Graphene oxide and its reduction: modeling and experimental progress
Shun
Mao
,
Haihui
Pu
and
Junhong
Chen
*
Department of Mechanical Engineering, University of Wisconsin-Milwaukee, 3200 N Cramer Street, Milwaukee, WI 53211, USA. E-mail: jhchen@uwm.edu; Fax: +1-414-229-6958; Tel: +1-414-229-2615
First published on 12th January 2012
Abstract
Graphene oxide (GO) has attracted intense interest for its use as a precursor material for the mass production of graphene-based materials, which hold great potential in various applications. Insights into the structure of GO and reduced GO (RGO) are of significant interest, as their properties are dependent on the type and distribution of functional groups, defects, and holes from missing carbons in the GO carbon lattice. Modeling the structural motifs of GO can predict the structural evolution in its reduction and presents promising directions to tailor the properties of RGO. Two general reduction approaches, chemical and thermal, are proposed to achieve highly reduced GO materials. This review introduces typical chemical oxidation methods to produce GO from pure graphite, then summarizes the modeling progress on the GO structure and its oxidation and reduction dynamics, and lastly, presents the recent progress of RGO preparation through chemical and thermal reduction approaches. By summarizing recent studies on GO structural modeling and its reduction, this review leads to a deeper understanding of GO morphology and reduction path, and suggests future directions for the scalable production of graphene-based materials through atomic engineering.
 Shun Mao | Shun Mao serves as a project director of NanoAffix Science LLC located in Milwaukee (USA). He received his M.S. degree in Materials from Wuhan University of Technology (China) in 2006 and Ph.D. degree in Mechanical Engineering from University of Wisconsin-Milwaukee (USA) in 2010. His research includes the synthesis of nanoparticle-, carbon nanotube-, and graphene-based nanomaterials and their applications for gas sensors and biosensors. |
 Haihui Pu | Haihui Pu is a Ph.D. candidate in Mechanical Engineering at the University of Wisconsin-Milwaukee. He obtained his M.S. degree in Optics Engineering from Fudan University in China in 2010, where he studied the holographic polymer dispersed liquid crystal (HPDLC) transmission grating. His current research includes the structural characterization of graphene oxide and its thermal reduction dynamics, interactions between graphene and various nanoparticles in hybrid nanostructures and their potential applications through density functional theory (DFT) modeling. |
 Junhong Chen | Junhong Chen received the B.E. degree in Thermal Engineering from Tongji University, Shanghai, China, in 1995 and M.S. and Ph.D. degrees in Mechanical Engineering from University of Minnesota, Minneapolis, MN, in 2000 and 2002, respectively. From 2002 to 2003, he was a postdoctoral scholar in Chemical Engineering at California Institute of Technology, where he worked on arc plasma synthesis of nanoparticles. He is currently a full Professor in the Department of Mechanical Engineering at the University of Wisconsin-Milwaukee. His current research interests include carbon nanotube- and graphene-based hybrid nanomaterials, plasma reacting flows, and nanotechnology for sustainable energy and environment. |
1. Introduction
Graphene, an atomic-thick layer of carbon atoms arranged in a honeycomb lattice, draws extensive attention from both the experimental and theoretical communities due to its prominent structural and electrical properties.1–16 Intrinsic graphene is a semi-metal or zero-gap semiconductor that has remarkably high electron mobility, around 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 V−1 s−1, arising from its linear energy dispersion with respect to wave vector near the Dirac point.17 The resistivity of graphene is on the order of 10−6 Ω cm and is known as the substrate with lowest resistivity at room temperature.18 In addition, graphene has large specific surface area (2630 m2 g−1),19 good chemical stability,20 and high sensitivity to electrical perturbations due to its ultra-small thickness.21 The unique transport properties and structure make graphene attractive for a variety of promising applications. For example, graphene can be used as anode materials to improve the efficiency of batteries22–26 and as transparent conductive electrodes in solar cell.27–31 It could also be a potential candidate for integrated circuits-the smallest transistor (one atom thick, 10 atoms wide) known so far was made of graphene.32 Other applications of graphene and graphene-based materials such as field emitters,33–35 hydrogen storage,36–42 supercapacitors,43–48 gas sensors,49–58 and bio-sensors59–65 have also been reported.
000 cm2 V−1 s−1, arising from its linear energy dispersion with respect to wave vector near the Dirac point.17 The resistivity of graphene is on the order of 10−6 Ω cm and is known as the substrate with lowest resistivity at room temperature.18 In addition, graphene has large specific surface area (2630 m2 g−1),19 good chemical stability,20 and high sensitivity to electrical perturbations due to its ultra-small thickness.21 The unique transport properties and structure make graphene attractive for a variety of promising applications. For example, graphene can be used as anode materials to improve the efficiency of batteries22–26 and as transparent conductive electrodes in solar cell.27–31 It could also be a potential candidate for integrated circuits-the smallest transistor (one atom thick, 10 atoms wide) known so far was made of graphene.32 Other applications of graphene and graphene-based materials such as field emitters,33–35 hydrogen storage,36–42 supercapacitors,43–48 gas sensors,49–58 and bio-sensors59–65 have also been reported.
A number of approaches have been employed to synthesize graphene, such as the micromechanical exfoliation of graphite,66 chemical vapor deposition (CVD),67–70 epitaxial growth,2,71,72 chemical intercalation,73 and the reduction of graphene oxide (GO).74–77 Among these methods, preparation of graphene from GO reduction stands out, since it is promising for the mass-production of graphene-based materials. On one hand, GO can be synthesized in large quantities by oxidizing inexpensive graphite powders using strong oxidants; on the other hand, GO can be reduced through different methods with tailored properties by controlling the reduction conditions. During the oxidation process, graphite powders are exfoliated and broken into layers with increased interlayer distance; the landscape of the sp2 carbon network is modified with oxygen-containing functional groups, defects, and holes. To date, the atomic structure of GO is still elusive due to its nonstoichiometry, and so far several models have been proposed to study the possible functional groups (epoxy, hydroxyl, carbonyl, and carboxyl) and their arrangements across the carbon plane. Hence, study on the GO atomic structure is crucial to understanding the basic physical properties of GO and RGO, and to improving properties of graphene-based nanomaterials. Meanwhile, to reduce GO, a number of chemical and thermal methods have been proposed and RGO could be readily obtained through these individual methods or their combinations. Although chemical and thermal reductions have their own limitations, the conductivity of RGO could reach as high as 5.7 × 104 S m−1,78 which is several orders of magnitude larger than that of GO (10−8–10−5 S m−1),79,80 on the same order of magnitude as that of graphite (8.4 × 104 S m−1),74 and about two orders of magnitude lower than that of graphene (5–6.4 × 106 S m−1).81
We note that many reviews have summarized the up-to-date achievements of graphene and graphene-based materials;6,7,19,82–91 however, no review has focused on the structural modeling of GO and its reduction. Here, in this review we offer insights into the structure of GO, its reduction dynamics, and outstanding properties due to specific reduction scheme. We first briefly introduce the GO synthesis methods, i.e., Hummer and Offeman, Staudenmaier, and Brodie methods, then review recent studies on the GO structure modeling, and finally end the review with discussion and summary of the recent progress on GO reduction through chemical and thermal methods.
2. Graphene oxide synthesis
GO can be synthesized in large quantities by oxidizing inexpensive graphite powders using strong oxidants (e.g., H2SO4, HNO3, KMnO4, KClO3, NaClO2) with individual sheets obtained by subsequent gentle exfoliation. Currently, there are three major methods used to synthesize GO from graphite: Hummer and Offeman,92 Staudenmaier,93 and Brodie.94Fig. 1 illustrates a schematic diagram of GO synthesis: graphite powders (black blocks) are first oxidized into graphite oxide (lighter coloured blocks) with functional groups spreading across the carbon skeleton, which increase the interlayer spacings and weaken the van der Waals force between adjacent layers; the as-prepared graphite oxide can then be further sonicated in water to obtain suspensions in which individual GO flakes are stabilized by mutual electrostatic attraction and repulsion.95![Scheme showing a chemical route to the synthesis of aqueous GO suspension: (1) oxidation of graphite (black blocks) to graphite oxide (lighter coloured blocks) with a greater interlayer distance; (2) exfoliation of graphite oxide in water by sonication to obtain GO suspensions that are stabilized by electrostatic repulsion. Reprinted with permission from Macmillan Publishers Ltd: [Nature Nanotechnology] (ref. 95), copyright (2008).](/image/article/2012/RA/c2ra00663d/c2ra00663d-f1.gif) | ||
| Fig. 1 Scheme showing a chemical route to the synthesis of aqueous GO suspension: (1) oxidation of graphite (black blocks) to graphite oxide (lighter coloured blocks) with a greater interlayer distance; (2) exfoliation of graphite oxide in water by sonication to obtain GO suspensions that are stabilized by electrostatic repulsion. Reprinted with permission from Macmillan Publishers Ltd: [Nature Nanotechnology] (ref. 95), copyright (2008). | ||
The Hummer and Offeman method is most commonly used to prepare GO. Typically, graphite oxide was prepared using ultra-pure graphite powder and sodium nitrate. The ingredients were mixed in sulfuric acid by stirring and cooled to 0 °C in an ice bath while maintaining vigorous agitation, then potassium permanganate was added. As the reaction progressed, the mixture gradually thickened and finally condensed into a brownish grey gel. Subsequently, excess water was slowly dripped, causing violent effervescence, and the gel was diluted into a brown suspension. This suspension was then further diluted with water and treated with hydrogen peroxide to reduce the residual permanganate and manganese dioxide to colourless soluble manganese sulfate. Upon treatment with the peroxide, the suspension turned bright yellow. To reduce the manganese sulfate and other residues, the suspension was then filtered and washed several times to obtain a yellow-brown graphite oxide residue, which was dispersed in water to prepare the GO suspension with the aid of sonication. GO powder could be obtained through the filtration and drying of the GO suspension. Alternatively, in the Staudenmaier method, graphite was mixed and reacted with concentrated sulfuric acid, fuming nitric acid, and potassium chlorate; in the Brodie method, graphite was mixed and reacted with fuming nitric acid and sodium chloride oxide. In all three methods, despite that the layers in graphite are extensively oxidized by oxidative treatment and the procedures to prepare GO suspension and GO powder are similar, the as-prepared GO structure and the residue contamination are distinct as a result of the different oxidants used. The Staudenmaier and the Hummers and Offeman methods are known to produce unstable GO with a high degree of contamination and degradation, while GO synthesized using the Brodie's method is very stable with a low degree of contamination.94
3. Structural modeling of graphene oxide
3.1 Experimental characterization
Despite its first experimental synthesis more than one and half centuries ago, the structure of GO is still elusive today due to its nonstoichiometry. So far, several models have been proposed to elucidate its structure, as shown in Fig. 2. Here, the overall features of these models are described and detailed characterization on these models can be found in another review.84 Hofmann84 first presented the atomic configuration of GO with only epoxies randomly distributed across the carbon plane. Ruess,84 however, proposed that hydroxyls are also common in GO, and moreover, the species of epoxies are not only limited to the ones sitting on the bridge sites of carbon atoms (1,2-ether); epoxies with O atoms connecting the 1,3 site C atoms (1,3-ether) also exist, and the skeleton of C is strongly distorted into three dimensions by hydroxyls and these 1,3-ethers. Scholz and Boehm84 removed the epoxies, and in their suggested structure, ribbons of conjugated carbon backbone and regular quinoidal species coexist. Another picture of structure proposed by Nakajima–Matso96 differed from the above ones in which the oxygen atoms in epoxies link adjacent layers. Szabo and Dekeny's model97 incorporated the features of both Sholz–Boehm and Ruess' models, which is shown in Fig. 2. In Lerf's model,98 which is now widely accepted by most researchers, epoxy (1,2-ether) and hydroxyl are the major functional groups that randomly distribute across the carbon layer, and at the edge are mainly carboxyls, lactones, and carbonyls.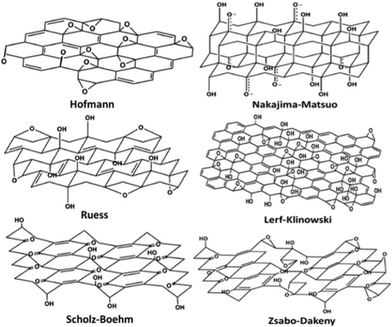 | ||
| Fig. 2 Proposed configurations for GO.84 Reproduced with permission of the Royal Society of Chemistry. | ||
The oxygen-containing groups and their arrangements across the carbon network are critical to obtaining a thorough view of GO structure. Microscopic techniques can provide deep insights into the types of oxygenated functional groups in GO and their distributions. By using the 13C and 1H nuclear magnetic resonance (NMR) spectra, Lerf et al.98 revisited the structure of GO in 1998 and critically assigned the chemical shift line around 60 ppm to 1,2-ethers instead of 1,3-ethers; they also confirmed the assignment of the line around 70 ppm to hydroxyls. Now the three major chemical shift peaks around 60, 70, and 130 ppm have been commonly accepted and assigned to epoxy (unless otherwise stated, epoxy refers to 1,2-ether hereafter), hydroxyl and sp2 carbon, respectively.77,97–104 Until then, epoxy and hydroxyl were determined to be two major functional groups across the basal plane in GO. In 2009, Gao and Ajayan et al.105 further assigned the peak around 101 ppm to five- or six-membered-ring lactol decorated on the edge of holes in GO flakes. To gain information about the distribution of major functional groups, two- and multi-dimensional NMR spectra conducted by Ruoff's group revealed that epoxy and hydroxyl were close to each other, with some tiny islands of pure epoxies or hydroxyls.100,101 The major peaks in the NMR mentioned above were related to the carbon atoms single-bonded to oxygen atoms. X-Ray photoelectron spectroscopy (XPS), however, showed evidence that sp2 carbon atoms are accompanied by three other carbons connected with epoxy or hydroxyl (single-bonded), carbonyl (double-bonded), and carboxyl (triple-bonded) groups, respectively.77,94,97,106,107
Although the consensus is that GO is decorated by epoxies and hydroxyls, which are randomly distributed across the carbon backbone, and carbonyls and carboxyls are mainly attached on the edge, some uncertainty still revolves around the morphology of GO, e.g., whether it has tiny islands of structural ordering. Near-edge X-ray absorption fine structure (NEXAFS) for the O–K edge suggested that the carbonyls in GO are arranged on the carbon layer on average, and there is locally ordered structure from some oxygenated functional groups.108 Again, this ordered structure is elusive until a scanning tunnelling microscope (STM) study on oxidized exfoliated graphene sheets showed an ordered structure exists from pure epoxies arranged in a rectangular lattice on both sides of the carbon layer,109 as shown in the inset of Fig. 3a. However, once hydroxyls are present, the oxidized regions in GO become amorphous (Fig. 3a) and the ordered structure can only be developed under specific conditions, which is discussed in the following text.
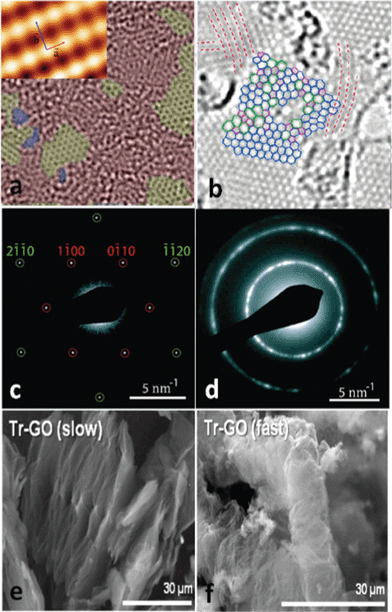 | ||
| Fig. 3 Morphology characterization of GO and reduced GO. (a) Aberration corrected TEM image of a single suspended sheet of GO, red area represents the oxygen functionalities, graphite region and holes are indicated in yellow and blue, respectively. Reprinted with permission from ref. 110. Copyright (2010) John Wiley and Sons. Inset is the high-resolution UHV STM image of exfoliated oxidized graphene revealing a rectangular lattice with a = (0.273 ± 0.008) nm and b = (0.406 ± 0.013) nm. Reprinted from ref. 109, copyright (2008), with permission from Elsevier. (b) Atomic resolution image of reduced GO. Carbon pentagons, hexagons, and heptagons are indicated by magenta, blue, and green, respectively. Red dashed lines represent the directions with strong lattice deformations. Reprinted with permission from ref. 111. Copyright (2010) American Chemical Society. (c) and (d) are selected area electron diffraction (SAD) patterns for single-layer and multi-layer of GO with the spots labeled with Miller–Bravais indices. Reprinted with permission from ref. 112. Copyright (2009) American Chemical Society. (e) and (f) are the representative SEM micrographs of GO thermally reduced at slow and fast rates. Reprinted with permission from ref. 113. Copyright (2010) American Chemical Society. | ||
3.2 Theoretical characterization
Apart from microscopic techniques, theoretical studies render vital perspectives into the structural characterization of GO. Since the major functional groups in GO are epoxies and hydroxyls, the existing density functional theory (DFT) for modeling GO focused mainly on the decoration of these groups on the basal plane of graphene. To identify the 1,3-ether in GO, Lahaye et al.114 showed that even if the 1,3-ether could exist with the hydroxyl pulling the carbon out of the plane, the system is still much less stable than the structure of 1,2-ether stabilized by hydroxyls (Fig. 4a and b). The authors also compared the system energy of a 2 × 2 cell deposited with one epoxy and one hydroxyl, and results showed that only the structure with epoxy and hydroxyl in close proximity (Fig. 4a) is stable, which is consistent with the NMR and other theoretical results.77,97–104 Moreover, the joint binding energy for epoxy and hydroxyl in such a structure is 0.8 eV (in absolute value) higher than the total binding energy of isolated epoxy and hydroxyl. Therefore, it can be concluded that 1,3-ether does not exist in GO and that epoxy and hydroxyl are mutually stabilized with each other nearby.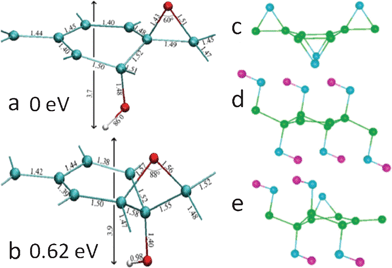 | ||
| Fig. 4 (a) and (b): detailed geometric configurations of GO with one ether oxygen and one hydroxyl in a unit cell with eight carbon atoms. The interatomic distances are in angstrom. Carbon, oxygen, and hydrogen atoms are shown in green, red, and white, respectively. (a) and (b) have 1,2-ether and 1,3-ether oxygens, respectively. Reprinted with permission from ref. 114. Copyright (2009) by the American Physical Society. (c)–(e): the most stable structures of GO with only epoxies (c), hydroxyls (d), and mixed coverage (e). Carbon, oxygen, and hydrogen atoms are shown in green, blue, and violet, respectively. Reprinted with permission from ref. 99. Copyright (2008) American Chemical Society. | ||
Besides its amorphous nature, GO is also nonstoichiometric and its chemical composition is dependent on its specific synthesis conditions. To look for possible building blocks of GO with different ratios of epoxy and hydroxyl representing nonstoichiometry, which are expected to exhibit a rough landscape of GO, the most commonly used method in the DFT calculation is to fix the number of epoxies and hydroxyls in a certain cell size and search for the most energetically possible configurations. Boukhvalov and Katsnelson99 carried out such calculations via variations of GO chemical composition in a 2 × 2 cell with 8 carbon atoms. The percentage of oxygen atoms to carbon atoms (oxygen coverage) was increased gradually from 12.5% to 100%, from which GO becomes nonconductive at an oxygen coverage greater than 25%. For GO with pure epoxies, the binding energy increases as the coverage goes up to the highest value of 50% (Fig. 4c); while with pure hydroxyls forming a chainlike structure, the highest coverage is 75% (Fig. 4d). In the mixed structure (oxygen coverage of 62.5%), the combination with one epoxy and four hydroxyls (Fig. 4e) is the most stable configuration. The structure in Fig. 4c shows the ordering the oxygen atoms in epoxies and is the candidate for the experimentally observed structural ordering in NEXAFS99 and STM.109 In addition, it could be used to explain the tiny islands of pure hydroxyls inferred from the multi-dimensional NMR.100,101 In Boukhvalov and Katsnelson's model, the two proposed chemical compositions of C(OH)0.25 and CO0.125(OH)0.25 could successfully explain the experimentally determined GO formula;97,115 however, since the interactions between the functional groups in nearby cells are not negligible; many other possible configurations of the groups are still excluded in their model due to the limited cell size.
Yan and Chou103 studied the structural characterization of GO in a 5 × 5 super cell with 50 carbon atoms and the number of oxygen atoms varied within 2 and 4 through the arrangements from pure epoxies (2O) or hydroxyls (2OH) to the mixed placements of 2O + 2OH. For sole epoxies and hydroxyls, the functional groups tend to locate near each other rather than forming isolated structures to reduce the tension generated in the basal plane of carbon atoms, and the most stable structure for epoxies at a coverage of 50% is dual with two epoxies either on the same carbon ring or on two nearby rings with oxygen atoms pointing up and down (not shown here). However, as epoxy and hydroxyl prefer to sit nearby, if one more hydroxyl (OH) is introduced into such a structure (O + OH), it turns out that the structure (O + 2OH) with epoxy close to both hydroxyls (Fig. 5a) is less energetically favourable than that with hydroxyls close to each other (Fig. 5b). Since local density approximation (LDA) was employed to treat the electron–electron interactions that usually overestimate the binding energies, Wang et al.42 found that the structure shown in Fig. 5a is comparable energetically with that in Fig. 5b. If one hydroxyl is introduced into the 2O structure, it is expected that epoxy and hydroxyl are in proximity, and the most stable structure (2O + OH) calculated is shown in Fig. 5c. Among the models discussed with two epoxies and two hydroxyls, the convincing one is that epoxies and hydroxyls form two mutually stabilized epoxy–hydroxyl complexes (2O + 2OH), as shown in Fig. 5d. Clearly, various combinations of these basic building blocks can be used to represent the morphology of GO to some degree; however, subjective arrangements of these blocks are unable to offer promising directions for future reduction or atomic engineering. The main difficulties in finding larger reliable blocks lie in both the nonstoichiometry of GO and the great computational effort taken by DFT calculations to search for such blocks. Other methods, such as Monte Carlo,116–119 molecular dynamics120–125 and generic algorithm,126 can be employed in combination with DFT to present more convincing results in developing GO structural blocks via adjusting the numbers of epoxy and hydroxyl in a fixed cell size.
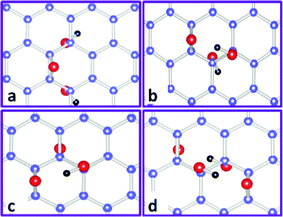 | ||
| Fig. 5 Some representative basic building blocks of GO with both epoxy and hydroxyl functional groups. Carbon, oxygen, and hydrogen atoms are indicated by blue, red, and black balls, respectively. (a) and (b): O + 2OH. (c): 2O + OH. (d): 2O + 2OH. Reprinted with permission from ref. 103 as follows: J. A. Yan et al., Physical Review B, 2010, 82, 125403. Copyright (2010) by the American Physical Society. | ||
As discussed previously, epoxy and hydroxyl are randomly deposited across the carbon skeleton; carbonyl and carboxyl are mainly on the edge sites. Both infrared (IR)127,128 and XPS97,127 have shown that the amount of carbonyls and carboxyls are much smaller than that of epoxies and hydroxyls. Jeong et al.94 assigned the peak at 289.0 eV in the XPS spectrum to carboxyl, and the integrated area of the peak is much larger than that of epoxy or hydroxyl (Fig. 6a). One possible reason is that it comes from the contributions of species of unknown oxygenated groups. Since 1,3-ethers are excluded from the energetic point of view and only the epoxy sits above the bridge site (1,2-ether) in the current models, these new functional groups could be related to the regular ones. Computational spectroscopy results extended this regular epoxy to the new one with the carbon bond underneath broken, since the discrepancy of the simulated XPS peak values for these two kinds of epoxies are within 0.8 eV104 and the binding energy for the new epoxy is greater than the regular one.129,130 This new epoxy exists only when regular epoxies line up under certain conditions on the same side of the carbon plane by getting over the energy barrier heights,130,131 and the amount of the new epoxy is small in GO. Fig. 6b shows the calculated binding energies of different labeled functional groups from the experimentally observed curve in Fig. 6a. Obviously, the results suggest that the binding energies of carboxyl, epoxy pair (Fig. 6c), epoxy, and hydroxyl complex (Fig. 6d) overlap. Note that the diffraction pattern of Jeong's samples94 shows graphitic stacking order, and this phenomenon disappears in XPS after further oxidization. This spectroscopic observation might occur during the transient oxidation stage when the precursor graphite powders are cut into smaller sizes and likely before they are peeled off into mono- to few-layer GO.
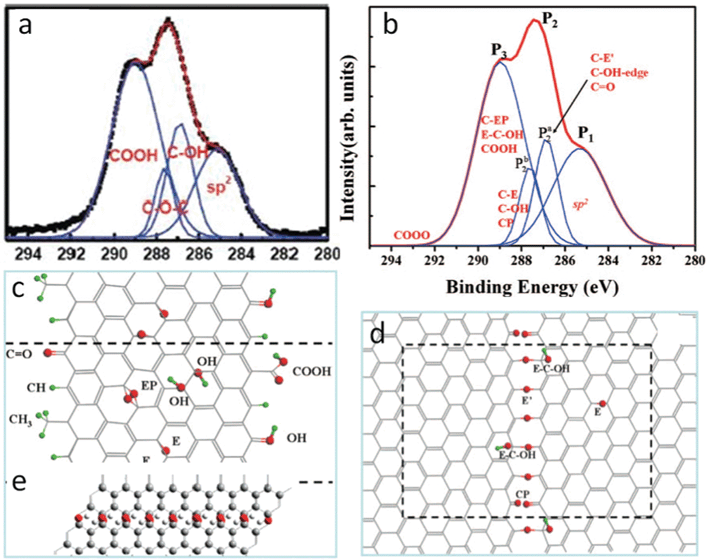 | ||
| Fig. 6 (a) XPS of GO oxidized for 24 h. Reprinted with permission from ref. 94. Copyright (2008) American Chemical Society. (b) Simulated experimental curve in (a) with assignments from DFT calculations; (c),(d) GO structure with functional groups responsible for C1s peaks in (b). Reprinted with permission from ref. 104. Copyright 2009, American Institute of Physics. (e) Nanoribbons of hydroxyl chain and graphene-like regions proposed for the observed blue shift of G band in the Raman spectrum. Reprinted with permission from ref. 132. Copyright (2008) American Chemical Society. | ||
Generally, the chemical composition of GO varies from C1O0.17H0.08 to C1O0.49H0.2 depending on the synthesis method and oxidation time,97,115 and hydroxyls are dominant among the functional groups. Considering that one epoxy is connected closely to two hydroxyls and epoxies usually are randomly spread across GO, the distributions of hydroxyls could develop into certain patterns. Theoretically, it is also found that hydroxyls forming two types of chainlike structures along the zigzag and armchair edges are energetically feasible.99,103 Nevertheless, a first-principle study of the NMR signatures of GO does not support this structural motif. The averaged chemical shift for carbon atoms connected to these hydroxyls in the zigzag (armchair) chainlike structure is about 6.8 (8.5) ppm greater than the general ones,102 but experimental NMR spectra does not show a broadened hydroxyl peak.100,105 Hence, this kind of chainlike structure is not present in a great amount, at least in GO.
Actually, Kudin and Car et al. proposed another pattern of chainlike structure (Fig. 6e) that could explain the G band blue shift132 in the Raman spectrum of GO compared with that of graphite.132–136 With regard to all potential causes for this frequency shift,136–139 the presence of isolated conjugate bonds leads to the observed phenomenon. The authors finally came up with the structure in the alternating regions of single and double carbon bond nanoribbons. In the regions of single bond, hydroxyls line up and sit on the bridge sites pointing up and down, respectively. Epoxies decorate sparsely in the graphene like region without destroying its entire conjugate carbon bond network. Indeed, for the partially reduced GO, the evolution of Raman modes undergoes the opposite effect; hence, the greatest finding is the alternating patterns of single-double bonds developed from the Scholz–Boehm model.97 Within the scope of our literature survey, despite the few studies available to gain more information about this pattern, it at least can provide us with some insights into the evolution of functional groups in GO during the oxidation process.
3.3 Morphology characterization during oxidation
As graphite is chemically exfoliated into mono- and few-layer graphene oxide, the calculated exfoliation energy showed that at least two layers of graphene should be oxidized to peel off a monolayer of GO.140 Although graphite is composed of layers of graphene piled in an AB stacking order and the electron diffraction pattern of monolayer GO (Fig. 3c) is similar to that of pure graphene, the diffracted rings of multilayered GO demonstrate the turbostratic stacking (Fig. 3d).112,141 An X-ray diffraction (XRD) spectrum indicated that interlayer spacing increases from 3.4 Å for graphite to as high as 10 Å for GO77,97,142 due to the intercalated water molecules; and the content of intercalated water plays a crucial role in determining the stacking order of GO. A DFT study suggested that the anhydrous GO retains a graphitic AB stacking order with an interlayer distance of 5.1 Å and 5.8 Å for low and high oxidation level, respectively. The spacing between layers increases up to 7.3 Å with one layer of water molecules, and for the case of high oxidation level, the stacking order is broken.143 Jeong et al.94 observed that the AB stacking order is preserved even when the graphite is oxidized for 48 h, as the layer spacing increases from 5.62 Å (1 h) to 7.37 Å (24 h) and stays as a plateau up to 48 h. Elemental analysis (EA) showed that the ratio of water molecules to carbon atoms up to 1:10 could be achieved at an oxidation level of 30%, from which the entropic disorder due to water molecules intercalated in highly oxidized GO would destroy the AB stacking order.143
Besides the broken stacking order, the morphologies of GO also display defects and holes (Fig. 3a).110,112,141 Moreover, the size of GO flake is much smaller than that of the graphite seed,142 suggesting that the graphite precursor is cut into smaller pieces associated with certain functional groups during the oxidation process. One explanation originates from Car's work, in which epoxy is demonstrated to migrate with an energy barrier height about 0.83 eV and get aligned.129 Then carbon bonds under the epoxy chain are unzipped (Fig. 7a) through getting over the potential barrier about 0.58 eV dependent on the distance between two nearest chains.130 This type of epoxies can further transform into epoxy pairs with the broken carbon bonds (Fig. 7b) and finally evolve into carbonyl pairs (Fig. 7c), when the energy difference between the transition state and the epoxy pair state decreases from 0.76 eV for the first epoxy pair to 0.26 eV for the second pair;131 in this case, GO is split from the edge to the center. This epoxy pair-induced unzipping mechanism could explain the smaller size of GO flakes and the large holes introduced in GO.
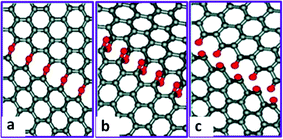 | ||
| Fig. 7 Mechanism for the epoxy chain-induced unzipping of GO during the oxidation process. (a) Epoxy chain formed when the epoxies lined up with the broken carbon bonds underneath. (b) Epoxy pair chain. (c) Carbonyl chain formed when the epoxies got over the potential energy barrier. Reprinted with permission from ref. 131. Copyright (2009) American Chemical Society. | ||
In summary, major functional groups in GO are epoxy, hydroxyl, and carbonyl, which locate on the basal plane of the carbon skeleton, and carboxyl, which sits on the edge. Epoxies are energetically favourable to sit above the bridge site and are randomly distributed in close proximity with hydroxyl to stabilize the structure, while carbonyls lie in the plane on average. Tiny islands of pure epoxies and hydroxyls also exist in the carbon network. In addition, graphene-like regions and holes are also common in GO. The AB stacking order of graphite can be retained in a low oxidation level with a small amount of intercalated water molecules between adjacent layers. Small GO flakes and holes formed during the oxidation process are due to the epoxy chain-induced unzipping of carbon rings. Despite its nonstoichiometry, the basic building blocks of GO can be obtained by DFT modeling; however, the existing modeling of the atomic structure of GO cannot be used to predict the formation of gas molecules during its thermal reduction process. On one hand, the building blocks are too small and expanded periodically in space to represent the configuration of GO; on the other hand, the blocks are selected by manually arranging the functional groups with specific numbers in a fixed cell size. To better understand the GO structure and its evolution during the thermal reduction process, larger building blocks, which can cast insight into the development of gas molecules, are highly desirable.
4. Graphene oxide reduction
To prepare RGO, many chemical and thermal reduction approaches have been proposed. At present, the chemical reduction of GO is fulfilled by using a wide range of reducing agents, such as hydrazine,33,75,76,95,107,144,145 alcohol,146,147 sodium borohydride,80,105,148,149 hydriodic acid with acetic acid,74,150 sodium/potassium hydroxide,151,152 iron/aluminium powder,153,154 ammonia,155,156 hexylamine,157 sulfur-containing compounds (NaHSO3, Na2SO3, Na2S2O4, Na2S2O3, Na2S·9H2O, SOCl2, and SO2),158,159 hydroxylamine hydrochloride,160 urea,161 lysozyme,162 vitamin C,163N-methyl-2-pyrrolidinone (NMP),78 poly(norepinephrine),164 BSA,165 TiO2 nanoparticles,166,167 manganese oxide,168 and bacteria respiration.169 On the other hand, the thermal reduction methods generally rely on heating the GO in various atmospheres (ultra-high vacuum, Ar, H2, NH3),77,79,106,170–173 or with different heating sources like microwave,174,175 flash light,176 laser,177 plasma,178 electric current,179 or heated tip atomic force microscopy.180 In general, chemical and thermal reduction approaches have their own advantages. With chemical methods, GO can be reduced in an aqueous phase and the resulting RGO could bear various organic functional groups or nanostructures, which may be used to tune RGO properties. For thermal reduction, the degree of the RGO reduction can be controlled by heating temperature, duration, and gaseous environment. Foreign atoms, e.g., N, also could be injected into the sp2 carbon network for tuning the electronic properties of RGO. In the following sections, based on the different reduction approaches, we generally summarize the current status of graphene oxide reduction into the two categories: chemical reduction of GO and thermal reduction of GO.4.1 Chemical reduction of graphene oxide
Among various chemical reducing agents, the first and most commonly used was hydrazine (N2H4), which produced highly reduced graphene oxide under low temperature. In 2007, Ruoff's group107 reported the first reduction of a colloidal suspension of exfoliated GO sheets in water with hydrazine hydrate, resulting in the aggregation and subsequent formation of a high-surface-area carbon material that consists of thin graphene-based sheets. In their method, the reduction of graphene oxide was achieved by adding hydrazine hydrate into the GO suspension and heating in an oil bath at 100 °C under a water-cooled condenser for 24 h. During the hydrazine reduction of GO suspension, the brown-coloured suspension turned black and the reduced sheets aggregated and eventually precipitated. The precipitation of the reduced sheets occurred, presumably due to their becoming less hydrophilic as a result of oxygen removal, and thus increased incompatibility with the aqueous medium. To investigate the structure change and prove that the oxygen groups were removed after reduction, the prepared RGO was characterized and the results from both elemental analysis and XPS clearly showed that reduction of the exfoliated GO resulted in considerable removal of oxygen. And the MAS NMR data additionally suggested that the reduction/de-oxygenation of GO also resulted in significant restoration of the sp2 carbon sites. The restoration of the sp2 carbon was also proved through the electrical conductivity measurements since the observed increase in conductivity upon reduction of GO required that conductive pathways of conjugated carbon atoms be re-established.
After the very first study on the hydrazine reduction of GO, many groups reported the GO reduction with hydrazine both experimentally and theoretically (Fig. 8).181 In general, GO sheets could be well dispersed in water but not in solvents with high polarity indices, e.g., N,N-dimethylformamide (DMF); however, RGO powders could be easily dispersed in high polarity solvents. Based on Ruoff's research, additional study was carried out to prepare RGO suspensions in different solvents. In Park's work,75 the chemical reduction of the suspension of graphene oxide sheets was conducted with hydrazine monohydrate and colloidal suspensions of RGO in various organic solvents were prepared. The solubility study indicated that the DMF/H2O seemed to be the best system for producing RGO suspension. Experimentally, highly reduced GO could be prepared with hydrazine; however, theoretically, the possible mechanisms for the chemical reduction of GO by hydrazine were still elusive, which is discussed in section 4.1.2.
![Chemically converted RGO suspensions. Photographs of (a) 15 mg of GO paper in a glass vial and (b) the resultant hydrazinium RGO dispersion after addition of hydrazine. Below each vial is a three-dimensional computer-generated molecular model of GO (carbon in grey, oxygen in red and hydrogen in white) and chemically converted RGO, respectively, suggesting that removal of –OH and –COOH functionalities upon reduction restores a planar structure. Reprinted with permission from Macmillan Publishers Ltd: [Nature Nanotechnology] (ref. 76), copyright (2009).](/image/article/2012/RA/c2ra00663d/c2ra00663d-f8.gif) | ||
| Fig. 8 Chemically converted RGO suspensions. Photographs of (a) 15 mg of GO paper in a glass vial and (b) the resultant hydrazinium RGO dispersion after addition of hydrazine. Below each vial is a three-dimensional computer-generated molecular model of GO (carbon in grey, oxygen in red and hydrogen in white) and chemically converted RGO, respectively, suggesting that removal of –OH and –COOH functionalities upon reduction restores a planar structure. Reprinted with permission from Macmillan Publishers Ltd: [Nature Nanotechnology] (ref. 76), copyright (2009). | ||
RGO could be successfully prepared with hydrazine; however, the main drawback of this method is that hydrazine is toxic, which prevents its use in mass production of RGO. To address this challenge, many other "non-toxic" reducing agents were used to reduce GO, e.g., alcohol, sodium/potassium hydroxide, sodium borohydride, hydriodic acid, and acetic acid. For the alcohol (C2H6O) reduction, Su et al.147 showed that RGO with highly graphitic structures and excellent electrical conductivity could be prepared by high-temperature alcohol vapor reduction. In this report, GO was heated in 20% H2/Ar with alcohol under high temperatures (600–1000 °C). The sheet resistance of transparent RGO films was as low as 15 kΩ/□ (>96% transparency at 550 nm). Based on the field-effect transistor (FET) measurement, the RGO sheets exhibited high field-effect hole mobility up to 210 cm2 V−1 s−1. In addition, Raman spectroscopic studies revealed that the conductivity enhancement in the low mobility regime was attributed to the removal of chemical functional groups and the formation of six-fold rings; while in the high mobility regime, the growth of the graphitic domain size became dominant for enhancing its electrical conductivity.
GO could also be reduced with sodium/potassium hydroxide (NaOH/KOH). Fan et al.151 showed that RGO suspension could be quickly prepared by simply heating a GO suspension under strongly alkaline conditions at moderate temperatures (50–90 °C). Careful experiments revealed that exfoliated GO can undergo fast deoxygenation in strongly alkaline solutions, resulting in stable aqueous graphene suspensions. The 13C NMR spectrum showed that after the reaction there was a significant reduction in the amount of epoxide and hydroxyl groups present in RGO. In addition, plentiful sp2 carbon atoms were introduced, suggesting the formation of graphene-based materials. The results were confirmed by XPS analysis, which showed that the O/C ratio in the exfoliated GO decreased remarkably after the reaction, and that most of the epoxide and hydroxyl functional groups were successfully removed. From Park's work,152 GO was reduced sequentially with potassium hydroxide and hydrazine. Adding KOH to the GO suspension produced a slightly darker suspension (“KMG”), which was caused by the reaction between KOH and oxygen functional groups in the graphene oxide sheets, such as carboxylic acid, hydroxyl, and epoxy groups, resulting in extensive decoration of the sheets with negative charges and with K+ ions. A black and homogeneous suspension of KMG sheets was then obtained by the addition of hydrazine monohydrate and reacting at 35 °C for 6 h. The aqueous “hydrazine-reduced KMG suspension (hKMG)” could be prepared with concentrations as high as 7 mg ml−1 and was stable for more than 4 months.
Similar to sodium/potassium hydroxide, sodium borohydride (NaBH4) was also used in the reduction of GO. Ajayan's group105 has developed a two-step reduction process-deoxygenation with NaBH4, followed by dehydration with concentrated sulfuric acid. Fig. 9 shows the schematic representation of the GO reduction procedure and characterization of the obtained products. Electrical conductivity of the RGO is an important criterion to evaluate how the sp2 carbon network has been restored in this structure. Original GO is an insulator, with a conductivity of around ∼10−8–10−5 S m−1. After reduction with NaBH4, concentrated sulfuric acid and thermal annealing, the conductivity of the prepared RGO increased to 20200 S m−1, an increase of more than nine orders of magnitude, clearly indicating an efficient restoration of the sp2 carbon network in RGO, induced by the deoxygenation and dehydration.
![Schematic representation of the GO reduction procedure and characterization of the products obtained. (a) The two-step reduction process, followed by the annealing treatment, is an effective method to convert GO sheets into graphene-based materials (where CCG stands for chemically converted graphene). (b) and (c) XPS and TEM characterization indicate good restoration of the graphene structure, as well as little sulfur remaining in the final product. (b) Left: XPS analysis of C1s in different samples. Right: XPS signal of S2p from 156 to 176 eV. (c) Left: TEM image of CCG2. Right: corresponding selected area electron diffraction (SAD) pattern taken at the relatively flat edge of the RGO sheet. (d) Solubility tests. From left to right: GO in deionized water; GO in DMF; CCG3 in deionized water; CCG3 in DMF after 50 min water bath sonication. A red laser beam was directed through the dispersions to show the Tyndall effect of these colloidal solutions. Although GO is more soluble in water than in DMF, after reduction the product CCG3 is more soluble in DMF than in water. Reprinted with permission from Macmillan Publishers Ltd: [Nature Chemistry] (ref. 105), copyright (2009).](/image/article/2012/RA/c2ra00663d/c2ra00663d-f9.gif) | ||
| Fig. 9 Schematic representation of the GO reduction procedure and characterization of the products obtained. (a) The two-step reduction process, followed by the annealing treatment, is an effective method to convert GO sheets into graphene-based materials (where CCG stands for chemically converted graphene). (b) and (c) XPS and TEM characterization indicate good restoration of the graphene structure, as well as little sulfur remaining in the final product. (b) Left: XPS analysis of C1s in different samples. Right: XPS signal of S2p from 156 to 176 eV. (c) Left: TEM image of CCG2. Right: corresponding selected area electron diffraction (SAD) pattern taken at the relatively flat edge of the RGO sheet. (d) Solubility tests. From left to right: GO in deionized water; GO in DMF; CCG3 in deionized water; CCG3 in DMF after 50 min water bath sonication. A red laser beam was directed through the dispersions to show the Tyndall effect of these colloidal solutions. Although GO is more soluble in water than in DMF, after reduction the product CCG3 is more soluble in DMF than in water. Reprinted with permission from Macmillan Publishers Ltd: [Nature Chemistry] (ref. 105), copyright (2009). | ||
Most of the chemical reduction methods are used to reduce GO sheets in suspensions; however, some methods could reduce GO paper or film in a reducing agent vapor. In 2010, Ruoff and Lee's group74 reported a novel reducing agent system (hydriodic acid with acetic acid (HI–AcOH)) that allows for an efficient, one-pot reduction of a solution-phased RGO powder and vapor-phased RGO paper and thin film. To prepare an RGO paper, the GO paper was placed inside a jar containing HI and acetic acid. The cover of the jar was sealed with vacuum grease and placed over an oil bath at 40 °C for 24 h. The color of the RGOHI–AcOH papers changed from brown to metallic grey after treatment with HI vapor, indicating the reduction of the materials. Fig. 10a and b show the digital image of flexible RGOHI–AcOH papers and films with GO reduced in the gas phase. The conductivity of the air-dried RGOHI–AcOH powder pellets was 30400 S m−1, which is the highest literature RGO value (air-dried), and on the same order of magnitude as that of graphite (84500 S m−1). The one-pot chemical reduction method of GO using HI–AcOH through gas phase demonstrates the possibility of producing RGO paper and film with high conductivity that is on the same order of magnitude of as that of graphite. As opposed to the reduction of GO to the extreme extent, partially reduced GO is also appealing with its own advantages. Li et al. reported that CCG by reduction of GO in water could form stable aqueous colloids without any surfactant.95 The CCG sheets were not flat but corrugated due to the sp3 carbon atoms connected to the unreduced functional groups. Through controlled reduction, the as-produced CCG sheets hold great potential for applications such as nanofiltration,182 in supercapacitors,183 and in conductive hydrogel films.184
![Flexible RGOHI–AcOH paper and thin film with GO reduced in the gas phase. (a) Flexible RGOHI–AcOH paper. (b) Flexible GO (left) and RGOHI–AcOH thin films on a PET substrate. (c) XPS analysis of the C1s region in RGOHI–AcOH and RGONH2–NH2 papers. A large loss of oxygen-functional groups after exposure to gas-phase reductants is evident. (d) SEM image of a cross-section and Raman spectra (inset) of RGOHI–AcOH paper. (e) Electrical properties of RGOHI–AcOH (blue) and RGONH2–NH2 (red) papers. RGOHI–AcOH paper shows significantly higher electrical conductivity compared with the RGONH2–NH2 paper. (f) Water contact angles: (left) GO (62.8°), (middle) RGOHI–AcOH (78.5°) and (right) RGONH2–NH2 (69.0°) papers. Reprinted by permission from Macmillan Publishers Ltd: [Nature Communications] (ref. 74), copyright (2010).](/image/article/2012/RA/c2ra00663d/c2ra00663d-f10.gif) | ||
| Fig. 10 Flexible RGOHI–AcOH paper and thin film with GO reduced in the gas phase. (a) Flexible RGOHI–AcOH paper. (b) Flexible GO (left) and RGOHI–AcOH thin films on a PET substrate. (c) XPS analysis of the C1s region in RGOHI–AcOH and RGONH2–NH2 papers. A large loss of oxygen-functional groups after exposure to gas-phase reductants is evident. (d) SEM image of a cross-section and Raman spectra (inset) of RGOHI–AcOH paper. (e) Electrical properties of RGOHI–AcOH (blue) and RGONH2–NH2 (red) papers. RGOHI–AcOH paper shows significantly higher electrical conductivity compared with the RGONH2–NH2 paper. (f) Water contact angles: (left) GO (62.8°), (middle) RGOHI–AcOH (78.5°) and (right) RGONH2–NH2 (69.0°) papers. Reprinted by permission from Macmillan Publishers Ltd: [Nature Communications] (ref. 74), copyright (2010). | ||
The chemical reducing agents are mostly based on inorganic chemical compounds; however, methods that rely on the metal, e.g., iron and aluminum, have also been developed for the reduction of GO. In Fan's work,153 synthesis of RGO sheets was conducted based on iron reduction of exfoliated graphite oxide. In a typical experiment, Fe powder and HCl were directly added into a GO suspension at ambient temperature. The mixture was stirred for 30 min and then maintained for a period of time (Fig. 11a). After reduction, more HCl was added into the above solution in order to fully remove excess Fe powder and the resulting RGO was collected with filtration and wash. The conductivity of the produced RGO was measured as 2300 S m−1. The reduction mechanism was proposed in which, after the introduction of H+, Fe powders reacted with H+ to produce Fe2+, which adsorbed on the surface of Fe particles so that GO sheets with negative charges were absorbed onto the surface of the positive charged Fe particles to form spherical structures (Fig. 11d and e). As a result, the GO sheets closely covering the surface of Fe particles facilitate the reduction of GO due to the fast electron transport from Fe/Fe2+ to GO sheets, in which the reduction process can be expressed as follows:
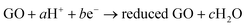 | (1) |
 | ||
| Fig. 11 (a) Photographs of aqueous dispersions (0.5 mg ml−1) of GO before and after being reduced via Fe for different reduction times. (b) AFM image of GO dispersed on mica and (c) corresponding line profile. (d) Photograph and (e) SEM image of RGO for 30 min without acid treatment. (f) SEM and (g, h) TEM images of RGO for 360 min at different magnifications. The inset of panel (f) shows the pressed RGO slice. Reprinted with permission from ref. 153. Copyright (2010) American Chemical Society. | ||
Because iron is a common and cheap metal on earth and a safe and green material, this method has great potential for the mass production of RGO. In another report from the same group,154 aluminum was also used to prepare RGO, and the resulting RGO has a conductivity at 2100 S m−1.
To summarize, RGO suspension, film, or paper could be successfully prepared by various reducing agents, ranging from inorganic agents to metals and from toxic to non-toxic agents. Most reducing agents show sufficient ability in the reduction of GO, and the highest conductivity obtained in the produced RGO is 30400 S m−1 (air dried), which is close to that of pristine graphite (84500 S m−1). However, no method so far has proved applicable for the mass production of RGO, since the chemical methods are limited by the toxicity of the reducing agents, multiple steps (reaction, washing, filtration, and dispersion), long reaction time (a few hours to a few days), and incomplete removal of the oxygen-containing groups in GO (relative low conductivity). In addition, since some of the carbon atoms are missing in the sp2 carbon network of GO,111 it is difficult if not impossible, to fully recover the carbon network by using chemical reduction methods alone. Thermal methods may provide some advantages in the reduction of GO, which is reviewed in section 4.2.
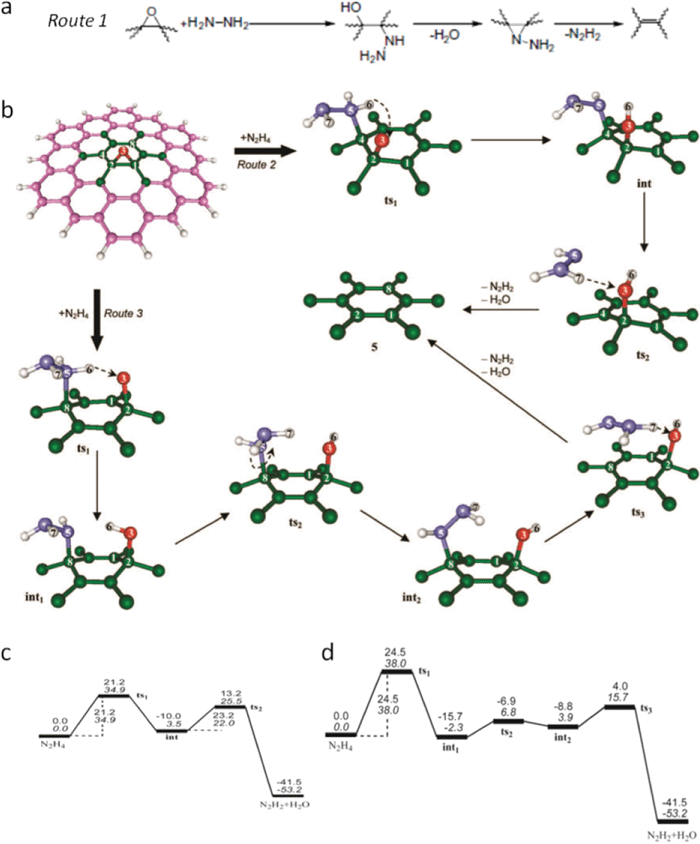 | ||
| Fig. 12 (a) Proposed reaction route 1 for epoxy reduction with hydrazine. Reprinted from S. Stankovich et al., Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide, Carbon, 2007, 45, 1558–1565.107 Copyright (2007), with permission from Elsevier. (b) Schematic evolution of local atomic structure of epoxy reduced with hydrazine via routes 2 and 3, the magenta (and green), red, violet, and white balls represent C, O, N, and H atoms, respectively. (c) and (d) reaction energy profiles for routes 2 and 3 in (b) under vacuum, values in upright and italic type are relative enthalpies (Hrel, in kcal mol−1) and relative Gibbs free energies (Grel, in kcal mol−1) at room temperature (25 °C). Reprinted with permission from ref. 181. Copyright (2010) American Chemical Society. | ||
In the theoretical work from Gao and Nagase et al.,181 nitrogen in the hydrazine derivatives (–N2H3 and –NNH2) sits at the atop site and two feasible routes are proposed, as shown in Fig. 12b depending on the nitrogen bonded with the nearest or meta-site carbon atom. In route 2, starting with the structure (not shown here) of physisorption of N2H4 adjacent to epoxy stabilized by hydrogen bond (Gibbs free energy is referred to as 0.0 kcal mol−1 in Fig. 12c), by getting over the potential energy barrier height of 34.9 kcal mol−1 indicated as transition structure 1 (ts1), in which N2H4 comes closer to the carbon plane within the bonding range of nitrogen and carbon, one H is spontaneously transferred from N2H4 into the atop oxygen and forms hydroxyl; in this way the nitrogen in N2H3 is bonded to nearest carbon. Again after getting over a slightly lower barrier height of 22.0 kcal mol−1 represented as transition structure 2 (ts2), in which the derivative (N2H3) rotates and forms a hydrogen bond with this hydroxyl, another hydrogen is dissociated from this derivative and bonds with hydroxyl to form a water molecule with the remaining nitrogen containing moiety, developing into N2H2. In route 3, the reaction mechanism is similar to that in route 2, except that the nitrogen is bonded to the meta-site carbon and the energy barrier height for ts1 is 3.1 kcal mol−1 greater (Fig. 12d). In reality, both routes exist during the reduction process and either can be dominant depending on the coverage extent and distribution of epoxies. At the beginning stage of reduction, as the epoxy coverage is high, it was found that route 3 is dominant since its energy barrier height in ts1 is 2.8 kcal mol−1 less than that of ts2 in route 2, while as the reduction proceeds, the coverage decreases and route 2 becomes dominant. However, no matter which route prevails, the reduction gets more and more difficult as epoxies are removed. Also, for the epoxies deposited at the edges, it turns out that they can only be transformed into hydroxyls.
Instead of this Langmuir–Hinshelwood mechanism, the results from Kim et al.186 showed that Eley–Rideal mechanism is predominant in which the nitrogen in hydrazine derivatives does not prefer to form any bonds with the carbon plane throughout the reduction process; the derivatives just fly away and could also react with another epoxy to transform it into hydroxyl (Fig. 13a). In fact, this reaction is more favourable since this derivative is less stable than hydrazine. In contrast to the results that hydrazine cannot serve as the reducing agent, Kim's calculation demonstrates that hydroxyl can indeed be reduced by hydrazine with a smaller barrier height than that of epoxy (Fig. 13b). From these results, it is proposed that both hydrazine and its derivatives act as reducing agents in the reduction of GO. Another noticeable phenomenon is that nitrogen can be incorporated into the GO during its reduction with hydrazine, but the current modeling of chemical reduction dynamics does not cover this direction. One interpretation is that the carbonyl containing functional groups (lactones, anhydrides, and quinones) in GO react with hydrazine to form hydrazides and hydrazones but only hydrazone results in the removal of oxygen.107
![Optimized configurations for the initial (IS), transition (TS), and final (FS) states of epoxy ring opening via H abstraction from NHNH2 (a) and OH hydrogenation via H abstraction from N2H4 (b), together with the corresponding activation energy and exothermicity (in kcal mol−1). For each state, both top and side views are presented. The gray, red, cyan, and white balls represent C, O, N, and H atoms, respectively. Selected bond lengths (in angstroms) and bond angles (in degrees) are also shown (hydrogen bonds are indicated by a dashed line "---"). Reprinted with permission from ref. 186. Copyright [2009], American Institute of Physics.](/image/article/2012/RA/c2ra00663d/c2ra00663d-f13.gif) | ||
| Fig. 13 Optimized configurations for the initial (IS), transition (TS), and final (FS) states of epoxy ring opening via H abstraction from NHNH2 (a) and OH hydrogenation via H abstraction from N2H4 (b), together with the corresponding activation energy and exothermicity (in kcal mol−1). For each state, both top and side views are presented. The gray, red, cyan, and white balls represent C, O, N, and H atoms, respectively. Selected bond lengths (in angstroms) and bond angles (in degrees) are also shown (hydrogen bonds are indicated by a dashed line "---"). Reprinted with permission from ref. 186. Copyright [2009], American Institute of Physics. | ||
4.2 Thermal reduction of graphene oxide
Chemical reductions of GO are successful with many different reducing agents; however, the reduction level of the RGO cannot be easily controlled and the conductivity of the RGO is relatively low when compared with graphite or graphene. To address these challenges, thermal reduction of GO is carried out by heating the GO in various atmospheres (ultra-high vacuum, Ar, H2, NH3), or with different heating sources like microwave, flash light, laser, plasma, electric current, or heated AFM tip; the results show that high conductivity could be achieved by heating GO at a high temperature. In the following text, thermal reduction of GO is discussed from both the theoretical and experimental point of view.:1. Since GO platelets are suspended in the solution and drop-cast onto the substrate, mass loss and pressure developed before 100 °C are generated by the evaporated water molecules intercalated initially between GO layers. The pressure peaks around the same temperature strongly suggest, however, that the production of these gas molecules occurs concurrently and even synergistically.
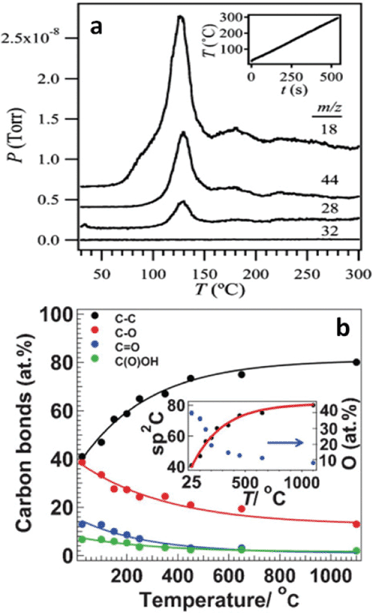 | ||
| Fig. 14 (a) Temperature programmed desorption spectra of GO film thermally reduced at heating rate of 30 °C min−1; m/z = 18, 44, 28, 32 indicate H2O, CO2, CO, and O2, respectively. The inset is the heating rate curve. Reprinted with permission from ref. 188. Copyright (2009) American Chemical Society. (b) The atomic percentage of different carbon atoms identified by XPS with respect to the annealing temperatures. The inset indicates the oxygen percentage evolution during the reduction process. Reprinted with permission from ref. 106. Copyright (2009) John Wiley and Sons. | ||
To elucidate the mass loss observed in TGA and simultaneously developed gas pressures, deep insight into the mechanism about how these gas molecules evolve is needed, not only because it is key to fully understanding thermal reduction process of GO, but also because it is prerequisite to tailor properties of thermally-reduced GO and to further atomically engineering TRGO-based materials for various applications. During the reduction process, the picture of the evolution of water molecule is clear, in which hydrogen atoms dissociate from hydroxyls and combine with other nearby hydroxyls to form water molecules, leaving the atop oxygen atoms to form epoxies.189 XRD spectra reveal some transition state structure with an interlayer distance of 5 Å corresponding to the desorption of hydrogen atoms.187 Nevertheless, the mechanism of CO2 and CO formation is still elusive at this time. One reason is that few efforts are devoted to this area, as limited conclusions can be found in the literature. Within the scope of our survey, Car et al.79 related the evolution of CO2 to the epoxy with the broken carbon bond underneath, but they did not provide any path for the formation process. By modeling the functional groups deposited on the basal plane of carbon to the highest oxidation level of 50% using the Monte Carlo method, Paci et al.119 proposed a mechanism on the CO release. In their study, epoxy could become an atop oxygen stabilized by the hydrogen bonding with nearby hydroxyl and even migrate across the carbon plane with the assistance of hydrogen transferred from hydroxyl. When two atop oxygens are close enough after a series of migrations, two of the three carbon bonds near an atop oxygen are broken, thereafter the third bond is broken to release the gas molecule of CO. The authors further proposed the formation of CO2 as a secondary reaction of CO with other oxygen-containing groups. Overall, the story about the mechanism of these gas molecules is far from clear and satisfactory, and demands future attention.
Considering that the carbon lattice in GO is conserved during the oxidation process and that transport properties of graphene is totally ruined by the functional groups, the purpose of thermal reduction is to remove the oxygen groups and restore the transport properties of graphene. Here, it is worth noting that although “defected” graphene loses some electrical conductivity/mobilities, the defects also offer extra functions to graphene to enable many interesting applications that are impossible for pure graphene. For example, "defects" make it possible to make surfactant-free graphene colloids,95 and they also bring advantages in specific applications like fuel cells, which are discussed in section 4.2.2. Despite great efforts, the atomic ratio of C1s and O1s obtained by XPS spectrum showed that oxygen could not be completely removed, even at a heating temperature up to 1100 °C in ultra-high vacuum,77,106 and close inspections manifest there are sp3-like carbon atoms in the thermally-reduced GO (TRGO) (Fig. 14b).106 A comparison of the binding energies of different functional groups190 and XPS peak assignments of oxygen atoms77,106,191 indicated that remaining oxygen could be in the form of cyclic ether, suggesting that planting oxygen atoms into the carbon backbone is relevant to the carbon loss resulting from the release of CO2 and/or CO. These ethers can either sit at the edge of TRGO platelets or spread across it. As a result of the latter case, two possible scenarios could develop: first, there is at least one C atom lost nearby the oxygen atom in the ether, leaving two sp2-like C atoms unsaturated; second, there is no carbon loss near this ether type of oxygen; however, in order to prevent forming any bond between one of the three nearest carbon atoms, the oxygen and one such atom are repulsed out of the carbon backbone oppositely, and, at this point, this outward carbon is off the sp2 one, and possibly close to the sp3 one.
The morphology of TRGO is different from that of pure graphene in several respects. With the carbon loss and some oxygen atoms remaining after thermal reduction, the line defects, holes, and lattice mismatch-induced crumples111 are prominent in the carbon skeleton of TRGO. In addition, tight binding molecular dynamics (TBMD) simulations under high temperature revealed that mono vacancies left by carbon atoms could migrate to coalesce into a pentagon–octagon–pentagon double vacancy at 3000 K, which could further evolve into the Stone Wales type defect with three alternating pentagon–heptagons forming into a cyclic structure.192 HRTEM images of a single-layer TRGO exhibited clear regions aggregated by such structures, as shown in Fig. 3b. For multilayer GO, the macroscopic appearance could be governed to some extent by the heating rates. SEM images showed that the thermally-reduced multi-layered GO has an accordion-like structure at a slow heating rate while the appearance becomes very fluffy at a fast heating rate (Fig. 3e and f).113 The GO flakes could be pushed far away from each other by the rapidly increasing pressure due to the release of gas molecules in the reduction process that overcomes the van der Waals force between adjacent layers. And for thermal exfoliation, McAllister et al.142 calculated the diffusion coefficients for the gases based on the Knudsen diffusion model and compared the results with the Arrhenius dependence of reaction rates. The study indicated that 550 °C is the threshold temperature for the thermal exfoliation to take place.
4.2.2 Thermal reduction in various atmospheres
GO could be thermally reduced in different atmospheres such as in Ar, H2, NH3 and hydrazine.77,155,190 Similar to that in UHV, GO thermally reduced in Ar prevents foreign adsorbates or contaminants. After the addition of H2, interestingly and beneficially, the outcome of the reduction differs. Both experimental and theoretical results provided evidence that the reduction level is higher for GO reduced in mixed H2 and Ar than in pure Ar.77,121 With an initial C1s/O1s atomic ratio of 2.8, the C1s/O1s ratio of RGO increases to 7.3 (6.8) and 12.4 (11.4) in the H2 and Ar mixture (pure Ar) under different temperatures (500 °C and 1000 °C), respectively.77 Simulated results showed that the reduction efficiency could be enhanced by 3.3–7.4% under all considered conditions with different initial oxygen percentages and epoxy to hydroxyl ratios at temperatures of 1000 K and 1500 K.121 A possible cause is that the thermally dissociated hydrogen atoms from hydrogen molecules could transform the carbonyls121 and epoxies193 into hydroxyls, which further develop into water molecules. This transformation is significant, as discussed in the prior section, as the outlet of oxygen in the form of CO or CO2 through epoxies is related to the loss of carbon and implantation of oxygen; however, oxygen released via H2O does not introduce any oxygen into the carbon backbone, and the exterior hydrogen atoms serve to transform these epoxies into hydroxyls.Not surprisingly but noteworthily, water also offers path to providing hydrogen. As a matter of fact, water in GO can be categorized into intercalated and bound ones that are constrained in the carbon vacancies and small holes in the carbon lattice of GO.194 Usually, the former can easily evaporate by heating, while the latter is hard to remove entirely since the water molecules are strongly physisorbed to the carbon atoms in vacancies and holes. To remove this water molecule, normally it takes about several days to obtain anhydrous GO in desiccators. Moreover, as opposed to a single-layer GO, by both theoretical calculation and experimental observation, Acik et al.195 revealed that such water molecules play a significant role in the formation of carbonyls and cyclic ethers. It is believed that water molecules could be dissociated into hydroxyls, carbonyls, and C–H groups via interacting with active carbon atoms at the edge of the holes after getting over the energy barrier of 0.692 eV.196 It is also noteworthy that even CO2 can be trapped in the multilayer GO during the thermal reduction due to the limited nature of diffusion,195 not to mention water molecules, as the carbon atoms are removed from the carbon plane to leave vacancies with the formation and release of CO2 and CO.
Foreign atoms like nitrogen could be introduced into TRGO in nitrogen-containing atmospheres such as NH3 and hydrazine.155,162 In 2009, Dai's group reported a simple method to obtain N-doped TRGO sheets through thermal annealing of GO in ammonia.155 N-doping was accompanied by the reduction of GO with decreases in oxygen levels from ∼28% in as-made GO down to ∼2% in 1100 °C NH3 treated RGO. To investigate the electronic properties of the N-doped RGO, an FET device (Fig. 15a) based on the RGO was fabricated and electrical measurements of individual RGO sheet devices demonstrate that GO annealed in NH3 exhibits an n-type semiconducting behaviour in vacuum (Fig. 15b and c), while GO reduced in H2 shows an p-type behaviour (Fig. 15d). Due to the introduced nitrogen in the carbon lattice, which increases the density of states (DOS) around the Fermi level, GO annealed in NH3 also shows higher conductivity than those annealed in H2 (Fig. 15e). Since GO reduced in NH3 shows superior conductivity, thermal reduction in NH3 was used to further reduce RGO that was pre-reduced by N2H4;33,95 and the resulting RGO has very high conductivity (35,000 S m−1 at 500 °C). The nitrogen-doped TRGO could be a potential candidate in fuel cells since both the introduced nitrogen22,197–201 and the carbon defects202–205 are active sites for oxygen-reduction reactions.
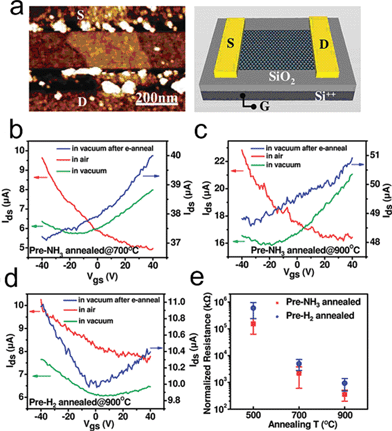 | ||
| Fig. 15 Electrical properties of single RGO sheet annealed in NH3vs. H2. (a) A typical AFM image of a N-doped RGO sheet device. (b) Current–gate voltage (Ids–Vgs) curves (recorded at Vds = 1 V) of a single RGO device fabricated with an NH3-annealed (700 °C) GO sheet. Red solid line: device measured in air. Green solid line: device measured in vacuum. Blue solid line: device measured in vacuum after electrical annealing. (c) Current–gate voltage (Ids–Vgs) curves of a single RGO device fabricated with an NH3-annealed (900 °C) GO sheet. (d) Current–gate voltage (Ids--Vgs) curves of a single RGO device fabricated with a H2-annealed (900 °C) GO sheet. (e) Statistics of normalized sheet resistance of devices fabricated with single RGO sheets annealed in NH3 and H2 at different temperatures. Normalized resistance is defined as RW/L, where R is resistance of device and W and L are the RGO sheet width and channel length, respectively. Reprinted with permission from ref. 155. Copyright (2009) American Chemical Society. | ||
The carbon loss in N-doped GO treated with hydrazine is based on the reaction between hydrazine and carbonyl containing functionalities,107 while the carbon loss in N-doped GO annealed in ammonia is due to the reduction of epoxies and hydroxyls. XPS analysis of the N binding configurations of doped GO presents two types of nitrogen: pyridinic N (N that is situated at the edge of holes or RGO flakes and forms two bonds with carbon atoms in 6-membered ring) and quaternary N (N that replaced the carbon atoms in the graphene plane). The evolution of nitrogen from XPS revealed that N-doping occurs at temperatures as low as 300 °C, and at this stage the quaternary N is dominant, while the highest doping level of ∼5% N is achieved at 500 °C and pyridinic N becomes dominant at higher temperatures (likely beyond 700 °C)155. The quaternary N develops at the initial stage of thermal reduction is due to the monovacancies left by CO or CO2. The reduction of carbonyls and carboxyls, the reduction induced cyclic ethers, and the possible carbon gasification206–208 give rise to the formation of pyridinic N at higher temperatures.
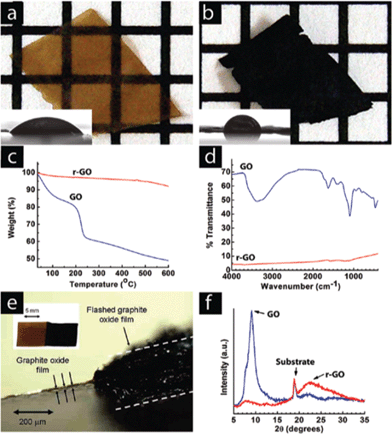 | ||
| Fig. 16 A GO paper (a) can be instantaneously reduced (b) upon exposure to a photographic camera flash. The grids in the background are 1 mm × 1 mm. The flash reduction of GO was evident by the dramatic changes in color (a, b), water contact angle (insets), (c) TGA, and (d) FTIR. In (c), (d) and (f) the blue lines correspond to GO and the red lines correspond with the flash-reduced GO. (e) Cross-sectional view of a RGO paper showing large thickness expansion after flash reduction. Only the right half of the GO sample was flashed. The left part of the picture shows the cross-sectional view of the light brown colored GO film. The thickness increased by almost 2 orders of magnitude, resulting in a very fluffy and potentially high surface area film. (f) The lack of a graphitic peak in the XRD pattern of the reduced material suggests disordered packing of the RGO sheets, consistent with the large volume expansion observed in (e). Reprinted with permission from ref. 176. Copyright (2009) American Chemical Society. | ||
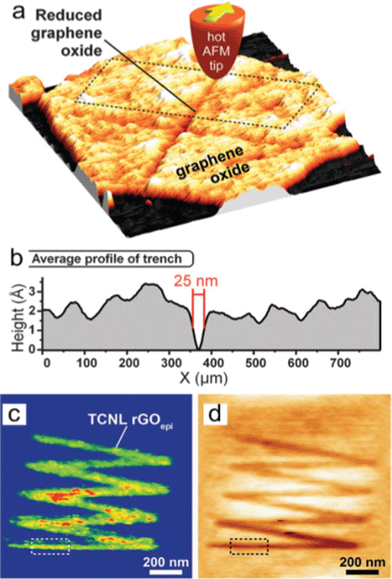 | ||
| Fig. 17 Locally thermal reduction of a single-layered GO flake. (a) Topography of a cross shape of RGO formed after an AFM tip heats the contact to 330 °C scanned across the GO sheet at 2 μm s−1. (b) The averaged profile of the trench outlined in (a) shows that the width of the line can be as narrow as 25 nm. (c) Room-temperature AFM current image (taken with a bias voltage of 2.5 V between tip and substrate) of a zigzag-shaped nanoribbon fabricated by TCNL on GO at Theater ∼ 1060 °C with a linear speed of 0.2 μm s−1 and a load of 120 nN. (d) Corresponding topography image taken simultaneously with (c). From ref. 180. Reprinted with permission from AAAS. | ||
To summarize, thermal reduction of GO is widely studied in the preparation of highly reduced GO, and the reduction of GO already occurs at moderate temperatures (150–180 °C).170,172 Compared with chemical methods, the reduction level of the RGO could be precisely controlled by heating temperature, gas environment, and duration. In addition, by heating the GO with N-containing gases like ammonia and hydrazine, RGO could be prepared as n-type doped. To achieve a high reduction level in RGO, i.e., the least amount of oxygen groups, many reports combined the chemical reduction with thermal reduction, e.g., reducing the GO with chemical reducing agents under high temperature or thermal annealing the chemically-reduced RGO. Table 1 summarizes the GO reduction approaches, the C/O ratio, and conductivity in as-prepared RGOs. So far, the RGO with the largest C/O ratio (>246) was prepared by reducing GO using NaBH4 and further thermal annealing at 1100 °C. Among these methods, the RGO with highest conductivity (57300 S m−1) was prepared by reduction of GO with NMP and further thermal annealing at 1000 °C. However, it is necessary to mention that the conductivity of a RGO sheet depends on various factors, such as the reduction level, defects, doping concentration, charge mobility, and so on.
| Sample | Forms | C:O (atomic ratio) |
Conductivity (S m−1) | Ref. |
|---|---|---|---|---|
| Graphite | pellet | 20 | 84500 |
74, 80, 209 |
| 72.5 | 1160–140000 |
|||
| Graphene | film | unknown | 5–6.4 × 106 | 81 |
| GO | film | 2.6 | 6 × 10−5 | 79, 80 |
| 2.2 | 6.8 × 10−8 | |||
| GO/N2H4, RT | pellet | 10.3 | 200 | 107 |
| GO/N2H4, RT | film | 12.5 | 9960 | 163 |
| GO/N2H4, RT | paper | 11 | 1700 | 75 |
| 150 °C | 16000 | |||
| GO/N2H4, 1100 °C | film | unknown | 55000 | 106 |
| GO/N2H4 + NH3, RT | paper | unknown | 7200 | 33, 95 |
| 220 °C | 11800 |
|||
| 500 °C | 35000 |
|||
| GO/N2H4 + KOH, RT | paper | 3.1 | 690 | 152 |
| GO/N2H4 + C16H10, RT | paper | unknown | 200 | 145 |
| GO/N2H4 + C6H15N, 500 °C | paper | >2.6 | 1700 | 157 |
| GO/N2H4+NaBH4, RT | film | unknown | 1250 | 148 |
| GO/NaBH4, RT | pellet | 4.8 | 82 | 105 |
| 180 °C | 8.6 | 1660 | ||
| 1100 °C | >246 | 20200 |
||
| GO/NaBH4, RT | film | 8.6 | 45 | 80 |
| GO/Na2S2O4+NaOH, 80 °C | film | unknown | 1377 | 159 |
| GO/NaHSO3, RT | paper | 6.48–7.89 | 6500 | 158 |
| GO/HI and AcOH, RT | pellet | 6.7 | 30400 |
74 |
| GO/HI, RT | film | 12 | 29800 |
150 |
| GO/NMP, RT | paper | 5.15 | 374 | 78 |
| 250 °C | unknown | 1380 | ||
| 500 °C | unknown | 5330 | ||
| 1000 °C | 6.03 | 57300 |
||
| GO/C4H6O3, 250 °C | paper | 6.8–8.3 | 2640–5230 | 173 |
| GO/C7H8O, RT | pellet | 30 | 4600 | 147 |
| GO/Fe, RT | paper | 7.9 | 2300 | 153 |
| GO/Al, RT | paper | 18.6 | 2100 | 154 |
| GO/vitamin C, RT | film | 12.5 | 7700 | 163 |
| GO/150 °C | paper | 4.7 | 230 | 170 |
| GO/1050 °C | film | 9.7 | 1000–2300 | 79 |
| GO/1100 °C | film | unknown | 55000 |
106 |
| GO/Flash | film | 4.2 | 1000 | 176 |
| GO/Microwave | paper | 5.46 | 200 | 174 |
| GO/Microwave | pellet | 2.75 | 274 | 175 |
5. Conclusions and perspectives
Various GO synthesis methods, i.e., Hummer and Offeman, Staudenmaier, and Brodie methods, were introduced, among which the Hummer and Offeman method is the most commonly used method to produce GO. Major functional groups in GO are epoxy, hydroxyl, and carbonyl, which locate on the basal plane of carbon skeleton, and carboxyl, which sits on the edge. Tiny islands of pure epoxies and hydroxyls, as well as graphene-like region and holes, co-exist in the GO carbon network. Although studies are available on GO structure modeling, the existing models fail to elucidate the formation dynamics of gas molecules released during their thermal reduction process. To better understand the GO structure and its evolution during the thermal reduction process, it is essential to introduce the mechanism that functional groups are competing with each other on bonding sites energetically in large blocks with different ratios and amounts.RGO could be successfully prepared by various reducing agents; however, so far, no method is proved applicable in the mass production of RGO, since the chemical methods are limited by the toxicity of the reducing agents, multiple steps, and long reaction time. On the other hand, thermal reduction of GO already occurs at moderate temperatures (150–180 °C); and compared with chemical methods, the reduction level of the RGO could be precisely controlled by the heating temperature, the gas environment, and the duration. To achieve a high reduction level, approaches are proposed to combine chemical reduction with thermal reduction. So far, the RGO paper with the largest conductivity (57300 S m−1) is prepared by reducing GO with NMP and further thermal heating at 1000 °C. Defects such as missing carbon atoms or holes in the GO carbon network could not be fixed by chemical reduction methods; however, with thermal reduction of GO with a carbon-containing gas source, it might be possible to repair the carbon network in GO. In addition, by heating the GO with N-containing gas-like ammonia and hydrazine, RGO could be prepared as n-type doped.
Future directions in RGO preparation are to develop a simple, green, and efficient method for the mass production of RGO. Based on the applications, e.g., the anode materials in the lithium ion batteries, field emitters, and hydrogen storage substrates, the mass production of RGO is needed because the amount of RGO used in these applications is large compared with RGO sensors and transistors. Meanwhile, for conductive electrodes used in energy conversion applications, RGO film or paper is desirable and the direct and one-step fabrication and reduction of RGO film or paper from GO is a promising direction. In general, to avoid the sophisticated GO reduction process and possible environmental pollution from the reducing agents, simple, green, and low-cost approaches are highly desirable. Another future direction in RGO preparation is to tune the structural or electrical properties of RGO by labeling the RGO sheets with functional groups or metallic/semiconducting nanoparticles. Chemical reduction methods are more suitable for this treatment and the resulting hybrid nanostructures may bring additional advantages towards RGO applications. Last, but not least, RGO structure is different from pristine graphene, even if all oxygen-containing groups are completely removed from the carbon network. The inherent structure differences and the dependent properties variations in RGO and graphene are crucial for graphene-based materials applications, and require more investigations both in modeling and experimental aspects in the future.
Acknowledgements
Financial support for this work was provided by the US NSF (CMMI-0900509) and the DOE (DE-EE0003208).References
- C. W. J. Beenakker, Rev. Mod. Phys., 2008, 80, 1337–1354 CrossRef CAS.
- C. Berger, Z. M. Song, X. B. Li, X. S. Wu, N. Brown, C. Naud, D. Mayou, T. B. Li, J. Hass, A. N. Marchenkov, E. H. Conrad, P. N. First and W. A. de Heer, Science, 2006, 312, 1191–1196 CrossRef CAS.
- A. Bostwick, T. Ohta, T. Seyller, K. Horn and E. Rotenberg, Nat. Phys., 2007, 3, 36–40 CrossRef CAS.
- J. S. Bunch, A. M. van der Zande, S. S. Verbridge, I. W. Frank, D. M. Tanenbaum, J. M. Parpia, H. G. Craighead and P. L. McEuen, Science, 2007, 315, 490–493 CrossRef CAS.
- X. Du, I. Skachko, F. Duerr, A. Luican and E. Y. Andrei, Nature, 2009, 462, 192–195 CrossRef CAS.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS.
- M. I. Katsnelson, K. S. Novoselov and A. K. Geim, Nat. Phys., 2006, 2, 620–625 CrossRef CAS.
- J. Martin, N. Akerman, G. Ulbricht, T. Lohmann, J. H. Smet, K. Von Klitzing and A. Yacoby, Nat. Phys., 2008, 4, 144–148 CrossRef CAS.
- J. L. McChesney, A. Bostwick, T. Ohta, T. Seyller, K. Horn, J. Gonzalez and E. Rotenberg, Phys. Rev. Lett., 2010, 104, 4 CrossRef.
- J. C. Meyer, A. K. Geim, M. I. Katsnelson, K. S. Novoselov, T. J. Booth and S. Roth, Nature, 2007, 446, 60–63 CrossRef CAS.
- D. L. Miller, K. D. Kubista, G. M. Rutter, M. Ruan, W. A. de Heer, P. N. First and J. A. Stroscio, Science, 2009, 324, 924–927 CrossRef CAS.
- R. R. Nair, P. Blake, A. N. Grigorenko, K. S. Novoselov, T. J. Booth, T. Stauber, N. M. R. Peres and A. K. Geim, Science, 2008, 320, 1308–1308 CrossRef CAS.
- N. Tombros, C. Jozsa, M. Popinciuc, H. T. Jonkman and B. J. van Wees, Nature, 2007, 448, 571–574 CrossRef CAS.
- Y. X. Xing, J. Wang and Q. F. Sun, Phys. Rev. B, 2010, 81, 10 Search PubMed.
- Y. B. Zhang, Y. W. Tan, H. L. Stormer and P. Kim, Nature, 2005, 438, 201–204 CrossRef CAS.
- K. I. Bolotin, K. J. Sikes, Z. Jiang, M. Klima, G. Fudenberg, J. Hone, P. Kim and H. L. Stormer, Solid State Commun., 2008, 146, 351–355 CrossRef CAS.
- J. H. Chen, C. Jang, S. D. Xiao, M. Ishigami and M. S. Fuhrer, Nat. Nanotechnol., 2008, 3, 206–209 CrossRef CAS.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752–7777 CrossRef CAS.
- L. R. Radovic and B. Bockrath, J. Am. Chem. Soc., 2005, 127, 5917–5927 CrossRef CAS.
- J. T. Robinson, F. K. Perkins, E. S. Snow, Z. Q. Wei and P. E. Sheehan, Nano Lett., 2008, 8, 3137–3140 CrossRef CAS.
- M. H. Liang and L. J. Zhi, J. Mater. Chem., 2009, 19, 5871–5878 RSC.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS.
- D. H. Wang, D. W. Choi, J. Li, Z. G. Yang, Z. M. Nie, R. Kou, D. H. Hu, C. M. Wang, L. V. Saraf, J. G. Zhang, I. A. Aksay and J. Liu, ACS Nano, 2009, 3, 907–914 CrossRef CAS.
- G. X. Wang, X. P. Shen, J. Yao and J. Park, Carbon, 2009, 47, 2049–2053 CrossRef CAS.
- E. Yoo, J. Kim, E. Hosono, H. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8, 2277–2282 CrossRef CAS.
- G. Eda, Y. Y. Lin, S. Miller, C. W. Chen, W. F. Su and M. Chhowalla, Appl. Phys. Lett., 2008, 92, 233305 CrossRef.
- W. J. Hong, Y. X. Xu, G. W. Lu, C. Li and G. Q. Shi, Electrochem. Commun., 2008, 10, 1555–1558 CrossRef CAS.
- X. Wang, L. J. Zhi and K. Mullen, Nano Lett., 2008, 8, 323–327 CrossRef CAS.
- J. B. Wu, H. A. Becerril, Z. N. Bao, Z. F. Liu, Y. S. Chen and P. Peumans, Appl. Phys. Lett., 2008, 92, 3 Search PubMed.
- Z. Y. Y. Z. Y. Yin, S. X. Wu, X. Z. Zhou, X. Huang, Q. C. Zhang, F. Boey and H. Zhang, Small, 2010, 6, 307–312 CrossRef CAS.
- L. A. Ponomarenko, F. Schedin, M. I. Katsnelson, R. Yang, E. W. Hill, K. S. Novoselov and A. K. Geim, Science, 2008, 320, 356–358 CrossRef CAS.
- H. Chen, M. B. Muller, K. J. Gilmore, G. G. Wallace and D. Li, Adv. Mater., 2008, 20, 3557–3561 CrossRef CAS.
- S. G. Wang, J. J. Wang, P. Miraldo, M. Y. Zhu, R. Outlaw, K. Hou, X. Zhao, B. C. Holloway, D. Manos, T. Tyler, O. Shenderova, M. Ray, J. Dalton and G. McGuire, Appl. Phys. Lett., 2006, 89, 183103 CrossRef.
- S. S. Yu and W. T. Zheng, Nanoscale, 2010, 2, 1069–1082 RSC.
- Z. M. Ao, Q. Jiang, R. Q. Zhang, T. T. Tan and S. Li, J. Appl. Phys., 2009, 105, 074307 CrossRef.
- C. Ataca, E. Akturk, S. Ciraci and H. Ustunel, Appl. Phys. Lett., 2008, 93, 043123 CrossRef.
- Y. Lin, F. Ding and B. I. Yakobson, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 041402 CrossRef.
- S. Patchkovskii, J. S. Tse, S. N. Yurchenko, L. Zhechkov, T. Heine and G. Seifert, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10439–10444 CrossRef CAS.
- C. Sealy, Nano Today, 2009, 4, 6–6 CrossRef.
- J. O. Sofo, A. S. Chaudhari and G. D. Barber, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 153401 CrossRef.
- L. Wang, K. Lee, Y. Y. Sun, M. Lucking, Z. F. Chen, J. J. Zhao and S. B. B. Zhang, ACS Nano, 2009, 3, 2995–3000 CrossRef CAS.
- S. Biswas and L. T. Drzal, Chem. Mater., 2010, 22, 5667–5671 CrossRef CAS.
- C. G. Liu, Z. N. Yu, D. Neff, A. Zhamu and B. Z. Jang, Nano Lett., 2010, 10, 4863–4868 CrossRef CAS.
- Y. Wang, Z. Q. Shi, Y. Huang, Y. F. Ma, C. Y. Wang, M. M. Chen and Y. S. Chen, J. Phys. Chem. C, 2009, 113, 13103–13107 CAS.
- A. P. Yu, I. Roes, A. Davies and Z. W. Chen, Appl. Phys. Lett., 2010, 96, 253105 CrossRef.
- K. Zhang, L. L. Zhang, X. S. Zhao and J. S. Wu, Chem. Mater., 2010, 22, 1392–1401 CrossRef CAS.
- L. L. Zhang, R. Zhou and X. S. Zhao, J. Mater. Chem., 2010, 20, 5983–5992 RSC.
- S. Hadlington, Chem. World, 2007, 4, 29–29 Search PubMed.
- B. Huang, Z. Y. Li, Z. R. Liu, G. Zhou, S. G. Hao, J. Wu, B. L. Gu and W. H. Duan, J. Phys. Chem. C, 2008, 112, 13442–13446 CAS.
- A. Kaniyoor, R. I. Jafri, T. Arockiadoss and S. Ramaprabhu, Nanoscale, 2009, 1, 382–386 RSC.
- U. Lange, T. Hirsch, V. M. Mirsky and O. S. Wolfbeis, Electrochim. Acta, 2011, 56, 3707–3712 CrossRef CAS.
- G. H. Lu, L. E. Ocola and J. H. Chen, Nanotechnology, 2009, 20, 445502 CrossRef.
- G. H. Lu, L. E. Ocola and J. H. Chen, Appl. Phys. Lett., 2009, 94, 083111 CrossRef.
- G. H. Lu, S. Park, K. H. Yu, R. S. Ruoff, L. E. Ocola, D. Rosenmann and J. H. Chen, ACS Nano, 2011, 5, 1154–1164 CrossRef CAS.
- F. Schedin, A. K. Geim, S. V. Morozov, E. W. Hill, P. Blake, M. I. Katsnelson and K. S. Novoselov, Nat. Mater., 2007, 6, 652–655 CrossRef CAS.
- K. H. Yu, P. X. Wang, G. H. Lu, K. H. Chen, Z. Bo and J. H. Chen, J. Phys. Chem. Lett., 2011, 2, 537–542 CrossRef CAS.
- L. S. Zhang, W. D. Wang, X. Q. Liang, W. S. Chu, W. G. Song, W. Wang and Z. Y. Wu, Nanoscale, 2011, 3, 2458–2460 RSC.
- B. G. Choi, H. Park, M. H. Yang, Y. M. Jung, J. Y. Park, S. Y. Lee, W. H. Hong and T. J. Park, Nanoscale, 2010, 2, 2692–2697 RSC.
- K. J. Huang, D. J. Niu, J. Y. Sun, C. H. Han, Z. W. Wu, Y. L. Li and X. Q. Xiong, Colloids Surf., B, 2011, 82, 543–549 CrossRef CAS.
- Z. X. Jiang, J. J. Wang, L. H. Meng, Y. D. Huang and L. Liu, Chem. Commun., 2011, 47, 6350–6352 RSC.
- S. Mao, G. H. Lu, K. H. Yu, Z. Bo and J. H. Chen, Adv. Mater., 2010, 22, 3521–3526 CrossRef CAS.
- L. L. Wei, J. Borowiec, L. H. Zhu and J. D. Zhang, Microchim. Acta, 2011, 173, 439–443 CrossRef CAS.
- X. C. Dong, Y. M. Shi, W. Huang, P. Chen and L. J. Li, Adv. Mater., 2010, 22, 1649 CrossRef CAS.
- N. Mohanty and V. Berry, Nano Lett., 2008, 8, 4469–4476 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS.
- Y. X. Huang, X. C. Dong, Y. M. Shi, C. M. Li, L. J. Li and P. Chen, Nanoscale, 2010, 2, 1485–1488 RSC.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, J. H. Ahn, P. Kim, J. Y. Choi and B. H. Hong, Nature, 2009, 457, 706–710 CrossRef CAS.
- S. Lee, K. Lee and Z. H. Zhong, Nano Lett., 2010, 10, 4702–4707 CrossRef CAS.
- P. W. Sutter, J. I. Flege and E. A. Sutter, Nat. Mater., 2008, 7, 406–411 CrossRef CAS.
- K. V. Emtsev, F. Speck, T. Seyller, L. Ley and J. D. Riley, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 155303 CrossRef.
- H. Huang, W. Chen, S. Chen and A. T. S. Wee, ACS Nano, 2008, 2, 2513–2518 CrossRef CAS.
- S. Malik, A. Vijayaraghavan, R. Erni, K. Ariga, I. Khalakhan and J. P. Hill, Nanoscale, 2010, 2, 2139–2143 RSC.
- I. K. Moon, J. Lee, R. S. Ruoff and H. Lee, Nat. Commun., 2010, 1, 1 CrossRef.
- S. Park, J. H. An, I. W. Jung, R. D. Piner, S. J. An, X. S. Li, A. Velamakanni and R. S. Ruoff, Nano Lett., 2009, 9, 1593–1597 CrossRef CAS.
- V. C. Tung, M. J. Allen, Y. Yang and R. B. Kaner, Nat. Nanotechnol., 2009, 4, 25–29 CrossRef CAS.
- D. Yang, A. Velamakanni, G. Bozoklu, S. Park, M. Stoller, R. D. Piner, S. Stankovich, I. Jung, D. A. Field, C. A. Ventrice and R. S. Ruoff, Carbon, 2009, 47, 145–152 CrossRef CAS.
- S. Dubin, S. Gilje, K. Wang, V. C. Tung, K. Cha, A. S. Hall, J. Farrar, R. Varshneya, Y. Yang and R. B. Kaner, ACS Nano, 2010, 4, 3845–3852 CrossRef CAS.
- H. C. Schniepp, J. L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K. Prud'homme, R. Car, D. A. Saville and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535–8539 CrossRef CAS.
- H. J. Shin, K. K. Kim, A. Benayad, S. M. Yoon, H. K. Park, I. S. Jung, M. H. Jin, H. K. Jeong, J. M. Kim, J. Y. Choi and Y. H. Lee, Adv. Funct. Mater., 2009, 19, 1987–1992 CrossRef CAS.
- J. Krupka and W. Strupinski, Appl. Phys. Lett., 2010, 96, 082101 CrossRef.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS.
- M. F. Craciun, S. Russo, M. Yamamoto and S. Tarucha, Nano Today, 2011, 6, 42–60 CrossRef CAS.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- D. R. Dreyer, R. S. Ruoff and C. W. Bielawski, Angew. Chem., Int. Ed., 2010, 49, 9336–9344 CrossRef CAS.
- X. T. Jia, J. Campos-Delgado, M. Terrones, V. Meunier and M. S. Dresselhaus, Nanoscale, 2011, 3, 86–95 RSC.
- L. M. Malard, M. A. Pimenta, G. Dresselhaus and M. S. Dresselhaus, Phys. Rep., 2009, 473, 51–87 CrossRef CAS.
- R. Mas-Balleste, C. Gomez-Navarro, J. Gomez-Herrero and F. Zamora, Nanoscale, 2011, 3, 20–30 RSC.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS.
- F. Schwierz, Nat. Nanotechnol., 2010, 5, 487–496 CrossRef CAS.
- Y. W. Zhu, S. Murali, W. W. Cai, X. S. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- J. R. Lomeda, C. D. Doyle, D. V. Kosynkin, W. F. Hwang and J. M. Tour, J. Am. Chem. Soc., 2008, 130, 16201–16206 CrossRef CAS.
- H. K. Jeong, Y. P. Lee, R. Lahaye, M. H. Park, K. H. An, I. J. Kim, C. W. Yang, C. Y. Park, R. S. Ruoff and Y. H. Lee, J. Am. Chem. Soc., 2008, 130, 1362–1366 CrossRef CAS.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS.
- T. Nakajima, A. Mabuchi and R. Hagiwara, Carbon, 1988, 26, 357–361 CrossRef CAS.
- T. Szabo, O. Berkesi, P. Forgo, K. Josepovits, Y. Sanakis, D. Petridis and I. Dekany, Chem. Mater., 2006, 18, 2740–2749 CrossRef CAS.
- A. Lerf, H. Y. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- D. W. Boukhvalov and M. I. Katsnelson, J. Am. Chem. Soc., 2008, 130, 10697–10701 CrossRef CAS.
- W. W. Cai, R. D. Piner, F. J. Stadermann, S. Park, M. A. Shaibat, Y. Ishii, D. X. Yang, A. Velamakanni, S. J. An, M. Stoller, J. H. An, D. M. Chen and R. S. Ruoff, Science, 2008, 321, 1815–1817 CrossRef CAS.
- L. B. Casablanca, M. A. Shaibat, W. W. W. Cai, S. Park, R. Piner, R. S. Ruoff and Y. Ishii, J. Am. Chem. Soc., 2010, 132, 5672–5676 CrossRef.
- N. Lu, Y. Huang, H. B. Li, Z. Y. Li and J. L. Yang, J. Chem. Phys., 2010, 133, 034502 CrossRef.
- J. A. Yan and M. Y. Chou, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 125403 CrossRef.
- W. H. Zhang, V. Carravetta, Z. Y. Li, Y. Luo and J. L. Yang, J. Chem. Phys., 2009, 131, 244505 CrossRef.
- W. Gao, L. B. Alemany, L. J. Ci and P. M. Ajayan, Nat. Chem., 2009, 1, 403–408 CrossRef CAS.
- C. Mattevi, G. Eda, S. Agnoli, S. Miller, K. A. Mkhoyan, O. Celik, D. Mostrogiovanni, G. Granozzi, E. Garfunkel and M. Chhowalla, Adv. Funct. Mater., 2009, 19, 2577–2583 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- D. Pacile, J. C. Meyer, A. F. Rodriguez, M. Papagno, C. Gomez-Navarro, R. S. Sundaram, M. Burghard, K. Kern, C. Carbone and U. Kaiser, Carbon, 2011, 49, 966–972 CrossRef CAS.
- D. Pandey, R. Reifenberger and R. Piner, Surf. Sci., 2008, 602, 1607–1613 CrossRef CAS.
- K. Erickson, R. Erni, Z. Lee, N. Alem, W. Gannett and A. Zettl, Adv. Mater., 2010, 22, 4467–4472 CrossRef CAS.
- C. Gomez-Navarro, J. C. Meyer, R. S. Sundaram, A. Chuvilin, S. Kurasch, M. Burghard, K. Kern and U. Kaiser, Nano Lett., 2010, 10, 1144–1148 CrossRef CAS.
- N. R. Wilson, P. A. Pandey, R. Beanland, R. J. Young, I. A. Kinloch, L. Gong, Z. Liu, K. Suenaga, J. P. Rourke, S. J. York and J. Sloan, ACS Nano, 2009, 3, 2547–2556 CrossRef CAS.
- F. Barroso-Bujans, A. Alegria and J. Colmenero, J. Phys. Chem. C, 2010, 114, 21645–21651 CAS.
- R. Lahaye, H. K. Jeong, C. Y. Park and Y. H. Lee, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 125435 CrossRef.
- T. Cassagneau, F. Guerin and J. H. Fendler, Langmuir, 2000, 16, 7318–7324 CrossRef CAS.
- K. Haug and N. K. N. Do, Phys. Rev. B: Condens. Matter, 1999, 60, 11095–11101 CrossRef CAS.
- M. Hloucha and U. K. Deiters, Mol. Phys., 1997, 90, 593–597 CrossRef CAS.
- A. Olivieri, D. Eisenberg, J. Soller, J. Eisenberg, R. Cooper, G. Tchobanoglous, R. Trussell and P. Gagliardo, Water Sci. Technol., 1999, 40, 223–233 CrossRef CAS.
- J. T. Paci, T. Belytschko and G. C. Schatz, J. Phys. Chem. C, 2007, 111, 18099–18111 CAS.
- S. Abe, Y. Nagoya, F. Watari and H. Tachikawa, Jpn. J. Appl. Phys., 2010, 49, 01AH07 CrossRef.
- A. Bagri, C. Mattevi, M. Acik, Y. J. Chabal, M. Chhowalla and V. B. Shenoy, Nat. Chem., 2010, 2, 581–587 CrossRef CAS.
- S. K. Chien, Y. T. Yang and C. K. Chen, Appl. Phys. Lett., 2011, 98, 033107 CrossRef.
- W. Qin, X. Li, W. W. Bian, X. J. Fan and J. Y. Qi, Biomaterials, 2010, 31, 1007–1016 CrossRef CAS.
- H. Tachikawa, T. Iyama and H. Kawabata, Jpn. J. Appl. Phys., 2010, 49, 01AH06 CrossRef.
- Q. D. Wang, J. B. Wang, J. Q. Li, N. X. Tan and X. Y. Li, Combust. Flame, 2011, 158, 217–226 CrossRef CAS.
- H. J. Xiang, S. H. Wei and X. G. Gong, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 035416 CrossRef.
- H. K. Jeong, H. J. Noh, J. Y. Kim, M. H. Jin, C. Y. Park and Y. H. Lee, Europhys. Lett., 2008, 82, 67004 CrossRef.
- T. Szabo, O. Berkesi and I. Dekany, Carbon, 2005, 43, 3186–3189 CrossRef CAS.
- J. L. Li, K. N. Kudin, M. J. McAllister, R. K. Prud'homme, I. A. Aksay and R. Car, Phys. Rev. Lett., 2006, 96, 176101 CrossRef.
- Z. P. Xu and K. Xue, Nanotechnology, 2010, 21, 045704 CrossRef.
- Z. Y. Li, W. H. Zhang, Y. Luo, J. L. Yang and J. G. Hou, J. Am. Chem. Soc., 2009, 131, 6320–6321 CrossRef CAS.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud'homme, I. A. Aksay and R. Car, Nano Lett., 2008, 8, 36–41 CrossRef CAS.
- C. Faugeras, A. Nerriere, M. Potemski, A. Mahmood, E. Dujardin, C. Berger and W. A. de Heer, Appl. Phys. Lett., 2008, 92, 011914 CrossRef.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS.
- D. Graf, F. Molitor, K. Ensslin, C. Stampfer, A. Jungen, C. Hierold and L. Wirtz, Nano Lett., 2007, 7, 238–242 CrossRef CAS.
- L. G. Cancado, M. A. Pimenta, B. R. A. Neves, M. S. S. Dantas and A. Jorio, Phys. Rev. Lett., 2004, 93, 247401 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter, 2000, 61, 14095–14107 CrossRef CAS.
- T. Livneh, T. L. Haslett and M. Moskovits, Phys. Rev. B: Condens. Matter, 2002, 66, 195110 CrossRef.
- R. Rasuli and A. I. Zad, Appl. Surf. Sci., 2010, 256, 7596–7599 CrossRef CAS.
- K. A. Mkhoyan, A. W. Contryman, J. Silcox, D. A. Stewart, G. Eda, C. Mattevi, S. Miller and M. Chhowalla, Nano Lett., 2009, 9, 1058–1063 CrossRef CAS.
- M. J. McAllister, J. L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car, R. K. Prud'homme and I. A. Aksay, Chem. Mater., 2007, 19, 4396–4404 CrossRef CAS.
- D. L. Duong, G. Kim, H. K. Jeong and Y. H. Lee, Phys. Chem. Chem. Phys., 2010, 12, 1595–1599 RSC.
- H. L. Wang, J. T. Robinson, X. L. Li and H. J. Dai, J. Am. Chem. Soc., 2009, 131, 9910–9911 CrossRef CAS.
- Y. X. Xu, H. Bai, G. W. Lu, C. Li and G. Q. Shi, J. Am. Chem. Soc., 2008, 130, 5856–5857 CrossRef CAS.
- D. R. Dreyer, S. Murali, Y. W. Zhu, R. S. Ruoff and C. W. Bielawski, J. Mater. Chem., 2011, 21, 3443–3447 RSC.
- C. Y. Su, Y. P. Xu, W. J. Zhang, J. W. Zhao, A. P. Liu, X. H. Tang, C. H. Tsai, Y. Z. Huang and L. J. Li, ACS Nano, 2010, 4, 5285–5292 CrossRef CAS.
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679–1682 CrossRef CAS.
- R. R. Wang, J. Sun, L. A. Gao, C. H. Xu, J. Zhang and Y. Q. Liu, Nanoscale, 2011, 3, 904–906 RSC.
- S. F. Pei, J. P. Zhao, J. H. Du, W. C. Ren and H. M. Cheng, Carbon, 2010, 48, 4466–4474 CrossRef CAS.
- X. B. Fan, W. C. Peng, Y. Li, X. Y. Li, S. L. Wang, G. L. Zhang and F. B. Zhang, Adv. Mater., 2008, 20, 4490–4493 CrossRef CAS.
- S. Park, J. H. An, R. D. Piner, I. Jung, D. X. Yang, A. Velamakanni, S. T. Nguyen and R. S. Ruoff, Chem. Mater., 2008, 20, 6592–6594 CrossRef CAS.
- Z. J. Fan, W. Kai, J. Yan, T. Wei, L. J. Zhi, J. Feng, Y. M. Ren, L. P. Song and F. Wei, ACS Nano, 2010, 5, 191–198 CrossRef.
- Z. J. Fan, K. Wang, T. Wei, J. Yan, L. P. Song and B. Shao, Carbon, 2010, 48, 1686–1689 CrossRef CAS.
- X. L. Li, H. L. Wang, J. T. Robinson, H. Sanchez, G. Diankov and H. J. Dai, J. Am. Chem. Soc., 2009, 131, 15939–15944 CrossRef CAS.
- D. H. Long, W. Li, L. C. Ling, J. Miyawaki, I. Mochida and S. H. Yoon, Langmuir, 2010, 26, 16096–16102 CrossRef CAS.
- O. C. Compton, D. A. Dikin, K. W. Putz, L. C. Brinson and S. T. Nguyen, Adv. Mater., 2010, 22, 892–896 CrossRef CAS.
- W. F. Chen, L. F. Yan and P. R. Bangal, J. Phys. Chem. C, 2010, 114, 19885–19890 CAS.
- T. N. Zhou, F. Chen, K. Liu, H. Deng, Q. Zhang, J. W. Feng and Q. A. Fu, Nanotechnology, 2011, 22, 045704 CrossRef.
- S. Mao, K. H. Yu, S. M. Cui, Z. Bo, G. H. Lu and J. H. Chen, Nanoscale, 2011, 3, 2849–2853 RSC.
- S. Wakeland, R. Martinez, J. K. Grey and C. C. Luhrs, Carbon, 2010, 48, 3463–3470 CrossRef CAS.
- F. Yang, Y. Q. Liu, L. A. Gao and J. Sun, J. Phys. Chem. C, 2010, 114, 22085–22091 CAS.
- M. J. Fernandez-Merino, L. Guardia, J. I. Paredes, S. Villar-Rodil, P. Solis-Fernandez, A. Martinez-Alonso and J. M. D. Tascon, J. Phys. Chem. C, 2010, 114, 6426–6432 CAS.
- S. M. Kang, S. Park, D. Kim, S. Y. Park, R. S. Ruoff and H. Lee, Adv. Funct. Mater., 2011, 21, 108–112 CrossRef CAS.
- J. B. Liu, S. H. Fu, B. Yuan, Y. L. Li and Z. X. Deng, J. Am. Chem. Soc., 2010, 132, 7279–7281 CrossRef CAS.
- O. Akhavan, M. Abdolahad, A. Esfandiar and M. Mohatashamifar, J. Phys. Chem. C, 2010, 114, 12955–12959 CAS.
- G. Williams, B. Seger and P. V. Kamat, ACS Nano, 2008, 2, 1487–1491 CrossRef CAS.
- Y. Qian, S. B. Lu and F. L. Gao, Mater. Lett., 2011, 65, 56–58 CrossRef CAS.
- E. C. Salas, Z. Z. Sun, A. Luttge and J. M. Tour, ACS Nano, 2010, 4, 4852–4856 CrossRef CAS.
- W. F. Chen and L. F. Yan, Nanoscale, 2010, 2, 559–563 RSC.
- A. Vollmer, X. L. Feng, X. Wang, L. J. Zhi, K. Mullen, N. Koch and J. P. Rabe, Appl. Phys. A: Mater. Sci. Process., 2009, 94, 1–4 CrossRef CAS.
- X. S. Wu, M. Sprinkle, X. B. Li, F. Ming, C. Berger and W. A. de Heer, Phys. Rev. Lett., 2008, 101, 026801 CrossRef.
- Y. W. Zhu, M. D. Stoller, W. W. Cai, A. Velamakanni, R. D. Piner, D. Chen and R. S. Ruoff, ACS Nano, 2010, 4, 1227–1233 CrossRef CAS.
- W. F. Chen, L. F. Yan and P. R. Bangal, Carbon, 2010, 48, 1146–1152 CrossRef CAS.
- Y. W. Zhu, S. Murali, M. D. Stoller, A. Velamakanni, R. D. Piner and R. S. Ruoff, Carbon, 2010, 48, 2118–2122 CrossRef CAS.
- L. J. Cote, R. Cruz-Silva and J. X. Huang, J. Am. Chem. Soc., 2009, 131, 11027–11032 CrossRef CAS.
- D. A. Sokolov, K. R. Shepperd and T. M. Orlando, J. Phys. Chem. Lett., 2010, 1, 2633–2636 CrossRef CAS.
- M. Baraket, S. G. Walton, Z. Wei, E. H. Lock, J. T. Robinson and P. Sheehan, Carbon, 2010, 48, 3382–3390 CrossRef CAS.
- P. P. Yao, P. L. Chen, L. Jiang, H. P. Zhao, H. F. Zhu, D. Zhou, W. P. Hu, B. H. Han and M. H. Liu, Adv. Mater., 2010, 22, 5008–5012 CrossRef CAS.
- Z. Q. Wei, D. B. Wang, S. Kim, S. Y. Kim, Y. K. Hu, M. K. Yakes, A. R. Laracuente, Z. T. Dai, S. R. Marder, C. Berger, W. P. King, W. A. de Heer, P. E. Sheehan and E. Riedo, Science, 2010, 328, 1373–1376 CrossRef CAS.
- X. F. Gao, J. Jang and S. Nagase, J. Phys. Chem. C, 2010, 114, 832–842 CAS.
- L. Qiu, X. Zhang, W. Yang, Y. Wang, G. P. Simon and D. Li, Chem. Commun., 2011, 47, 5810–5812 RSC.
- X. Yang, J. Zhu, L. Qiu and D. Li, Adv. Mater., 2011, 23, 2833–2838 CrossRef CAS.
- X. Yang, L. Qiu, C. Cheng, Y. Wu, Z.-F. Ma and D. Li, Angew. Chem., Int. Ed., 2011, 50, 7325–7328 CrossRef CAS.
- Z. Zalan, L. Lazar and F. Fulop, Curr. Org. Chem., 2005, 9, 357–376 CrossRef CAS.
- M. C. Kim, G. S. Hwang and R. S. Ruoff, J. Chem. Phys., 2009, 131, 064704 CrossRef.
- H. K. Jeong, Y. P. Lee, M. H. Jin, E. S. Kim, J. J. Bae and Y. H. Lee, Chem. Phys. Lett., 2009, 470, 255–258 CrossRef CAS.
- I. Jung, D. A. Field, N. J. Clark, Y. W. Zhu, D. X. Yang, R. D. Piner, S. Stankovich, D. A. Dikin, H. Geisler, C. A. Ventrice and R. S. Ruoff, J. Phys. Chem. C, 2009, 113, 18480–18486 CAS.
- N. Ghaderi and M. Peressi, J. Phys. Chem. C, 2010, 114, 21625–21630 CAS.
- M. Acik, G. Lee, C. Mattevi, M. Chhowalla, K. Cho and Y. J. Chabal, Nat. Mater., 2010, 9, 840–845 CrossRef CAS.
- C. D. Zangmeister, Chem. Mater., 2010, 22, 5625–5629 CrossRef CAS.
- G. D. Lee, C. Z. Wang, E. Yoon, N. M. Hwang, D. Y. Kim and K. M. Ho, Phys. Rev. Lett., 2005, 95, 205501 CrossRef.
- A. Jelea, F. Marinelli, Y. Ferro, A. Allouche and C. Brosset, Carbon, 2004, 42, 3189–3198 CrossRef CAS.
- A. Buchsteiner, A. Lerf and J. Pieper, J. Phys. Chem. B, 2006, 110, 22328–22338 CrossRef CAS.
- M. Acik, C. Mattevi, C. Gong, G. Lee, K. Cho, M. Chhowalla and Y. J. Chabal, ACS Nano, 2010, 4, 5861–5868 CrossRef CAS.
- P. Cabrera-Sanfelix and G. R. Darling, J. Phys. Chem. C, 2007, 111, 18258–18263 CAS.
- A. Morozan, B. Jousselme and S. Palacin, Energy Environ. Sci., 2011, 4, 1238–1254 CAS.
- S. R. Stoyanov, A. V. Titov and P. Kral, Coord. Chem. Rev., 2009, 253, 2852–2871 CrossRef CAS.
- A. Titov, P. Zapol, P. Kral, D. J. Liu, H. Iddir, K. Baishya and L. A. Curtiss, J. Phys. Chem. C, 2009, 113, 21629–21634 CAS.
- P. Wang, Z. K. Wang, L. X. Jia and Z. L. Xiao, Phys. Chem. Chem. Phys., 2009, 11, 2730–2740 RSC.
- L. P. Zhang and Z. H. Xia, J. Phys. Chem. C, 2011, 115, 11170–11176 CAS.
- M. Kendig and P. Kinlen, J. Electrochem. Soc., 2007, 154, C195–C201 CrossRef CAS.
- B. Shan and K. Cho, Chem. Phys. Lett., 2010, 492, 131–136 CrossRef CAS.
- S. M. Unni, V. M. Dhavale, V. K. Pillai and S. Kurungot, J. Phys. Chem. C, 2010, 114, 14654–14661 CAS.
- Y. H. Xu and X. Q. Lin, Electrochim. Acta, 2007, 52, 5140–5149 CrossRef CAS.
- L. B. Avdeeva, T. V. Reshetenko, V. B. Fenelonov, A. L. Chuvilin and Z. R. Ismagilov, Carbon, 2004, 42, 2501–2507 CrossRef CAS.
- S. Dong, P. Alvarez, N. Paterson, D. R. Dugwell and R. Kandiyoti, Energy Fuels, 2009, 23, 1651–1661 CrossRef CAS.
- F. Kapteijn, J. A. Moulijn, S. Matzner and H. P. Boehm, Carbon, 1999, 37, 1143–1150 CrossRef CAS.
- N. Deprez and D. S. McLachlan, J. Phys. D: Appl. Phys., 1988, 21, 101–107 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |