
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Experimental investigation of anion–π interactions – applications and biochemical relevance
M.
Giese
*a,
M.
Albrecht
*b and
K.
Rissanen
*c
aInstitut für Organische Chemie, Universität Duisburg Essen, Universitätsstraße 7, 45141 Essen, Germany. E-mail: michael.giese@uni-due.de
bInstitut für Organische Chemie, RWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany. E-mail: markus.albrecht@oc.rwth-aachen.de
cUniversity of Jyvaskyla, Department of Chemistry, Nanoscience Center, P.O. Box. 35, FI-40014 University of Jyvaskyla, Finland. E-mail: kari.t.rissanen@jyu.fi
First published on 23rd December 2015
Abstract
Anion–π interactions, intuitively repulsive forces, turned from controversial to a well-established non-covalent interaction over the past quarter of a century. Within this time frame the question “Anion–π interactions. Do they exist?” could be answered and even more importantly its functional relevance was proven. The present feature article summarizes the experimental findings of anion–π studies in the gas phase, solution and in the solid state and highlights the application of anion–π interactions in anion recognition, sensing and transport as well as in catalysis. Moreover, the biochemical relevance of this weak intermolecular force is comprehensively reviewed.
 M. Giese | Michael Giese studied Chemistry at the RWTH Aachen University (Germany), where he obtained his PhD in 2011 with Prof. M. Albrecht. After his postdoctoral stay at the University of British Columbia (Vancouver, Canada) with Prof. Mark MacLachlan he returned to Germany. Since 2014 he is appointed Juniorprofessor at the University of Duisburg-Essen (Germany). His research interests are supramolecular chemistry, materials science and functional systems. |
 M. Albrecht | In 1992 Markus Albrecht obtained his Dr rer. nat. with Prof. G. Erker in Münster. From 1992 until 1993 he performed some postdoctoral studies in the laboratories of Prof. K. N. Raymond in Berkeley. After the habilitation at the Universität Karlsruhe he moved in 2002 to Aachen. His interests are in the field of Supramolecular Chemistry focusing on metallosupramolecular assemblies and anion recognition. Markus Albrecht is editor-in-chief of the Journal of Inclusion Phenomena and Macrocyclic Chemistry. |
 K. Rissanen | Kari Rissanen did his PhD at the University of Jyväskylä, Finland. After his work as Research Fellow and then a Senior Research Fellow of The Academy of Finland at the University of Jyväskylä, he became an Associate Professor of Organic Chemistry at the University of Joensuu, Finland. Since 1995 he has been professor and Head of the Laboratory of Organic Chemistry, University of Jyväskylä. He is also an Academy Professor of the Academy of Finland from 2008 to the present. His research topics include structural and synthetic supramolecular, organic and nanochemistry, X-ray crystallography, and crystal engineering. |
Introduction
Weak non-covalent interactions are highly relevant in chemical and biological processes. Both, chemical as well as biochemical processes rely on the well-orchestrated interplay of intermolecular forces.1 In order to develop novel functional materials a comprehensive understanding of these interactions is vital. Supramolecular chemistry deals with this issue and is nowadays one of the most interdisciplinary fields in science.2 While non-covalent interactions such as hydrogen bonds, ion pairing, hydrophobic effects as well as π–π and cation–π interactions can be considered as well established and were already employed in various functional assemblies, the rather “unconventional” σ/π-hole interactions3 such as halogen bonding4 and anion–π interactions5 were ignored for a long time. However, a number of recent studies recognized the functional relevance of the interaction between electron-deficient arenes and anions in both chemical2a as well as biological systems.6 This feature article intends to summarize the recent results in the field of anion–π interactions with the focus on the experimental findings. Nevertheless theoretical studies will be shortly described for integrity reasons. In addition, the seminal findings from the last few years, proving the relevance of anion–π interactions in biology as well as their functional significance, will be discussed.Early findings and the anion–π controversy
What now seems to be generally accepted and well established in the chemical community started as scientific curiosity and was considered truly controversial. The under-appreciation of anion–π interactions relied on the intuitive assumption of repulsive forces between anions and the seemingly electron-rich π-system of an arene. Although experimental results already indicated the existence of an attractive interaction between anions and electron-deficient arenes, it was ignored for a long time. The seminal computational work by Mascal, Alkorta and Deyà served as turning point and directed the scientific interest on this elusive intermolecular force by asking “Anion–π interactions. Do they exist?”.7 This report in 2002 was the starting point for many scientific publications in the field of anion–π studies and led to debate about its existence. Five years later Hay et al. responded in a study “Anion–π interactions in crystal structures: commonplace or extraordinary? and found not a single crystallographic evidence for anion–π interactions in the solid state by applying very strict evaluation criteria.8 This view to anion–π interactions started a controversial discussion among theoretical and experimental chemists. However, Frontera et al. set anion–π interactions into perspective in 2011 and critically reviewed the recent findings in the field.9 Beside the summary of key studies in the field of anion–π interactions, the authors provided insight into the directionality of the interaction between anions and electron-poor arenes.Nevertheless, even if the strength of the anion–π interactions remains controversial, the recent studies strongly support the functional relevance in ion transport,10 biomolecular structures5b,6a,b or catalysis.11
One of the first experimental observations for anion–π interactions was given by Hiraoka et al. in 1987.12 They investigated complexes of halide anions (F−, Cl−, Br−, I−) with hexafluorobenzene in the gas-phase by pulsed electron-beam high-pressure mass spectrometry. Interestingly, they found significant differences between the fluoride and other halides in the hexafluorobenzene complex in the gas phase. The fluoride was found to be bound via a Meisenheimer-type complex to the C6F6 moiety.12a In contrast, the other halides were only weakly bound by a non-covalent interaction.12b Supported by theoretical calculations a C6v-symmetric anion–π complex was postulated, with halide anions located above the centre of the hexafluorobenzene moiety.
A few years later Schneider and his group recognized a weak interaction between negatively charged groups and polarized aryl groups in solution.13 The calculated association constants for the dimerization of diphenylamine with dications as well as with dianions were found to be similar.13b
However, only since 2002 the term “anion–π interaction” was introduced into literature by Mascal,14 Alkorta15 und Deyà.7,16 These reports brought the non-covalent interaction between anions and arenes out of obscurity and started a still growing interest in this intermolecular force.
Anion–π interactions – definition, properties and interplay with other non-covalent forces
A large number of computational studies have been performed in order to investigate the nature of the anion–π interactions.14,15,17 These results revealed that the interactions of anions with π-systems relies mainly on two effects – the electrostatic attraction and the ion-induced polarization. The electrostatic term is well represented by the quadrupole moment of the arene Qzz, which describes the charge distribution of the aromatic system in respect to the z-axis (perpendicular to the arene plane). For benzene Qzz is negative (−8.5 B) and therefore repulsive in respect to anions. However, arenes with strongly electronegative substituents such as fluorine (Qzz(C6F6) = +9.5 B) or heteroarenes such as trifluorotriazine (Qzz = +8.2 B) have positive quadrupole moments and interact attractively with negatively charged species. The differences in the charge distributions are visualized by charge density simulations, where the red colour represents excess of electrons and the blue colour the electron-deficiency (Fig. 1).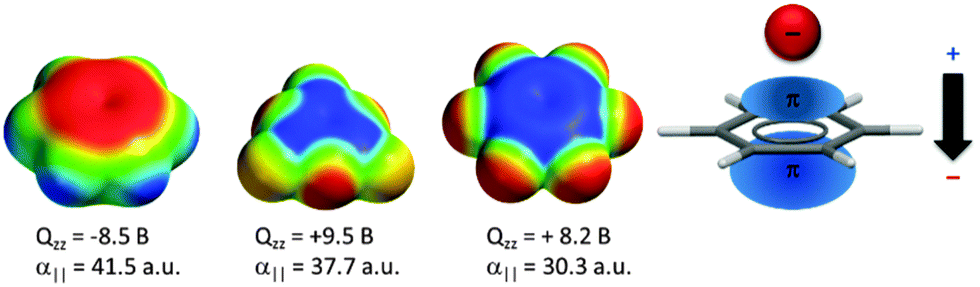 | ||
| Fig. 1 Charge density simulations of benzene, trifluorotriazine and hexafluorobenzene as well as a schematic representation of the anion-induced polarization. | ||
The ion-induced polarization is correlated with the molecular polarizability α∥ of the arene. Molecules like s-tetrazine (α∥ = 58.7 a.u.)18 have a high molecular polarizability and therefore the ion-induced polarisation contributes significantly to the total interaction energy of the corresponding anion–π complexes.
The following section will summarize systematically the experimental studies investigating the nature of anion–π interactions. These seminal investigations together with detailed computational studies are fundamental for the applications based on anion–π interactions.
The role of the π-system
Anion–π interactions have been investigated for a multitude of arenes.14–16,17b,18,19 While theoretical investigations suggest attractive interactions between most of the aromatic systems and anions, obtaining experimental proof has been challenging.Mascal,14 Alkorta7,15,16 and Deyà reported attractive interactions for a series of electron-deficient arenes ranging from 1,3,5-triazine via hexafluorobenzene to 1,3,5-trinitrobenzene (Fig. 2). Their results indicated that the increasing quadrupole moment leads to increased interactions energies supporting the significance of the electrostatic term in the anion–π interactions.
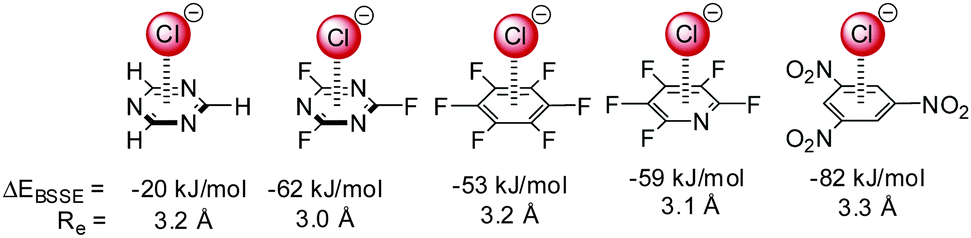 | ||
| Fig. 2 Interaction energy with basis set superposition error correction (EBSSE) and equilibrium distances (Re) of selected anion–π complexes from the MP2/6-31++G** level of theory.7,15,16 | ||
Experimentally the role of the arenes in the anion–π complexes was studied in solution and in the solid state by Kochi et al.20 The addition of tetrabutylammonium halides to solutions of the neutral, electron-deficient aromatic π-acceptors 1–3 in acetonitrile yielded a significant change in colour. The association constants for the corresponding anion–π complexes were determined by UV-Vis titration experiments and revealed stronger binding with increasing acceptor strength. The obtained solid state structures support their findings in solution and showed the anions to be surrounded by the electron-deficient arenes, exclusively interacting with each other by anion–π interactions (Fig. 3).
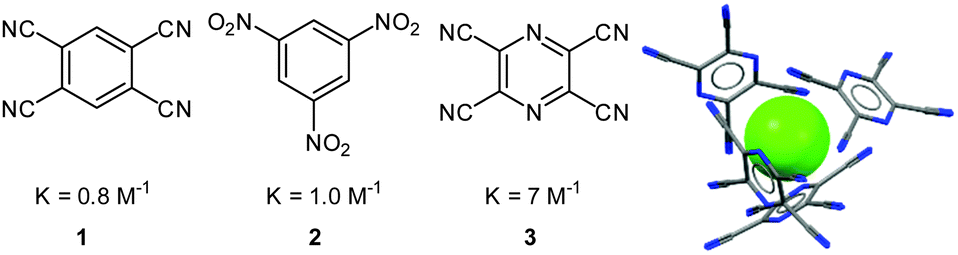 | ||
| Fig. 3 Neutral organic π-acceptors and their complexation constants with bromide as determined by Kochi and coworkers. The crystal structure of the tetracyanopyrazine with chloride shows the anion surrounded by four electron-deficient arenes interacting via anion–π contacts with the central anion (counter cation omitted for clarity).20 | ||
Gotz et al. investigated anion–π interactions in the solid state structure of 2-(N,N-di(pyridine-2-yl))-4-(perfluorophenyoxy)-6-phenoxy[1,3,5]triazine (4) copper(II)-complex (Fig. 4).21 This system offers four concurrent π-binding sites to the perchlorate anion. Interestingly anion–π contacts are only found for the central triazine unit (centroid⋯O = 3.20, 3.36, 3.19, 2.94 Å) and the pentafluorophenyl group (C⋯O = 3.65, 3.60 Å). These findings were supported by computational studies, which confirm the contribution of anion–π interactions to the observed binding.
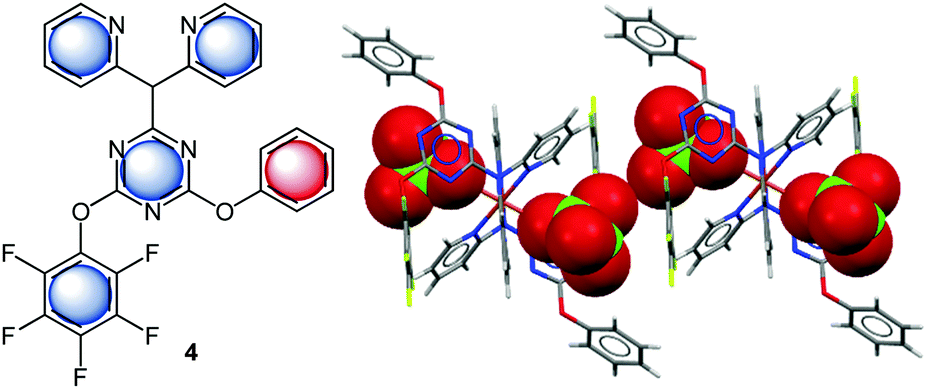 | ||
| Fig. 4 Molecular structure of the 2-(N,N-di(pyridine-2-yl))-4-(perfluorophenyoxy)-6-phenoxy[1,3,5]triazine (4) and a representative view of the crystal structure of the corresponding Cu(II)-complex showing anion–π interactions between the pentafluorophenyl and triazine units with the perchlorate anions.21 | ||
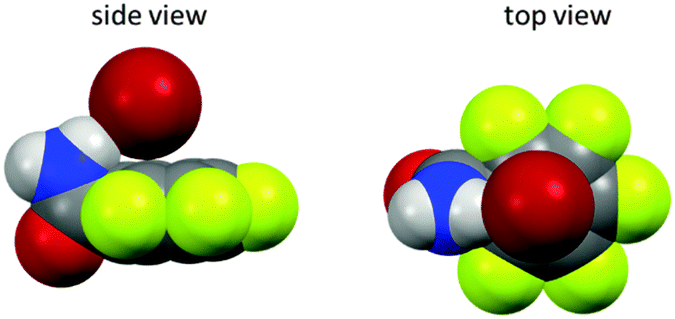
We have also investigated the role of the electron-density of the π-system in the solid state (Fig. 5).22 Therefore, a series of 1-(fluorobenzyl-ammonium)-4-aza-1-azoniabicyclo[2.2.2] octane salts (fluorobenzyl substituted DABCO, 5–9) were synthesized and crystallized. Comparison of the molecular structures showed that the attraction between the pentafluoroarene and the anion turns into a repulsion when less than four fluorine atoms are attached to the phenyl group. In this series the distance between the centroid and the bromide was increased from 3.68 to 6.03 Å clearly indicating the significance of the electrostatic component in anion–π interactions. Remarkably, structures with an ortho-hydrogen atom (like 9) do not show anion–π interactions at all. In this case the anions were found to interact with a side-on position fixed by non-classical hydrogen bonds. The accompanying computational study confirmed the experimental findings.
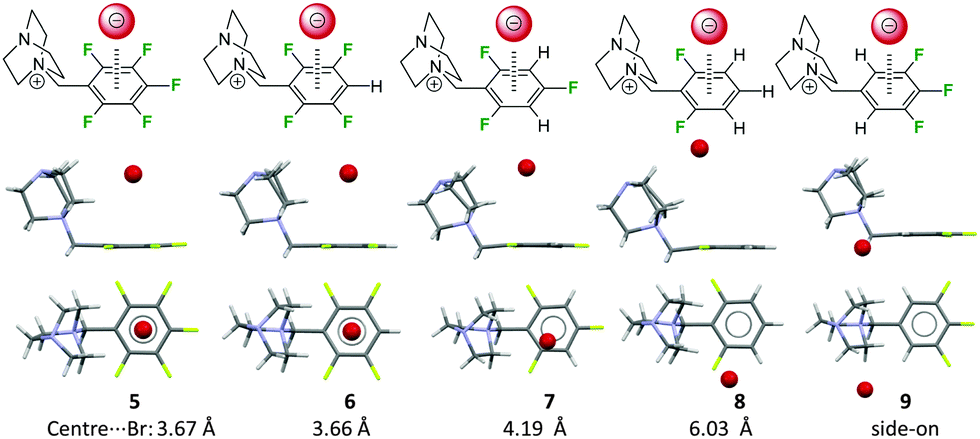 | ||
| Fig. 5 Side and top view of the ion pairs 5–9 as observed in the solid state. The distance between the centre of the fluoroarene and the anion rises in dependence of the degree of fluorine substitution at the aromatic unit (5–8). ortho-Hydrogen atoms (9) form non-classical CH–hydrogen bonds to the anion and fixes it in the side-on position.22 | ||
As already mentioned above anion–π interactions are dominated by the electrostatics and the ion-induced polarizability. These opposing effects rule out each other, which leads to net attractive anion–π interactions in many π-systems. Thus, arenes with negligible quadrupole moments but significant polarizabilities are able to interact with both, cations and anions.18b,23
Frontera et al. investigated these opposing effects in a systematic study.19c They investigated a series of cyanuric acid derivatives in which they successively replaced the oxygen atoms with sulphur atoms. This leads to a substantial decrease in the quadrupole moment values (from 6.96 B to 5.15 B) and an increase in the molecular polarizability (from 35.15 a.u to 63.75 a.u.) of the π-system. Interestingly, their calculations revealed that the interaction energies for the corresponding anion–π complexes remained almost constant (∼15 kcal mol−1), supporting the compensation of the opposing effects. However, crystal structures of the corresponding ethylene ammonium-substituted chloride salts of the thio- and dithiocyanuric acids 10 and 11 show significant differences in their centroid-to-anion distances (Fig. 6).
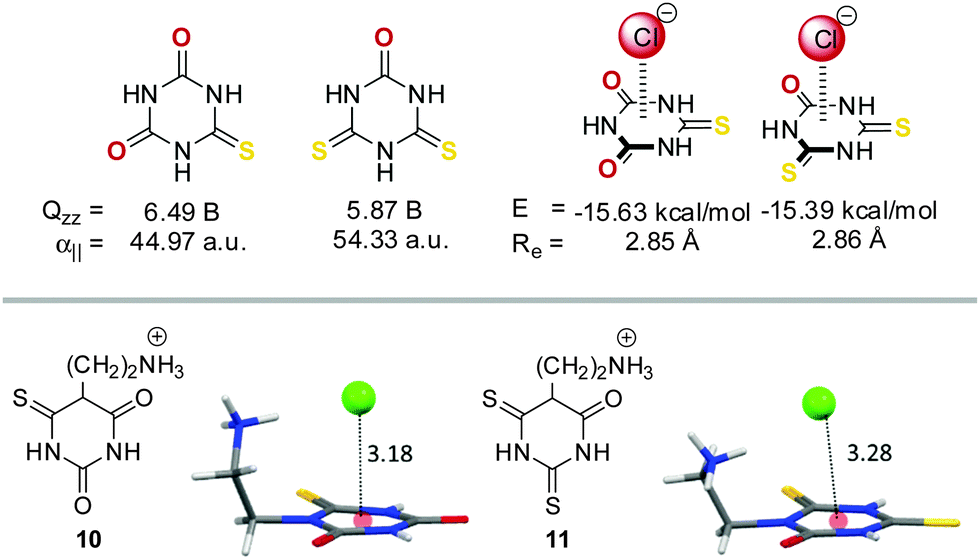 | ||
| Fig. 6 Molecular structures, quadrupole moments and molecular polarizabilities of selected thio cyanuric acid derivatives and their corresponding anion–π complexes with chloride (including the calculated interaction energies and equilibrium distances). Additionally, the ion pairs of the ethylene ammonium-substituted chloride salts of the thio- (10) and dithiocyanuric acids (11) are shown as observed in the solid state.19c | ||
The role of the anion
Also the nature of the anion is relevant in respect to its interaction with π-systems. Generally both terms, the electrostatic attraction as well as the polarizability depend on and influence the anion–arene distance.7,17b,c,18b Theoretical studies show that small anions such as fluoride or chloride are strongly polarizing. They are closely located to the arenes and exhibit therefore strong interaction energies. In addition, anions like CO32−, NO3− or N3− exhibit π–π-interactions with arenes. Accordingly, these complexes exhibit extraordinary high interaction energies.In order to investigate the anion effect in anion–π complexes we have performed a series of studies.24 First the anion size effect was studied by employing the already established pentafluorobenzyl substituted DABCO system. Crystallization with ammonium chloride, bromide and iodide yielded the solid state complexes of the salts, showing the halide anions to be located on top of the aromatic moieties. As expected the centroid-to-anion distances increased in the order chloride (3.60 Å) < bromide (3.67 Å) < iodide (3.78 Å) (Fig. 7).
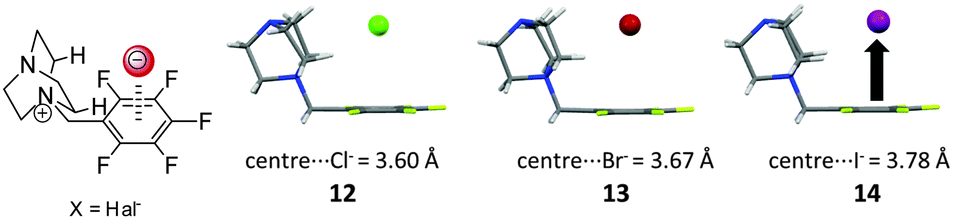 | ||
| Fig. 7 Molecular structure and representation of the ion pairs of the investigated pentafluorobenzyl substituted DABCO halides 12–14 as observed in the solid state.24 | ||
In order to prove that the weak interaction between anions and pentafluorophenyl groups has a significant influence on the crystal structure, we investigated a series of 1,3-bis(diphenyl-pentafluorobenzylphosphonium)propane dications and crystallized them with various anions.24b Interestingly, anions such as bromide (15) did not efficiently fill the space between the two adjacent electron-deficient arenes, thus two independent anion–π contacts were observed on opposite sides of the phosphonium backbone. However, increasing the size of the anion led to a ditopic anion–π interaction, where the anion is panelled by two C6F5 units. The ditopic arrangement was found for iodide (16), tetrafluoroborate (17) and hexafluorophosphate (18) (Fig. 8).
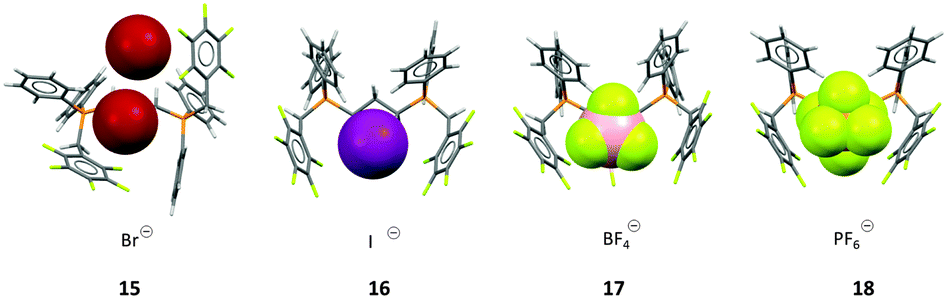 | ||
| Fig. 8 View of the ion pairs of the 1,3-bis(diphenylpenta-fluorobenzylphosphonium) propane salts 15–18 as observed in the solid state showing the individual or simultaneous interaction of the C6F5 units with the anion.24b | ||
These results raised the question about the role of the anion geometry. The modulation of the anion geometries is generally discussed as a major issue in anion-recognition and templation studies.25 Therefore, the DABCO-system was crystallized with spherical (Br−), linear (BrIBr−), planar (NO3−), tetrahedral (BF4−) and octahedral (PF6−) counter ions (Fig. 9). In all crystal structures the anions are located on top of the electron-deficient arene. However, not all anions were located exactly above the centre of the pentafluorophenyl group. The spherical bromide and the linear interhalide Br–I–Br− is located on top of the centre of the C6F5 moiety exhibiting several anion–π contacts. In contrast the solid state structure of the nitrate (20) and the tetrafluoroborate (21) showed two different ion pairs in the asymmetric unit with different relative orientations of the anions in respect to the π-system (Fig. 9). The crystal structure of the nitrate salt revealed one anion oriented parallel to the π-system (face-to-face, Fig. 9), suggesting π–π-interactions and a tilted orientation (edge-to-face) with weak anion–π interaction between one of the oxygen atoms and two carbon atoms of the pentafluorophenyl group (η2-type: C5⋯ONO2 = 3.15 Å; C6⋯ONO2 = 3.30 Å). Also the tetrafluoroborate structure showed two different ion pairs in the asymmetric unit (Fig. 9). The anion is located on top of the C6F5 unit for both subunits. The first ion pair showed close anion–π contacts between the fluorine atom of tetrafluoroborate pointing directly to the centre of the arene (C⋯F–BF3: 2.879–3.399 Å, centroid⋯FBF3: 2.829 Å). Due to the short anion-to-arene distances this structure is described as η6 anion–π interaction. In the other ion pair the tetrafluoroborate anion is slightly rotated with three fluorine atoms close to the pentafluorophenyl group (C⋯F3BF: 3.314–3.885 Å), whereas the anion–π interactions are only to the two or three carbons of the π-system (η2–η3). These structures support the non-directionality of anion–π interactions, which is in line with the fact that the anion–π interactions are mainly based on electrostatic attraction.
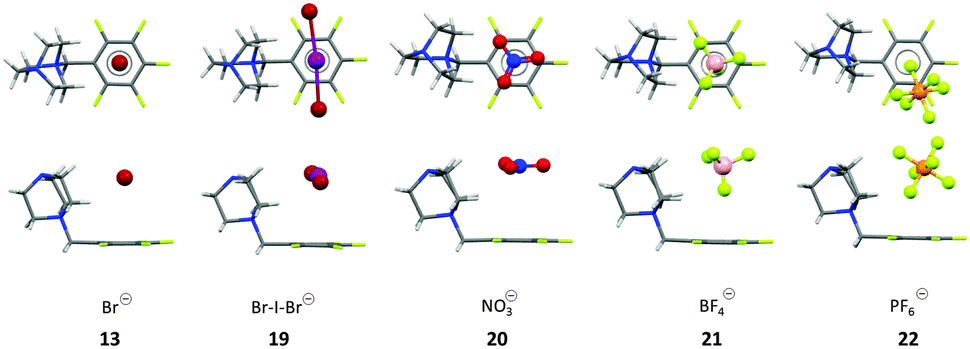 | ||
| Fig. 9 Top and side view of the ion pairs as observed in the crystal structures of the pentafluorobenzyl substituted DABCO salts with Br− (13), BrIBr− (19), NO3− (20), BF4− (21) and the PF6− (22) salts. For BF4− and the NO3− salt only one of the two orientations of the anion in the asymmetric cell is shown.25 | ||
Also the interaction between giant negatively charged polyoxometalates (POMs) and N,N′-di(4-pyridyl)-1,4,5,8-naphthalene-diimide (DPNDI) were studied in hydrogen- or coordination-bonded supramolecular frameworks.26 The authors suggest a significant contribution of anion–π interactions in the templation of the nanostructure.
Hapticity of anion–π interactions
The interaction of anions with arenes is versatile and different interaction geometries are both predicted and observed experimentally (Fig. 10). Firstly, anions can be located to the “side-on” position at the plane of the arene. In this case the anionic species interact via non-classical CH-hydrogen bonding (I/II) and hydrogen bonding (III) to e.g. a protonated heterocyclic arene or via halogen bonding (IV) with the arene. Secondly, the anion can be located “on-top” of the arene (V–VIII) interacting directly with the π-system by anion–π interactions. A series of crystallographic studies revealed a pronounced structural variation in the anion–π complexes and showed that the theoretically predicted (η6) C6-π-complex is rarely found. Generally anions show close anion–π contacts to certain parts of the π-system defined as η1 η2 and η3 (V–VII).27 In addition, anions can be covalently bound to one of the carbon atoms of the arene resulting in a Meisenheimer-complex (IX).28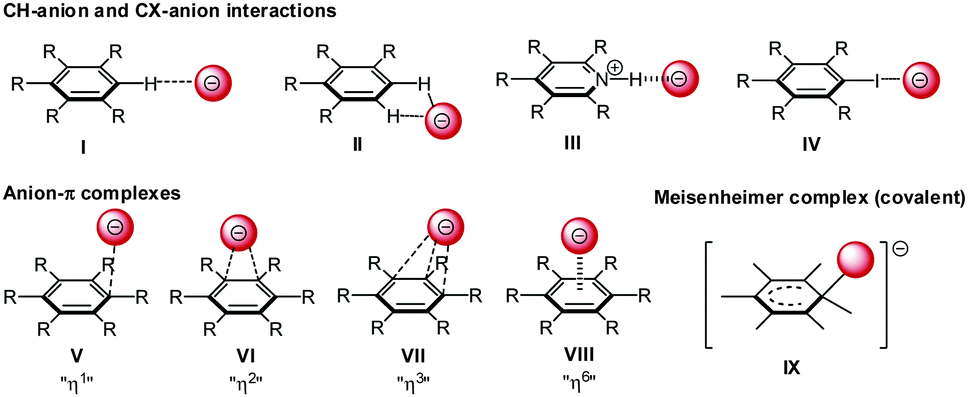 | ||
| Fig. 10 Possible interactions of anions with arenes: for anions in the side-on position mono- or ditopic CH–anion interaction (I and II), hydrogen (III) and halogen bonding (IV) is possible. In addition anion–π complexes with different hapticities (V–VIII) and the covalent Meisenheimer complex (IX) are possible. | ||
Experimental evidence for different binding modes between anions and arenes was found by Hiraoka et al.12 They investigated complexes of fluoride and hexafluorobenzene in the gas phase via pulsed electron-beam high-pressure mass spectrometry. The results indicated a covalent binding between the arene and the anion corresponding to a Meisenheimer complex (Fig. 10, I). In contrast, their extended study from 1987 found a non-covalent interactions in gas phase clusters of hexafluorobenzene and various halides (C6F6⋯X−; X = Cl, Br, I). Supported by computational results a η6-type anion–π complex was (Fig. 10, VIII) was proposed, whereby the anion resides on top of the centroid of the hexafluorobenzene.12b
In 2007, Schneider et al. investigated a series of anion–arene complexes by infrared photodissociation spectroscopy.29 The mass-selected complexes of chloride, iodide and hexafluorosulphate with various fluoroarenes (C6FnH6−n⋯X−) were studied. Interestingly, they found, that the η6-type anion–π complex is exclusively found for perfluorinated arenes (figure, H). In all other cases the anion is found in the “side-on” position bound by non-classical CH-anion hydrogen bonds (Fig. 10, I and II).
Computational chemists commonly underline their findings by analyzing crystallographic data bases.9,30 One of the first studies of this kind on anion–π interactions was reported by Quiñonero et al.7 Since then many of these structural studies have been published. Surprisingly, the initial evidence for anion–π interactions in the solid state was supported by a large number of hits in the CSD7,16,31 turned into to nearly non-existent by changing the search criteria applied in the data mining studies.8
Initially, Quiñonero et al. analyzed the CSD and reported 1944 hits for anion–π and lone-pair-π interactions of perfluorinated arenes.7 Responding to this study Hay and his group repeated the CSD analysis with slightly different criteria reducing the number of hits to a few hundreds27a and finally the very tight search criteria in 2009 resulted in zero anion–π interactions in the solid state.8
Very recently we reported a structural analysis of fluorophenyl substituted anion-receptors.32 In total the position of 101 anions (8 chloride, 58 bromide, 17 iodide and 17 others such as tetrafluoroborate, hexafluorophosphate or nitrate) from 80 crystal structures were extracted and plotted against a normalized C6F5 unit with the xy plane centered in the origin As shown in Fig. 11 most of the anions are located above or below the π-system (91 hits, 89%) with close contacts to the carbon atoms of the arene unit. Only 11 anions were found at the periphery of the aromatic moiety.
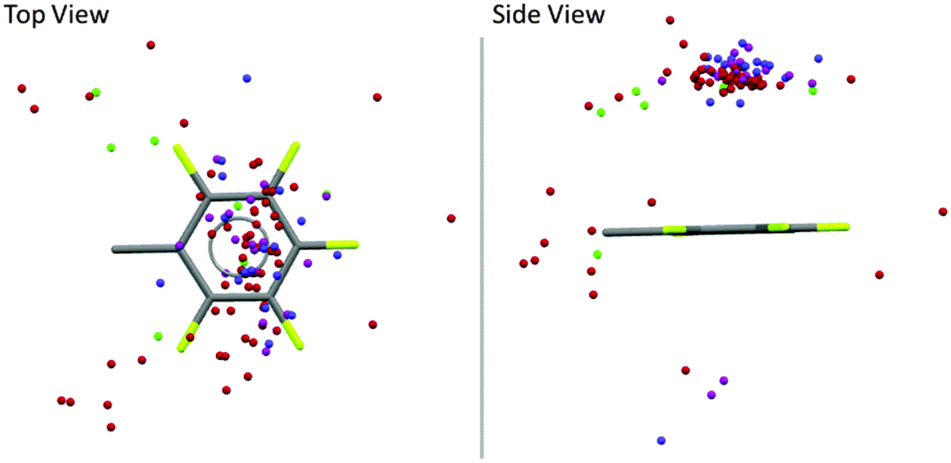 | ||
| Fig. 11 Graphical analysis of the anion positions in respect to a normalized fluoroarene (Cl−: green, Br−: red, I−: pink, other anions (BF4−, PF6−, NO3−, I3−, BrIBr−, I42−): blue).32 | ||
In contrast to the theoretical predictions,7 our study showed a broad distribution of the anions above the arene plane, which is in line with the structural versatility of anion–π interactions in the solid state as reported by Hay27a and us.27b The large structural variability of anion–π interactions can be attributed to their non-directed electrostatic character. However, these results together with the discussion started by Frontera and Hay demonstrated the need for a more general evaluation criteria and a tool to describe the structural versatility of the anion–π contacts. Experimental findings show that the limiting only to the C6v-symmetric systems with an anion located exactly on top of the arene is insufficient. Furthermore, the restriction to the exact sum of van der Waals radii appears too harsh taking into account that the anion–π interactions are based on weak non-covalent forces. Therefore, we proposed new search criteria based on our own experimental observations from a multitude of single crystal structures. Attractive anion–π interactions exist when the anion is located on top of an arene (criterion 1) in distance to the plane of the arene defined as ∑vdW + 0.4 Å (criterion 2). The tolerance of 0.4 Å was selected according to the recent adjustment of the tabulated vdW radii of halide anions as reported by Deyà and coworkers in 2011.31 In order to consider the structural versatility of anion–π interactions the hapticity concept of cation–π interaction was adapted. The number of carbon atom-anion distances in the range of ∑vdW + 0.4 Å yields the hapticity of the corresponding anion–π interaction.
Our results initiated a discussion on the versatility of anion–π interactions.27b A series of penta-fluorobenzyl substituted ammonium, iminium and amidinium salts was crystallized and showed significant differences in the relative position of the anion in respect to the electron-deficient arene. The anions in the solid state structures of the pentafluorobenzyl amidinium chloride and bromide salts (23, Fig. 12) revealed distinct anion–π contacts, either η6 or η2 interaction (C⋯Cl = 3.34, 3.41 Å; C⋯Br = 3.66, 3.73 Å) to the pentafluorophenyl group of an adjacent ion pair. An extended study on related ammonium compounds demonstrated the crucial role of the interfering CH–anion interactions allowing to control the position of the anion in the crystalline lattice.33 Accordingly, systems with a highly symmetric backbone such as the pentafluorobenzyl substituted tetramethylethylenediamine (TMEDA) derivative (24) directed the bromide anion above the centre of the arene (centre⋯Br− = 3.48 Å, η6) by fixing the anion via two CH–anion interactions (CH⋯Br− = 2.98, 3.04 Å). In contrast, a less symmetric backbone, dimethylphenylpentafluorobenzyl ammonium bromide (25), showed the fixation of the anion above one side of the arene. The reason for this is the higher sterical demand of the phenyl group which “pushes” the anion towards the rim of the π-system leading to a η2 interaction with the arene.
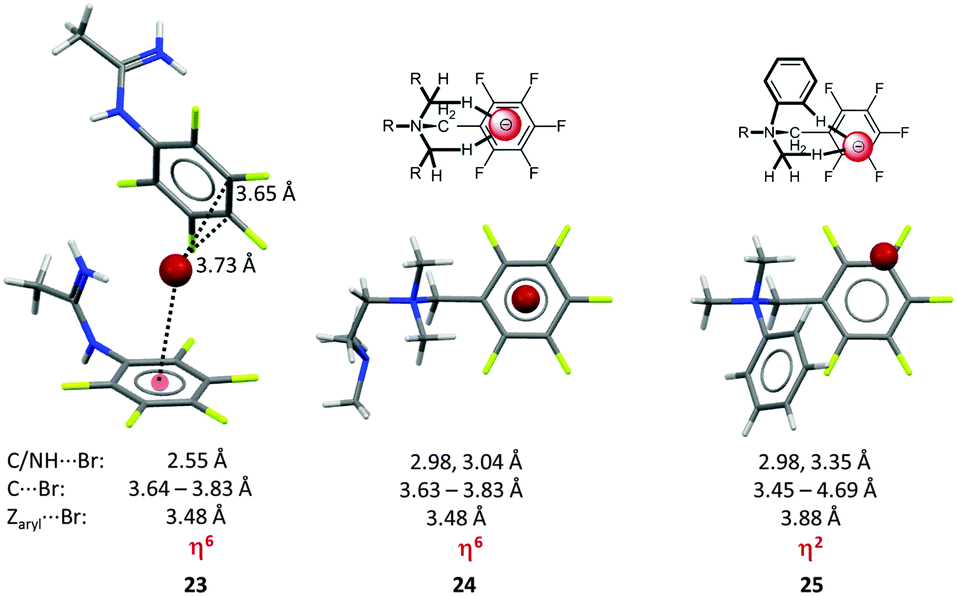 | ||
| Fig. 12 Molecular structure of the amidinium bromide (23) TMEDA (24) and the dimethylphenyl pentafluoro-benzyl (25) systems demonstrating the directing effect of the substituents in the backbone of the receptor.33 | ||
The extensive structural analysis in 201532 demonstrated, that this observation can be generalized. While a broad anion distribution above the normalized fluoroarene was found, limiting the structural data to systems with a rigid and symmetric backbone (e.g. the DABCO or TMEDA systems) revealed that the anions accumulate above the centre of the electron-deficient π-system. For the anions in solid state structures with a less symmetric backbone a significantly broader positional distribution was observed (Fig. 13).
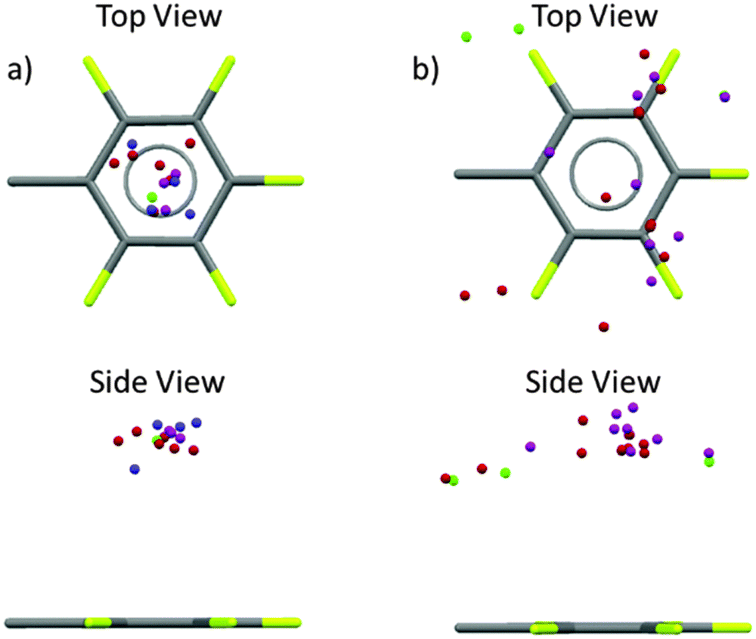 | ||
| Fig. 13 Analysis of the crystal structures of the π-acceptors for a symmetrical, rigid scaffold without co-crystallized solvent molecules (a) and for less rigid and symmetrical scaffolded acceptors (b) as reported by Albrecht's group. | ||
In conclusion, the theoretically proposed η6 anion–π complex represents an exceptional, very rarely observed structure in the solid state, which can be attributed to other interfering non-covalent interactions yielding an off-set of the anion from the centre of the arene.8,34
Quantification of anion–π interactions in solution
So far we have discussed that anion–π interactions may be attractive in nature and interfere with a number of other non-covalent forces. This interplay makes the experimental quantification of anion–π interaction a challenging task, especially in solution. Interfering interactions and solvent effects complicate or even prevent the specific quantification of the absolute interaction energy for the anion–π interaction. Nevertheless a number of studies have been performed and gave first insights in the strength and relevance of anion–π interactions in solution.35Two receptor designs have been employed in anion–π studies in solution, either charged receptors based on e.g. ammonium or phosphonium backbones24a,36,37 or charge neutral receptor systems.20,38–46
Gosh et al.36 and us24a reported anion–π studies of pentafluorobenzyl substituted ammonium (26) and phosphonium compounds (27), respectively. Both groups started their NMR-titration experiments with the corresponding ammonium or phosphonium compounds with weakly coordinating anions (such as tetrafluoroborate) and added successively tetraalkylammonium halides to these solutions. By comparing the association constants to the corresponding non-fluorinated systems, significant differences were found for the system reported by Gosh in DMSO-d6, while our study did not show notably differences between the receptors in CDCl3. However, both Ghosh and we found weak anion–π interactions in the solid state structures of the corresponding halides (Fig. 14). We attributed the small differences in the association constants to the interference by the stronger CH–anion interactions. Very similar results were found for similar pyridinium receptors.37
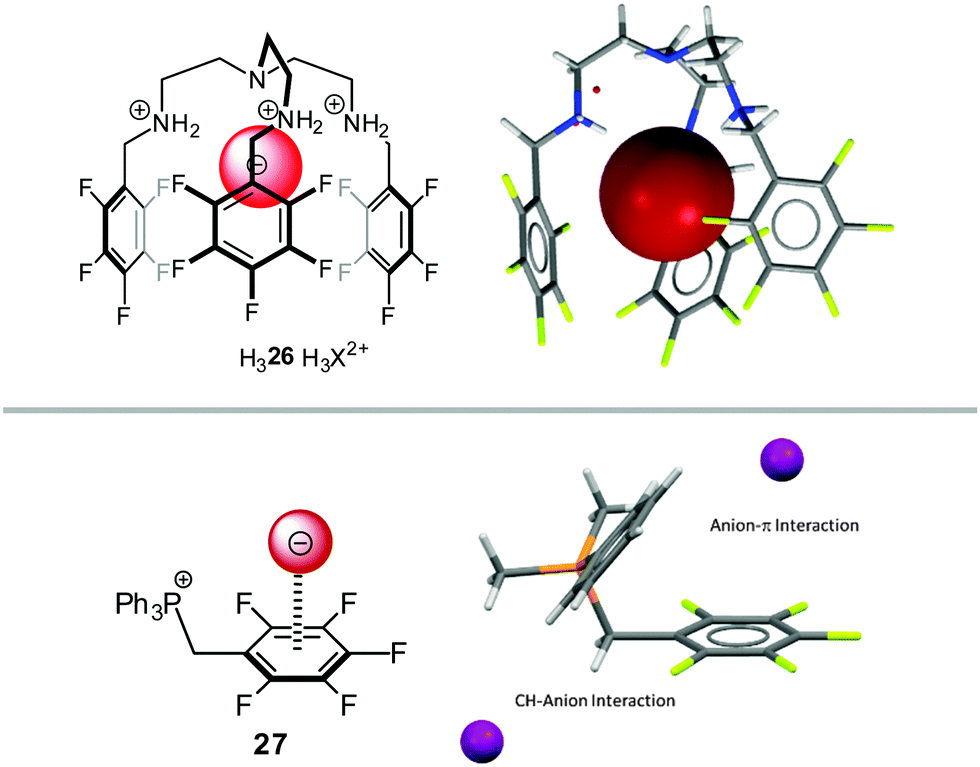 | ||
| Fig. 14 Molecular structures and representative views of the receptor systems in the solid state as employed by Ghosh's36 and us.24a | ||
One of the first anion–π studies in solution with charge-neutral π-acceptors was reported by Kochi et al. and was already described above.20 Structurally related to the tetracarbonitrile pyrazine compounds investigated by Kochi is the 1,4,5,8,9,12-hexaazatriphenylenehexacarbonitrile 28 as investigated by the Dunbar's group (Fig. 15).38 The association constants for the anion–π complexes were studied in THF by UV-vis spectroscopy via addition of tetrabutylammonium halides. Mulliken correlation20b and Job plot analysis supported the formation of a charge transfer complex with stoichiometry of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (receptor:guest), which was further supported by the co-crystal structure of 28 with tetrabutylammonium bromide. Surprisingly, the authors determined the association constants by applying a 1:1 model to the obtained titration curves and found decreasing binding affinities in the order Cl− > Br− > I−, which can be attributed to the decreasing Lewis basicity in the same order.
:2 (receptor:guest), which was further supported by the co-crystal structure of 28 with tetrabutylammonium bromide. Surprisingly, the authors determined the association constants by applying a 1:1 model to the obtained titration curves and found decreasing binding affinities in the order Cl− > Br− > I−, which can be attributed to the decreasing Lewis basicity in the same order.
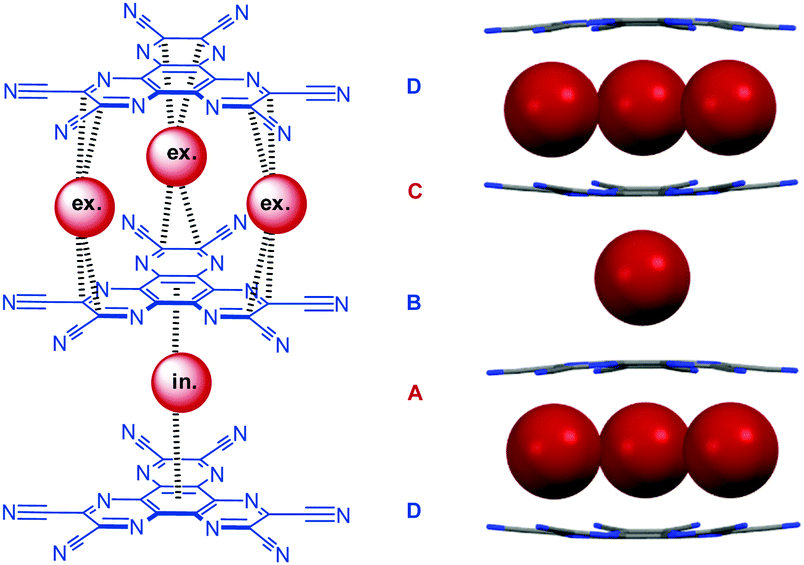 | ||
| Fig. 15 Molecular structure of the hexacarbonitrile π-acceptor 28 as well as the corresponding CT complex with bromide as obtained by co-crystallisation with tetrabutylammonium bromide (tetrabutylammonium cations omitted for clarity).38a | ||
Furuta and his group chose pentafluorophenyl decorated and metallated (M(II) = Ni, Pd and Cu) N-confused porphyrins (NCP) to investigate anion–π interactions in dichloromethane.39 By UV-vis titration experiments they found somewhat higher affinities for the fluorinated derivative 29 when compared to the phenyl substituted model compound 30 (Fig. 16). Job plot analysis revealed 1:1 complexes with chloride, bromide and iodide with decreasing interaction affinity in the order Cl− (5.7 ± 0.4 M−1) > Br− (0.8 ± 0.04 M−1) > I− (0.1 ± 0.002 M−1). For the reference compound 30 the binding constant was found to be significantly smaller (<0.0001 M−1). These observations were explained by the enhanced acidity of the NH-group due to the electron withdrawing effects and anion–π interactions, which were concluded from 19F-NMR spectra. Extending their study to NCP derivatives 31–33 with three pentafluorophenyl units in different positions (Fig. 16) yielded slightly smaller binding constants for the nickel(II) complexes with chloride in respect to the tetra-substituted porphyrin 29.39b To the authors' surprise, the highest association constant was found for 31, which lacks the C6F5-unit close to the outer NH-group. Concluding from this anion–π interactions play a minor role in the anion binding ability of this receptor system and changes in the 19F-NMR spectra could be better attributed to the shielding of the negative charge rather than to the presence of anion–π interactions.
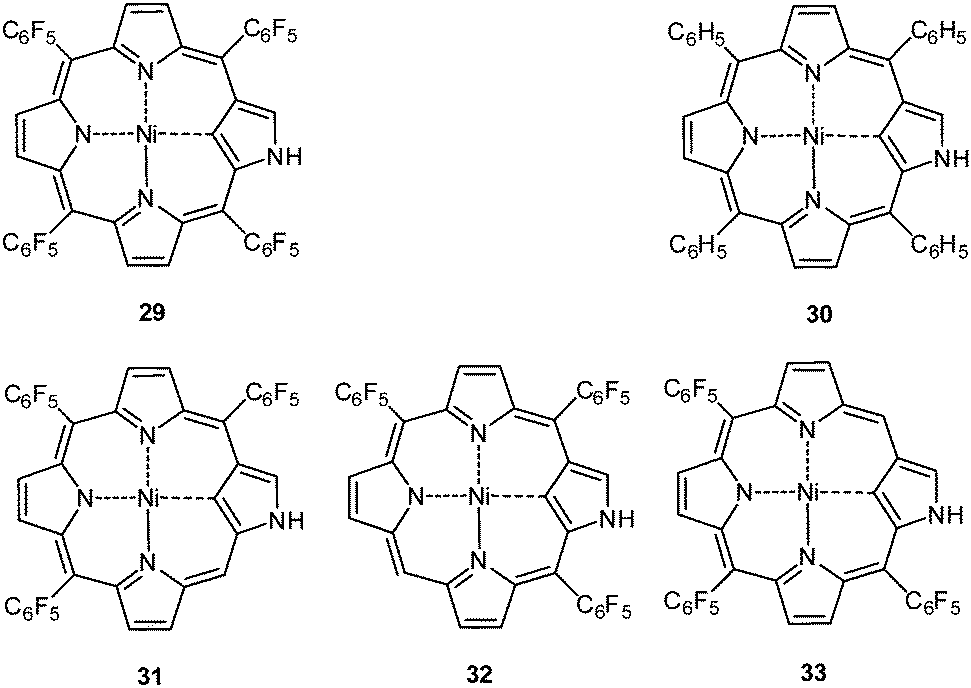 | ||
| Fig. 16 Molecular structures of the N-confused porphyrin receptors 29–33 as used in the studies of Maeda et al.39 | ||
Berryman et al. designed a receptor system for cooperative binding of an anion by NH–anion interactions to the amide and anion–π bonding to the pentafluorophenyl group.40 The 1H-NMR-titration experiments with comparison to a non-fluorinated analogue revealed enhanced binding of 34a in respect to 34b (Fig. 17) in chloroform for Cl−, Br− and I−. The enhanced binding was partly attributed to anion–π interactions. However, the crystal structure of 34a·Cl− did not support anion–π interactions in the solid state.
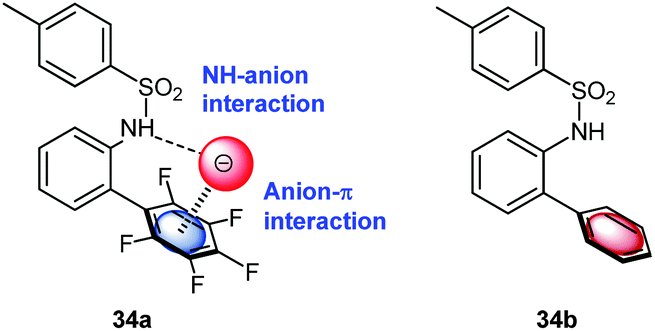 | ||
| Fig. 17 Schematic representation of the anion receptor design as employed by Johnson and coworkers for anion–π studies in solution. | ||
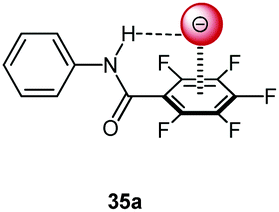
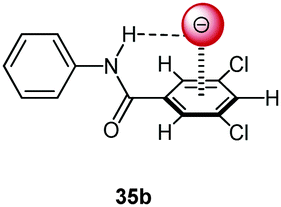
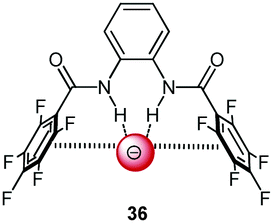
Following cooperative anion binding by hydrogen bonding and anion–π interactions we studied the anion binding of benzamide receptors 35a and 35b (Table 1).41 The Job plot analysis revealed 1:1 complexes and 1H-NMR titration in CDCl3 yielded slightly higher association constant for 35a when compared to 35b. The binding affinity decreased for both systems in the order of Cl− > Br− > I−. In addition, the ditopic receptor 36 (Table 1) was studied via NMR titrations in CD3CN and shows significantly stronger interactions than 35a or b. Supported by computational studies a contribution from anion–π interaction was proposed for the anion binding. Experimental evidence was given by the crystal structure of pentafluorobenzamide with tetraethylammonium bromide 37TEABr (Fig. 18) co-crystal, which showed short distances between the carbon atoms of the C6F5 unit and the bromide (C⋯Br = 3.65–4.16 Å) with a distance of 3.67 Å to the center of the arene unit.
:1 complexes of 35a, b and 36 with various anions (TBAX, X = Cl−, Br−, I−, BF4−, PF6−) in CDCl3 or CD3CN41
|
|
|
|
|
|---|---|---|---|
| Cl− | 237 | 163 | 3345 |
| Br− | 173 | 114 | 637 |
| I− | 99 | 82 | 182 |
| BF4− | 135 | 102 | — |
| PF6− | 89 | 74 | — |
 | ||
| Fig. 18 Anion–π complex of pentafluorobenzamide (37) as observed in the solid state structure of the cocrystal of pentafluorobenzamide with TBABr (TBA cation removed for clarity).41 | ||
Closely related 1,3-bis(pentafluorophenylimino)-isoindoline (38) and 3,6-di-tert-butyl-1,8-bis(pentafluoro-phenyl)-9H-carbazole (39) (Fig. 19)42 were designed to trap the anion in-between the two pentafluorophenyl rings by simultaneous NH-to-anion binding. The shift of the signals in the 1H-NMR confirm the binding of halide anions by 38 and 39 in solution. Only the carbazole 39 yielded reliable titration data in different solvents (CDCl2, CD3CN, C6D6 and THF-d8).42 The association constants were found to be in the range of 2.8 to 500 M−1, which is somewhat lower than previously reported for a related system by us.41 However, qualitatively the trend for the association constants following the order Cl− > Br− > I− was confirmed and also the superiority of the perfluoro over the simple phenyl system was observed. Accompanying computational studies suggest a significant contribution of anion–π interactions in the anion binding, this was also supported by the crystal structures of 38·Cl and 382·Br (Fig. 19).42
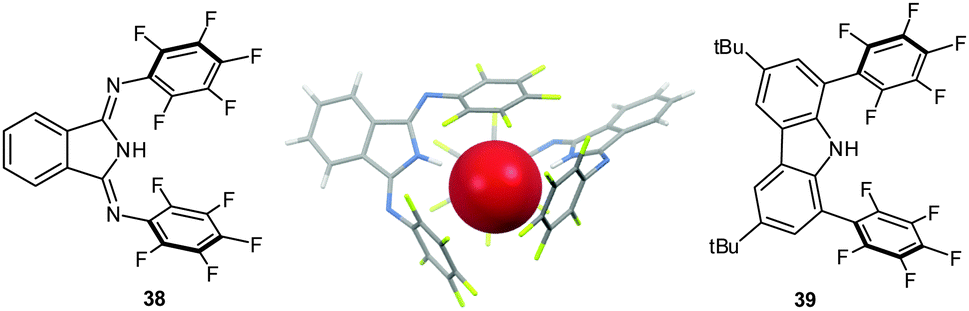 | ||
| Fig. 19 Molecular structures of the 1,3-bis(pentafluorophenylimino)isoindoline (38) and 3,6-di-tert-butyl-1,8-bis(pentafluoro-phenyl)-9H-carbazole (39) as well as representative crystal structure of 382·Br (co-crystallized solvent molecules and counter cations are omitted for clarity).42 | ||
Taylor and his group have been very active in quantifying halogen bonding to anions in solution and also performed a series of experiments giving first insights in the strength of anion–π interactions. For their studies they chose urea-based receptor capable in trapping anions by a combination of hydrogen and halogen bonds and anion–π interactions (Fig. 20).43 The differences in the associating energies of receptor 40a with two attached pentafluorophenyl groups and the benzoate 40b were determined in acetonitrile showing a slightly more stronger interaction between chloride and bromide with 40a over 40b. Interestingly, the titration with benzoate and tosylate anions revealed a more clear difference between the two receptors, which was attributed to the stronger interactions of the oxoanions with the arene and auxiliary π–π interactions.
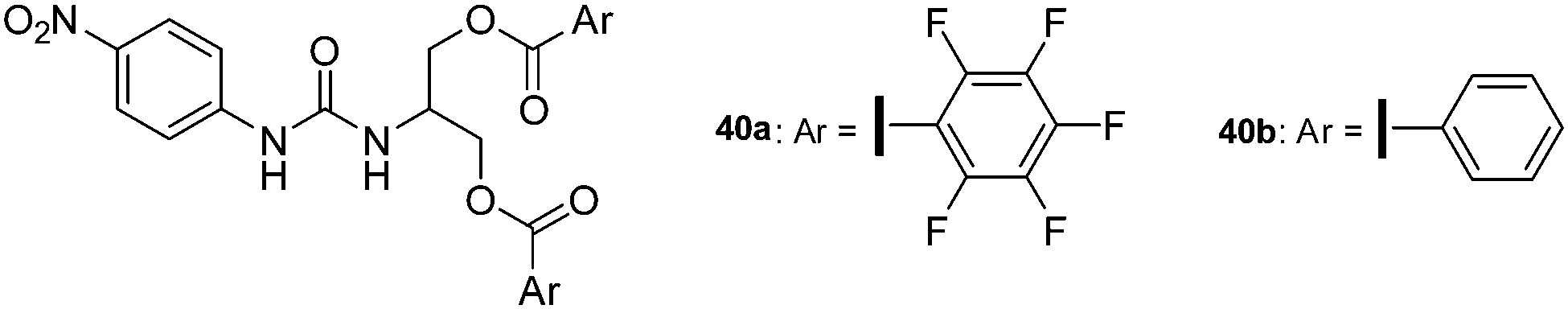 | ||
| Fig. 20 Molecular structure of the urea-based receptors as used by Taylor for studying anion–π and halogen bonding in solution.43 | ||
Wang and coworkers reported a macrocyclic receptor system based on tetraoxacalix[2]arene[2]-triazine decorated with pentafluorophenyl groups (41a and 41b) (Fig. 21).44 Fluorescence titration experiments in acetonitrile yielded association affinities of 3500–1300 M−1 for the addition of TBAF and TBAN3 salts. The obtained results were determined by following the changes of the emission bands at 322 and 365 nm and were in agreement with earlier findings.45
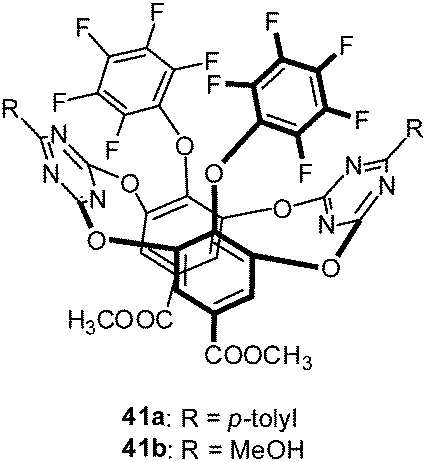 | ||
| Fig. 21 Molecular structure of the tetraoxacalix[2]arene[2]-triazine receptor as employed by Li et al. in their solution studies. | ||
The Ballester group performed comprehensive studies on anion–π interactions by employing calix[4]pyrroles-based receptors and added highly relevant new information about the anion–π interactions in solution.46 The two (42) or four-walled receptors (43) (Fig. 22) were used to quantify anion–π interactions in solution. A series of crystal structures of the substituted calix[4]pyrroles with tetraalkylammonium salts confirmed the efficient anion trapping in their receptors. The anions are fixed by the NH-groups in the centre of the macrocycle close to the electron-deficient arenes. The two walled system with pentafluorophenyl attached to the calix[4]pyrroles with various halides showed close contacts to the electron-deficient arenes ranging from 3.90–4.12 Å and also the four-walled receptor with p-nitrophenyl moieties with chloride showed the anion to be located in the centre of the receptor between the paneling arenes. However, due to the long distances between the anion and the carbon-atoms of the arenes (4.10–4.39 Å) they were not considered as anion–π interactions.
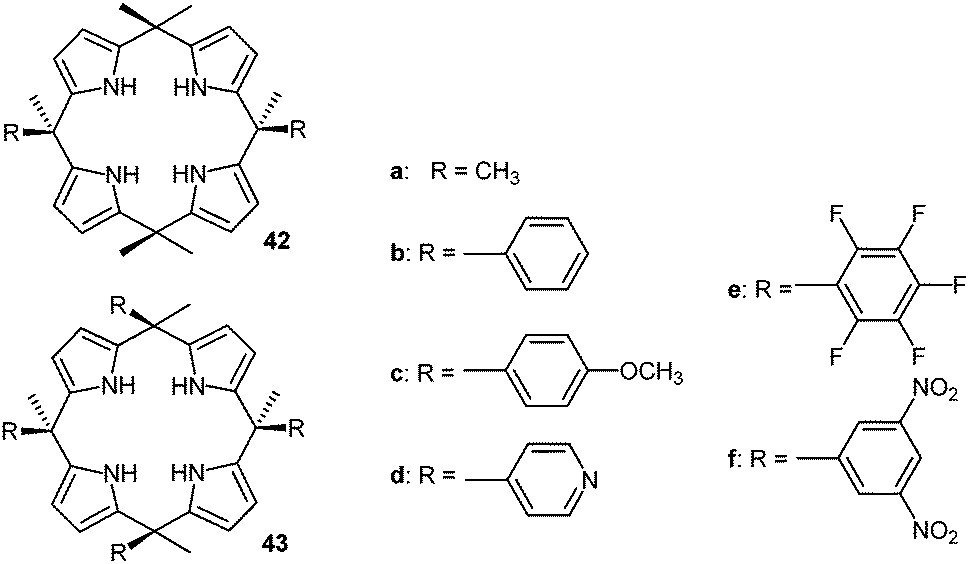 | ||
| Fig. 22 Molecular structures of the calix[4]pyrrole-based anion receptors as used by Ballester's group for their quantification of anion–π interactions in solution.46 | ||
The association constants of the anion–receptor complexes were obtained by 1H-NMR and isothermal calorimetry (ITC). The “two-walled” systems showed an increasing attraction to the anions with increasing electron-withdrawing character of the substituents on the aromatic unit. By comparing their results to the association energies of octamethylcalix-[4]pyrrole (42a) the anion–π contribution to the total interaction energy was estimated. The free Gibbs energies indicated that only systems with electron-deficient arenes such as the dinitrophenyl or pentafluorophenyl interact attractively with anions. The binding strengths for chloride and bromide are almost the same (−0.6 to −0.8 kcal mol−1), while iodide showed slightly increased binding ability (−1.0 kcal mol−1), which was explained by the increased size and polarizability of the anion.
Since the thermodynamic data showed that the enthalpy is almost constant for the receptor–anion complexes in acetonitrile, entropy was suspected to generate the differences in the Gibbs free energies.46b However, the opposite was observed for weakly solvating/coordinating/interacting solvents such as chloroform. In this case changes in the Gibbs free energy are mainly attributed to enthalpy.
According to the findings of Ballester and coworkers the anion–π interactions are weakly attractive in polar as well as in non-polar solvents (−0.7 to −1.0 kcal mol−1).35a The authors stated that the free binding energies were obtained by comparison with the octamethyl calix[4]pyrrole possessing itself CH⋯anion interactions and therefore the anion–π contribution to the total interaction energy might be slightly underestimated.5b,46b,47
The relevance of anion–π interactions in functional materials
So far we have summarized the theoretical and experimental evidence for the existence of anion–π interactions. Traditionally Supramolecular Chemistry has been focusing on concepts and proof of principles:1,25,48 Nowadays the research in this field is focused on efficient approaches to novel functional materials.2c,49 Recent studies in the field of anion–π interactions clearly demonstrate the potential in the utilization of this weak intermolecular force as an important feature in novel functional materials for potential applications such as anion sensing,19b,50 ion transport10a,e,46a,b and catalysis.11,51 Although nucleophilic aromatic substitution involve the reaction of electron-deficient arenes with anionic species and therefore the formation of a potential anion–π complex is reasonable. However, since this aspect of anion–π interactions is not studied in detail so far it will not be discussed in the following.Previous chapters have already introduced a number of anion receptor systems for the quantification of the strength of anion–π interactions and some of these studies already manifested efficient anion binding and some selectivity. However, the following chapter will summarize the more sophisticated anion receptors and their use in ion transport. Furthermore, anion–π interactions have been recognized in the field of biochemistry and a number of publications suggest their relevance in biochemical processes.6,52
Anion recognition, sensing and transport
An important field with increasing interest in anion–π interactions is the anion recognition and sensing.53 Several concepts have been available for the design of anion receptors which employ the classical non-covalent interactions such as coulombic attractions (ion pairing) and Lewis acid–base pairing (metal coordination), dipole–anion interactions, and hydrogen bonding. However, most recent anion binding research has focused on the utilization of the less established interactions like halogen bonding,4a–c,54 CH–anion or anion–π interactions.5a,c,9,55 The goal is to optimize the existing anion receptor systems towards higher selectivity and stimuli-control by taking benefit from the cooperative interplay of various non-covalent forces.Mascal et al. reported an anion receptor cage based on triazine linked by trialkylamines, which shows high selectivity for fluoride ions.19b,50a By protonation of the amine linkers the receptor becomes an ideal host for binding anions by combination of charge-assisted hydrogen bonds and anion–π interactions. The selectivity for fluoride anions was attributed to the tight steric fit of the anion between the two electron-deficient arenes. Complexation studies for chloride with the host system 44 by electrospray mass spectrometry revealed no complexation. The crystal structure of the host–guest complex confirmed the binding mode predicted by theoretical calculations and the complexation studies, whereas the fluoride anion is fixed by strong charge-assisted hydrogen bonds in the centre of the cavity in between the triazine units (Fig. 23).
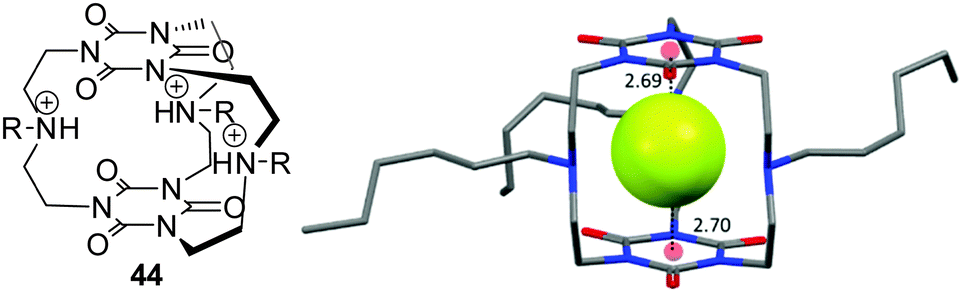 | ||
| Fig. 23 Molecular Structure and crystal structure of the anion receptor cage as reported by Mascal et al. The crystal structure demonstrates the selectivity for fluoride anions by size fitting between the two triazine units.19b,50a | ||
A highly selective nitrate receptor was recently reported by Johnson and coworkers (Fig. 24).50b The 1H-NMR studies were performed and showed decreasing anion affinities for the fluorinated receptor 45a in the order of NO3− > Cl− > Br− > I−. Remarkably, the nitrate selectivity is lost in absence of the fluorine atoms (45b), which was attributed to attractive interactions between the nitrate and the central trifluorophenyl group. The accompanying computational study suggests nitrate binding by the urea–hydrogen bonds for both receptors 45a and 45b. The presence of the fluorine atoms at the central unit enables additional anion–π interactions to the nitrate causing the higher association constant. For 45b anion binding occurs exclusively by NH-to-anion hydrogen bonds and is therefore more selective for the more Lewis basic chloride anion.
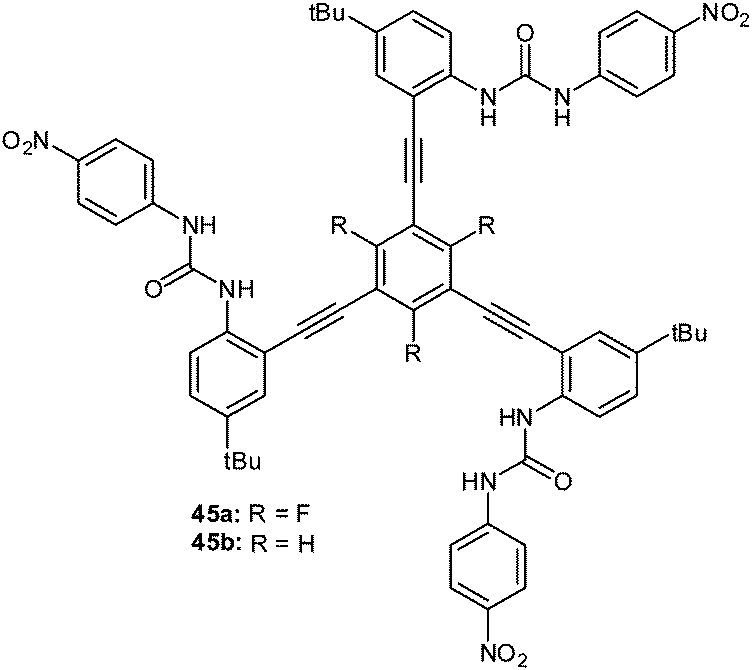 | ||
| Fig. 24 Molecular structures of the tripodal urea receptors 45a and b employed by Watt et al. for selective binding of nitrate.50b | ||
Another remarkable example for the involvement of anion–π interactions in the sensing of fluoride anions in solution was reported by Saha et al.50c,56 They observed the electron transfer processes from certain anions to NDIs (naphthalene diimides) via either thermal or photochemical pathways. In both cases an anion-radical NDI (NDI˙−) is produced via, as proposed by the authors, an anion–π stabilized intermediate (Fig. 25). By UV-Vis spectroscopy applying the Benesi–Hildebrand method 1:1 complexes of fluoride and acetate were found for NDI 46 with decreasing association constants of 1225 and 70 M−1 in ortho-dichlorobenzene (ODCB). The 1H-NMR studies gave evidence that the thermal electron transfer process is turned off in more polar solvents such as acetonitrile. While the radical anion of 46 is formed in the presence of fluoride and acetate in acetonitrile, with chloride does not occur. In contrast, irradiation of the same sample generated the 46˙− ion. Less basic anions such as bromide or iodide did not produce 46˙− even under irradiation. Due to the colorimetric response of the receptor system, the authors propose the use of the NDIs as fluoride sensors and demonstrated it by detecting fluoride from commercially available toothpaste.
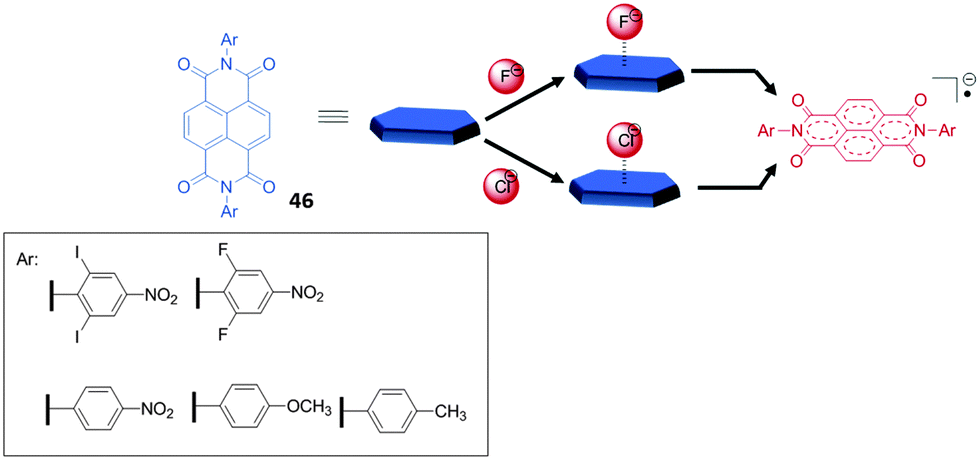 | ||
| Fig. 25 Molecular structures of the NDI systems (46) involved in the thermal (upper path way) and the photochemical (lower pathway) electron transfer processes with anions.50c | ||
Another remarkable study on anion–π interactions in the gas phase was reported by Matile, Schalley and coworkers.2a Their electrospray ionization Fourier-transform ion cyclotron resonance tandem mass spectrometric (FT-ICR) studies showed anion–π complexes of naphthalene diimides (Fig. 26) with different anions sprayed from solutions of acetonitrile. All NDI derivatives 47–51 showed stable complexes with chloride, bromide and nitrate in the gas phase. Additionally, the macrocyclic NDI 51 revealed complexes with iodide, dihydrogenphosphate, triflate and chlorate. Competition experiments supported by computational studies revealed decreasing complex stability in the order of 50 > 49 > 48 > 47 suggesting a loss of attraction between anion and the NDI with decreasing π-acidity of the arene.
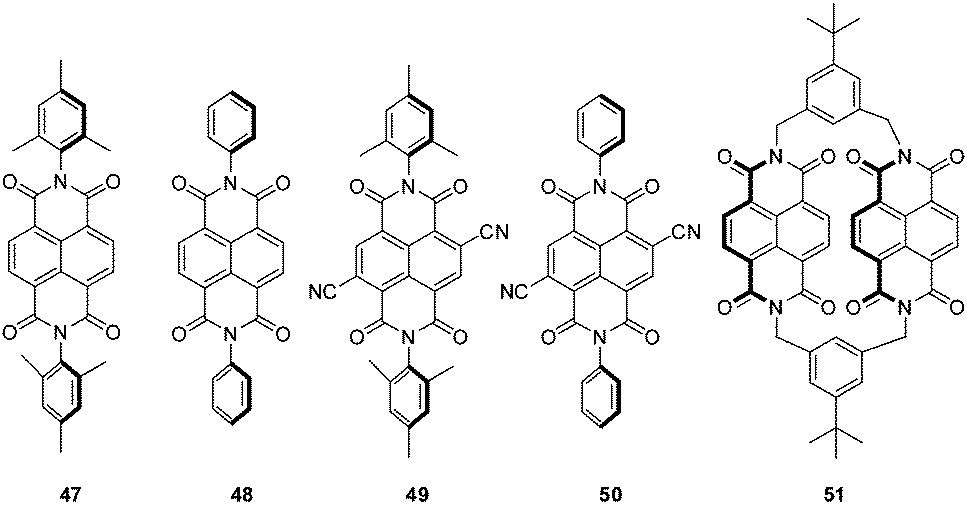 | ||
| Fig. 26 NDI derivatives investigated in the study of Matile, Schalley and coworkers.2a | ||
Matile and coworkers were able to put the NDI-system to work in a biomimetic application.2a,10e,57 Selective ion transport is of crucial relevance in biological systems.57f,58 In this context, anions are usually transported by hydrogen bonds, ion pairing or dipole interactions through the transport channels. So far an anion transport supported by anion–π interactions was not observed in natural systems. Matile's group was able to prove the selective transmembrane anion transport by utilizing NDIs. The challenge in these processes is that too tight binding of anions hinders the movement of the anions, while too weak interactions prevent the transport. Nature overcomes this problem by multiple cooperative binding sites leading to an “ion hopping” in the channels.10e,57a,b,d,e,59 In order to employ the anion–π interactions, which are weak enough to guarantee ion mobility, multiple electron-deficient π-systems have to be available along the channel for an efficient ion transport to be mediated by anion–π binding. Therefore, Matile and coworkers designed a series of shape-persistent oligonaphthalenediimides (Fig. 27) and proved their suitability for chloride-selective transmembrane transport by multi-ion hopping. They showed that the design of the end-groups is essential for the functionality of the “anion–π slides”. The end-group needs to ensure the self-assembly of the NDIs into bundles which are intercalated in the membranes of the lipid bilayers to be active in the ion transport.
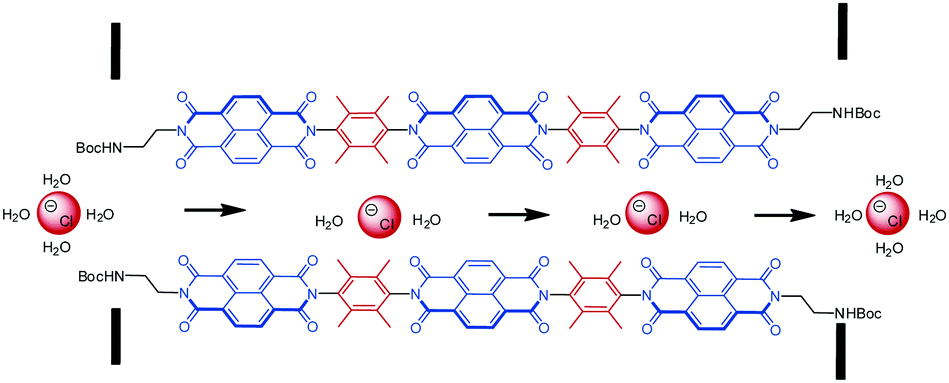 | ||
| Fig. 27 Schematic representation of the NDI-based anion–π slide for transmembranal anion transport. | ||
An extended study of the same group introduced sulfur redox chemistry into the NDIs showing an enhanced π-acidity.60 Oxidation of the thioether substituted NDI 52 by meta-chloroperbenzoic acid (mCPBA) leads to the chiral sulfoxide 53, which might be useful in anion–π actuated asymmetric catalysis. Further oxidation to the corresponding tetrasulfone 54 yielded the most π-acidic compound reported so far as proven by optoelectronic properties and computational studies (Fig. 28).60
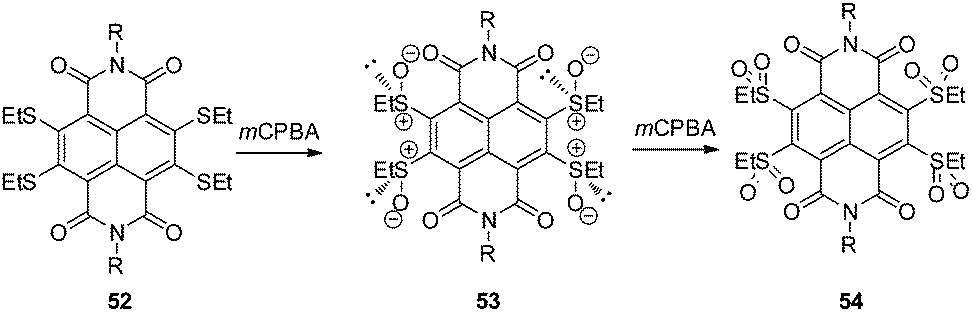 | ||
| Fig. 28 Molecular structures of the thioether, sulfoxide and sulfone substituted NDIs as reported by Matile and coworkers.60 | ||
In respect to functional materials a NDI-prism 55 was reported by Stoddart et al. (Fig. 29).61 Experiment as well as theoretical calculations revealed that the through-space orbital interactions and electron sharing phenomena of the triangular NDI prisms is the cause of the redox-activity in the system. The electronic communication between the naphthalenediimide units provides the system an unusually large number of redox states, which are individually accessible and therefore are of interest for potential applications in the field of molecular electronics.62 The three NDIs create an electron deficient cavity, which is able to host negatively charged species and indeed the authors were able to obtain a crystal structure showing the encapsulation of linear triiodide within the trigonal prism. Remarkably, the inclusion of triiodide induces π–π stacking of the chiral prisms into supramolecular helices.
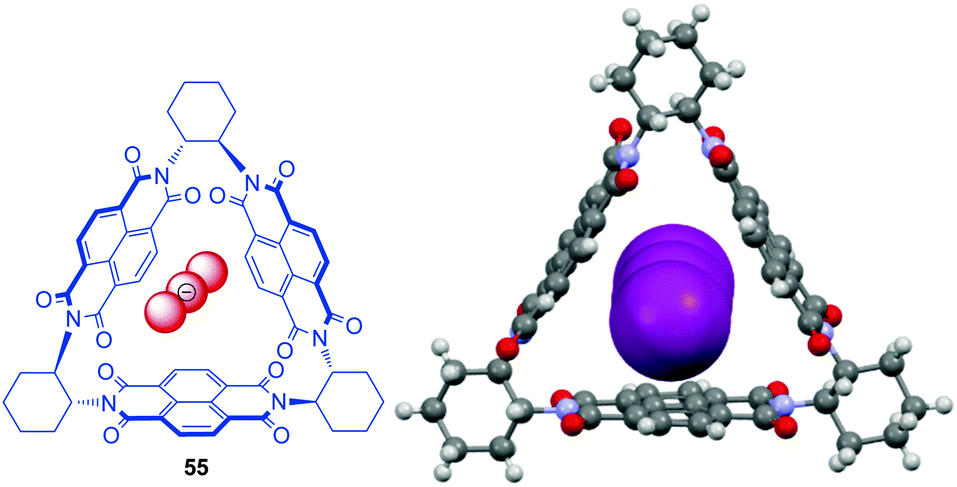 | ||
| Fig. 29 Molecular structure and crystal structure of Stoddart's triangular NDI prism 55 demonstrating the encapsulation of the triiodide anions (counter ions and solvent molecules omitted for clarity).61 | ||
In 2010, we discovered that anion–π interactions can be involved in the stabilization of rather unstable anionic species.63 By serendipity the crystal structure of tetraiodide dianion I42− was obtained by oxidation from the corresponding iodide of the bi-substituted phosphonium salt. This anionic species is rarely found in the solid state and DFT calculation suggest a high reactivity and tendency to degradation.63 However, in the solid state structure of 56, the dianionic charge transfer complex of the I42− is trapped in a channel by CH–anion interactions, whereas the negatively charged ends showed anion–π interactions to the pentafluorophenyl caps (C⋯I = 3.93–4.13 Å, Fig. 30). Supported by theoretical studies the observed anion–π interactions are envisaged to stabilize highly reactive anions or intermediates. Thus based on this stabilizing effect of anion–π interactions we were able to synthesize a series of ammonium and phosphonium salts encapsulating the sensitive tetraiodide dianion simply by mixing the corresponding iodide salts with iodine.64
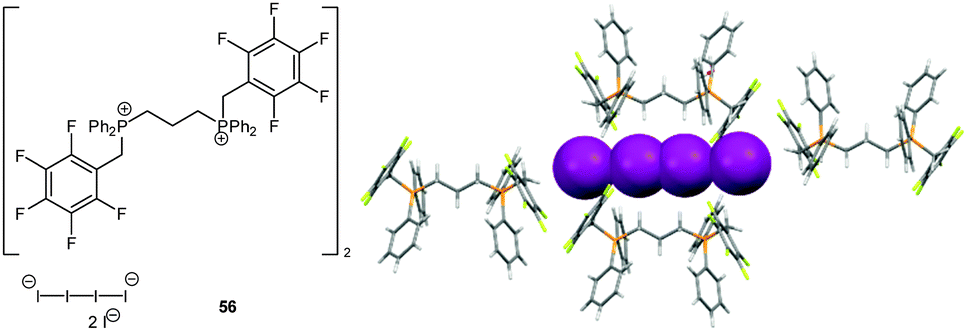 | ||
| Fig. 30 Molecular structure and representative part of the crystal structure of 56 showing the unstable I42− anion stabilized by CH–anion and anion–π interactions. | ||
In 2015, Stoddart and his group reported the synthesis of a macrobicyclic cyclophane derivative 57 (Fig. 31), denoted as blue cage6+.65 The cage was synthesized by the addition of phenanthrene as a template and possesses a remarkably electron-deficient cavity consisting of six pyridinium rings fused with two central triazines bridged by three para-xylene units. The blue cage is a strong receptor for polycylic arenes and anions. The binding of the guest species by anion–π as well as π–π interactions is further supported by the obtained crystal structures.
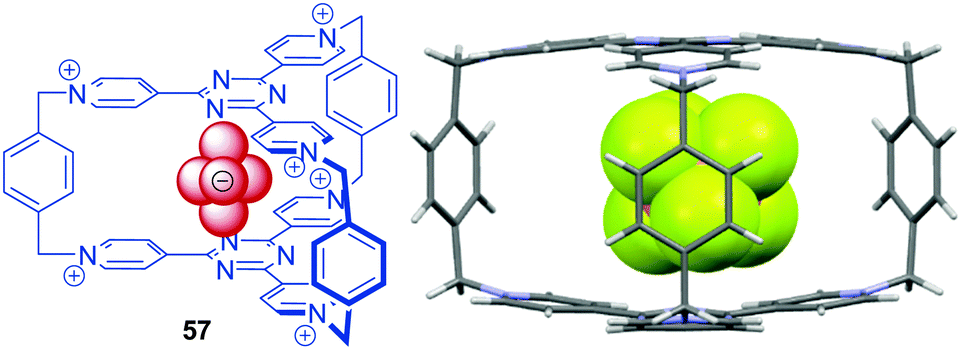 | ||
| Fig. 31 Molecular structure and solid state structure of the Blue-Cage6+ encapsulating an PF6− anion as reported by Stoddart and co-workers (disordered anions and solvent molecules omitted for clarity).65 | ||
A series of ditopic calix[4]arene receptors 58a–f (Fig. 32) were reported by Metrangelo, Resnati, Matile and coworkers investigating ion transport by the use of anion–π interactions, hydrogen or halogen bonding.10a Remarkably, for receptor 58a, where anion binding is expected to be dominated by anion–π interactions, the highest transport activity was found. The participation of anion–π interactions in the transport of anions by 58a was confirmed by theoretical calculations. The transport activity significantly decreased for 58b and the hydrogen/halogen bonded complexes 58c–f showed an intermediate activity. Detailed NMR studies in methanol-d4 revealed 1:1 complexes of 58b and 58a with chloride. Moderate association constants of 55 M−1 were found for 58b, while no detectable interactions occurred under comparable conditions with 58a. Thus, the weaker binding affinity of 58a is crucial for the more efficient anion transport, due to a higher mobility of the ions. These results represents the first example that weak non-covalent interactions such as anion–π can be of importance in biological systems.
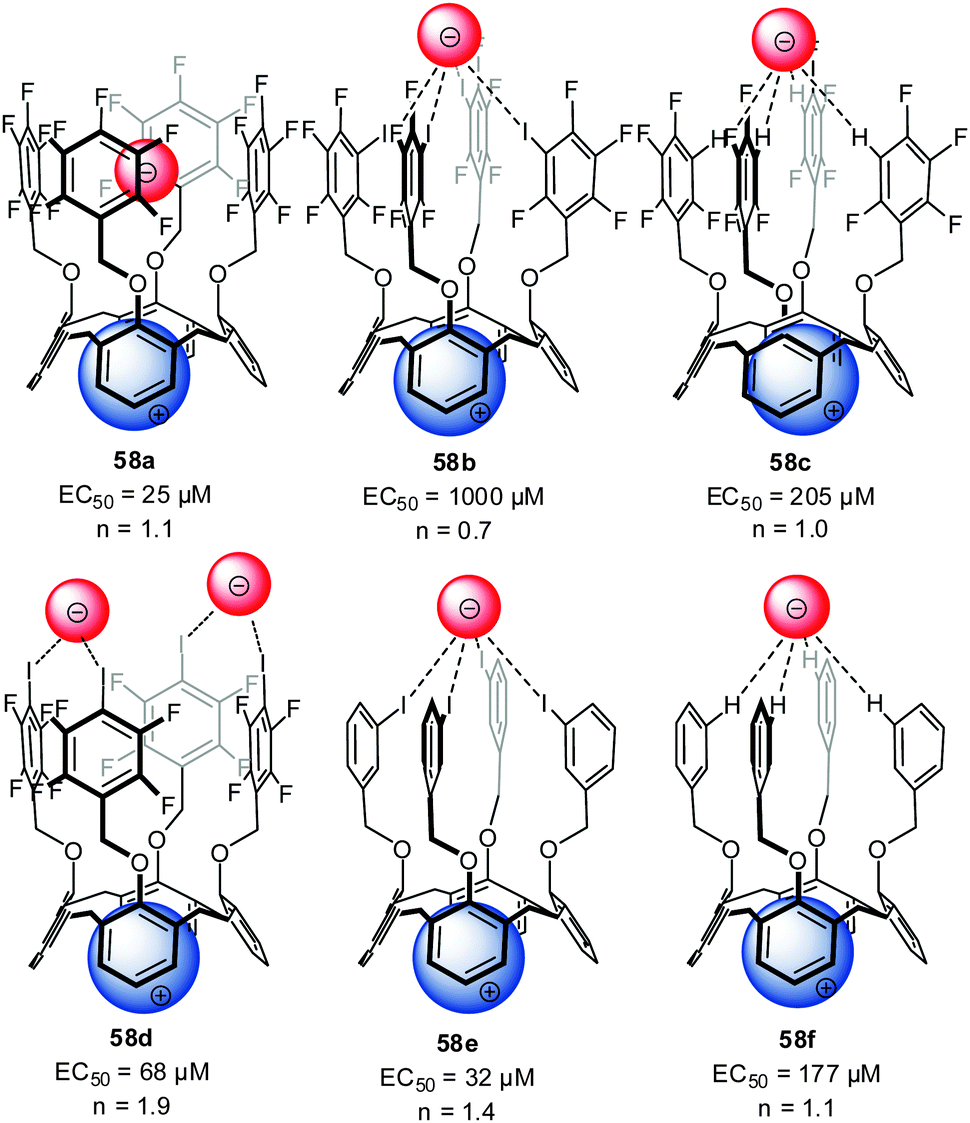 | ||
| Fig. 32 Molecular structure of the investigated ion pair receptors 58 for transport studies by Metrangolo, Resnati and Matile et al.10a | ||
The Wang group investigated anion–π interactions of a series of macrocyclic calix[2]arene[2]triazine receptors 59 in solution.45a UV-Vis and NMR spectra did not show significant changes upon addition of chloride and bromide salts to the solution of 59. The authors used small changes in the emission band intensities to estimate the binding ability for chloride and found the highest affinity constants for the dichloro-substituted macrocycle 59. A series of crystal structures support the attractive interaction of anions with the electron-deficient triazine moiety (Fig. 33).45a
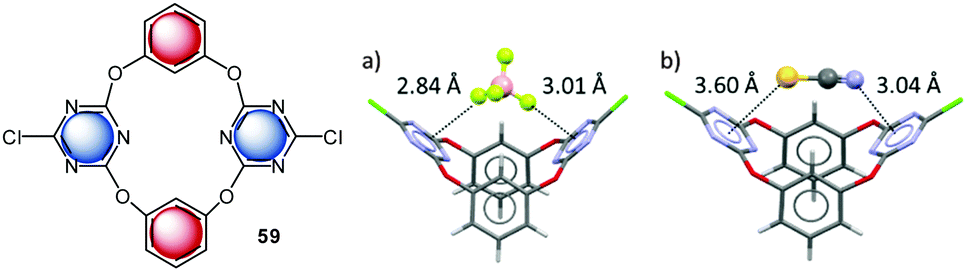 | ||
| Fig. 33 Molecular structure of the anion receptor as investigated by Wang's group. The solid state structures of the co-crystals with corresponding tetraalkyammonium salts demonstrate anion–π interactions between the anions and the electron deficient triazine moiety. | ||
Very recently the same group investigated the anion–π induced self-assembly and disassembly of anionic amphiphiles.66 Their study showed that amphiphiles such as dodecylsulphate or sodium laurate form stable 1:1 complexes with tetracalix[2]arene[2]chlorotriazine by anion–π interactions, which induces the self-assembly of vesicles in water (Fig. 34). The formation of the anion–π complexes was confirmed by HRMS, spectroscopic titrations, X-ray diffraction and DFT calculations and the morphology of the vesicles was studied by SEM, TEM and DLS. To confirm the functional relevance of anion–π interactions in this systems the authors performed control experiments by adding sodium nitrate to the vesicles in aqueous solution. As proven by SEM images the morphology of the vesicles changes dramatically after the addition of three equivalents of sodium nitrate (Fig. 34).
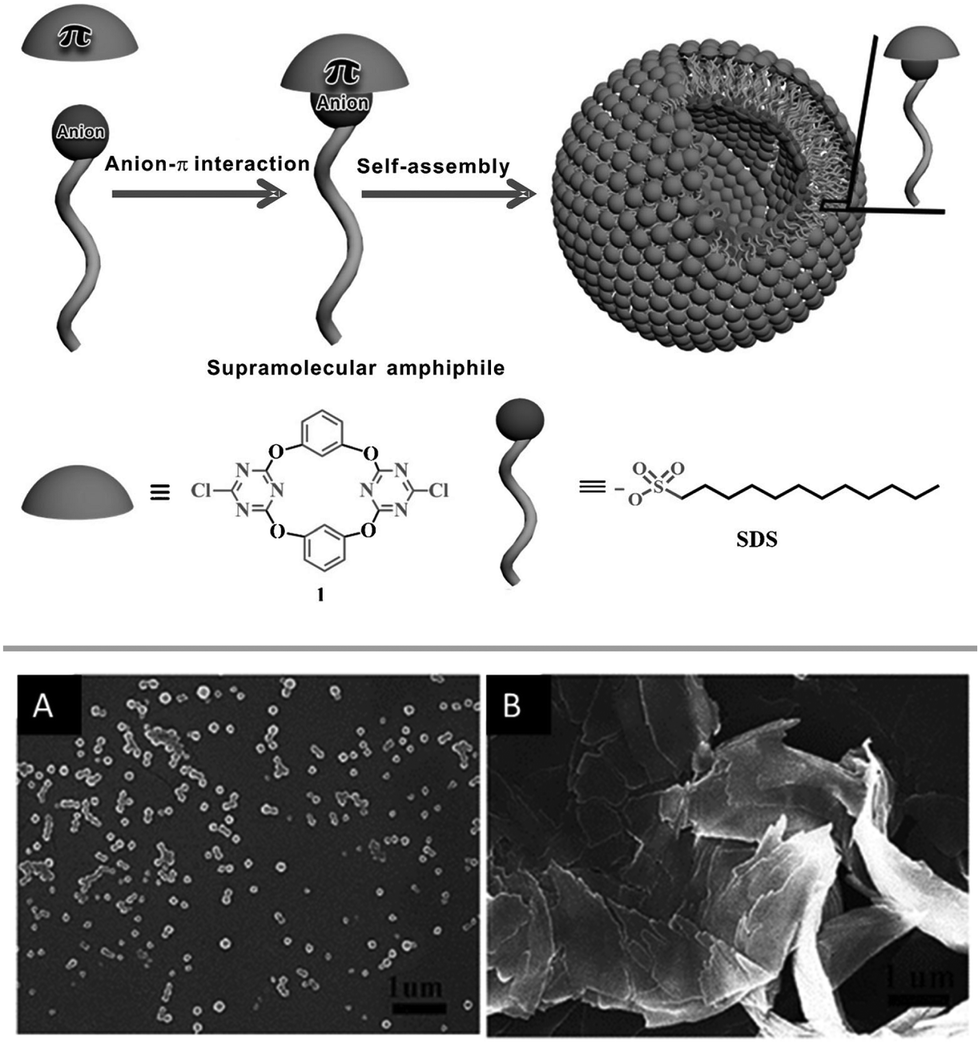 | ||
| Fig. 34 Schematic representation of the anion–π induced self-assembly forming the vesicle as well as SEM images before (A) and after addition of 3 eq. of NaNO3 (B). Reproduced with permission from John Wiley and Sons.66 | ||
Another interesting study on the contribution of anion–π binding in self-assembly processes was reported in 2014.10c A series of new amphiphilic anion receptors based on tetraoxacalix[2]arene[2]triazine (60) was investigated in respect to its self-assembling into stable vesicles in a mixture of THF and water (Fig. 35). The surface of the vesicles bears electron-deficient cavities, which interact with anions. Dynamic light scattering data revealed the size-regulation of the assembled vesicles by anions, increasing in the order of F− < ClO4− < SCN− < BF4− < Br− < Cl− < NO3−. The observed selectivity is in line with the complex stabilities of the tetraoxacalix[2]arene[2]triazine anion complexes. The authors suggest a significant contribution of anion–π interaction to the observed binding behavior. In addition, the chloride transport across the membrane of the vesicles was investigated by fluorescence experiments, demonstrating the use of the heterocalixarenes as novel models for functional vesicles 1–2.
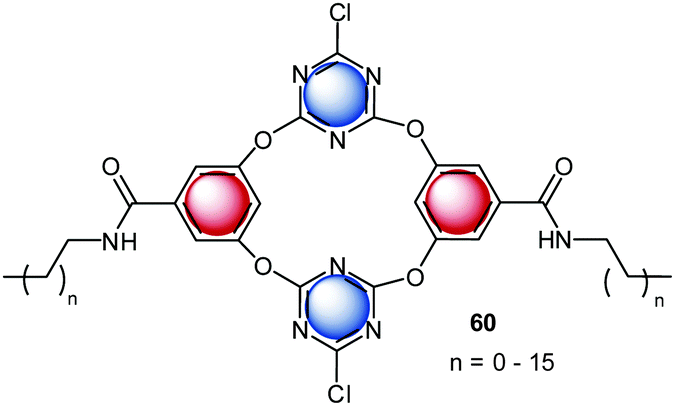 | ||
| Fig. 35 Molecular Structure of the tetraoxacalix[2]arene[2]triazine amphiphiles, which form vesicles by self-assembly.10c,d | ||
Another ion transport system for nitrate ions was reported recently by Ballester et al.46a The system is based on the already described calix[4]pyrroles.57f,58,67 Nitrate anions play a crucial role in biological systems and nature has developed efficient methods to capture or discard/expel nitrate.68 Ballester's group demonstrated that calix[4]pyrroles67a,b with electron-deficient arenes effectively transport ions across lipid membranes, where the anion–π interactions are expected to have of crucial role. Their study indicated that the “two-wall” aryl extended calix[4]pyrroles are active in ion transport, whereas the activity could be related to the electronic nature of the “aromatic walls”. The best transporter system, bearing the mono- and dinitro-substituted arenes, showed high selectivity for nitrate, which was attributed to complementary geometry of the calix[4]pyrrole system and the nitrate anion. The presence of the anion–π interactions in their systems was also confirmed by the crystal structure of 42fNO3−. The anion is fixed by four NH–anion interactions to one of the oxygen atoms of the nitrate (Fig. 36) and the two remaining oxygen atoms interact with the electron-deficient arenes via anion–π interactions.
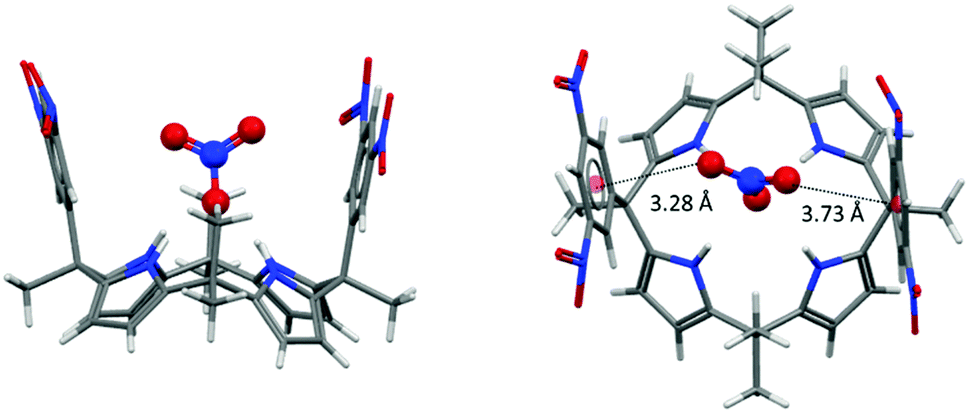 | ||
| Fig. 36 Crystal structure of the calix[4]pyrrole nitrate complex showing the interactions of the nitrate–oxygen atoms with the dinitrophenyl groups of 42f.46a,b | ||
Catalysis
While cation–π interactions69 are well established in catalysis and organocatalysis70 the use of the complementary anion–π interaction in catalysis is rarely described.11a,c However, recent examples of ion-pair catalysis demonstrates the significance of these moderately attractive intermolecular forces in synthesis.71 The following chapter summarizes the recent advances in anion–π enhanced catalysis.The first experimental evidence of the relevance of anion–π interactions in catalytic reactions was reported by Zhao et al.11a They studied the Kemp's elimination supported by π-acidic naphthalene diimides (Fig. 37). The investigated systems fix the substrate by CH–anion and π–π-interactions above the electron-deficient NDI, which is able to stabilize the anionic transition state. Experimental and theoretical evidence was found that by increasing the π-acidity of the NDI results in a more efficient stabilization of the transition state.11c This pioneering work introduced a new strategy for organo catalysis by employing anion–π interactions. Very recently, the same group was able to provide experimental evidence for an asymmetric version of the anion–π catalysis with related systems.51a
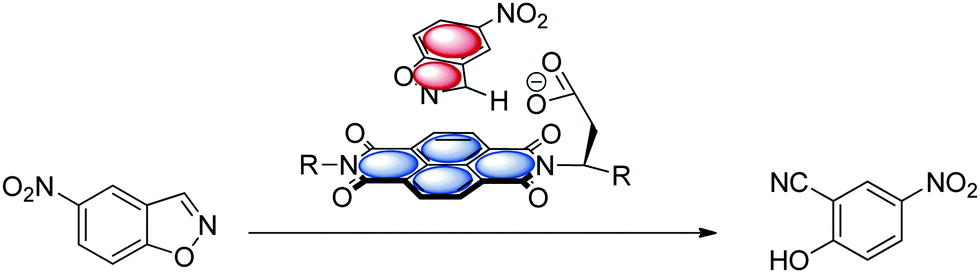 | ||
| Fig. 37 Schematic representation of the anion–π supported Kemp's elimination as reported by Zhao et al.11a,c | ||
Another important study was reported by the same group employing anion–π interactions in enolate chemistry (Fig. 38).11b,51b,72 The NMR studies have proven that the acidity of a malonate unit covalently bound to NDI shows a significant up-field shift compared to the free diethylmalonate, suggesting an enhanced acidity of the attached malonate group. The authors explain this effect by the stabilization of the enolate by anion–π interactions. Thorough 1H-NMR titrations revealed an enhancement of the acidity by almost two pKa units. Furthermore, the data showed that the reactions of the anion–π stabilized enolate with enones and nitroolefines have significantly stabilized transition states (up to 11.0 kJ mol−1). Since enolate chemistry is of crucial relevance in various biological and chemical processes the findings of Matile group will have a profound impact in the field of organocatalysis.
 | ||
| Fig. 38 Schematic representation of anion–π supported enolate formation as reported by Matile and coworkers.11b | ||
Biological relevance
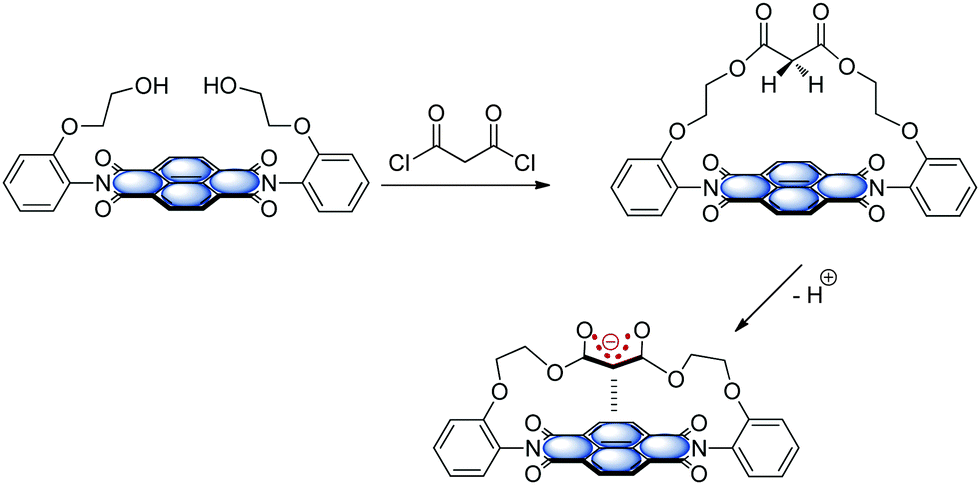
The functionality of biological systems is based on the complex interplay of intermolecular forces.48a,73 In this context, interactions involving arenes are highly relevant74 and control the processes like drug-receptor recognition, protein folding, enzyme inhibition or the π-stacked structure of DNA and RNA75 and their interaction with proteins.76 The interaction of arenes with cations is generally recognized as important and already well established.77 Recently, rather unconventional σ/π-hole interactions3 such as CH–π,78 lone pair-π,79 salt-bridge-π,80 halogen bonding4a,b and anion–π9 have gained considerable attention in the field of supramolecular chemistry and crystal engineering.A nice overview exploiting the relevance of anion–π interactions in purine and pyrimidine bases is given by Fiol et al.81 Their review focusses on anion–π interactions in purely organic systems therefore most of the points discussed therein are outside the scope of this feature article. However, the remarkable crystal structure reported by Garcia-Raso et al.82 showing rather short anion–π contacts between N6,N6′-ethylenebisadenium and chloride is discussed (Fig. 39). The chloride is fixed by two strong NH–anion interactions. The analysis of the crystalline packing shows that the chloride anions are located above the center of the six-membered ring with a distance of 3.42 Å. This crystal structure already gives a first indication of the importance of anion–π interactions in biological systems viz. in polynucleic acids or proteins. The following chapter will summarize some of the theoretical and experimental findings that suggest the importance of the interaction of arenes with anionic species for the functionality and structure of biomolecules.
 | ||
| Fig. 39 Crystal structure of N6,N6′-ethylenebisadenium chloride showing the anion–π contacts between the chloride and the electron-deficient six-membered ring of the adenium moiety.82 | ||
Indirect evidence for the rising interest in anion–π interactions in biological systems is given by the development of a software tool analyzing non-covalent interactions involving arenes in the protein data base (PDB).83 This program was already used to demonstrate the relevance of anion–π interactions in bimolecular structures.
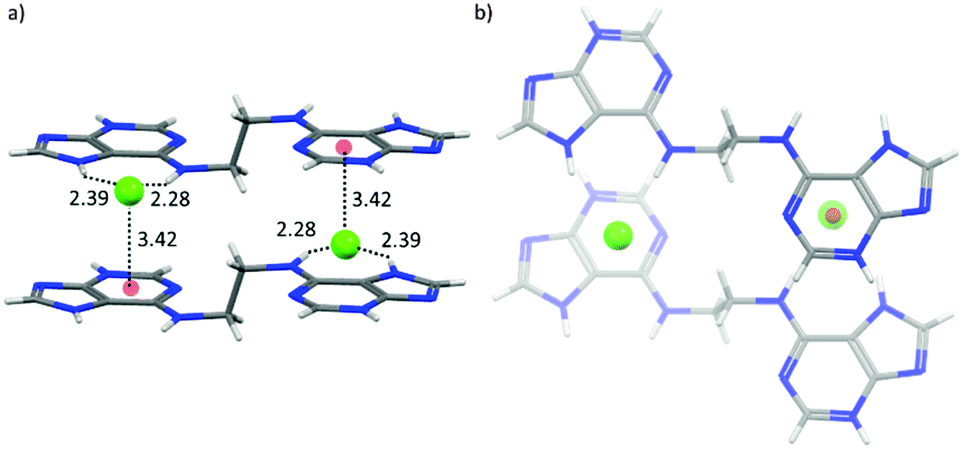
One of the first studies investigating anion–π interactions in biomolecular processes was reported by Chakravarty et al.52a They examined high-resolution structures of proteins and nucleic acids and found that anion–π interactions are crucial in the formation and recognition of biomolecular structures. The crystallographic evidence was confirmed by computational results. However, anion–π interactions occur less frequently than their counter-part cation–π interactions. Nevertheless, unambiguous evidence was found and seems to occur often in protein and nucleic acid loops. A representative example is the interaction of the phosphate in the nucleic acid backbone with the guanine-nucleotide as shown (Fig. 40). One of the oxygens of the phosphate group showed distinct anion–π contacts to the nucleotide, while the other oxygen atoms of the phosphate backbone face outwards. This binding motif was found in many RNA hairpins indicating that anion–π interactions might be a feature of this structural arrangement of nucleic acid loops.52a
 | ||
| Fig. 40 Features of the “η6”-type anion–π contacts as observed in RNA loops of Eschirichia coli 4.5S (right) or the ribozyme stem C (left). Reproduced and adapted with permissions from Elsevier.52a | ||
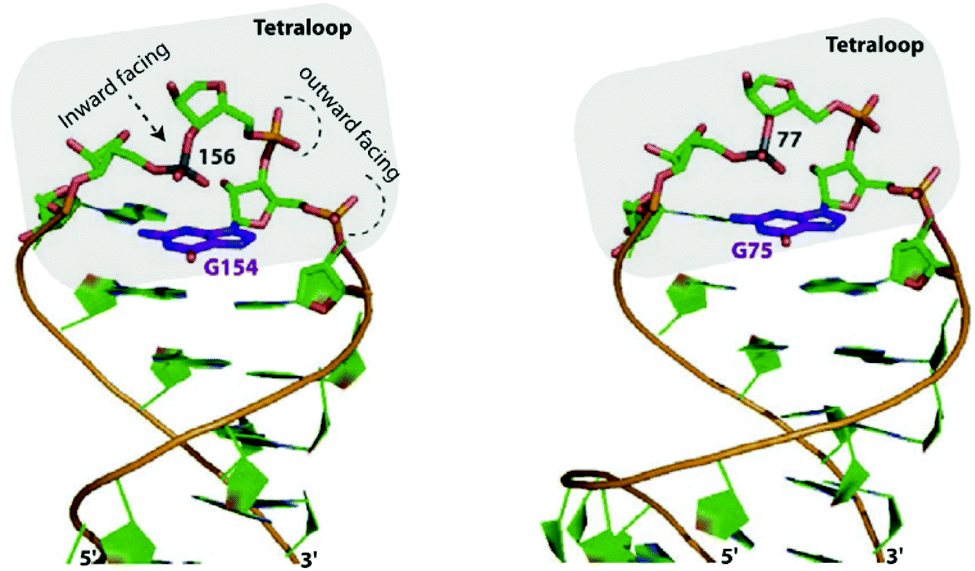
In 2011, Estarellas et al. investigated the relevance of anion–π interactions in the active site of the urate oxidase enzyme.6a This enzyme is inhibited by cyanide ions. Cyanide anions bound in the active site of the enzyme showed strong interactions between the anion and the electron-deficient uric acid moiety. Computational results support the attractive nature of the anion–π contacts as found in the crystal structure of the enzyme and furthermore suggest a cooperative effect between the π–π stacking of phenylalanine residue PHE159 and the analyzed anion–π interaction. The authors state that this study is the first showing the crucial relevance of anion–π interactions in the inhibition of an enzyme (Fig. 41).
 | ||
| Fig. 41 Representation view demonstrating anion–π interactions between the uric acid and cyanide (left) or chloride (right) as observed in the crystal structures of the active site of urate oxidase. Reproduced and adapted with permissions from John Wiley and Sons.6a | ||
Estarellas et al. also investigated anion–π interactions in flavoproteins.6b A structural survey of the PDB yielded a significant number of hits exhibiting anion–π contacts. Since many of these structures are reductases and show high affinities to biologically relevant anions such as acetate, chloride and thiocyanate, the authors propose a high relevance of anion–π interactions for the functionality of these enzymes. A representative example is found in the crystal structure of the flavine mononucleotide (FMN) dependent nitroreductase 1ylU.84 The acetate-inhibited form of the oxidized enzyme revealed an anion located in the active site interacting by a bifurcated hydrogen bond, a CH–π and an anion–π interaction with the FMN enzyme complex. The distance between the anion and the pyrimidinic ring of the FMN were found to be 3.0 Å, which indicates strong interactions (Fig. 42).
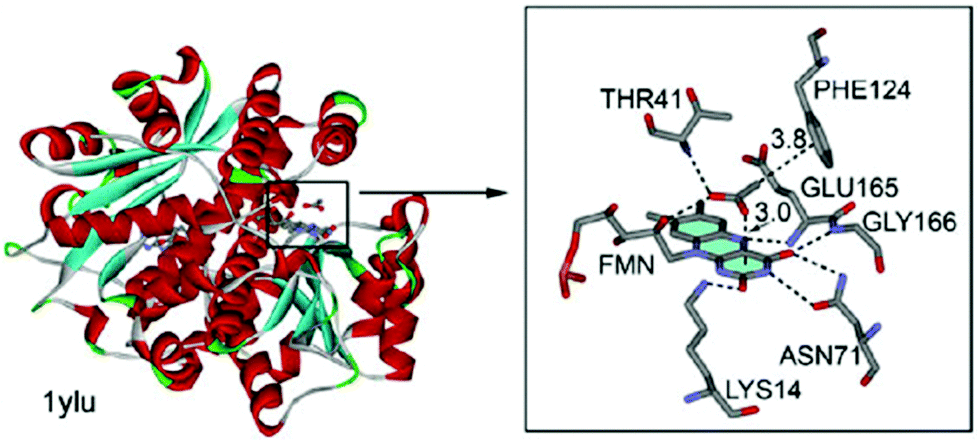 | ||
| Fig. 42 Crystal structure of FMN dependent nitroreductase 1ylU showing the anion–π contact between the acetate and the FMN unit. Reproduced and adapted with permissions from John Wiley and Sons.6b | ||
In turn Frontera and coworkers investigated the relevance of anion–π interactions for the functionality of oxidoreductase. This Flavin-dependent enzyme is active in the sulfide detoxification85 and sulfide-dependent respiration and anaerobic photosynthesis.86 The discussed mechanisms explaining the activity of this enzymes involve the stabilization of an anionic intermediate.6c Bauzá et al. analyzed the X-ray structures of the enzyme and performed thorough DFT calculations. Their results indicated the participation of anion–π interactions in the stabilization of the anionic intermediate by the interaction with the π-system of the flavine adenine dinucleotide (Fig. 43).
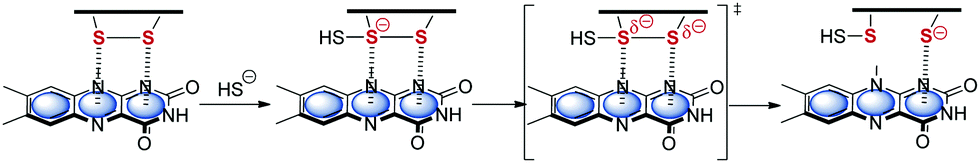 | ||
| Fig. 43 Mechanism of the addition and elimination process showing the anion–π stabilized intermediate as proposed by Bauzà et al.6c | ||
Another important study by Bauzà et al. demonstrated the long-range effect of anion–π interactions and their influence on the inhibition of the mycobacterium tuberculosis malate synthase.52b Since the enzyme plays a crucial role in the tuberculosis virulence of the mycobacterium, thus exploiting its inhibition might yield in an antitubercular therapeutics. An effective anti-tuberculosis drug based on phenyldiketo acids (PDKDA) was reported by Krieger et al.87 The crystal structure of the inhibitor complexed by the malate synthase exhibited close contacts between the carboxylate of Asp633 and the phenyl ring of the PDKA. Interestingly, no inhibition is found for the corresponding derivative with an alkyl group instead of the phenyl unit. Thus, the arene anion interaction appears of crucial importance in the inhibition process. However, the anion–π interaction between the phenyl group and the carboxylate is expected to be either very weakly attractive or repulsive, since phenyl is not electron-deficient. Computational results by Bauzá et al. suggested a long-range effect of the Mg2+ ion present in the active site, enhancing the anion–π interaction by coordinating to the PDKA (Fig. 44).
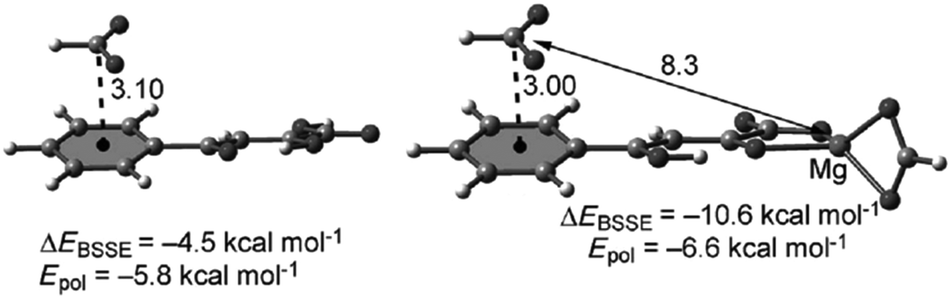 | ||
| Fig. 44 Molecular structures of the formate complexes with PKDA and interaction energies (ΔEBSSE) and the polarization contributions as calculated by Bauzà et al. (distances given in Å). Reproduced and adapted with permissions from John Wiley and Sons.52b | ||
Conclusions
The research on anion–π interactions has had many turns and surprises. While initially the counterintuitive interactions between negatively charged species and π-systems was carefully introduced by asking “Anion–π interactions. Do they exist?”7 it transformed into a well-established and comprehensively investigated intermolecular interaction. Nevertheless, there are still questions which need to be addressed, most critically those concerning the absolute quantification of anion–π interactions in solution and their functional importance. Many investigations have shed light on the strength of anion–π interaction in solution, but none of the reported systems have been able to ascribe the binding events clearly enough to exclusively show the interactions only between an anion and an electron-deficient arene. However, important progress has been made towards the employment of the anion–π interactions in functional systems as demonstrated by Ballester,46a,88 Matile2a,10e,11,60 and Wang.10c,d In addition a series of combined investigations discussed in this feature article suggest a crucial role of these weak non-covalent interactions in biochemical processes as nicely pioneered by Frontera and Deyà.6,52b Starting with these promising results the challenge is now to the design and utilization of novel materials and receptors systems by the smart use of subtle intermolecular interactions like the anion–π interaction.Acknowledgements
We kindly acknowledge the Prof. Werdelmann Stiftung (MG), the Fonds der Chemischen Industrie (MG, MA) and the Academy of Finland (KR, proj. no. 263256 and 265328) for financial support.Notes and references
- J.-M. Lehn, Supramolecular Chemistry. Concepts and Perspectives, Wiley-VCH, Weinheim, 1995 Search PubMed.
- (a) R. E. Dawson, A. Hennig, D. P. Weimann, D. Emery, V. Ravikumar, J. Montenegro, T. Takeuchi, S. Gabutti, M. Mayor, J. Mareda, C. A. Schalley and S. Matile, Nat. Chem., 2010, 2, 533 CrossRef CAS PubMed; (b) F. Meyer and P. Dubois, CrystEngComm, 2013, 15, 3058 RSC; (c) K. Liu, Y. Kang, Z. Wang and X. Zhang, Adv. Mater., 2013, 25, 5530 CrossRef CAS PubMed.
- A. Bauzá, T. J. Mooibroek and A. Frontera, ChemPhysChem, 2015, 16, 2496 CrossRef PubMed.
- (a) P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114 CrossRef CAS PubMed; (b) K. Rissanen, CrystEngComm, 2008, 10, 1107 RSC; (c) M. Erdelyi, Chem. Soc. Rev., 2012, 41, 3547 RSC; (d) G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711 CrossRef CAS.
- (a) B. L. Schottel, H. T. Chifotides and K. R. Dunbar, Chem. Soc. Rev., 2008, 37, 68 RSC; (b) A. Bauzá, P. M. Deyà and A. Frontera, in Noncovalent Forces, ed. S. Scheiner, Springer International Publishing, 2015, pp. 471–500 Search PubMed; (c) M. Giese, M. Albrecht and K. Rissanen, Chem. Rev., 2015, 115, 8867 CAS.
- (a) C. Estarellas, A. Frontera, D. Quiñonero and P. M. Deyà, Angew. Chem., Int. Ed., 2011, 50, 415 CrossRef CAS PubMed; (b) C. Estarellas, A. Frontera, D. Quiñonero and P. M. Deyà, Chem. – Asian J., 2011, 6, 2316 CrossRef CAS PubMed; (c) A. Bauzá, D. Quiñonero, P. M. Deyà and A. Frontera, Chem. – Asian J., 2013, 8, 2708 CrossRef PubMed.
- D. Quiñonero, C. Garau, C. Rotger, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, Angew. Chem., Int. Ed., 2002, 41, 3389 CrossRef.
- B. P. Hay and R. Custelcean, Cryst. Growth Des., 2009, 9, 2539 CAS.
- A. Frontera, P. Gamez, M. Mascal, T. J. Mooibroek and J. Reedijk, Angew. Chem., Int. Ed., 2011, 50, 9564 CrossRef CAS PubMed.
- (a) A. Vargas Jentzsch, D. Emery, J. Mareda, P. Metrangolo, G. Resnati and S. Matile, Angew. Chem., Int. Ed., 2011, 50, 11675 CrossRef CAS PubMed; (b) R. Dutta and P. Ghosh, Chem. Commun., 2015, 51, 9070 RSC; (c) Q. He, Y. Han, Y. Wang, Z.-T. Huang and D.-X. Wang, Chem. – Eur. J., 2014, 20, 7486 CrossRef CAS PubMed; (d) Q. He, Z.-T. Huang and D.-X. Wang, Chem. Commun., 2014, 50, 12985 RSC; (e) J. Mareda and S. Matile, Chem. – Eur. J., 2009, 15, 28 CrossRef CAS PubMed.
- (a) Y. Zhao, Y. Domoto, E. Orentas, C. Beuchat, D. Emery, J. Mareda, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2013, 52, 9940 CrossRef CAS PubMed; (b) Y. Zhao, N. Sakai and S. Matile, Nat. Commun., 2014, 5 Search PubMed; (c) Y. Zhao, C. Beuchat, Y. Domoto, J. Gajewy, A. Wilson, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2014, 136, 2101 CrossRef CAS PubMed.
- (a) K. Hiraoka, S. Mizuse and S. Yamabe, J. Chem. Phys., 1987, 86, 4102 CrossRef CAS; (b) K. Hiraoka, S. Mizuse and S. Yamabe, J. Phys. Chem., 1987, 91, 5294 CrossRef CAS.
- (a) H.-J. Schneider, Angew. Chem., Int. Ed., 1991, 30, 1417 CrossRef; (b) H.-J. Schneider, F. Werner and T. Blatter, J. Phys. Org. Chem., 1993, 6, 590 CrossRef CAS.
- M. Mascal, A. Armstrong and M. D. Bartberger, J. Am. Chem. Soc., 2002, 124, 6274 CrossRef CAS PubMed.
- I. Alkorta, I. Rozas and J. Elguero, J. Am. Chem. Soc., 2002, 124, 8593 CrossRef CAS PubMed.
- D. Quiñonero, C. Garau, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, Chem. Phys. Lett., 2002, 359, 486 CrossRef.
- (a) D. Kim, E. C. Lee, K. S. Kim and P. Tarakeshwar, J. Phys. Chem. A, 2007, 111, 7980 CrossRef CAS PubMed; (b) D. Kim, P. Tarakeshwar and K. S. Kim, J. Phys. Chem. A, 2004, 108, 1250 CrossRef CAS; (c) D. Y. Kim, N. J. Singh and K. S. Kim, J. Chem. Theory Comput., 2008, 4, 1401 CrossRef CAS PubMed; (d) D. Y. Kim, N. J. Singh, J. W. Lee and K. S. Kim, J. Chem. Theory Comput., 2008, 4, 1162 CrossRef CAS PubMed.
- (a) C. Garau, D. Quiñonero, A. Frontera, A. Costa, P. Ballester and P. M. Deyà, Chem. Phys. Lett., 2003, 370, 7 CrossRef CAS; (b) C. Garau, A. Frontera, D. Quiñonero, P. Ballester, A. Costa and P. M. Deyà, J. Phys. Chem. A, 2004, 108, 9423 CrossRef CAS.
- (a) C. Garau, A. Frontera, D. Quiñonero, P. Ballester, A. Costa and P. M. Deyà, ChemPhysChem, 2003, 4, 1344 CrossRef CAS PubMed; (b) M. Mascal, Angew. Chem., Int. Ed., 2006, 45, 2890 CrossRef CAS PubMed; (c) A. Frontera, F. Saczewski, M. Gdaniec, E. Dziemidowicz-Borys, A. Kurland, P. M. Deyà, D. Quiñonero and C. Garau, Chem. – Eur. J., 2005, 11, 6560 CrossRef CAS PubMed; (d) C. Garau, D. Quiñonero, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, J. Phys. Chem. A, 2005, 109, 9341 CrossRef CAS PubMed; (e) C. Garau, A. Frontera, P. Ballester, D. Quiñonero, A. Costa and P. M. Deyà, Eur. J. Org. Chem., 2005, 179 CrossRef CAS; (f) D. Quiñonero, C. Garau, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, J. Phys. Chem. A, 2005, 109, 4632 CrossRef PubMed; (g) C. Garau, A. Frontera, D. Quiñonero, P. Ballester, A. Costa and P. M. Deyà, Chem. Phys. Lett., 2004, 392, 85 CrossRef CAS.
- (a) B. Han, J. Lu and J. K. Kochi, Cryst. Growth Des., 2008, 8, 1327 CrossRef CAS; (b) Y. S. Rosokha, S. V. Lindeman, S. V. Rosokha and J. K. Kochi, Angew. Chem., Int. Ed., 2004, 43, 4650 CrossRef CAS PubMed.
- R. J. Gotz, A. Robertazzi, I. Mutikainen, U. Turpeinen, P. Gamez and J. Reedijk, Chem. Commun., 2008, 3384 RSC.
- M. Giese, M. Albrecht, C. Bannwarth, G. Raabe, A. Valkonen and K. Rissanen, Chem. Commun., 2011, 47, 8542 RSC.
- C. Garau, D. Quiñonero, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, Org. Lett., 2003, 5, 2227 CrossRef CAS PubMed.
- (a) M. Muller, M. Albrecht, J. Sackmann, A. Hoffmann, F. Dierkes, A. Valkonen and K. Rissanen, Dalton Trans., 2010, 39, 11329 RSC; (b) M. Giese, M. Albrecht, A. Valkonen and K. Rissanen, Eur. J. Org. Chem., 2013, 3247 CrossRef CAS; (c) M. Giese, M. Albrecht, G. Ivanova, A. Valkonen and K. Rissanen, Supramol. Chem., 2011, 24, 48 CrossRef.
- A. Bianchi, K. Bowman-James and E. Garcia, Supramolecular Chemistry of Anions, Wiley-VCH, New York, 1997 Search PubMed.
- (a) J.-Z. Liao, X.-J. Dui, H.-L. Zhang, X.-Y. Wu and C.-Z. Lu, CrystEngComm, 2014, 16, 10530 RSC; (b) J.-J. Liu, Y. Wang, Y.-J. Hong, M.-J. Lin, C.-C. Huang and W.-X. Dai, Dalton Trans., 2014, 43, 17908 RSC; (c) J.-J. Liu, Y.-F. Guan, C. Jiao, M.-J. Lin, C.-C. Huang and W.-X. Dai, Dalton Trans., 2015, 44, 5957 RSC; (d) J.-J. Liu, Y. Wang, M.-J. Lin, C.-C. Huang and W.-X. Dai, Dalton Trans., 2015, 44, 484 RSC.
- (a) O. B. Berryman, V. S. Bryantsev, D. P. Stay, D. W. Johnson and B. P. Hay, J. Am. Chem. Soc., 2006, 129, 48 CrossRef PubMed; (b) M. Albrecht, C. Wessel, M. de Groot, K. Rissanen and A. Lüchow, J. Am. Chem. Soc., 2008, 130, 4600 CrossRef CAS PubMed.
- J. Meisenheimer, Liebigs Ann. Chem., 1902, 323, 205 CrossRef CAS.
- H. Schneider, K. M. Vogelhuber, F. Schinle and J. M. Weber, J. Am. Chem. Soc., 2007, 129, 13022 CrossRef CAS PubMed.
- T. J. Mooibroek and P. Gamez, CrystEngComm, 2012, 14, 3902 RSC.
- C. Estarellas, A. Bauza, A. Frontera, D. Quinonero and P. M. Deya, Phys. Chem. Chem. Phys., 2011, 13, 5696 RSC.
- M. Giese, M. Albrecht, A. Valkonen and K. Rissanen, Chem. Sci., 2015, 6, 354 RSC.
- M. Albrecht, M. Müller, O. Mergel, K. Rissanen and A. Valkonen, Chem. – Eur. J., 2010, 16, 5062 CrossRef CAS PubMed.
- R. Custelcean, Chem. Soc. Rev., 2010, 39, 3675 RSC.
- (a) P. Arranz-Mascarós, C. Bazzicalupi, A. Bianchi, C. Giorgi, M.-L. Godino-Salido, M.-D. Gutiérrez-Valero, R. Lopez-Garzón and M. Savastano, J. Am. Chem. Soc., 2012, 135, 102 CrossRef PubMed; (b) P. Ballester, Acc. Chem. Res., 2012, 46, 874 CrossRef PubMed.
- P. S. Lakshminarayanan, I. Ravikumar, E. Suresh and P. Ghosh, Inorg. Chem., 2007, 46, 4769 CrossRef CAS PubMed.
- M. Giese, M. Albrecht, T. Repenko, J. Sackmann, A. Valkonen and K. Rissanen, Eur. J. Org. Chem., 2014, 2435 CrossRef CAS.
- (a) H. T. Chifotides, B. L. Schottel and K. R. Dunbar, Angew. Chem., Int. Ed., 2010, 49, 7202 CrossRef CAS PubMed; (b) P. S. Szalay, J. R. Galán-Mascarós, B. L. Schottel, J. Bacsa, L. M. Pérez, A. S. Ichimura, A. Chouai and K. R. Dunbar, J. Cluster Sci., 2004, 15, 503 CrossRef CAS.
- (a) H. Furuta and H. Maeda, J. Porphyrins Phthalocyanines, 2004, 08, 67 CrossRef; (b) H. Maeda, T. Morimoto, A. Osuka and H. Furuta, Chem. – Asian J., 2006, 1, 832 CrossRef CAS PubMed; (c) H. Maeda, A. Osuka and H. Furuta, J. Inclusion Phenom. Macrocyclic Chem., 2004, 49, 33 CrossRef CAS.
- O. B. Berryman, F. Hof, M. J. Hynes and D. W. Johnson, Chem. Commun., 2006, 506 RSC.
- M. Giese, M. Albrecht, T. Krappitz, M. Peters, V. Gossen, G. Raabe, A. Valkonen and K. Rissanen, Chem. Commun., 2012, 48, 9983 RSC.
- A. Bretschneider, D. M. Andrada, S. Dechert, S. Meyer, R. A. Mata and F. Meyer, Chem. – Eur. J., 2013, 19, 16988 CrossRef CAS PubMed.
- M. G. Chudzinski, C. A. McClary and M. S. Taylor, J. Am. Chem. Soc., 2011, 133, 10559 CrossRef CAS PubMed.
- S. Li, S.-X. Fa, Q.-Q. Wang, D.-X. Wang and M.-X. Wang, J. Org. Chem., 2012, 77, 1860 CrossRef CAS PubMed.
- (a) D.-X. Wang, Q.-Y. Zheng, Q.-Q. Wang and M.-X. Wang, Angew. Chem., Int. Ed., 2008, 47, 7485 CrossRef CAS PubMed; (b) D.-X. Wang, Q.-Q. Wang, Y. Han, Y. Wang, Z.-T. Huang and M.-X. Wang, Chem. – Eur. J., 2010, 16, 13053 CrossRef CAS PubMed; (c) D.-X. Wang and M.-X. Wang, J. Am. Chem. Soc., 2013, 135, 892 CrossRef CAS PubMed.
- (a) L. Adriaenssens, C. Estarellas, A. Vargas Jentzsch, M. Martinez Belmonte, S. Matile and P. Ballester, J. Am. Chem. Soc., 2013, 135, 8324 CrossRef CAS PubMed; (b) L. Adriaenssens, G. Gil-Ramírez, A. Frontera, D. Quiñonero, E. C. Escudero-Adán and P. Ballester, J. Am. Chem. Soc., 2014, 136, 3208 CrossRef CAS PubMed; (c) A. Bauzá, D. Quiñonero, A. Frontera and P. Ballester, Int. J. Mol. Sci., 2015, 16, 8934 CrossRef PubMed.
- A. Bauzá, R. Ramis and A. Frontera, Comput. Theor. Chem., 2014, 1038, 67 CrossRef.
- (a) H.-J. Y. Schneider, Anatoly Principles and Methods in Supramolecular Chemistry, Wiley, Chichester, 2000 Search PubMed; (b) P. A. Gale, Chem. Soc. Rev., 2007, 36, 141 RSC.
- S. I. Stupp and L. C. Palmer, Chem. Mater., 2014, 26, 507 CrossRef CAS.
- (a) M. Mascal, I. Yakovlev, E. B. Nikitin and J. C. Fettinger, Angew. Chem., Int. Ed., 2007, 46, 8782 CrossRef CAS PubMed; (b) M. M. Watt, L. N. Zakharov, M. M. Haley and D. W. Johnson, Angew. Chem., Int. Ed., 2013, 52, 10275 CrossRef CAS PubMed; (c) S. Guha and S. Saha, J. Am. Chem. Soc., 2010, 132, 17674 CrossRef CAS PubMed.
- (a) Y. Zhao, Y. Cotelle, A.-J. Avestro, N. Sakai and S. Matile, J. Am. Chem. Soc., 2015, 137, 11582 CrossRef PubMed; (b) Y. Zhao, S. Benz, N. Sakai and S. Matile, Chem. Sci., 2015, 6, 6219 RSC.
- (a) S. Chakravarty, Z.-Z. Sheng, B. Iverson and B. Moore, FEBS Lett., 2012, 586, 4180 CrossRef CAS PubMed; (b) A. Bauzá, D. Quiñonero, P. M. Deyà and A. Frontera, Chem. – Eur. J., 2014, 20, 6985 CrossRef PubMed.
- (a) A. Caballero, F. Zapata and P. D. Beer, Coord. Chem. Rev., 2013, 257, 2434 CrossRef CAS; (b) M. Cametti and K. Rissanen, Chem. Commun., 2009, 2809 RSC; (c) M. Cametti and K. Rissanen, Chem. Soc. Rev., 2013, 42, 2016 RSC; (d) P. A. Gale, N. Busschaert, C. J. E. Haynes, L. E. Karagiannidis and I. L. Kirby, Chem. Soc. Rev., 2014, 43, 205 RSC; (e) M. Wenzel, J. R. Hiscock and P. A. Gale, Chem. Soc. Rev., 2012, 41, 480 RSC.
- (a) T. M. Beale, M. G. Chudzinski, M. G. Sarwar and M. S. Taylor, Chem. Soc. Rev., 2013, 42, 1667 RSC; (b) L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118 CrossRef CAS PubMed.
- P. Gamez, Inorg. Chem. Front., 2014, 1, 35 RSC.
- S. Saha, B. Akhuli, I. Ravikumar, P. S. Lakshminarayanan and P. Ghosh, CrystEngComm, 2014, 16, 4796 RSC.
- (a) V. Gorteau, G. Bollot, J. Mareda and S. Matile, Org. Biomol. Chem., 2007, 5, 3000 RSC; (b) V. Gorteau, G. Bollot, J. Mareda, A. Perez-Velasco and S. Matile, J. Am. Chem. Soc., 2006, 128, 14788 CrossRef CAS PubMed; (c) N. Sakai, J. Mareda, E. Vauthey and S. Matile, Chem. Commun., 2010, 46, 4225 RSC; (d) V. Gorteau, M. D. Julliard and S. Matile, J. Membr. Sci., 2008, 321, 37 CrossRef CAS; (e) A. Perez-Velasco, V. Gorteau and S. Matile, Angew. Chem., Int. Ed., 2008, 47, 921 CrossRef CAS PubMed; (f) J. T. Davis, O. Okunola and R. Quesada, Chem. Soc. Rev., 2010, 39, 3843 RSC.
- A. P. Davis, D. N. Sheppard and B. D. Smith, Chem. Soc. Rev., 2007, 36, 348 RSC.
- (a) D. A. Doyle, J. M. Cabral, R. A. Pfuetzner, A. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait and R. MacKinnon, Science, 1998, 280, 69 CrossRef CAS PubMed; (b) R. Dutzler, E. B. Campbell and R. MacKinnon, Science, 2003, 300, 108 CrossRef CAS PubMed.
- J. Míšek, A. Vargas Jentzsch, S.-I. Sakurai, D. Emery, J. Mareda and S. Matile, Angew. Chem., Int. Ed., 2010, 49, 7680 CrossRef PubMed.
- S. T. Schneebeli, M. Frasconi, Z. Liu, Y. Wu, D. M. Gardner, N. L. Strutt, C. Cheng, R. Carmieli, M. R. Wasielewski and J. F. Stoddart, Angew. Chem., Int. Ed., 2013, 52, 13100 CrossRef CAS PubMed.
- (a) A. P. H. J. Schenning and E. W. Meijer, Chem. Commun., 2005, 3245 RSC; (b) M. Ratner, Nat. Nanotechnol., 2013, 8, 378 CrossRef CAS PubMed; (c) A. Jain and S. J. George, Mater. Today, 2015, 18, 206 CrossRef CAS.
- M. Müller, M. Albrecht, V. Gossen, T. Peters, A. Hoffmann, G. Raabe, A. Valkonen and K. Rissanen, Chem. – Eur. J., 2010, 16, 12446 CrossRef PubMed.
- M. Giese, M. Albrecht, C. Bohnen, T. Repenko, A. Valkonen and K. Rissanen, Dalton Trans., 2014, 43, 1873 RSC.
- N. Hafezi, J. M. Holcroft, K. J. Hartlieb, E. J. Dale, N. A. Vermeulen, C. L. Stern, A. A. Sarjeant and J. F. Stoddart, Angew. Chem., Int. Ed., 2015, 54, 456 CAS.
- Q. He, Y.-F. Ao, Z.-T. Huang and D.-X. Wang, Angew. Chem., Int. Ed., 2015, 54, 11785 CrossRef CAS PubMed.
- (a) P. A. Gale, C. C. Tong, C. J. E. Haynes, O. Adeosun, D. E. Gross, E. Karnas, E. M. Sedenberg, R. Quesada and J. L. Sessler, J. Am. Chem. Soc., 2010, 132, 3240 CrossRef CAS PubMed; (b) C. C. Tong, R. Quesada, J. L. Sessler and P. A. Gale, Chem. Commun., 2008, 6321 RSC; (c) J. P. Behr, M. Kirch and J. M. Lehn, J. Am. Chem. Soc., 1985, 107, 241 CrossRef CAS.
- (a) M. G. Guerrero, J. M. Vega and M. Losada, Annu. Rev. Plant Physiol., 1981, 32, 169 CrossRef CAS; (b) T. Omata, Plant Cell Physiol., 1995, 36, 207 CAS.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2013, 113, 2100 CrossRef CAS PubMed.
- (a) D. A. Dougherty, Acc. Chem. Res., 2013, 46, 885 CrossRef CAS PubMed; (b) K. U. Wendt, G. E. Schulz, E. J. Corey and D. R. Liu, Angew. Chem., Int. Ed., 2000, 39, 2812 CrossRef CAS; (c) S. Yamada and J. S. Fossey, Org. Biomol. Chem., 2011, 9, 7275 RSC; (d) R. R. Knowles, S. Lin and E. N. Jacobsen, J. Am. Chem. Soc., 2010, 132, 5030 CrossRef CAS PubMed.
- (a) M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518 CrossRef CAS PubMed; (b) J. Lacour and D. Moraleda, Chem. Commun., 2009, 7073 RSC; (c) K. Fujisawa, C. Beuchat, M. Humbert-Droz, A. Wilson, T. A. Wesolowski, J. Mareda, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2014, 53, 11266 CrossRef CAS PubMed; (d) K. Fujisawa, M. Humbert-Droz, R. Letrun, E. Vauthey, T. A. Wesolowski, N. Sakai and S. Matile, J. Am. Chem. Soc., 2015, 137, 11047 CrossRef CAS PubMed.
- F. N. Miros, G. Huang, Y. Zhao, N. Sakai and S. Matile, Supramol. Chem., 2015, 27, 303 CrossRef CAS.
- H.-J. Schneider, Angew. Chem., Int. Ed., 2009, 48, 3924 CrossRef CAS PubMed.
- (a) E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210 CrossRef CAS PubMed; (b) L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808 CrossRef CAS PubMed.
- (a) S. Burley and G. Petsko, Science, 1985, 229, 23 CAS; (b) S. Li, V. R. Cooper, T. Thonhauser, B. I. Lundqvist and D. C. Langreth, J. Phys. Chem. B, 2009, 113, 11166 CrossRef CAS PubMed.
- L. R. Rutledge, L. S. Campbell-Verduyn and S. D. Wetmore, Chem. Phys. Lett., 2007, 444, 167 CrossRef CAS.
- (a) S. Tsuzuki, M. Yoshida, T. Uchimaru and M. Mikami, J. Phys. Chem. A, 2001, 105, 769 CrossRef CAS; (b) J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303 CrossRef CAS PubMed; (c) P. B. Crowley and A. Golovin, Proteins: Struct., Funct., Bioinf., 2005, 59, 231 CrossRef CAS PubMed; (d) J. P. Gallivan and D. A. Dougherty, J. Am. Chem. Soc., 2000, 122, 870 CrossRef CAS.
- (a) M. H. Nishio, M. Minoru and Y. Umezawa, The CH/π Interaction: Evidence, Nature, and Consequences, Wiley-VCH, New York, 1998 Search PubMed; (b) M. Nishio, Tetrahedron, 2005, 61, 6923 CrossRef CAS.
- (a) T. J. Mooibroek, P. Gamez and J. Reedijk, CrystEngComm, 2008, 10, 1501 RSC; (b) M. Egli and S. Sarkhel, Acc. Chem. Res., 2007, 40, 197 CrossRef CAS PubMed.
- M. Mitra, P. Manna, S. K. Seth, A. Das, J. Meredith, M. Helliwell, A. Bauza, S. R. Choudhury, A. Frontera and S. Mukhopadhyay, CrystEngComm, 2013, 15, 686 RSC.
- J. J. Fiol, M. Barceló-Oliver, A. Tasada, A. Frontera, À. Terrón and Á. García-Raso, Coord. Chem. Rev., 2013, 257, 2705 CrossRef CAS.
- A. Garcia-Raso, F. M. Albertí, J. J. Fiol, A. Tasada, M. Barceló-Oliver, E. Molins, D. Escudero, A. Frontera, D. Quiñonero and P. M. Deyà, Inorg. Chem., 2007, 46, 10724 CrossRef CAS PubMed.
- D. D. Jenkins, J. B. Harris, E. E. Howell, R. J. Hinde and J. Baudry, J. Comput. Chem., 2013, 34, 518 CrossRef CAS PubMed.
- P. R. Race, A. L. Lovering, R. M. Green, A. Ossor, S. A. White, P. F. Searle, C. J. Wrighton and E. I. Hyde, J. Biol. Chem., 2005, 280, 13256 CrossRef CAS PubMed.
- M. M. Cherney, Y. Zhang, M. Solomonson, J. H. Weiner and M. N. G. James, J. Mol. Biol., 2010, 398, 292 CrossRef CAS PubMed.
- V. H. Pham, J.-J. Yong, S.-J. Park, D.-N. Yoon, W.-H. Chung and S.-K. Rhee, Microbiology, 2008, 154, 3112 CrossRef CAS PubMed.
- I. V. Krieger, J. S. Freundlich, V. B. Gawandi, J. P. Roberts, V. B. Gawandi, Q. Sun, J. L. Owen, M. T. Fraile, S. I. Huss, J.-L. Lavandera, T. R. Ioerger and J. C. Sacchettini, Chem. Biol., 2012, 19, 1556 CrossRef CAS PubMed.
- J. Sabek, L. Adriaenssens, T. Guinovart, E. J. Parra, F. X. Rius, P. Ballester and P. Blondeau, Chem. – Eur. J., 2015, 21, 448 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |