Catalytic asymmetric synthesis of 3-hydroxyoxindole: a potentially bioactive molecule
Akshay
Kumar
and
Swapandeep Singh
Chimni
*
Department of Chemistry, U.G.C. Centre of Advance Studies in Chemistry, Guru Nanak Dev University, Amritsar, 143005, India. Fax: (+)91-183-2258820; E-mail: sschimni@yahoo.com; sschimni.chem@gndu.ac.in
First published on 7th August 2012
Abstract
The recent emergence of biological activities of chiral 3-substituted-3-hydroxy-2-oxindoles has inspired synthetic chemists to develop new methodologies for their synthesis. Both chiral organocatalysts and organometallic catalysts have provided an important platform for their synthesis and in recent years, great achievements have been made in their catalytic asymmetric synthesis. This review summarizes the catalytic strategies for enantioselective synthesis of targeted frameworks.
 Akshay Kumar | Akshay Kumar was born in 1983 at Maslana Kalan, a small village in Hamirpur District of Himachal Pradesh in India. He obtained his B.Sc. From Himachal Pradesh University in 2004. After completing his M.Sc. from Guru Nanak Dev University, Amritsar, in 2007, he moved to the R&D centre of Ind. Swift. Company Mohali. In February 2008, he joined the group of Prof. Swapandeep Singh Chimni at the Department of Chemistry, Guru Nanak Dev University, Amritsar and started his Ph.D. His current research is the development of chiral organocatalysts for carbon–carbon bond-forming reactions. |
 Swapandeep Singh Chimni | Swapandeep Singh Chimni was born in 1962 at Amritsar, India. He received his M.Sc. (Hons. Sch.) in Chemistry in 1985 and Ph.D. in 1991 from Guru Nanak Dev University, Amritsar. After two years as a lecturer at Regional Engineering College (now NIT) Jalandhar, he joined the Department of Chemistry, Guru Nanak Dev University as Lecturer in 1992. He is presently working as a Professor in the same department. He has 22 years of research experience and published over 80 publications. He works in synthetic organic chemistry with emphasis on asymmetric organocatalysis, biocatalysis and phase-transfer catalysis as well as green chemistry. |
1. Introduction
3-Substituted-3-hydroxy-2-oxindole is one of the key structural units found in a large variety of natural products, such as convolutamydines, donaxaridines, maremycins, dioxibrassinines, celogentin K, 3′-hydroxyglucoisatisin, TMC-95A, etc. showing a wide spectrum of biological activities (Fig. 1).1 This structural moiety, especially 3-aryl/alkenyl-3-hydroxy-2-oxindole, constitutes the core of many drug candidates which have been used in a number of recent pharmaceutical studies.2 For example, SM-130686 (A) is a potent growth hormone secretagogue,2a compound (−)-B has been identified as an effective activator of maxi-K channels2b and compound C possesses improved anti-HIV properties compared to the FDA-approved NNRTI drug efavirenz2c (Fig. 2). Structure–activity relationship studies have shown that the biological activities of these compounds are greatly affected both by the configuration of the C3 carbinol carbon and its substituent pattern.2 In recent times, chiral 3-substituted-3-hydroxy-2-oxindole molecules have been favourably placed in new drug discovery programs.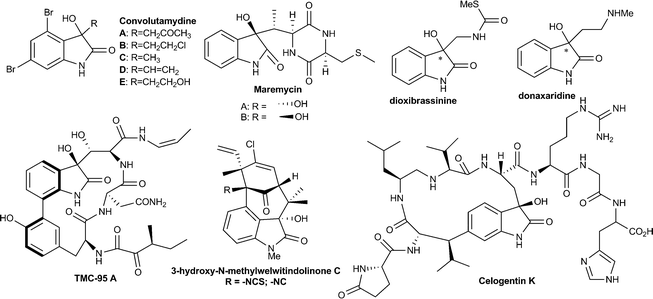 | ||
| Fig. 1 Some example of bioactive molecule containing the tetrasubstituted 3-hydroxyoxindole structural motif. | ||
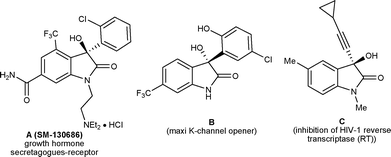 | ||
| Fig. 2 Representative example of some drug candidates. | ||
Consequently, the chiral 3-substituted-3-hydroxy-2-oxindole moiety has attracted the attention of synthetic chemists for developing synthetic methods for the synthesis of these molecules possessing advanced pharmaceutical values.
Owing to the importance of natural and synthetic chiral 3-substituted-3-hydroxy-2-oxindoles in medicinal chemistry, a significant effort has continuously been devoted to develop new methods for their preparation. The asymmetric hydroxylation of enolates employing chiral oxaziridine developed by Davis et al. and use of chiral auxiliaries are some of the old and uncommon strategies utilized for the synthesis of chiral 3-substitited 3-hydroxy-2-oxindoles.3 With the advancement in the field of asymmetric catalysis, a large number of strategies for the synthesis of a diverse range of chiral 3-substituted-3-hydroxy-2-oxindoles have been developed. Organometallic catalysis4 and organocatalysis5 have played an important role in their synthesis. Metal catalysts, in particular transition metal complexes, have been shown to catalyze many reactions such as the nucleophilic addition to the C3 carbonyl of isatins, hydroxylation reaction of 3-substituted 2-oxindoles and anilide cyclization to procure chiral 3-substituted-3-hydroxy-2-oxindole derivatives.
Many organocatalytic reactions such as the aldol reaction, Michael reaction, Morita–Baylis–Hillman reaction, Henry reaction and Friedel–Crafts reaction have been used effectively for the synthesis of chiral 3-substituted-3-hydroxy-2-oxindole derivatives.
In recent times, due to the biological activities associated with oxindole derivatives they have been the subject of many reviews.6 The majority of these reviews discuss the synthetic strategies for the synthesis of spirooxindoles and 3,3′-disubstituted oxindoles, but none of these have focused on the catalytic asymmetric synthesis of chiral 3-substituted-3-hydroxy-2-oxindoles. In this review we present the strategies developed for the asymmetric synthesis of 3-substituted-3-hydroxy-2-oxindoles using chiral catalysts (Fig. 3).
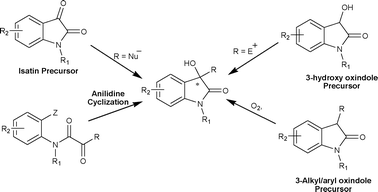 | ||
| Fig. 3 Strategies for catalytic asymmetric synthesis of 3-substituted-3-hydroxy-2-oxindoles. | ||
For simplicity in presentation and understanding, the review has been divided into two classes, based on the catalytic approach used in the synthesis of chiral 3-substituted-3-hydroxy-2-oxindoles: 1) organocatalysis and 2) organometallic catalysis. Each of these have been further discussed based on the substrate employed in the reaction.
2. Organocatalysis
2.1 Isatin as an electrophile
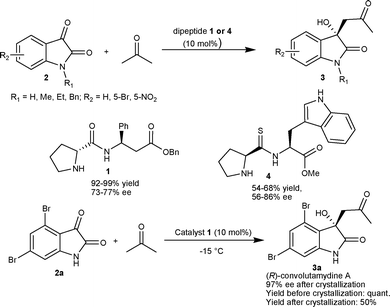 | ||
| Scheme 1 Dipeptide-catalyzed aldol reaction of acetone with isatins. | ||
Recently, Juaristi et al.10 developed a solvent-free high speed ball-milling (HSBM) procedure for direct asymmetric aldol reaction of acetone to isatins (2) using thio-dipeptides as organocatalysts. The thio-peptide 4 catalyzed the formation of 3-acetonyl-3-hydroxy-2-oxindoles (3) in moderate yield (54–68%) and moderate to good enantioselectivity (56–86% ee) (Scheme 1). The thio-dipeptides proved to be better organocatalysts relative to their analogous amides for this reaction using a HSBM procedure.
In 2007, Malkov and co-workers11 reported highly enantioselective synthesis of 3a catalyzed by primary amino alcohols derived from natural α-amino acids. L-Leucinol (5) catalyzes the aldol reaction between acetone and 4,6-dibromoisatin (2a) affording 3a in excellent yield (98%) with an enantiomeric excess of 95% (Scheme 2). It was established with the help of designed experiments that the hydrogen bonding between the hydroxyl group of the catalyst and the keto group of the isatin is a prerequisite for highly enantioselective aldol reaction, since the reaction performed in methanol provided a racemic adduct. Further, the catalysis by O-methylleucinol and O-trimethylsilylleucinol organocatalysts gave product with low enantioselectivity (50% ee) (TS 1, Scheme 2).
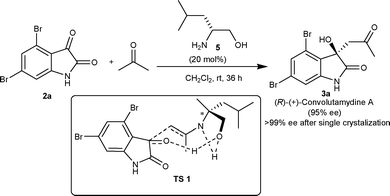 | ||
| Scheme 2 Synthesis of (R)-convolutamydine A. | ||
The aldol reaction of ketones with variety of isatins (2) catalyzed by carbohydrate-derived amino alcohol 7 provides the corresponding aldol adducts 3 in good to excellent yield (80–99%) and moderate to good enantioselectivity (55–75% ee).12 The heterogeneous organocatalyst chitosan 8, a renewable feedstock material, has been successfully used to catalyze the aldol reaction of ketones with isatins (2), providing aldol adducts (3) in high yield, albeit with low enantioselectivity.13 The recyclability was an important asset for this catalytic protocol (Scheme 3).
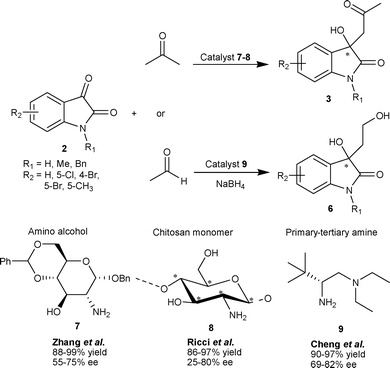 | ||
| Scheme 3 Primary amine based organocatalyst 7–9 catalyzed aldol reaction of ketones with isatins. | ||
The primary amino acid derived primary–tertiary diamine organocatalyst was developed for catalyzing the direct aldol reaction of isatins with acetaldehyde.14 The L-tert-leucine 9 in conjugation with (H4SiW12O40)0.25 was identified as the optimal catalytic system which gave aldol products 6 in excellent yield (90–97%) and good enantioselectivity (69–82% ee) (Scheme 3). Nakamura and co-workers have developed the two-point coordinate heteroarylsulfonylprolinamide organocatalyst for aldol reaction of isatins with aldehyde/ketone as a donor.15,16 The direct aldol reaction between acetone and isatins (2) catalyzed by 10 under neat conditions with a small amount of water (10 eq.) afforded aldol adduct 3 in moderate to excellent yield (59–99%) and good to excellent enantioselectivity (77–97% ee) (Scheme 4).15 The enantioselectivity of all derivatives was enriched to >99% after single crystallization. Hydrogen bonding between the amidic proton and sulfur atom of thiophene in the organocatalyst plays an important role in orienting the reactants for high enantioselectivity. The preferential formation of the R enantiomer has been attributed to anti–trans-TS-2, since syn–trans-TS-3 leads to the formation of the S enantiomer, which is destabilized by a steric repulsion between 4-bromo and 2-thienyl groups (Scheme 4).
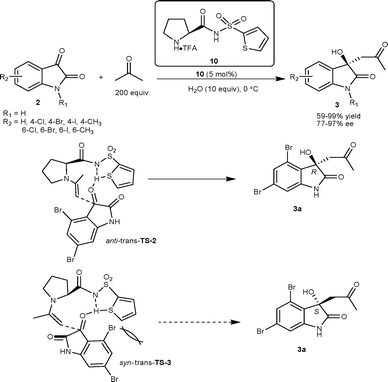 | ||
| Scheme 4 Heteroarylsulfonylprolinamide 10 catalyzed aldol reaction of acetone with isatins. | ||
The organocatalyst 10 have also been used to catalyze the aldol reaction of linear aldehydes (11) with isatins (2) in THF to provide aldol adducts (6) with good to excellent yield (73–99%) and excellent enantioselectivity (89–98% ee).16 This process was used for the first time for the enantioselective synthesis of (R)-convolutamydine E (6a) and (R)-convolutamydine B (12) (Scheme 5). The (S)-pyrrolidine tetrazole organocatalyst 13 catalyzes the aldol reaction of α-branched aldehyde (11) with isatins (2) providing aldol product 6 with two contiguous stereocentres in moderate to good yield (46–92%) and moderate to good enantioselectivity (49–93% ee) (Scheme 5).17 Under the reaction conditions studied by the authors, the catalyst 13 was highly efficient for both α-branched and linear aldehydes, whereas catalyst 10 only works efficiently with linear aldehydes.
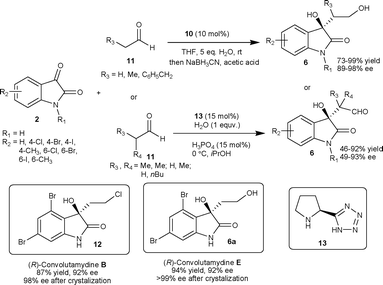 | ||
| Scheme 5 Aldol reaction of aldehydes to isatins catalyzed by organocatalysts 10 and 13. | ||
Hayashi and co-workers18 have studied the aldol reaction of acetaldehyde (11a) with isatins (2) catalyzed by 4-hydroxydiarylprolinol 14 (30 mol%) as an organocatalyst in the presence of 60 mol% of chloroacetic acid as an additive. The aldol adducts 6 were isolated in moderate to good yield (55–86%) and good enantioselectivity (81–86% ee). The resulting aldol products 6 have been synthetically transformed to valuable products such as ent-convolutamydine E (6a), CPC-1 (15a) and a half fragment of madindoline A and B (15b) (Scheme 6).
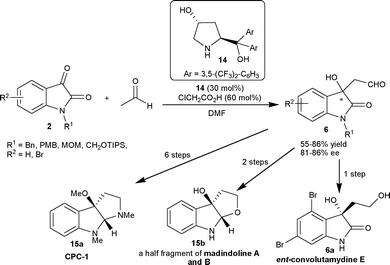 | ||
| Scheme 6 Prolinol 14 catalyzed aldol reaction of acetaldehyde with isatins. | ||
Yuan et al. have also studied the similar transformation with wide substrate scope using the catalyst 14 without acid additive.19 The aldol adducts 6 was isolated after reduction with sodium borohydride in 65–95% yield and 77–98% enantiomeric excess (Scheme 7). This approach was utilized for the first enantioselective synthesis of optically active (−)-donaxaridine (16a) and (R)-chimonamidine (16b) besides its application in the concise stereoselective synthesis of enantiopure 6a and 12. The aldol addition of acetaldehyde to isatins has also been shown to be catalyzed by 9-amino-(9-deoxy)-epi-qunine (9-NH2-epiQN) to provide aldol adducts in high yield (up to 96%) and good enantioselectivity (up to 93% ee).20
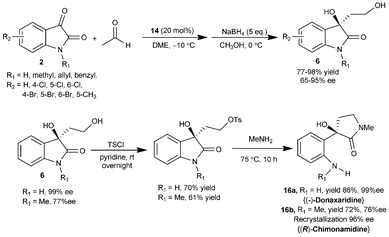 | ||
| Scheme 7 Aldol reaction of acetaldehyde with isatins catalyzed by 14. | ||
Xiao et al. have developed a proline-derived bifunctional organocatalyst for direct aldol reaction of isatin derivatives with acetone and 2-butanone.21 The 10 mol% of catalyst 17 in combination with acetic acid (20 mol%) catalyzes the direct aldol reaction between ketones (acetone and 2-butanone) and isatins (2) to provided aldol products 3 in high yield (85–99%) with good enantioselectivity (70–90% ee) and with regioselectivity of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 favouring the linear isomer (Scheme 8). The methodology has been used in the simple synthesis of (S)-convolutamydine A in 45% yield and 87% enantiomeric excess. The enantiofacial discrimination in the transition state was governed by the catalyst which controls the enamine geometry and double hydrogen bonds with isatin which fixes its orientation in the transition state (TS 4, Scheme 8).
:1 favouring the linear isomer (Scheme 8). The methodology has been used in the simple synthesis of (S)-convolutamydine A in 45% yield and 87% enantiomeric excess. The enantiofacial discrimination in the transition state was governed by the catalyst which controls the enamine geometry and double hydrogen bonds with isatin which fixes its orientation in the transition state (TS 4, Scheme 8).
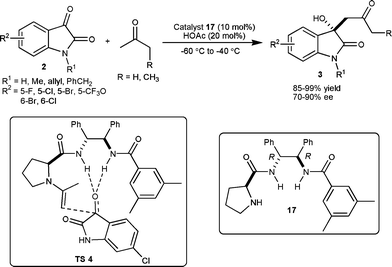 | ||
| Scheme 8 Proline-derived catalyst 17 catalyzed aldol reaction of ketone with isatins. | ||
A systematic study on the aldol reaction of isatin and acetone catalyzed by simple N-aryl and N-heteroaryl pyrrolidine amide organocatalysts (18a–h) has been reported.22 After screening of various N-pyridylprolinamide and N-quinolinylprolinamide organocatalysts, the prolinamide 18a in combination with AcOH at −20 °C provided the S-isomer of the product 3d with 72% enantiomeric excess. In the case of aminopyridine (18a–c) and aminoquinoline (18d–e) based catalysts, the relative position of the amino group and the pyridine nitrogen atom have a major impact on the enantioselectivity of the reaction. In order to analyze the effect of the pyridine nitrogen, the reaction was performed with the simple prolinamide catalyst 18f, which provided the product with 65% ee; however the reaction was slow. Further, to assess the role of the amidic proton, the same reaction was performed with organocatalyst 18g, which gave trace amounts of product. These experiments highlighted the importance of the amidic proton for high enantioselectivity and yield of the product (Scheme 9).
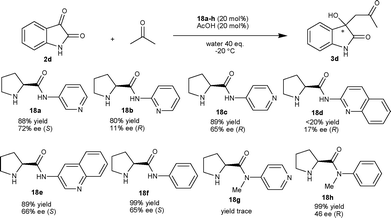 | ||
| Scheme 9 L-Prolinamide organocatalysts catalyzed aldol reaction of 2d with acetone. | ||
L-Proline has also been shown to catalyze the direct aldol reaction of acetone with isatin derivatives (2) to provide 3-acetonyl-3-hydroxy-2-oxindoles (3) in high yield (76–96%) with moderate to good enantioselectivity (39–79% ee).23
Zhao et al. has revised the old protocol of the aldol reaction, involving an enolate mechanism, in the light of enamine-mediated modern aldol reactions.24 The quinidine thiourea 20 efficiently catalyzes the aldol reaction of unactivated ketones 19 and isatins 2via a noncovalent catalytic mechanism involving an enolate intermediate, providing biologically important 3-alkyl-3-hydroxyindolin-2-ones (3) in good yield and enantioselectivity (Scheme 10). This approach was utilized for the first enantioselective synthesis of 3-hydroxyindolin-2-one 3f, which is the lead compound for treating Ewing's sarcoma discovered most recently by Toretsky and co-workers.25 The mechanism of reaction was proposed to be initiated by initial deprotonation of acetone by the tertiary amine of the quinidine thiourea catalyst backbone. After deprotonation, the enolate associates closely with quinuclidine nitrogen of the catalyst through ionic interactions (i), while thiourea moiety of the quinidine thiourea catalyst forms two hydrogen bonds with isatin carbonyl groups (ii), which activate the ketone group for the enolate attack and also direct the approach of isatin in the transition state. Among the two possible transition states, TS-5 is favoured over TS-6, since the unfavourable interaction between the aromatic ring of isatin and the enolate is avoided (Scheme 10). The key importance of this methodology has been realized in those cases of aldol reactions where the formation of enamine was difficult.
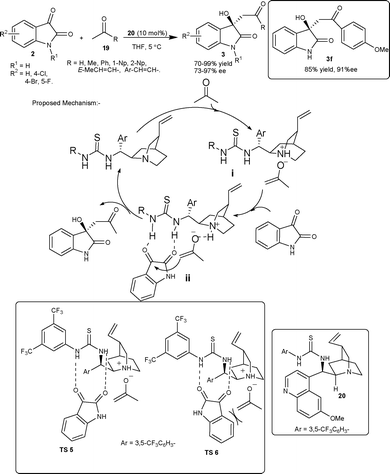 | ||
| Scheme 10 Thiourea 20 catalyzed aldol reaction of ketone with isatins. | ||
Recently, Wang and co-workers26 extended the above methodology to various α,β-unsaturated ketones as an aldol donor. The quinidine thiourea 20 and its epimer catalyzes the aldol reaction of 19 with 2 to provide both enantiomers of the aldol adduct 3 in low to excellent yield (18–98%) and moderate to excellent enantioselectivity (30–97% ee).
Singh et al. developed the first enantioselective aldol reaction of cyclohexanone with isatins for procuring chiral 3-cycloalkane-3-hydroxyoxindoles, which are potentially bioactive molecules.27 The organocatalyst 24 in the presence of TFA as an additive efficiently catalyzes the aldol reaction affording adducts 22 in quantitative yield, dr of 99:1 (syn/anti) with 99% enantiomeric excess of the syn isomer (Scheme 11). The prolinamide catalyst failed to catalyze this reaction, in contrast to their successful use in the aldol reaction of cyclohexanone with aldehydes,28 thus highlighting the utility of primary amine organocatalysts in the catalysis of sterically demanding molecules.
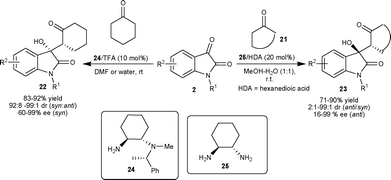 | ||
| Scheme 11 Cyclohexyl diamine-based organocatalysts (24 and 25) catalyzed aldol reaction of ketones with isatins. | ||
1,2-Diaminocyclohexane–hexanedioic acid (25–HDA) efficiently catalyzes the anti-selective direct aldol reaction of cycloketones (21) and various isatin derivatives (2) in MeOH–H2O providing the corresponding products 23 in good yield (71–90%) with high diastereoselectivity (up to 99:1 anti/syn) and enantioselectivity of up to 99% ee for the anti isomer (Scheme 11).29
Later, the authors used Cinchona alkaloid-derived organocatalysts to extend the scope of the above reaction to various acyclic aldol donors.30 The 10 mol% of the catalytic mixture of 26–TFA (1:1) catalyzes the aldol reaction of acetophenone with isatin to provide the aldol product in ten days with good enantiomeric excess of 90%, and yield of 58% (Scheme 12). Further, they explored Cinchona alkaloid-derived urea organocatalysts (27) for a variety of acetophenone derivatives (19) employing a noncovalent catalysis mechanism. The 3-substituted-3-hydroxy-2-oxindoles 3 were obtained in good yield (71–93%) and good enantioselectivity (72–92% ee) (Scheme 12). The application of the catalyst 27 has been successfully extended to different aldol donors such as acetone and acetaldehyde. The methodology finds its application in the synthesis of 3a in 92% enantiomeric excess.
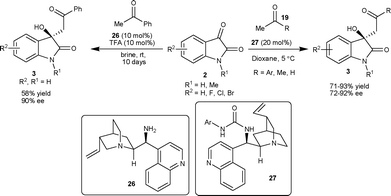 | ||
| Scheme 12 Cinchona alkaloid-derived catalyst 26 and 27 catalyzed aldol reaction of ketone and aldehyde with isatins. | ||
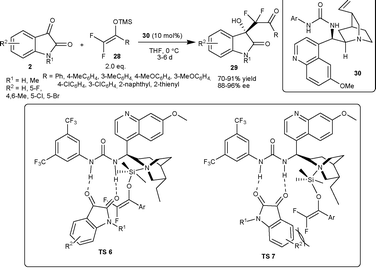 | ||
| Scheme 13 Aldol reaction of silyl enol ethers to isatins. | ||
 | ||
| Scheme 14 Phospho-aldol reaction of isatins. | ||
 | ||
| Scheme 15 β-Isocupreidine catalyzed MBH reaction of acrolein to isatins. | ||
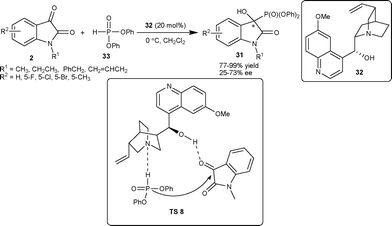
Shi and co-workers34 have also reported the application of β-isocupreidine as a catalyst for asymmetric MBH reaction of acrylate 38 with isatin derivatives (2) to generate a quaternary hydroxylated stereocenter on the oxindole (39) with moderate to excellent yield (17–99%) and good enantioselectivity (46–94% ee) in dichloromethane at room temperature. Lu et al.35 also reported the similar transformation in good yield (61–96%) and high enantioselectivity (86–96% ee) catalyzed by 10 mol% of β-isocupreidine in chloroform with 4 Å molecular sieves (MS) as additives at room temperature.
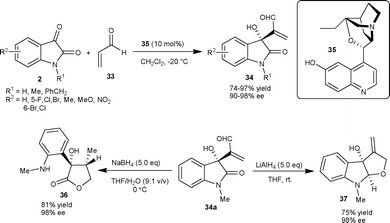
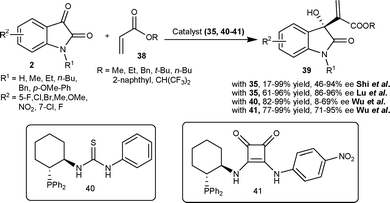
Wu et al.36 have also studied the MBH reaction of acrylates 38 with isatins (2) employing bifunctional phosphinothiourea organocatalyst based on a chiral cyclohexane scaffold. The reaction proceeded smoothly in the presence of 10 mol% of catalyst 40 to provide 3-hydroxy-2-oxindoles 39 in good to excellent yield (82–99%) and low to moderate enantioselectivity (8–69% ee) (Scheme 16). In order to increase the enantioselectivity of the reaction, the authors have recently developed a new bifunctional phosphine organocatalyst bearing squaramide as H-bond donor.37 The phosphine–squaramide 41 catalyzed the MBH reaction of 38 with 2 in high yield (77–99%) and good enantioselectivity (71–95% ee) (Scheme 16). The squaramide catalyst (41) proved to be superior to the thiourea catalyst (40) for this transformation.
 | ||
| Scheme 16 MBH reactions of acrylate to isatins. | ||
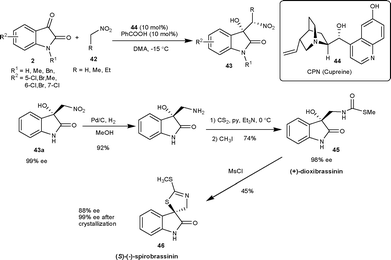 | ||
| Scheme 17 Cupreine (44) catalyzed Henry reaction of isatins. | ||
Two independent reports have been published on a similar transformation catalyzed by 9-O-protected cupreine. The 9-O-benzyl cupreine39 (47) and 9-O-3,5-trifluoromethylbenzoyl cupreine40 (48) have been shown to catalyze the Henry reaction of nitromethane with isatin derivatives to provide the nitro aldol adduct in high yield (up to 98%) and moderate to good enantioselectivity (up to 95% ee) (Fig. 4).
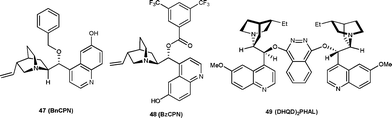 | ||
| Fig. 4 Organocatalysts (47–49) used to catalyze the Henry reaction of isatins. | ||
Very recently, Rao and co-workers41 have developed the biscinchona alkaloid 49 catalyzed Henry reactions of isatins with nitromethane. The resulting Henry adducts bearing a C3 quaternary stereocentre were obtained in high yield (79–94%) and with a good level of enantioselectivity (64–97% ee) (Fig. 4).
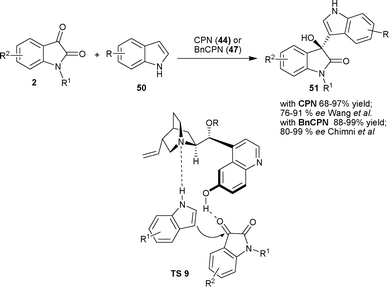 | ||
| Scheme 18 Friedel–Crafts reaction of indoles with isatins. | ||
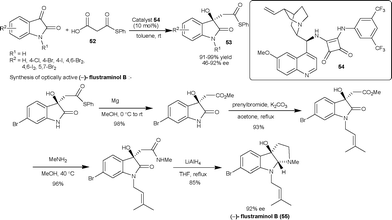 | ||
| Scheme 19 Squaramide 54 catalyzed decarboxylative aldol reaction of malonic acid half thioesters (52) with isatins. | ||
2.2 Oxindole as a nucleophile
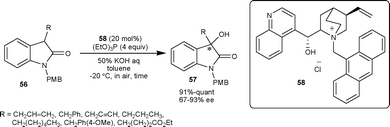 | ||
| Scheme 20 Hydroxylation reaction of isatins catalyzed by PTC 58. | ||
:1 dr, 94% ee), while minimizing the formation of isatide. The potential synthetic usefulness of this methodology has been shown in the straightforward preparation of compound 63 which bears the hexahydropyrrolo[2,3-b]indole unit found in many natural molecules (Scheme 21).
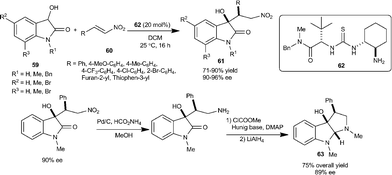 | ||
| Scheme 21 Michael addition of dioxindole to nitroalkenes. | ||
2.3 Miscellaneous
Barbas and co-workers47 have described a novel aminooxygenation of oxindoles 56 with nitrosobenzene catalyzed by a newly designed quinidine dimer 65 that affords the desired product 64 in good yield (65–86%) and good to high enantioselectivity (73–96% ee). Interestingly, in most of the cases aminooxygenation is O-selective. The reaction mechanism proceeds initially through enolate (i), which add to nitrosobenzene in an O-selective fashion in a chiral environment provided by the Cinchona catalyst. Finally, the proton transfer from the protonated amine catalyst to species (ii) provides the desired product (Scheme 22).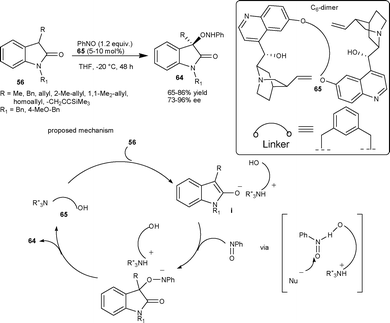 | ||
| Scheme 22 Aminooxygenation of oxindoles. | ||
Melchiorre et al.48 developed a new catalytic strategy for synthesis of 3-substituted-3-hydroxy-2-oxindoles. The prolinol 68 promotes the nucleophilic addition of dioxindole 59 to unsaturated aldehyde 66 by imminium ion activation. The spiro oxindole γ-butyrolactones (67 and 67′) were obtained after direct oxidation of crude reaction mixture in good yield and optical purity. 3-Substituted-3-hydroxy-2-oxindole was easily obtained after simple chemical manipulation of the product 67 (Scheme 23). The utility of this methodology was highlighted by the total synthesis of maremycine A.
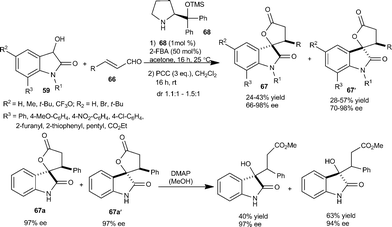 | ||
| Scheme 23 Prolinol 68 catalyzed Michael addition of dioxindole to unsaturated aldehydes. | ||
Scheidt et al.49 developed an enantioselective NHC(69)–Lewis acid catalyzed homoenolate annulation of enals 66 with isatins 2 to provide spiro oxindole γ-butyrolactone 67 in moderate to high yield (36–93%), good diastereomeric ratio (up to 20:1) and low to high enantioselectivity (13–98% ee) (Scheme 24). This approach was utilized in the concise total synthesis of maremycin B, which posseses anticancer activity.
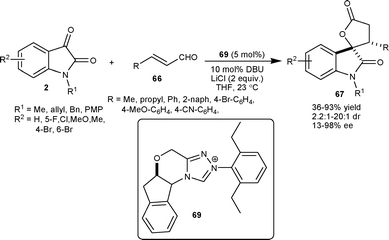 | ||
| Scheme 24 NHC 69 catalyzed homoenolate annulation of enals with isatins. | ||
3. Organometallic catalysts
3.1 Isatins as electrophiles
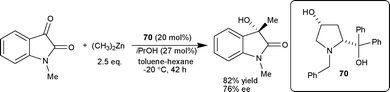 | ||
| Scheme 25 Addition of diethyl zinc to N-methylisatin. | ||
:2 catalyzes the reaction to furnish 3-aryl-3-hydroxy-2-oxindoles (72) in good yield (79–98%) and enantioselectivity (72–93% ee) (Scheme 26). The Rh–(R)-MeO-mop complex also catalyzes the addition of alkenylboronic acids to give tertiary allylic alcohol derivatives (72) in good yield (91–93%) and enantioselectivity (88–93% ee) (Scheme 26).
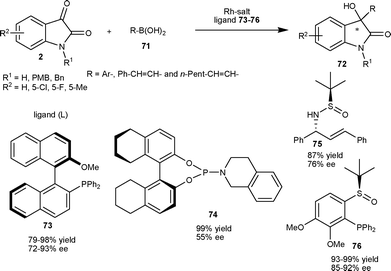 | ||
| Scheme 26 Organoboronic acids (71) addition to isatins (2) catalyzed by a Rh complex. | ||
Almost concurrently, another report on rhodium-catalyzed asymmetric addition of phenylboronic acid to isatin employing phosphoramidite ligand appeared in the literature. The catalyst generated from 3 mol% of [(C2H4)2Rh(acac)] and 9 mol% of 74 provided 3-phenyl-3-hydroxy-2-oxindole in virtually quantitative yield and 55% ee, which can be enantioenriched up to 94% ee with 59% yield after a single crystallization in 2-propanol.52 Recently, a new report on Rh-catalyzed asymmetric arylation of N-PMB-5-chloroisatin with phenylboronic acid, employing simple sulfur–alkene hybrid ligand 75, provided the desired product in good yield (87%) and moderate enantioselectivity (76% ee) (Scheme 26).53
This methodology is normally limited to N-protected isatins,51–53 while N–H isatins afford products with low enantioselectivity and low yield. Very recently, a rhodium-catalyzed asymmetric variant of this reaction employing chiral sulfoxide phosphine as ligand (76) has been reported for the addition of arylboronic acid (71) to N–H isatins as well as N-protected isatins to provided chiral 3-aryl-3-hydroxy-2-oxindoles (72) in excellent yield (93–99%) and good enantioselectivity (85–92% ee) (Scheme 26).54
In 2009, Qin et al.55 prepared an enantiopure tetra-ortho-substituted phosphinoimine ligand with a biphenyl backbone, which finds application in the asymmetric arylation of N-benzylisatin at the C3 position. 3-Aryl-3-hydroxy-2-oxindoles (72) were obtained in moderate to good yield (36–78%) and moderate to good enantioselectivity (38–73% ee) via nucleophilic addition of arylboronic acid (71) to N-benzylisatin catalyzed by a catalytic mixture of Pd(OAc)2 and ligand 77 in molar ratio of 1:2 (Scheme 27). In 2011, Shi and co-workers56 developed the enantioselective arylation of isatins with arylboronic acids by using chiral C2-symmetric cationic NHC-Pd2+ diaqua complex (78) as the catalyst under mild conditions. The 3-aryl-3-hydroxy-2-oxindoles (72) were obtained in good yield (79–92%) and moderate to good enantioselectivity (60–80% ee) (Scheme 27).
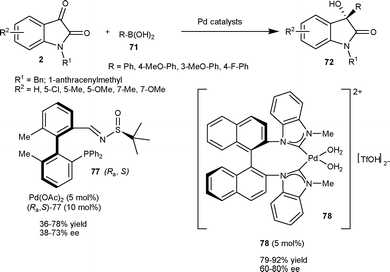 | ||
| Scheme 27 Arylboronic acid addition to isatins catalyzed by a Pd complex. | ||
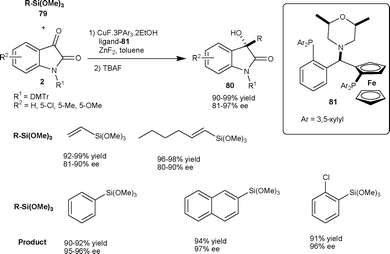 | ||
| Scheme 28 Reaction of organotrimethoxysilanes with isatins. | ||
 | ||
| Scheme 29 Friedel–Crafts reaction of arene/heteroarenes with isatins. | ||
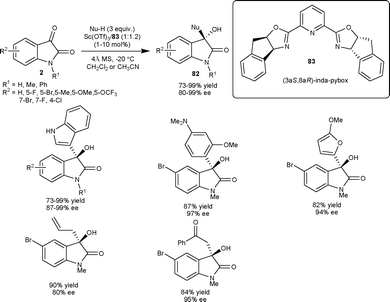
Later in 2011, they extended the scope of this methodology for the nucleophilic addition of pyrrole derivatives to isatins catalyzed by indium(III)-inda-pybox complex (10 mol%) to provide products in moderate to good yield (45–98%) with excellent level of enantioselectivity (94 to >99% ee).59
 | ||
| Scheme 30 Hetero-ene reaction of isatins. | ||
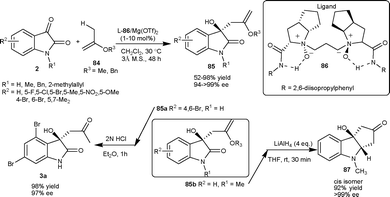
Chiral dicationic Pd-complex catalyzed enantioselective hetero-ene and Mukaiyama-aldol reactions of isatins result in the formation of optically active tertiary alcohols (Scheme 30).61 In the hetero-ene reaction, the isopropenyloxy(triisopropyl)silane 84c reacted with isatin derivatives (2) in the presence of 10 mol% of Pd complex 90 to provide the corresponding products (85) in good yield (72–98%) and good to excellent enantioselectivity (80–>99% ee). The methodology was applied in the synthesis of 3a in good yield (87%) and excellent enantioselectivity (99% ee). Additionally, the ene product could be transformed into other valuable products. After the success of hetero-ene reaction, the author extended the application of the Pd complex catalyst 90 to the aldol reaction of isatin derivatives (2) with trimethylsilyl ketene thioacetal (88). The aldol adducts 89 were isolated in good yield and enantioselectivity and could be used as synthons for the synthesis of a variety of natural products (Scheme 31).
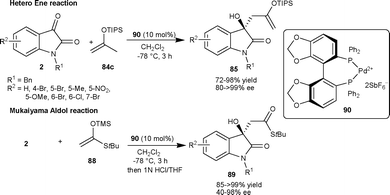 | ||
| Scheme 31 Hetero-ene and Mukaiyama-aldol reaction of isatins. | ||
Zhou and co-workers63 have developed the palladium-catalyzed asymmetric allylation of isatins (2) with allylic alcohols (91) as an allyl donor using chiral spiro phosphoramidite ligand (93). The chiral tertiary homoallylic alcohols (92) were obtained in excellent yields (up to 99%) and with moderate enantioselectivity (48–71%) (Scheme 32).
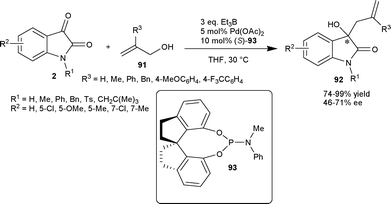 | ||
| Scheme 32 Allylation of isatins with allylic alcohols. | ||
Krische and co-workers64 reported the highly enantioselective allylations, crotylations, and prenylations of isatin catalyzed by an Ir complex in the absence of stoichiometric amounts of allylmetal reagents, unlike conventional allylation methodologies which employ stoichiometric quantities of allyl–metal reagents. The allylations, crotylations, and prenylations of N-benzylisatin derivatives have been performed using cyclometalated C,O-benzoate complex 96 generated in situ from [{Ir(cod)Cl}2], cth-(R)-p-phos (ligand) and 3-nitrobenzoic acid or 4-cyano-3-nitrobenzoic acid (Scheme 32). Allylation of isatin using allyl acetate produce tertiary homoallyllic alcohols 92 in good yields (65–92%) with uniformly high levels of optical enrichment (91–96% ee). The crotylation of isatins employing α-methyl allyl acetate as the crotyl donor afford 94 in good yield (64–87%) and good enantioselectivity (80–92% ee). The reverse prenylation of isatins has been performed using 1,1-dimethylallene to provide 95 with two contiguous quaternary carbon centers in good yield (70–90%) and high levels of optical enrichment (90–96% ee) under mild reaction conditions. The mechanism for reverse prenylation is depicted in Scheme 33. The deprotonation of complex (i) in the presence of Cs2CO3 to furnish the anionic iridium C,O-benzoate, which undergo oxidative addition to 1,2-dimethylallene leads to the formation of (E)-σ-crotonyl iridium intermediate (ii). Isatin addition to the intermediate (ii) occurs through a chair-like transition structure, which delivers the homoallylic iridium alkoxide (iii). The enantiofacial selectivity of carbonyl addition is opposite to crotylation which occurs by way of the transition structure A, whereas isatin prenylation occurs by way of the transition structure B. The basis of this partitioning may arise from nonbonding interactions of the axial methyl group of the σ-prenyl iridium intermediate with the amide π-bond of isatin, which is presumably more destabilizing than nonbonding interactions of the axial methyl group with the electron-deficient rim of the arene (Scheme 33).
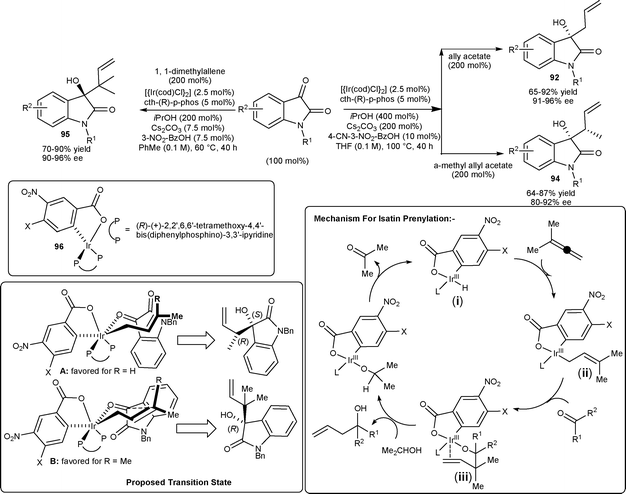 | ||
| Scheme 33 Allylations, crotylations and prenylations of isatins. | ||
Very recently, Franz et al.65 developed the enantioselective Hosomi–Sakurai allylation of isatins (2) with substituted allylic silanes (97) catalyzed by Sc(OTf)3-indapybox catalyst with a TMSCl activator and additive such as NaSbF6. The 3-substituted-3-hydroxy-2-oxindoles (98) were obtained in good to excellent yield (72–99%) and enantioselectivity (80–99% ee) (Scheme 34). The important features of the methodology are low catalyst loading of 0.05 mol%, wide substrate scope of isatins, and use of various substituted allylic silanes and application to gram scale preparation.
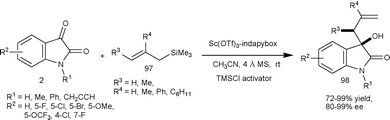 | ||
| Scheme 34 Hosomi–Sakurai allylation of isatins. | ||
3.2 Oxindole as a nucleophile
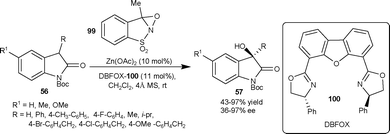 | ||
| Scheme 35 Hydroxylation reaction of isatins using oxaziridine 99. | ||
Feng and co-workers67 have developed the enantioselective hydroxylation of 3-methyl-2-oxindole using TMPO as the oxygen source. The 3-methyl-3-hydroxy-2-oxindole was obtained in moderate yield (63%) and enantioselectivity (43% ee) by using Sc(OTf)3/L-86 complex as a catalyst.
3.3 Anilide cyclization
Shibasaki et al.57 have successfully developed the highly enantioselective intermolecular arylation of isatins leading to 3-hydroxy-3-aryl-2-oxindoles. The methodology found its limitation in the case of isatin derivatives with a bulky CF3 group at the C4 position, which dramatically decreases the reactivity and enantioselectivity of the reaction. To overcome this limitation, they developed a catalytic enantioselective intramolecular arylation of α-keto amides 101 as an alternative strategy.57 After screening of different ligands, the Ph-BPE 105 was found to be an optimum ligand for the intramolecular cyclization reaction providing product 102 with high yield and enantioselectivity. This strategy was applied in the total synthesis of SM-130686, involving the enantioselective intramolecular arylation of 103 in the presence of 10 mol% CuF-(R,R)-105 catalyst, to obtain 104 in 90% yield with 85% ee, which could be synthetically transformed to SM-130686 (Scheme 36).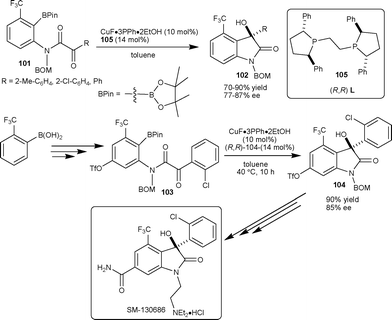 | ||
| Scheme 36 Total synthesis of SM-130686 by anilide cyclization. | ||
Recently Shibasaki et al.68 described the palladium–difluorphos complex catalyzed intramolecular arylation of α-keto amides, leading to the formation of enantiomerically enriched 3-substituted-3-hydroxy-2-oxindoles. The chiral catalyst derived from (R)-DifluorPhos (108) and [Pd(CH3CN)4](BF4)2 could efficiently promote the intramolecular cyclization of triflate 106 to furnish tetrasubstituted-3-hydroxyoxindoles 107 in good yield (53–92%) and enantioselectivity (83–99% ee). This methodology was applied to the total synthesis of ECi8, which is a potent antimicrobial lead drug69 (Scheme 37).
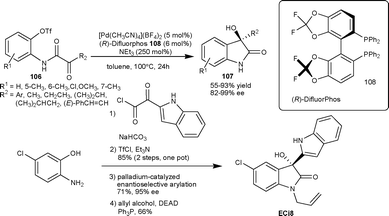 | ||
| Scheme 37 Palladium–difluorphos complex catalyzed intramolecular arylation of α-keto amides. | ||
3.4 Miscellaneous
Kündig et al.70 have developed an asymmetric Pd-catalyzed intramolecular arylation of α-alkoxy amide (109) using chiral N-heterocyclic carbene ligand 111. The 3-alkoxy-3-aryl-2-oxindoles (110) were obtained in moderate to excellent yield (45–99%) and good enantioselectivity (79–97% ee) (Scheme 38).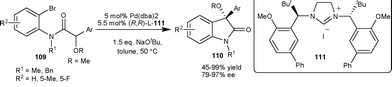 | ||
| Scheme 38 Intramolecular arylation of α-alkoxy amide (109). | ||
Antilla et al.71 have reported the asymmetric benzoyloxylation of 3-aryl-2-oxindoles (56) using readily available benzoyl peroxide (112) as a benzoyloxylation reagent catalyzed by a chiral VAPOL calcium phosphate salt 114. A series of 3-aryl-3-benzoyl-2-oxindoles (113) were obtained in good yield (60–96%) and excellent enantioselectivity (91 to >99% ee). The potential synthetic utility of this methodology has been demonstrated in the two step synthesis of 3-aryl-3-hydroxy-2-oxindole in good yield (88%) and enantioselectivity (95% ee) (Scheme 39).
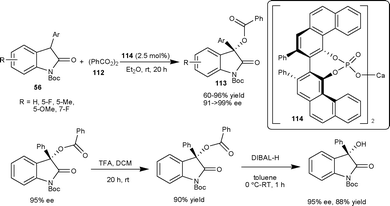 | ||
| Scheme 39 Benzoyloxylation of oxindoles. | ||
Trost and Hirano72 developed a highly enantioselective tandem Michael addition–transesterification process for synthesis of spirooxindole using ligand 116. The zinc-ProPhenol complex catalyzes the Michael addition of dioxindole (59) to α,β-unsaturated esters (115) and subsequent transesterification gave spirooxindole γ-butyrolactone (67) in moderate to good yield (38–89%) with good diastereoselectivity (up to 86%) and moderate to excellent enantioselectivity (54–99% ee) (Scheme 40). Spirocyclic oxindole products (67) serve as an important synthetic building blocks for synthesis of various 3-alkyl-3-hydroxy-2-oxindoles.
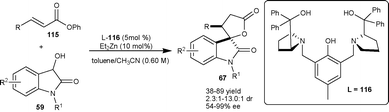 | ||
| Scheme 40 Zinc-ProProlinol catalyzed tandem Michael addition–transestrification. | ||
Conclusions
Inspired by the biological activities associated with the chiral 3-hydroxy-2-oxindole framework, considerable achievements have been made in their synthesis, especially in last few years. Both chiral organocatalysts and metal complex catalysts play an important role in the asymmetric synthesis of 3-substituted-3-hydroxyoxindoles. Since the first report on the organocatalytic enantioselective aldol reaction of isatins with acetone, a number of new enantioselective reactions catalyzed by a variety of organocatalysts have been reported. Metal catalysts have also proved to be useful in catalyzing many reactions. So, with the advancement in catalytic approaches, an impressive progress has been made in the synthesis of natural products and drug candidates containing this structural moiety. Biocatalysts still wait their turn in their synthesis which may add an important tool in the portfolio of catalysts.Isatins are an ideal substrate employed in the majority of the reactions in comparison with 3-substituted 2-oxindoles. Dioxindole has very recently emerged as an important precursor in the synthesis of targeted frameworks. There is still enough room for the nucleophilic addition of dioxindole to variety of electrophiles, which may lead to new 3-substituted-3-hydroxy-2-oxindole derivatives with improved bioactivities.
This area has recently blossomed and become more mature as new catalysts, new substrates and reaction conditions are being explored and shall be explored in the future. We envision that new chiral 3-substituted-3-hydroxy-2-oxindoles having enhanced medicinal properties will be synthesized in the future.
Acknowledgements
We are thankful to CSIR for the SRF fellowship to A. K. Our research work was supported by the research project (SR/SI/OC35/2011) sanctioned to S. S. C. by the DST, India. The financial support to Department of Chemistry by the Department of Science and Technology (DST), India under FIST program and UGC, India, under CAS-I is gratefully acknowledged.References
- (a) S. Peddibhotla, Curr. Bioact. Compd., 2009, 5, 20 CrossRef CAS; (b) S. Hibino and T. Choshi, Nat. Prod. Rep., 2001, 18, 66–87 RSC; (c) Y.-Q. Tang, I. Sattler, R. Thiericke, S. Grabley and X.-Z. Feng, Eur. J. Org. Chem., 2001, 261 CrossRef CAS; (d) Y. Koguchi, J. Kohno, M. Nishio, K. Takahashi, T. Okuda, T. Ohnuki and S. Komatsubara, J. Antibiot., 2000, 53, 105 CrossRef CAS; (e) R. B. Labroo and L. A. Cohen, J. Org. Chem., 1990, 55, 4901 CrossRef CAS.
- (a) T. Tokunaga, W. E. Hume, T. Umezome, K. Okazaki, Y. Ueki, K. Kumagai, S. Hourai, J. Nagamine, H. Seki, M. Taiji, H. Noguchi and R. Nagata, J. Med. Chem., 2001, 44, 4641 CrossRef CAS; (b) P. Hewawasam, N. A. Meanwell, V. K. Gribkoff, S. I. Dworetzky and C. G. Boissard, Bioorg. Med. Chem. Lett., 1997, 7, 1255 CrossRef CAS; (c) N. Boechat, W. B. Kover, V. Bongertz, M. M. Bastos, N. C. Romeiro, M. L. G. Azavedo and W. Wollinger, Med. Chem., 2007, 3, 533 CrossRef CAS; (d) P. Hewawasam, M. Erway, S. L. Moon, J. Knipe, H. Weiner, C. G. Boissard, D. J. Post-Munson, Q. Gao, S. Huang, V. K. Gribkoff and N. A. Meanwell, J. Med. Chem., 2002, 45, 1487 CrossRef CAS.
- (a) For review on asymmetric hydroxylation using chiral oxaziridines, see F. A. Davis and B.-C. Chen, Chem. Rev., 1992, 92, 919 CrossRef CAS; (b) For synthesis of enantioenriched 3-hydroxyoxindoles using chiral reagent, chiral substrate, HPLC resolution and chiral auxiliary, see S. Barroso, G. Blay, L. Cardona, I. Fernandez, B. Garcia and J. R. Pedro, J. Org. Chem., 2004, 69, 6821 CrossRef CAS; (c) T. Nakamura, S.-i. Shirokawa, S. Hosokawa, A. Nakazaki and S. Kobayashi, Org. Lett., 2006, 8, 677 CrossRef CAS , also references cited in 2a, 2b and 2d.
- (a) For selected reviews on asymmetric metallocatalysis, see F. Wang, L.-j. Liu, W. Wang, S. Li and M. Shi, Coord. Chem. Rev., 2012, 256, 804 CrossRef CAS; (b) D. Zhao and R. Wang, Chem. Soc. Rev., 2012, 41, 2095 RSC; (c) J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Soc. Rev., 2012, 41, 4126 RSC; (d) J. F. Larrow and E. N. Jacobsen, Top. Organomet. Chem., 2004, 6, 123 CAS; (e) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS; (f) Z. Lu and S. Ma, Angew. Chem., Int. Ed., 2008, 47, 258 CrossRef CAS; (g) S. Lühr, J. Holz and A. Börner, ChemCatChem, 2011, 3, 1708 CrossRef.
- (a) For selected reviews on asymmetric organocatalysis, see P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2001, 40, 3726 CrossRef CAS; (b) B. List, Tetrahedron, 2002, 58, 5573 CrossRef CAS; (c) B. List, Synlett, 2001, 1675 CrossRef CAS; (d) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138 CrossRef CAS; (e) W. Notz, F. Tanaka and C. F. Barbas III, Acc. Chem. Res., 2004, 37, 580 CrossRef CAS; (f) J. Seayad and B. List, Org. Biomol. Chem., 2005, 3, 719 RSC; (g) M. Marigo and K. A. Jorgensen, Chem. Commun., 2006, 2001 RSC; (h) C. M. Kleiner and P. R. Schreiner, Chem. Commun., 2006, 4315 RSC; (i) A. Erkkila, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416 CrossRef; (j) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS; (k) M. J. Gaunt, C. C. C. Johansson, A. McNally and N. T. Vo, Drug Discovery Today, 2007, 12, 8 CrossRef CAS; (l) R. M. de Figueiredo and M. Christmann, Eur. J. Org. Chem., 2007, 2575 CrossRef CAS; (m) D. Enders, C. Grondal and M. R. M. Huttl, Angew. Chem., Int. Ed., 2007, 46, 1570 CrossRef CAS; (n) H. Pellisier, Tetrahedron, 2007, 63, 9267 CrossRef; (o) A. Dondoni and A. Massi, Angew. Chem., Int. Ed., 2008, 47, 4638 CrossRef CAS; (p) P. Melchiorre, M. Marigo, A. Carlone and G. Bartoli, Angew. Chem., Int. Ed., 2008, 47, 6138 CrossRef CAS; (q) P. Chauhan and S. S. Chimni, RSC Adv., 2012, 2, 737 RSC; (r) P. Chauhan and S. S. Chimni, RSC Adv., 2012, 2, 6117 RSC; (s) For leading books on organocatalysis, see books: A. Berkessel and H. Groger, Metal-Free Organic Catalysis in Asymmetric Synthesis, Wiley-VCH, Weinheim, 2004 Search PubMed; (t) A. Berkessel and H. Groger, Asymmetric Organocatalysis–From Biomimetic Concepts to Applications in Asymmetric Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2005 Search PubMed; (u) P. I. Dalko, Enantioselective Organocatalysis, Wiley-VCH, Weinheim, 2007 Search PubMed; (v) B. List, Asymmetric Organocatalysis, Springer, 2009 Search PubMed.
- (a) N. R. Ball-Jones, J. J. Badillo and A. K. Franz, Org. Biomol. Chem., 2012, 10, 5165 RSC; (b) R. Rios, Chem. Soc. Rev., 2012, 41, 1060 RSC; (c) K. Shen, X. Liu, L. Lin and X. Feng, Chem. Sci., 2012, 3, 327 RSC; (d) J. E. M. N. Klein and R. J. K. Taylor, Eur. J. Org. Chem., 2011, 6821 CrossRef CAS; (e) J. J. Badillo, H. V. Hanhan and A. K. Franz, Curr Opin Drug Discov Devel., 2010, 13, 758 CAS; (f) F. Zhou, Y.-L. Liu and J. Zhou, Adv. Synth. Catal., 2010, 352, 1381 CrossRef CAS; (g) B. M. Trost and M. K. Brennan, Synthesis, 2009, 3003 CrossRef CAS; (h) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748 CrossRef CAS; (i) C. Marti and E. M. Carreira, Eur. J. Org. Chem., 2003, 2209 CrossRef CAS , also reference 1a.
- F. Braude and H. G. Lindwall, J. Am. Chem. Soc., 1933, 55, 325 CrossRef CAS.
- G. Luppi, P. G. Cozzi, M. Monari, B. Kaptein, Q. B. Broxterman and C. Tomasini, J. Org. Chem., 2005, 70, 7418 CrossRef CAS.
- G. Luppi, M. Monari, R. J. Corrêa, F. A. Violante, A. C. Pinto, B. Kaptein, Q. B. Broxterman, S. J. Garden and C. Tomasini, Tetrahedron, 2006, 62, 12017 CrossRef CAS.
- J. G. Hernández, V. G-López and E. Juaristi, Tetrahedron, 2012, 68, 92 CrossRef.
- A. V. Malkov, M. A. Kabeshov, M. Bella, O. Kysilka, D. A. Malyshev, K. Pluháčková and P. Kočovský, Org. Lett., 2007, 9, 5473 CrossRef CAS.
- C. Shen, F. Shen, H. Xia, P. Zhang and X. Chen, Tetrahedron: Asymmetry, 2011, 22, 708 CrossRef CAS.
- A. Ricci, L. Bernardi, C. Gioia, S. Vierucci, M. Robitzer and F. Quignard, Chem. Commun., 2010, 46, 6288 RSC.
- S. Hu, L. Zhang, J. Li, S. Luo and J.-P. Cheng, Eur. J. Org. Chem., 2011, 3347 CrossRef CAS.
- S. Nakamura, N. Hara, H. Nakashima, K. Kubo, N. Shibata and T. Toru, Chem.–Eur. J., 2008, 14, 8079 CrossRef CAS.
- N. Hara, S. Nakamura, N. Shibata and T. Toru, Chem.–Eur. J., 2009, 15, 6790 CrossRef CAS.
- F. Xue, S. Zhang, L. Liu, W. Duan and W. Wang, Chem.–Asian J., 2009, 4, 1664–1667 CrossRef CAS.
- T. Itoh, H. Ishikawa and Y. Hayashi, Org. Lett., 2009, 11, 3854 CrossRef CAS.
- W.-B Chen, X.-L Du, L.-F. Cun. X.-M. Zhang and W.-C. Yuan, Tetrahedron, 2010, 66, 1441 CrossRef CAS.
- Q. Guo and J. C.-G. Zhao, Tetrahedron Lett., 2012, 53, 1768 CrossRef CAS.
- J.-R. Chen, X.-P. Liu, X.-Y. Zhu, L. Li, Y.-F. Qiao, J.-M. Zhang and W.-J. Xiao, Tetrahedron, 2007, 63, 10437 CrossRef CAS.
- M. Kinsella, P. G. Duggan and C. M. Lennon, Tetrahedron: Asymmetry, 2011, 22, 1423 CrossRef CAS.
- G. Chen, Y. Wang, H.-P. He, S. Gao, X.-S. Yang and X.-J. Hao, Heterocycles, 2006, 68, 2327 CrossRef CAS.
- Q. Guo, M. Bhanushali and C.-G. Zhao, Angew. Chem., Int. Ed., 2010, 49, 9460 CrossRef CAS.
- V. Erkizan, Y. Kong, M. Merchant, S. Schlottmann, J. S. Barber-Rotenberg, L. Yuan, O. D. Abaan, T.-H. Chou, S. Dakshanamurthy, M. L. Brown, A. Uren and J. A. Toretsky, Nat. Med., 2009, 15, 750 CrossRef.
- G.-G. Liu, H. Zhao, Y.-B. Lan, B. Wu, X.-F. Huang, J. Chen, J.-C. Tao and X.-W. Wang, Tetrahedron, 2012, 68, 3843 CrossRef CAS.
- M. Raj, N. Veerasamy and V. K. Singh, Tetrahedron Lett., 2010, 51, 2157 CrossRef CAS.
- (a) M. Raj, V. Maya, S. K. Ginotra and V. K. Singh, Org. Lett., 2006, 8, 4097 CrossRef CAS; (b) V. Maya, M. Raj and V. K. Singh, Org. Lett., 2007, 9, 2593 CrossRef CAS.
- Y. Liu, P. Gao, J. Wang, Q. Sun, Z. Ge and R. Li, Synlett, 2012, 23, 1031 CrossRef CAS.
- S. Allu, N. Molleti, R. Panem and V. K. Singh, Tetrahedron Lett., 2011, 52, 4080 CrossRef CAS.
- Y.-L. Liu and J. Zhou, Chem. Commun., 2012, 48, 1919 RSC.
- L. Peng, L.-L. Wang, J.-F. Bai, L.-N. Jia, Q.-C. Yang, Q.-C. Huang, X.-Y. Xu and L.-X. Wang, Tetrahedron Lett., 2011, 52, 1157 CrossRef CAS.
- Y.-L. Liu, B.-L. Wang, J.-J. Cao, L. Chen, Y.-X. Zhang, C. Wang and J. Zhou, J. Am. Chem. Soc., 2010, 132, 15176 CrossRef CAS.
- X.-Y. Guan, Y. Wei and M. Shi, Chem.–Eur. J., 2010, 16, 13617 CrossRef CAS.
- F. Zhong, G.-Y. Chen and Y. Lu, Org. Lett., 2011, 13, 82 CrossRef CAS.
- C.-C. Wang and X.-Y. Wu, Tetrahedron, 2011, 67, 2974 CrossRef CAS.
- J.-Y. Qian, C.-C. Wang, F. Sha and X.-Y. Wu, RSC Adv., 2012, 2, 6042 RSC.
- L. Liu, S. Zhang, F. Xue, G. Lou, H. Zhang, S. Ma, W. Duan and W. Wang, Chem.–Eur. J., 2011, 17, 7791 CrossRef CAS.
- M. Q. Li, J.-X. Zhang, X.-F. Huang, B. Wu, Z.-M. Liu, J. Chen, X.-D. Li and X.-W. Wang, Eur. J. Org. Chem., 2011, 5237 CrossRef CAS.
- Y. Zhang, Z. J. Li, H. S. Xu, Y. Zhang and W. Wang, RSC Adv., 2011, 1, 389 RSC.
- P. S. Prathima, K. Srinivas, K. Balaswamy, R. Arundhathi, G. N. Reddy, B. Sridhar, M. M. Rao and P. R. Likhar, Tetrahedron: Asymmetry, 2011, 22, 2099 CrossRef.
- J. Deng, S. Zhang, P. Ding, H. Jiang, W. Wang and J. Li, Adv. Synth. Catal., 2010, 352, 833 CrossRef CAS.
- P. Chauhan and S. S.Chimni, Chem.–Eur. J., 2010, 16, 7709 CrossRef CAS.
- N. Hara, S. Nakamura, Y. Funahashi and N. Shibata, Adv. Synth. Catal., 2011, 353, 2976 CrossRef CAS.
- D. Sano, K. Nagata and T. Itoh, Org. Lett., 2008, 10, 1593 CrossRef CAS.
- M. Retini, G. Bergonzini and P. Melchiorre, Chem. Commun., 2012, 48, 3336 RSC.
- T. Bui, N. R. Candeias and C. F. Barbas III, J. Am. Chem. Soc., 2010, 132, 5574 CrossRef CAS.
- G. Bergonzini and P. Melchiorre, Angew. Chem., Int. Ed., 2012, 51, 971 CrossRef CAS.
- J. Dugal-Tessier, E. A. O'Bryan, T. B. H. Schroeder, D. T. Cohen and K. A. Scheidt, Angew. Chem., Int. Ed., 2012, 51, 4963 CrossRef CAS.
- K. Funabashi, M. Jachmann, M. Kanai and M. Shibasaki, Angew. Chem., Int. Ed., 2003, 42, 5489 CrossRef CAS.
- R. Shintani, M. Inoue and T. Hayashi, Angew. Chem., Int. Ed., 2006, 45, 3353 CrossRef CAS.
- P. Y. Toullec, R. B. C. Jagt, J. G. de Vries, B. L. Feringa and A. J. Minnaard, Org. Lett., 2006, 8, 2715–2718 CrossRef CAS.
- X. Feng, Y. Nie, J. Yang and H. Du, Org. Lett., 2012, 14, 624 CrossRef CAS.
- J. Gui, G. Chen, P. Cao and J. Liao, Tetrahedron: Asymmetry, 2012, 23, 554 CrossRef CAS.
- H. Lai, Z. Huang, Q. Wu and Y. Qin, J. Org. Chem., 2009, 74, 283 CrossRef CAS.
- Z. Liu, P. Gu, M. Shi, P. McDowell and G. Li, Org. Lett., 2011, 13, 2314 CrossRef CAS.
- D. Tomita, K. Yamatsugu, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 6946 CrossRef CAS.
- N. V. Hanhan, A. H. Sahin, T. W. Chang, J. C. Fettinger and A. K. Franz, Angew. Chem., Int. Ed., 2010, 49, 744 CrossRef CAS.
- E. G. Gutierrez, C. J. Wong, A. H. Sahin and A. K. Franz, Org. Lett., 2011, 13, 5754 CrossRef CAS.
- K. Zheng, C. Yin, X. Liu, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2011, 50, 2573 CrossRef CAS.
- K. Aikawa, S. Mimura, Y. Numata and K. Mikami, Eur. J. Org. Chem., 2011, 62 CrossRef CAS.
- M. Kitajima, I. Mori, K. Arai, N. Kogure and H. Takayama, Tetrahedron Lett., 2006, 47, 3199 CrossRef CAS.
- X.-C. Qiao, S.-F. Zhu and Q.-L. Zhou, Tetrahedron: Asymmetry, 2009, 20, 1254 CrossRef CAS.
- J. Itoh, S. B. Han and M. J. Krische, Angew. Chem., Int. Ed., 2009, 48, 6313 CrossRef CAS.
- N. V. Hanhan, Y. C. Tang, N. T. Tran and A. K. Franz, Org. Lett., 2012, 14, 2218 CrossRef CAS.
- T. Ishimaru, N. Shibata, J. Nagai, S. Nakamura, T. Toru and S. Kanemasa, J. Am. Chem. Soc., 2006, 128, 16488 CrossRef CAS.
- K. Shen, X. Liu, G. Wang, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2011, 50, 4684 CrossRef CAS.
- L. Yin, M. Kanai and M. Shibasaki, Angew. Chem., Int. Ed., 2011, 50, 7620 CrossRef CAS.
- Y. Shen, J. Liu, G. Estiu, B. Isin, Y. Y. Ahn, D.-S. Lee, A.-L. Barabási, V. Kapatral, O. Wiest and Z. N. Oltvai, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1082 CrossRef CAS.
- Y.-X. Jia, J. M. Hillgren, E. L. Watson, S. P. Marsden and E. P. Kündig, Chem. Commun., 2008, 4040 RSC.
- Z. Zhang, W. Zheng and J. C. Antilla, Angew. Chem., Int. Ed., 2011, 50, 1135 CrossRef CAS.
- B. M. Trost and K. Hirano, Org. Lett., 2012, 14, 2446 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |