
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceiSERS microscopy guided by wide field immunofluorescence: analysis of HER2 expression on normal and breast cancer FFPE tissue sections†
Xin-Ping
Wang
a,
Yuying
Zhang
a,
Matthias
König
a,
Evanthia
Papadopoulou
a,
Bernd
Walkenfort
a,
Sabine
Kasimir-Bauer
b,
Agnes
Bankfalvi
c and
Sebastian
Schlücker
*a
aPhysical Chemistry I, Department of Chemistry and Center for Nanointegration Duisburg-Essen (CENIDE), University of Duisburg-Essen, Universitätsstr. 5, Essen 45141, Germany. E-mail: sebastian.schluecker@uni-due.de
bDepartment of Gynecology and Obstetrics, University Hospital Essen, University of Duisburg-Essen, Hufelandstrasse 55, D-45122 Essen, Germany
cInstitute of Pathology, University Hospital Essen, Hufelandstrasse 55, D-45122 Essen, Germany
First published on 6th June 2016
Abstract
Surface-enhanced Raman scattering (SERS) microscopy is an emerging imaging technique for tissue-based cancer diagnostics. Specifically, immuno-SERS (iSERS) microscopy employs antibodies labelled by molecularly functionalized noble metal colloids for antigen localization on tissue specimen. Spectrally resolved iSERS acquisition schemes are typically rather time-consuming when large tissue areas must be scanned. Here, we demonstrate the application of iSERS imaging guided by wide field immunofluorescence (IF) for localization of the human epidermal growth factor receptor 2 (HER2) on breast tissue sections. The addition of unlabelled anti-HER2 primary antibodies to the tissue is followed by the incubation with secondary antibodies labelled with both Alexa-647 (for IF) and hydrophilically stabilized gold nanostars coated with aromatic thiols (for iSERS). False-color iSERS images clearly reveal the different HER2 expression levels on normal and breast cancer tissue, respectively. A series of negative controls confirms that the binding specificity of the secondary antibody is maintained after conjugation to the SERS nanoparticles.
1 Introduction
Immunohistochemistry is widely used in diagnosis, prognostication, individualized therapeutic decisions and investigations into the pathogenesis of diseases. Current pathology practice utilizes chromogenic immunohistochemistry (IHC) by bright field microscopy. Immunofluorescence (IF) is preferably used to visualize several antigens simultaneously both in research and diagnostics. In addition to these established techniques, surface-enhanced Raman scattering (SERS) is employed as an emerging optical technique in biomedical diagnostics.1–3 In immuno-SERS (iSERS) microscopy, the corresponding antibodies are labelled by molecularly functionalized noble metal colloids (SERS labels or nanotags), i.e., gold or silver metal nanoparticles (NPs) with organic Raman reporter molecules chemisorbed on their surface.4,5 The reporter molecules provide the characteristic “molecular fingerprint” for the identification of the label, while the metal NP provides the necessary signal enhancement upon excitation of a localized surface plasmon resonance (LSPR).A central advantage of Raman/SERS-based detection schemes is their enormous spectral multiplexing capacity for multi-colour IHC due to the small line width of vibrational Raman bands and no or only minimal spectral overlap.6–8 Autofluorescence, a major drawback in fluorescence microscopy including IF, can be minimized in iSERS by red to near-infrared laser excitation and also easily be separated from the narrow Raman peaks via data processing.
IHC is generally performed in a wide field configuration for fast, global analysis of large tissue areas. In contrast, iSERS is typically performed in the Raman mapping mode for a spectrally resolved detection at each pixel. Although acquisition times per pixel have been systematically lowered from 1 s in the first iSERS experiments on FFPE tissue in 2006![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9 to the millisecond regime in 201410 due to the development of brighter SERS labels, iSERS inherits the disadvantages of normal Raman microscopy in the mapping mode for scanning large areas. A time-efficient approach to iSERS therefore needs to combine a fast global analysis by wide field IHC or IF with a subsequent local iSERS analysis. One elegant strategy is based on fluorescent SERS NPs (F-SERS dots)11,12 conjugated to unlabelled antibodies. Alternatively, SERS NPs can be conjugated to fluorophore-labelled antibodies. Overall, such combined IF-iSERS approaches pave the way for future studies exploring the full potential of multiplexed iSERS for personalized medicine.
9 to the millisecond regime in 201410 due to the development of brighter SERS labels, iSERS inherits the disadvantages of normal Raman microscopy in the mapping mode for scanning large areas. A time-efficient approach to iSERS therefore needs to combine a fast global analysis by wide field IHC or IF with a subsequent local iSERS analysis. One elegant strategy is based on fluorescent SERS NPs (F-SERS dots)11,12 conjugated to unlabelled antibodies. Alternatively, SERS NPs can be conjugated to fluorophore-labelled antibodies. Overall, such combined IF-iSERS approaches pave the way for future studies exploring the full potential of multiplexed iSERS for personalized medicine.
In this contribution, such a dual-mode IF/iSERS imaging approach is applied to the analysis of breast tissue sections. Breast cancer is by far the most common cancer in women in the world and the second cause of cancer death in women in more developed regions after lung cancer.13 This heterogeneous disease is categorized into three basic therapeutic groups: estrogen receptor (ER)-positive, human epidermal growth factor receptor 2 (HER2)-enriched and triple negative breast carcinomas.14,15 Since the first report from Slamon et al.,16 who revealed that HER2 is a significant diagnostic biomarker for breast, many studies investigating the HER2 status have been conducted. HER2, encoded by proto-oncogene ErbB2, is a member of the epidermal growth factor receptor family having tyrosine kinase activity receptors and is involved in the regulation of cellular signalling in cell growth and development.17 Due to its overexpression in 10–20% of breast cancers, HER2 is used as a critical biomarker in clinical diagnosis and therapy with trastuzumab (Herceptin®).18
Recently, a range of techniques in addition to conventional IHC has been proposed for the detection of HER2 in solution, cells and tissue biopsies utilizing electrochemistry,19–21 fluorescence22 and also SERS.23,24 For example, Wu et al.22 reported the use of quantum dots (QDs) conjugated to antibodies for the detection of HER2 and other cancer markers on fixed live cells by measuring the fluorescence signal from the QDs. Previous SERS studies have reported on the use of NP-labelled antibodies23 on breast cancer cells. However, in none of these studies FFPE breast tissue specimens, the gold standard in clinical diagnosis by pathologists, were employed.
Fig. 1 shows the experimental approach to dual mode IF/iSERS imaging for HER2 localization on FFPE breast tissue employed in this study. Gold nanostars (AuNS) are used because of their high plasmonic activity (Fig. 1A).25–33 Hydrophilic stabilization of the metal colloid is achieved by a mixture of Raman reporter molecules conjugated to ethylene glycol spacers (4-NTB-MEG-OH = 4-nitrothiobenzoate-monoethylene glykole-hydroxy; 4-NTB-TEG-COOH = 4-nitrothio-benzoate-triethyleneglykole-carboxy).34,35 For bioconjugation, the COOH group at the terminus of the Raman reporter with the longer TEG chain is activated for covalent binding to primary amines (lysine residues) of the Alexa647-labelled secondary antibody. The AuNS-labelled secondary antibody is therefore both fluorescence- and Raman/SERS-active. After deparaffinization and enzymatic antigen retrieval, the breast tissue section is first incubated with an unlabelled anti-HER2 antibody (Fig. 1B). Then, the glass slides are washed for removing unbound free primary antibodies. Finally, the Alexa-647/AuNS-labelled secondary antibody is added for HER2 localization on the tissue section. Wide field fluorescence microscopy allows the pathologist to quickly inspect a large field of view, followed by SERS mapping on selected smaller areas.
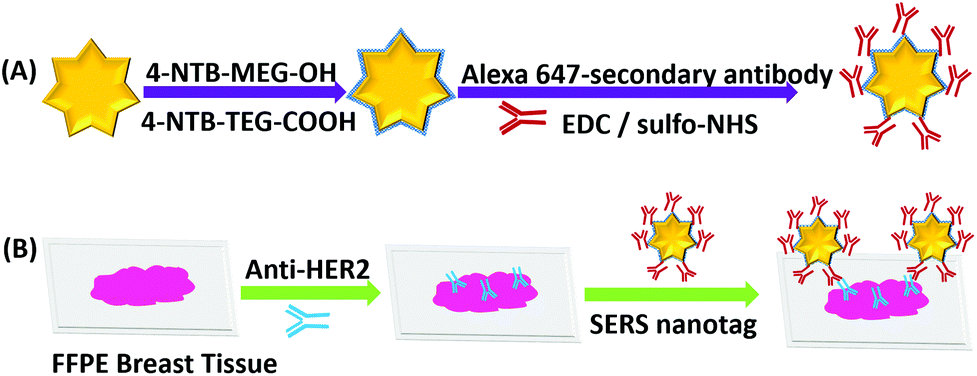 | ||
| Fig. 1 (A) Bioconjugation route to Alexa 647- and AuNS-labelled secondary antibodies. (B) Tissue incubation with unlabelled primary Anti-HER2 antibody and the doubly labelled secondary antibodies for wide field IF-guided local analysis by iSERS mapping. | ||
2 Experimental methods
2.1 Reagents and instruments
Tetrachloroauric acid (HAuCl4·3H2O), bovine serum albumin (BSA), ethanolamine, N-hydroxysulfosuccinimide sodium salt (sulfo-NHS), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid (HEPES) and silver nitrate were purchased from Sigma Aldrich, Germany. L(+)-ascorbic acid was obtained from AppliChem, Germany. Sodium chloride and hydrochloric acid were obtained from Bernd Kraft, Germany. Disodium monohydrogen phosphate (Na2HPO4) and potassium dihydrogen phosphate (KH2PO4) were obtained from Carl Roth, Germany. Phosphate-buffered saline (PBS) was prepared in house and freshly sterilized. Monoclonal anti-HER2 antibody (TAB250), a fast enzyme for antigen retrieval, and a ZytoChem plus HRP polymer kit were purchased from Zytomed, Germany. Alexa-647 conjugated goat anti-mouse and goat anti-sheep antibodies were purchased from Thermofisher, Germany. FFPE breast tissue sections (1.5 μm thick) were provided by the Institute of Pathology, University Hospital Essen, Germany. Ultrapure water (18.2 MΩ cm, Millipore) was used in the experiments.SERS and fluorescence images were recorded by an in-house modified confocal Raman microscope (WITec Alpha 300R, 30 cm focal length and 600 grooves per mm grating spectrometer) equipped with an EM-CCD (Andor Newton DU970N–BV-353). A high- pressure mercury lamp (LEJ LQ-HXP 120 VIS) was used for wide field excitation and coupled by a liquid waveguide to the illumination port of the microscope. Bandpass filters centered at 628 nm and 692 nm with a bandwidth of 40 nm (Semrock BrightLine 628/40 HC and 692/40 HC) were placed in the excitation and emission path, respectively. The fibre coupler to the grating spectrometer in the original configuration (Raman only) was replaced by a T-coupler (Zeiss) including a transfer optic. The two ports at the end of the T-coupler were used for coupling to the grating spectrometer for Raman microscopy and to a CCD (Zeiss AxioCam ICm1), respectively, for fluorescence microscopy.
Radiation from the 632.8 nm line of a HeNe laser was focused onto the sample using a 40× objective (Olympus) with a numerical aperture of 0.6. The integration time per pixel in the Raman mapping experiments was 50 ms and the laser power at the sample was 4.9 mW. A 40× objective (Olympus) was used in the correlative iSERS and IF measurements.
Extinction spectra were recorded with a Jasco V600 UV/vis absorption spectrometer. SEM images were obtained with a field emission scanning electron microscope (FESEM) from Jeol (JSM 7500F).
Immuno-fluorescence images were recorded with a Leica DMI6000 microscope. All correlative images were recorded with the WITec setup.
2.2 Synthesis of Au nanostars and preparation of SERS active labels
Au nanostars were synthesised utilizing a seed-mediated36 technique. 20 nm Au seeds were synthesized by citrate reduction. Then, 400 μL of 20 nm seed NPs were added into a 15 ml Falcon tube. Afterwards, a mixture of 300 μL of 2 mM AgNO3, 150 μL of 100 mM L(+)-ascorbic acid, 100 μL of 2% w/v SDS, and 30 μL of HCl were added into the gold seed suspension. Subsequently, 10 mL of 750 μM HAuCl4 was added to the mixture. The colour of the suspension immediately changed from red to dark blue due to the formation of gold nanostars. The mixture was left to react for 15 minutes at room temperature and then washed twice by centrifugation at 2200 rpm for 20 min and re-dispersion in ultrapure water. The concentration of the nanostars (8 × 1010 particles per mL) was determined by a light scattering based tracking system (Nanosight, model LM10).Gold nanostars (1500 μL) were further modified with a mixture of the hydrophilically stabilized Raman reporter molecules 4-nitrothiobenzoate-monoethylene glykole-hydroxy (4-NTB-MEG-OH, 7.5 μL, 10 mM in ethanol) and 4-nitrothio-benzoate-triethylene-glykole-carboxy (4-NTB-TEG-COOH, 7.5 μL, 10 mM in ethanol).34 After incubation overnight, the colloid was centrifuged and redispersed in 750 μL HEPES buffer.
2.3 Functionalization of Au nanostars with secondary antibody (goat anti mouse-Alexa 647)
Bioconjugation of the SERS NPs was achieved by covalent coupling of the carboxylic acid moieties from the Raman reporter 4-NTB-TEG-COOH and primary amines (-NH2) from the secondary antibody. For activation of COOH groups, freshly prepared 0.1 mM EDC and 0.25 mM sulfo-NHS were added to the suspension of SERS labels. After shaking at RT for 25 min, the remaining excess of EDC and NHS was removed by centrifugation and the NPs were suspended in fresh HEPES buffer. Subsequently, 8.1 μg of goat anti mouse-Alexa 647 was added and allowed to react in darkness at RT for 2.5 hours. Then, 10% of ethanolamine was added to the particle suspension to block unreacted carboxylic acids. The mixture was kept at RT for 30 min while shaking. Finally, the SERS-labelled secondary antibodies were washed 4 times with 2% BSA-PBS buffer to remove non-conjugated antibodies and stored in 2% BSA-PBS buffer.2.4 Deparaffinization of FFPE tissue sections and antigen retrieval
FFPE breast tissue was cut into 1.5 μm thick sections and mounted on SuperFrost TM glass slides (Fisher Scientific GmbH, Schwerte, Germany). Slides were heated at 60 °C overnight. Then, the sections were deparaffinized first in Xylol for 20 min, rehydrated in ethanol and a series of ethanol/water mixtures (100%, 96%, 70%, 1 min for each), respectively, rinsed in water and finally with PBS.A proteolytic antigen retrieval method was employed. Briefly, several drops of fast enzyme were added onto tissue areas and incubated for 5 min at RT. After that, slides were washed with PBS buffer three times and incubated with 2% BSA-PBS for protein blocking.
2.5 Staining of breast tissue sections
:150 dilution) was added onto breast tissue slides and incubated for 1 h at RT. HRP polymer was added after washing slides with PBS (incubation time: 20 min). Diaminobenzidine (DAB) was chosen as the chromogen (incubation time: 10 min). After that, sections were washed with water and stained with hematoxylin as the nuclear counterstaining agent. Finally, the sections were dehydrated with three ethanol/water mixtures (70%, 96%, 100%, 1 min for each), followed by adding mounting medium.
2.5.2 Immunofluorescence (IF)
After deparaffinization, rehydration and enzymatic antigen retrieval, the tissue sections were blocked with 2% BSA-PBS and then incubated with the primary mouse anti-HER2 antibody (1:150 dilution). Then, the tissue sections were incubated at RT for 1 h. Afterwards, the tissue slides were thoroughly washed with PBS for at least three times. Finally, the tissue sections were incubated with the fluorophore-labelled secondary antibody (1:300 dilution) for 30 min at RT. Then the tissue sections were washed with PBS and treated by DAPI as a nuclear counterstain.
3 Results and discussion
Fig. 2 shows a SEM image of the synthesized Au nanostars. They are relatively monodisperse (average size ≈ 75 nm), however, the core diameter and the number of tips per star differ from NP to NP. Upon resonant excitation by light, the tips of the Au nanostars exhibit very high local electric fields, which leads to a very strong enhancement of Raman signals from molecules located at the tips. Since we employ a self-assembled monolayer on the surface of the Au nanostars, Raman reporter molecules are present at all tips and thereby provide the maximum possible SERS signal. Furthermore, the LSPR peak of the Au nanostars has a maximum in the red (λmax ≈ 666 nm) in order to maximize the SERS signal upon illumination with red laser light (Fig. S1†). | ||
| Fig. 2 SEM image of Au nanostars. | ||
IHC including IF are widely used techniques in clinical diagnostics for the detection of protein expression on FFPE tissue sections. In this study, HER2 was used as a biomarker to differentiate between human normal and breast cancer tissue. First, IHC images were recorded as a gold standard for checking the quality and performance of staining procedures, tissue slides and both the primary mouse anti-HER2 and the goat anti mouse-HRP secondary antibody. As shown in Fig. 3(A), most cell membranes on the HER2 (3+) tissue section are stained in dark brown (left), whereas no staining of cell membranes on normal breast tissue is observed (right). These positive control experiments confirm that the HER2 antigen is well preserved after deparaffinization and proteolytic antigen retrieval. Second, IF images were recorded, as shown in Fig. 3(B) left. In this case, normal (bottom) and breast cancer (top) tissue specimen were first incubated with the primary anti-HER2 antibody after deparaffinization and proteolytic antigen retrieval, followed by the addition of the Alexa 647-labelled secondary antibody for localization by wide field fluorescence microscopy. Fluorescence from the Alexa 647-labelled secondary antibody is only observed in the HER2 (3+) breast cancer tissue. In addition to the IF contrast, the cell nuclei were stained with DAPI as shown in Fig. 3(B) middle. The merged Alexa 647-IF/DAPI images in Fig. 3(B) show that almost all membranes of tumour cells on HER2 (3+) breast tissue were stained with the fluorophore-labelled secondary antibody (top right). In contrast, nearly no staining of membranes is observed for the epithelial cells in normal breast tissue (bottom left and right). The IF images obtained by wide field fluorescence microscopy served as a starting point for selecting smaller regions on the tissue for analysis by iSERS microscopy in the mapping mode.
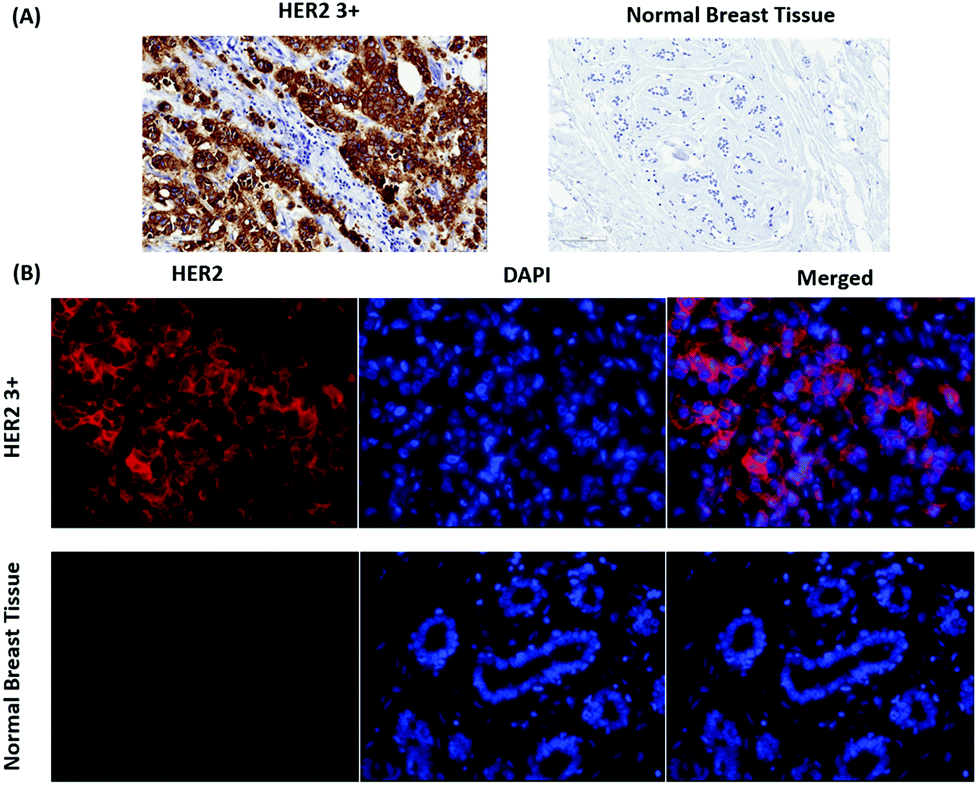 | ||
| Fig. 3 Localization of HER2 on FFPE breast tissue by IHC and IF. (A) IHC staining of HER2 (3+) breast cancer and normal breast tissue (B) IF staining of HER2 (3+) and DAPI staining of cell nuclei on both breast cancer and normal breast tissue. | ||
For iSERS microscopy, hydrophilically stabilized Au nanostars with bright SERS signals (Fig. S2†) were covalently conjugated to the Alexa 647-labelled secondary antibody for correlative wide field IF/mapping iSERS microscopy on the same tissue section. Due to the conjugation of the fluorophore-labelled secondary antibody to the SERS-active nanostars, both fluorescence (Alexa 647) and Raman signals (Raman reporter) can be detected after binding of the fluorescence- and SERS-labelled secondary antibody to the primary anti-HER2 antibody. Fig. 4 (top row) displays the results from correlative bright field/iF/iSERS microscopy for the localization of HER2 on normal and breast cancer tissue (HER2 3+). The fluorescence image of HER2 (3+) tissue in Fig. 4(B) top shows the selective staining of the tumour cells when compared with the bright field image in Fig. 4(A) left. Specifically, only the cell membranes are stained with the Alexa 647-labelled secondary antibody which is in consistent with results reported in the literature.37 Based on the IF images, a smaller rectangular region (box with blue lines) was chosen for iSERS analysis by confocal Raman mapping. The contrast in the corresponding SERS false-colour image in Fig. 4(C) is based on the integrated intensity of the most intense Raman marker band at 1340 cm−1, which is assigned to the symmetric nitro stretching vibration of 4-NTB. Fig. 4(D) shows representative spectra recorded at three distinct cellular regions: cell membrane, cytoplasm and the extracellular region, as indicated by circles in the corresponding SERS false-colour image. The characteristic Raman signature of the SERS label is observed only on the cell membrane (Fig. 4D, top). This indicates that iSERS microscopy is capable of selectively localizing the cell membrane of HER2-positive tumour cells and that the SERS NPs do not notably influence the binding specificity of the secondary antibody to the primary antibody. As a negative control, the same experiments were performed on normal breast tissue. As shown in Fig. 4 (middle row), there are neither fluorescence (B) nor Raman signals (C) observed.
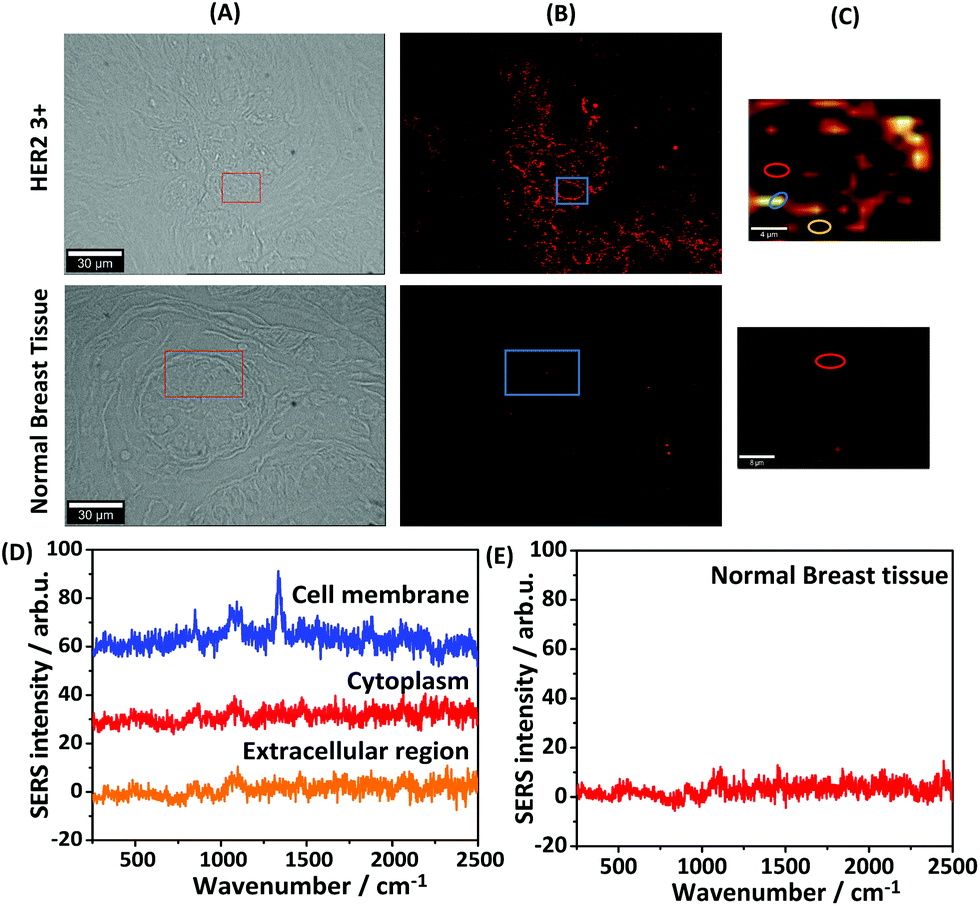 | ||
| Fig. 4 Dual-mode imaging microscopy for normal breast tissue and breast cancer tissue by staining with antibody-conjugated Au nanostars. (A) Bright field. (B) Immunofluorescence. (C) iSERS false-colour image. (colour scale: 30–300 CCD cts) (D) Raman spectra of three different regions on HER2 3+ breast tissue. (E) Spectrum from normal breast tissue. | ||
The central advantage of this dual-mode IF/iSERS imaging approach is that a fast global analysis by wide field IF guides the slower local analysis by Raman mapping. The latter has the potential of a highly multiplexed analysis of cancer biomarkers on the same tissue section.
Furthermore, two additional negative controls were performed in order to confirm the specificity for staining HER2 in iSERS imaging experiments. The first negative control was performed without using the primary antibody. Since the secondary antibody only recognizes the primary antibody, but not directly the HER2 antigen itself, the fluorophore/SERS-labelled antibodies should ideally be completely removed from the tissue by washing. Fig. 5(A) shows the staining result without the presence of the primary antibody. As expected, neither fluorescence nor Raman signals are observed. The second negative control was performed by using a primary antibody (mouse), but in combination with a secondary antibody not directed against mouse (goat anti-sheep). The employed goat anti sheep Alexa-647 labelled secondary antibody does not bind to the mouse anti-HER2 primary antibody and should therefore not bind to the tissue section. Fig. 5(B) shows the staining results with the “wrong” fluorophore/SERS-labelled secondary antibody. As expected, neither fluorescence nor Raman signals are observed. These negative control experiments additionally demonstrate the binding specificity of the fluorophore/SERS-labelled secondary antibody.
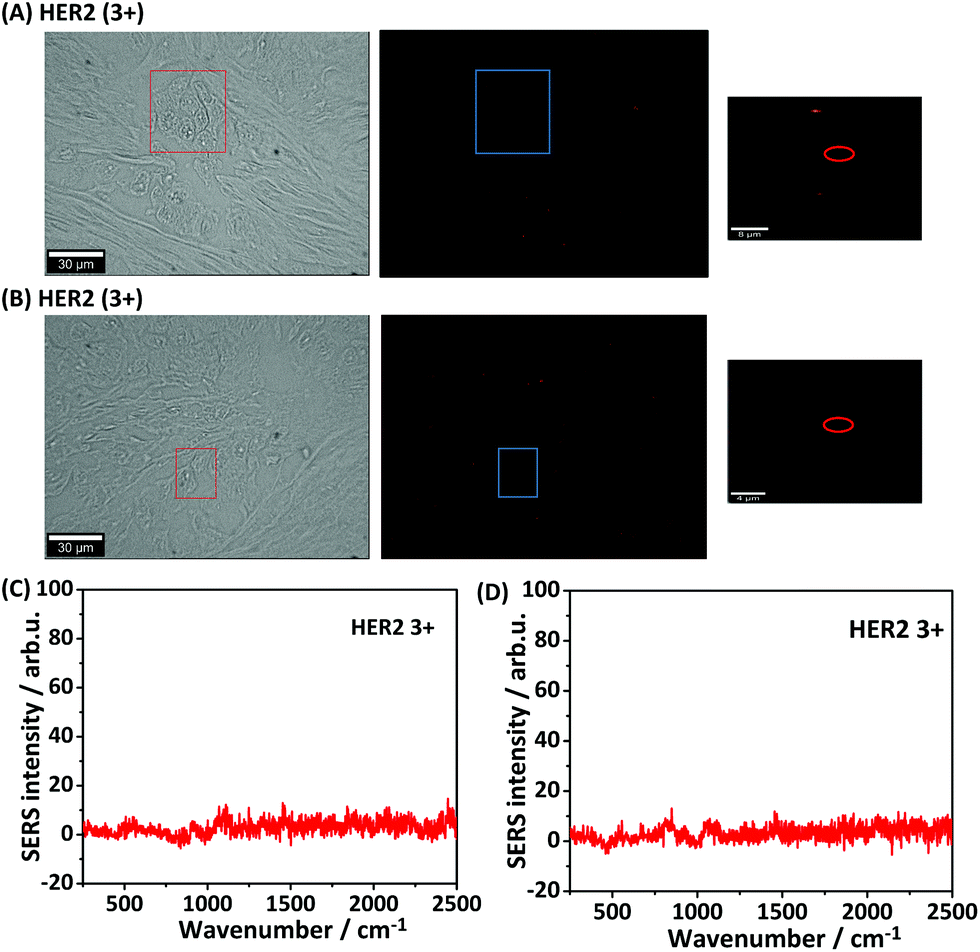 | ||
| Fig. 5 Negative control experiments (A) HER2 (3+) breast tissue stained with Alexa 647 labelled goat anti mouse secondary antibody without adding primary antibody. From left to right: bright-field, immunofluorescence and iSERS false-colour images. (B) HER2 (3+) breast tissue stained first with primary antibody anti-HER2 followed with Alexa 647-labelled goat anti-sheep secondary antibody. From left to right: bright-field, immunofluorescence and iSERS false-colour images. (C) SERS spectra measured on HER2 (3+) without primary antibody staining. (D) SERS spectra measured on HER2 (3+) with the Alexa 647-labelled goat anti-sheep secondary antibody staining. The colour scale in all SERS false-colour images is 30–300 CCD counts. | ||
Finally, we also successfully recorded iSERS images after 51 days (Fig. S3†), demonstrating the capability of the technique for a potential re-inspection by the pathologist after some storage time.
Conclusions and outlook
In summary, we have demonstrated the use of iSERS for selective staining of HER2 on FFPE breast cancer tissue sections, the gold standard in routine clinical pathology. Negative controls confirm that the binding specificity of the antibody is maintained after covalent binding to the SERS NPs. Highly SERS-active Au nanostars in conjugation with a fluorophore-labelled secondary antibody provide both fluorescence and Raman contrast. The initial global analysis by rapid wide field fluorescence imaging directs a subsequent local analysis by iSERS mapping microscopy. The same tissue specimen may also be investigated multiple times or re-inspected after a longer storage time because photobleaching is not critical in iSERS microscopy. Finally, iSERS offers a unique multiplexing potential due to the small line width of vibrational Raman band, enabling multi-colour imaging of several diagnostic biomarkers. This is particularly important for the analysis of single tumour cells because one reason for developing recurrent carcinoma in breast cancer is tumour cell dissemination into distant organs, preferentially bone marrow, which often occurs prior to surgery. Disseminated tumour cells in the bone marrow as well as circulating tumour cells in blood are suggested as potential surrogate markers for minimal residual disease, the precursor of metastatic disease, and their independent prognostic significance has already been demonstrated.38,39 These cells are highly heterogeneous and their characterization is important not only to confirm their malignant origin but also to follow phenotypic changes with tumour progression and to identify diagnostically and therapeutically relevant targets which will help to select cancer patients for individual therapies. In this regard, HER-2 as well as ER/PR was shown to be differentially expressed between the primary tumour and corresponding metastases and/or single cells in blood and bone marrow.40–43 Based on these data we can assume that targeting these cells could have important clinical implications and that multi-marker analysis by iSERS may help to match the right drug to the right patient.Acknowledgements
We thank for Ms Andrea Kutritz from the Institute of Pathology at the University Hospital Essen for IHC staining and the preparation of sections from breast tissue. We thank Prof Matthias Gunzer from UDE and his group for access to their Leica wide field fluorescence microscopy. Thanks also to Aleksandar Radojcic for the synthesis of the Au nanostars. Xin-ping Wang thanks the China Scholarship Council (CSC) for a PhD stipend. Matthias König was supported by a PhD stipend from the Fonds der Chemischen Industrie (FCI). Evanthia Papadopoulou acknowledges support from the EU under Horizon 2020 via a Marie-Curie postdoctoral fellowship.References
- Surface Enhanced Raman Spectroscopy: Analytical, Biophysical and Life Science Applications, ed. S. Schlücker, Wiley VCH, 2010 Search PubMed.
- X. Qian, X. H. Peng, D. O. Ansari, Q. Yin-Goen, G. Z. Chen, D. M. Shin, L. Yang, A. N. Young, M. D. Wang and S. Nie, Nat. Biotechnol., 2008, 26, 83–90 CrossRef CAS PubMed.
- S. McAughtrie, K. Faulds and D. Graham, J. Photochem. Photobiol., C, 2014, 21, 40–53 CrossRef CAS.
- Y. Wang, B. Yan and L. Chen, Chem. Rev., 2013, 113, 1391–1428 CrossRef CAS PubMed.
- Y. Wang and S. Schlücker, Analyst, 2013, 138, 2224–2238 RSC.
- C. L. Zavaleta, B. R. Smith, I. Walton, W. Doering, G. Davis, B. Shojaei, M. J. Natan and S. S. Gambhir, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13511–13516 CrossRef CAS PubMed.
- S. Schlücker, ChemPhysChem, 2009, 10, 1344–1354 CrossRef PubMed.
- L. Rodriguez-Lorenzo, L. Fabris and R. A. Alvarez-Puebla, Anal. Chim. Acta, 2012, 745, 10–23 CrossRef CAS PubMed.
- S. Schlücker, B. Küstner, A. Punge, R. Bonfig, A. Marx and P. Ströbel, J. Raman Spectrosc., 2006, 37, 719–721 CrossRef.
- M. Salehi, D. Steinigeweg, P. Ströbel, A. Marx, J. Packeisen and S. Schlücker, J. Biophotonics, 2013, 6, 785–792 CAS.
- M. A. Woo, S. M. Lee, G. Kim, J. Baek, M. S. Noh, J. E. Kim, S. J. Park, A. Minai-Tehrani, S. C. Park, Y. T. Seo, Y. K. Kim, Y. S. Lee, D. H. Jeong and M. H. Cho, Anal. Chem., 2009, 81, 1008–1015 CrossRef CAS PubMed.
- K. N. Yu, S. M. Lee, J. Y. Han, H. Park, M. A. Woo, M. S. Noh, S. K. Hwang, J. T. Kwon, H. Jin, Y. K. Kim, P. J. Hergenrother, D. H. Jeong, Y. S. Lee and M. H. Cho, Bioconjugate Chem., 2007, 18, 1155–1162 CrossRef CAS PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, CA-Cancer J. Clin., 2016, 66, 7–30 CrossRef PubMed.
- L. A. Carey, C. M. Perou, C. A. Livasy, L. G. Dressler, D. Cowan, K. Conway, G. Karaca, M. A. Troester, C. K. Tse, S. Edmiston, S. L. Deming, J. Geradts, M. C. Cheang, T. O. Nielsen, P. G. Moorman, H. S. Earp and R. C. Millikan, J. Am. Med. Assoc., 2006, 295, 2492–2502 CrossRef CAS PubMed.
- T. Sorlie, C. M. Perou, R. Tibshirani, T. Aas, S. Geisler, H. Johnsen, T. Hastie, M. B. Eisen, M. van de Rijn, S. S. Jeffrey, T. Thorsen, H. Quist, J. C. Matese, P. O. Brown, D. Botstein, P. E. Lonning and A. L. Borresen-Dale, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10869–10874 CrossRef CAS PubMed.
- D. J. Slamon, W. Godolphin, L. A. Jones, J. A. Holt, S. G. Wong, D. E. Keith, W. J. Levin, S. G. Stuart, J. Udove and A. Ullrich, et al. , Science, 1989, 244, 707–712 CAS.
- Y. Yarden and M. X. Sliwkowski, Nat. Rev. Mol. Cell Biol., 2001, 2, 127–137 CrossRef CAS PubMed.
- A. C. Wolff, M. E. Hammond, J. N. Schwartz, K. L. Hagerty, D. C. Allred, R. J. Cote, M. Dowsett, P. L. Fitzgibbons, W. M. Hanna, A. Langer, L. M. McShane, S. Paik, M. D. Pegram, E. A. Perez, M. F. Press, A. Rhodes, C. Sturgeon, S. E. Taube, R. Tubbs, G. H. Vance, M. van de Vijver, T. M. Wheeler and D. F. Hayes, Arch. Pathol. Lab. Med., 2007, 131, 18–43 CAS.
- L. Loo, J. A. Capobianco, W. Wu, X. Gao, W. Y. Shih, W. H. Shih, K. Pourrezaei, M. K. Robinson and G. P. Adams, Anal. Chem., 2011, 83, 3392–3397 CrossRef CAS PubMed.
- L. Chun, S. E. Kim, M. Cho, W. S. Choe, J. Nam, D. W. Lee and Y. Lee, Sens. Actuators, B, 2013, 186, 446–450 CrossRef CAS.
- Q. A. M. Al-Khafaji, M. Harris, S. Tombelli, S. Laschi, A. P. F. Turner, M. Mascini and G. Marrazza, Electroanalysis, 2012, 24, 735–742 CrossRef CAS.
- X. Wu, H. Liu, J. Liu, K. N. Haley, J. A. Treadway, J. P. Larson, N. Ge, F. Peale and M. P. Bruchez, Nat. Biotechnol., 2003, 21, 41–46 CrossRef CAS PubMed.
- S. Lee, H. Chon, M. Lee, J. Choo, S. Y. Shin, Y. H. Lee, I. J. Rhyu, S. W. Son and C. H. Oh, Biosens. Bioelectron., 2009, 24, 2260–2263 CrossRef CAS PubMed.
- H. Park, S. Lee, L. Chen, E. K. Lee, S. Y. Shin, Y. H. Lee, S. W. Son, C. H. Oh, J. M. Song, S. H. Kang and J. Choo, Phys. Chem. Chem. Phys., 2009, 11, 7444–7449 RSC.
- C. L. Nehl, H. Liao and J. H. Hafner, Nano Lett., 2006, 6, 683–688 CrossRef CAS PubMed.
- F. Hao, C. L. Nehl, J. H. Hafner and P. Nordlander, Nano Lett., 2007, 7, 729–732 CrossRef CAS PubMed.
- C. G. Khoury and T. Vo-Dinh, J. Phys. Chem. C, 2008, 112, 18849–18859 CAS.
- C. Hrelescu, T. K. Sau, A. L. Rogach, F. Jäckel and J. Feldmann, Appl. Phys. Lett., 2009, 94, 153113 CrossRef.
- S. K. Dondapati, T. K. Sau, C. Hrelescu, T. A. Klar, F. D. Stefani and J. Feldmann, ACS Nano, 2010, 4, 6318–6322 CrossRef CAS PubMed.
- W. Ma, M. Sun, L. Xu, L. Wang, H. Kuang and C. Xu, Chem. Commun., 2013, 49, 4989–4991 RSC.
- H. Yuan, Y. Liu, A. M. Fales, Y. L. Li, J. Liu and T. Vo-Dinh, Anal. Chem., 2013, 85, 208–212 CrossRef CAS PubMed.
- A. S. Indrasekara, R. Thomas and L. Fabris, Phys. Chem. Chem. Phys., 2015, 17, 21133–21142 RSC.
- L. Rodriguez-Lorenzo, R. A. Alvarez-Puebla, I. Pastoriza-Santos, S. Mazzucco, O. Stephan, M. Kociak, L. M. Liz-Marzan and F. J. G. de Abajo, J. Am. Chem. Soc., 2009, 131, 4616–4618 CrossRef CAS PubMed.
- C. Jehn, B. Küstner, P. Adam, A. Marx, P. Ströbel, C. Schmuck and S. Schlücker, Phys. Chem. Chem. Phys., 2009, 11, 7499–7504 RSC.
- M. Schütz, D. Steinigeweg, M. Salehi, K. Kömpe and S. Schlücker, Chem. Commun., 2011, 47, 4216–4218 RSC.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- C. Chen, J. Peng, H. S. Xia, G. F. Yang, Q. S. Wu, L. D. Chen, L. B. Zeng, Z. L. Zhang, D. W. Pang and Y. Li, Biomaterials, 2009, 30, 2912–2918 CrossRef CAS PubMed.
- A. D. Hartkopf, F. A. Taran, M. Wallwiener, M. Hahn, S. Becker, E. F. Solomayer, S. Y. Brucker, T. N. Fehm and D. Wallwiener, Eur. J. Cancer, 2014, 50, 2550–2559 CrossRef PubMed.
- B. Rack, C. Schindlbeck, J. Juckstock, U. Andergassen, P. Hepp, T. Zwingers, T. W. Friedl, R. Lorenz, H. Tesch, P. A. Fasching, T. Fehm, A. Schneeweiss, W. Lichtenegger, M. W. Beckmann, K. Friese, K. Pantel, W. Janni and S. S. Group, J. Natl. Cancer. Inst., 2014, 106 Search PubMed.
- C. Simmons, N. Miller, W. Geddie, D. Gianfelice, M. Oldfield, G. Dranitsaris and M. J. Clemons, Annu. Oncol., 2009, 20, 1499–1504 CrossRef CAS PubMed.
- T. Fehm, O. Hoffmann, B. Aktas, S. Becker, E. F. Solomayer, D. Wallwiener, R. Kimmig and S. Kasimir-Bauer, Breast Cancer Res., 2009, 11, R59 CrossRef PubMed.
- B. Aktas, V. Muller, M. Tewes, J. Zeitz, S. Kasimir-Bauer, C. R. Loehberg, B. Rack, A. Schneeweiss and T. Fehm, Gynecol. Oncol., 2011, 122, 356–360 CrossRef CAS PubMed.
- S. Kasimir-Bauer, O. Hoffmann, D. Wallwiener, R. Kimmig and T. Fehm, Breast Cancer Res., 2012, 14, R15 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6an00927a |
| This journal is © The Royal Society of Chemistry 2016 |