
Two solvent-dependent porous coordination polymers with –OH decorated ligands: unusual non-crystallographic net and fsh topology†
Jingui
Duan
*a,
Masakazu
Higuchi
bc,
Changchang
Zou
a,
Wanqin
Jin
a and
Susumu
Kitagawa
*b
aState Key Laboratory of Materials-Oriented Chemical Engineering, College of Chemistry and Chemical engineering, Nanjing Tech University, Nanjing, 210009, China. E-mail: duanjingui@njtech.edu.cn
bInstitute for Integrated Cell-Material Sciences (WPI-iCeMS), Kyoto University, Yoshida, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: kitagawa@icems.kyoto-u.ac.jp
cJapan Science and Technology Agency, PRESTO 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 24th June 2015
Abstract
Two new porous coordination polymers (PCPs), ([Cu6(L)4·(H2O)6]·10DMA·4EtOH, (1) and [Cu5(L)2 (OH)2·(H2O)2·DMA2]·2DMA·EtOH·2H2O (2), were solvothermally synthesized and structurally characterized. Interestingly, their variable architectures controlled by solvent system exhibit a structural progression from an unusual non-crystallographic (NC) net to a (4,6)-connected framework with fsh topology. Moreover, the combination of 3D channels of about 3.0 × 7.2, 4.7 × 9.5, and 6.3 × 7.8 Å2, and functional –OH groups in 2′ lead to good selectivity of CO2 over CH4 (26-55 by IAST) at 273 K.
Porous coordination polymers (PCPs), with the tunable nature of their structure and properties, are excellent rivals to other porous materials, such as zeolites and activated carbon, for gas storage and separation.1 However, the rational control of coordination assemblies remains a challenge.1a,g,2 Because, many factors (reactant stoichiometry, temperature, pH, solvent, reaction time and template agents) may affect the final architectures.3 Among them, solvent systems have been found to produce more dramatic effects on self-assembly.4 This is because, by acting as a template or a second ligand, the traditional coordination geometry that is preferred by the metal center could be changed.5 The generated structure cannot follow the rules of reticular chemistry, even based on well-designed ligands. Thus, further investigation of solvent systems in the formation of coordination polymers is very essential.
In addition, exposed functional sites and an optimized pore size play important roles in selective gas capture, due to the enhanced ability of host–guest interactions and also maximum size exclusion effects.6 The immobilization of functional sites (such as open metal sites and alkylamines) into PCPs was considered as rational design.7 Moreover, by introducing some specific substituent groups, e.g. –NH2, –COOH on the pore surface also worked well.8 However, there is a dearth of research on the investigation of the impact of open –OH groups in PCPs, because of the difficulty of getting open –OH sites after coordination assembly.9
We are interested in the design and construction of porous coordination polymers with functional pores for the recognition of energy gas molecules, and identifying a series of water and chemical stable frameworks for expected feasible gas storage and separation.10 Here, continuing our work, we report the syntheses of two new PCPs, ([Cu6(L)4·(H2O)6]·10DMA·4EtOH, (1, the desolvated solid is named as 1′), and [Cu5(L)2(OH)2·(H2O)2·DMA2]·2DMA·EtOH·2H2O (2, the desolvated solid is named as 2′), (H6L = 1,3,5-tris(3-carboxy-4-hydroxylphenyl)benzene), based upon a ligand with an –OH functional group. Interestingly, their variable architectures controlled by solvent system exhibit a structural progression from an unusual NC net to a (4,6)-connected framework with fsh topology. In addition, PCP 2 bearing a suitable pore size, as well as an open –OH site shows high selectivity of CO2 over CH4 (25–55) at 273 K (Scheme 1).
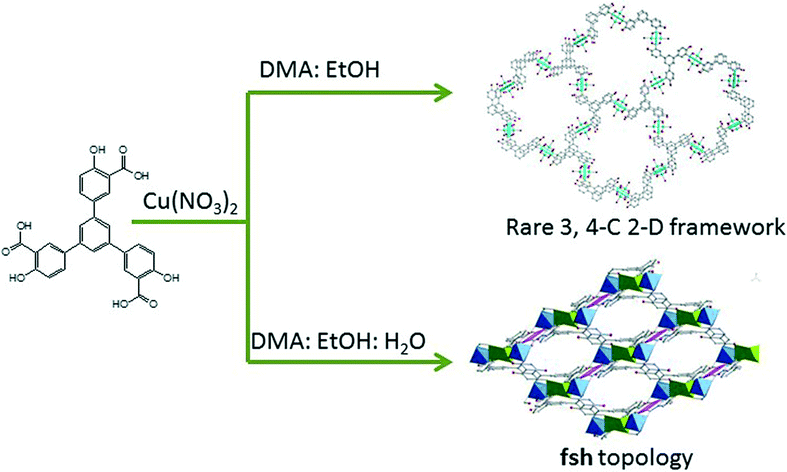 | ||
| Scheme 1 Synthetic procedures of PCP 1 and 2. | ||
A solvothermal reaction of H6L and Cu(NO3)2·6H2O in DMA/EtOH afforded green crystals of ([Cu6(L)4·(H2O)6]·10DMA·4EtOH. The phase purity of them was confirmed by comparing their experimental powder X-ray diffraction pattern to that calculated based on the single-crystal structures. X-ray diffraction analysis reveals that the L3− ligand in 1 is connected by three copper paddlewheels, and each cluster is bridged by four carboxylate groups from four different ligands, forming a highly symmetrical fan-like sub-structure (Fig. 1). The double layers of fan-like structures share their edges with each other to generate a 1D channel with a diameter of 15.6 Å. In addition, all of the inserted –OH groups are well-aligned inside the channels of 1 (Fig. 1d). For the easy understanding of the connection of this structure, we simply assign the Cu2(ArCOO)4 units as 4-connected nodes, and L3− units as 3-connected linkers. The 3-c nodes come in pairs in which each of them is connected to the same three 4-c nodes. Clearly there is a net automorphism that simply involves interchanging those two vertices, leaving the rest fixed.11 Thus, the generated topology of 1 is an example of a non-crystallographic (NC) net that is different to the another two (3,4)-connected nets of HKUST-1(tbo)12 and MOF-14 (pto).13 This significant change can be explained as being due to the shift of the coordination site from the 4- to 3-position of the benzene ring. The total accessible volume of desolvated 1 is ca. 71.1%, calculated using the PLATON program.14 Additionally, the powder X-ray diffraction (PXRD) pattern showed that the peak positions of the as-synthesized phase are the same as the simulated data, but the peak intensity, especially for the [2 0 0] peak, is different. This is because 1 exhibits a stronger preferential orientation along the [2 0 0] direction.15 Meanwhile, with very good reliability factors (Rp = 0.0252 and Rwp = 0.0553), Le Bail analysis of the PXRD pattern shows that the refined parameters are very close to the data from the single crystal, reflecting good phase purity and also a well-defined structure (Fig. S8†).
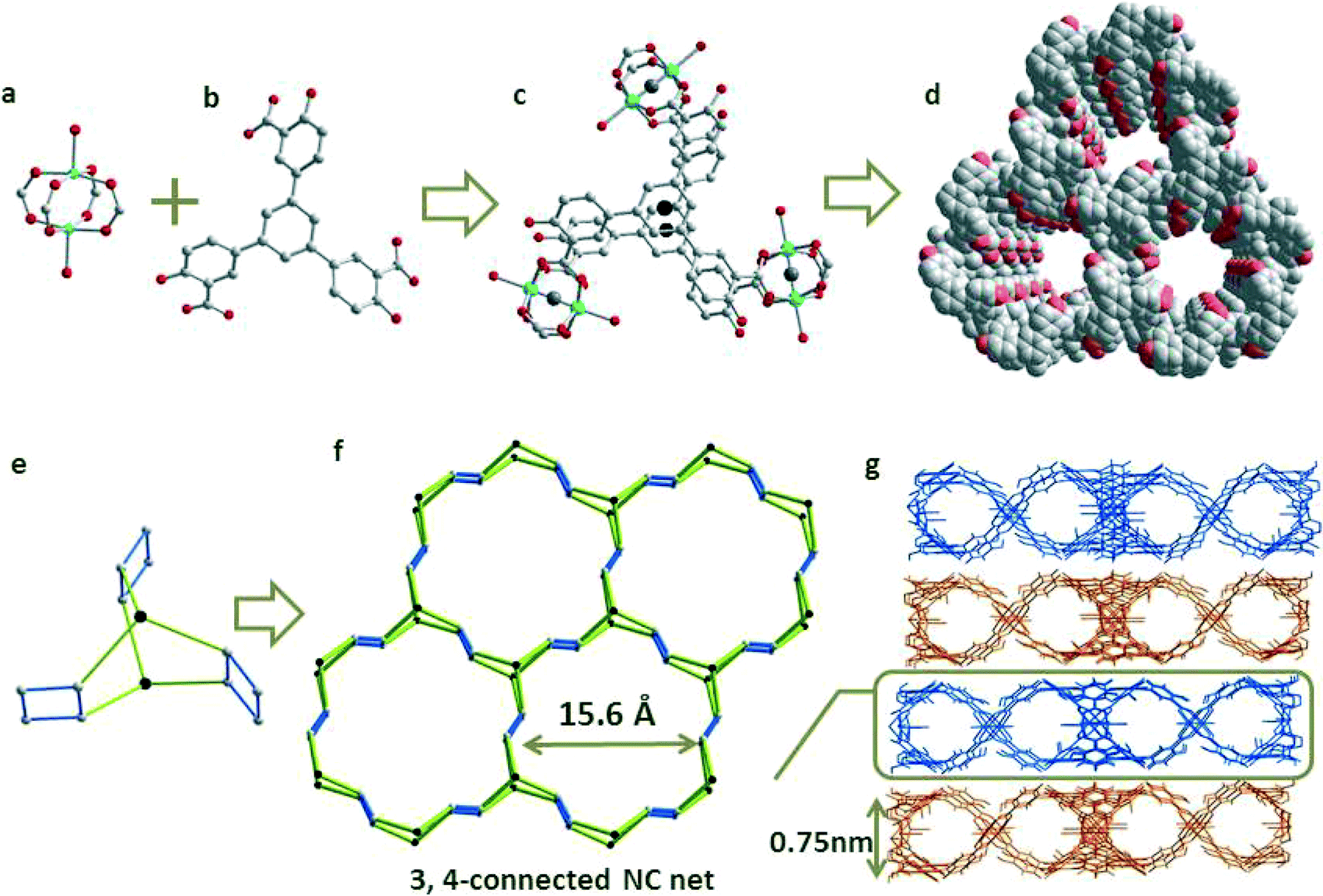 | ||
| Fig. 1 The structure of 1: (a) the dicopper cluster; (b) H6L ligand; (c) double layered fan-like structure; (d) functional and free –OH groups aligned inside the 1D channel (O atoms from the –OH groups are highlighted in a red color); (e) the simplification method; (f) unusual 3,4-connected NC net; (g) the packing of the 2D framework with a 0.75 nm layer height. | ||
[Cu5(L)2(OH)2·(H2O)2·DMA2]·2DMA·EtOH·2H2O (2) was harvested from a DMA/EtOH/H2O solvent system. Interestingly, crystallographic analysis revealed that 2 crystallizes in P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group (Fig. 2). The asymmetric unit of 2 includes one half Cu(II) atom (Cu1) on an inversion centre, two other Cu(II) atoms in general positions, one partially deprotonated L ligand, one –OH group, one coordinated water molecule and one coordinated DMA molecule (Fig. 2a). The coordination numbers of these three copper atoms are 4, 5 and 5, respectively. The coordination geometry of Cu1 is a slightly distorted pentahedron, and is completed by two oxygen atoms from bridged –OH groups, two oxygen atoms from the carboxylate groups, and one coordinated water molecule. Meanwhile, the coordination geometry of Cu2 is completed by one oxygen atom from a bridged –OH group, three oxygen atoms from the carboxylate groups, and one coordinated DMA molecule. Last but not least, a rare and planar coordination square around Cu3 is finished by two phenols oxygen atoms and two carboxylate oxygen atoms. In addition, one ligand is connected by five Cu atoms, forming a porous 3D framework. Thus, a high density of open metal sites can be expected in 2, due to the existence of the original open metal site and also the sites from the removal of coordinated DMA and water. In addition, free –OH groups can also be found inside the channel as shown in Fig. 2d. Compared with the significant channel in 1, the structure of 2 with 3D intercrossed channels of about 3.0 × 7.21, 4.72 × 9.52, and 6.37 × 7.81 Å2 indicates a suitable pore size for CO2 separation (kinetic parameter of CO2: 3.3 Å). The total accessible volume of desolvated 2 is ca. 41.5%, calculated using the PLATON program.14 In addition, after soaking 2 in water for 1 day, the PXRD profiles show the integrity of the framework (Fig. S9†), which is a rare result for carboxylate- and Cu-based PCPs. In order to further understand the connection of 2, the Cu4(ArCOO)4(OH)2 and Cu(ArCOO)2(ArO)2 clusters were simplified as 4- and 2-connected nodes, where the L4− ligands are 4-connected linkers (Fig. 2c). However, it is usual to subsume 2-connected vertices into a link, and then the generated basic unit of 2 can be assigned as a hexatopic carboxylate linker that joins 4-connected nodes. Considering the 4-c branch points of the L4− unit, the framework shows a (4,6)-c net with fsh topology (Fig. 2e).
space group (Fig. 2). The asymmetric unit of 2 includes one half Cu(II) atom (Cu1) on an inversion centre, two other Cu(II) atoms in general positions, one partially deprotonated L ligand, one –OH group, one coordinated water molecule and one coordinated DMA molecule (Fig. 2a). The coordination numbers of these three copper atoms are 4, 5 and 5, respectively. The coordination geometry of Cu1 is a slightly distorted pentahedron, and is completed by two oxygen atoms from bridged –OH groups, two oxygen atoms from the carboxylate groups, and one coordinated water molecule. Meanwhile, the coordination geometry of Cu2 is completed by one oxygen atom from a bridged –OH group, three oxygen atoms from the carboxylate groups, and one coordinated DMA molecule. Last but not least, a rare and planar coordination square around Cu3 is finished by two phenols oxygen atoms and two carboxylate oxygen atoms. In addition, one ligand is connected by five Cu atoms, forming a porous 3D framework. Thus, a high density of open metal sites can be expected in 2, due to the existence of the original open metal site and also the sites from the removal of coordinated DMA and water. In addition, free –OH groups can also be found inside the channel as shown in Fig. 2d. Compared with the significant channel in 1, the structure of 2 with 3D intercrossed channels of about 3.0 × 7.21, 4.72 × 9.52, and 6.37 × 7.81 Å2 indicates a suitable pore size for CO2 separation (kinetic parameter of CO2: 3.3 Å). The total accessible volume of desolvated 2 is ca. 41.5%, calculated using the PLATON program.14 In addition, after soaking 2 in water for 1 day, the PXRD profiles show the integrity of the framework (Fig. S9†), which is a rare result for carboxylate- and Cu-based PCPs. In order to further understand the connection of 2, the Cu4(ArCOO)4(OH)2 and Cu(ArCOO)2(ArO)2 clusters were simplified as 4- and 2-connected nodes, where the L4− ligands are 4-connected linkers (Fig. 2c). However, it is usual to subsume 2-connected vertices into a link, and then the generated basic unit of 2 can be assigned as a hexatopic carboxylate linker that joins 4-connected nodes. Considering the 4-c branch points of the L4− unit, the framework shows a (4,6)-c net with fsh topology (Fig. 2e).
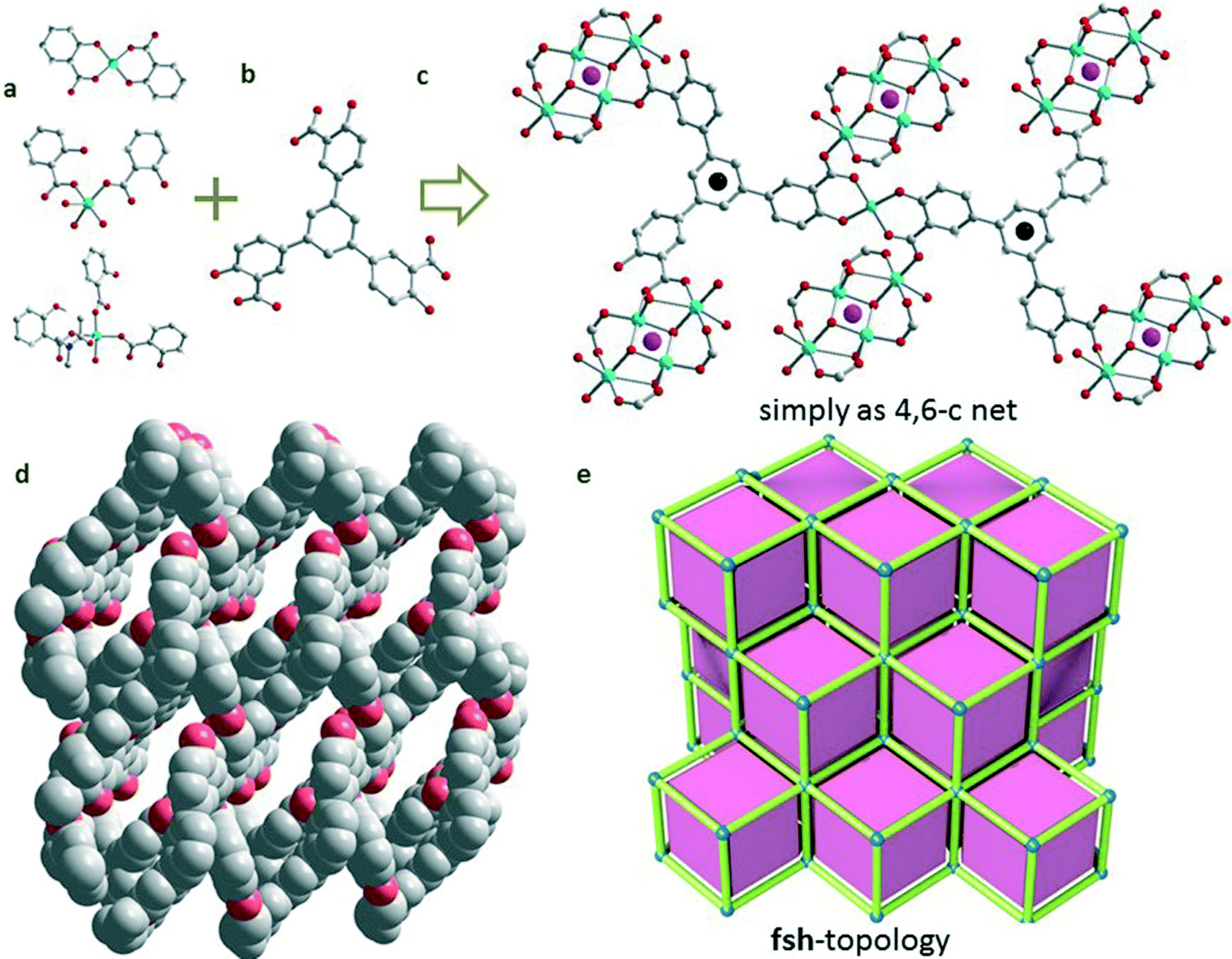 | ||
| Fig. 2 The structure of 2: (a) the three different coordination geometries of the Cu2+ atoms; (b) the H6L ligand; (c) the simple method; (d) functional –OH groups aligned inside the 1-D channel (O atoms from –OH groups are highlighted in red); (f) fsh topology with one kind of tiling. | ||
Interestingly, comparing the synthesis conditions of these two PCPs (changing the solvent systems, while the other conditions were intentionally held constant), we found that the solvent systems induced the changed coordination of the ligand, as well as the structural progression. In order to confirm this point, the following experiments were conducted and the observations from PXRD (Fig S7 and S11†) are detailed in Table 1: (1) when DMA/EtOH (2 mL, 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ) is used as a solvent system, 1 with phase purity can be obtained at 65 °C, but not at 90 °C and 130 °C; (2) when a very small amount of water was added to the DMA/EtOH system (2 mL, DMA:EtOH:H2O = 4:2:0.5), we got purified 2 at 65 °C; (3) when we increased the amount of water in the solvent system from 4:2:0.5 to 4:2:2, only 2 could be generated at 65 and 90 °C and (4) when the amount of water increased to a very high level (4:2:12), no PCPs could be formed at the three temperatures. Thus, different solvent systems regulate the formation of different environments for the assembly of Cu(II) and the linking modes of the H6L ligand. In 1, only the carboxylate groups coordinated to the Cu(II) paddlewheel nodes, whereas, in 2, three different coordination geometries were bridged by the carboxylate groups and also an –OH group from the ligand. Therefore, the ligand can be simplified as three- and four-connected linkers in 1 and 2, respectively. Then, a structural progression from an unusual NC net to a (4,6)-connected framework with fsh topology was well observed (Fig. 3). Herein, more theoretical and experimental efforts are required for finding the role of water in influencing the coordination self-assembly, but we can make a preliminary conclusion: the volumetric ratio of water in a mixed solvent system may influence the solvation process and coordination kinetics in solution. This is maybe due to water being a kind of special ligand that can result in a new balance of the kinetics and thermodynamics of the final product.4,16
:2 ) is used as a solvent system, 1 with phase purity can be obtained at 65 °C, but not at 90 °C and 130 °C; (2) when a very small amount of water was added to the DMA/EtOH system (2 mL, DMA:EtOH:H2O = 4:2:0.5), we got purified 2 at 65 °C; (3) when we increased the amount of water in the solvent system from 4:2:0.5 to 4:2:2, only 2 could be generated at 65 and 90 °C and (4) when the amount of water increased to a very high level (4:2:12), no PCPs could be formed at the three temperatures. Thus, different solvent systems regulate the formation of different environments for the assembly of Cu(II) and the linking modes of the H6L ligand. In 1, only the carboxylate groups coordinated to the Cu(II) paddlewheel nodes, whereas, in 2, three different coordination geometries were bridged by the carboxylate groups and also an –OH group from the ligand. Therefore, the ligand can be simplified as three- and four-connected linkers in 1 and 2, respectively. Then, a structural progression from an unusual NC net to a (4,6)-connected framework with fsh topology was well observed (Fig. 3). Herein, more theoretical and experimental efforts are required for finding the role of water in influencing the coordination self-assembly, but we can make a preliminary conclusion: the volumetric ratio of water in a mixed solvent system may influence the solvation process and coordination kinetics in solution. This is maybe due to water being a kind of special ligand that can result in a new balance of the kinetics and thermodynamics of the final product.4,16
| Temperature, time and solvent | DMA:EtOH:H2O |
|||
|---|---|---|---|---|
| 4:2:0 |
4:2:0.5 |
4:2:2 |
4:2:12 |
|
| 65 °C, 48 h | 1 | 2 | 2 | × |
| 90 °C, 48 h | × | × | 2 | × |
| 130 °C, 48 h | × | × | × | × |
 | ||
| Fig. 3 The coordination of metals and ligands in PCP 1 (a) and 2 (b). | ||
Encouraged by the porous and functional sites in the PCPs, the gas adsorption of them was measured. Before the gas sorption experiments, the activation of these two structures was explored. Unfortunately, desolvated 1′ collapsed, and did not show any gas uptake. Meanwhile, compound 2′ showed a little CO2 gas uptake (25 cm3 g−1) at 1 bar and 195 K. The PXRD pattern of desolvated 2′ indicated structural change after the activation. However, interestingly, 2′ did not show any uptake of O2, CH4, C2H4 and C2H6 (Fig. S14†). Thus, the unique gas adsorption isotherms show the possibility of the selective capture of CO2 by 2′. The adsorption isotherms of CO2 and CH4 in 2′ were collected at 273 K to 9 bar. The maximum uptakes of them reached 1.82 mmol g−1 and 0.35 mmol g−1, respectively. In order to explore the selectivity of 2′, ideal adsorbed solution theory was employed to predict multi-component adsorption behaviors from the experimental pure-gas isotherms. The predicted adsorption selectivity for equimolar CO2/CH4 mixtures in 2′ as a function of bulk pressure is presented in Fig. 4. The selectivity of CO2 over CH4 is very sensitive to loading and shows two steps in the changes of selectivity: a quick decrease of CO2 selectivity in the low pressure region, and a slow increase at high pressure (22–55). This high selectivity can be explained as a result of the good combination of suitable pore size, open metal sites and functionalized –OH groups in 2′. In addition, based on the IAST model and similar conditions, these selectivities are higher than those of the reported adsorbent materials (Zn2(NDC)2(DPNI) (30),17 Zn5(BTA)6(TDA)2 (37),18 Cu-BTC (6–10),19 zeolites 13× (2–24)20 and comparable with the performance of our previously reported amide-functionalized materials of NJU-Bai3 (25–60),10b indicating a selective removal of CO2 from natural gas.
 | ||
| Fig. 4 High pressure gas adsorption isotherms and the Dual-site Langmuir–Freundlich fit lines of CO2 and CH4 in 2′ at 273 K. The green lines show the IAST predicted selectivity of CO2 over CH4. | ||
Conclusions
In summary, we have demonstrated the water-controlled assembly of two new porous coordination polymers originating from the variable coordination of the ligands around the metal center. Interestingly, their variable architectures controlled by solvent systems exhibit a structural progression from an unusual non-crystallographic net to a (4, 6)-connected 3D framework with fsh topology. PCP 2, possessing a suitable pore size and functional –OH groups, exhibits good gas selectivity of CO2 (SCO2/CH4: 22–55). Therefore, we believe that our results could further the perspective of the chemistry of structural control and progression in the area of PCPs.Acknowledgements
The authors gratefully acknowledge support from the Natural Science Foundation of China (no. 21301148), Talent Development Program of Nanjing Tech University (39801116), Advanced Catalytic Transformation Program for Carbon Utilization (ACT-C), the PRESTO Program of the Japan Science and Technology Agency (JST), ENEOS Hydrogen Trust Fund. The Institute for Integrated Cell-Material Sciences (iCeMS) is supported by the World Premier International Research Initiative (WPI). We thank Dr. Mawlin Foo for discussion.Notes and references
- (a) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed; (b) R. Matsuda, R. Kitaura, S. Kitagawa, Y. Kubota, R. V. Belosludov, T. C. Kobayashi, H. Sakamoto, T. Chiba, M. Takata, Y. Kawazoe and Y. Mita, Nature, 2005, 436, 238–241 CrossRef CAS PubMed; (c) H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. O. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS PubMed; (d) O. K. Farha, A. O. Yazaydin, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, Nat. Chem., 2010, 2, 944–948 CrossRef CAS PubMed; (e) D. Q. Yuan, D. Zhao, D. F. Sun and H. C. Zhou, Angew. Chem., Int. Ed., 2010, 49, 5357–5361 CrossRef CAS PubMed; (f) L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC; (g) J. P. Zhang, Y. B. Zhang, J. B. Lin and X. M. Chen, Chem. Rev., 2012, 112, 1001–1033 CrossRef CAS PubMed; (h) H. L. Jiang and Q. Xu, Chem. Commun., 2011, 47, 3351–3370 RSC; (i) M. Hirscher, Angew. Chem. Int. Ed., 2011, 50, 581–582 CrossRef CAS PubMed; (j) R. Vaidhyanathan, S. S. Iremonger, G. K. H. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Science, 2010, 330, 650–653 CrossRef CAS PubMed.
- (a) G. Ferey, C. Mellot-Draznieks, C. Serre and F. Millange, Acc. Chem. Res., 2005, 38, 217–225 CrossRef CAS PubMed; (b) B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629–1658 CrossRef CAS PubMed.
- (a) Y. T. Wang, H. H. Fan, H. Z. Wang and X. M. Chen, Inorg. Chem., 2005, 44, 4148–4150 CrossRef CAS PubMed; (b) J. G. Duan, B. S. Zheng, J. F. Bai, Q. A. Zhang and C. Y. Zuo, Inorg. Chim. Acta, 2010, 363, 3172–3177 CrossRef CAS PubMed; (c) L. Carlucci, G. Ciani, D. M. Proserpio and S. Rizzato, New J. Chem., 2003, 27, 483–489 RSC; (d) B. Zheng, J. F. Bai and Z. X. Zhang, CrystEngComm, 2010, 12, 49–51 RSC; (e) D. M. Shin, I. S. Lee, D. Cho and Y. K. Chung, Inorg. Chem., 2003, 42, 7722–7724 CrossRef CAS PubMed; (f) B. Zheng, H. Dong, J. F. Bai, Y. Z. Li, S. H. Li and M. Scheer, J. Am. Chem. Soc., 2008, 130, 7778–7779 CrossRef CAS PubMed.
- P. P. Cui, J. L. Wu, X. L. Zhao, D. Sun, L. L. Zhang, J. Guo and D. F. Sun, Cryst. Growth Des., 2011, 11, 5182–5187 CAS.
- I. S. Lee, D. M. Shin and Y. K. Chung, Chem. – Eur. J., 2004, 10, 3158–3165 CrossRef CAS PubMed.
- (a) J. R. Li, Y. G. Ma, M. C. McCarthy, J. Sculley, J. M. Yu, H. K. Jeong, P. B. Balbuena and H. C. Zhou, Coord. Chem. Rev., 2011, 255, 1791–1823 CrossRef CAS PubMed; (b) D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- (a) D. Britt, H. Furukawa, B. Wang, T. G. Glover and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20637–20640 CrossRef CAS PubMed; (b) A. Demessence, D. M. D'Alessandro, M. L. Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131, 8784–8785 CrossRef CAS PubMed.
- A. Torrisi, R. G. Bell and C. Mellot-Draznieks, Cryst. Growth Des., 2010, 10, 2839–2841 CAS.
- (a) H. X. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gandara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018–1023 CrossRef CAS PubMed; (b) Z. X. Chen, S. C. Xiang, H. D. Arman, P. Li, D. Y. Zhao and B. L. Chen, Eur. J. Inorg. Chem., 2011, 2227–2231 CrossRef CAS PubMed.
- (a) J. G. Duan, M. Higuchi, S. Horike, M. L. Foo, K. P. Rao, Y. Inubushi, T. Fukushima and S. Kitagawa, Adv. Funct. Mater., 2013, 23, 3525–3530 CrossRef CAS PubMed; (b) J. G. Duan, Z. Yang, J. F. Bai, B. S. Zheng, Y. Z. Li and S. H. Li, Chem. Commun., 2012, 48, 3058–3060 RSC; (c) J. G. Duan, M. Higuchi, R. Krishna, T. Kiyonaga, Y. Tsutsumi, Y. Sato, Y. Kubota, M. Takata and S. Kitagawa, Chem. Sci., 2014, 5, 660–666 RSC; (d) B. S. Zheng, J. F. Bai, J. G. Duan, L. Wojtas and M. J. Zaworotko, J. Am. Chem. Soc., 2011, 133, 748–751 CrossRef CAS PubMed; (e) J. G. Duan, W. Q. Jin and R. Krishna, Inorg. Chem., 2015, 54, 4279–4284 CrossRef CAS PubMed; (f) J. G. Duan, M. Higuchi and S. Kitagawa, Inorg. Chem., 2015, 54, 1645–1649 CrossRef CAS PubMed.
- O. Delgado-Friedrichs, S. T. Hyde, S. W. Mun, M. O'Keeffe and D. M. Proserpio, Acta Crystallogr., Sect. A: Found. Crystallogr., 2013, 69, 535–542 CAS.
- S. S. Y. Chui, S. M. F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS.
- B. L. Chen, M. Eddaoudi, S. T. Hyde, M. O'Keeffe and O. M. Yaghi, Science, 2001, 291, 1021–1023 CrossRef CAS PubMed.
- A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, 2001 Search PubMed.
- K. Rao, M. Higuchi, K. Sumida, S. Furukawa, J. Duan and S. Kitagawa, Angew. Chem., Int. Ed., 2014, 53, 8225–8230 CrossRef CAS PubMed.
- (a) S. R. Caskey, A. G. Wong-Foy and A. J. Matzger, Inorg. Chem., 2008, 47, 7751–7756 CrossRef CAS PubMed; (b) D. Frahm, F. Hoffmann and M. Froba, Cryst. Growth Des., 2014, 14, 1719–1725 CrossRef CAS; (c) Y. Wang, S. X. Cui, B. Li, J. P. Zhang and Y. Zhang, Cryst. Growth Des., 2009, 9, 3855–3858 CrossRef CAS.
- Y. S. Bae, K. L. Mulfort, H. Frost, P. Ryan, S. Punnathanam, L. J. Broadbelt, J. T. Hupp and R. Q. Snurr, Langmuir, 2008, 24, 8592–8598 CrossRef CAS PubMed.
- Z. J. Zhang, S. C. Xiang, Y. S. Chen, S. Q. Ma, Y. Lee, T. Phely-Bobin and B. L. Chen, Inorg. Chem., 2010, 49, 8444–8448 CrossRef CAS PubMed.
- Q. Y. Yang and C. L. Zhong, J. Phys. Chem. B, 2006, 110, 17776–17783 CrossRef CAS PubMed.
- S. Cavenati, C. A. Grande and A. E. Rodrigues, J. Chem. Eng. Data, 2004, 49, 1095–1101 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of PCPs, PXRD, TGA, IR, and sorption isotherms. CCDC for the two PCPs: 1023464–1023465. For ESI and crystallographic data in CIF or other electronic format. See DOI: 10.1039/c5ce00762c |
| This journal is © The Royal Society of Chemistry 2015 |