
High stabilization of pentavalent uranium on magnetite nanoparticles evidenced by high-energy-resolution X-ray absorption spectroscopy
Takumi
Yomogida
 *abc,
Jaimy
Scaria
d,
Laura
Fablet
d,
Kohei
Tokunaga
e,
Syuntaro
Dei
f,
Kotaro
Higashi
g,
Naomi
Kawamura
g,
Yoshio
Takahashi
c and
Rémi
Marsac
*b
*abc,
Jaimy
Scaria
d,
Laura
Fablet
d,
Kohei
Tokunaga
e,
Syuntaro
Dei
f,
Kotaro
Higashi
g,
Naomi
Kawamura
g,
Yoshio
Takahashi
c and
Rémi
Marsac
*b
aNuclear Safety Research Center, Japan Atomic Energy Agency, 2-4 Shirakata, Naka, Tokai, Ibaraki 319-1195, Japan
bUniversité Paris Cité, Institut de physique du globe de Paris, CNRS, F-75005 Paris, France
cDepartment of Earth and Planetary Science, The University of Tokyo, Hongo 7-3-1, Bunkyo, Tokyo 113-0033, Japan. E-mail: yomogida.takumi@jaea.go.jp
dUniv Rennes, CNRS, Géosciences Rennes – UMR 6118, F-35000 Rennes, France
eNingyo-toge Environmental Engineering Center, Japan Atomic Energy Agency, Tomata-gun, Okayama 708-0601, Japan
fHoronobe Underground Research Center, Japan Atomic Energy Agency, 432-2 Hokushin, Horonobe, Hokkaido 098-3224, Japan
gCenter for Synchrotron Radiation Research, Japan Synchrotron Radiation Research Institute (JASRI), Sayo, Hyogo 679-5198, Japan
First published on 14th October 2025
Abstract
We observed that U(V) is more stable on magnetite nanoparticles than expected, across a wide range of pH and redox conditions and even after 10 days of reaction, as revealed by high-energy-resolution fluorescence-detection X-ray absorption spectroscopy combined with multivariate curve resolution.
Uranium(U), widely used as a fuel for nuclear power generation, is a key element in actinide chemistry, nuclear technology, and environmental science due to its usefulness as an energy resource and its chemical toxicity.1–3 In the environment, it has traditionally been considered to exist as U(IV) and U(VI). Although long overlooked,4,5 recent studies have highlighted the significance of U(V), notably when U(VI) interacts with Fe(II)-bearing minerals, such as magnetite.6–11 Magnetite is a ubiquitous mineral in natural settings and may also form in artificial barriers for nuclear waste disposal as a result of steel corrosion.12 However, the role of U(V) remains unresolved as it is either considered as a metastable reaction intermediate during U(VI) reduction to U(IV) at magnetite surfaces9 or as a stable species when incorporated via coprecipitation with magnetite.11 As a result, the retention of U by magnetite has been intensively studied in recent years.9,11,13–15 Nonetheless, the mechanisms controlling U interaction with magnetite in aqueous solution relevant to natural systems remain poorly understood. In particular, many studies have employed high U concentrations, in which the precipitation of UO2(s) nanoparticles is often observed. In contrast, single-atom surface species formation may be a more relevant mechanism to investigate in many environments where U occurs at trace levels,5 as was also recently shown for rhenium.16
Recently, high-energy-resolution fluorescence detection (HERFD)-XANES has been applied to a variety of U compounds as a more sensitive technique for analysing electronic structures.17–27 The spectral features of HERFD-XANES are more distinct than those of conventional XANES spectra. For example, our group observed a specific peak splitting of U(V) in FeUO4 with U L3-edge HERFD-XANES, due to the strong crystal field splitting of the 6d orbital, which could not be detected in normal XANES spectra.28 This specific peak splitting is not observed in HERFD-XANES spectra of U(IV) in UO2 because of its weaker crystal-field splitting than that of U(V) in FeUO4. Therefore, these promising results suggest that HERFD-XANES spectra could be a powerful technique to unravel the role played by of U(V) when interacting with Fe-bearing minerals such as magnetite nanoparticles.
This paper presents insights into the influence of magnetite stoichiometry (0 ≤ R = Fe(II)/Fe(III) ≤ 0.5) on the surface reduction of U(VI) to U(V) and U(IV), as a key parameter controlling U redox speciation in natural settings. Although R can readily change due to the oxidation of structural Fe(II) or proton/ligand-promoted dissolution,29–31 prior studies have not quantified U(V) when assessing these effects.4,5 To address this gap, we employed U L3-edge HERFD–XANES spectroscopy to investigate the electronic structure of U on magnetite with varying stoichiometries and observed a peak splitting in the HERFD–XANES spectrum of U on magnetite, an effect not previously detected using conventional XANES. In this study, we explore whether U(V) stabilization correlates with structural Fe(II) in magnetite by comparing U reference materials of U(IV)O2, FeU(V)O4, and U(VI)O2(NO3)2(H2O)6, for which HERFD-XANES analyses have already been reported.28
Stock suspensions of 10 nm-sized stoichiometric (R = 0.5) and non-stoichiometric (R = 0.1, 0.2, 0.3 and 0.4) magnetite were synthesized and characterized as described in previous studies.31,32 The U stock solution (0.5 mM) was prepared from uranyl acetate and dissolved in 0.01 M HCl solution. The uranium concentration (10 μM, 2.3 ppm), background electrolyte concentration (10 mM NaCl), and total Fe concentration in the suspension (20 mM; equivalent to 1.1 g L−1 for stoichiometric magnetite) were set equal for all experiments. As the surface area of 10 nm-sized magnetite nanoparticles is approximately 100 m2 g−1, the maximum surface loading corresponds to 0.1 μmol m−2 (0.06 atom nm−2), which ensures that surface complexation was the predominant mechanism over U precipitation.5 Batch experiments were conducted in 50 mL polypropylene tubes. Dilute HCl or NaOH solutions were used to adjust the solution pH. Adsorption and reduction experiments of U(VI) were performed (i) for 6 distinct pH values (4, 5, 6, 7, 8, and 9) onto stoichiometric magnetite (R0.5) and (ii) at pH 6 and 8 onto non-stoichiometric magnetite (R0.3 and R0.2) at room temperature under anaerobic condition (N2-glovebox, JACOMEX, O2 < 10 ppm). In addition, interaction of U(VI) with R0.1 was studied under ambient atmospheric conditions (i.e. in presence of O2(g)); a reference system in which U(VI) is expected stable. After 10 days of reaction time, the solid phase was separated from the liquid phase by magnetic separation and dried in the glovebox. The dried samples were triple sealed using 100 μm high-barrier film (AP-1522, ISO). The HERFD-XANES experiments were performed at beamline BL39XU, SPring-8, as previously reported.28 A combined energy resolution of ∼1.5 eV was achieved in the experiments, as determined by measuring the full width at half-maximum (FWHM). Data processing was carried out using the Fastosh software33 and Athena software.34 Detailed experimental conditions are described in the SI.
The HERFD-XANES spectra at the U L3 edge of U interacting with R0.5 at pH 6 (denoted R0.5 pH6), collected by monitoring the Lα1 emission line, and the corresponding XANES spectrum collected in the conventional fluorescence mode are compared in Fig. 1a. The white line at approximately 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 176 eV in the conventional XANES spectrum of R0.5pH6 mainly originates from the electron transition from U 2p3/2 to unoccupied 6d states. The HERFD-XANES spectrum of the same sample, the red line in Fig. 1a, revealed more distinct features: the white line was split into two peaks in the HERFD-XANES spectra. The HERFD-XANES spectra at the U L3 edge of R0.5 pH6, UO2, FeUO4, and UO2(NO3)2(H2O)6 are compared in Fig. 1b. Peak splitting was observed in the HERFD-XANES spectrum of FeUO4, whereas no peak splitting was observed in the HERFD-XANES spectra of UO2 and UO2(NO3)2(H2O)6, indicating that a significant fraction of U is stabilized as U(V) species on magnetite.
176 eV in the conventional XANES spectrum of R0.5pH6 mainly originates from the electron transition from U 2p3/2 to unoccupied 6d states. The HERFD-XANES spectrum of the same sample, the red line in Fig. 1a, revealed more distinct features: the white line was split into two peaks in the HERFD-XANES spectra. The HERFD-XANES spectra at the U L3 edge of R0.5 pH6, UO2, FeUO4, and UO2(NO3)2(H2O)6 are compared in Fig. 1b. Peak splitting was observed in the HERFD-XANES spectrum of FeUO4, whereas no peak splitting was observed in the HERFD-XANES spectra of UO2 and UO2(NO3)2(H2O)6, indicating that a significant fraction of U is stabilized as U(V) species on magnetite.
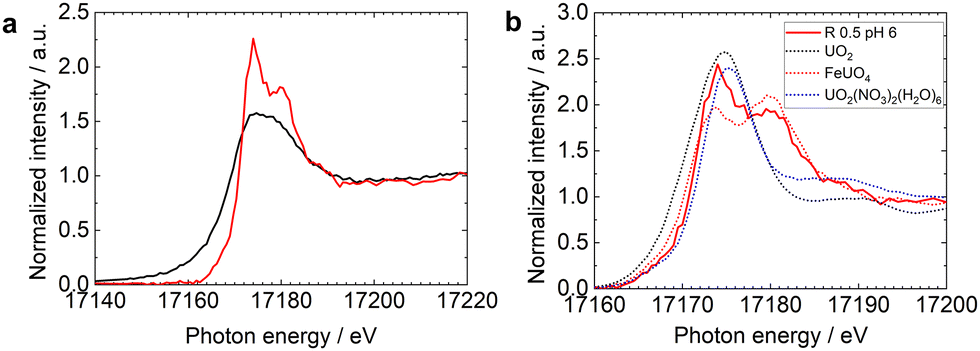 | ||
| Fig. 1 (a) U L3 edge HERFD-XANES spectrum (red line) and XANES spectrum (black line) of U-magnetite samples prepared at pH 6. (b) (Solid line) U L3 edge HERFD-XANES spectrum of U-magnetite samples prepared at pH 6. (Dotted line) HERFD-XANES spectra of UO2(black), FeUO4 (red), and UO2(NO3)2·6H2O(bule), previously published by Yomogida et al.28 | ||
Fig. 2a shows the HERFD-XANES spectra of U-R0.5 samples prepared at different pH values. Only one peak was observed at 17175 eV at pH 4. As the pH increased, the intensity of the first peak decreased, while the second peak around 17180 eV increased. Fig. 2b and c show the HERFD-XANES spectra of U-magnetite samples with different stoichiometries at pH 6 and 8, respectively. HERFD-XANES analysis revealed a gradual decrease in the second peak intensity (17180 eV) with decreasing stoichiometry. This trend was observed at both pH 6 (Fig. 2b) and pH 8 (Fig. 2c). These observations confirm previous observations that the U redox speciation is influenced by R, with higher extent of U(VI) reduction in Fe(II)-rich system.4 The effect of pH, which jointly affects U redox speciation in solution and R with the redox potential (Eh),30 will be discussed in more detail later in this manuscript.
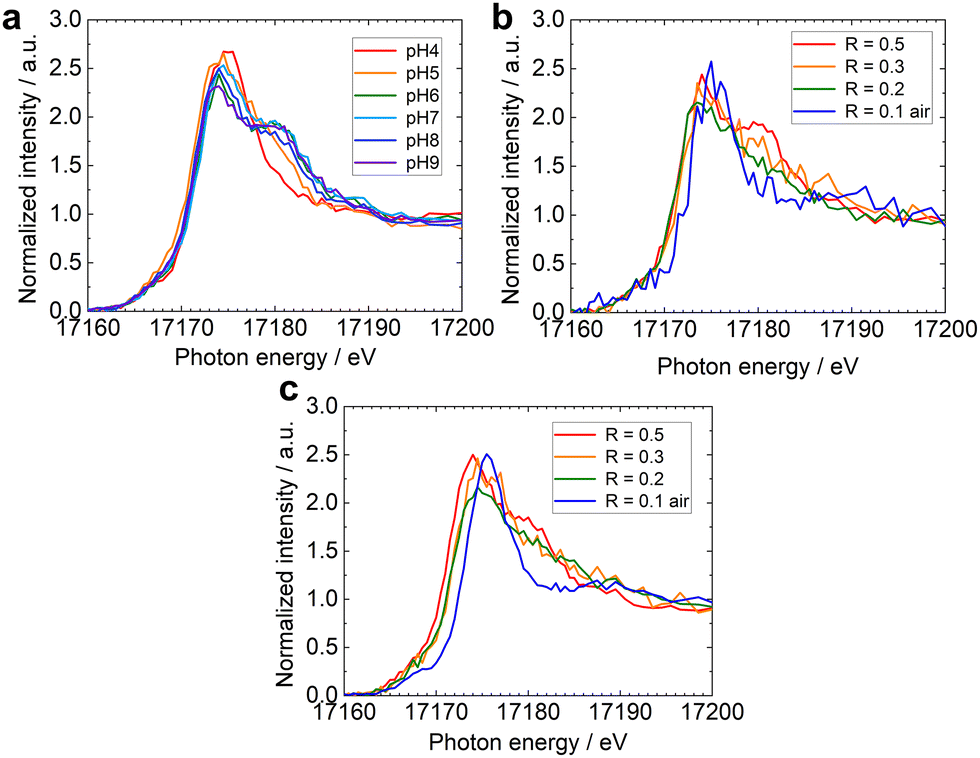 | ||
| Fig. 2 U L3-edge HERFD-XANES spectra of U reacted with (a) stoichiometric magnetite (R = 0.5) nanoparticles at different pH, (b) magnetites with different stoichiometries at pH 6 and (c) pH 8. | ||
To quantify the number of U species on stoichiometric and non-stoichiometric magnetite, singular value decomposition (SVD) analysis was applied to HERFD-XANES spectra of U on magnetites using FASTOSH software.33 Fig. S1 shows the dependence of the singular values on the number of components (n), obtained by the SVD of the data matrices. Because the singular values were nearly zero for n > 4, we considered that 3 components were sufficient to analyze the experimental data. Fig. 3a–c show the pure XANES spectra of components 1–3, obtained by multivariate curve resolution with alternating least squares (MCR-ALS) analysis of the data.35Fig. 3a shows the XANES of component 1, which displays a broad white line peak. The features of the XANES of the component 1 are consistent with those of the UO2. Fig. 3b shows the XANES the component 2, whose main peak is split and agrees with that of FeUO4. Fig. 3c shows the XANES spectrum of component 3, whose main peak is the narrowest but agrees relatively well with that of UO2(NO3)2(H2O)6. Thus, we assign the U surface species on magnetite to the 3 relevant oxidation states of U.
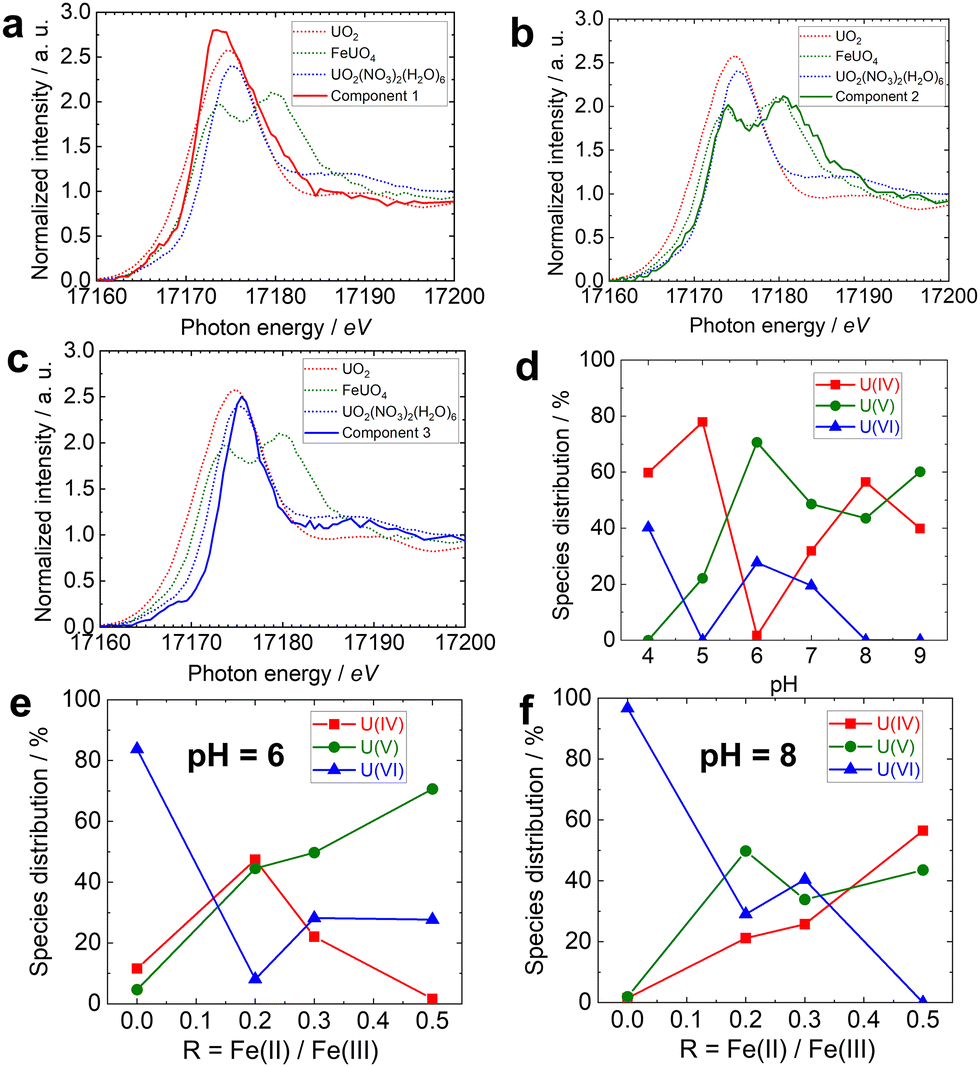 | ||
| Fig. 3 Pure XANES spectra of (a) component 1, (b) component 2, and (c) component 3 in MCR-ALS analysis of the data matrix using the PureS method. (Dotted line) HERFD-XANES spectra of UO2(black), FeUO4 (red), and UO2(NO3)2·6H2O(bule), as previously published in Yomogida et al. Percentage of fractions of U(IV), U(V), and U(VI) versus (d) different pH, (e) different magnetite stoichiometry at pH 6, and (f) different magnetite stoichiometry at pH 8. | ||
Fig. 3d–f show the distribution (%) of the 3 components obtained by MCR-ALS. Linear combination fitting (LCF) of XANES spectra of U on magnetite, using reference spectra of UO2, FeUO4 and UO2(NO3)2(H2O)6, also provided results similar to MCR-ALS (Fig. S2). On stoichiometric magnetite, approximately half of the U bound to magnetite at pH > 6 was U(V) (Fig. 3d). Thermodynamic speciation calculations in solution may support this observation, EH values determined in R0.5 suspensions30 approach the predominant field of U(V) for pH 5.5–7.5 (Fig. S3). However, such calculations do not account for the formation of surface species, which generally favour tetra- and hexavalent oxidation states of actinides (An = U, Np, Pu) over the pentavalent ones. Indeed, actinides(V) weakly sorb to iron oxide surfaces because of the smaller effective charge of cation (U(V)O2+ < U(VI)O22+ < U4+).36,37 In addition, for pH > 7.5, the theoretical prevalence of aqueous U(IV) contrasts with the observed prevalence of U(V) at the magnetite surface. Therefore, this unexpectedly high stability of U(V) is difficult to explain solely by surface complexation processes. This may instead result from its favorable incorporation into the magnetite structure. Indeed, spectroscopic studies evidenced that U(V) was highly stable when coprecipitated with Fe during magnetite synthesis,6 or that other ions such as Ni(II),38 Co(II),39 or Cr(III)32 incorporated into magnetite during sorption experiments. The XANES spectra of U(V) (Fig. 3b) is almost consistent with U(V) in FeUO4, U(V) may partially substitute octahedral site in magnetite.6 At pH < 6, magnetite can partially reduce U(VI) to U(IV), but U(V) is absent. In fact, significant amounts of structural Fe(II) are released from magnetite as Fe2+ ion to solution bellow pH 6,30 leading to a decrease in the magnetite stoichiometry. Since non-stoichiometric magnetites are less efficient at incorporating metal ions (e.g. Ni(II),38 Co(II)39 and Cr(III)32), this process may explain the absence of U(V) stabilisation. At pH 6 (Fig. 3e), the presence of U(IV) at intermediate stoichiometries, but its absence replaced by U(V) under the most reducing conditions (R = 0.5), supports this interpretation. At pH 8 (Fig. 3f), strong interaction of U(IV) with magnetite, combined with unfavourable redox conditions for U(V) aqueous species, allows its occurrence. However, these mechanisms and conditions are not sufficient to overcome U(V) stabilisation, which occurs in similar proportions to U(IV). These results indicate that the stabilization of U(V) species on magnetite is strongly related to Fe(II) in magnetite structures, not only for the reduction process, but also for the U(V)-magnetite binding mechanism.
In a previous study involving conventional XANES, Latta et al.4 reported that reduction of U(VI) to UO2 occurred only for R > 0.38 at pH 7.2. Our results show the presence of U(V) species on magnetite and that the U(VI) to U(V) reduction occurred at lower magnetite stoichiometry (R > 0.2). Besides the more difficult detection of U(V) by using normal XANES spectroscopy when U(VI) and U(IV) co-occur, this difference might be caused by the experimental conditions, such as the use of pH-buffer (50 mM MOPS or NaHCO3) that might limit U(V) adsorption, and the higher U/Fe ratio (5 g L−1 magnetite and 500 μM U) that may favour the prevalence of U(IV) in the solid phase due to the enhanced precipitation of UO2. Our results suggest that the chemical species of U and its concentration in solution may also contribute to differences in the U adsorption mechanism on magnetite.
In conclusion, the present study provides new insights into interactions between U and magnetite nanoparticles regarding the influence of magnetite stoichiometry and pH on the U speciation. HERFD-XANES spectra of U(V) on magnetites show a peak splitting that has not been observed in conventional XANES, which enable us to demonstrate the high stability of U(V) species under a wide range of conditions, after 10 days on magnetite by the combination of MCR-ALS analysis. A key finding is that structural Fe(II), whose abundance depends on pH and redox conditions, plays a critical role in the stabilization of U(V) on magnetite. Compared with previous studies using conventional XANES, our results with the novel application of HERFD-XANES reveal that the reported U(VI) reduction of UO2 only occurs for R > 0.38 at pH 7.2,4 highlighting the importance of high-resolution spectroscopy in accurately determining complex redox interactions at mineral surfaces. Revealing U(V) species, potentially acting a meta-stable species such as temporary sink or a reservoir for slow re-oxidation, on a widespread mineral like magnetite may be important to better predict the fate of U in reducing environments.
This work was also supported by the Grants-in-Aid for Scientific Research (KAKENHI) from JSPS (Grant No. 25K08534). The study was conducted with the approval of JASRI/SPring-8 (Proposal No. 2021B1821, 2022A1733, 2024B1496, and 2024B1905). Additional support was provided by the COLOSSAL project funded by ANR (project number ANR-23-CE01-0001; PI: R.M.), as well as the French Brittany Region SAD program COLORED.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included in the IPGP Research Collection See DOI: https://doi.org/10.18715/IPGP.2025.mgb3600v.Supplementary information (SI): details on sample preparation, XANES data acquisition, data analysis, and speciation modeling results. See DOI: https://doi.org/10.1039/d5cc04540a.
Notes and references
- P. L. Smedley and D. G. Kinniburgh, Appl. Geochem., 2023, 148, 105534 CrossRef
.
- A. Y. Romanchuk, I. E. Vlasova and S. N. Kalmykov, Front. Chem., 2020, 8, 630 CrossRef
-
Y. Takahashi, A. Yamaguchi and T. Yomogida, in Treatise on Geochemistry, ed A. Anbar and D. Weis, Elsevier, Oxford, 3rd edn, 2025, pp. 105–150 Search PubMed
- D. E. Latta, C. A. Gorski, M. I. Boyanov, E. J. O’Loughlin, K. M. Kemner and M. M. Scherer, Environ. Sci. Technol., 2012, 46, 778–786 CrossRef
- Z. Wang, K.-U. Ulrich, C. Pan and D. E. Giammar, Environ. Sci. Technol. Lett., 2015, 2, 227–232 CrossRef
- H. E. Roberts, K. Morris, G. T. W. Law, J. F. W. Mosselmans, P. Bots, K. Kvashnina and S. Shaw, Environ. Sci. Technol. Lett., 2017, 4, 421–426 CrossRef
- O. Stagg, K. Morris, A. Lam, A. Navrotsky, J. M. Velázquez, B. Schacherl, T. Vitova, J. Rothe, J. Galanzew, A. Neumann, P. Lythgoe, L. Abrahamsen-Mills and S. Shaw, Environ. Sci. Technol., 2021, 55, 16445–16454 CrossRef PubMed
- S. Kushwaha, B. Sreedhar and P. Padmaja, Langmuir, 2012, 28, 16038–16048 CrossRef PubMed
- K. Yuan, E. S. Ilton, M. R. Antonio, Z. Li, P. J. Cook and U. Becker, Environ. Sci. Technol., 2015, 49, 6206–6213 CrossRef PubMed
- C. Dewey, D. Sokaras, T. Kroll, J. R. Bargar and S. Fendorf, Environ. Sci. Technol., 2020, 54, 6021–6030 CrossRef PubMed
- Z. Pan, B. Bartova, T. LaGrange, S. M. Butorin, N. C. Hyatt, M. C. Stennett, K. O. Kvashnina and R. Bernier-Latmani, Nat. Commun., 2020, 11, 4001 CrossRef PubMed
- C. J. Dodge, A. J. Francis, J. B. Gillow, G. P. Halada, C. Eng and C. R. Clayton, Environ. Sci. Technol., 2002, 36, 3504–3511 CrossRef PubMed
- K. Yuan, D. Renock, R. C. Ewing and U. Becker, Geochim. Cosmochim. Acta, 2015, 156, 194–206 CrossRef
- H. Veeramani, D. S. Alessi, E. I. Suvorova, J. S. Lezama-Pacheco, J. E. Stubbs, J. O. Sharp, U. Dippon, A. Kappler, J. R. Bargar and R. Bernier-Latmani, Geochim. Cosmochim. Acta, 2011, 75, 2512–2528 CrossRef
- Y. Wang, J. Wang, P. Li, H. Qin, J. Liang and Q. Fan, Environ. Tech. Innov., 2021, 23, 101615 CrossRef
- R. Ding, C. Guida, C. I. Pearce, E. Arenholz, J.-M. Grenèche, A. Gloter, A. C. Scheinost, K. O. Kvashnina, K. Wang, A. Fernandez-Martinez, Y. Mu, K. M. Rosso and L. Charlet, Sci. Adv., 2025, 11, eadq3650 CrossRef PubMed
- T. Vitova, K. O. Kvashnina, G. Nocton, G. Sukharina, M. A. Denecke, S. M. Butorin, M. Mazzanti, R. Caciuffo, A. Soldatov, T. Behrends and H. Geckeis, Phys.
Rev. B: Condens. Matter Mater. Phys., 2010, 82, 235118 CrossRef
- K. O. Kvashnina, S. M. Butorin, P. Martin and P. Glatzel, Phys. Rev. Lett., 2013, 111, 253002 CrossRef PubMed
- K. O. Kvashnina and F. M. F. de Groot, J. Electron Spectrosc. Relat. Phenom., 2014, 194, 88–93 CrossRef CAS
- K. O. Kvashnina, Y. O. Kvashnin and S. M. Butorin, J. Electron Spectrosc. Relat. Phenom., 2014, 194, 27–36 CrossRef CAS
- J. G. Tobin, S. W. Yu, C. H. Booth, T. Tyliszczak, D. K. Shuh, G. van der Laan, D. Sokaras, D. Nordlund, T. C. Weng and P. S. Bagus, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 035111 CrossRef
- R. Bes, M. Rivenet, P. L. Solari, K. O. Kvashnina, A. C. Scheinost and P. M. Martin, Inorg. Chem., 2016, 55, 4260–4270 CrossRef CAS PubMed
- G. Leinders, R. Bes, J. Pakarinen, K. Kvashnina and M. Verwerft, Inorg. Chem., 2017, 56, 6784–6787 CrossRef CAS
- I. Pidchenko, K. O. Kvashnina, T. Yokosawa, N. Finck, S. Bahl, D. Schild, R. Polly, E. Bohnert, A. Rossberg, J. Gottlicher, K. Dardenne, J. Rothe, T. Schafer, H. Geckeis and T. Vitova, Environ. Sci. Technol., 2017, 51, 2217–2225 CrossRef CAS
- L. Desfougeres, E. Welcomme, M. Ollivier, P. M. Martin, J. Hennuyer, M. Hunault, R. Podor, N. Clavier and L. Favergeon, Inorg. Chem., 2020, 59, 8589–8602 CrossRef CAS PubMed
- R. Bes, G. Leinders and K. Kvashnina, J. Synchrotron Radiat., 2022, 29, 21–29 CrossRef CAS PubMed
- K. O. Kvashnina, Y. O. Kvashnin, J. R. Vegelius, A. Bosak, P. M. Martin and S. M. Butorin, Anal. Chem., 2015, 87, 8772–8780 CrossRef CAS PubMed
- T. Yomogida, D. Akiyama, K. Ouchi, Y. Kumagai, K. Higashi, Y. Kitatsuji, A. Kirishima, N. Kawamura and Y. Takahashi, Inorg. Chem., 2022, 61, 20206–20210 CrossRef CAS
- P. Jungcharoen, M. Pédrot, F. Choueikani, M. Pasturel, K. Hanna, F. Heberling, M. Tesfa and R. Marsac, Environ. Sci.: Nano, 2021, 8, 2098–2107 RSC
- P. Jungcharoen, M. Pédrot, F. Heberling, K. Hanna, F. Choueikani, C. Catrouillet, A. Dia and R. Marsac, Environ. Sci.: Nano, 2022, 9, 2363–2371 RSC
- P. Jungcharoen, R. Marsac, F. Choueikani, D. Masson and M. Pédrot, Nanoscale Adv., 2023, 5, 4213–4223 RSC
- J. Scaria, M. Pédrot, L. Fablet, T. Yomogida, T. T. Nguyen, Y. Sivry, C. Catrouillet, A. E. Pradas Del Real, F. Choueikani, D. Vantelon, A. Dia, A. Groleau and R. Marsac, Environ. Sci. Technol., 2025, 59, 5747–5755 CrossRef CAS PubMed
- G. Landrot and E. Fonda, J. Synchrotron Radiat., 2025, 1085–1094 CrossRef
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 537–541 CrossRef CAS
- W. Windig and D. A. Stephenson, Anal. Chem., 1992, 64, 2735–2742 CrossRef CAS
- H. Geckeis, J. Lützenkirchen, R. Polly, T. Rabung and M. Schmidt, Chem. Rev., 2013, 113, 1016–1062 CrossRef CAS PubMed
- R. Marsac, C. Catrouillet, M. Pédrot, M. F. Benedetti, A. Dia, E. D. van Hullebusch, M. Davranche, Y. Sivry, A.-C. Pierson-Wickmann, M. Tharaud and F. Heberling, Curr. Opin. Colloid Interface Sci., 2024, 72, 101820 CrossRef CAS
- L. Fablet, M. Pédrot, F. Choueikani, I. Kieffer, O. Proux, A.-C. Pierson-Wickmann, V. Cagniart, T. Yomogida and R. Marsac, Environ. Sci.: Nano, 2025, 12, 2815–2827 RSC
- L. Fablet, F. Choueikani, M. Pédrot and R. Marsac, Environ. Sci.: Nano, 2024, 11, 2036–2048 RSC
| This journal is © The Royal Society of Chemistry 2025 |