | 24 | 63 | 46 (2100, 1.3) | 85 | 79/21 |
The selectivity towards the polycarbonate increased up to 84% by decreasing the temperature reaction to 25 °C at longer reaction time (entries 2 and 3, Table 3). At this temperature the Mn of the poly(cyclohexene carbonate) increased up to 2900. Similarly, when the CO2 pressure decreased to 10 bar at 45 °C the reaction proceeded at 63% conversion although the % CO2 incorporation decreased to 85% and the cyclic carbonate increased due to decrease of the CO2 insertion29 (entry 4, Table 3). The conversions obtained with 1/PPNCl were higher than the ones reported for Al(III) Schiff base ligands with a similar propyl diamine skeleton when NEt4OAc was used as a cocatalyst.52 Instead, the polymers obtained by the 1/PPNCl catalytic system possessed a lower molecular weight than the one reported by Inoue and coworkers with salophen aluminum Schiff base complexes. Still, the polydispersity obtained was higher (2.47, 20 bar, 80 °C) compared with the 1/PPNCl catalytic system (1.3, 10 bar, 45 °C). It is also remarkable for the use of solvent-free conditions.
Analyses of the polycarbonates by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF) revealed two distributions in the polycarbonates formed (Fig. 6). The major distribution was attributed to a chain with the presence of –Cl as well as –OH as end groups (a + K in Fig. 6) and the second distribution fitted with chains with two –OH terminal groups (b + K in Fig. 6). This observation suggests that chain a (Fig. 6) was formed by an initiation step involving the opening of the epoxide by a nucleophilic attack of the chloride anion, which, presumably, comes from the PPNCl co-catalyst, to the coordinated epoxide. On the other hand, chain b (Fig. 6), which contains two terminal –OH, suggests that the initiation step involves the opening of the epoxide by a nucleophilic attack with –OH originating from traces of water present in the reactor. The termination step for both polymer chains may proceed by hydrolysis.
 | ||
| Fig. 6 MALDI-TOF mass spectrum of polycarbonate from entry 4, Table 3. | ||
Mechanistic studies for the cycloaddition of CO2 to styrene oxide
In order to elucidate the mechanism of the cycloaddition of CO2 to styrene oxide 27Al NMR spectra and kinetic studies were undertaken. 27Al NMR spectrum in CDCl3 of 1 exhibited a single broad and strong resonance centred at δ 50 ppm with a width of 65–35 ppm (a, Fig. 7), which probably collapses with the probe background53 and can be attributed to a pentacoordinated species.54–56 When an equimolar amount of styrene oxide was added into the NMR tube containing 1 two defined signals appeared at δ 40.01 ppm and δ 3.70 ppm (b, Fig. 7). According to the literature data we assigned the downfield broad signal to the pentacoordinate complex 1, whereas the up field signal could be ascribed to a hexacoordinated complex: this compound was formed by the coordination of styrene oxide to the aluminium metal center.57–59 At this point it was proposed that in an initial step an equilibrium between the pentacoordinated 1 and hexacoordinated 1-SO takes place (Scheme 3). To continue with the investigation of the role of the aluminium complex, the co-catalyst and the CO2 pressure in the reaction mechanism, a kinetic study of the formation of cyclic styrene carbonate catalyzed by the 1/TBAB system was undertaken. Solvent-free conditions were chosen using a neat substrate to work in a similar environment to that in the catalytic studies. The reaction kinetics was monitored by sampling and subsequent analyses by 1H NMR spectroscopy to determine the conversion of epoxide to cyclic carbonate. The approximations taken into consideration were those reported in the literature for similar studies.60–63 It was assumed that the concentration of 1 and TBAB does not change during the reaction since they both act as catalysts, and CO2 is present in large excess due to the semi-batch operation. Thus, the general rate law (eqn (1)) can be transformed in eqn (2).| rate = k[SO]a[CO2]b[1]c[TBAB]d | (1) |
| rate = kobs[SO]a | (2) |
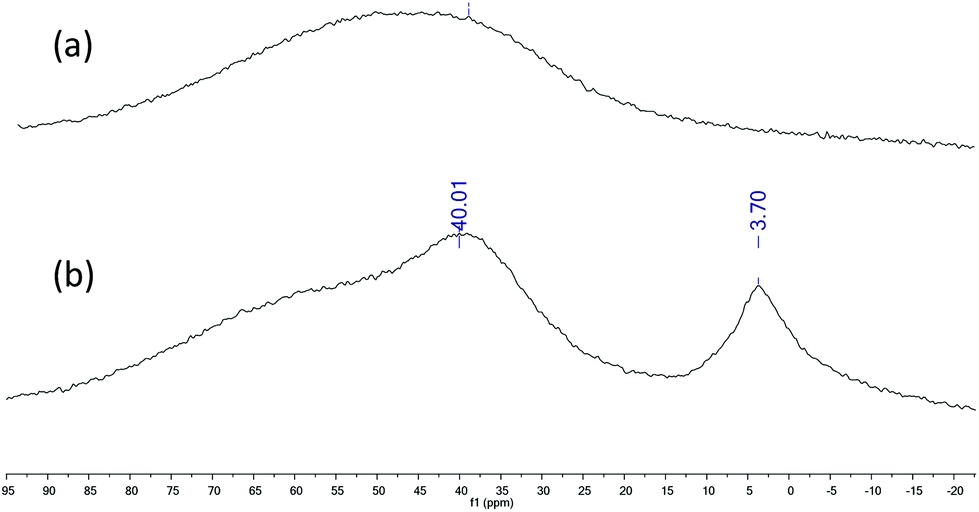 | ||
| Fig. 7
27Al NMR spectra of complex 1 (a) and a mixture 1/styrene oxide (1:1) (b) in CDCl3. | ||
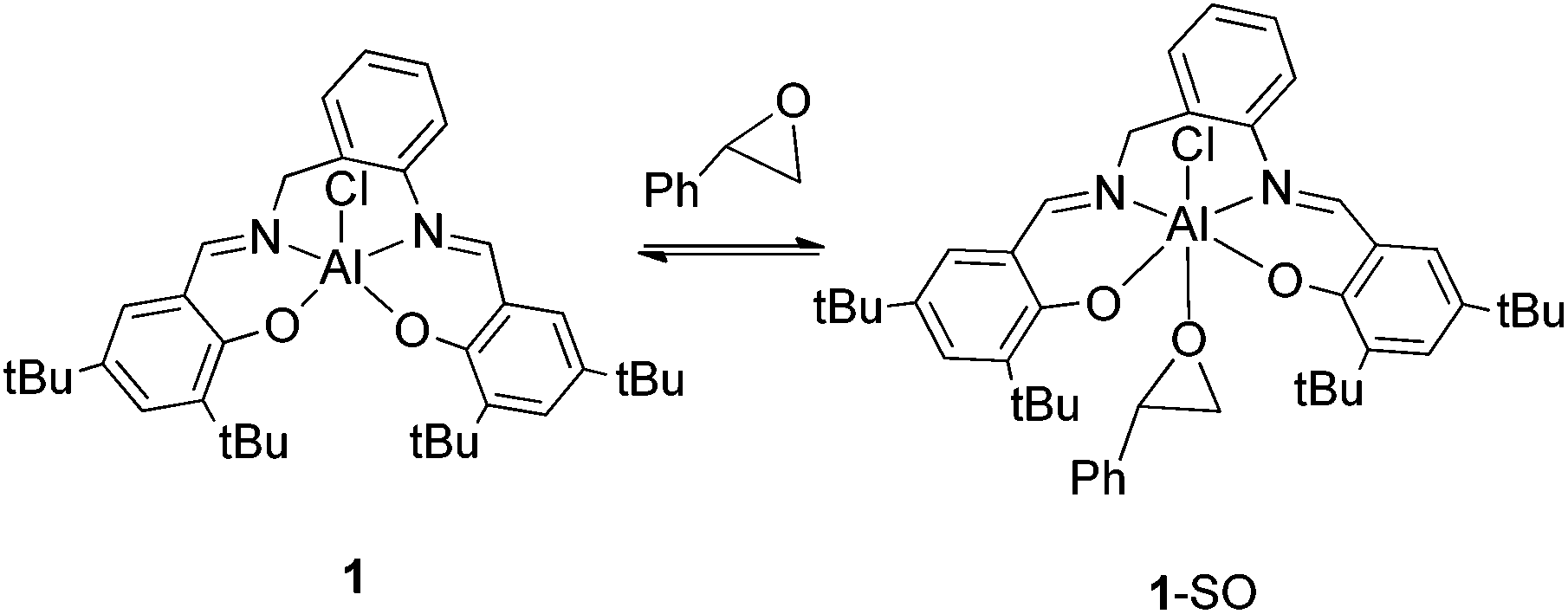 | ||
| Scheme 3 Equilibrium between five and six-coordinated aluminium species. | ||
The representation of styrene oxide (SO) in front of time suggested a pseudo-first order dependence with respect to the substrate since there is a linear dependence of ln[SO] as a function of time (a = 1, eqn (2)) (Fig. S28†). Contrary to the observations by North and co-workers using catalyst B/TBAI (Chart 1) in the synthesis of styrene carbonate, no induction period was detected using the catalytic system 1/TBAB.64 The same authors showed that the reaction under solvent-free conditions was pseudo-zero order whereas in solvent it was first order with respect to the starting material.65
To determine the order with respect to the catalyst 1 and the co-catalyst TBAB, the reactions were performed maintaining constant the reaction temperature and working in the presence of an excess of CO2. Under these conditions its concentration may be considered pseudo constant at the initial stage of the reaction. Thus, the natural logarithm of the rate law (kobs = k[CO2]b[1]c[TBAB]d) results in lnkobs = lnk + b ln[CO2] + cln[1] + dln[TBAB], from which it is possible to afford the orders b–d with respect to the catalyst, co-catalyst and CO2 concentration by examination of the double logarithmic plot for each case. Thus, initially, the pressure of CO2 and the amount of TBAB was fixed at 0.2 mol% while the concentration of 1 was varied between 0.1 and 0.4 mol%. Similarly, the pressure of CO2 and the concentration of 1 were maintained at 0.2 mol% and TBAB concentration was changed from 0.2–1.0 mol%. The double logarithmic plot of the initial rates against the catalyst or the co-catalyst concentration (see Fig. S29–S34 in ESI†) shows a linear dependence with a slope of 0.6618 and 1.1127, respectively, suggesting that the reaction was first order in the concentration of catalyst 1 and TBAB. This pointed to a mechanism in which only one molecule of monometallic Al complex and also one molecule of TBAB were involved, before or during the rate-determining step of the catalytic cycle. Comparing with the previously reported kinetic analysis of cyclic carbonate synthesis, similar results were reported by Castro-Osma et al. and co-workers, with a bimetallic aluminium complex,65 and by Kleij and co-workers with a binary zinc(salen)-based complex.63 It is then assumed that the role of the ammonium halide is to ring-open the coordinated epoxide to form a halo-alkoxide species. However a second order with respect to the concentration of TBAB was found for the dinuclear catalytic system B/TBAI (Chart 1).64 In this case it was attributed to the formation of NBu3, which reacted to CO2 in a bimetallic mechanism.
The role of the CO2 pressure in the catalytic cycle was analysed at four different CO2 pressures between 10 and 40 bar, maintaining 1/TBAB concentration and temperature constant (80 °C). A first-order dependence was observed at low CO2 pressures, between 10 and 30 bar, (slope of 0.6049 including points between 10 and 30 bar, Fig. 8) suggesting that one CO2 molecule is involved in the catalytic cycle as reported by North.64 However, at a higher pressure (40 bar) a decrease of the rate constant was observed, which may be related with the low solubility of the catalytic system in a more dense solvent.47
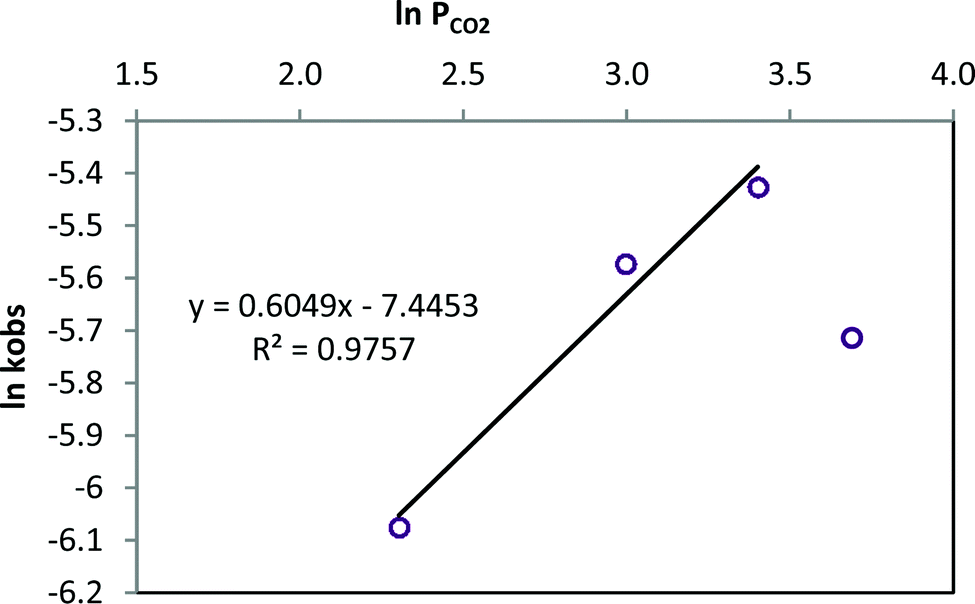 | ||
| Fig. 8 Styrene carbonate synthesis at four different CO2 pressures (10–40 bar). Reaction conditions: T = 80 °C, 1 0.2 mol%, TBAB 0.2 mol%. | ||
The activation energy (Ea) of the formation of styrene carbonate using the catalytic system 1/TBAB was calculated using the Arrhenius equation from the relationship between the observed rate constant (kobs) and the reaction temperature (Fig. 8 and S39†). The temperature range analysed was 40–100 °C at 10 bar pressure CO2. The activation energy was calculated to be 38.0 kJ mol−1. Styring and co-workers obtained a lower energetic activation barrier of 23 kJ mol−1 using [Al(salacen)]/TBAB and a similar value using the [Al(salacen)] catalyst alone when they studied the synthesis of styrene carbonate.61 Higher values (up to 78 kJ mol−1) were reported using Mg–Al mixed metal oxides.66
According to 27Al NMR experiments and the kinetic studies we propose a catalytic cycle (Scheme 4), which initiates with the activation of the epoxide by coordination to the metal center forming a hexacoordinated aluminum complex. The second step is the formation of a reactive aluminum-alkoxide species through a nucleophilic attack of the bromide anion from TBAB to the epoxide. This metal–alkoxide bond is known to react easily with CO2 forming a carbonate Al(III) species. This intermediate can either form a polycarbonate through further alternating insertions of epoxide and CO2 or a cyclic carbonate monomer via intramolecular rearrangement and leaving group liberation.
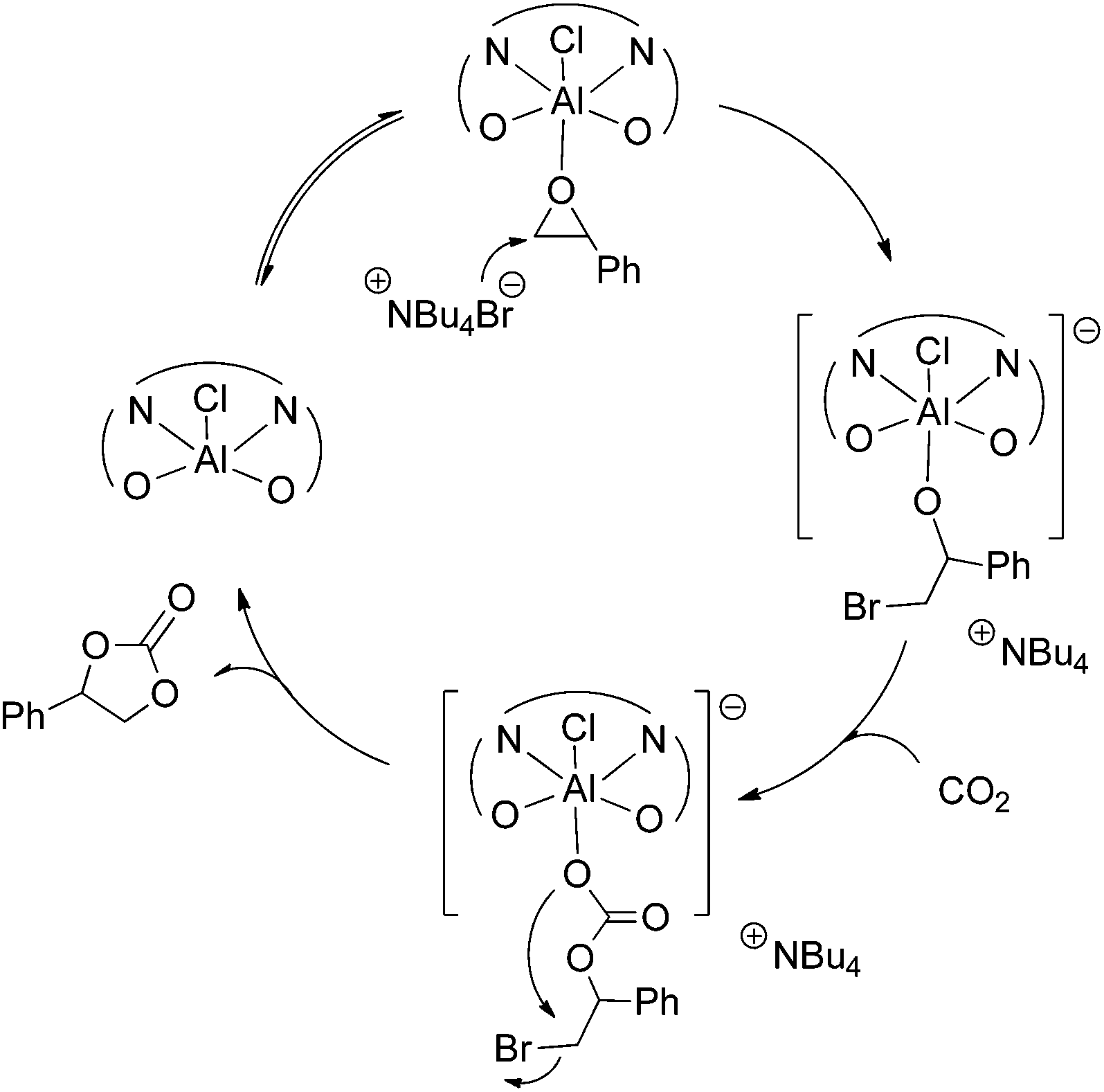 | ||
| Scheme 4 Proposed reaction mechanism for the cycloaddition of epoxides into CO2 with the catalyst system 1/TBAB. | ||
Conclusion
In this work we reported new catalytic systems based on tetradentate N2O2 salabza metal catalysts. In particular, aluminium complex 1, was found to be very stable and easy to synthesize from a simple aluminium trichloride salt. This mononuclear aluminium complex, combined with TBAB, formed an active binary catalytic system for cycloaddition of CO2 and epoxides. This catalytic system provides cyclic carbonates selectively with excellent conversions even at low pressures of CO2 (up to 94% at 10 bar). Higher catalytic activities were obtained with functionalized terminal epoxides such 1-chloro-2,3-epoxypropane with a maximum TOF of 3434 h−1. However, a more sterically hindered substrate such as methyl epoxyoleate was also transformed selectively in the cyclic carbonate product although under harsher reaction conditions. Also a detailed kinetic analysis of styrene carbonate synthesis catalysed by the 1/TBAB system was carried out. As a result of the observed first order dependence of the reaction rate on the catalyst, co-catalyst and CO2 concentration, we propose a catalytic cycle, which explains the role of each component. Furthermore, using the catalytic system 1/PPNCl in the reaction of cyclohexene oxide and CO2 produces poly(cyclohexene carbonate) as a main product. The polymer formed contains high chain incorporation of CO2 and a molecular weight up to 2900 g mol−1 and low polydispersity (1.3). MALDI-TOF analysis of the polycarbonates obtained indicated that the initiating step involved the opening of the epoxide by the Cl– anion and OH– (from water traces).Experimental
Synthesis of [N,N′-bis(3,5-di-tert-butylsalicylene)-2-aminobenzyl-amino]chloridoaluminium(III) (1)
Anhydrous AlCl3 (144.1 mg, 1.08 mmol) was added to a solution of H2L (400 mg, 0.72 mmol) in 15 mL of dry THF. The yellow solution was stirred for 4 h at room temperature under an inert atmosphere, filtered over Celite and the filtrate was evaporated under vacuum. The resulting solid was washed with acetonitrile, pentane and dried again. Bright yellow solid, 377.4 mg, (yield 85%). 1H NMR (400 MHz, CDCl3, ppm): δ 1.28 (s, 9H, tBu), 1.33 (s, 9H, tBu), 1.52 (s, 9H, tBu), 1.57 (s, 9H, tBu), 4.80 (br, 2H, ArCH2N), 7.04 (d, 1H, CH-phenol, J = 2.4 Hz), 7.20 (d, 1H, CH-phenol, J = 2.4 Hz), 7.22–7.27 (m, 3H, ArH), 7.38 (m, 1H, ArH), 7.54 (d, 1H, CH-phenol, J = 2.4 Hz), 7.66 (d, 1H, CH-phenol, J = 2.4 Hz), 8.35 (s, 1H, CHN), 8.42 (s, 1H, CHN); 13C NMR (75.43 MHz, CDCl3, ppm): δ 29.78, 29.83 (CH3, tBu), 31.37, 31.40 (CH3, tBu), 34.09, 34.20 (C, tBu), 35.56, 35.67 (C, tBu), 62.63 (CH2), 118.08, 119.25, 123.15, 126.78, 127.41, 127.57, 128.51, 130.08, 131.57, 131.68, 132.95, 138.96, 139.55, 140.94, 141.44, 148.28, 162.84, 164.02, 171.63, 171.87. UV-vis (CH3CN, 2.5 × 10−5 M): λ (nm) (ε, L mol−1 cm−1): 226.0 (116100), 282.0 (41884), 369.0 (18752). Anal. calcd for C37H48AlClN2O2·2H2O: C, 68.24; H, 8.05; N, 4.30. Found: C, 68.11; H, 8.21; N, 4.03. HRMS (ESI, m/z) calculated for [M − Cl]+: 579.3531, found 579.3503.
Synthesis of [N,N′-bis(3,5-di-tert-butylsalicylene)-2-aminobenzyl-amino]chloridoiron(III) (2)
A 10 mL MeOH solution of FeCl3 (90.5 mg, 0.56 mmol) was added dropwise to a 10 mL CH3CN suspension containing H2L (309.4 mg, 0.56 mmol) and Et3N (0.15 ml, 1.12 mmol). The resulting solution changed color to dark purple and was gently refluxed for 2 h. Then the solution was filtered while warm and concentrated to one-third of the original volume. The filtrate was dissolved with CH2Cl2, filtered again over Celite and the volatiles removed. The solid was further washed with hexane and dried under vacuum. Black solid, 302 mg, (yield 85%). UV-vis (CH3CN, 2.5 × 10−5 M): λ (nm) (ε, L mol−1 cm−1): 220.0 (86576), 240.0 (31304), 276.0 (23496), 332.0 (11528), 532.0 (3948). μeff (25 °C) = 5.90μB. Anal. calcd for C37H48ClFeN2O2: C, 69.00; H, 7.51; N 4.35. Found: C, 69.29; H, 7.99; N, 4.40. HRMS (ESI, THF, m/z) calculated for [M − Cl]+: 608.3065, found: 608.3038.
Synthesis of (acetato-κ2O,O)[N,N′-bis(3,5-di-tert-butylsalicylene)-2-aminobenzyl-amino]chloridocobalt(III) (3)
To a stirred solution of H2L (300 mg, 0.54 mmol) in THF (10 mL) at room temperature under an inert atmosphere, an ethanol solution (10 mL) containing 1.0 equiv. of Co(OAc)2·2H2O (134.7 mg, 0.54 mmol) was added. The reaction mixture was refluxed for 1 h under an inert atmosphere, cooled down to room temperature and was further stirred under an air stream for 6 h. The resultant solution was concentrated and hexane was added to precipitate the product, which was filtered off, washed with diethyl ether and hexane, and dried under vacuum. Dark red solid, 180.3 mg, (yield: 49%). 1H NMR (400 MHz, CDCl3, ppm): δ 1.16 (br, 9H, tBu), 1.25 (br, 9H, tBu), 1.30 (br, 9H, tBu), 1.45 (br, 9H, tBu), 1.59 (br, 3H, CH3–OAc), 4.17 (d, 1H, ArCH2N, J = 13.6 Hz), 4.54 (d, 1H, ArCH2N, J = 12.4 Hz), 6.99 (br, 1H, ArH), 7.08 (br, 1H, ArH), 7.16 (br, 1H, ArH), 7.34–7.36 (br, 2H, ArH), 7.44 (br, 1H, ArH), 7.75 (br, 1H, CHN), 7.85 (br, 1H, CHN). UV-vis (CH3CN, 2.5 × 10−5 M): λ (nm) (ε, L mol−1 cm−1): 217.0 (78980), 230.0 (35624), 258.0 (36144), 416.0 (6088). μeff (25 °C) = 0.05μB. Anal. calcd for C37H48CoN2O2·H2O·CH3CH2OH: C, 68.36; H, 8.02; N 3.99. Found: C, 68.42; H, 7.83; N, 3.79. HRMS (ESI, THF, m/z) calculated for [M − OAc]+: 611.3048, found: 611.3060.
Synthesis of aqua[N,N′-bis(3,5-di-tert-butylsalicylene)-2-aminobenzyl-amino]chloridochromium(III) (4)
To a stirred solution of H2L (300.0 mg, 0.5407 mmol) in THF (15 mL) anhydrous CrCl2 (66.5 mg, 0.5407 mmol) was added. The resulting mixture was stirred under nitrogen at room temperature for 3 h. Then, it was further stirred under air for 3 h. The solution was filtered over Celite and the filtrate was evaporated under vacuum. Cold hexane was added to the brown mixture. The suspension was filtered off and the solid was washed with hexane and dried under vacuum. Brown solid, 193.7 mg, (yield 55%). Anal. calcd for C37H50ClCrN2O3·H2O·OC4H8: C, 65.80; H, 8.08; N 3.74. Found: C, 65.99; H, 8.02; N, 3.86. HRMS (ESI, THF, m/z) calculated for [M − Cl − H2O]+: 604.3121, found: 604.3206.X-ray crystallography
Diffraction data for the structures reported were collected on a Smart CCD 1000 Bruker diffractometer system with Mo Kα radiation (λ = 0.71073 Å). Cell refinement, indexing and scaling of the data sets were carried out using programs Bruker Smart and Bruker Saint. All the structures were solved by SIR9767 and refined by SHELXL968 and the molecular graphics with ORTEP-3 for Windows.69 All the calculations were performed using the WinGX publication routines.70 Crystallographic data are collected in Table S1 (ESI†).Standard procedure for the synthesis of cyclic carbonates
The catalytic tests were carried out in a 100 mL Berghof reactor, which was previously kept for 4 hours under vacuum at 100 °C. After cooling, a solution under an inert atmosphere containing the catalyst dissolved in a neat distilled substrate and the co-catalyst, when indicated, was injected into the reactor. The autoclave was pressurized with CO2, and then heated to the specific temperature to reach the desired pressure. After the reaction time, the reactor was cooled with an ice bath and slowly depressurized (with PO a dichloromethane trap was used). The conversion was determined by 1H NMR of the crude mixture by the integral ratio between the alkene oxide and cyclic carbonate. The yield was determined by 1H NMR using mesitylene as the internal standard.Standard procedure for copolymerization with cyclohexene oxide
Using the same procedure for the synthesis of cyclic carbonates the conversion was also determined by 1H NMR of the crude mixture by the integral ratio between the alkene oxide with the copolymer and cyclic carbonate. The yield was determined by 1H NMR using mesitylene as the internal standard. The final mixture was dissolved in dichloromethane, the solvent was evaporated and the residue dried under vacuum at 100 °C for 3 hours to remove excess of cyclohexene oxide. The final residue was washed several times with hexane to purify the poly(carbonate) and was analysed by 1H NMR spectroscopy. The CO2 content was calculated from 1H NMR data by the integral ratio between copolymer carbonate linkages (δ = 4.65 ppm) with respect to ether linkage signals (δ = 3.45 ppm).Standard kinetic experiment procedure
A Parr 477 autoclave equipped with a proportional-integral-derivative (PID) temperature controller and gas reservoir was used for kinetic experiments in the reaction of styrene oxide with CO2. In a typical experiment, the autoclave was charged with the catalyst and co-catalyst in neat distilled styrene oxide, heated and pressurized with CO2. Samples were taken at the determined time and the conversion of the product was determined by 1H NMR spectroscopy. After the reaction time the autoclave was depressurized.Acknowledgements
We gratefully acknowledge the Ministerio de Economia y Competitividad (CTQ2013-43438-R) and the Departament d'Economia i Coneixement (Generalitat de Catalunya, 2014SGR670) for financial support.Notes and references
- Y. Li, K. Junge and M. Beller, ChemCatChem, 2013, 5, 1072 CrossRef CAS.
- Z.-M. Cui, Z. Chen, C.-Y. Cao, W.-G. Song and L. Jiang, Chem. Commun., 2013, 49, 6093 RSC.
- M. Fleischer, H. Blattmann and R. Mülhaupt, Green Chem., 2013, 15, 934 RSC.
- B. Nohra, L. Candy, J.-F. Blanco, C. Guerin, Y. Raoul and Z. Mouloungui, Macromolecules, 2013, 46, 3771 CrossRef CAS.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243 CAS.
- C. Beattie, M. North and P. Villuendas, Molecules, 2011, 16, 3420 CrossRef CAS PubMed.
- B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554 CrossRef PubMed.
- A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951 CrossRef CAS PubMed.
- G. W. Coates and R. C. Jeske, Handbook of Green Chemistry-Green Catalysis Vol. 1: Homogeneous Catalysis, ed. P. T. Anastas and R. H. Crabtree, WILEY-VCH, Weinheim, Deutschland, 2009, p. 343 Search PubMed.
- M. Aresta, Carbon Dioxide as Chemical Feedstock, Wiley-VCH, Weinheim, 2010 Search PubMed.
- D. J. Darensbourg and M. W. Holtcamp, Coord. Chem. Rev., 1996, 153, 155 CrossRef CAS.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514 RSC.
- P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169 CAS.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388 CrossRef CAS PubMed.
- S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460 CrossRef CAS.
- W. J. Kruper and D. V. Dellar, J. Org. Chem., 1995, 60, 725 CrossRef CAS.
- H. V. Babu and K. Muralidharan, Dalton Trans., 2013, 42, 1238 RSC.
- S. Iksi, A. Aghmiz, R. Rivas, M. D. González, L. Cuesta-Aluja, J. Castilla, A. Orejón, F. El Guemmout and A. M. Masdeu-Bultó, J. Mol. Catal. A: Chem., 2014, 383–384, 143 CrossRef CAS.
- (a) N. Takeda and S. Inoue, Bull. Chem. Soc. Jpn., 1978, 51, 3564 CrossRef CAS; (b) T. Aida and S. Inoue, J. Am. Chem. Soc., 1983, 105, 1304 CrossRef CAS.
- (a) T. Aida and S. Inoue, Macromolecules, 1982, 15, 682 CrossRef CAS; (b) T. Aida, M. Ishikawa and S. Inoue, Macromolecules, 1986, 19, 8 CrossRef CAS; (c) S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2861 CrossRef CAS.
- (a) C. J. Whiteoak, N. Kielland, V. Laserna, E. C. Escudero-Adán, E. Martin and A. W. Kleij, J. Am. Chem. Soc., 2013, 135, 1228 CrossRef CAS PubMed; (b) C. J. Whiteoak, N. Kielland, V. Laserna, V. Castro-Gómez, E. Martin, E. C. Escudero-Adán, C. Bo and A. W. Kleij, Chem. – Eur. J., 2014, 20, 2264 CrossRef CAS PubMed.
- (a) D. J. Darensbourg and J. C. Yarbrough, J. Am. Chem. Soc., 2002, 124, 6335 CrossRef CAS PubMed; (b) D. J. Darensbourg and A. I. Moncada, Macromolecules, 2010, 43, 5996 CrossRef CAS.
- J. Meléndez, M. North and R. Pasquale, Eur. J. Inorg. Chem., 2007, 3323 CrossRef.
- W. Clegg, R. Harrington, M. North and R. Pasquale, Chem. – Eur. J., 2010, 16, 6828 CrossRef CAS PubMed.
- X.-B. Lu, X.-J. Feng and R. He, Appl. Catal., A, 2002, 234, 25 CrossRef CAS.
- D. Tian, B. Liu, Q. Gan, H. Li and D. J. Darensbourg, ACS Catal., 2012, 2, 2029 CrossRef CAS.
- R. Luo, X. Zhou, S. Chen, Y. Li, L. Zhou and H. Ji, Green Chem., 2014, 16, 1496 RSC.
- Y. Ren, O. Jiang, H. Zeng, Q. Mao and H. Jiang, RSC Adv., 2016, 3243 RSC.
- D. J. Darensbourg and D. R. Billodeaux, Inorg. Chem., 2005, 44, 1433 CrossRef CAS PubMed.
- H.-L. Chen, S. Dutta, P.-Y. Huang and C.-C. Lin, Organometallics, 2012, 31, 2016 CrossRef CAS.
- M. Sasaki, K. Manseki, H. Horiuchi, M. Kumagai, M. Sakamoto, H. Sakiyama, Y. Nishida, M. Sakai, Y. Sadaoka, M. Ohbad and H. Okawa, J. Chem. Soc., Dalton Trans., 2000, 259 RSC.
- G. J. Clarkson, V. C. Gibson, P. K. Y. Goh, M. L. Hammond, P. D. Knight, P. Scott, T. M. Smit, A. J. P. White and D. J. Williams, Dalton Trans., 2006, 5484 RSC.
- L. Salmon, P. Thuery, E. Riviere and M. Ephritikhine, Inorg. Chem., 2006, 45, 83 CrossRef CAS PubMed.
- M. Asadi, M. Mohammadikish and K. Mohammadi, Cent. Eur. J. Chem., 2010, 8, 291 CAS.
- Y. Niu and H. Li, Colloid Polym. Sci., 2013, 291, 2181 CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. V. Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
- R. A. Pascal Junior, R. P. L'Esperance and D. Van Engen, private communication, 2014, CCDC 990458.
- K. Oyaizu and E. Tsuchida, Inorg. Chim. Acta, 2003, 355, 414 CrossRef CAS.
- L. Dyers Jr., S. Y. Que, D. VanDerveer and X. R. Bu, Inorg. Chim. Acta, 2006, 359, 197 CrossRef.
- H. L. Shyu, H. H. Wei, G. H. Lee and Y. Wang, J. Chem. Soc., Dalton Trans., 2000, 915 RSC.
- K. M. Szécsényi, V. M. Leovac, Z. K. Jacimovic, V. I. Cesljevic, A. M. Kovacs, G. Pokol and S. Gal, J. Therm. Anal. Calorim., 2001, 63, 723 CrossRef.
- (a) K. Nakamoto, Infrared and Raman spectra of inorganic and coordination compounds, Wiley & Sons, New York, 1998 Search PubMed; (b) G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227 CrossRef CAS.
- X. Zhuang, K. Oyaizu, Y. Niu, K. Koshika, X. Chen and H. Nishide, Macromol. Chem. Phys., 2010, 211, 669 CrossRef CAS.
- S. Elmas, M. Subhani, M. Harrer, W. Leitner, J. Sundermeyer and T. E. Müller, Catal. Sci. Technol., 2014, 4, 1652 CAS.
- D. J. Darensbourg, A. I. Moncada and S.-H. Wei, Macromol., 2011, 44, 2568 CrossRef CAS.
- X.-B. Lu, Y.-J. Zhang, B. Liang, X. Li and H. Wang, J. Mol. Catal. A: Chem., 2004, 210, 31 CrossRef CAS.
- J. Langanke, L. Greiner and W. Leitner, Green Chem., 2013, 15, 1173 RSC.
- A. Buchard, M. R. Kember, K. G. Sandeman and C. K. Williams, Chem. Commun., 2011, 47, 212 RSC.
- K. Matsumoto, Y. Sato, M. Shimojo and M. Hatanaka, Tetrahedron: Asymmetry, 2000, 11, 1965 CrossRef CAS.
- D. J. Darensbourg, J. C. Yarbrough, C. Ortiz and C. C. Fang, J. Am. Chem. Soc., 2003, 125, 7586 CrossRef CAS PubMed.
- Y. Qin, X. Sheng, S. Liu, G. Ren, X. Wang and F. Wang, J. CO2 Util., 2015, 11, 3 CrossRef CAS.
- H. Sugimoto, H. Ohtsuka and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4172 CrossRef CAS.
- S. N. Azizi and S. Ehsani-Tilami, J. Chin. Chem. Soc., 2009, 56, 898 CrossRef CAS.
- N. Ropson, P. Dubois, R. Jerome and P. Teyssie, Macromolecules, 1993, 26, 6378 CrossRef CAS.
- A. Dhammani, R. Bohra and R. C. Mehrotra, Polyhedron, 1998, 17, 163 CrossRef CAS.
- D. A. Atwood and M. J. Harvey, Chem. Rev., 2001, 101, 37 CrossRef CAS PubMed.
- D. Tian, B. Liu, L. Zhang, X. Wang, W. Zhang, L. Han and D.-W. Park, J. Ind. Eng. Chem., 2012, 18, 1332 CrossRef CAS.
- H. Haraguchi and S. Fujiwara, J. Phys. Chem., 1969, 10, 3467 CrossRef.
- D. Tian, B. Liu, Q. Gan, H. Li and D. J. Darensbourg, ACS Catal., 2012, 2, 2029 CrossRef CAS.
- J. E. Dengler, M. W. Lehenmeier, S. Klaus, C. E. Anderson, E. Herdtweck and B. Rieger, Eur. J. Inorg. Chem., 2011, 336 CrossRef CAS.
- S. Supasitmongkol and P. Styring, Catal. Sci. Technol., 2014, 4, 1622 CAS.
- R. Luo, X. Zhou, W. Zhang, Z. Liang, J. Jiang and H. Ji, Green Chem., 2014, 16, 4179 RSC.
- C. Martín and A. W. Kleij, Beilstein J. Org. Chem., 2014, 10, 1817 CrossRef PubMed.
- M. North and R. Pasquale, Angew. Chem., Int. Ed., 2009, 48, 2946 CrossRef CAS PubMed.
- J. A. Castro-Osma, A. Lara-Sánchez, M. North, A. Otero and P. Villuendas, Catal. Sci. Technol., 2012, 2, 1021 CAS.
- K. Yamaguchi, K. Ebitani, T. Yoshida, H. Yoshida and K. Kaneda, J. Am. Chem. Soc., 1999, 121, 4526 CrossRef CAS.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115 CrossRef CAS.
- G. M. Sheldrick, SHELXS97 and SHELXL97, University of Göttingen, Germany, 1997 Search PubMed.
- Ortep-3 for Windows - A Version of ORTEP-III with a Graphical User Interface (GUI) CrossRef CAS; L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- WinGX Suite for Single Crystal Small Molecule Crystallography CrossRef CAS; L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1455054 (2), 1455055 (3) and 1455056(4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt01069e |
| This journal is © The Royal Society of Chemistry 2016 |