
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoanodic and photocathodic behaviour of La5Ti2CuS5O7 electrodes in the water splitting reaction†
Guijun
Ma
ab,
Yohichi
Suzuki
ac,
Rupashree Balia
Singh
c,
Aki
Iwanaga
a,
Yosuke
Moriya
ab,
Tsutomu
Minegishi
ab,
Jingyuan
Liu
a,
Takashi
Hisatomi
ab,
Hiroshi
Nishiyama
bd,
Masao
Katayama
ab,
Kazuhiko
Seki
c,
Akihiro
Furube
c,
Taro
Yamada
bd and
Kazunari
Domen
*ab
aDepartment of Chemical System Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: domen@chemsys.t.u-tokyo.ac.jp; Fax: +81-3-5841-8838; Tel: +81-3-5841-1148
bJapan Technological Research Association of Artificial Photosynthetic Chemical Process (ARPChem), 5-1-5 Kashiwanoha, Kashiwa-shi, Chiba 277-8589, Japan
cNational Institute of Advanced Industrial Science and Technology (AIST), Tsukuba Central 5, 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan
dDepartment of Chemical System Engineering, The University of Tokyo, 5-1-5 Kashiwanoha, Kashiwa-shi, Chiba 277-8589, Japan
First published on 11th June 2015
Abstract
The particulate semiconductor La5Ti2CuS5O7 (LTC) with a band gap energy of 1.9 eV functioned as either a photocathode or a photoanode when embedded onto Au or Ti metal layers, respectively. By applying an LTC/Au photocathode and LTC/Ti photoanode to, respectively, photoelectrochemical (PEC) water reduction and oxidation concurrently, zero-bias overall water splitting was accomplished under visible light irradiation. The band structures of LTC/Au and LTC/Ti calculated using a semiconductor device simulator (AFORS-HET) confirmed the critical role of the solid/solid junction of the metal back contact in the charge separation and PEC properties of LTC photoelectrodes. The prominently long lifetime of photoexcited charge carriers in LTC, confirmed by transient absorption spectroscopy, allowed the utilization of both photoexcited electrons and holes depending on the band structure at the solid/solid junction.
Introduction
The exploration and development of solar energy related technologies has attracted much interest in recent years because of the global concern for environmental problems and depletion of fossil energy resources. Photoelectrochemical (PEC) water splitting is one of the ideal approaches to converting solar energy into H2 as a clean and renewable chemical resource. To fabricate a photoelectrode for PEC water splitting, semiconductor thin films are deposited on a conductive layer. Because of the band bending caused by charge carrier diffusion between the semiconductor and the electrolyte at the solid/liquid junction to establish thermal equilibrium, in general, n-type semiconducting materials function as photoanodes for water oxidation, while p-type semiconducting materials function as photocathodes for water reduction.1,2 In addition, the backside conductive layers are chosen so as to form an ohmic contact for the majority carriers of semiconductors at the backside solid/solid junction in order to avoid undesirable rectification of carrier transport.2,3 To achieve unassisted PEC water splitting, also referred to as artificial photosynthesis, combinations of photoelectrodes and photovoltaic cells have also been studied recently.4La5Ti2CuS5O7 (LTC) is an oxysulfide semiconductor material.5–9 Diffuse reflectance spectroscopy reveals that the light absorption edge of LTC is 650 nm, which is equal to a band gap energy of 1.9 eV.6 Our group has reported that LTC exhibited photocatalytic activity for both water reduction and oxidation under visible light irradiation in the presence of sacrificial reagents,6,7 which implied that the conduction band of LTC was more negative than the hydrogen evolution potential, E(H+/H2), and that the valence band was more positive than the oxygen evolution potential, E(O2/H2O). Besides, the abundances of the elements Ti, Cu, and La in the earth's crustal rocks are 6320, 68, and 35 ppm, respectively, which are much higher than those of the elements widely used in semiconducting materials for photovoltaic and PEC applications, such as Ga (19 ppm) and In (0.24 ppm).10 Thus, LTC is a prospective candidate for solar-driven PEC water splitting. Our recent study revealed that the photocathodic current of LTC photoelectrodes fabricated by the particle transfer (PT) method was improved by a factor of eight by doping 1 mol% Sc into the LTC powder precursor.8
In the present work, we studied the PEC properties of LTC photoelectrodes prepared by the PT method using different metal back contacts in the water splitting reaction. By exploiting the PT method, a monolayer of LTC particles was embedded into Au or Ti metal layers.3,11 It was found that LTC photoelectrodes with Au and Ti as back contact layers showed photocathodic and photoanodic current under light irradiation attributable to PEC hydrogen and oxygen evolution, respectively. Furthermore, visible light irradiation of LTC/Ti and LTC/Au connected with each other drove PEC overall water splitting at a moderate Faradaic efficiency (∼80%) without the aid of an external voltage or photovoltaic cells. The unique behaviour of LTC photoelectrodes can be explained by the relative barrier heights at the LTC/electrolyte and LTC/metal junctions.
Experimental
LTC powder was prepared by a solid state reaction as described in the ESI.†5,6 X-ray diffraction patterns (Fig. S1 in ESI†) and scanning electron micrographs (Fig. S2 in ESI†) revealed that the produced LTC was a well-crystallized rod-shaped material. The diameter and length of LTC rods ranged from 0.7 to 1.4 μm and 2 to 6 μm, respectively. To fabricate photoelectrodes by the PT method, the LTC powder was densely dispersed on glass plates (Scheme S1 in ESI†). A thin (∼2.5 μm) metallic layer of Au or Ti was deposited on top of stacked LTC particles by evaporation (for Au) or radio-frequency (RF) magnetron sputtering (for Ti). In this process, the top layer of LTC particles was embedded into the Au or Ti film, which ensured an intimate electrical and physical contact between the semiconductor particles and the metal layer. The metal layer was bonded to a second glass plate by double-face tape and then peeled off of the primary glass plate. Thus, photoelectrodes of LTC/metal (Au or Ti)/tape/glass plate were fabricated. The LTC/Au electrode was loaded with Pt (1 nm) by sputtering and the LTC/Ti electrode was loaded with cobalt phosphate (CoPi) by electrodeposition12 (see ESI†) to enhance PEC H2 or O2 evolution on the surface. Current–potential curves for the PEC water splitting reaction were acquired in an aqueous solution containing 0.1 M Na2SO4 with a pH value of 12 adjusted by NaOH. A 300 W xenon (Xe) lamp equipped with a mirror module was used to irradiate visible light (420 nm < λ < 800 nm). The power spectra of the Xe lamp and the standard AM1.5G irradiation are shown in Fig. S1.† The photon flux of the Xe lamp was 2.1 × 1018 photon s−1 cm−2 in the wavelength region shorter than 650 nm, the absorption edge wavelength of LTC, and was roughly 19 times stronger than that of sunlight.Results
Fig. 1 shows the current–potential curves of LTC/Au and LTC/Ti photoelectrodes during PEC water splitting under chopped light irradiation in an aqueous solution at pH 12. The photoresponses of the two electrodes were different from each other: the LTC/Au photoelectrode showed a photocathodic current typical of p-type semiconductor photoelectrodes (Fig. 1a), while the LTC/Ti photoelectrode exhibited a photoanodic current characteristic of n-type semiconductor photoelectrodes (Fig. 1c). The onset potentials of the photocathodic current on bare LTC/Au and the photoanodic current on bare LTC/Ti were 0.6 and 0.1 V vs. RHE, respectively. In addition, photoanodic and photocathodic currents were observed on LTC/Au and LTC/Ti when the electrode potentials were more positive than 0.6 V vs. RHE and more negative than 0.1 V vs. RHE, respectively. The photocathodic and photoanodic currents observed on the LTC/Au and the LTC/Ti photoelectrodes, respectively, suggest the occurrence of PEC H2 and O2 evolution on the surface viaeqn (1) and (2):| 4H2O + 4e− → 2H2 + 4 OH− (on LTC/Au) | (1) |
| 4OH− + 4h+ → O2 + 2H2O (on LTC/Ti) | (2) |
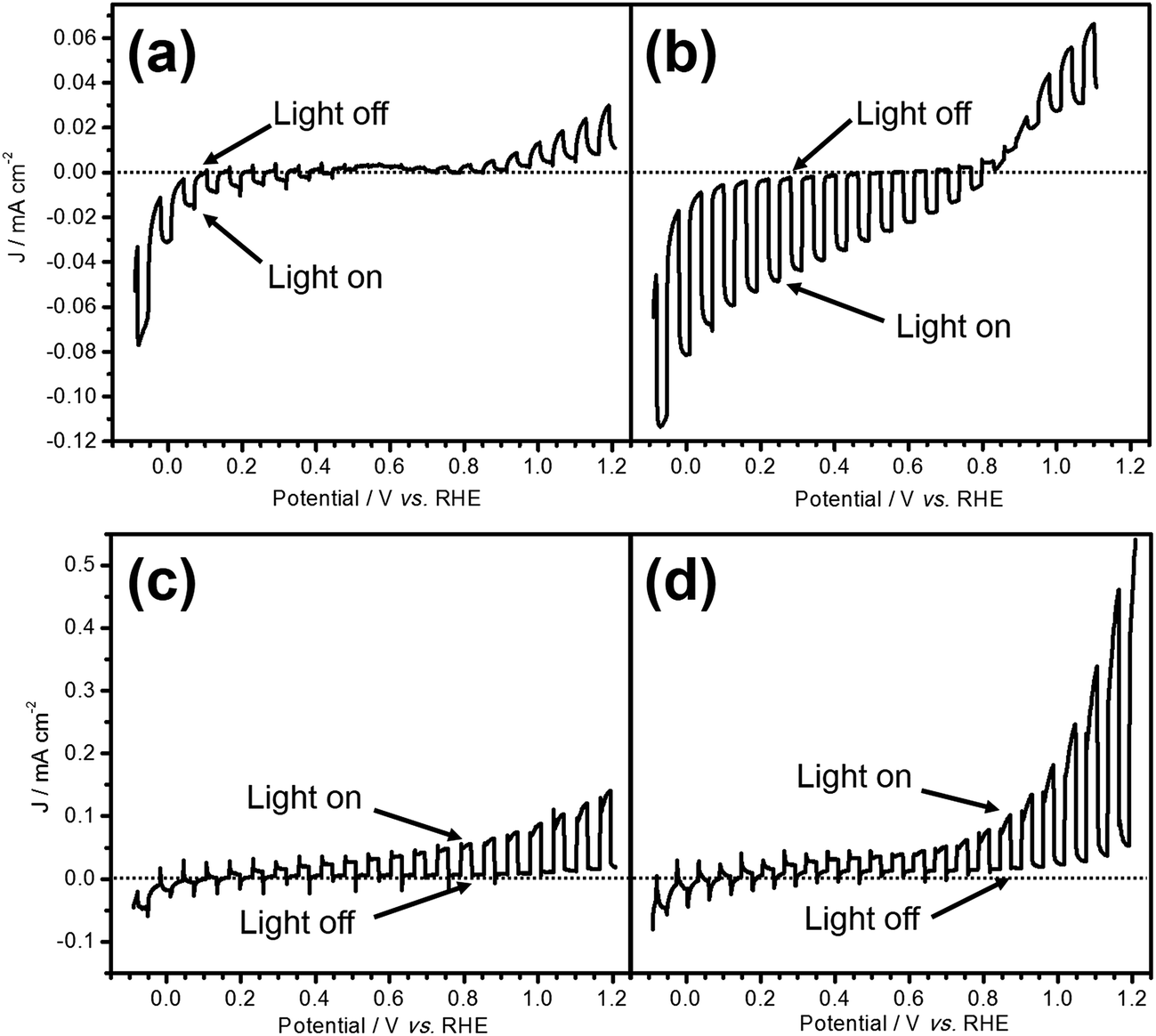 | ||
| Fig. 1 Potential–current curves of (a) LTC/Au, (b) Pt/LTC/Au, (c) LTC/Ti, and (d) CoPi/LTC/Ti electrodes under visible light (λ > 420 nm) irradiation. The reference and counter electrodes used were an Ag/AgCl electrode and Pt wire, respectively. The potential was scanned in the positive direction. | ||
Loading Pt as a H2 evolution catalyst and CoPi as an O2 evolution catalyst on the surface of LTC/Au and LTC/Ti increased the photocathodic and photoanodic currents, respectively, as shown in Fig. 1b and d. As a result, the difference between the photocathodic and photoanodic currents was increased.
To confirm PEC gas production on the respective photoelectrodes, a sealed two-electrode system was constructed using Pt/LTC/Au or CoPi/LTC/Ti as the working electrode and Pt wire as the counter electrode. A half and a quarter of the total charge passing as photocathodic and photoanodic current correspond to the amounts of H2 and O2 generated during PEC water splitting, respectively, given that the Faradaic efficiency is unity. A comparison of the measured amount of gas generated on a Pt/LTC/Au photocathode and the amount calculated from the total charge is shown in Fig. 2a, along with the current–time profile in the inset. A photocathodic current was generated continuously on the Pt/LTC/Au photoelectrode under light irradiation at −0.9 V vs. counter electrode and stopped immediately when the illumination was turned off. The Faradaic efficiency of the Pt/LTC/Au photocathode was 97% for PEC H2 production over 20 h. Fig. 2b shows the results of gas detection on a CoPi/LTC/Ti photoanode in PEC water oxidation at 0.8 V vs. counter electrode. After a 23 h reaction, the Faradaic efficiency of the CoPi/LTC/Ti photoanode for O2 production was estimated to be 79%. The comparatively lower efficiency of the CoPi/LTC/Ti photoanode was most likely due to the partial photooxidation of sulphide ions in LTC.
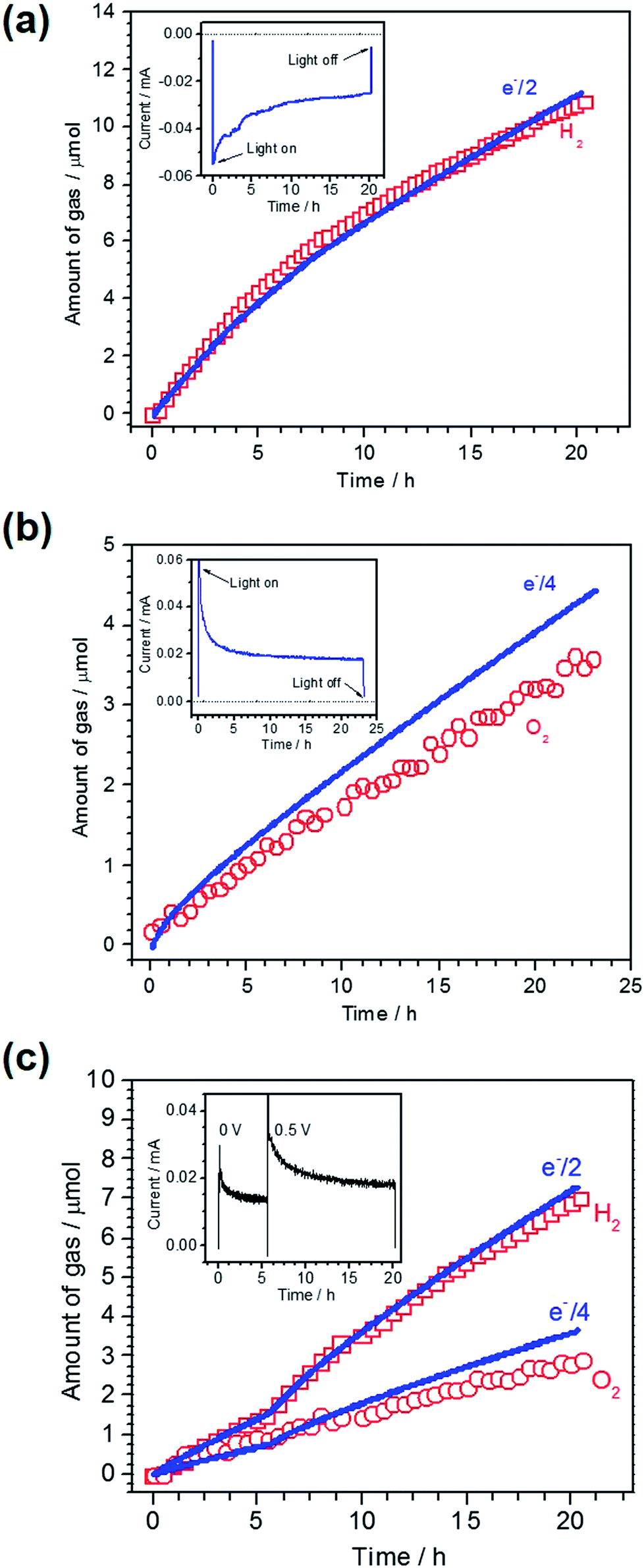 | ||
| Fig. 2 Evolution of hydrogen/oxygen from the two-electrode system under visible light (λ > 420 nm) irradiation. (a) Pt/LTC/Au (3.5 cm2) – Pt wire under a bias of −0.9 V, (b) CoPi/LTC/Ti (4.3 cm2) – Pt wire under a bias of 0.8 V and (c) CoPi/LTC/Ti (4.5 cm2) – Pt/LTC/Au (2 cm2) under a bias of 0 and 0.5 V. The inset shows current–time curves recorded during the respective measurements. | ||
As shown in Fig. 1b and d, the onset potentials of the photocathodic and photoanodic currents were 0.8 and 0.1 V vs. RHE for Pt/LTC/Au and CoPi/LTC/Ti photoelectrodes, respectively. Therefore, unassisted PEC water splitting was feasible using these two photoelectrodes connected in series. Fig. 2c shows the time course of H2 and O2 evolution and the current–time curve (in the inset) for Pt/LTC/Au and CoPi/LTC/Ti photoelectrodes connected in series. During the first 5 h, a photocurrent was generated without any additional external voltage under visible light irradiation. Subsequently, an external voltage of 0.5 V was applied to increase the photocurrent and to observe gas production more easily. As shown in Fig. 2c, the evolution of H2 and O2 occurred concurrently with the observation of photocurrent. After 20 h of illumination, the Faradaic efficiencies for PEC H2 and O2 evolution on Pt/LTC/Au and CoPi/LTC/Ti photoelectrodes were calculated to be 95% and 78%, respectively. These values were close to the Faradaic efficiencies observed when either of the photoelectrodes and a Pt counter electrode were used.
Discussion
It was found that the PEC behaviour of LTC photoelectrodes varied depending on the type of back metal conductor, even though the LTC powder used was the same. Specifically, the use of Au and Ti as the back metal contacts resulted in the generation of photocathodic and photoanodic currents at relatively positive (0.8 V vs. RHE) and negative (0.1 V vs. RHE) potentials, respectively. Moreover, because the potential regions where the photocathodic and photoanodic LTC electrodes could generate photocurrents overlapped, Pt/LTC/Au and CoPi/LTC/Ti photoelectrodes connected in series were capable of splitting water into H2 and O2 at a moderate Faradaic efficiency (∼80%) for oxysulfide photoelectrodes. The importance of the back metal conductor in the PEC properties such as the photocurrent and the onset potential of photoanodic current has been discussed in terms of the formation of Ohmic, Schottky, and Bardeen-type junctions in earlier studies on titanium oxide photoanodes.13,14 However, the switching of the photoanodic response to photocathodic response was not observed although metal conductors with various work functions were used. In nanoparticulate systems such as PbS and copper zinc indium sulphide (ZCIS) quantum dots, in which the development of a depletion layer is not generally considered, both photoanodic and photocathodic are sometimes observed depending on the electrode potential.15,16 A photocathodic response was observed for PbS photoelectrodes when the electrode potential was once held at a positive potential, presumably because the surface trap states were unfilled with electrons and vice versa.15 Electrochemical stripping of Cu+ ions occurred in CZIS quantum dots when the electrode potential was held at a positive potential. Because of the formation of Cu vacancies, the electrochemically-treated CZIS photoelectrode showed enhanced photocathodic response.16 However, the situation is likely different for photoelectrodes consisting of micrometre-sized LTC particles in which the band bending at the interface generally rectifies migration of carriers. In the following section, we will discuss the characteristic PEC behaviour and the onset potential on the basis of band diagrams, taking account of the properties of the back metal contact.For a qualitative understanding of the effect of metal substrates on the PEC properties of LTC, the band structures of electrolyte/LTC/Au and electrolyte/LTC/Ti were analysed using a semiconductor device simulator (AFORS-HET17). The material parameters for LTC are shown in Fig. 3a: band gap Eg = 1.9 eV6 and the top of the valence band Ev = 5.7 eV (see Fig. S3†). The work functions of Au and Ti were reported to be 5.1 and 4.33 eV, respectively.18 The electrolyte can be treated as a hypothetical metal with a work function between 4.4 eV (the hydrogen evolution potential) and 5.67 eV (the oxygen evolution potential).19 In the following discussion, only the case of an electrolyte potential of 4.4 eV is described for simplicity because, qualitatively, the same conclusions are reached regardless of the potential difference between 4.4 and 5.67 eV. The other parameters used in the calculations are listed in Table S1.†
 | ||
| Fig. 3 Energy diagrams for the LTC electrodes. (a) Energy diagrams for the electrolyte, LTC, and metal substrates before contacts. (b and c) The equilibrium band structures for (b) electrolyte/LTC/Au and (c) electrolyte/LTC/Ti. The black, red, and blue curves represent the conduction band, valence band, and Fermi level, while the solid, dotted, and dashed curves correspond to the cases in which the LTC is assumed to be a p-, intrinsic-, and n-type semiconductor, respectively. (d and e) The band diagrams for (d) electrolyte/LTC/Au and (e) electrolyte/LTC/Ti under AM1.5 illumination at varying electrode potentials (E) for the case of LTC as a p-type semiconductor. | ||
Fig. 3b and c show the equilibrium band diagrams of LTC in contact with the electrolyte on one side and Au or Ti on the other. Although LTC was previously studied as a photocathode,8,9 the LTC photoelectrodes function as both photocathode and photoanode depending on the kinds of back contact metals. Therefore, three types of conductivity should be considered for the LTC semiconductor, viz., n-type, intrinsic-type, and p-type, with, respectively, the following Fermi levels (EF): −4.0, −4.75, and −5.4 eV (relative to the vacuum level). The black, red, and blue curves represent the conduction band, valence band, and the Fermi level; the solid, dotted, and dashed curves correspond to the cases in which LTC is assumed to be a p-, intrinsic-, and n-type semiconductor, respectively. Note that the barrier height, ϕb, at interfaces was assumed to be ϕb = ϕ − χ, where ϕ is the work function of the contact material and χ is the electron affinity of the semiconductor.20 Depending on the energy difference between the Fermi level and the work function of the contact materials, the band structure of LTC is expected to rearrange as shown in Fig. 3b and c. Taking the case of the p-type (n-type) LTC as an example, a two-sided downward (upward) bend is formed because the majority carriers in LTC diffuse into both the electrolyte and the metal layer. However, because of the different work functions of the Au and Ti substrates, the difference in band-bending height between the LTC/electrolyte and LTC/Au junctions is larger than the difference between the LTC/electrolyte and LTC/Ti junctions.
Fig. 3d and e plot the band diagrams of LTC/Au and LTC/Ti electrodes under AM1.5G illumination at varying applied biases with the conditions that the photons having energies larger than the band gap of LTC are all absorbed and LTC is a p-type semiconductor. The solid and dotted curves in Fig. 3d and e depict the band structures at the potential with no bias (E = 0) and at the potential where no photocurrent is generated, which corresponds to the onset potential (E = Eonset). The pale black and red lines indicate, respectively, the conduction and valence bands for E = Eonset ± 0.5 V. For the LTC/Au electrode (see Fig. 3d), the cathodic current would be expected to occur at E = 0 because the electron (hole) energy level at the electrolyte side is more positive than that at the metal substrate side. As the electrode potential shifts positively, the electron energy level on the metal substrate side also shifts positively. The cathodic current vanishes at E = Eonset, and the photoanodic current eventually starts to flow with a further anodic shift in the electrode potential. On the other hand, for the LTC/Ti electrode shown in Fig. 3e, Eonset should be very close to zero because the electron (hole) energy level at the metal substrate side is hardly more negative than that of the electrolyte. The plots in Fig. 3d and e match well with the PEC behaviour shown in Fig. 1.
In summary, the relative barrier heights at the LTC/electrolyte and LTC/metal junctions dominate the directions of the photogenerated charge migration, which eventually leads to the photocathodic and photoanodic response on LTC/Au and LTC/Ti, respectively. Note that, although the present study is based on the p-type LTC, the dependence of Eonset on the metal substrate type can be explained in a similar manner for intrinsic-type and n-type LTC.
It is generally not believed that photoexcited electrons and holes generated in the same semiconductor are both used for PEC reactions, because the semiconductor/liquid junction rectifies the direction of charge migration. The unique properties of LTC are possibly due to the special band and crystal structures that allow charge migration over a long range. The density functional theory (DFT) calculations in our previous study suggested that the valence band and the conduction band of LTC were spatially localized around one-dimensional chains of CuS4 tetrahedra and Ti(O,S)6 octahedra, respectively.6 Such a structure was considered to be favourable for charge separation and transport. In fact, it was recently suggested that the charge migration distance in LTC was on the order of microns in the case of LTC/Au photocathodes, because PEC deposition of noble metal nanoparticles occurred only onto the tip of rod-like LTC particles a few micrometres in length.9 In order to further investigate the nature of photogenerated charge carriers in LTC powder, we employed transient absorption spectroscopy (TAS), which has been successfully used in monitoring carrier dynamics in many semiconductor photocatalysts21,22 (see ESI† for experimental details). Since the powder was opaque, we collected and analyzed the diffuse reflected light from the sample, instead of transmitted light, to obtain information on the kinetics of transient species; femtosecond time resolved diffuse reflectance (fs-TRDR) spectroscopy was employed. The transient time profile of the LTC powder probed at 900 nm (λexc = 535 nm) shown in Fig. 4 suggests the presence of long-lived charge carriers. The experiment was performed under weak excitation conditions (pump power ≈ 0.1 μJ) to rule out the second-order electron–hole recombination processes. As is evident from Fig. 4, even at a reasonably long delay time of 2000 ps, ca. 78% of photogenerated carriers survived. Thus, recombination was much slower in LTC than in other visible-light-driven photocatalysts such as GaN:ZnO (66% at 500 ps), TaON (28% at 500 ps), Ta3N5 (12% at 100 ps), and LaTiO2N (50% at 270 ps).21,22 Even though at this preliminary stage of investigation we cannot conclusively assign the observed transient absorption to electrons or holes, such a long photoexcited carrier lifetime hints toward the validity of the long charge migration distance estimated in our previous study9 and possibly also explains the sensitivity of the PEC properties of LTC electrodes to the backside solid/solid junction.
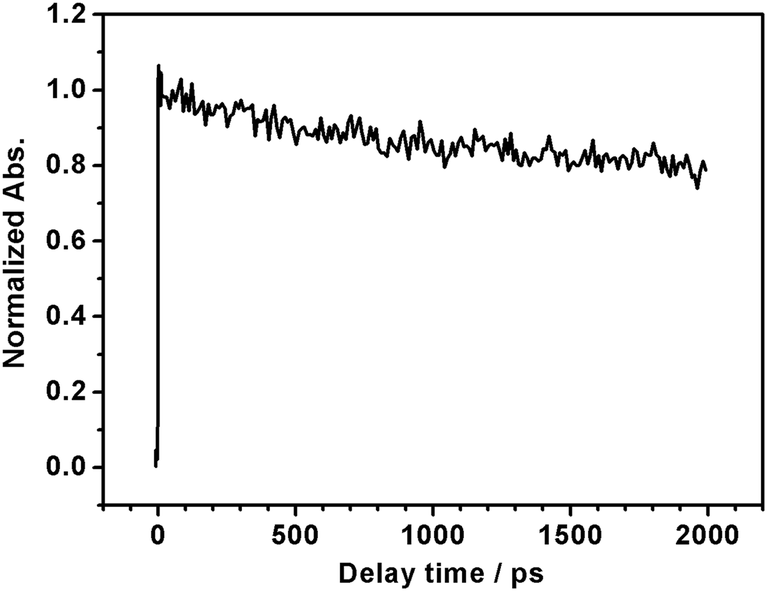 | ||
| Fig. 4 Normalized femtosecond transient time profile of LTC powder probed at 900 nm (λexc = 535 nm, pump power ≈ 0.1 μJ). | ||
Despite the long lifetime of the photoexcited state and the crystal and the band structure being favourable for charge separation and transport, the photocurrent observed in this study was not high or stable. Therefore, significant advancement in the PEC properties of LTC photoelectrodes is needed to make the best of the unique properties of the material. It was found that the photocathodic current could be increased almost by an order of magnitude by p-type doping of LTC.8 Significant enhancement in the photocathodic and the photoanodic responses can be thus expected by appropriate compositional modifications. On the other hand, the decrease of the photocurrent due to photocorrosion was more critical for the CoPi/LTC/Ti photoanode. Therefore, upgrading the stability of LTC photoanodes is demanded. Recently, surface modifications by amorphous titanium oxide layers were reported to make some non-oxide photoanodes durable during the PEC water oxidation reaction.23 Advancement in such surface modification techniques for LTC photoanodes could offer a solution to the stability issue.
Conclusion
The PEC properties of LTC/Au and LTC/Ti photoelectrodes fabricated by the PT method were discussed in connection with water splitting. Under light irradiation, LTC/Au acted as a photocathode for H2 evolution, while LTC/Ti acted as a photoanode for O2 evolution. By combining the LTC/Au photocathode and LTC/Ti photoanode, zero-bias overall water splitting was achieved under visible light irradiation. Using a semiconductor device simulator (AFORS-HET), the p-type and n-type characteristics observed for LTC/Au and LTC/Ti photoelectrodes, respectively, were ascribed to the difference in barrier height between the LTC/electrolyte and the LTC/metal junctions under light irradiation. As already mentioned above, LTC exhibited a carrier lifetime much longer than some of the other commonly investigated visible-light-driven photocatalysts. This may account for the uniqueness of this material. Our work on LTC opens up a new possibility, namely, that the conductivity type of a semiconductor photoelectrode can be controlled by engineering the barrier heights not only at the semiconductor/liquid junction, but also at the semiconductor/back metal junction.Acknowledgements
This work was supported by the Artificial Photosynthesis Project of the Ministry of Economy, Trade and Industry (METI) of Japan and Grants-in-Aid for Specially Promoted Research (no. 23000009) and for Young Scientists (A) (no. 15H05494) of the Japan Society for the Promotion of Science (JSPS).References
- (a) A. J. Bard, Science, 1980, 207, 139 CAS; (b) F. Williams and A. J. Nozik, Nature, 1984, 312, 21 CrossRef CAS PubMed; (c) M. Grätzel, Nature, 2001, 414, 338 CrossRef PubMed.
- M. G. Walter, E. L. Warren, J. R. Mckone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446 CrossRef CAS PubMed.
- T. Minegishi, N. Nishimura, J. Kubota and K. Domen, Chem. Sci., 2013, 4, 1120 RSC.
- (a) J. R. Mckone, N. S. Lewis and H. B. Gray, Chem. Mater., 2014, 26, 407 CrossRef CAS; (b) F. F. Abdi, L. Han, A. H. M. Smets, M. Zeman, B. Dam and R. Krol, Nat. Commun., 2013, 4, 2195 Search PubMed; (c) C. Liu, J. Tang, H. M. Chen, B. Liu and P. Yang, Nano Lett., 2013, 13, 2989 CrossRef CAS PubMed; (d) X. Wang, K. Peng, Y. Hu, F. Zhang, B. Hu, L. Li, M. Wang, X. Meng and S. Lee, Nano Lett., 2014, 14, 18 CrossRef CAS PubMed; (e) C. R. Cox, J. Z. Lee, D. G. Nocera and T. Buonassisi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14057 CrossRef CAS PubMed.
- V. Meignen, L. Cario, A. Lafond, Y. Moelo, C. Guillot-Deudon and A. Meerschaut, J. Solid State Chem., 2004, 177, 2810 CrossRef CAS PubMed.
- T. Suzuki, T. Hisatomi, K. Teramura, Y. Shimodaira, H. Kobayashi and K. Domen, Phys. Chem. Chem. Phys., 2012, 14, 15475 RSC.
- M. Katayama, D. Yokoyama, Y. Maeda, Y. Ozaki, M. Tabata, Y. Matsumoto, A. Ishikawa, J. Kubota and K. Domen, Mater. Sci. Eng., B, 2010, 173, 275 CrossRef CAS PubMed.
- J. Liu, T. Hisatomi, G. Ma, A. Iwanaga, T. Minegishi, Y. Moriya, M. Katayama, J. Kubota and K. Domen, Energy Environ. Sci., 2014, 7, 2239 CAS.
- G. Ma, J. Liu, T. Hisatomi, T. Minegishi, Y. Moriya, M. Iwase, H. Nishiyama, M. Katayama, T. Yamada and K. Domen, Chem. Commun., 2015, 51, 4302 RSC.
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, 2nd edn, 1997 Search PubMed.
- (a) K. Ueda, T. Minegishi, J. Clune, M. Nakabayashi, T. Hisatomi, H. Nishiyama, M. Katayama, N. Shibata, J. Kubota, T. Yamada and K. Domen, J. Am. Chem. Soc., 2015, 137, 2227 CrossRef CAS PubMed; (b) H. Urabe, T. Hisatomi, T. Minegishi, J. Kubota and K. Domen, Faraday Discuss., 2014, 176, 213 RSC; (c) H. Kumagai, T. Minegishi, Y. Moriya, J. Kubota and K. Domen, J. Phys. Chem. C, 2014, 118, 16386 CrossRef CAS.
- M. W. Kanan, Y. Surendranath and D. G. Nocera, Chem. Soc. Rev., 2009, 38, 109 RSC.
- A. Shiga, A. Tsujiko, T. Ide, S. Yae and Y. Nakato, J. Phys. Chem. B, 1998, 102, 6049 CrossRef CAS.
- M. Kitano, K. Tsujimaru and M. Anpo, Appl. Catal., A, 2006, 314, 179 CrossRef CAS PubMed.
- S. Ogawa, K. Hu, F.-R. F. Fan and A. J. Bard, J. Phys. Chem. B, 1997, 101, 5707 CrossRef CAS.
- N. Guijarro, T. Lana-Villarreal and R. Gómez, Chem. Commun., 2012, 48, 7681 RSC.
- R. Stangl, C. Leendertz and J. Haschke, Solor Energy, InTech, Croatia, 2010 Search PubMed.
- H. B. Michaelson, J. Appl. Phys., 1977, 48, 4729 CrossRef CAS PubMed.
- T. Hisatomi, T. Minegishi and K. Domen, Bull. Chem. Soc. Jpn., 2012, 85, 647 CrossRef CAS.
- K. C. Kao, Dielectric Phenomena in Solids, Elsevier Academic Press, San Diego, 2004 Search PubMed.
- A. Furube, K. Maeda and K. Domen, Proc. SPIE, Solar Hydrogen Nanotechnol. VI, 2011, vol. 8109, p. 810904 Search PubMed.
- R. B. Singh, H. Matsuzaki, Y. Suzuki, K. Seki, T. Minegishi, T. Hisatomi, K. Domen and A. Furube, J. Am. Chem. Soc., 2014, 136, 17324 CrossRef CAS PubMed.
- S. Hu, M. R. Shaner, J. A. Beardslee, M. Lichterman, B. S. Brunschwig and N. S. Lewis, Science, 2014, 344, 1005 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Power spectra of light source, preparation process and SEM images of LTC, schematic of the PT method, UV photoelectron spectroscopy, parameters for calculation, and other experimental procedures. See DOI: 10.1039/c5sc01344e |
| This journal is © The Royal Society of Chemistry 2015 |