DOI:
10.1039/D5LC00856E
(Paper)
Lab Chip, 2026,
26, 257-272
DCMiC: a double-cylinder micro-chamber platform for high-throughput drug screening and modeling of microenvironmental resistance in Ewing sarcoma
Received
9th September 2025
, Accepted 27th November 2025
First published on 9th December 2025
Abstract
We fabricated a double-cylinder micro-chamber (DCMiC) platform using stereolithography-printed master molds, followed by PDMS replica molding and integration into a 96-well plate format for scalable and reproducible generation of Ewing sarcoma spheroids. The simple yet novel DCMiC design stabilizes spheroids during media exchange, enabling reliable long-term culture and high-throughput drug screening. Using this platform, we screened 11 small-molecule compounds previously shown to target vulnerabilities relevant to Ewing sarcoma, including epigenetic regulators, DNA damage response, growth signaling and metabolic pathways. As a result, we identified Torin 2, talazoparib, and trabectedin as top 3 candidates with potent anti-Ewing sarcoma activity. To more accurately model the metastatic tumor microenvironment, we incorporated human lung fibroblasts to generate heterotypic spheroids, which consistently conferred resistance to all 3 compounds. Transcriptomic profiling revealed that fibroblasts reprogram Ewing sarcoma cells by activating pro-survival NFκB and TGF-β1/SMAD signaling while repressing tumor-suppressive programs, highlighting how stromal cues promote therapy resistance. Mechanistically, exogenous TGF-β1 was sufficient to induce resistance in tumor-only spheroids, whereas pharmacological inhibition of TGF-β1 signaling restored drug sensitivity in heterotypic spheroids. These findings establish the DCMiC platform as a low-cost, physiologically relevant system for modeling tumor–stroma interactions and enabling scalable drug discovery in clinically relevant contexts for Ewing sarcoma and other solid tumors.
1. Introduction
Sarcomas represent a rare and heterogeneous group of malignant tumors derived from mesenchymal tissues, characterized by significant histological diversity and aggressive clinical behavior.1,2 Among these, Ewing sarcoma predominantly affects children and young adults, with long-term survival rates below 30% in metastatic or relapsed cases, highlighting the urgent need for effective therapeutic strategies.3–5 To address this therapeutic gap, the field has developed numerous human preclinical in vitro models for Ewing sarcoma over the past few decades.2,6,7 Among these, conventional two-dimensional (2D) cell cultures, while widely used, fail to accurately replicate essential features of tumor in vivo, including tissue architecture, extracellular matrix interactions, nutrient gradients, and drug penetration barriers.8,9 In contrast, three-dimensional (3D) spheroid models offer improved physiological relevance by more closely recapitulating in vivo tumor architecture, cellular heterogeneity, and drug penetration, offering enhanced physiological relevance.10,11 Such models establish physiochemical gradients and microenvironmental interactions that critically modulate therapeutic responses, thereby providing more clinically predictive drug response profiles.12,13 However, traditional spheroid-forming methods such as hanging-drop cultures,14,15 ultra-low attachment plates,16,17 and droplet microfluidics18,19 face limitations including inconsistent spheroid size, variable morphology, limited scalability, or the need for specialized equipment.
To address these limitations, recent advances in stereolithography (SLA)-based 3D printing have transformed tumor model engineering by enabling rapid and precise fabrication of customizable micro-chamber arrays at a fraction of the cost of traditional manufacturing.20 Unlike plastic injection molding or other fixed tooling methods, which involve significant upfront costs and long lead times that make iterative design changes impractical during the optimization phase,21 SLA printing allows researchers to quickly redesign, fabricate, and test multiple geometries within hours. Also, SLA-based 3D printing offers high-resolution fabrication, enabling precise reproduction of patterns ranging from tens to several hundreds of micrometers, which allows the creation of micro-chamber arrays tailored to spheroid size and geometry for diverse tumor models.22 These features enable researchers to customize the dimensions and geometrical properties of micro-chambers according to their specific objectives, while precisely and significantly accelerating the process of achieving optimization.23
Here, we present a scalable and physiologically relevant 3D screening platform, the double-cylinder micro-chamber (DCMiC), designed for uniform Ewing sarcoma spheroid formation and high-throughput drug testing. Using this platform, we screened a focused panel of 11 small-molecule compounds and identified Torin 2, talazoparib, and trabectedin as the most potent agents. Incorporation of human lung fibroblasts into the spheroids consistently induced drug resistance, recapitulating key features of the metastatic tumor microenvironment. Transcriptomic profiling revealed fibroblast-driven activation of the TGF-β1 pathway in tumor cells, and pharmacologic inhibition of TGF-β1 signaling restored drug sensitivity in co-culture. These findings are consistent with the known role of cancer-associated fibroblasts in promoting therapeutic resistance through paracrine signaling.24–26 Together, our study establishes the DCMiC as a practical preclinical platform for identifying effective therapeutics and uncovering mechanisms of microenvironment-mediated resistance.
Experimental
More detailed information on the materials and methods used can be found in the SI.
2. Materials and methods
2.1 Fabrication of the master mold for the double-cylinder micro-chamber and preparation of its PDMS insert
The master mold was designed using SolidWorks CAD design software (Dassault Systèmes, Vélizy-Villacoublay, France) and fabricated using an Elegoo Saturn 4 Ultra 3D printer (Elegoo Inc., Shenzhen, China), as shown in Fig. 1a and S1a. The schematic in Fig. S1b illustrates the stereolithography (SLA)-based printing principle, in which a 405 nm light source selectively cures photopolymer resin layer by layer to achieve high-resolution microstructures. The mold was printed with an Elegoo Water-Washable Photopolymer Resin 2.0 (Elegoo Inc., Shenzhen, China) and the detailed procedure is described in Fig. S1c. To ensure reliable and reproducible fabrication of the micro-chambers, we optimized the printing conditions, focusing particularly on the exposure time for photopolymerization and layer thickness (Table S1). This optimization enabled the stable and uniform generation of micro-chambers, which is crucial for subsequent biological experiments (see sub-optimization in Fig. S1d). Following printing, the mold was thoroughly rinsed under running water to remove uncured resin. Incomplete washing may leave uncured resin on the mold surface, which can generate unintended structural artifacts during post-UV exposure and interfere with polydimethylsiloxane (PDMS) curing due to residual photo-initiators (Fig. S1e).27 The mold was then dried using an air blower and further cured under 405 nm UV light at room temperature for 20 minutes to enhance its mechanical strength. After UV curing, the mold was placed in a drying oven at 80 °C for 24 hours. To facilitate demolding of PDMS inserts, surface silanization was performed by applying chlorotrimethylsilane (Cat# 92361, Sigma-Aldrich, MO, USA) to the mold surface, followed by vacuum incubation for 30 minutes. This silanization step was repeated 4 times to enhance release. Without silane treatment, residual photo-initiators from the resin inhibited proper PDMS curing and caused the PDMS to adhere strongly to the mold surface (Fig. S1f).27 After the final treatment, the mold was thoroughly dried in a vacuum chamber for an hour. The PDMS (Dow Corning, MI, USA) prepolymer and curing agent were mixed at a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (w/w) ratio, poured onto the mold, and degassed using a vacuum chamber to eliminate air bubbles. The PDMS mixture was cured in a drying oven at 80 °C for 2 hours. After curing, individual PDMS inserts were punched using a 6 mm biopsy punch and attached to a standard 96-well plate using an uncured PDMS mixture. To ensure stable attachment within the 96-well plate, the DCMiC insert was designed with a slightly smaller diameter (∼6.0 mm) than the well bottom (∼6.6 mm), leaving a ∼0.3 mm clearance for easy assembly. A minimal amount (∼3 μL) of uncured PDMS was used as an adhesive, forming a thin (∼10 μm) bonding layer without affecting flatness or imaging quality. The assembled plate was leveled at room temperature before curing at 80 °C to ensure optical uniformity. During cell seeding, cells were allowed to settle for 20 minutes, followed by medium change to remove any that might enter the side gap. For sterilization, the assembled plates were exposed to UV light for 1 hour, followed by the addition of 70% ethanol (EtOH; Cat# E7023, Sigma-Aldrich, MO, USA) to each well. The wells were then washed 3 times with phosphate-buffered saline (PBS, Gibco™, Cat# 10010023, Thermo Fisher Scientific, MA, USA). To prevent cell adhesion to the PDMS surface, 2% Pluronic® F-127 (Cat# P2443, Sigma-Aldrich, MO, USA) in deionized water was added to each well for surface coating. Air bubbles trapped within the micro-chambers were removed by vacuum treatment at room temperature for 2 hours, followed by overnight incubation at 4 °C to eliminate any remaining small bubbles. Prior to cell seeding, the Pluronic® F-127 solution was completely removed, and the wells were washed 3 times with the cell culture medium. This treatment not only prevents cell adhesion to the PDMS surface, but also minimizes nonspecific drug adsorption.28
:1 (w/w) ratio, poured onto the mold, and degassed using a vacuum chamber to eliminate air bubbles. The PDMS mixture was cured in a drying oven at 80 °C for 2 hours. After curing, individual PDMS inserts were punched using a 6 mm biopsy punch and attached to a standard 96-well plate using an uncured PDMS mixture. To ensure stable attachment within the 96-well plate, the DCMiC insert was designed with a slightly smaller diameter (∼6.0 mm) than the well bottom (∼6.6 mm), leaving a ∼0.3 mm clearance for easy assembly. A minimal amount (∼3 μL) of uncured PDMS was used as an adhesive, forming a thin (∼10 μm) bonding layer without affecting flatness or imaging quality. The assembled plate was leveled at room temperature before curing at 80 °C to ensure optical uniformity. During cell seeding, cells were allowed to settle for 20 minutes, followed by medium change to remove any that might enter the side gap. For sterilization, the assembled plates were exposed to UV light for 1 hour, followed by the addition of 70% ethanol (EtOH; Cat# E7023, Sigma-Aldrich, MO, USA) to each well. The wells were then washed 3 times with phosphate-buffered saline (PBS, Gibco™, Cat# 10010023, Thermo Fisher Scientific, MA, USA). To prevent cell adhesion to the PDMS surface, 2% Pluronic® F-127 (Cat# P2443, Sigma-Aldrich, MO, USA) in deionized water was added to each well for surface coating. Air bubbles trapped within the micro-chambers were removed by vacuum treatment at room temperature for 2 hours, followed by overnight incubation at 4 °C to eliminate any remaining small bubbles. Prior to cell seeding, the Pluronic® F-127 solution was completely removed, and the wells were washed 3 times with the cell culture medium. This treatment not only prevents cell adhesion to the PDMS surface, but also minimizes nonspecific drug adsorption.28
 |
| | Fig. 1 Schematic overview of the fabrication and application of the DCMiC platform. (a) Design and fabrication of the DCMiC master mold using stereolithography (SLA)-based 3D printing from a custom drawing file. PDMS inserts were fabricated using a DCMiC master mold, and the resulting inserts were placed into a standard 96-well plate to enable scalable and parallelized culture of spheroids. (b) Workflow for spheroid formation. Tumor cells are seeded into the DCMiC array, allowed to settle for 20 minutes, and cultured for 24 hours to form compact spheroids (scale bar: 100 μm). (c) Schematic illustration of the effect of chamber geometry on spheroid stability during media exchange and representative bright-field and fluorescence images of spheroids (green) within single- and double-cylinder microchambers before and after washing. Scale bar: 600 μm. (d) Quantification of the average number of spheroids per well before and after washing. Data were collected from 3 technical replicates (n = 3) with 75 total spheroids analyzed. (e) Ewing sarcoma spheroids were generated in the DCMiC platform and exposed to a panel of 11 inhibitor candidates. High-throughput drug screening identified the effective candidates, which were subsequently evaluated for drug resistance in heterotypic spheroids composed of Ewing sarcoma cells and fibroblasts, thereby mimicking in vivo-like microenvironmental conditions. (f) Heterotypic spheroids, composed of Ewing sarcoma cells and lung fibroblasts, exhibit increased viability and drug resistance. To investigate the underlying mechanisms, RNA sequencing was performed to identify up- and down-regulated genes, highlighting pathways such as TGF-β1 and NFκB. Sequencing results were subsequently validated, and functional assays were carried out using the top candidate drugs, incorporating, respectively, with TGF-β1 treatment of Ewing sarcoma spheroids or TGF-β1 receptor inhibition in heterotypic spheroids. | |
2.2 Cell culture and spheroid formation
A673 cells (ATCC, CRL-1598) were cultured in Dulbecco's modified Eagle medium (DMEM), high glucose (4.5 g L−1, with pyruvate; Gibco™, Cat# 11995065, Thermo Fisher Scientific, MA, USA) supplemented with 10% fetal bovine serum (FBS; Cat# A5256701, Thermo Fisher Scientific), 1% penicillin–streptomycin (P/S; Cat# 15140163, Thermo Fisher Scientific), 100 μM GlutaMAX™ L-glutamine supplement (Cat# 35050061, Thermo Fisher Scientific), and 1% sodium pyruvate (Thermo Fisher Scientific, Cat# 11360070). TC-71 cells (DSMZ, ACC-516) were cultured in IMDM (Iscove's modified Dulbecco's medium; Gibco™, Cat# 12440053, Thermo Fisher Scientific, MA, USA) supplemented with 15% FBS, 1% penicillin–streptomycin, and 100 μM GlutaMAX™. RH-1 cells (DSMZ, ACC-493) were maintained in RPMI supplemented with 10% FBS and 1% penicillin–streptomycin. To facilitate imaging-based analysis, the 3 Ewing sarcoma cell lines (A673, TC-71, and RH-1) were established to stably express GFP via lentiviral transduction. GFP was inserted into the pLV-EF1a-IRES-Blast vector (Addgene plasmid #85133), and lentivirus was produced and transduced in-house. Normal human lung fibroblasts (NHLFs, Cat# CC-2512, Lonza, MD, USA) were cultured in FGM™-2 BulletKit™ fibroblast growth medium (Cat# CC-3132, Lonza, MD, USA), according to the manufacturer's instructions. A549 cells (ATCC, CCL-185) and HeLa cells (ATCC, CCL-2) were cultured in DMEM high-glucose supplemented with 10% FBS and 1% P/S. All cells were incubated at 37 °C in a humidified atmosphere with 5% CO2. Subculturing was performed either when cells reached approximately 90% confluency or every 3 days, using 0.25% trypsin–EDTA (Gibco™, Cat# 25300054). To optimize seeding density, cells (1–5 × 105 cells per mL in 200 μL) were seeded into triplicate DCMiC wells pre-treated with 2% Pluronic® F-127 and cultured for 3 days. Of note, after 20 minutes of incubation to allow initial cell settling, the medium was gently aspirated from the edge of each well to remove excess cells remaining on the PDMS surface and replaced with a fresh medium, thereby promoting the formation of uniformly sized spheroids. On the first day, the area of the spheroid models was measured, and after 3 days of incubation, both the spheroid area and cell viability were assessed to determine the optimal seeding density. Detailed methods for the size measurement and area analysis of spheroids are provided in Fig. S2. For spheroid formation under the optimal conditions, cells were seeded at a concentration of 1 × 105 cells per mL onto the DCMiC platform. Heterotypic spheroids composed of Ewing sarcoma cells and fibroblasts were generated by diluting each cell type to 2 × 105 cells per mL in their respective culture media, mixing them at a 1:1 ratio, and seeding the mixture into the DCMiC platform.
2.3 Immunofluorescence staining
To assess cellular organization and cell–cell adhesion within spheroids, immunofluorescence staining for CD99 and E-cadherin was performed. Spheroids cultured in the DCMiC platform were gently washed twice with PBS, and fixed using 4% paraformaldehyde (Sigma-Aldrich, Cat# 158127, St. Louis, MO, USA) for 30 minutes at room temperature. Following fixation, spheroids were washed 3 times with PBS and permeabilized using 0.1% Triton X-100 (Sigma-Aldrich, Cat# T9284, St. Louis, MO, USA) in PBS for 15 minutes at room temperature. After permeabilization, samples were blocked with 5% bovine serum albumin (BSA; Sigma-Aldrich, Cat# A7906, St. Louis, MO, USA) in PBS for 1 hour at room temperature to prevent nonspecific antibody binding. Following blocking, spheroids were incubated at room temperature for 2 hours with a mixture of APC-conjugated anti-CD99 antibody (Cat# 371308, BioLegend, San Diego, CA, USA) and anti-E-cadherin primary antibody (rat monoclonal, Cat# 13-1900, Thermo Fisher Scientific, Waltham, MA, USA) diluted in PBS containing 1% BSA. After incubation, samples were washed 3 times with PBS (5 minutes each) and, for E-cadherin staining, subsequently incubated with Alexa Fluor 555-conjugated goat anti-rat IgG (Cat# A21434, Thermo Fisher Scientific, Waltham, MA, USA) at a 1:500 dilution in PBS containing 1% BSA for 1 hour at room temperature in the dark. To visualize nuclei, spheroids were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Thermo Fisher Scientific, Cat# D1306, Waltham, MA, USA) at a concentration of 1 μg mL−1 in PBS for 10 minutes at room temperature in the dark. After DAPI staining, spheroids were rinsed 3 times with PBS. Fluorescence imaging was performed using a Leica TCS-SP8-AOBS confocal inverted microscope (Leica Microsystems, Wetzlar, Germany), and images were processed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
2.4 Scanning electron microscopy
To prepare the spheroid samples for scanning electron microscopy (SEM), the culture medium was removed directly from the DCMiC platform containing the spheroids. The spheroids were then fixed with 4% formaldehyde solution for 1 hour at room temperature. After fixation, samples were washed 3 times with PBS, followed by post-fixation in 1% osmium tetroxide (Cat# 75632, Sigma-Aldrich, MO, USA) for 30 minutes at room temperature. A graded EtOH series (30%, 50%, 70%, 80%, 90%, and 100%; E7023, Sigma-Aldrich, MO, USA) was used for dehydration, with each step lasting 10 minutes and dehydrated spheroids were air-dried. Final drying was performed using 100% hexamethyldisilazane (HMDS; 440191, Sigma-Aldrich, MO, USA). The dried samples were coated with a 10 nm platinum layer and imaged using a Sigma VP scanning electron microscope (Carl Zeiss Inc., White Plains, NY, USA).
2.5 Reusability test
To evaluate the reusability of the DCMiC-based spheroid culture platform, plates previously used for cell culture were cleaned and sterilized following a standardized procedure. First, all residual culture media were carefully aspirated from each well. Then, 200 μL of 0.25% trypsin–EDTA solution (Gibco™, Cat# 25200056, Thermo Fisher Scientific, MA, USA) was added to each well and incubated at 37 °C for 5 minutes to detach any residual cells. The wells were pipetted to dislodge adherent cells, which were then removed by suction. Subsequently, each well was washed 3 times with PBS to eliminate remaining cell residues. For sterilization, the entire plate was exposed to UV light for 1 hour. After UV treatment, 70% EtOH was added to each well, incubated briefly, and removed. Wells were then rinsed 3 times with PBS. To re-establish a non-adhesive surface, 2% Pluronic® F-127 was added to each well and incubated for 1 hour at room temperature. As air bubbles had already been removed during the initial preparation, no additional vacuum treatment or cold incubation was required at this stage. Prior to reseeding, the Pluronic® F-127 solution was fully removed, and the wells were washed 3 times with a fresh cell culture medium.
2.6 Drug screening
Drug screening was performed using 11 compounds (Table 1) obtained from Cayman Chemical (MI, USA): RGFP966 (Cat# 16948), linsitinib (Cat# 11595), Torin 2 (Cat# 14796), palbociclib (Cat# 16276), talazoparib (Cat# 19155), BML281 (Cat# 16794), JQ1 (Cat# 11187), apitolisib (Cat# 16664), TK216 (Cat# 31138), olaparib (Cat# 10621), and trabectedin (Cat# 20417). Each drug was dissolved with dimethyl sulfoxide (DMSO; Cat# D2650, Sigma-Aldrich, MO, USA) and tested at 6 concentrations: 0 nM (control), 1 nM, 10 nM, 100 nM, 1 μM, and 10 μM. Working concentrations were prepared by serial dilution starting from a 10 mM stock solution to keep the final DMSO concentration below 0.1%. Each drug screening experiment was independently repeated 3 times to ensure reproducibility. For drug treatment, the culture medium in each well containing Ewing sarcoma spheroids was carefully aspirated and replaced with 200 μL of the corresponding drug solution at the designated concentration. Following drug treatment, the spheroids were maintained in the incubator for 48 hours to allow drug exposure under standard cell culture conditions (37 °C, 5% CO2). After the 48 hour incubation period, the media in each well was completely removed and replaced with fresh culture medium containing 1 μg mL−1 of propidium iodide (PI, Cat# P4170, Sigma-Aldrich, MO, USA). The spheroids were then incubated for an additional 30 minutes under standard cell culture conditions to allow for staining of non-viable cells. After the incubation, PI solution was completely replaced with a fresh culture medium to prevent additional toxic response. The double-cylinder architecture design minimizes spheroid displacement during medium exchange, thereby enhancing reproducibility and reducing experimental variability across repeated treatments. Fluorescence and brightfield imaging were performed using a motorized inverted microscope (Nikon Eclipse Ti2, Nikon Instruments Inc., Tokyo, Japan), and images were processed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Table 1 Drug information
| Drug name |
Primary target(s) |
Mechanism of action |
Ref. |
| RGFP966 |
HDAC3 |
Selective inhibitor of HDAC3; modulates gene expression through histone deacetylation |
46
|
| Linsitinib |
IGF-1R, insulin receptor (IR) |
Dual inhibitor of IGF-1R and IR; blocks signaling pathways involved in cell growth and survival |
48
|
| Torin 2 |
mTOR, ATM, ATR, DNA-PK |
Potent inhibitor of mTOR with additional activity against DNA damage response kinases |
43
|
| Palbociclib |
CDK4, CDK6 |
Inhibits CDK4/6, leading to G1 cell cycle arrest and inhibition of proliferation |
50
|
| Talazoparib |
PARP1, PARP2 |
PARP inhibitor; prevents DNA repair, especially effective in BRCA-mutated cancers |
51
|
| BML281 |
HDAC6 |
Selective HDAC6 inhibitor; affects protein acetylation and cell motility |
56
|
| JQ1 |
BRD4 (BET family) |
Inhibits bromodomain and extraterminal domain (BET) proteins; suppresses oncogene expression |
53
|
| Apitolisib |
PI3K, mTOR |
Dual PI3K/mTOR inhibitor; disrupts PI3K–AKT–mTOR signaling involved in tumor growth |
49
|
| TK216 |
EWS-FLI1 fusion protein |
Specifically targets the EWS-FLI1 oncoprotein, found in Ewing sarcoma |
57
|
| Olaparib |
PARP1, PARP2 |
PARP inhibitor that exploits synthetic lethality in BRCA-deficient cells |
52
|
| Trabectedin |
DNA minor groove |
Alkylating agent that binds to the minor groove of DNA, interfering with transcription and repair |
54
|
2.7 Analysis of remission and viability
Prior to drug treatment, each spheroid was imaged using both bright-field and GFP fluorescence channels to establish the baseline tumor size. Following 48 hours of drug exposure, the extent of tumor remission was quantified based on changes in GFP-expressing area, calculated using the following eqn (1):| | 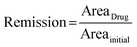 | (1) |
Here, AreaDrug represents the GFP fluorescence area measured after 48 hours of drug treatment and Areainitial corresponds to the GFP fluorescence area prior to drug treatment.
Cell viability was determined based on the relative areas of live and dead cells using the following eqn (2):
| |  | (2) |
Here, Area
Live was calculated from the GFP fluorescence area (green fluorescence) corresponding to viable cells, whereas Area
Dead was calculated from the PI fluorescence area (red fluorescence) representing dead cells.
2.8 RNA-seq and data analysis
Heterotypic or monoculture spheroids were digested into single cells using trypsin. GFP positive Ewing sarcoma cells were sorted using BD Symphony S6 (CWRU Cytometry & Imaging Microscopy Core), and subjected to total RNA extraction using TRIzol (Cat# 15596026, Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's instructions. 250 ng of total RNA was used to construct an RNA-seq library using a NEBNext UltraExpress RNA Library Prep Kit (Cat# E3330, New England Biolabs, Ipswich, MA, USA) following manufacturer's instructions. Briefly, RNA with polyA was enriched using a NEBNext Poly(A) mRNA magnetic isolation module (Cat# E7490, New England Biolabs, Ipswich, MA, USA) and enzymatically fragmented. cDNA was synthesized followed by end repair, adapter ligation, and PCR enrichment with indexed primers. The library was paired end sequenced for 76 bp using an Illumina NovaSeq platform. Sequencing reads were mapped to reference genome hg38 using RNA STAR with default parameters.29 Read count tables were created using FeatureCounts30 with a custom GTF file containing protein coding genes only. Differentially expressed genes were analyzed using DESeq2 (ref. 31) with 2 independent replicates using default parameters. Transcripts per million (TPM) values were calculated using Kallisto (version: 0.44.0).32 All the datasets generated in this study have been deposited in Gene Expression Omnibus (GEO) under accession number GSE303862.
2.9 TGF-β1 and vactosertib treatment
Following the protocol of spheroid formation described in Methods 2.2, the freshly seeded cells were incubated for 20 minutes to allow initial cell settling. The medium was then carefully aspirated from the edge of each well and replaced with a fresh medium containing either 10 ng mL−1 TGF-β1 (Fisher Scientific, Cat# 240B002, MA, USA) or 1 μM TGF-β1 receptor inhibitor (Vactosertib, MedChemExpress, Cat# HY-19928, NJ, USA). Spheroids were allowed to form for 24 hours under standard culture conditions. Following the protocol of drug testing in Methods 2.6, the medium was then replaced with a fresh medium containing the top 3 drugs together with TGF-β1 or vactosertib. After a 48 hour incubation period, cell viability was assessed with a GFP signal and PI staining. Following staining, spheroids were imaged and images were processed using ImageJ software to quantify the remission and viability of spheroids after drug treatment following the protocol described in Methods 2.7.
2.10 Statistical analysis
Half-maximal inhibitory concentration (IC50) values for each compound were determined using dose-response data obtained from the drug screening assays (Fig. 4d). The normalized viability data were fitted to a nonlinear regression model using a standard 4-parameter logistic (4PL) curve in OriginPro 2024 (OriginLab Corporation, Northampton, MA, USA). The fitted curves were used to interpolate the IC50 values, defined as the drug concentration that reduces cell viability by 50% relative to the untreated control. All curve fitting and IC50 determinations were performed with default convergence criteria and weighting options provided in OriginPro. All quantitative data are expressed as mean ± standard error of the mean. Statistical comparisons between two independent groups, including Ewing sarcoma-only spheroids and Ewing sarcoma-fibroblast heterotypic spheroids, were performed using unpaired two-tailed Student's t-tests. All statistical analyses and graph generation were performed using OriginPro 2024.
3. Results
3.1 Overview of the study
Fig. 1 provides an overview of the workflow for developing and applying our DCMiC platform. The master mold was fabricated using cost-effective SLA-based 3D printing and the resulting PDMS inserts integrated into standard 96-well plates, enabling scalable spheroid formation (Fig. 1a). Ewing sarcoma cells, alone or co-cultured with fibroblasts, were seeded into the DCMiC wells, where stable and uniform spheroids were formed within 24 hours, supported by the double-cylinder design that minimizes shear-induced loss of spheroids from the culture platform during media exchange (Fig. 1b). To further improve the stability of spheroids during media exchange, we compared single- and double-cylinder microchamber geometries. As illustrated in Fig. 1c, the single-cylinder design often caused spheroid displacement and sample loss due to shear flow generated during washing, whereas the double-cylinder structure effectively prevented such disturbances by positioning spheroids deeper within the chamber. Quantitative analysis confirmed that nearly all spheroids were retained after washing in the double-cylinder platform (100%), compared with only 21.8 ± 2.6% remaining in the single-cylinder wells (Fig. 1d), demonstrating the superior stability of our DCMiC design. While the double-cylinder configuration could theoretically attenuate convective flow, our experimental assessment revealed that this geometric constraint did not exert a measurable effect on nutrient diffusion or drug penetration within the chamber (Fig. S3). These spheroids were subsequently subjected to a panel of mechanistically diverse small-molecule inhibitors for therapeutic screening (Fig. 1e). Finally, comparative transcriptomic profiling between monoculture and heterotypic spheroids revealed fibroblast-driven pathways such as TGF-β1 and NFκB underlying drug resistance, underscoring stromal niche co-targeting as a potential therapeutic strategy (Fig. 1f).
3.2 Design and fabrication of double-cylinder micro-chamber (DCMiC)
To fabricate micro-chamber structures for Ewing sarcoma spheroid formation, we employed SLA-based 3D printing, which provides sufficient precision for rapid and cost-effective prototyping.33,34 Conventional single-cylinder micro-chambers are widely used to form spheroids, yet they frequently suffer from spheroid loss during media exchange, limiting their reliability for drug testing. To address this limitation, we developed a double-cylinder micro-chamber (DCMiC) design with tailored dimensions (top cylinder: 300 μm diameter × 300 μm depth; bottom cylinder: 500 μm diameter × 300 μm depth; bottom gap: 300 μm) (Fig. S1a). This geometry shields them from flow-induced disturbances during media changes and minimizes interactions between neighboring spheroids. The protective effect of the double-cylinder structure was first confirmed by in silico simulation (Fig. S4). Beyond improving stability, the DCMiC design offers convenient handling during culture and screening. Moreover, the use of SLA 3D printing enables straightforward customization of chamber dimensions and provides a substantially more cost-effective fabrication route compared with traditional photolithography (Table S2).
3.3 Robust generation of uniform Ewing sarcoma spheroids using the reusable DCMiC platform
To establish a robust and reproducible platform for drug screening, we first optimized spheroid formation by evaluating the effect of initial cell seeding density. Ewing sarcoma cells (TC-71) seeded at 5 different densities (1–5 × 105 cells per mL) formed spheroids with diameters that increased proportionally with the initial cell number (Fig. 2a and b). Live/dead staining revealed that higher seeding densities led to a greater proportion of non-viable cells by day 3, whereas spheroids seeded at 1 × 105 cells per mL maintained uniform size, compact morphology, and sustained proliferation without early necrosis (Fig. 2c). Growth curves further confirmed that spheroids at this density exhibited continuous expansion without an early compacting phase (Fig. 2d), while viability assays showed reduced survival at higher densities across all 3 Ewing sarcoma models (Fig. 2e). From the perspective of spheroid size increase, at a cell concentration of 1 × 105 cells per mL, the diameters of A673, TC-71, and RH-1 spheroids on day 3 were 1.66-, 1.94-, and 1.75-fold larger, respectively, compared to those on day 1. This concentration exhibited the greatest degree of spheroid growth among all tested conditions. Notably, a greater extent of size increase is expected to enhance the detectability and dynamic range of drug-induced responses. Additionally, at a cell concentration of 1 × 105 cells per mL on day 3 of culture, the viabilities of A673, TC-71, and RH-1 spheroids were 98.20 ± 0.23%, 99.11 ± 0.09%, and 99.51 ± 0.09%, respectively. In contrast, at a higher concentration of 5 × 105 cells per mL, the viabilities decreased to 80.16 ± 1.03%, 84.97 ± 0.74%, and 97.03 ± 0.32%, respectively. Based on these findings, 1 × 105 cells per mL was selected as the optimal condition, providing compact spheroids with long-term viability and allowing drug treatment to begin as early as 24 hours post-seeding without misinterpreting natural size reduction due to compacting as a treatment effect.17,35,36
 |
| | Fig. 2 Design and characterization of a reusable DCMiC array-based spheroid culture platform. (a) Representative bright-field images of Ewing sarcoma spheroids generated at different initial cell seeding densities (1–5 × 105 cells per mL) on day 1 and day 2 of culture. Representative images are from 3 independent experiments (n = 3) with similar results. (b) Quantification of spheroid diameters at day 1 (orange) and day 2 (green) across the indicated seeding densities. Data are presented as box-and-whisker plots (min to max) with median values. Data were collected from 3 independent experiments (n = 3) with 24 images for day 1 and 15 images for day 2, respectively. (c) Viability analysis of spheroids formed at varying cell densities. Live (GFP signal, green) and dead (PI stained, red) cells show an increased dead cell population at higher seeding densities. Representative images are from 3 independent experiments (n = 3) with 10 total images analyzed. (d) Growth patterns of Ewing sarcoma cell lines (A673, TC-71, and RH-1) under varying seeding densities. Despite differences in initial cell numbers, no significant compacting of spheroids was observed across all cell lines. Data were collected from 3 independent experiments (n = 3) with 45 total images analyzed. (e) Assessment of cell viability in relation to seeding density. Live/dead staining and representative fluorescence images show a progressive increase in dead cells with higher seeding densities on day 3 in all 3 Ewing sarcoma models. Data were collected from 3 independent experiments (n = 3) with 45 total images analyzed. (f) Immunofluorescence staining of spheroids cultured in the DCMiC array for biological validation. The spheroids exhibited positive expression of CD99, a known Ewing sarcoma-associated marker. Representative images are from 3 independent experiments (n = 3) with similar results. (g) Scanning electron microscopy (SEM) images of Ewing sarcoma spheroids cultured in the DCMiC array, demonstrating the morphological characteristics and surface texture of the 3D Ewing sarcoma spheroids. (h) Evaluation of device reusability. Brightfield images of spheroids generated through repeated use of the same DCMiC platform confirm consistent spheroid morphology and integrity across multiple cycles. Data were collected from 5 independent experiments (n = 5) with 75 total images analyzed. | |
We next assessed the biological features of Ewing sarcoma spheroids generated using our platform, with TC-71 as a representative model (Fig. 2f). DAPI staining and constitutive GFP expression confirmed even cell distribution across the spheroid. E-cadherin, typically expressed in epithelial cells, was undetectable, which is consistent with the mesenchymal origin of Ewing sarcoma.37,38 In contrast, CD99, a highly expressed and diagnostically relevant surface marker for Ewing sarcoma, was robustly and uniformly expressed across the spheroid, confirming preservation of lineage identity.39 Merged and Z-stack projections revealed uniform organization and compact 3D architecture. In parallel, scanning electron microscopy (SEM) imaging provided high-resolution visualization of spheroids within the micro-chambers, revealing smooth outer contours, with cells closely attached to one another (Fig. 2g). These features collectively ensure that the spheroids are robust and reproducible, making them suitable for downstream drug testing. Furthermore, the platform demonstrated similar robustness with non-Ewing sarcoma cell lines such as A549 (lung cancer) and HeLa (cervical cancer), which consistently formed uniform and growing spheroids (Fig. S5), underscoring the versatility of the platform beyond Ewing sarcoma models. Lastly, we evaluated the reusability of the DCMiC platform. Through multiple wash-and-reseed cycles (see Methods 2.5), spheroid formation remained consistent across 5 consecutive uses, with minimal variation in diameter, confirming the structural reliability of the platform for repeated applications (Fig. 2h and S6). These results indicate that the DCMiC platform can reliably produce spheroids of consistent quality over multiple uses, thereby reducing overall experimental costs and improving operational efficiency.
3.4 High-throughput drug screening in Ewing sarcoma spheroids
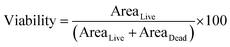
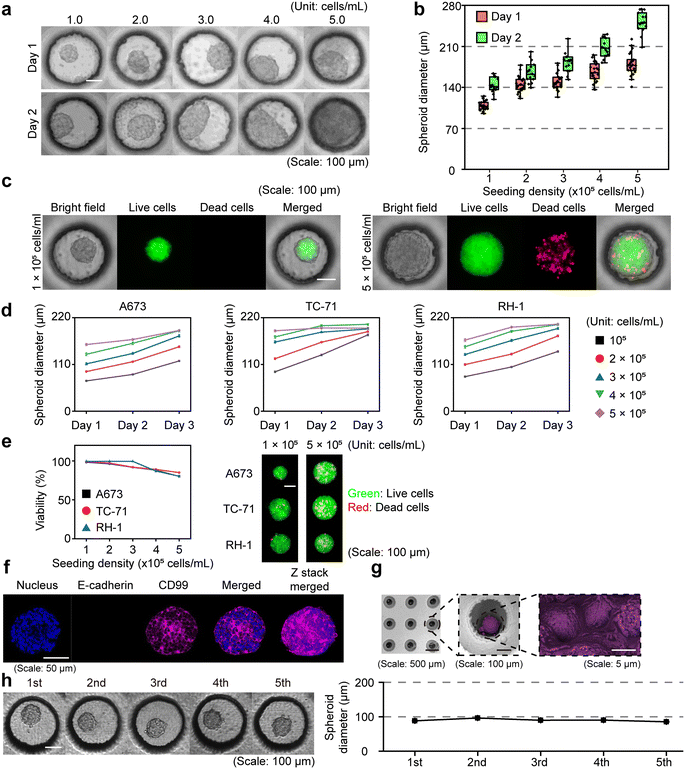
To assess the therapeutic sensitivity in 3D Ewing sarcoma spheroids, we conducted a drug screening assay using 11 small-molecule compounds (Table 1), selected for their relevance to Ewing sarcoma pathobiology, including epigenetic regulation, growth factor signaling, cell cycle control, DNA repair, and metabolic pathways.40–45 Histone deacetylase (HDAC) inhibitors (RGFP966 (ref. 46) and BML281 (ref. 47)) were included due to the prominent role of epigenetic dysregulation in Ewing sarcomas. The insulin-like growth factor 1 receptor (IGF-1R) and mammalian target of rapamycin (mTOR) pathway inhibitors (linsitinib,48 Torin 2,43 and apitolisib49) were selected based on the frequent activation of PI3K/AKT/mTOR and IGF signaling cascades in Ewing sarcoma progression and therapy resistance. Cyclin-dependent kinase 4 and 6 (CDK4/6) inhibition (palbociclib50) reflects the high prevalence of retinoblastoma (RB) pathway alterations in certain Ewing sarcoma subtypes. Poly (ADP-ribose) polymerase (PARP) inhibitors (talazoparib51 and olaparib52) address DNA repair vulnerabilities, particularly in tumors with BRCA-like deficiencies. Bromodomain and extra-terminal domain (BET) inhibition (JQ1 (ref. 53)) targets transcriptional regulators involved in Ewing sarcoma oncogenesis. Trabectedin,54 a clinically approved DNA-binding agent, was included as a benchmark with proven efficacy in soft tissue Ewing sarcomas. Finally, TK216 specifically targets EWS-FLI1 fusion, making it highly relevant for Ewing sarcoma. Collectively, this drug panel enables a systematic evaluation of Ewing sarcoma cell responses across a diverse range of mechanistic targets and supports the identification of therapeutic vulnerabilities relevant to Ewing sarcoma as shown in Fig. 3a. The screening was carried out using the DCMiC platform formatted into a standard 96-well plate, as described in Fig. 3b. Briefly, Ewing sarcoma cell lines were seeded at an optimal density of 1 × 105 cells per mL and treated with the drug at concentrations of 0 (control), 1 nM, 10 nM, 100 nM, 1 μM, and 10 μM (see Methods 2.6). After 48 hours of incubation, viability was assessed using GFP and PI based analysis, and remission was defined by the decrease in spheroid size (see Methods 2.7). This dual-analysis approach was adopted because many compounds reduce both cell survival and proliferation, resulting in decreased spheroid viability and size.55 As a result, A673 spheroids treated with the mTOR inhibitor Torin 2 at 1 μM showed marked decreases in both viability (based on GFP/PI analysis) and spheroid size (based on the area of GFP) (Fig. 3c). Among the panel of 11 compounds, Torin 2, talazoparib, and trabectedin showed significant efficacy at relatively low concentrations, consistently reducing spheroid size and viability across 3 Ewing sarcoma cell lines (Fig. 3d). For example, upon treatment with trabectedin (100 nM), cell viability was markedly reduced, with A673, TC-71, and RH-1 spheroids showing 58.03%, 68.21%, and 62.85% decreases, respectively, compared with the control group. Correspondingly, the spheroid diameters were reduced by 76.86%, 57.96%, and 35.45%, respectively.
 |
| | Fig. 3 High-throughput drug screening of Ewing sarcoma spheroids using the DCMiC platform. (a) Schematic representation of the 11 small-molecule compounds tested, categorized by their primary mechanisms of action, including epigenetic modulators (RGFP966, BML281, and JQ1), growth factor pathway inhibitors (linsitinib, apitolisib, and TK216), DNA damage repair pathway inhibitors (talazoparib, olaparib, and Torin 2), a cell cycle inhibitor (palbociclib), and a DNA-binding agent (trabectedin). (b) Experimental timeline for drug screening. Spheroids were seeded on day 0, drugs were administered on day 1, and live/dead assays and imaging were performed after 48 hours of drug treatment (day 3). (c) Representative images of A673 spheroids cultured under control (DMSO) conditions or treated with 1 μM Torin 2 for 48 hours, shown in bright-field, GFP (live), PI (dead), and merged fluorescence channels. (d) Heatmaps showing dose-dependent effects of the 11 compounds on viability (top) and remission (bottom), across 3 Ewing sarcoma cell lines (A673, TC-71, and RH-1). To ensure consistency, we used uniformly sized spheroids generated within a defined diameter range, and all quantifications were performed on the same spheroids before and after drug treatment. Drug concentrations range from control (C, 0 nM) to 10 μM (log10 scale, 0–4). Viability is expressed as a percentage, and remission is expressed as normalized size change (arbitrary units, AU). Torin 2, talazoparib, and trabectedin exhibited the most pronounced effects on both viability and remission across all cell lines. Data were collected from 3 independent experiments (n = 3) with 40 total images analyzed. | |
3.5 Therapeutic evaluation in tumor-fibroblast heterotypic spheroids
Ewing sarcoma frequently metastasizes to the lungs, where interactions between tumor cells and resident stromal fibroblasts can significantly influence disease progression and treatment outcomes.58 To model this metastatic niche, we incorporated human lung fibroblasts into our spheroid platform, enabling evaluation of how stromal components modulate drug sensitivity. We focused on the top 3 compounds (Torin 2, talazoparib, and trabectedin) from our monoculture drug screening, each of which exhibited potent anti-tumor activity across Ewing sarcoma cell lines. We first confirmed that fibroblast alone can form spheroids, and treatment of the selected drugs has no significant effect on fibroblast spheroids (Fig. S7). Next, we established the heterotypic co-culture (Fig. 4a) and treated each compound at its IC50 concentration (Fig. 4b), as determined from the initial screening (Fig. 3). We found that heterotypic spheroids exhibited significantly higher viability following treatment compared to monoculture counterparts across all 3 Ewing sarcoma cell lines, indicating robust and consistent fibroblast-induced drug resistance (Fig. 4c). In the heterotypic model, A673 cells exhibited higher viability than their monoculture counterparts following treatment with Torin 2, talazoparib, and trabectedin, showing average increases of 18.18%, 10.56%, and 19.52%, respectively. Similarly, the heterotypic TC-71 model showed 31.59%, 26.21%, and 31.65% higher viability compared with the monoculture under the same treatments. For RH-1, the heterotypic spheroids also demonstrated 30.13%, 25.77%, and 22.61% greater viability, respectively. These findings are consistent with the well-established role of cancer-associated fibroblasts in promoting therapy resistance and demonstrate the utility of our DCMiC platform for modeling stromal influences on drug response in a scalable and physiologically relevant format.
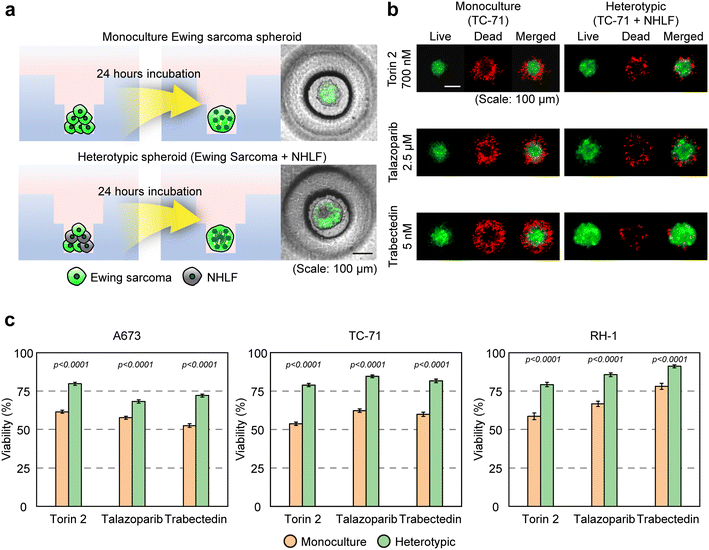 |
| | Fig. 4 Evaluation of the top 3 compounds in Ewing sarcoma–fibroblast heterotypic spheroids. (a) Generation of heterotypic Ewing sarcoma spheroids by co-culturing GFP-labeled Ewing sarcoma cells with unlabeled fibroblasts in the DCMiC platform. Non-fluorescent zones represent fibroblast-rich regions after 24 hours of incubation. (b) Representative fluorescence images of monoculture (TC-71) and heterotypic spheroids (TC-71 + fibroblast) are shown following treatment with each drug (Torin 2, talazoparib, and trabectedin) at IC50 concentrations, which were determined from the dose-response curves generated using the drug screening data presented in Fig. 3d. Green: live cells (GFP); red: dead cells (PI staining). (c) Quantification of spheroid viability (%) for monoculture and heterotypic spheroids under IC50 treatment. (IC50 for A673, Torin 2: 72.9 nM, talazoparib: 46.9 nM, trabectedin: 0.7 nM; IC50 for TC-71, Torin 2: 700 nM, talazoparib: 2.5 μM, trabectedin: 5 nM; IC50 for RH-1, Torin 2: 200 nM, talazoparib: 3 μM, trabectedin: 1 nM). Data were collected from 3 independent experiments (n = 3) with 45 total images analyzed. Significance was assessed by two-tailed unpaired Student's t-test (detail statistical results in Table S3). | |
3.6 Dissecting the chemoresistance mechanisms contributed by fibroblasts
To dissect how fibroblasts remodel Ewing sarcoma cells to confer therapy resistance, we performed transcriptomic profiling of tumor cells isolated from either monoculture or heterotypic spheroids co-cultured with fibroblasts. Across the 3 independent Ewing sarcoma cell lines, we identified a conserved set of differentially expressed genes induced by co-culture with fibroblasts (Fig. 5a). To investigate the upstream regulators of these gene expression changes, we performed transcription factor enrichment analysis using TRRUST59 (transcriptional regulatory relationships unraveled by sentence-based text mining). Notably, genes upregulated in Ewing sarcoma cells by the presence of fibroblasts were significantly enriched for targets of NFκB family transcription factors, suggesting activation of NFκB signaling by the heterotypic co-culture (Fig. 5b). We also observed enrichment for SMAD3 targets, implicating the activation of the transforming growth factor-beta (TGF-β1) pathway. These findings were corroborated by gene set enrichment analysis (GSEA), which revealed strong correlations between fibroblast-induced gene expression changes and canonical NFκB and TGF-β1 signaling pathways (Fig. 5c). Functionally, fibroblast co-culture led to the upregulation of classical pro-survival genes and the repression of tumor-suppressive or pro-differentiation genes in Ewing sarcoma (Fig. 5d). These results suggest that fibroblasts remodel the tumor transcriptome by activating stress response and immune signaling pathways, particularly NFκB and TGF-β1, thereby promoting a transcriptional state associated with reduced drug sensitivity and therapeutic resistance (Fig. 5e). In addition, gene ontology (GO) enrichment analysis revealed that genes upregulated in tumor cells from the heterotypic spheroids were enriched in processes related to tumor adaptation and resistance, including response to nutrient levels, blood vessel development, and cytokine signaling. Conversely, downregulated genes were enriched in pathways such as amino acid catabolism and modulation of synaptic signaling, indicating a metabolic reprogramming consistent with fibroblast-driven resistance mechanisms (Fig. 5f). These transcriptional changes indicate that fibroblasts shift Ewing sarcoma cells toward a state characterized by enhanced nutrient sensing, angiogenesis, cytokine signaling, and reduced metabolic and differentiation programs. Alongside the observed NFκB and TGF-β1 pathway activation, these findings indicate that fibroblasts extensively reprogram the tumor transcriptome by upregulating pro-survival and stress-responsive pathways and suppressing tumor-suppressive programs, changes that are consistent with reduced drug sensitivity in heterotypic spheroids.
 |
| | Fig. 5 Transcriptomic profiling reveals fibroblast-induced transcriptional remodeling and activation of pro-survival pathways in Ewing sarcoma heterotypic spheroids. (a) Heatmaps of differentially expressed genes across 3 Ewing sarcoma cell lines (A673, TC-71, and RH-1) cocultured with fibroblasts compared to those of monoculture spheroids. Normalized gene expression (TPM, z-score) of upregulated (red) and downregulated (blue) gene clusters identified by DESeq2 analysis (log2 fold change >0.5 or <−0.5, heterotypic vs. monoculture conditions). (b) Transcription factor enrichment analysis (TRRUST) for genes upregulated in heterotypic spheroids, showing significant enrichment of NFκB family members (NFKB1, RELA) and SMAD3 targets. Bubble size represents the percentage of input genes regulated by each transcription factor, and color indicates −log10(p-value). (c) Transcription factor enrichment for downregulated genes, highlighting GATA3 as a key upstream regulator. (d) Gene set enrichment analysis (GSEA) demonstrating significant upregulation of canonical NFκB and TGFβ/SMAD pathway target genes in heterotypic spheroids relative to monocultures. (e) Heatmaps of selected pro-survival genes (e.g., BCL3, STAT3, and JUN) and tumor-suppressive or pro-differentiation genes (e.g., FOXP3, CBX7, and GATA3), showing fibroblast-induced upregulation or repression, respectively. (f) Gene ontology (GO) enrichment of fibroblast-induced gene expression changes is shown; upregulated genes are associated with nutrient response, blood vessel development, and cytokine-mediated signaling, whereas downregulated genes are linked to fear response, amino acid metabolism, and synaptic transmission. | |
3.7 Evaluating the impact of TGF-β1 modulation on drug response in monoculture and heterotypic spheroids
We observed strong activation of pro-survival pathways, including NFκB and TGF-β1 signaling, in heterotypic spheroids.60 Given that cancer-associated fibroblasts (CAFs) are known to secrete TGF-β1,25,61 we hypothesized that fibroblast-derived TGF-β1 contributes to the observed drug resistance (Fig. 6a). To test whether TGF-β1 activation is sufficient to induce resistance, we treated Ewing sarcoma monoculture spheroids with the recombinant TGF-β1 ligand prior to drug exposure (see Methods 2.9). Across all 3 Ewing sarcoma cell lines (A673, TC-71 and RH-1), TGF-β1 treatment led to significantly increased viability following Torin 2, talazoparib, or trabectedin exposure, indicating that activation of this pathway alone can blunt therapeutic efficacy (Fig. 6b). In A673 spheroids treated with TGF-β1, cell viability increased by an average of 11.67%, 7.35%, and 8.63% following treatment with Torin 2, talazoparib, and trabectedin, respectively, compared with the monoculture without TGF-β1 treatment. Likewise, the TGF-β1-treated TC-71 spheroids exhibited 6.37%, 9.09%, and 13.62% higher viability than the corresponding monoculture controls. In RH-1 spheroids, TGF-β1 supplementation resulted in 14.05%, 9.53%, and 10.65% increases in viability, respectively. We next asked whether TGF-β1 signaling is required for fibroblast-mediated resistance in sarcoma cells. Strikingly, pharmacologic inhibition of the TGF-β1 receptor in heterotypic spheroids restored drug sensitivity across all 3 models, resulting in markedly decreased viability after drug treatment (Fig. 6c). In the heterotypic A673 spheroids treated with the TGF-β1 receptor inhibitor, cell viability decreased by an average of 12.19%, 9.18%, and 11.99% upon treatment with Torin 2, talazoparib, and trabectedin, respectively, compared with that of the heterotypic control. Similarly, in TC-71 spheroids, the TGF-β1 receptor inhibition group exhibited 9.45%, 10.85%, and 15.85% reductions in viability relative to the heterotypic condition. For the heterotypic RH-1 spheroids, the cell viability decreased by 8.45%, 9.54%, and 14.91%, respectively, upon inhibition of the TGF-β1 receptor. These findings establish TGF-β1 as a functional driver of fibroblast-induced chemoresistance in Ewing sarcoma and nominate TGF-β1 blockade as a promising strategy to overcome stromal protection. More broadly, they highlight the utility of our DCMiC platform for uncovering microenvironmental mechanisms of resistance and enabling rational therapeutic interventions.
 |
| | Fig. 6 Modulation of TGF-β1 signaling alters drug response in monoculture and heterotypic Ewing sarcoma spheroids. (a) Schematic illustration of TGF-β1 paracrine signaling from fibroblasts to Ewing sarcoma. Fibroblast-derived TGF-β1 was hypothesized to act on neighboring Ewing sarcoma cells, leading to increased proliferation, survival, and drug resistance. (b) Viability analysis of monoculture spheroids showing that exogenous TGF-β1 supplementation reduces drug sensitivity across all 3 Ewing sarcoma cell lines. Data were collected from 3 independent experiments (n = 3) with 45 total images analyzed. Statistical significance was determined by two-tailed unpaired Student's t-test (detailed results in Table S4). (c) Viability analysis of heterotypic spheroids showing that pharmacologic inhibition of TGF-β1 signaling partially restored drug sensitivity compared to untreated heterotypic controls. Data were collected from 3 independent experiments (n = 3) with 45 total images analyzed. Statistical significance was determined by two-tailed unpaired Student's t-test (detailed results in Table S4). | |
4. Discussion
Recent studies have increasingly indicated a role for TGF-β1 signaling in Ewing sarcoma progression and therapy resistance.62,63 For instance, TGF-β1 activation has been associated with metastasis, immune exclusion, and poor patient outcomes in clinical cohorts, and inhibition of TGF-β1 signaling during radiotherapy was shown to enhance immune infiltration and reduce metastasis.62 However, most of these studies rely on subcutaneous xenografts, irradiated tumors, or indirect transcriptomic inferences from patient biopsies. Unlike many adult carcinomas, Ewing sarcoma lacks a genetically engineered mouse model (GEMM) due to its fusion-driven biology and developmental origins, fundamentally limiting causal dissection of stromal contributions to resistance in a human, tumor-intrinsic context. Our study addresses this gap by developing a scalable, low-cost, and highly controlled co-culture system that enables parallelized testing of drug response and signaling modulation within defined stromal environments. Using the double-cylinder micro-chamber (DCMiC) platform, we demonstrate that fibroblast-derived TGF-β1 blunts drug efficacy, and that pharmacologic inhibition of this pathway restores sensitivity, providing mechanistic evidence of paracrine-mediated resistance in a physiologically relevant and experimentally tractable setting. The ability to perform such high-content mechanistic studies at <$1 per well in a standard 96-well format enables scalable exploration of resistance pathways that would be prohibitively expensive or infeasible in animal models. Beyond serving as a tool for robust spheroid formation, the DCMiC platform offers several methodological advantages that extend its utility. The defined double-cylinder geometry ensures positional stability during sequential manipulations (e.g., drug treatment, washout, genetic perturbation, or fluorescence labeling), supporting dynamic tracking of signaling responses without sample loss. The matrix-free design allows efficient recovery of viable tumor cells, making the platform compatible with downstream analyses such as RNA-seq, flow cytometry, or proteomics, which are often challenging in matrix-embedded systems. Furthermore, reusability and compatibility with automated imaging significantly reduce cost and inter-experimental variability, making the system ideal for both academic and translational screening applications. Mechanistically, our findings align with the established role of cancer-associated fibroblasts in driving resistance via NFκB and TGF-β1 signaling,25,26,64 and extend this paradigm into a fusion-driven pediatric sarcoma context. Notably, our transcriptomic analysis revealed coordinated activation of NFκB and SMAD3 target genes in fibroblast-exposed tumor cells, confirming conserved pro-survival signaling programs across multiple Ewing sarcoma lines. While NFκB was not functionally tested here, our previous work in leukemia65 demonstrated that co-targeting NFκB enhances chemotherapeutic response, suggesting potential synergy with TGF-β1 pathway inhibition in future combinatorial regimens.
Looking forward, the unique DCMiC design is particularly helpful in addressing technical limitations seen in conventional spheroid/organoid culture systems, including loss of samples during manipulation or inconsistent spheroid positioning. Although the application of spheroids to uncover new biological insights is beyond the scope of the present study, the platform provides a strong foundation for future work. We are now planning to introduce immune cells into spheroids, along with vasculature components, to evaluate their impact on immunotherapeutics such as CAR T-cell therapy in Ewing sarcoma. Moreover, the DCMiC system can be further extended to integrate other stromal and immune subsets, such as myeloid-derived suppressor cells or T cells, to model immune-tumor-stroma crosstalk under defined conditions. Incorporating patient-derived xenograft (PDX)-derived cells or primary sarcoma specimens would enable personalized profiling of resistance mechanisms. Collectively, these future directions position the DCMiC as a versatile preclinical platform for both mechanistic dissection and therapeutic discovery, particularly valuable in cancer types such as Ewing sarcoma where standard 3D and animal models remain limited.
5. Conclusion
We developed a scalable and compatible 3D screening platform, which enables robust spheroid formation, long-term culture, and high-throughput drug testing in Ewing sarcoma. Incorporating lung fibroblasts into the system revealed consistent stromal-induced drug resistance, driven in part by activation of TGF-β1 signaling. Functional validation confirmed TGF-β1 as a key mediator, and its inhibition restored drug sensitivity in co-culture. These findings highlight the DCMiC platform not only as a practical tool for scalable drug testing, but also as a powerful system for exploring microenvironment-driven mechanisms of resistance and facilitating the identification of potential therapeutic targets. With its broad adaptability across tumor types, the DCMiC platform offers a transformative approach for accelerating the discovery of more effective, context-informed cancer therapies.
Author contributions
J. H. L. and M. Ye contributed equally to this work. J. H. L., and M. Ye conducted the experiment and collected the data. J. H. L. drafted the manuscript. C. M. conceptualized the project. Y. G. and C. M. revised the manuscript and supervised the work. All authors commented on the manuscript.
Conflicts of interest
The authors declare no competing financial interest.
Data availability
The data supporting this article, including reagents and relevant supplementary data, have been included as part of the supplementary information (SI). All the datasets generated in this study have been deposited in Gene Expression Omnibus (GEO) under accession number GSE303862.
Supplementary information is available. See DOI: https://doi.org/10.1039/d5lc00856e.
Acknowledgements
C. M. is supported by start-up funds from the Cleveland Clinic Research, Grant#22-148-27IRG to the Case Comprehensive Cancer Center from the American Cancer Society, and Cleveland Clinic Catalyst Grant. M. Ybarra is supported by the VeloSano Graduate Student Grant. Y. G. is supported by start-up funds from the Case Western Reserve University School of Medicine and a grant from the NCI (R00 CA273523). The funders had no role in the design, data collection, data analysis, or reporting of this study. We thank Madelyn Dirrim for editing our manuscript. We also acknowledge the assistance of the Cleveland Clinic Imaging Core for their support with confocal microscopy and scanning electron microscopy.
References
- B. A. Nacev, F. Sanchez-Vega, S. A. Smith, C. R. Antonescu, E. Rosenbaum, H. Shi, C. Tang, N. D. Socci, S. Rana and R. Gularte-Merida, Nat. Commun., 2022, 13, 3405 CrossRef CAS
 .
.
- L. De Cock, I. Palubeckaite, F. Bersani, T. Faehling, S. Pasquali, S. Umbaugh, M. T. Meister, M. R. Danks, P. Manasterski, R. Miallot, M. Krumbholz, S. Roohani, D. Heymann, F. Cidre-Aranaz, A. Wozniak, P. Schoffski, J. Bovee, A. Merlini and S. Venneker, Neoplasia, 2025, 65, 101171 CrossRef CAS .
- T. G. Grünewald, F. Cidre-Aranaz, D. Surdez, E. M. Tomazou, E. de Álava, H. Kovar, P. H. Sorensen, O. Delattre and U. Dirksen, Nat. Rev. Dis. Primers, 2018, 4, 5 CrossRef .
- N. Riggi and I. Stamenkovic, Cancer Lett., 2007, 254, 1–10 CrossRef CAS PubMed .
- Y. Gao, X. Y. He, X. S. Wu, Y. H. Huang, S. Toneyan, T. Ha, J. J. Ipsaro, P. K. Koo, L. Joshua-Tor, K. M. Bailey, M. Egeblad and C. R. Vakoc, Nat. Cell Biol., 2023, 25, 298–308 CAS .
- M. S. Merchant, C. W. Woo, C. L. Mackall and C. J. Thiele, J. Natl. Cancer Inst., 2002, 94, 1673–1679 CrossRef CAS .
- E. L. Fong, S. E. Lamhamedi-Cherradi, E. Burdett, V. Ramamoorthy, A. J. Lazar, F. K. Kasper, M. C. Farach-Carson, D. Vishwamitra, E. G. Demicco, B. A. Menegaz, H. M. Amin, A. G. Mikos and J. A. Ludwig, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6500–6505 CrossRef CAS .
- W. H. Abuwatfa, W. G. Pitt and G. A. Husseini, J. Biomed. Sci., 2024, 31, 7 CrossRef .
- C. Jubelin, J. Muñoz-Garcia, L. Griscom, D. Cochonneau, E. Ollivier, M.-F. Heymann, F. M. Vallette, L. Oliver and D. Heymann, Cell Biosci., 2022, 12, 155 CrossRef PubMed .
- H. Peterziel, N. Jamaladdin, D. ElHarouni, X. F. Gerloff, S. Herter, P. Fiesel, Y. Berker, M. Blattner-Johnson, K. Schramm and B. C. Jones, npj Precis. Oncol., 2022, 6, 94 CrossRef CAS PubMed .
- G. Mehta, A. Y. Hsiao, M. Ingram, G. D. Luker and S. Takayama, J. Controlled Release, 2012, 164, 192–204 CrossRef CAS PubMed .
- J. Friedrich, C. Seidel, R. Ebner and L. A. Kunz-Schughart, Nat. Protoc., 2009, 4, 309–324 CrossRef CAS PubMed .
- R. Fevre, G. Mary, N. Vertti-Quintero, A. Durand, R. F.-X. Tomasi, E. Del Nery and C. N. Baroud, iScience, 2023, 26(5), 106651 CrossRef CAS PubMed .
- O. Frey, P. M. Misun, D. A. Fluri, J. G. Hengstler and A. Hierlemann, Nat. Commun., 2014, 5, 4250 CrossRef CAS PubMed .
- Y. C. Tung, A. Y. Hsiao, S. G. Allen, Y. S. Torisawa, M. Ho and S. Takayama, Analyst, 2011, 136, 473–478 RSC .
- L. B. Weiswald, D. Bellet and V. Dangles-Marie, Neoplasia, 2015, 17, 1–15 CrossRef PubMed .
- J. Friedrich, C. Seidel, R. Ebner and L. A. Kunz-Schughart, Nat. Protoc., 2009, 4, 309–324 CrossRef CAS .
- B. Kwak, Y. Lee, J. Lee, S. Lee and J. Lim, J. Controlled Release, 2018, 275, 201–207 CrossRef CAS .
- J. M. Lee, J. W. Choi, C. D. Ahrberg, H. W. Choi, J. H. Ha, S. G. Mun, S. J. Mo and B. G. Chung, Microsyst. Nanoeng., 2020, 6, 52 CrossRef CAS PubMed .
- E. Steinberg, R. Friedman, Y. Goldstein, N. Friedman, O. Beharier, J. A. Demma, G. Zamir, A. Hubert and O. Benny, Commun. Biol., 2023, 6, 1157 CrossRef PubMed .
- T. D. Ngo, A. Kashani, G. Imbalzano, K. T. Nguyen and D. Hui, Composites, Part B, 2018, 143, 172–196 CrossRef CAS .
- F. P. Melchels, J. Feijen and D. W. Grijpma, Biomaterials, 2010, 31, 6121–6130 CrossRef CAS PubMed .
- A. P. Zhang, X. Qu, P. Soman, K. C. Hribar, J. W. Lee, S. Chen and S. He, Adv. Mater., 2012, 24, 4266–4270 CrossRef CAS PubMed .
- F. Wu, J. Yang, J. Liu, Y. Wang, J. Mu, Q. Zeng, S. Deng and H. Zhou, Signal Transduction Targeted Ther., 2021, 6, 218 CrossRef CAS .
- H. Masuda, Cell Death Discovery, 2025, 11, 341 CrossRef PubMed .
- D. Yang, J. Liu, H. Qian and Q. Zhuang, Exp. Mol. Med., 2023, 55, 1322–1332 CrossRef CAS PubMed .
- B. Venzac, S. Deng, Z. Mahmoud, A. Lenferink, A. Costa, F. Bray, C. Otto, C. Rolando and S. Le Gac, Anal. Chem., 2021, 93, 7180–7187 CrossRef CAS PubMed .
- Z. Wu and K. Hjort, Lab Chip, 2009, 9, 1500–1503 RSC .
- A. Dobin, C. A. Davis, F. Schlesinger, J. Drenkow, C. Zaleski, S. Jha, P. Batut, M. Chaisson and T. R. Gingeras, Bioinformatics, 2013, 29, 15–21 CrossRef CAS .
- Y. Liao, G. K. Smyth and W. Shi, Bioinformatics, 2014, 30, 923–930 CrossRef CAS .
- M. I. Love, W. Huber and S. Anders, Genome Biol., 2014, 15, 550 CrossRef .
- N. L. Bray, H. Pimentel, P. Melsted and L. Pachter, Nat. Biotechnol., 2016, 34, 525–527 CrossRef CAS .
- M. J. Lerman, J. Lembong, G. Gillen and J. P. Fisher, Appl. Phys. Rev., 2018, 5, 041109 Search PubMed .
- F. Barbosa, P. Coutinho, M. P. Ribeiro, A. F. Moreira, L. M. Lourenço and S. P. Miguel, Mater. Des., 2025, 114254 CrossRef .
- M. Vinci, S. Gowan, F. Boxall, L. Patterson, M. Zimmermann, W. Court, C. Lomas, M. Mendiola, D. Hardisson and S. A. Eccles, BMC Biol., 2012, 10, 29 CrossRef CAS PubMed .
- J. Lee, Y. Kim, J. Lim, H.-I. Jung, G. Castellani, F. Piccinini and B. Kwak, BioChip J., 2024, 18, 160–169 CrossRef CAS .
- V. Damerell, M. S. Pepper and S. Prince, Signal Transduction Targeted Ther., 2021, 6, 246 CrossRef CAS .
- A. N. Schuetz, B. P. Rubin, J. R. Goldblum, B. Shehata, S. W. Weiss, W. Liu, M. R. Wick and A. L. Folpe, Mod. Pathol., 2005, 18, 1403–1410 CrossRef CAS .
- E. L. S. Fong, S.-E. Lamhamedi-Cherradi, E. Burdett, V. Ramamoorthy, A. J. Lazar, F. K. Kasper, M. C. Farach-Carson, D. Vishwamitra, E. G. Demicco and B. A. Menegaz, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6500–6505 CrossRef CAS .
- J. C. Brenner, F. Y. Feng, S. Han, S. Patel, S. V. Goyal, L. M. Bou-Maroun, M. Liu, R. Lonigro, J. R. Prensner, S. A. Tomlins and A. M. Chinnaiyan, Cancer Res., 2012, 72, 1608–1613 CrossRef CAS .
- L. M. Guenther, N. V. Dharia, L. Ross, A. Conway, A. L. Robichaud, J. L. Catlett, 2nd, C. S. Wechsler, E. S. Frank, A. Goodale, A. J. Church, Y. Y. Tseng, R. Guha, C. G. McKnight, K. A. Janeway, J. S. Boehm, J. Mora, M. I. Davis, G. Alexe, F. Piccioni and K. Stegmaier, Clin. Cancer Res., 2019, 25, 1343–1357 CrossRef CAS PubMed .
- M. L. Harlow, M. H. Chasse, E. A. Boguslawski, K. M. Sorensen, J. M. Gedminas, S. M. Kitchen-Goosen, S. B. Rothbart, C. Taslim, S. L. Lessnick, A. S. Peck, Z. B. Madaj, M. J. Bowman and P. J. Grohar, Clin. Cancer Res., 2019, 25, 3417–3429 CrossRef CAS PubMed .
- S. L. Koppenhafer, K. L. Goss, W. W. Terry and D. J. Gordon, Mol. Cancer Ther., 2018, 17, 2676–2688 CrossRef CAS PubMed .
- N. Sen, A. M. Cross, P. L. Lorenzi, J. Khan, B. E. Gryder, S. Kim and N. J. Caplen, Mol. Carcinog., 2018, 57, 1342–1357 CrossRef CAS PubMed .
- A. M. van Maldegem, J. V. Bovee, E. F. Peterse, P. C. Hogendoorn and H. Gelderblom, Eur. J. Cancer, 2016, 53, 171–180 CrossRef CAS PubMed .
- Y. Ma, M. Baltezor, L. Rajewski, J. Crow, G. Samuel, V. S. Staggs, K. M. Chastain, J. A. Toretsky, S. J. Weir and A. K. Godwin, J. Mol. Med., 2019, 97, 957–972 CrossRef CAS PubMed .
- D. J. García-Domínguez, N. Hajji, S. Sánchez-Molina, E. Figuerola-Bou, R. M. de Pablos, A. M. Espinosa-Oliva, E. Andrés-León, L. C. Terrón-Camero, R. Flores-Campos and G. Pascual-Pasto, Oncogene, 2021, 40, 5843–5853 CrossRef .
- H. Sun, D.-C. Lin, Q. Cao, X. Guo, H. Marijon, Z. Zhao, S. Gery, L. Xu, H. Yang and B. Pang, Cancer Res., 2016, 76, 2687–2697 CrossRef CAS PubMed .
- A. Moein, J. Y. Jin, M. R. Wright, B. Alicke and H. Wong, Drugs R&D, 2024, 24, 155–167 CrossRef CAS .
- D. S. Shulman, P. Merriam, E. Choy, L. M. Guenther, K. L. Cavanaugh, P. C. Kao, A. Posner, K. Bhushan, G. Fairchild, E. Barker, K. Klega, K. Stegmaier, B. D. Crompton, W. B. London and S. G. DuBois, Cancer Med., 2023, 12, 15207–15216 CrossRef CAS PubMed .
- V. Del Pozo, A. J. Robles, S. D. Fontaine, Q. Liu, J. E. Michalek, P. J. Houghton and R. T. Kurmasheva, iScience, 2022, 25, 103725 CrossRef CAS .
- F. Engert, C. Schneider, L. M. Weibeta, M. Probst and S. Fulda, Mol. Cancer Ther., 2015, 14, 2818–2830 CrossRef CAS .
- P. N. Gollavilli, A. Pawar, K. Wilder-Romans, R. Natesan, C. G. Engelke, V. L. Dommeti, P. M. Krishnamurthy, A. Nallasivam, I. J. Apel and T. Xu, Cancer Res., 2018, 78, 4760–4773 CrossRef CAS .
- J. Hernando-Cubero, P. Sanz-Moncasi, A. Hernandez-Garcia, I. Pajares-Bernard and J. Martinez-Trufero, Oncol. Lett., 2016, 12, 2936–2941 CrossRef .
- M. Liu, X. Zhang, C. Long, H. Xu, X. Cheng, J. Chang, C. Zhang, C. Zhang and X. Wang, RSC Adv., 2018, 8, 8910–8919 RSC .
- S. Sanchez-Molina, E. Figuerola-Bou, V. Sanchez-Margalet, L. de la Cruz-Merino, J. Mora, E. de Alava Casado, D. J. Garcia-Dominguez and L. Hontecillas-Prieto, Cancers, 2022, 14(21), 5473 CrossRef CAS PubMed .
- P. A. Meyers, N. Federman, N. Daw, P. M. Anderson, L. E. Davis, A. Kim, M. E. Macy, A. Pietrofeso, R. Ratan, R. F. Riedel, M. Trucco, J. B. Breitmeyer, J. A. Toretsky and J. A. Ludwig, J. Clin. Oncol., 2024, 42, 3725–3734 CrossRef CAS PubMed .
- B. Feng, J. Wu, B. Shen, F. Jiang and J. Feng, Cancer Cell Int., 2022, 22, 166 CrossRef PubMed .
- H. Han, J. W. Cho, S. Lee, A. Yun, H. Kim, D. Bae, S. Yang, C. Y. Kim, M. Lee, E. Kim, S. Lee, B. Kang, D. Jeong, Y. Kim, H. N. Jeon, H. Jung, S. Nam, M. Chung, J. H. Kim and I. Lee, Nucleic Acids Res., 2018, 46, D380–D386 CrossRef CAS PubMed .
- F. Wu, J. Yang, J. Liu, Y. Wang, J. Mu, Q. Zeng, S. Deng and H. Zhou, Signal Transduction Targeted Ther., 2021, 6, 218 CrossRef CAS .
- Z. Fang, Q. Meng, J. Xu, W. Wang, B. Zhang, J. Liu, C. Liang, J. Hua, Y. Zhao, X. Yu and S. Shi, Cancer Commun., 2023, 43, 3–41 CrossRef PubMed .
- J. D. Daley, E. Mukherjee, D. Ferraro, A. C. Tufino, N. Bailey, S. Bhaskar, N. Periyapatna, I. MacFawn, S. Hartwick and S. Kunning, Cancer Res. Commun., 2025, 5, 1441–1457 CrossRef PubMed .
- J. D. Daley, S. L. Whiteway, C. M. Heske, F. S. Dela Cruz, S. B. Whittle, S. Cohen-Gogo, A. B. Collier, A. Gupta, J. Ohm and A. Clark, Pediatr. Blood Cancer, 2025, 72, e31917 CrossRef PubMed .
- M. E. Fiori, S. Di Franco, L. Villanova, P. Bianca, G. Stassi and R. De Maria, Mol. Cancer, 2019, 18, 70 CrossRef PubMed .
- C. Ma, M. T. Witkowski, J. Harris, I. Dolgalev, S. Sreeram, W. Qian, J. Tong, X. Chen, I. Aifantis and W. Chen, Sci. Adv., 2020, 6(44), eaba5536 CrossRef CAS PubMed .
Footnote |
| † These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ac,
Joy
Fei
b,
Yuan
Gao
*be and
Chao
Ma
ac,
Joy
Fei
b,
Yuan
Gao
*be and
Chao
Ma







