 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and properties of ZnTe and ZnTe/ZnS core/shell semiconductor nanocrystals†
Christophe
Lincheneau
a,
Matteo
Amelia
a,
Marek
Oszajca
ab,
Alice
Boccia
c,
Fabio
D'Orazi
c,
Mattia
Madrigale
c,
Robertino
Zanoni
*c,
Raffaello
Mazzaro
ad,
Luca
Ortolani
d,
Vittorio
Morandi
d,
Serena
Silvi
ae,
Konrad
Szaciłowski
bf and
Alberto
Credi
*ae
aNanosciences Laboratory, Dipartimento di Chimica “G. Ciamician”, Università di Bologna, Via Selmi 2, 40126 Bologna, Italy. E-mail: alberto.credi@unibo.it; Fax: +39 051 2099456; Tel: +39 051 2099540
bFaculty of Chemistry, Jagiellonian University, Ingardena 3, 30-060 Kraków, Poland
cDipartimento di Chimica, Università La Sapienza, P.le Aldo Moro 5, 00185 Roma, Italy. E-mail: robertino.zanoni@uniroma1.it; Fax: +39 06 49913328; Tel: +39 06 490324
dCNR-IMM, via Gobetti 101, 40129 Bologna, Italy
eInteruniversity Center for the Chemical Conversion of Solar Energy, Bologna Unit, Via Selmi 2, 40126 Bologna, Italy
fFaculty of Non-Ferrous Metals, AGH University of Science and Technology, al. Mickiewicza 30, 30-060 Kraków, Poland
First published on 27th January 2014
Abstract
We report the synthesis of spherical ZnTe nanocrystals and the successive coating with a ZnS shell to afford core/shell quantum dots. These nanocrystals can represent alternatives to cadmium-based quantum dots but their preparation and properties are challenging and relatively unexplored. The effect of various synthetic parameters on the reaction outcome was investigated, and the resulting nanocrystals were characterized by TEM, EDX, XPS, and spectroscopic measurements. The optical data indicate that these core/shell quantum dots belong to type I, i.e., both the electron and the hole are confined within the ZnTe core. Both the ZnTe core and ZnTe/ZnS core/shell quantum dot samples absorb in the visible region and are not luminescent. The ZnS shell preserves the optical properties of the core and improves the chemical and photochemical stability of the nanoparticles in air equilibrated solution, whereas they appear to be quite fragile in the solid state. XPS results have evidenced the distinct nature of core and core/shell QDs, confirming the formation of QDs with shells of different thicknesses and their evolution due to oxidation upon air exposure. Anodic photocurrent generation was observed when an ITO electrode functionalized with ZnTe/ZnS nanocrystals was irradiated in the visible region in a photoelectrochemical cell, indicating that the quantum dots perform spectral sensitization of the electron injection into the ITO electrode. Conversely, cathodic photocurrent generation was not observed; hence, the QD-modified electrode performs electrical rectification under a photon energy input.
Introduction
Semiconductor quantum dots (QDs)1–3 are nanocrystals that exhibit peculiar size-dependent optical and electronic properties, owing to the quantum confinement of the charge carriers.4 Because of these properties, QDs are promising substitutes for photoactive molecular species,5 and are being extensively investigated for applications that include chemo- and bio-sensing,6–9 biological imaging,10 medical therapy,11 opto-electronic devices,12 and solar cells.13,14QDs are generally prepared1–3 by reacting inorganic precursors in the presence of organic ligands, which eventually form a molecular coating around the QD core and stabilize the nanoparticles against aggregation.15 Chemical modification of this coating allows the regulation of the physicochemical properties of the QDs.16–18 Moreover, the nanocrystal surface can be passivated with a protective layer of another inorganic material, yielding core/shell QDs.19,20
The most commonly used core/shell QDs are certainly CdSe/ZnS; the type I band structure of these QDs, in which the exciton is confined to the core material, brings about valuable optical properties with excellent size-tunability across the visible region, together with a high chemical and photochemical stability. Nowadays, CdSe/ZnS QDs are commercially available in a variety of diameters and surface functionalization. The intrinsic toxicity of cadmium, however, poses concerns for real-world applications of such systems.21 QDs for practical technological uses should ideally be environmentally friendly and, in the case of biologic and medical applications, should not be toxic. The choice of II–VI semiconductors that do not contain harmful elements and possess a band gap energy suitable for visible absorption and emission appears to be limited to ZnTe and ZnSe.19,20,22,23 The size-controlled synthesis of colloidal ZnTe nanoparticles24–32 is particularly challenging and has received much less attention compared to that of other II–VI compounds. ZnTe nanocrystals have been used to make type II core/shell QDs, often in combination with Cd-based semiconductors.20,25,28,31,33 In such systems, however, either the electron or the hole is localized in the shell, yielding QDs that are more sensitive to the environment compared to type I systems. Cadmium-free QDs with interesting optical properties have also been obtained by doping binary semiconductors (e.g., ZnSe) with transition metals (e.g., Mn)34 but their emission wavelength is not finely tunable, or from multinary semiconductors.35,36
ZnTe is a semiconducting material with a direct band gap of 2.26 eV, corresponding to a wavelength of about 550 nm.37 ZnS has a substantially larger band gap (3.61 eV) and band edge levels that enable the construction of luminescent type I or type I1/2 core/shell nanocrystals (Fig. 1).19,20,22 Moreover, these two semiconductors crystallize in the same structure (zinc blende) and exhibit a not too large mismatch of the lattice parameters (≈11%), making ZnS a candidate for the role of shell materials for ZnTe.39
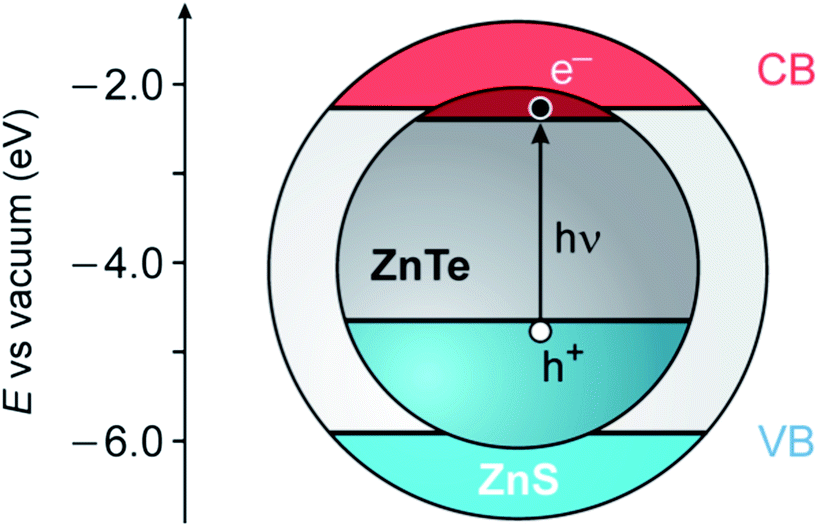 | ||
| Fig. 1 Schematic representation of the possible energy-level alignment of the band edges in ZnTe/ZnS core/shell QDs, based on bulk values, together with the photogeneration of an exciton in the ZnTe core (VB, valence band; CB, conduction band). The VB offset (1.26 eV) is obtained from ref. 38. | ||
Here we describe the size-controlled preparation and characterization of ZnTe and ZnTe/ZnS core/shell QDs, and we report the results of a systematic study aimed at understanding the effect of various parameters on both the core and shell syntheses. The resulting nanocrystals were characterized by TEM, XPS, EDX, and spectroscopic measurements. Photocurrent measurements on ITO electrodes covered with ZnTe/ZnS QDs were also carried out.
Experimental section
Materials
Zinc chloride (ZnCl2·2H2O, analytical grade) was purchased from Merck. Zinc oxide (ZnO, 99.999%), oleic acid (OA, 90%), stearic acid (SA, 98%), 1-octadecene (ODE, 90%), tris(n-butyl)phosphine (TBP), tris(n-octyl)phosphine (TOP), tris(n-octyl)phosphine oxide (TOPO), hexadecylamine (HDA, 98%), octadecylamine (ODA, 99.0%) and tetrabutylammonium hexafluorophosphate were purchased from Aldrich. 1,1′-Dimethyl-4,4′-bipyridinium (methylviologen) dihexafluorophosphate was available from previous investigations. Elemental tellurium (325 mesh, 99.99%) was purchased from Alfa Aesar. Chloroform, methanol and hexane of HPLC grade were purchased from Aldrich and used for synthesis and purification. The chloroform employed for photophysical studies was purchased from Merck Uvasol. The acetonitrile used in the photoelectrochemical experiments was purchased from Romil SPS. Indium-tin oxide (ITO) coated PET transparent photoelectrodes (1 cm2, surface resistivity 60 Ω sq−1) were purchased from Sigma-Aldrich and were used as received.Synthetic procedures
The general methodologies followed for the preparation of the ZnTe core nanocrystals and for the growth of the ZnS shell are described in the Results and discussion section. Synthetic details are reported in the ESI.† In a typical synthesis of the ZnTe core (protocol 4 in Table 1), 8.2 mg (0.1 mmol) of ZnO, 129 mg of stearic acid and 7.9 g (10 mL) of ODE were loaded in a 100 mL three-neck flask. The mixture was stirred under vacuum for 1 h at 70 °C and for 1 h at 120 °C; it was then heated at 270 °C under an argon atmosphere until a clear solution was obtained. The Te solution was prepared in a dry box (N2 inert atmosphere, humidity <0.5 ppm, oxygen <5 ppm) by mixing 25.7 mg of Te (0.2 mmol) with 2.0 mL of TBP and 2.0 mL of ODE. The suspension was stirred and gradually heated up to 170 °C to afford a maximum dissolution of the chalcogen, and then maintained at the injection temperature (80 °C). The Zn-containing mixture was heated up to 300 °C and the Te solution was rapidly injected with a syringe from a rubber sealed vial to the reaction vessel. After the Te injection the nanocrystals were allowed to grow at 270 °C; the reaction was monitored by taking aliquots at different time intervals, and recording UV/vis absorption spectra for each aliquot. The reaction was eventually quenched by cooling down to room temperature; chloroform was added at ca. 60 °C to prevent solidification of the mixture. The QDs were isolated by precipitation with methanol or acetone; after centrifugation the supernatant was discarded and the solid nanocrystals were redissolved in chloroform. This procedure was repeated five times by adding chloroform to dissolve the nanocrystals, and methanol or acetone to trigger their precipitation.| Protocol 1 | Protocol 2 | Protocol 3 | Protocol 4 | Protocol 5 | Protocol 6 | Protocol 7 | |
|---|---|---|---|---|---|---|---|
| a Only one parameter at a time, italicized, was modified upon changing the protocol from 2 to 6. b Time required to reach the saturation regime (no further changes observed in the exciton absorption band). c Wavelength of the exciton absorption band at saturation. d Average diameter of the particle, estimated from TEM images; size range in parentheses. e No nanocrystals formed. | |||||||
| Zn precursor (Zn) | ZnCl2 | ZnO | ZnO | ZnO | ZnO | ZnO | ZnO |
| Zn activation (A) | OA | SA | SA | SA | SA | SA | SA |
| Passivating ligand | — | — | — | ODA (15 eq.) | ODA (15 eq.) | ODA (15 eq.) | ODA (15 eq.), TOPO (15 eq.) |
| Phosphine (P) | TOP | TBP | TBP | TBP | TBP | TBP | TBP |
| Zn/Te/A/P | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:30:30 :1:30:30 |
1:2:4:80 |
1:2:5:80 |
1:2:4:80 |
1:2:4:80 |
1:2:4:80 |
1:5:4:150 |
| T nucleation (°C) | 285 | 300 | 300 | 300 | 300 | 320 | 320 |
| T growth (°C) | 250 | 270 | 270 | 270 | 285 | 285 | 290 |
| t saturation (min) | 90 | 10 | 20 | 10 | 10 | ||
| λ exciton (nm) | 503 | 502 | 510 | 508 | 514 | ||
| Sized (nm) | 5.0 (4.5–5.4) | 4.9 (4.4–5.5) | 6.1 (5.5–6.7) | 6.0 (5.7–6.2) | 8.3 (7.5–9.0) | ||
For the preparation of ZnTe/ZnS core/shell QDs, a SILAR procedure was employed.40 The solution of the Zn precursor was obtained by mixing ZnO (32.6 mg, 0.4 mmol) and OA (0.9 g, 3.2 mmol) with 7.09 g (9 mL) of ODE; the resulting suspension was stirred and heated at 230 °C for about one hour, and then maintained at 80 °C. The S solution was prepared dissolving elemental sulphur (12.8 mg, 0.4 mmol) in 7.9 g (10 mL) of ODE at 80 °C. The ZnTe nanocrystals were prepared according to protocol 4 (Table 1) and were allowed to grow for 20 min. The temperature of the reaction mixture was then decreased to 230 °C, a solution containing 1 g of ODA in 4 mL of ODE was added, and the SILAR procedure was started. A predetermined amount (0.43 mL) of Zn precursor solution was added dropwise and the mixture was stirred at 230 °C for 20 min. The sulphur solution (0.43 mL) was then slowly injected and the mixture was kept at 230 °C for 20 min. The procedure was repeated for the deposition of more ZnS layers, adjusting the added volumes of the precursors considering the increase of the nanocrystal size. The number n of shells deposited is hereafter indicated as nsh.
The CdSe nanocrystals (4.7 nm diameter, HDA/TOPO capped) used for the photocurrent measurements were prepared following a previously published procedure41 very similar to protocol 4 in Table 1 (Tinjection = 250 °C and Tnucleation = 280 °C).
Measurements
TEM measurements were carried out with a Philips CM 100 transmission electron microscope operating at 80 kV, whereas HRTEM observations were performed with a FEI Tecnai F20 TEM equipped with a Schottky emitter and operating at 200 kV. Energy dispersive X-ray (EDX) spectra were recorded with a Philips 515 SEM operating at 15 kV equipped with an EDAX DX4 system and a backscattered detector. A drop of the nanocrystal solution diluted with hexane was deposited on a 400 mesh copper support grid coated with formvar (TAAB Ltd), which was then dried under vacuum or air to remove any solvent trace. The analysis of the morphology and size of the nanocrystals was performed using TEM images with a resolution of 0.25 nm per pixel. From 5 to 10 different pictures were analyzed for each sample, and at least 100 particles were considered to calculate the diameter distribution. The error on the diameter is estimated to be <1 nm. For the EDX experiments, HOPG (a gift from V. Palermo, ISOF-CNR Institute, Bologna) was also employed as the support. Photoelectron spectra were acquired using a modified Omicron NanoTechnology MXPS system equipped with various photon sources and an Omicron EA-127-7 energy analyzer. The solid compounds were homogeneously spread over a graphite tip attached to the XPS sample holder. Excitation was carried out with MgKα photons (1253.6 eV), generated by operating the anode at 14 kV, 16 mA. XPS atomic ratios were estimated from the experimentally determined area ratios of the relevant core lines, corrected for the corresponding theoretical atomic cross-sections and for a square-root dependence of the kinetic energies of the photoelectrons. Absorption and luminescence spectra were recorded with a Perkin-Elmer Lambda 45 spectrophotometer and a Perkin-Elmer LS50B spectrofluorimeter equipped with a Hamamatsu R928 phototube, respectively.In photoelectrochemical experiments, the nanocrystals were deposited onto an ITO-coated PET electrode by dropcasting 200 μL of a core/shell QD solution in CHCl3 (5 μM). Only ZnTe/ZnS (2sh) QDs were used because the amount of the other samples was insufficient to perform meaningful experiments. After the solvent was gently evaporated, the electrode was placed in a quartz cuvette filled with a deoxygenated CH3CN solution of tetrabutylammonium hexafluorophosphate (10−2 M) as the electrolyte, and connected to a Bio-logic SP150 potentiostat. For the cathodic photocurrent experiments, either O2 (1.9 mM) or methylviologen dihexafluorophosphate (0.78 mM) were added to the electrolyte solution as electron acceptors. A three-electrode cell was set up with the modified ITO–PET slide as the working electrode, an Ag/AgCl electrode as the reference, and a platinum wire as the counter electrode. The working electrode was set at the established potential value, and conditioned for 30 s before starting each measurement. The ITO electrode was illuminated with a 150 W high pressure xenon lamp, equipped with a computer-controlled monochromator and a shutter. To avoid any influence of the thickness of the QD layer, the electrode was illuminated through the transparent, conducting support. In this way only the few layers of nanoparticles closer to the electrode can absorb light and hence generate a photocurrent, while the contribution of the film bulk is negligible.
Results and discussion
ZnTe nanocrystals
The synthesis of the ZnTe core was based on the hot injection method,42 which has been widely applied for the preparation of, e.g., CdSe and CdTe QDs. Generally, the active Zn species was produced in situ by reaction of an appropriate Zn precursor with a fatty acid in 1-octadecene (ODE) as a high boiling solvent. The solution was then degassed and heated up to the injection temperature. The tellurium solution was prepared under an inert atmosphere by dissolving Te powder in ODE in the presence of a trialkylphosphine as a complexing agent. The nucleation was initiated by swiftly injecting the Te solution in the Zn-containing reaction mixture at high temperature. The growth of the nanocrystals was then allowed to occur at a fixed temperature, and eventually stopped by cooling down the reaction mixture.In hot injection methodologies the clear temporal separation of the nucleation and growth phases, which is an essential requirement to afford monodisperse colloids, is achieved by rapid mixing of the reagents in a solvent at high temperature. The injection leads to a sudden supersaturation of the solution and subsequent formation of nanocrystal seeds; such a nucleation is quickly quenched by fast cooling of the reaction mixture when the room temperature reactants are added to the hot solvent, and by the decrease of saturation caused by the nucleation burst. Ideally, all the crystal nuclei should be formed at the same time and undergo an identical growth. The nanocrystal growth implies the diffusion of the reactants to the particle and their successive reaction with the surface. Usually, the latter process controls the growth kinetics only at a very high concentration of reactants, that is, in the very first stage of the synthesis. Later, when the reservoir of reactants is partially depleted, the rate-determining step is the diffusion of the reactants. Clearly, any synthetic parameters affecting these processes can influence the crystallinity, size and morphology of the final nanoparticles.
Table 1 summarizes the experimental conditions employed in the syntheses, together with the characteristics of the nanocrystals obtained. Only one parameter at a time (italicized for clarity in Table 1) was modified upon changing the protocol from 2 to 6.
Zn(Et)2 is frequently used as a precursor in the synthesis of ZnTe nanocrystals.25,28,30 This reactant, while affording a facile activation of the metal, is pyrophoric and thus difficult to handle. Hence, in our protocols we used ZnCl2 or ZnO as the zinc sources. The activation of ZnCl2 with OA in ODE was difficult: the metal salt was fully dissolved only upon raising the temperature to 270 °C. The subsequent injection of either Te(TOP) or Te(TBP) solutions did not produce nanocrystals (Table 1, protocol 1).
Conversely, when ZnO/SA was used (protocol 2), the formation of nanocrystals was detected upon the fast injection of a solution of Te(TBP) (2 eq.) at 300 °C. The obtained QDs had a mean diameter of 5.0 nm (from TEM data), and exhibited a broad exciton absorption band at about 503 nm (Fig. 2A). Under these conditions the nucleation process was found to be relatively slow, as the first nanocrystal formation was detected only after 40 minutes.
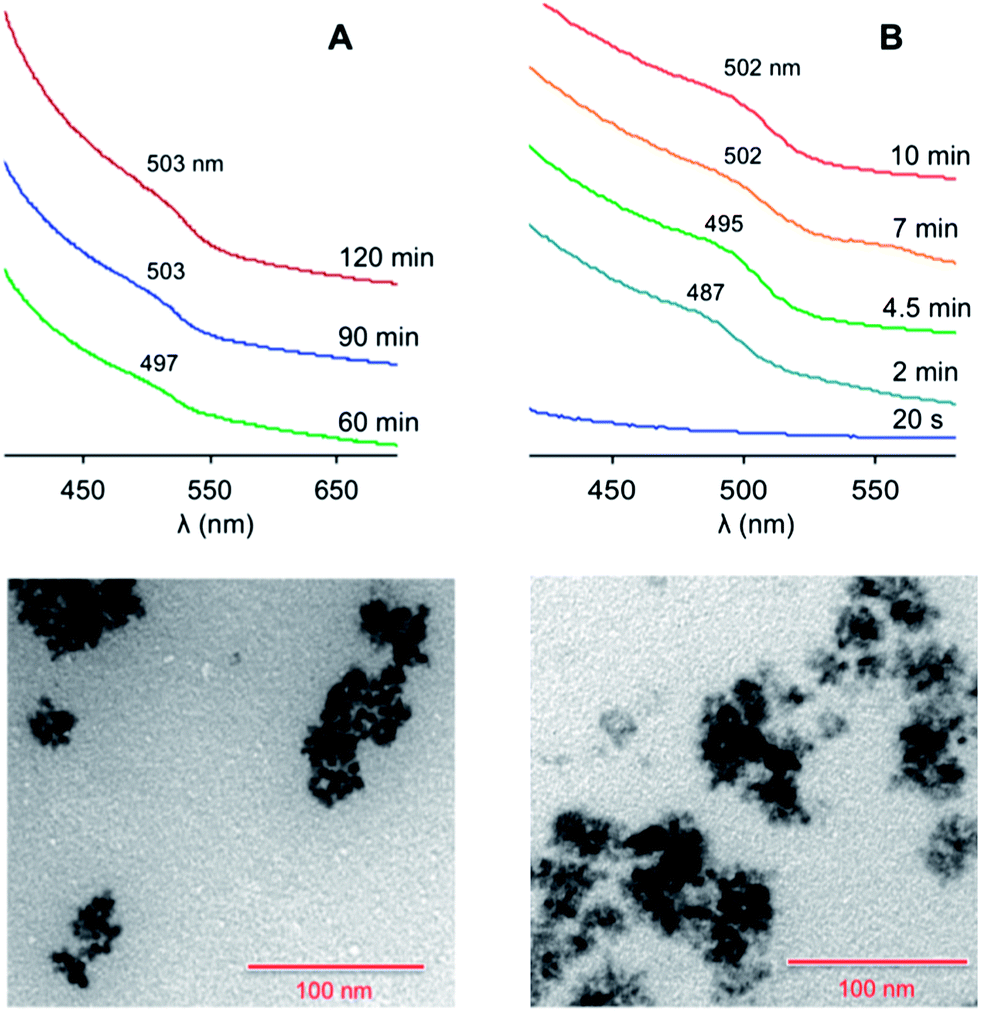 | ||
| Fig. 2 Top: absorption spectra (CHCl3, room temperature) of the ZnTe nanocrystals at different growth times, prepared following (A) protocol 2 or (B) protocol 3 in Table 1. The spectra successive to the first one are vertically shifted for clarity. Bottom: TEM images of the nanocrystals obtained at saturation in the case of (A) protocol 2 and (B) protocol 3. Scale bar, 100 nm. | ||
An increase in the SA concentration (Table 1, protocol 3) made the nucleation rate much faster, and saturation was reached within 10 minutes. The average diameter (4.9 nm) and size distribution remained essentially unchanged. The exciton absorption band was slightly more defined (Fig. 2B), suggesting a better quantum confinement, possibly determined by a higher structural quality of the particles.
The addition of ODA – a good ligand for Zn ions – to the Zn-containing solution (protocol 4 in Table 1) also caused a faster nucleation. The coloration of the reaction mixture, with the appearance of an exciton band at 490 nm, took place within one minute (Fig. 3A). We observed that the effect of ODA on the growth rate is less pronounced than that of SA: the saturation regime for protocol 4 was reached in 20 minutes, while it occurred after 10 minutes for protocol 3. Conversely, the presence of ODA influenced the final size of the particles, and quasi-spherical larger QDs (average diameter, 6.1 nm) were produced. Similar results were obtained by adding ODA to the chalcogen solution.
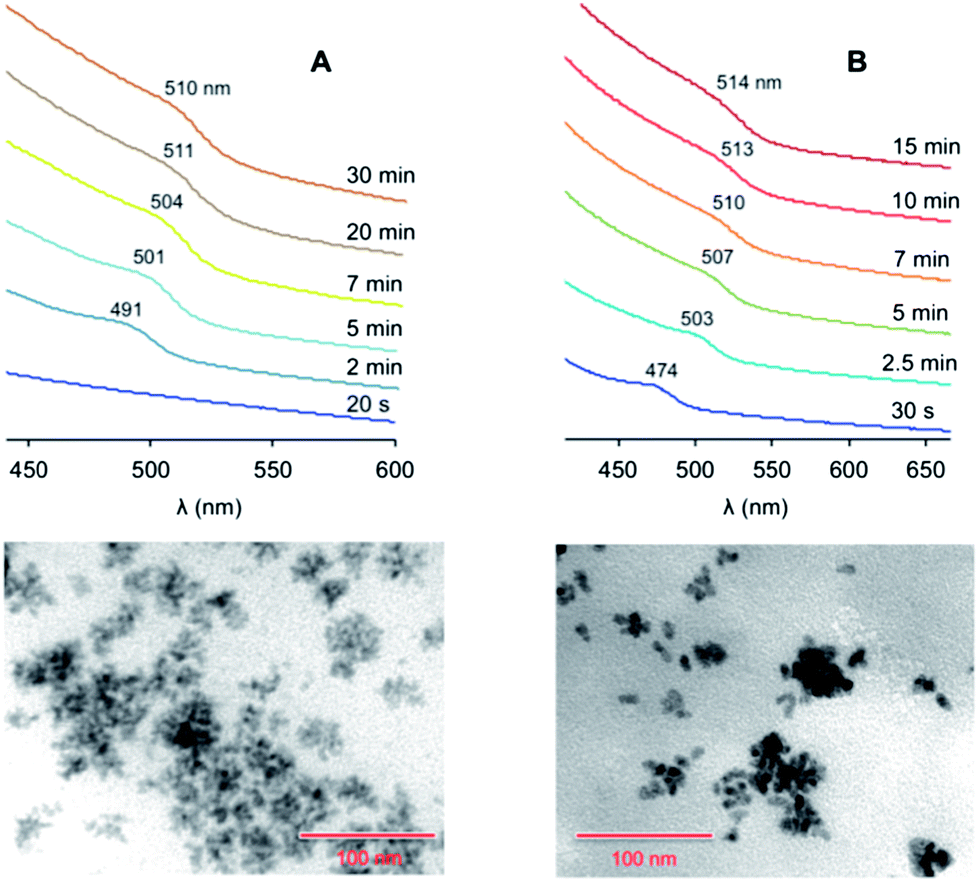 | ||
| Fig. 3 Top: absorption spectra (CHCl3, room temperature) of the ZnTe nanocrystals at different growth times, prepared following (A) protocol 4 or (B) protocol 6 in Table 1 (the spectra are vertically offset for clarity). Bottom: TEM images of the nanocrystals obtained at saturation in the case of (A) protocol 4 and (B) protocol 6. Scale bar, 100 nm. | ||
TOPO is a surfactant ligand that affords excellent results in the synthesis of CdSe and CdTe QDs.1 However, when TOPO was employed as an additional surfactant besides ODA in protocol 7 (Table 1), no nanocrystals were obtained.
Protocol 5, in which the growth temperature was raised to 285 °C (Table 1), yielded QDs of a similar average size than in the case of protocol 4. However, the size distribution was narrower (Δd < 1 nm) than for the other protocols (Δd ≥ 0.9–1.2 nm), indicating a more efficient size focusing at the employed temperature. The exciton band started to appear at 484 nm and reached 508 nm at saturation (Fig. S4 in the ESI†).
In protocol 6 the injection temperature was raised to 320 °C (reflux conditions in ODE). The increase in the nucleation temperature had a somewhat modest influence on the nucleation rate, whereas the mean diameter of the resulting QDs (8.3 nm) was substantially affected by this parameter. An exciton peak at 474 nm showed up 30 seconds after the injection (Fig. 3B); the peak underwent a substantial red shift during growth, ending up at 514 nm at saturation. TEM data showed that the quality of the QDs was similar to that obtained with the other protocols, suggesting that the injection temperature as well as the nature of the surfactants have a greater influence on the size of the particles rather than on the homogeneity of their size and shape distribution. HRTEM measurements confirmed that these nanoparticles are spherical and reasonably monodisperse (see ESI†). The average diameter (7.5 ± 1.7 nm) is consistent with that determined from conventional TEM images (Table 1). Moreover, selected area diffraction (SAD) patterns indicate that the nanoparticles are completely crystalline and are made of ZnTe in the cubic (sphalerite) structure.
The literature reports on UV-vis absorption24–27,29–31 and luminescence24,26,30,31 properties of ZnTe QDs are relatively scarce and often not in line with one another. This is certainly because of the difficult synthesis and poor chemical stability (mainly due to Te oxidation) of these colloids.28,32,44 As a matter of fact, our samples were completely decomposed after less than one hour in an air equilibrated solution at room temperature, while they could survive for at least 5 months in the dry box.
As shown in Fig. 2 and 3, the nanocrystals exhibit an exciton absorption shoulder at ca. 500 and 510 nm for average diameters of 5 and 6 nm, respectively. These values are consistent with the band gap energy of ZnTe in the presence of a moderate quantum confinement. Earlier studies reported room temperature luminescence for nanocrystals of 4.2, 5.4 and 14 nm in size,24,26 whereas no luminescence from ZnTe QDs was detected in more recent studies.30,31 In our case, none of the prepared samples exhibited photoluminescence upon excitation at various wavelengths across their absorption spectrum. Luminescence quenching could arise from trapping of the photoexcited electron or hole by surface states, as commonly observed for semiconductor nanocrystals. Additionally, the electronic passivation of the surface by the organic ligands is made difficult by the relatively high energy of the ZnTe conduction band edge: the photoexcited electron may be transferred to a capping ligand, thus preventing the radiative recombination of the exciton.
ZnTe/ZnS core/shell nanocrystals
The SILAR methodology was used to coat the ZnTe QDs with a ZnS layer.40 Briefly, this approach consists in serial injections of Zn and S precursors, separated by a certain period of time to allow the growth of the ZnS layers. Any attempt to perform SILAR on solutions of purified ZnTe cores resulted in the formation of a black precipitate, which turned colorless upon further heating, and the disappearance of the typical absorption features of the ZnTe cores. The same results were obtained even with freshly prepared nanocrystals kept in the dry box. This behaviour, which was recently observed by other researchers,32 indicates that the ZnTe QDs are unstable under the conditions ordinarily employed for SILAR.We therefore attempted to carry out the deposition of the ZnS shell quickly after the core synthesis, in the same reaction medium and without purification of the ZnTe QDs. When the ZnTe saturation regime was reached, the temperature of the mixture was decreased to 230 °C and the successive Zn and S injections were performed. In this manner no black precipitate was observed and up to 5 ZnS layers could be deposited on the ZnTe cores prepared following either protocols 4 or 6 (Table 1). TEM analyses (Fig. 4 and ESI†) showed that nanocrystals with an average diameter of 9.3 nm were obtained starting from ZnTe cores prepared with protocol 4. Considering that each ZnS monolayer exhibits an average thickness of 0.31 nm,43 the observed size is consistent with core nanocrystals of 6.1 nm coated with 5 ZnS layers. Size distribution analyses obtained from the TEM images (see ESI†) are in full agreement with the formation of the expected core–shell particles. Unfortunately, due to the strong influence of the surfactant residues on the grid and the high degree of aggregation of the QDs, no insightful observations could be made from HRTEM experiments.
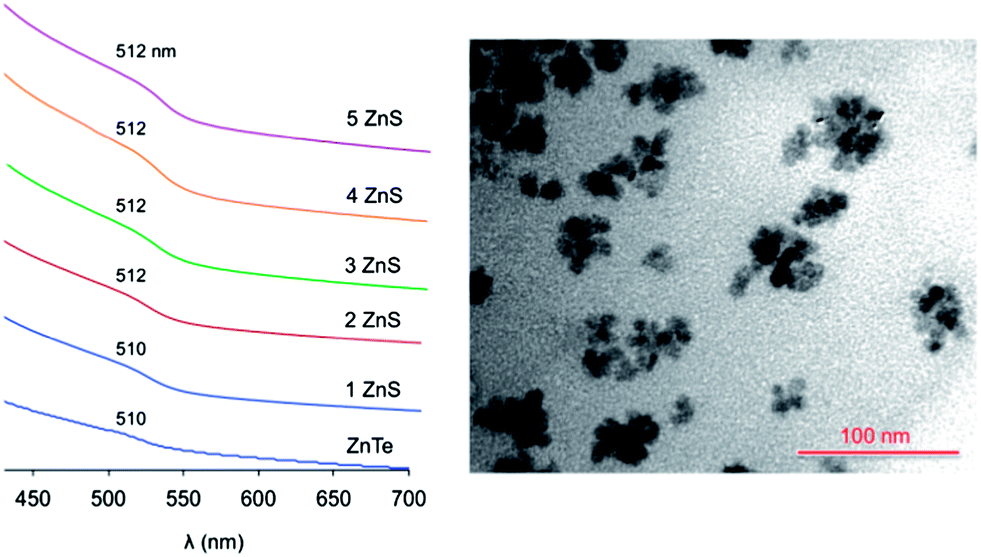 | ||
| Fig. 4 Left: absorption spectra (CHCl3, room temperature) of the ZnTe nanocrystals (6.1 nm diameter) upon the successive deposition of ZnS layers (the spectra are vertically offset for clarity). Right: TEM image of the ZnTe/ZnS QDs having 5 shell layers. | ||
The absorption spectral changes upon deposition of successive ZnS layers onto the ZnTe core are displayed in Fig. 4. As observed for the core QDs, the spectra show a negligible scattering background, indicating that the particles are not aggregated in the solution. The exciton band energy remains essentially unchanged upon the shell growth. This behavior indicates that the exciton produced upon visible light excitation is effectively confined within the ZnTe core, in agreement with a type I heterostructure. In fact, the alignment of the band energy-levels of ZnTe and ZnS (Fig. 1) suggests that, while the hole is trapped in the core, the excited electron could be delocalized over the whole nanocrystal, yielding a type I1/2 (or quasi-type II) behavior.20,44–46 Type I1/2 QDs are characterized by a marked red shift of the exciton band upon shell growth, while the band shape is essentially unchanged.46 In our case, however, the exciton band occurs practically at the same energy for ZnTe QDs with no shell and with 5 ZnS layers, in which the shell constitutes about 70% of the nanocrystal volume.47
The presence of the ZnS shell greatly improves the stability of the nanoparticles: an air equilibrated solution of ZnTe/ZnS (5sh) QDs maintains its colloidal stability and optical properties for more than one week. Also a shell of 2 ZnS layers brings about stabilization of the QDs which, however, is lower than that exerted by a thicker 5-layer shell. These results indicate that (i) the ZnS coating can protect the core from Te oxidation,48,49 and (ii) a thin shell may not afford a complete coverage of the core,50 as also suggested by the TEM images (see ESI†) and the XPS data (see below).
No photoluminescence was detected for any of the core/shell nanoparticles, suggesting that the ZnS coating is not able to remove all the charge traps responsible for luminescence quenching. In the case of the ZnTe/ZnS (2sh) particles, as noticed above, the shell could not completely coat the ZnTe surface. On the other hand, owing to the 11% lattice mismatch between ZnTe and ZnS, a thicker shell may exhibit defects (e.g., dangling bonds)50 that can provide radiationless deactivation routes for excitons.
XPS characterization
The XPS results obtained on a series of QDs representative of the full sequence produced – ZnTe, ZnTe/ZnS (2sh), ZnTe/ZnS (5sh) – are shown in Table 2 and Fig. 5. The ZnTe core is a reference sample for the more complex core/shell QDs. To investigate the effect of air exposure on the nanocrystals, a solution of purified ZnTe/ZnS (2sh) QDs was evaporated and the solid material was left in air for two months before submitting it to XPS analysis. This sample is referred to as ZnTe/ZnS (2sh) ‘aero’.| Sample | Zn 2p3/2 | Te 3d5/2 | N 1s | Zn/Te | Zn/N | Te/N | |
|---|---|---|---|---|---|---|---|
| Te2− | TeO2 | ||||||
| ZnTe | 1022.0 | 572.6 | 399.5 | 2.50 | 0.40 | 0.14 | |
| ZnTe/ZnS (2sh) | 1022.2 | 572.6 | 399.3 | 3.50 | 0.49 | 0.14 | |
| ZnTe/ZnS (2sh) ‘aero’ | 1022.1 | 572.4 | 576.0 | 399.3 | 2.20 | 0.37 | 0.17 |
| ZnTe/ZnS (5sh) | 1022.2 | 572.4 | 400.0 | 9.00 | 0.50 | 0.050 | |
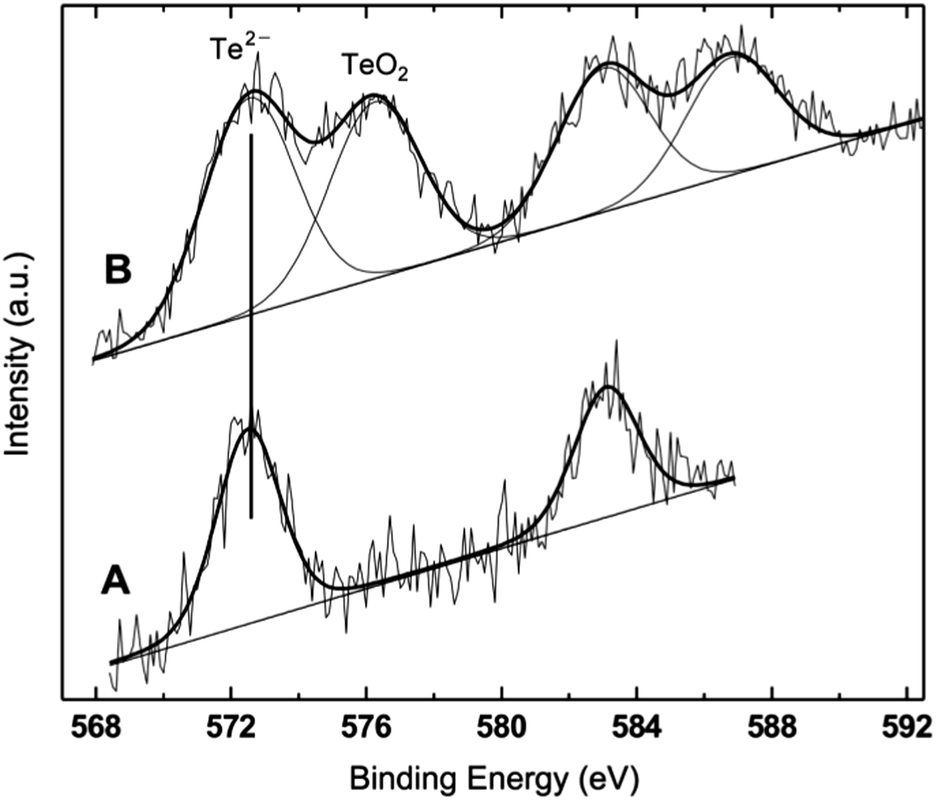 | ||
| Fig. 5 Te 3d photoemission region from ZnTe/ZnS (2sh) (A) and ZnTe/ZnS (2sh) ‘aero’ (B). Notice the presence of Te oxide in addition to telluride in sample B. The two minor peaks at higher binding energies are the 1/2 spin–orbit components of the two major peaks, which represent the 3/2 components of the Te 3d signal. | ||
The analyzed QDs present the characteristic signals from Zn, Te, N, C and O photoemission regions. The first notable overall result is the absence of a sulphur peak in all samples. This observation is confirmed by the results of EDX analyses performed on the core/shell QDs deposited on either copper grids or HOPG (see the ESI†). Only in one sample – ZnTe/ZnS (2sh) – the S signal was detected in the EDX spectrum. The result is not fully unexpected, since in the relevant literature on ZnS-based QDs it has been already reported that the formation of the volatile sulphur oxides can result in a total lack of the corresponding XPS signals, within the inherent sensitivity limits of XPS, estimated at 0.1%.51 The loss of sulphur has been ascribed to the exposure of the QDs to a humid oxygen atmosphere.51 Also for CdSe/ZnS QDs there is a literature report on the effect of exposing the nanocrystal surface to air for extended periods of time for a series of samples differing in the thickness of the ZnS shell.52 The authors showed the progressive formation of Se oxide with the exposure time for samples with 0.65 and 1.3 nm of ZnS, and a substantial loss of Se in both samples.
The binding energy (BE) values of Zn 2p, Te 3d and N 1s fall in restricted ranges, considering the experimental uncertainty of ±0.2 eV, which allow for a clear assignment. The Te peak is assigned to Te in telluride, and only in the case of the ‘aero’ sample a second component in the Te 3d photoemission region can be clearly detected (Fig. 5), and assigned to TeO2, based on its BE.53
As for zinc, in principle, each spin–orbit component of Zn 2p is the convolution of a largely attenuated peak associated with the QD core, and of a surface component from each ZnS shell; however, we could not resolve any fine structure in the Zn 2p signal. The Zn 2p3/2 BE is compatible with the presence of ZnO.54 However, the BE difference between ZnTe and ZnO amounts, in the bulk solids, to 0.5 eV (ref. 52) and may escape detection, while Zn oxide and hydroxide cannot be resolved in XPS from the analysis of Zn 2p3/2 BE. The Zn/Te XPS atomic ratio shows a surface enrichment in Zn for all the samples. The amount of Zn2+ in excess with respect to Te is likely associated with the presence of ZnO and Zn(OH)2 at the QD surface. It is noteworthy that a surface enrichment of Cd over Se was previously found for bare CdSe QDs.51
The second remarkable and general result is the low intensity of Zn and Te peaks, which we assign to the presence of a relevant quantity of ODA molecules at the surface on the basis of the low Zn/N and Te/N ratios. The N 1s signal exclusively comes from ODA and its relative intensity is further intensified over Zn and Te because the long chains of ODA are located on the QD surface. The constancy (within the associated experimental error, ±10%), of the Zn/Te atomic ratio in all samples suggests that the surface composition is invariably given by a comparable extent of the ODA overlayer on Zn oxide/hydroxide. We can take the attenuation exerted by ODA on the Te signals (and on its Zn counterpart in ZnTe, which remains, however, hard to be determined because of what was reported above) as a way of ascertaining the presence of a different number of outer shells in the corresponding QDs. The experimentally found large difference in the Te/N values obtained in the ZnTe/ZnS (5sh) with respect to both ZnTe cores and ZnTe/ZnS (2sh) can be explained by the much larger attenuation effect exerted on Te by the 5 ZnS layers and is an indication of the actual establishment of a multilayer shell in the ZnTe/ZnS (5sh) QDs. The constancy of the Te/N ratio for ZnTe/ZnS (2sh) and ZnTe cores can be ascribed to an incomplete coverage afforded by the 2-layer shell on the ZnTe nucleus, as also suggested by TEM images (see ESI†) and in agreement with the observed trends in the chemical stability of the samples (vide infra).
A further indication of the presence of a multilayer shell on ZnTe is given by the Zn/Te XPS ratio. Such a ratio (Table 2) increases from 2.5 to 3.5 and to 9 on going from ZnTe to ZnTe/ZnS (2sh) and to ZnTe/ZnS (5sh). It is noteworthy that the Zn/Te ratio of the ‘aero’ sample is comparable with that found for the ZnTe core. This observation could indicate that the generation of zinc oxide/hydroxide in the ‘aero’ sample does not give rise to a continuous surface coating but more likely to ‘patches’ of these species. The formation of such deposits may be thought of as a general phenomenon associated with the degradation of QDs upon exposure to air.
A few articles have been recently published on XPS quantitation in QDs and core/shell nanoparticles.55,56 The complex nature of poorly stable multishell QDs does not allow for a straightforward modelization of XPS atomic ratios into an exact structure, as instead reported in the literature for stable CdS-based QDs.57 We can, however, proceed to a more quantitative test for the presence of a 2- or 5-layer ZnS overcoating on the ZnTe core by comparing the values of the Te 3d5/2 XPS peak intensity (ITe3d) associated with the various QDs. The attenuation equation in the case of a continuous ZnS shell deposited over ZnTe is:
| ITe 3d = I°Te 3dexp(−z/λcosθ) |
3d intensities normalized to IN1s for both the 5- and 2-shell QDs (ITe3d/IN1s and I°Te 3d/I°N 1s). The use of normalized intensities is suggested by the fact that one cannot directly compare XPS peak intensity values for distinct compounds for which the increased thickness of the overlayer is due to a chemical reaction in solution and not due to a progressive evaporation on the same surface.
In the modified equation
| ITe3d/IN1s = (I°Te 3d/I°N 1s)exp(−z′/λcosθ) |
By taking the calculated value of IMFP for Te 3d photoelectrons travelling through ZnO as 1.50 nm (ref. 58) and introducing the experimental value of θ = 11°, a value z′ = 1.52 nm is obtained. This value is very close to the thickness of 5-layers of ZnS (each measuring 0.31 nm),43 and can be compared with the expected difference in thickness between 5-layer (5 × 0.31 nm) and 2-layer (2 × 0.31 nm) shells, that is, 0.93 nm. Notice that the same calculations performed in the case of a ZnS overlayer (λ = 1.596 nm) would lead to a value of z′ = 1.61 nm, which is also to be compared with the expected value. The smaller value obtained, apart from any approximations used, could be related to an imperfect 2-layer shell of ZnS, as suggested by XPS data reported above. Considering the crude approximations operated, we can take these results as qualitative evidence for the actual formation of 2- and 5-layer shells onto the ZnTe nanocrystals.
Photocurrent measurements
Since a promising application of QDs is as photosensitizers in solar cells,13,14 we investigated the behavior of ITO electrodes covered with ZnTe/ZnS (2sh) QDs in a photoelectrochemical cell. Fig. 6 shows the photocurrent spectrum measured for the sample (red circles), after subtraction of the signal generated by the bare ITO electrode (green triangles), at an applied potential of 0 V vs. Ag/AgCl (+0.197 V vs. NHE). An anodic photocurrent, indicative of an electron injection from the QD layer into the conducting electrode, was observed upon irradiation in the UV-visible region. It can be noticed that the onset of the photocurrent matches with that of the absorption spectrum of the QDs (Fig. 6, black line). The photocurrent started at 530 nm and gradually increased, reaching a value of 5 nA at 390 nm. A much steeper growth of the current intensity was observed for λ < 360 nm (33 nA at 350 nm), i.e., in the spectral region in which ZnS can also absorb light.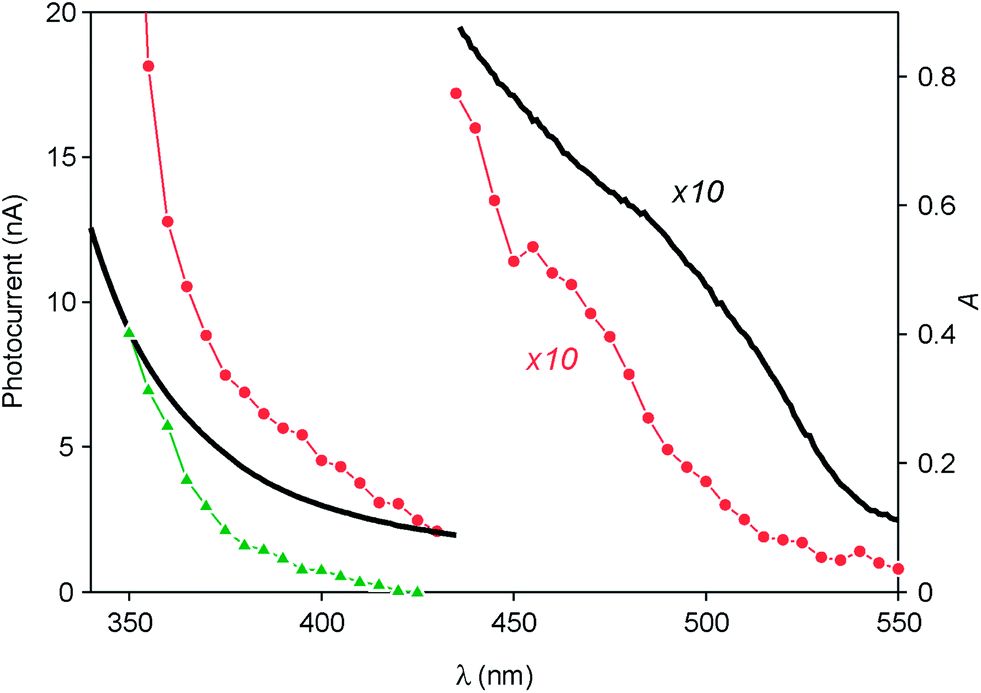 | ||
| Fig. 6 Photocurrent spectrum (left scale) of the ITO electrode functionalized with ZnTe/ZnS (2sh) nanocrystals (red circles) after subtraction of the photocurrent generated under the same conditions by the bare ITO (green triangles). The absorption spectrum of the QDs in CHCl3 solution (black line, right scale) is also shown. | ||
These results can be interpreted in terms of the energy-level diagrams shown in Fig. 7A and B. Visible light irradiation causes band gap excitation of the ZnTe core, resulting in an electron transfer to the ITO electrode, possibly mediated by the conduction levels of the ZnS shell (Fig. 7A). Upon UV irradiation, bandgap excitation of the ZnS layer may be obtained, causing electron injection into the ITO electrode and the migration of the hole within the ZnTe core (Fig. 7B). The relatively higher anodic photocurrent observed in the wavelength region of the ZnS band gap, compared to the visible region (Fig. 6), can be accounted for by the fact that the ZnS shell is in the direct contact with the electrode. This observation also indicates that the excited electron generated by visible irradiation is effectively confined in the ZnTe core, supporting the view that the ZnTe/ZnS heterojunction in these QDs is of type I.
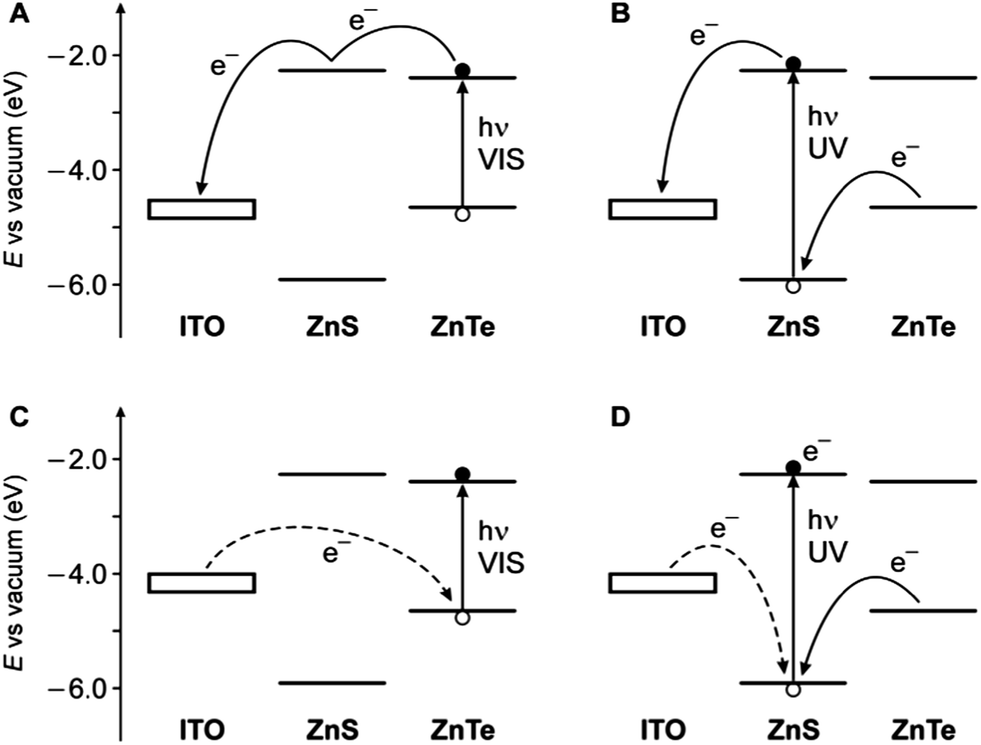 | ||
| Fig. 7 Schematic energy-level diagram for the ITO photoelectrode coated with the ZnTe/ZnS QDs and representation of the electron transfer processes that may occur under either visible light irradiation (A, anodic current; C, cathodic current) or UV irradiation (B, anodic current; D, cathodic current). The experimentally observed cases correspond to panels A and B. Dashed lines represent processes that are unlikely to occur. The electrode level is positioned considering an ITO work function of 4.4–4.7 eV,59,60 taking into account the potential applied in the experiments. | ||
In principle, considering that the valence band energies of both ZnTe and ZnS are lower than the Fermi level of the ITO, a cathodic photocurrent may also be observed in the investigated cell (Fig. 7C and D). No cathodic current, however, was observed upon UV-visible illumination of the photoelectrode in the presence of either oxygen or methylviologen as electron acceptors, even if a negative potential (up to −0.5 V vs. Ag/AgCl) was applied. Presumably, because of the barrier represented by the ZnS shell, electron transfer from the electrode to fill the photogenerated hole in the ZnTe VB is too slow to compete with the recombination of the exciton (Fig. 7C). On the other hand, band gap (UV) excitation of ZnS may result in an electron transfer from ITO to the VB of the semiconductor (dashed line in Fig. 7D); it is likely, however, that in these QDs the hole migrates quickly to the ZnTe core, thus preventing a cathodic photocurrent flow. Such an electrical rectification was already observed for surface-deposited QDs;61,62 in this particular case it may be interesting for optoelectronic applications, because it can be controlled by light.
Using the same experimental setup described above, we also performed photocurrent measurements on cadmium selenide QDs for comparison with the results obtained on the ZnTe/ZnS nanocrystals. We found that CdSe QDs also generate weak photocurrents within their UV-visible absorption spectrum (Fig. S17 in the ESI†). In contrast to the ZnTe/ZnS-based photoelectrodes, however, ITO electrodes coated with CdSe QDs did not show electrical rectification. Such a behavior indicates that, depending on the photoelectrode potential, both electrons and holes can be transferred to the electrolyte, as already observed for chalcogenide quantum dots.63–66 It is thus plausible that the rectification phenomenon observed with ZnTe/ZnS-modified photoelectrodes is a consequence of the core–shell structure and arises from hole trapping within the ZnTe core at negative applied potentials (Fig. 7C and D). Such hole trapping would be less effective in the case of cadmium selenide QDs because the valence band energy of CdSe is considerably lower than that of ZnTe.19,20
Conclusions
We have described the preparation of spherical ZnTe nanocrystals with good monodispersion and average diameters around 6 nm, and we have investigated the effect of a number of synthetic parameters on the reaction product. By adopting a SILAR methodology, we were able to grow ZnS layers onto the ZnTe nanocrystals, affording – to our best knowledge – the first examples of ZnTe/ZnS core/shell quantum dots.The UV-visible absorption data in solution indicate that the ZnTe/ZnS heterojunction in these QDs exhibits a type I behavior, i.e., both the electron and the hole are confined within the ZnTe core. The ZnS shell improves the chemical and photochemical stability of the nanoparticles in air equilibrated solution; both the ZnTe core and ZnTe/ZnS core/shell quantum dot samples, however, are not luminescent. The XPS results have evidenced the distinct nature of core and core/shell QDs and confirmed the formation of the expected 2- and 5-layer shell on the nanocrystal core. The XPS data also indicate the oxidation of the QDs upon air exposure, even if a multilayer shell is present.
Irradiation of an ITO electrode functionalized with ZnTe/ZnS nanocrystals results in the generation of an anodic current, suggesting that the QDs perform spectral sensitization of the electron injection into the ITO electrode. As similar cathodic photocurrent generation was not observed, the QD-modified electrode could perform electrical rectification upon a photon energy input. Although the preparation of high quality ZnTe/ZnS nanocrystals appears to be quite laborious, they can represent alternatives to cadmium-based quantum dots for specific optoelectronic applications.
Acknowledgements
This work was supported by the European Union (EU) (Hysens Project, grant no. 263091, ATOMIN project, grant no. POIG.02.01.00-12-023/08), the Ministero dell’Università e della Ricerca (MIUR) (PRIN 2010CX2TLM and 2010N3T9M4, FIRB RBAP11C58Y) and the University of Bologna. M. O. thanks The National Science Centre (grant no. UMO 2011/01/N/ST5/02550) and the Foundation for Polish Science for the MPD programme fellowship co-financed by the EU European Regional Development Fund. K. S. acknowledges financial support from The National Science Centre (grant no. UMO-2011/03/B/ST5/01495).Notes and references
- C. B. Murray, C. R. Kagan and M. G. Bawendi, Annu. Rev. Mater. Sci., 2000, 30, 545 CrossRef CAS.
- Semiconductor and Metal Nanocrystals, ed. V. I. Klimov, Dekker, New York, 2012 Search PubMed.
- A. L. Rogach, Semiconductor Nanocrystal Quantum Dots, Springer-Verlag, Wien, 2008 Search PubMed.
- A. L. Efros and M. Rosen, Annu. Rev. Mater. Sci., 2000, 30, 475 CrossRef CAS.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763 CrossRef CAS PubMed.
- J. Callan, A. P. De Silva, R. C. Mulrooney and B. McCaughan, J. Inclusion Phenom. Macrocyclic Chem., 2007, 58, 257 CrossRef CAS PubMed.
- R. C. Somers, M. G. Bawendi and D. G. Nocera, Chem. Soc. Rev., 2007, 36, 579 RSC.
- R. Gill, M. Zayats and I. Willner, Angew. Chem., Int. Ed., 2008, 47, 7602 CrossRef CAS PubMed.
- R. Freeman and I. Willner, Chem. Soc. Rev., 2012, 41, 4067 RSC.
- N. Hildebrandt, ACS Nano, 2011, 5, 5286 CrossRef CAS PubMed.
- T. L. Doane and C. Burda, Chem. Soc. Rev., 2012, 41, 2885 RSC.
- D. V. Talapin, J.-S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389 CrossRef CAS PubMed.
- P. V. Kamat, K. Tvrdy, D. R. Baker and J. G. Radich, Chem. Rev., 2010, 110, 6664 CrossRef CAS PubMed.
- S. Rühle, M. Shalom and A. Zaban, ChemPhysChem, 2010, 11, 2290 CrossRef PubMed.
- M. Green, J. Mater. Chem., 2010, 20, 5797 RSC.
- A. Credi, New J. Chem., 2012, 36, 1925 RSC.
- I. Yildiz, E. Deniz, B. McCaughan, S. F. Cruickshank, J. F. Callan and F. M. Raymo, Langmuir, 2010, 26, 11503 CrossRef CAS PubMed.
- G. Palui, T. Avellini, N. Zhan, F. Pan, D. Gray, I. Alabugin and H. Mattoussi, J. Am. Chem. Soc., 2012, 134, 16370 CrossRef CAS PubMed.
- P. Reiss, M. Protière and L. Li, Small, 2009, 5, 154 CrossRef CAS PubMed.
- C. de Mello Donegá, Chem. Soc. Rev., 2011, 40, 1512 RSC.
- M. Bottrill and M. Green, Chem. Commun., 2011, 47, 7039 RSC.
- S. Adachi, Optical constants of crystalline and amorphous semiconductors, Kluwer, Dordrecht, 1999, p. 473 Search PubMed.
- For a recent paper, see: H. Li, R. Brescia, R. Krahne, G. Bertoni, M. J. P. Alcocer, C. D'Andrea, F. Scotognella, F. Tassone, M. Zanella, M. De Giorgi and L. Manna, ACS Nano, 2012, 6, 1637 CrossRef CAS PubMed.
- Y.-W. Jun, C.-S. Choi and J. Cheon, Chem. Commun., 2001, 101 RSC.
- R. Xie, X. Zhong and T. Basché, Adv. Mater., 2005, 17, 2741 CrossRef CAS.
- S. H. Lee, Y. J. Kim and J. Park, Chem. Mater., 2007, 19, 4670 CrossRef CAS.
- J. Zhang, K. Sun, A. Kumbhar and J. Fang, J. Phys. Chem. C, 2008, 112, 5454 CAS.
- J. Bang, J. Park, J. H. Lee, N. Won, J. Nam, J. Lim, B. Y. Chang, H. J. Lee, B. Chon, J. Shin, J. B. Park, J. H. Choi, K. Cho, S. M. Park, T. Joo and S. Kim, Chem. Mater., 2010, 22, 233 CrossRef CAS.
- S. Xu, C. Wang, Q. Xu, H. Zhang, R. Li, H. Shao, W. Lei and Y. Cui, Chem. Mater., 2010, 22, 5838 CrossRef CAS.
- E. Groeneveld, S. van Berkum, A. Meijerink and C. de Mello Donegá, Small, 2011, 7, 1247 CrossRef CAS PubMed.
- S. M. Fairclough, E. J. Tyrrell, D. M. Graham, P. J. B. Lunt, S. J. O. Hardman, A. Pietzsch, F. Hennies, J. Moghal, W. R. Flavell, A. A. R. Watt and J. M. Smith, J. Phys. Chem. C, 2012, 116, 26898 CAS.
- R. Mahadevu, A. R. Yelameli, B. Panigrahy and A. Pandey, ACS Nano, 2013, 7, 11055 CrossRef CAS PubMed.
- C.-T. Cheng, C.-Y. Chen, C.-W. Lai, W.-H. Liu, S.-C. Pu, P.-T. Chou, Y.-H. Chou and H.-T. Chiu, J. Mater. Chem., 2005, 15, 3409 RSC.
- N. Pradhan, D. M. Battaglia, Y. Liu and X. Peng, Nano Lett., 2007, 7, 312 CrossRef CAS PubMed.
- D. Aldakov, A. Lefrancois and P. Reiss, J. Mater. Chem. C, 2013, 1, 3756 RSC.
- See, e.g.: L. Li, T. J. Daou, I. Texier, T. K. C. Tran, Q. L. Nguyen and P. Reiss, Chem. Mater., 2009, 21, 2422 CrossRef CAS.
- T. Tanaka, K. Hayashida, S. Wang, Q. Guo, M. Nishio and H. Ogawa, J. Cryst. Growth, 2003, 248, 43 CrossRef CAS.
- S.-H. Wei and A. Zunger, Appl. Phys. Lett., 1998, 72, 2011 CrossRef CAS PubMed.
- For a study of ZnTe quantum dots embedded in a ZnS matrix, see: M. C. Harris Liao, Y. H. Change, C. C. Tsai, M. H. Chieng and Y. F. Chen, J. Appl. Phys., 1999, 86, 4694 CrossRef PubMed.
- J. J. Li, Y. A. Wang, W. Guo, J. C. Keay, T. D. Mishima, M. B. Johnson and X. Peng, J. Am. Chem. Soc., 2003, 125, 12567 CrossRef CAS PubMed.
- M. Amelia, S. Impellizzeri, S. Monaco, I. Yildiz, S. Silvi, F. M. Raymo and A. Credi, ChemPhysChem, 2011, 12, 2280 CrossRef CAS PubMed.
- C. De Mello Donegá, P. Liljeroth and D. Vanmaekelbergh, Small, 2005, 1, 1152 CrossRef PubMed.
- D. Chen, F. Zhao, H. Qi, M. Rutherford and X. Peng, Chem. Mater., 2010, 22, 1437 CrossRef CAS.
- A. Pandey and P. Guyot-Sionnest, J. Chem. Phys., 2007, 127, 104710/1 CAS.
- S. A. Ivanov, A. Piryatinski, J. Nanda, S. Tretiak, K. R. Zavadil, W. O. Wallace, D. Werder and V. I. Klimov, J. Am. Chem. Soc., 2007, 129, 11708 CrossRef CAS PubMed.
- C. De Mello Donegá, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 165303 CrossRef.
- Indication of a type-I behavior in ZnSe/ZnS core/shell nanoobjects was recently observed. See ref. 23.
- S. K. Poznyak, N. P. Osipovich, A. Shavel, D. V. Talapin, M. Gao, A. Eychmüller and N. Gaponik, J. Phys. Chem. B, 2005, 109, 1094 CrossRef CAS PubMed.
- J. M. Tsay, M. Pflughoefft, L. A. Bentolila and S. Weiss, J. Am. Chem. Soc., 2004, 126, 1926 CrossRef CAS PubMed.
- J. McBride, J. Treadway, L. C. Feldman, S. J. Pennycook and S. J. Rosenthal, Nano Lett., 2006, 6, 1496 CrossRef CAS PubMed.
- K. Pechstedt, T. Whittle, J. J. Baumberg and T. Melvin, J. Phys. Chem. C, 2010, 114, 12069 CAS.
- B. O. Dabbousi, J. Rodriguez-Viejo, F. V. Mikulec, J. R. Heine, H. Mattoussi, R. Ober, K. F. Jensen and M. G. Bawendi, J. Phys. Chem. B, 1997, 101, 9463 CrossRef CAS.
- D. Briggs and M. P. Seah, Practical Surface Analysis, Wiley, Chichester, 2nd edn, 1990, vol. 1 Search PubMed.
- G. J. Schön, J. Electron Spectrosc. Relat. Phenom., 1973, 2, 75 CrossRef.
- G. Zorn, S. R. Dave, X. Gao and D. G. Castner, Anal. Chem., 2011, 83, 866 CrossRef CAS PubMed.
- A. G. Shard, J. Phys. Chem. C, 2012, 116, 16806 CAS.
- C. F. Hoener, K. A. Alla, A. J. Bard, A. Campion, M. F. Fox, T. E. Mallouk, S. E. Webber and J. M. White, J. Phys. Chem., 1992, 96, 3812 CrossRef CAS.
- C. J. Powell and A. Jablonski, Nucl. Instrum. Methods Phys. Res., Sect. A, 2009, 601, 54 CrossRef CAS PubMed.
- K. Sugiyama, H. Ishii and Y. Ouchi, J. Appl. Phys., 2000, 87, 295 CrossRef CAS PubMed.
- Y. Zhou, J. W. Shim, C. Fuentes-Hernandez, A. Sharma, K. A. Knauer, A. J. Giordano, S. R. Marder and B. Kippelen, Phys. Chem. Chem. Phys., 2012, 14, 12014 RSC.
- M. Amelia, C. Lincheneau, S. Silvi and A. Credi, Chem. Soc. Rev., 2012, 41, 5728 RSC.
- G. P. Kissling, C. Bunzli and D. J. Fermin, J. Am. Chem. Soc., 2010, 132, 16855 CrossRef CAS PubMed.
- S. Gawęda, A. Podborska, W. Macyk and K. Szaciłowski, Nanoscale, 2009, 1, 299 RSC.
- S. Ogawa, K. Hu, F. Fan and A. J. Bard, J. Phys. Chem. B, 1997, 101, 5707 CrossRef CAS.
- V. Pardo-Yissar, E. Katz, J. Wasserman and I. Willner, J. Am. Chem. Soc., 2003, 125, 622 CrossRef CAS PubMed.
- M. El Harakeh, L. Alawieh, S. Saouma and L. I. Halaoui, Phys. Chem. Chem. Phys., 2009, 11, 5962 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, TEM, EDX and photocurrent data. See DOI: 10.1039/c3tc32385d |
| This journal is © The Royal Society of Chemistry 2014 |