DOI:
10.1039/D5CE00132C
(Paper)
CrystEngComm, 2025,
27, 3664-3674
Structural modulations: unraveling the impact of benzothiazole positions on the optical properties of carbazole–cyanostilbenes†
Received
6th February 2025
, Accepted 15th April 2025
First published on 16th April 2025
Abstract
This study explores the influence of positional substitution of benzothiazole on the optical and solid-state properties of carbazole–cyanostilbene derivatives, focusing on two key positional isomers: 2-BTCZCS and 5-BTCZCS. These isomers exhibit remarkable aggregation-induced emission (AIE) with solid-state quantum yields reaching up to 24%. Despite anticipated variations in molecular geometry and electronic interactions due to positional substitution, both isomers display remarkably similar photophysical behaviors, underscoring the robustness and versatility of the carbazole–cyanostilbene framework. The isomers exhibit notable mechanofluorochromic (MFC) behavior, characterized by a subtle blue shift of 12 nm in emission upon mechanical grinding. This shift highlights their sensitivity to external mechanical stimuli. Powder X-ray diffraction (PXRD) analysis confirms that the grinding process does not disrupt the crystallinity of the materials, preserving their ordered structure. Furthermore, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) reveal excellent thermal stability, emphasizing their durability and suitability for practical applications. Morphological stability and aggregation behavior were further validated using scanning electron microscopy (SEM) and dynamic light scattering (DLS). Density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations validate the experimental findings, providing deeper insight into their electronic structures and excited-state properties. This work highlights the minimal impact of benzothiazole substitution positions on the optical properties of these materials, emphasizing their reliability and potential for high-performance optoelectronic applications. These findings open new avenues for the design of stable and predictable materials tailored for next-generation devices.
Introduction
Modern organic materials science heavily relies on donor–acceptor (D–A) systems, which serve as powerful tools for fine-tuning optical properties and achieving superior device performance through the strategic management of intramolecular charge transfer (ICT).1–4 These systems are built upon the strategic pairing of electron-rich donor moieties with electron-deficient acceptor units, enabling a rich diversity of photophysical phenomena. The interaction between donor and acceptor components governs essential properties, including efficient charge separation, adjustable emission spectra, and adaptable solid-state behavior. Consequently, D–A systems have become a cornerstone in advancing technologies such as organic light-emitting diodes (OLEDs),5–8 organic photovoltaics (OPVs),9–11 bioimaging,12–15 and nonlinear optical (NLO) devices.16–18 By tailoring the nature of the donor and acceptor groups, researchers have achieved remarkable control over optoelectronic properties, paving the way for next-generation materials with precision-tuned functionalities. A critical challenge in D–A systems lies in achieving efficient solid-state emission, which is vital for real-world applications. Organic emitters often face a significant drawback known as aggregation-caused quenching (ACQ), where their emission efficiency quenches in the aggregated state.19,20 To overcome this challenge, researchers have focused on aggregation-induced emission (AIE), a groundbreaking phenomenon discovered by Tang et al. in 2001. In AIE-active materials, fluorescence is enhanced in the aggregated state due to restricted intramolecular motions, making them ideal for achieving high solid-state quantum efficiencies.21 In parallel, the discovery of mechanofluorochromism (MFC) in certain donor–acceptor systems has opened new possibilities in material design. MFC refers to changes in emission properties-such as color or intensity, when subjected to mechanical forces like grinding or pressing.22–24 These properties hold tremendous potential for the development of smart materials, responsive sensors, and advanced optoelectronic devices.25–28 Together, AIE and MFC provide a powerful toolkit for creating materials with enhanced solid-state performance and dynamic optical properties.29–31 Carbazole has emerged as a prominent donor moiety in donor–acceptor (D–A) systems due to its excellent electron-donating ability, robust chemical stability, and versatile photophysical properties. As a tricyclic aromatic compound, it offers high thermal and photochemical stability, making it a preferred choice for various optoelectronic applications, including OLEDs and OPVs materials.32–35 Incorporating carbazole with complementary acceptor units has enabled the development of materials with improved charge-transfer efficiency and tunable optical properties. Among these, cyanostilbene has proven to be an exceptional acceptor.36–38 Its strong electron-withdrawing capabilities, coupled with its ability to facilitate ICT, make it ideal for achieving advanced photophysical effects such as solvatochromism, AIE, and MFC. To further refine the properties of carbazole–cyanostilbene systems, additional acceptor units can be integrated into the molecular framework. This approach allows for precise tuning of the electronic structure, offering enhanced control over emission behavior and broadening the scope for innovative applications in optoelectronic devices.
Our research group has been actively unlocking new horizons in the design and functionalization of carbazole–cyanostilbene-based systems to modulate optical properties. Earlier investigations have showcased the adaptability and promising performance of this framework in diverse structural arrangements. For example, carbazole–anthracene conjugated cyanostilbenes exhibited significant charge-transfer properties and solid-state emission modulations,39 while carbazole–cyanostilbene systems with π-spacers revealed the critical role of spacer units in influencing solid-state emissive behavior. Specifically, we found that the nature of the π-spacer significantly impacts aggregation, intramolecular charge transfer, and mechanofluorochromic responses.40 The carbazole–cyanostilbene framework has proven to be highly versatile, demonstrating its suitability for a wide range of applications. To build on these promising results, we are now exploring the introduction of additional acceptor units, specifically benzothiazole, into this system. This approach aims to strengthen charge-transfer interactions and optimize the photophysical properties of the resulting materials. Benzothiazole, a heterocyclic compound with strong electron-withdrawing capabilities, offers significant potential for enhancing molecular performance. Its ability to promote ICT and refine emission characteristics is well-documented, especially when strategically incorporated into the molecular framework.41–45 There are several reports which highlight the impact of halogenation on benzothiazole derivatives, demonstrating that halogen substituents can effectively modulate photophysical properties, intermolecular interactions, and solid-state packing.46 Such modifications not only fine-tune emission behavior but also enhance the responsiveness of these systems to external stimuli. Including these structural strategies provides valuable context for understanding the design rationale of our system and allows meaningful comparison with related stimuli-responsive luminophores. However, the impact of substituting benzothiazole at the 2- or 5-position on the photophysical behavior of carbazole–cyanostilbene derivatives remains underexplored, providing a compelling direction for future research. In this study, we investigate the effect of benzothiazole positional substitution on the optical and solid-state properties of carbazole–cyanostilbene systems by comparing two positional isomers: 2-BTCZCS and 5-BTCZCS (Chart 1). Despite the expected influence of positional variation on molecular geometry, charge distribution, and intermolecular interactions, our findings reveal that the optical properties of both isomers remain largely consistent. Both isomers exhibit remarkable solid-state quantum yields of up to 24% and demonstrate mechanofluorochromic behavior with a minor blue shift of approximately 12 nm upon grinding, highlighting the stability of the emission despite the structural changes. This blue shift in MFC indicates a subtle yet noteworthy response to mechanical stress, confirming the presence of a dynamic photophysical behavior while maintaining overall optical stability. The consistency in the optical properties of both isomers challenges the conventional assumption that positional changes in the acceptor unit would significantly affect emission behavior, suggesting that the carbazole–cyanostilbene framework is highly robust and capable of maintaining consistent performance across different structural variations. Our findings underscore the critical interplay between molecular structure, electronic interactions, and solid-state behavior in donor–acceptor (D–A) systems, offering valuable insights for designing stable, high-performance materials. The robustness of these carbazole–cyanostilbene derivatives, even with positional changes to the benzothiazole unit, underscores their potential as a versatile design platform. This work demonstrates the potential to create advanced materials with reliable and predictable performance without extensive structural optimization. By providing a deeper understanding of how structural changes influence behavior, this study paves the way for new advances in optoelectronics, smart materials, and responsive devices. These insights not only deepen our understanding of carbazole–cyanostilbene systems but also contribute broadly to the field of D-A systems, driving future innovations in high-performance, stable optoelectronic materials.
 |
| | Chart 1 Chemical structures of two isomers. | |
Results and discussions
Synthesis and characterisation
Two novel benzothiazole-conjugated carbazole–cyanostilbenes, with varying positions of the benzothiazole unit, were successfully designed and synthesized through slight modifications in the synthetic pathway, as depicted in Scheme 1. The synthesis of all precursors (1–7) followed established literature protocols, and their characterization data were consistent with reported values.47,48 The synthesis commenced with the protection of 9H-carbazole using 1-bromohexane in the presence of NaOH in DMF, yielding compound 1. Compound 1 underwent a Vilsmeier–Haack reaction with anhydrous DMF and POCl3 in dry dichloroethane to produce compound 2. Subsequent bromination of compound 2 using NBS in CHCl3 afforded 6-bromo-9-hexyl-9H-carbazole-3-carbaldehyde (3) in moderate yields. For 5-BTCZCS, compound 3 underwent Suzuki–Miyaura borylation using a palladium(II) catalyst to yield 9-hexyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9H-carbazole-3-carbaldehyde (4). Compound 4 was then subjected to a cross-coupling reaction with 5-bromobenzo[d]thiazole, producing the main intermediate for 5-BTCZCS. Finally, Knoevenagel condensation of this intermediate (5) with benzyl cyanide in the presence of tBuOK in methanol resulted in the target compound 5-BTCZCS. The synthesis of 2-BTCZCS, however, proved challenging using the above-mentioned synthetic pathway. Initial attempts to directly cross-couple the 2-bromo benzothiazole unit after Suzuki–Miyaura borylation were unsuccessful, necessitating modifications to the approach. Compound 3 was first subjected to Knoevenagel condensation with benzyl cyanide using NaOH in methanol, yielding (Z)-3-(6-bromo-9-hexyl-9H-carbazol-3-yl)-2-phenylacrylonitrile (6). Compound 6 then underwent Suzuki–Miyaura borylation to form the main intermediate (7), which was successfully cross-coupled with 2-bromobenzo[d]thiazole in the presence of a palladium catalyst to afford the target compound 2-BTCZCS. These modifications in the synthetic route proved crucial for obtaining the desired product in good purity. All newly synthesized isomers were thoroughly characterized using 1H and 13C NMR spectroscopy, as well as high-resolution mass spectrometry (HRMS) (Fig. S1–S17†). The 1H and 13C NMR spectra displayed well-defined and anticipated signals, with integration values precisely aligning with the expected molecular structures, thereby confirming their structural integrity. Additionally, ESI-HRMS analysis provided explicit evidence for the molecular identities of the target isomers, supporting the successful synthesis. Single crystals of 2-BTCZCS were obtained as greenish-yellow crystals via slow evaporation from a methanol/ether mixture at room temperature. The crystal structure of 2-BTCZCS was further elucidated via single-crystal X-ray diffraction (XRD) analysis, providing a definitive confirmation of its molecular architecture, as depicted in Fig. 1.
 |
| | Scheme 1 Synthesis route adopted for target isomers. | |
 |
| | Fig. 1 (A) Crystal structure of compound 2-BTCZCS, (B) interactions present in compound 2-BTCZCS and (C) ladder-like arrangement and packing diagram of compound 2-BTCZCS. | |
Single crystal structure
Crystals of 2-BTCZCS suitable for X-ray diffraction were successfully obtained through the slow evaporation of MeOH/Et2O solutions at room temperature. The crystal structure of 2-BTCZCS, as confirmed through single-crystal X-ray diffraction, reveals a unique molecular arrangement that significantly influences its solid-state properties. The compound crystallizes in the triclinic system with the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, indicating a lack of symmetry in the unit cell, which allows for a more complex molecular packing. Two molecules are present in each unit cell, aligned in a parallel fashion but exhibiting opposite configurations. This alternating configuration suggests a potential for specific intermolecular interactions that could contribute to the solid-state behavior of isomer. The carbazole unit, linked to the cyanostilbene group, adopts a twisted conformation as shown in Fig. 1(A), preventing close packing of molecules in the crystal lattice. This twist restricts π–π interactions, reducing aggregation and self-quenching, thereby enhancing solid-state emission. Significant intermolecular interactions further stabilize the crystal structure. These include –C
space group, indicating a lack of symmetry in the unit cell, which allows for a more complex molecular packing. Two molecules are present in each unit cell, aligned in a parallel fashion but exhibiting opposite configurations. This alternating configuration suggests a potential for specific intermolecular interactions that could contribute to the solid-state behavior of isomer. The carbazole unit, linked to the cyanostilbene group, adopts a twisted conformation as shown in Fig. 1(A), preventing close packing of molecules in the crystal lattice. This twist restricts π–π interactions, reducing aggregation and self-quenching, thereby enhancing solid-state emission. Significant intermolecular interactions further stabilize the crystal structure. These include –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N⋯H–C and C–S⋯π interactions, with bond distances of 2.694 Å and 3.499 Å, respectively (Fig. 1(B), Tables S5 and S6†). The –CN⋯H–C interaction likely plays a role in promoting molecular stability by maintaining a proper molecular orientation within the crystal lattice, while the C–S⋯π interaction introduces additional rigidity to the structure. These noncovalent forces contribute to the overall structural rigidity, effectively restricting intramolecular motions. The formation of a dimer with a slipped face-to-face arrangement further enhances the packing arrangement. This dimeric unit appears to form a ladder-like structure within the crystal lattice, which could be responsible for the separation of molecules along the a and c axes (as shown in Fig. 1(C) and S26†). The ladder-like arrangement suggests that the twisted molecular conformation leads to a loose stacking, resulting in minimized interactions between adjacent molecules.49 This spatial separation likely helps preserve the twisted conformation, preventing undesirable interactions that could affect the optical properties of the compound.
N⋯H–C and C–S⋯π interactions, with bond distances of 2.694 Å and 3.499 Å, respectively (Fig. 1(B), Tables S5 and S6†). The –CN⋯H–C interaction likely plays a role in promoting molecular stability by maintaining a proper molecular orientation within the crystal lattice, while the C–S⋯π interaction introduces additional rigidity to the structure. These noncovalent forces contribute to the overall structural rigidity, effectively restricting intramolecular motions. The formation of a dimer with a slipped face-to-face arrangement further enhances the packing arrangement. This dimeric unit appears to form a ladder-like structure within the crystal lattice, which could be responsible for the separation of molecules along the a and c axes (as shown in Fig. 1(C) and S26†). The ladder-like arrangement suggests that the twisted molecular conformation leads to a loose stacking, resulting in minimized interactions between adjacent molecules.49 This spatial separation likely helps preserve the twisted conformation, preventing undesirable interactions that could affect the optical properties of the compound.
Photophysical properties
The photophysical properties of the isomers 2-BTCZCS and 5-BTCZCS were conducted in dichloromethane (DCM) solution using absorption and emission spectroscopy, with the normalized absorption and emission spectra shown in Fig. S18.† The absorption spectra of both isomers reveal similar peak positions, indicating that the substitution position of benzothiazole has little influence on their overall photophysical characteristics. For 2-BTCZCS, the absorption spectrum displays bands at ∼240, ∼320, and ∼385 nm, whereas 5-BTCZCS exhibits at ∼250, ∼290, and ∼385 nm. Both isomers exhibit shorter-wavelength bands near 240 nm, associated with π–π* transitions within the aromatic system. The bands observed at approximately 315 nm for 2-BTCZCS and 290 nm for 5-BTCZCS arise from localized electronic transitions, which are modulated by the specific positions of benzothiazole substitution.50 For both 2-BTCZCS and 5-BTCZCS, the intramolecular charge transfer (ICT) bands consistently appear at ∼385 nm, demonstrating that the position of benzothiazole substitution has a negligible effect on the charge transfer process or donor–acceptor interactions. This observation highlights that the electronic environments of the two isomers remain largely unaffected by their structural variations. The subtle variations in the shorter-wavelength absorption bands, while noteworthy, do not influence the ICT behavior, which remains a dominant and position-independent feature. The results indicate that the benzothiazole substitution position has no influence on the absorption profiles of these isomers, highlighting their potential for applications requiring consistent ICT behavior. Fluorescence studies were conducted in dichloromethane by exciting the isomers at their ICT absorption band (∼385 nm). The emission spectrum of 2-BTCZCS displayed a peak at 447 nm with a quantum yield of 3.0%, while 5-BTCZCS exhibited a slightly red-shifted emission peak at 460 nm with a quantum yield of 3.7% (Fig. S18(B) and Table S1†). These results further demonstrate the minimal impact of benzothiazole substitution position on the photophysical properties, with both isomers showing similar emission profiles and modest quantum yields.
Solvatochromism
The ICT transition arising in D–A molecules is easily affected by changes in the solvent polarity. The solvent dependent photophysical studies of isomers (2-BTCZCS and 5-BTCZCS) were studied in different solvents by changing polarity from non-polar to polar (Hexane, toluene, DCM, Tetrahydrofuran (THF), acetonitrile (ACN) and dimethylformamide (DMF)) and the effect of the solvent polarity and positional change on the ICT transition was explored in detail by emission (Fig. 2) and absorption spectra (Fig. S19†). In this regard, the photophysical properties of the isomers 2-BTCZCS and 5-BTCZCS were carried out solvent-dependent studies to investigate the influence of solvent polarity on their ICT transitions. The absorption spectra (Fig. S19†) exhibit a slight red shift with increasing solvent polarity, indicating slight destabilization of the ground state in polar environments. This subtle red shift suggests that the ground-state electronic distribution experiences weak but measurable interactions with polar solvent molecules. In contrast, the emission spectra (Fig. 2) revealed a pronounced red shift with increasing solvent polarity, characteristic of ICT transitions where the excited state, having a higher dipole moment, is more stabilized in polar environments. For 2-BTCZCS, the emission maxima shifted from 425 nm in hexane (a non-polar solvent) to 465 nm in ACN, (a polar solvent), while 5-BTCZCS exhibited a shift from 430 nm to 488 nm under the same conditions (Table S2†).50 The slightly longer emission wavelengths observed for 5-BTCZCS across all solvents suggest marginally stronger ICT character or greater excited-state stabilization compared to 2-BTCZCS. Despite these subtle differences, the overall trends highlight that solvent polarity plays a dominant role in modulating the ICT transitions of both isomers, with the positional change of the benzothiazole unit exerting only a minimal influence. To understand the excited-state relaxation dynamics, time-resolved fluorescence measurements were carried out for both 2-BTCZCS and 5-BTCZCS in solvents of varying polarity (hexane, toluene, THF, DCM, ACN, and DMF) (Table S3 and Fig. S20†). The average fluorescence lifetimes (τavg) showed clear dependence on the solvent environment, reflecting the influence of polarity on radiative and non-radiative decay processes. For 2-BTCZCS, the fluorescence lifetime decreased significantly from 4.63 ns in toluene to 0.83 ns in DMF, indicating enhanced non-radiative decay in polar media. This trend is consistent with the positive solvatochromism observed in the emission spectra and suggests that polar solvents stabilize the excited state and facilitate faster non-radiative relaxation. However, an anomaly was observed in hexane, which, despite being non-polar, exhibited a short τavg of 0.66 ns, comparable to that in DCM (0.63 ns). This unexpected behavior may be attributed to the low viscosity and weak solvation capability of hexane, which can lead to increased intramolecular motion and thus promote non-radiative decay. Similarly, 5-BTCZCS exhibited the longest fluorescence lifetime in toluene (4.68 ns), while the values in polar solvents such as THF (1.70 ns), DCM (1.46 ns), ACN (1.56 ns), and DMF (1.53 ns) were significantly shorter. The lifetime in hexane was again the lowest (0.82 ns), reinforcing the notion that besides solvent polarity, other factors such as solvent viscosity, solute–solvent interaction strength, and conformational freedom play important roles in dictating the excited-state behavior. Overall, both compounds exhibit a general trend of decreasing fluorescence lifetime with increasing solvent polarity, which is in agreement with the observed red-shift in emission maxima and supports the hypothesis of enhanced non-radiative decay in polar environments.
 |
| | Fig. 2 Normalised emission spectra of (A) 2-BTCZCS, and (B) 5-BTCZCS in solvents of different polarity (Con. 10 μM; λex = 385 nm). Digital photos taken under UV 365 nm lamp are displayed on the top of the spectra. | |
Aggregation-induced emission properties
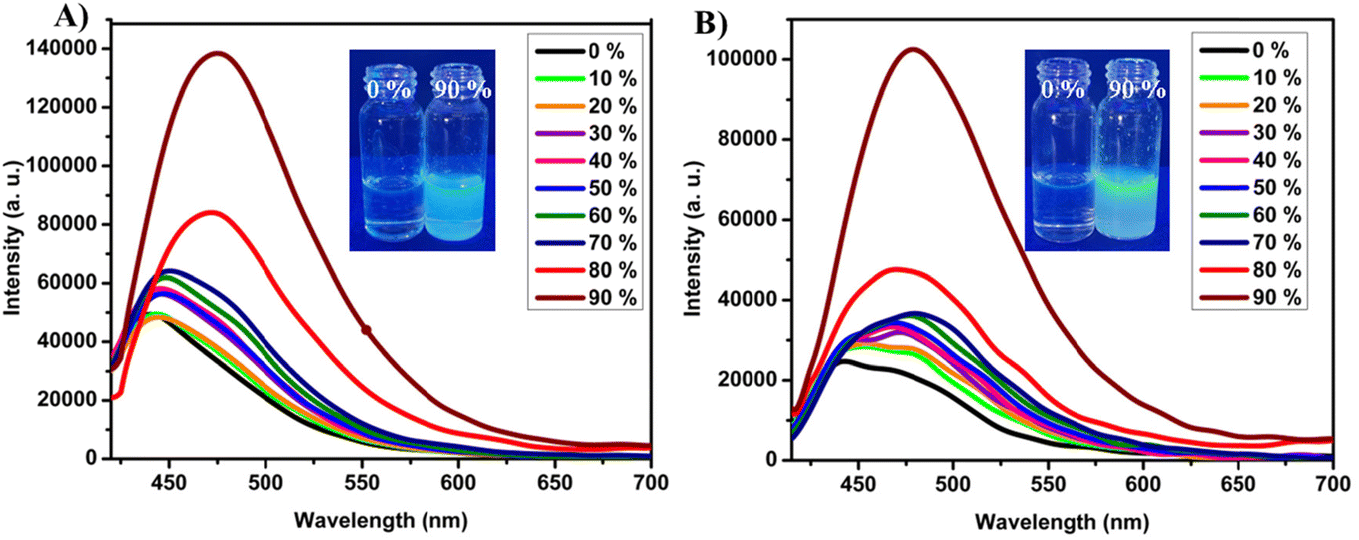
The aggregation-induced emission (AIE) behavior of 2-BTCZCS and 5-BTCZCS was investigated using emission spectroscopy in THF–water mixtures with varying water fractions (fw) (Fig. 3). Both isomers, highly soluble in THF but insoluble in water, showed distinct AIE characteristics due to their twisted structures, which inhibit π–π stacking as well as inter molecular motions, which promote enhanced emission in the aggregated state. As we discussed solvent dependent studies, the compound 2-BTCZCS exhibited poor emission maximum at 440 nm in pure THF, attributed to the exclusive ICT transition. Gradually increasing the water fraction up to 30% led to a moderate enhancement in emission intensity and a bathochromic shift, indicating stabilization of the ICT state in the polar medium. This observation aligns with findings from solvent-dependent studies. At 40% water fraction, the onset of aggregation was evident, accompanied by a notable increase in intensity and a red-shift in emission. Significant enhancement in emission intensity was noted at 80% water fraction. At 90% water content, the emission intensity reached its maximum, further confirming strong AIE characteristics, with the emission peak at 475 nm. Similarly, 5-BTCZCS exhibited an analogous AIE profile. The initial emission peak at 450 nm in pure THF shifted to 480 nm as the water fraction increased (90%), mirroring the behavior of 2-BTCZCS. The emission intensity followed a pattern similar to 2-BTCZCS, affirming consistent AIE behavior in both isomers. The shift in emission suggests the presence of weak intermolecular interactions in the excited state. However, the absence of a broad and featureless emission band typically characteristic of excimer formation indicates that excimer contribution is likely minimal. Thus, the observed aggregation-induced emission (AIE) behavior can be primarily attributed to the restriction of intramolecular motions (RIM), which suppresses non-radiative decay pathways and enhances fluorescence. Further, the photoluminescence quantum yields of 2-BTCZCS and 5-BTCZCS were examined in both dilute THF solutions and their aggregated states. Compound 2-BTCZCS displayed a modest quantum yield of 2.7% in solution, which improved to 8% upon aggregation. In comparison, 5-BTCZCS showed a quantum yield of 2.1% in solution that increased to 9% in the aggregated state, as summarized in Table S4.† The formation of nanoaggregates in THF–water mixtures (90% water) was confirmed by scanning electron microscopy (SEM) and dynamic light scattering (DLS) analyses. The DLS results indicated the presence of aggregates with average diameters of ∼220 nm for 2-BTCZCS and ∼204 nm for 5-BTCZCS, confirming nanoaggregate formation at high water content (Fig. S22†). Morphological differences were evident from SEM imaging, where 2-BTCZCS aggregates formed a spherical-like aggregates, while 5-BTCZCS displayed a spherical with flakes like aggregates (Fig. S21†). These findings strongly support the AIE characteristics of both isomers and demonstrate their ability to form distinct aggregate morphologies under similar conditions.
 |
| | Fig. 3 Emission spectra of (A) 2-BTCZCS and (B) 5-BTCZCS in THF–water mixtures with different water fractions. Inset shows the digital photos taken for the solution and aggregated states THF/water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9) under UV 365 nm lamp. :9) under UV 365 nm lamp. | |
Solid state emission and mechanofluorochromism
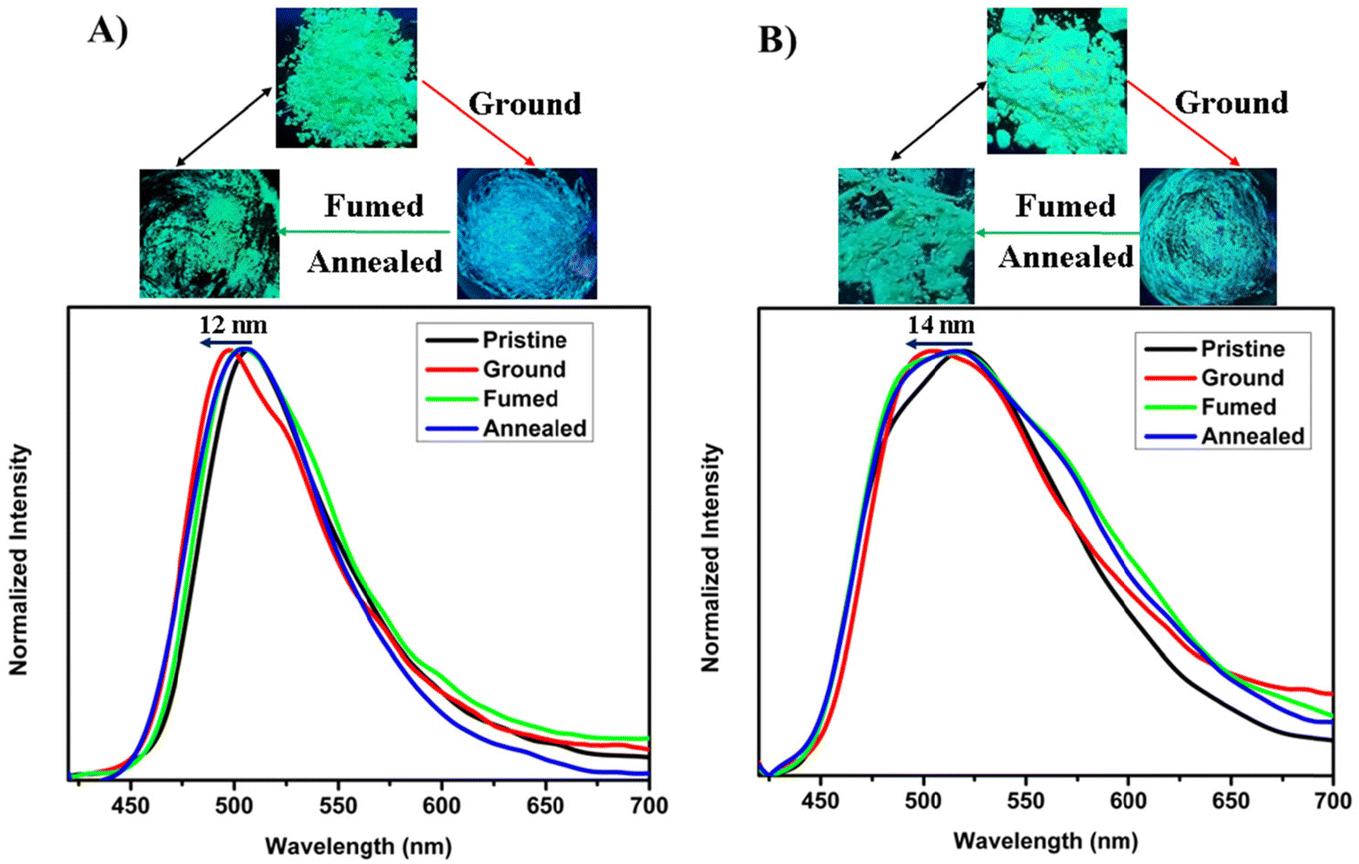
The solid-state emission (SSE) properties of 2-BTCZCS and 5-BTCZCS were explored, showing green color luminescence for both isomers. In their pristine state, 2-BTCZCS exhibited an emission peak at 508 nm, while 5-BTCZCS displayed a slightly red-shifted peak at 518 nm (Fig. S23†). Both isomers exhibited broad emission profiles, with 2-BTCZCS showing a narrower compared to the 5-BTCZCS. These results reflect subtle differences in their molecular packing and intermolecular interactions in the solid state. To investigate their mechanochromic fluorescence behavior, the photoluminescence of both isomers was analyzed under mechanical force. Upon grinding, both isomers exhibited blue shifts in their emission spectra: 2-BTCZCS shifted from 508 nm to 496 nm (12 nm), and 5-BTCZCS shifted from 518 nm to 504 nm (14 nm). These shifts were reversible upon exposure to most of the volatile solvents such as dichloromethane, acetone, chloroform, ethyl acetate vapors as well as annealing and could be cycled four to five times (Fig. 4, S24, S25† and Table 1). The blue-shifted emission upon grinding is an unusual phenomenon, suggesting specific structural changes induced by mechanical force.
 |
| | Fig. 4 Mechanochromic fluorescence responses of (A) 2-BTCZCS and (B) 5-BTCZCSλ = 380 nm for isomers. Digital images taken under a UV lamp 365 nm are shown above the spectra. | |
Table 1 Emission wavelengths of isomers in solid-state under various external stimuli and their absolute quantum yields
| Isomers |
Emission |
Φ
|
Φ
|
|
λ
Pristine (nm) |
λ
Grinded (nm) |
λ
Fumed/Annealed (nm) |
Δλa (nm) |
|
Grinding induced spectral shift.
Absolute quantum yield in solid-state in pristine state.
Absolute quantum yield in solid-state in ground state.
|
|
2-BTCZCS
|
508 nm |
496 nm |
504 nm |
−12 nm |
23.2 |
12.2 |
|
5-BTCZCS
|
518 nm |
504 nm |
516 nm |
−14 nm |
24.3 |
4.7 |
To gain insight into the structural changes responsible for the observed hypsochromic shifts, powder X-ray diffraction (PXRD) analysis was conducted on the pristine, ground, and solvent-fumed samples. The pristine powders exhibited sharp and intense diffraction peaks, indicative of high crystallinity and well-ordered molecular packing. Upon grinding, the PXRD patterns showed a noticeable reduction in peak intensity, suggesting partial disruption of the crystalline lattice. However, the persistence of some diffraction peaks indicates that the isomers did not undergo complete amorphization but retained partially crystalline in nature. This residual order likely contributes to the blue-shifted emission, as mechanical force fragments the larger crystalline domains into smaller ordered regions (Fig. 5).51 To further probe the molecular origin of this emission shift, the single-crystal structure of 2-BTCZCS was analyzed. The structure reveals a twisted conformation of the carbazole–cyanostilbene backbone, stabilized by key noncovalent interactions, including CN⋯H–C (2.694 Å) and C–S⋯π (3.499 Å) contacts (Table S3†). Upon grinding, this molecular arrangement is likely disturbed, weakening the intermolecular interactions and possibly altering the torsion angle between the donor (carbazole) and acceptor (cyanostilbene) units. This disruption likely perturbs the aggregated-state packing and reduces the extent of interactions, leading to a hypsochromic (blue) shift in the emission. Moreover, the ladder-like dimeric stacking observed in the single-crystal structure is expected to be diminished upon grinding, further influencing the excited-state properties and emission behavior. These findings highlight the key role of molecular conformation and packing in governing the mechanofluorochromic response of the system.
 |
| | Fig. 5 PXRD pattern of isomers (A) 2-BTCZCS and (B) 5-BTCZCS. | |
Theoretical calculations
Density functional theory (DFT) calculations were performed to analyse the geometries and the electronic distribution present in the ground state for the isomers by utilizing the B3LYP/6-311G(d,p) level of theory with full geometry optimization carried out without any imposed symmetry constraints.52 For 2-BTCZCS, the initial geometry was extracted from its crystal structure, and the optimized structure was found to align well with the experimental crystallographic data. To confirm the stability of the optimized geometries, vibrational frequency analyses were conducted at the same level of theory, and the absence of imaginary frequencies validated the optimization results. The HOMO of both isomers was observed to be delocalized on the entire system major part on the electron-donating carbazole moiety, indicating significant π-conjugation across this region. In contrast, the electron-withdrawing cyano groups, along with alkyl chains and peripheral phenyl groups, contributed minimally to the HOMO, demonstrating clear electronic segregation within the molecules. On the other hand, the LUMO was predominantly localized on the cyanostilbene unit, consistent with the strong electron-withdrawing nature of the cyano groups (Fig. 6). The calculated band gap for both isomers was approximately 3.48 eV. The twisted conformations of these molecules are anticipated to hinder tight molecular packing in the solid state, contributing to enhanced emission quantum yields.
 |
| | Fig. 6 Frontier molecular orbitals and energy gap of isomers A) 2-BTCZCS and B) 5-BTCZCS. | |
To gain a deeper understanding of their photophysical behavior, time-dependent density functional theory (TD-DFT) calculations were carried out using several functionals, including B3LYP, CAM-B3LYP, M06, M06-2X, and ωB97XD, paired with the 6-311G(d,p) basis set (Fig. S27 and S28†). The polarizable continuum model (PCM) was applied to simulate solvent effects, specifically for dichloromethane, to replicate experimental conditions. These calculations, performed using the Gaussian 09W software, provided insights into the electronic transitions and absorption spectra of the isomers. The results showed that CAM-B3LYP, M06-2X, and ωB97XD underestimated the absorption wavelengths by approximately 50 nm when compared to experimental values, highlighting their limitations for these systems (Table S10†). In contrast, both B3LYP and M06 provided predictions that closely matched the experimental absorption spectra, with deviations falling within acceptable ranges. Among these, M06 proved to be the most reliable, offering the most accurate reproduction of the spectral features observed experimentally. This finding establishes M06 as the preferred functional for accurately capturing the photophysical characteristics of the studied isomers (Fig. S29†). The calculated vertical electronic transitions, oscillator strengths, and the configurations of the major transitions, along with the computed frontier orbitals, are detailed in Tables S11 and S12.† These computational results align well with experimental data, reinforcing the proposed ICT mechanism and highlighting the reliabilty of the selected computational approaches in describing the electronic behavior of 2-BTCZCS and 5-BTCZCS. The oscillator strength (f) is significantly influenced by the degree of wavefunction overlap between the HOMO and LUMO during photoexcitation, where a weaker overlap results in a lower f value.53 For 5-BTCZCS, the notably low oscillator strength can be attributed to the diminished orbital interactions caused by the highly twisted geometry of the carbazole and phenyl groups. This reduced overlap impacts the intensity of the primary electronic transition associated with the absorption maximum. Moreover, the twisted conformation enhances charge separation, thereby promoting a stronger ICT character and greater stabilization of the excited state. These features are consistent with the slightly longer emission wavelengths observed for 5-BTCZCS and highlight the pivotal role of molecular geometry and electronic transitions in shaping the photophysical properties of the isomers. Despite the structural modulation, the optical properties of 2-BTCZCS and 5-BTCZCS remain similar, further emphasizing the influence of their electronic structures on their overall photophysical behavior.
Thermal properties
The thermal stability and phase transition behaviors of the isomers 2-BTCZCS and 5-BTCZCS were investigated using TGA and DSC (Fig. S30†). TGA analysis revealed distinct degradation patterns for the two isomers, highlighting differences in their molecular structures. 2-BTCZCS exhibited a single-step degradation process with a high decomposition temperature (Td) of 420 °C, reflecting excellent thermal stability and a uniform molecular composition. In contrast, 5-BTCZCS displayed a two-step degradation pattern with an onset Td of 392 °C, suggesting sequential degradation involving the cleavage of side chains followed by decomposition of the core framework. These findings emphasize the superior thermal robustness of 2-BTCZCS and the more complex thermal behavior of 5-BTCZCS, which can be attributed to structural variations between the isomers. DSC analysis provided further insights into the phase transition behaviors of these isomers. The sharp melting point (Tm) peaks observed at 150 °C in both isomers demonstrated their crystalline nature, indicative of high purity and well-defined crystal structures. Furthermore, the absence of significant exothermic crystallization peaks during cooling suggested stable thermal behavior without recrystallization. These results confirm that benzothiazole derivatives possess excellent thermal stability and phase transition characteristics, making them promising candidates for applications in optoelectronics and other high-performance materials.
Conclusions
This study explores the effect of benzothiazole positional substitution on the optical and solid-state properties of carbazole–cyanostilbene positional isomers, offering new insights into their structure–property relationships. The isomers 2-BTCZCS and 5-BTCZCS were synthesized to evaluate how acceptor positioning impacts photophysical properties in solution and solid state as well as molecular geometry, charge distribution, and intermolecular interactions. Despite these structural modifications, both isomers exhibit comparable optical properties, including strong AIE behavior, high solid-state quantum yields (up to 24%), and a minor mechanofluorochromic blue shift of 12 nm upon grinding. Crystallinity is preserved under mechanical stimuli, as evidenced by PXRD, while TGA and DSC analyses confirm robust thermal stability. SEM and DLS studies reveal consistent aggregation and morphological features, underscoring the structural integrity of these systems. Computational investigations using DFT and TD-DFT align closely with experimental data, confirming that positional substitution minimally affects molecular geometry and electronic properties. By elucidating the interplay between molecular structure, optical properties, and solid-state behavior, this work not only advances our understanding of donor–acceptor systems but also offers valuable guidance for the rational design of next-generation materials. These insights pave the way for the development of innovative applications in optoelectronics, smart materials, and other emerging technologies.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
CAS P. thanks, SERB/EEQ/2021/000180 for funding and support. A. A. thanks the National Institute of Technology, Calicut, for GATE-JRF fellowship. The authors acknowledge the Centre for Materials Characterisation NIT Calicut for NMR, PXRD facilities, HRMS (DST-FIST) and Centre for Computational Modelling and Simulations NIT Calicut. We would like to thank CLIF, IIT Palakkad, for SEM, lifetime and TGA facility. DST and SCXRD LAB, SAIF, IIT Madras is acknowledged for single-crystal XRD analysis.
References
- A. P. Kulkarni, X. Kong and S. A. Jenekhe, Adv. Funct. Mater., 2006, 16, 1057–1066 CrossRef CAS.
- Y. Li, J. Y. Liu, Y. D. Zhao and Y. C. Cao, Mater. Today, 2017, 20, 258–266 CrossRef.
- A. Slama-Schwok, M. Blanchard-Desce and J. M. Lehn, J. Phys. Chem., 1990, 94, 3894–3902 CrossRef CAS.
- M. Ahn, M. J. Kim, D. W. Cho and K. R. Wee, J. Org. Chem., 2020, 86, 403–413 CrossRef PubMed.
- X. Cao, D. Zhang, S. Zhang, Y. Tao and W. Huang, J. Mater. Chem. C, 2017, 5, 7699–7714 RSC.
- J. Tan, Y. Huo, N. Cai, S. Ji, Z. Li and L. Zhang, Chin. J. Org. Chem., 2017, 37, 2457 CAS.
- J. Jiang, D. Hu, M. Hanif, X. Li, S. Su, Z. Xie, L. Liu, S. Zhang, B. Yang and Y. Ma, Adv. Opt. Mater., 2016, 4, 2109–2118 CrossRef CAS.
- W. Z. Yuan, Y. Gong, S. Chen, X. Y. Shen, J. W. Lam, P. Lu, Y. Lu, Z. Wang, R. Hu, N. Xie and H. S. Kwok, Chem. Mater., 2012, 24, 1518–1528 CrossRef CAS.
- J. Lee, S. M. Lee, S. Chen, T. Kumari, S. H. Kang, Y. Cho and C. Yang, Adv. Mater., 2019, 31, 1804762 CrossRef PubMed.
- Y. Liu, Y. Wu, Y. Geng, E. Zhou and Y. Zhong, Adv. Funct. Mater., 2022, 32, 2206707 CrossRef CAS.
- J. Yu, Y. Zheng and J. Huang, Polymers, 2014, 6, 2473–2509 CrossRef.
- C. Chen and C. Fang, Chem. – Asian J., 2020, 15, 1514–1523 CrossRef CAS PubMed.
- S. Jaswal and J. Kumar, Mater. Today: Proc., 2020, 26, 566–580 CAS.
- K. W. Lee, Y. Wan, Z. Huang, Q. Zhao, S. Li and C. S. Lee, Adv. Mater., 2024, 36, 2306492 CrossRef CAS PubMed.
- C. Kalarikkal, A. Anjali, S. Bhattacharjee, K. Mapa and P. C. A. Swamy, J. Mater. Chem. B, 2025, 13, 1474–1486 RSC.
- P. N. Day, R. Pachter and K. A. Nguyen, J. Chem. Phys., 2014, 140, 184308 CrossRef PubMed.
- B. Traber, J. J. Wolff, F. Rominger, T. Oeser, R. Gleiter, M. Goebel and R. Wortmann, Chem. – Eur. J., 2004, 10, 1227–1238 CrossRef CAS PubMed.
- S. J. Sharma and N. Sekar, ChemistrySelect, 2022, 7, e202203262 CrossRef CAS.
- M. Huang, R. Yu, K. Xu, S. Ye, S. Kuang, X. Zhu and Y. Wan, Chem. Sci., 2016, 7, 4485–4491 RSC.
- J. Gierschner, J. Shi, B. Milia-Medina, D. Roca-Sanjuan, S. Varghese and S. Park, Adv. Opt. Mater., 2021, 9, 2002251 CrossRef CAS.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073–1095 CrossRef CAS PubMed.
- X. Huang, L. Qian, Y. Zhou, M. Liu, Y. Cheng and H. Wu, J. Mater. Chem. C, 2018, 6, 5075–5096 RSC.
- Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC.
- R. Yerushalmi, A. Scherz, M. E. van der Boom and H.-B. Kraatz, J. Mater. Chem., 2005, 15, 4480–4487 RSC.
- Y. Wang, X. Tan, Y.-M. Zhang, S. Zhu, I. Zhang, B. Yu, K. Wang, B. Yang, M. Li, B. Zou and S. X.-A. Zhang, J. Am. Chem. Soc., 2015, 137, 931–939 CrossRef CAS PubMed.
- G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160–2162 CrossRef CAS PubMed.
- C. Weder, J. Mater. Chem., 2011, 21, 8235–8236 RSC.
- J. Zhao, Z. Chi, Z. Yang, Z. Mao, Y. Zhang, E. Ubba and Z. Chi, Mater. Chem. Front., 2018, 2, 1595–1608 RSC.
- F. Khan, A. Ekbote, S. M. Mobin and R. Misra, J. Org. Chem., 2021, 86, 1560–1574 CrossRef CAS PubMed.
- J. Zhao, Z. Chi, Y. Zhang, Z. Mao, Z. Yang, E. Ubba and Z. Chi, J. Mater. Chem. C, 2018, 6, 6327–6353 RSC.
- H. Jiang, J. Sun and J. Zhang, Curr. Org. Chem., 2012, 16, 2014–2025 CrossRef CAS.
- S. Tan, Y. Yin, W. Chen, Z. Chen, W. Tian and S. Pu, Dyes Pigm., 2020, 177, 108302 CrossRef CAS.
- F. Zhao, Z. Chen, C. Fan, G. Liu and S. Pu, Dyes Pigm., 2019, 164, 390–397 CrossRef CAS.
- A. Afrin and P. C. A. Swamy, J. Mater. Chem. C, 2024, 12, 1923–1944 RSC.
- B. K. An, J. Gierschner and S. Y. Park, Acc. Chem. Res., 2012, 45, 544–554 CrossRef CAS PubMed.
- S. Sasaki, G. P. C. Drummen and G. Konishi, J. Mater. Chem. C, 2016, 4, 2731–2743 RSC.
- H.-J. Kim, J. Gierschner and S. Y. Park, J. Mater. Chem. C, 2020, 8, 7417–7421 RSC.
- A. Afrin and P. C. A. Swamy, New J. Chem., 2023, 47, 18919–18932 RSC.
-
(a) A. Afrin and P. C. A. Swamy, J. Org. Chem., 2024, 89, 7946–7961 CrossRef CAS PubMed;
(b) A. Afrin and P. C. A. Swamy, Chem. – Eur. J., 2025, 31, e202403644 CrossRef CAS PubMed.
- C. H. Chen, W. H. Lin, M. H. Hsieh and Y. T. Ke, J. Lumin., 2024, 269, 120501 CrossRef CAS.
- Y. Shen, P. Chen, J. Liu, J. Ding and P. Xue, Dyes Pigm., 2018, 150, 354–362 CrossRef CAS.
- K. S. Abou-Melha, Results Chem., 2024, 12, 101904 CrossRef CAS.
- A. Kundu, S. Karthikeyan, Y. Sagara, D. Moon and S. P. Anthony, ACS Omega, 2019, 4, 5147–5154 CrossRef CAS PubMed.
- Z. Su, H. Zhuang, H. Liu, H. Li, Q. Xu, J. Lu and L. Wang, J. Mater. Chem. C, 2014, 2, 5673–5680 RSC.
-
(a) J. Dhar, K. Swathi, D. P. Karothu, K. S. Narayan and S. Patil, ACS Appl. Mater. Interfaces, 2015, 7, 670–681 CrossRef CAS PubMed;
(b) R. B. K. Siram, D. P. Karothu, T. N. G. Row and S. Patil, Cryst. Growth Des., 2013, 13, 1045–1049 CrossRef CAS.
- A. Carella, R. Centore, F. Borbone, M. Toscanesi, M. Trifuoggi, F. Bella, C. Gerbaldi, S. Galliano, E. Schiavo, A. Massaro and A. B. Munoz-Garcia, Electrochim. Acta, 2018, 292, 805–816 CrossRef CAS.
- Q. Zhang, W. Zhu, M. Fang, F. Yin and C. Li, Spectrochim. Acta, Part A, 2015, 135, 379–385 CrossRef CAS PubMed.
- A. Kundu, S. Karthikeyan, D. Moon and S. P. Anthony, CrystEngComm, 2017, 19, 6979–6985 RSC.
- Q. Zhang, W. Zhu, M. Fang, F. Yin and C. Li, Spectrochim. Acta, Part A, 2015, 135, 379–385 CrossRef CAS PubMed.
- X. Feng, J. Zhang, Z. Hu, Q. Wang, M. M. Islam, J. S. Ni, M. R. Elsegood, J. W. Lam, E. Zhou and B. Z. Tang, J. Mater. Chem. C, 2019, 7, 6932–6940 RSC.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- J.-L. Bredas, J. E. Norton, J. Cornil and V. Coropceanu, Acc. Chem. Res., 2009, 42, 1691–1699 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR, HRMS, and characterization data for all the isomers, photophysical data, SCXRD, SEM, DLS, DFT and TD-DFT results (PDF). CCDC 2413279. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ce00132c |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*