
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Valorization of lignin for advanced material applications: a review
Rohan
Shorey
a,
Ayyoub
Salaghi
b,
Pedram
Fatehi
 b and
Tizazu H.
Mekonnen
*a
b and
Tizazu H.
Mekonnen
*a
aDepartment of Chemical Engineering, Institute of Polymer Research, Waterloo Institute of Nanotechnology, University of Waterloo, Waterloo, ON, Canada. E-mail: tmekonnen@uwaterloo.ca
bGreen Processes Research Centre and Chemical Engineering Department, Lakehead University, 955 Oliver Road, Thunder Bay, ON P7B SE1, Canada
First published on 8th March 2024
Abstract
Lignin is the second most abundant natural biopolymer after cellulose, constituting between 18 wt% and 35 wt% of lignocellulosic biomass. Its renewability, vast availability, and sustainability make it a desirable alternative and/or complementary macromolecule to conventional polymers. However, due to its relatively short and branched molecular size, high polarity and moisture absorption, lack of melt processability, and inferior dispersibility in nonpolar polymer matrices, its extensive application in polymers is limited. For effective and sizable application of lignin in polymers and composite materials, functionalization and modifications are necessary. This review paper intends to discuss the valorization of lignin as feedstock to produce alternative chemicals for converting lignin moieties into value-added products which can be used in advanced applications. A comprehensive overview of various physicochemical modification and functionalization routes of lignin is critically reviewed. Furthermore, the engineering of lignin bulk structures into nanomaterials with various morphologies for advanced material applications is covered. Certain lignin structures exhibit antibacterial and antifungal properties, thus fueling the potential employment of lignin in bioactive materials. The most significant advances in the valorization of lignin including lignin-incorporating sunscreen formulations, shape-memory applications, carbon fibers, additive manufacturing, pickering emulsion stabilizers, self-healing elastomers, etc. are critically reviewed. Lastly, perspectives for future development and opportunities for the effective valorization of lignin for material applications are discussed.
Sustainability spotlightTransitioning from the fossil fuel era towards a sustainable future necessitates the utilization of regenerative materials sourced from nature, characterized by a minimal environmental footprint. Sustainable macromolecules derived from biomass, such as lignin, offer a promising solution due to their abundance, cost-effectiveness, and adaptability to various modifications that serve as a feedstock for the development of sustainable materials. In this review, our primary focus is to showcase noteworthy achievements within a diverse research landscape dedicated to lignin valorization, modifications, and sustainable advanced materials. This work underscores the alignment with the UN SDGs, including clean water and sanitation (SDG 6), industry, innovation, and infrastructure (SDG 9), climate action (SDG 13), life below water (SDG 14), and life on land (SDG 15). |
1. Introduction
Conventional polymers have revolutionized human life to a great extent. From the polymer materials utilized in food packaging to running shoes and airplane parts, their applications are innumerable. Over the years, their fossil fuel origin, health and safety impacts, and the lack of environmental degradability that caused immense plastic pollution have spawned serious ecological concerns. Thus, to counter these increasing environmental concerns an inclination towards the development of renewable and sustainable material alternatives has been witnessed.1,2 Natural macromolecules, such as starch, lignin, and cellulose are favorable feedstocks for the design of biodegradable, multifunctional, and biocompatible polymers.3–5 Lignin, being the second most abundant natural biopolymer after cellulose that constitutes between 18 wt% and 35 wt% of biomass, with unique and complex aromatic and aliphatic backbone structures and abundant functional groups amenable for functionalization, is among the most appealing nature derived macromolecules for sustainable and functional material development.6–9Lignin's availability in the biosphere is estimated to be 300 billion tonnes, out of which 50 to 70 million tonnes of lignin exist in the black liquor generated from the paper and pulp industry.10–13 In a study by Dessbesell et al., the global production capacity of kraft lignin was estimated to be 265![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 t year−1.14 Currently, the vast majority of the lignin produced is incinerated as a low-cost fuel in power and heat generation facilities15 and a mere 2% is employed for the manufacture of chemicals and materials, such as dispersants, adhesives, and surfactants.10,16–18
000 t year−1.14 Currently, the vast majority of the lignin produced is incinerated as a low-cost fuel in power and heat generation facilities15 and a mere 2% is employed for the manufacture of chemicals and materials, such as dispersants, adhesives, and surfactants.10,16–18
Lignin is composed of three monomeric units (monolignols): p-coumaryl, coniferyl, and sinapyl alcohols (Fig. 1). In nature, these monolignol units undergo random radical polymerization to form complex three-dimensional (3D) structures.19 The 3D architecture, poor solubility in the majority of organic solvents, numerous functional groups and structural variability (due to the extraction method or the biomass source) make chemical characterization of lignin and its derivatives a complex task. Moreover, the employed extraction method dictates the sulfur content of the extracted lignin and thus it creates two major types of lignin, as illustrated in Table 1. Sulfur-containing lignins are composed of kraft lignin, lignosulfonates, and hydrolyzed lignin and their sulfur-free counterparts include soda lignin and organosolv lignin.
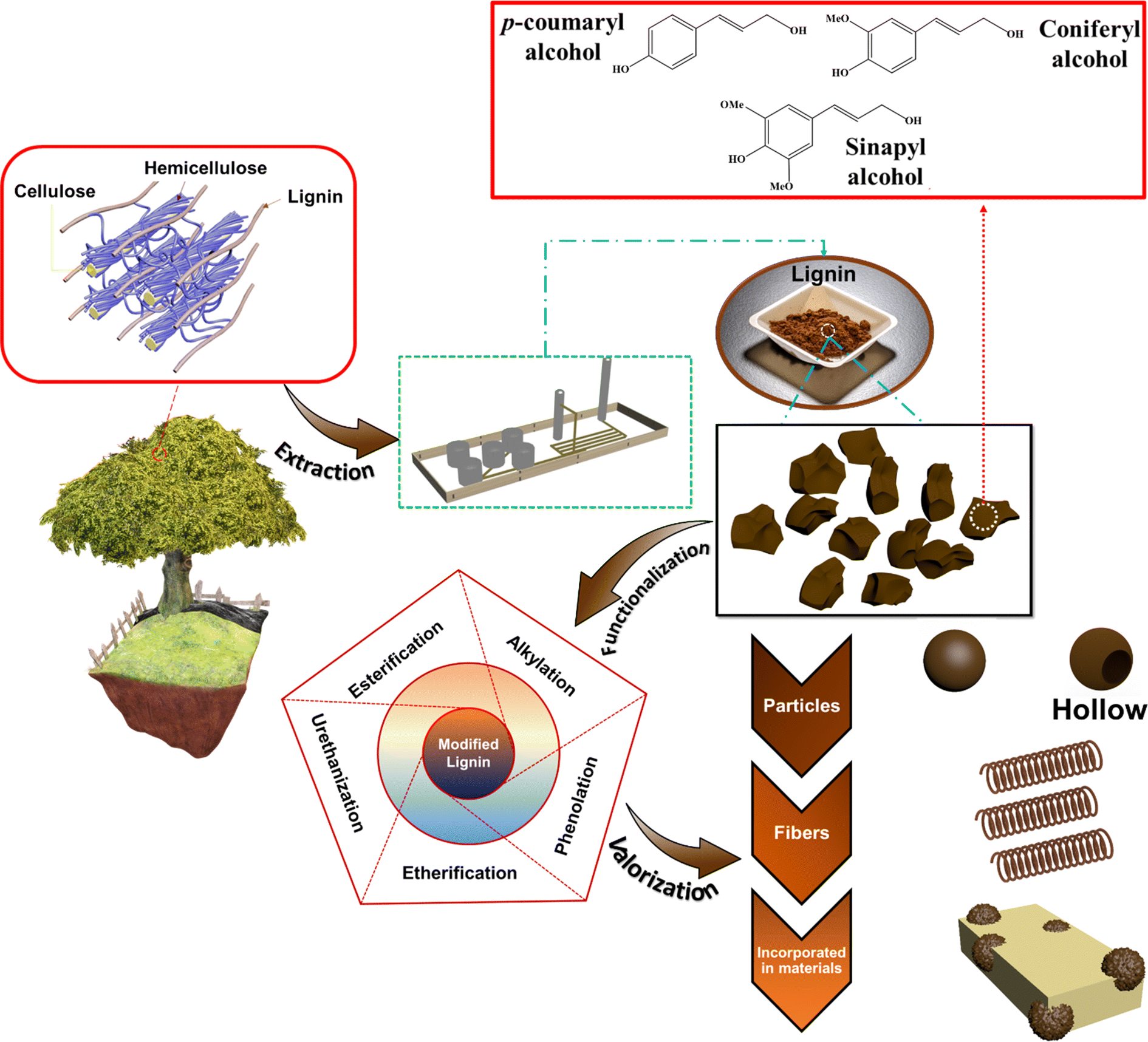 | ||
| Fig. 1 Valorization and functionalization of lignin. | ||
| Type of lignin | Sulfur-containing lignin | Sulfur-free lignin | ||
|---|---|---|---|---|
| Kraft lignin | Sulfonated lignin | Organosolv lignin | Soda lignin | |
| Alternate names | Kraft lignin powder and alkali lignin33 | Lignosulfonate | Acetosolv lignin | Alkaline lignin |
| Obtained from | Kraft pulping process | Sulfite pulping process | Organosolv pulping process | Soda pulping process |
| Appearance | Powder | Powder | Powder | Powder |
| Raw materials | Softwood and hardwood34,35 | Softwood and hardwood34,35 | Softwood, hardwood, and annual plants34,35 | Annual plants17,18,34,35 |
| Color | Light to dark brown | Brown/Black | Light brown | Black |
| Odor | Faint woody33 | Light odor | — | — |
| Mn (g mol−1) | 1000–3000 (ref. 34 and 35) | 15 000–50 000 (ref. 34 and 35) | 500–5000 (ref. 34 and 35) | 800–3000 (ref. 17, 34 and 35) |
| T g (°C) | 150–162 (ref. 33) | 130 (ref. 34 and 35) | 90–110 (ref. 34 and 35) | 140 (ref. 17, 34 and 35) |
| Sulfur content (%) | 1.2–2.4%33,34 | 3.5–8%34,36 | 0 (ref. 34 and 36) | 0 (ref. 17, 34 and 36) |
| PDI | 3.0–4.5 (ref. 33) | 6.0–8.0 (ref. 34 and 35) | 1.5–2.5 (ref. 34 and 35) | 2.5–3.5 (ref. 17, 34 and 35) |
| Solubility | Soluble in alkali, and low solubility in water33 | Completely soluble in water35 | Organic solvents35 | Alkali17,35 |
| Aliphatic OH (mmol OH g−1) | 1.5 (ref. 34 and 37) | 5.7 (ref. 34 and 37) | 2.9 (ref. 34 and 38) | 1.3 (ref. 34 and 37) |
| Phenolic OH (mmol OH g−1) | 1.3 (ref. 34 and 37) | 0.6 (ref. 34 and 37) | 2.8 (ref. 34 and 38) | 1.6 (ref. 18, 34 and 37) |
| Carboxylic acid (mmol g−1) | 0.6 (ref. 34 and 37) | 0.3 (ref. 34 and 37) | 0.48 (ref. 34 and 39) | 0.7 (ref. 18, 34 and 37) |
| Market leaders | West Fraser Timber Co. Ltd, Canada | Borregaard USA, Inc. (S/P) GREEN AGROCHEM, China | Nippon Paper Industries Co., Ltd, Japan | Biosynth group |
| WestRock Company, US | Nippon Paper Industries Co., Ltd, Japan | Northway Lignin Chemical, Canada | Northway Lignin Chemicals | |
| Domtar Corporation, US | The Dallas Group of America | Domtar Corporation, US | ||
| Stora Enso, Finland | Sappi Limited, South Africa | The Dallas Group of America | ||
| RISE, Sweden | Borregaard USA, Inc. (S/P) | |||
| Liquid Lignin Company, US | ||||
Kraft lignin is derived from lignocellulosic biomass via the kraft pulping process, thus making it the most abundantly available type of lignin. Brown in colour, kraft lignin is moderately soluble in water. As a byproduct of paper production, a vast majority of kraft lignin is burned to supplement the energy requirements of pulp mills.19 Lignosulfonates are typically sourced from the paper and pulp industry too and are extracted using the sulfite pulping process10,19 Contrary to Kraft lignin, sulphonated lignin is completely soluble in water. This is attributed to the presence of an extensive amount of sulfonate in its chemical structure.19 Sulfur-free organosolv lignins are the products of intentional biorefinery processes.10 In addition, soda lignin is another type of sulfur-free lignin. It is employed to extract lignin from annual plants employing the soda process, which is the oldest pulping method.19 Furthermore, wood could be hydrolyzed employing various acids, bases, or enzymes resulting in dissolution of sugars from cellulose and hemicellulose fractions. The dissolved sugars could be utilized for the production of value-added products (for instance, ethanol). Moreover, hydrolyzed lignin moieties (sulfur-free) remain undissolved and are often utilized as an economical fuel alternative.20,21
Due to the inherent limitations of lignin, such as high polarity, inferior compatibility with polymers, poor melt flow properties, inferior thermal stability, structural complexity, and the variability of chemical structure (due to differences in lignocellulosic feedstock and biorefining processes), lignin moieties have limited applicability in polymers.22 Although reports are available in the literature on lignin valorization, the focus of this review paper is on the use of lignin in advanced material applications via various modification pathways and its associated challenges.
Various companies have tried to develop commercial scale lignin-based products. A Norwegian chemical company, Borregaard ASA, has developed a range of lignin-based products that include emulsion stabilizers, binding agents, dispersing agents, crystal growth modifiers, and complexing agents.23 Hexion is also making substantial progress in the utilization of lignin as a partial replacement of phenols for insulating foams, molding compounds, and adhesives.24 Other major companies, such as Huntsman, China Petroleum & Chemical Corporation, and Rampf Holdings, have also described processes for the utilization of lignin as a partial polyol replacement agent and filed patents.25 In this work, we focused on the applications of lignin in polymers, enhancing its utility in this specific field and relevant functionalization reactions and routes.15,26–30 The effective employment of lignin in various value-added polymer system applications could aid in fulfilling some of the United Nations Sustainable Development Goals (UN SDGs), such as diverting from fossil resources, implementing new degradation pathways, and incorporating less toxic reagents and solvents in production processes, thus promoting a circular economy.31,32
2. Lignin functionalization routes
Lignin has abundant functional groups, such as hydroxyl (–OH), carboxyl (–COOH), methoxy (–OCH3), and carbonyl (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) moieties, making it amenable to various functionalization reactions. The functionalization platforms reported include electrochemical,27 biological,26 photochemical29 and chemical15,27,29,40 processes. The chemical functionalization of lignin has particularly attracted substantial interest and could be carried out by employing various approaches, including depolymerization and fractionation, chemical modification of lignin's active moieties, and polymer grafting.41 In a study by Laurichesse et al., an extensive overview of various functionalization routes of lignin was reviewed.40
O) moieties, making it amenable to various functionalization reactions. The functionalization platforms reported include electrochemical,27 biological,26 photochemical29 and chemical15,27,29,40 processes. The chemical functionalization of lignin has particularly attracted substantial interest and could be carried out by employing various approaches, including depolymerization and fractionation, chemical modification of lignin's active moieties, and polymer grafting.41 In a study by Laurichesse et al., an extensive overview of various functionalization routes of lignin was reviewed.40
The solubility of lignin macromolecules in appropriate solvents is a critical parameter for effective modification reactions.42,43 Conventional solvents for lignin include dimethyl sulfoxide (DMSO), toluene, acetone, ethanol, pyridine, methanol, dimethyl formamide (DMF), tetrahydrofuran (THF), chloroform, and dichloromethane.43–46 Also, lignin moieties are known to be soluble in hot alkaline solutions.44,46,47 Additionally, the solubility of lignin could be further improved by fractionation.43
In recent years, many studies focusing on the chemical functionalization of lignins have been conducted.40,48–50 As illustrated in Fig. 2, various chemical modification processes of lignin to functionalize its active moieties have been studied, including alkylation, esterification, etherification, urethanization, and phenolation.41,51 A few of the aforementioned functionalization routes have been further discussed.
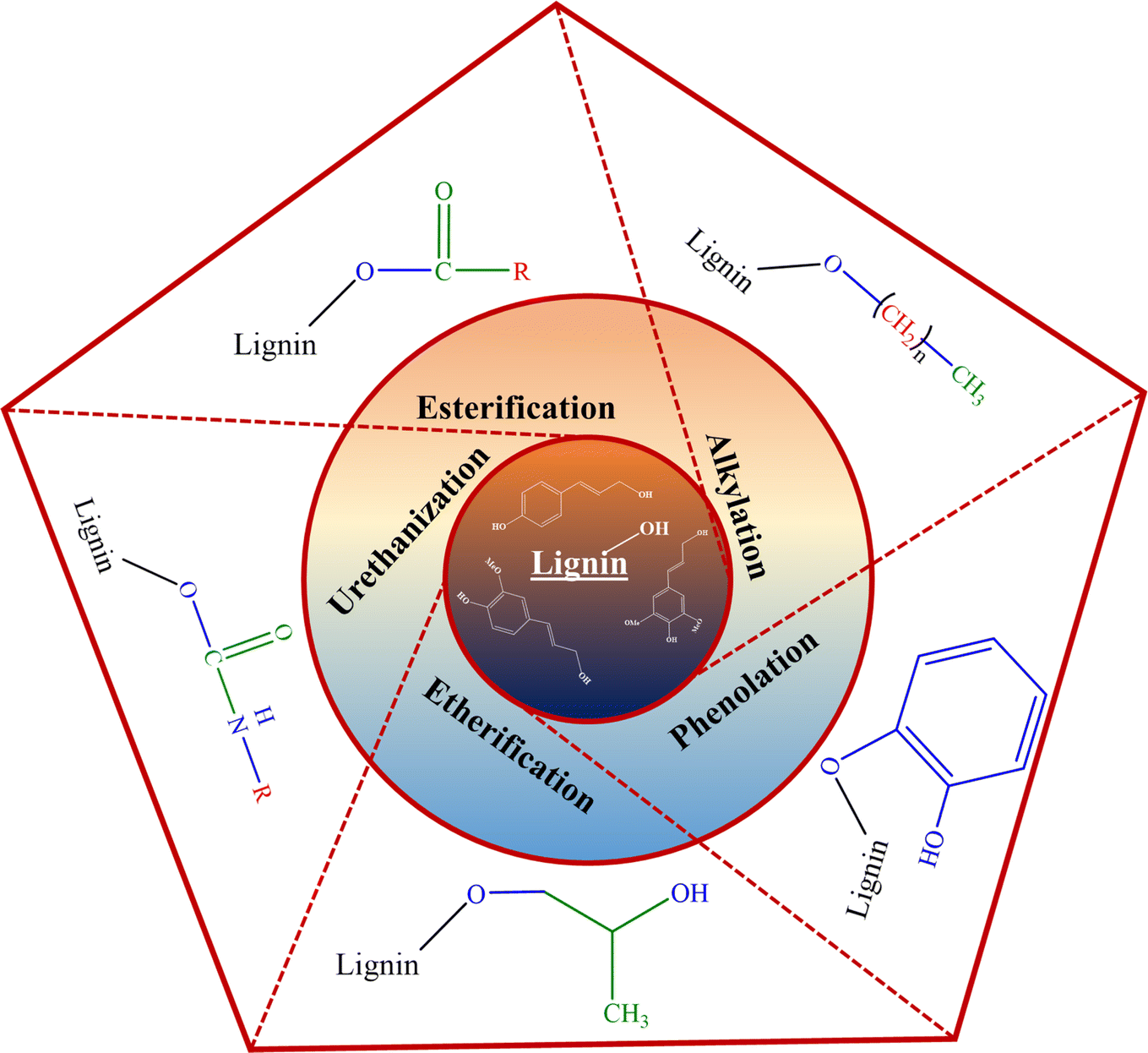 | ||
| Fig. 2 Chemical functionalization reactions of lignin. | ||
Esterification reactions involve the formation of an ester bond between the carboxylic acid functional group of the esterifying agent and the hydroxyl moieties of the lignin fraction. This modification route using fatty acids has been shown to produce modified lignin moieties with enhanced hydrophobicity and thermoplastic properties.44,45,48,51 In a study by Shorey et al., multiple activated fatty acids (with varying aliphatic carbon chain lengths) in the presence of pyridine as a reaction medium, a catalyst, and an acid scavenger were employed to functionalize kraft lignin. The resulting modified lignin samples exhibited enhanced hydrophobic behavior and better dispersion in nonpolar polymer matrices.44
Another functionalization route, the urethanization of lignin, translates to formation of a urethane bond between the hydroxyl functional groups of lignin and the employed modifying agent.46 One such functionalization (silylation) entails the substitution of lignin's hydroxyl moieties by silyl functionalities.46 In a separate study, the silylation of kraft lignin employing 3-(triethoxysilyl)propyl isocyanate (TEPI, 95%) as a silylating agent and tin(II) 2-ethyl hexanoate (92.5–100%) as a catalyst was undertaken. The modified lignin possessed enhanced thermal stability, lower glass transition temperature, enhanced solubility in organic solvents, and hydrophobicity.46 Similar results were observed in other silylation studies making it appealing for polymeric material applications.44,48
Although the various functionalization routes impart baseline lignin samples with numerous desirable properties, the key challenges in lignin functionalization (including the heterogeneous structure, the presence of impurities, varying solubility in organic solvents and unique chemical reactivity of various lignin types) render it a complicated endeavor.52
Moreover, the functionalization of lignin moieties can also be carried out by grafting or copolymerizing foreign monomeric units onto its chemical structure.44 Hence, various advanced composites can be fabricated. In such a study by Sun et al., lignin was functionalized by grafting multiple monomeric units (lauryl methacrylate (LMA) and tetrahydrofurfuryl methacrylate (THFMA)) onto its structure by employing free-radical polymerization.53 The functionalized lignin Lig-g-P(LMA-co-THFMA) was melt compounded with polylactic acid (PLA) to fabricate composites of different compositions. The 20 wt% functionalized lignin loaded composite recorded a significant increment (in excess of 190%) in the elongation at break value over the baseline PLA specimen.53
The functionalization of lignin moieties for their effective valorization is essential. However, there are several limitations associated with the functionalization routes. Primarily, the heterogeneity and variability of lignin's chemical structure limit the development of a controlled modification protocol. Furthermore, the high polarity and resulting hydrophilicity associated with the extensive hydroxyl groups result in inherent tendency to agglomerate in mostly non-polar polymer host-matrices, thus limiting its industrial application in polymer systems. Furthermore, the dark brown color imparted by the lignin to host matrices might influence consumer acceptance of some products. Hence, the functionalization of lignin and/or further valorization to counter the limitations is advantageous for its effective utilization.
The present study aimed to explore the eventual utilization of functionalized lignin in value-added applications. The incorporation of lignin in sunscreen formulations, carbon fibers, Pickering emulsions, shape-memory applications, self-healing elastomers, additive manufacturing processes, etc. has been meticulously discussed. Recent successes and limitations in commercializing these lignin incorporating products have been discussed. Lastly, the influence of chemical structure, performance of lignin-based alternatives compared to their conventional counterparts, and the sustainability of various lignin loaded formulations were reviewed and reported.
3. Lignin engineered for advanced material applications
In the past decade, the use of biopolymers in the design and manufacture of specialized materials with controlled structures has been witnessed. Herein, the literature on the engineering and functionalization of lignin into various structures, morphologies, shapes, and sizes for functional material applications is discussed.3.1. Lignin-polymer blends for value-added applications
A polymer blend of two or more polymers entails interdiffusion amongst the constituent polymer fractions.54 Based on the interaction (mutual miscibility) of the constituent polymers, the level of interdiffusion varies. While the polymer fractions with weak interactions have been witnessed to result in heterogeneous blends with agglomeration sites, the constituents with strong interactions exhibit homogeneity.54–60Lignin having a complex structure exhibits an inherent thermoplastic behavior caused by inter- and intramolecular hydrogen bonds.61,62 Moreover, at high temperature lignin presents thermosetting behavior which can be attributed to the high cross-linking reactions that lignin moieties undergo at elevated temperatures.61,63,64 This results in inefficient blending of lignin specimens with other polymer materials. Effective methods of overcoming these processing limitations include addition of plasticizers and chemical modification of lignin. Blends of pristine lignin and their functionalized counterparts with various polymer materials have been reported in the literature.54
The blends of lignin with conventional polymers have been extensively researched.65,66 In a review article by Kun et al., various avenues of lignin-polymer blends were extensively discussed.66 In a study by Sailaja et al., lignin phthalates were melt blended with low density polyethylene (LDPE) fractions.67 Amongst various formulations, maleic anhydride grafted LDPE was added to improve interfacial adhesion, thus, acting as a compatibilizer. The resulting blend exhibited excellent thermal, mechanical, and morphological properties. Moreover, the presence of interlocked surfaces resulting from superior adhesion between the lignin moieties and the LDPE fractions in the maleic anhydride grafted LDPE compatibilized blends was recorded.67
In another study exploring the physical properties of lignin-polypropylene (Lig-PP) blends, the feasibility of bromododecane modified lignin was studied.68 It was worth noting that the addition of lignin to PP enhanced its fire retardancy and toughness. For a 20 wt% lignin loaded formulation, an LOI value of 25.2 was recorded. A further increase in lignin content resulted in specimens exhibiting enhanced damping effect.
The use of lignin as a reinforcing agent in synthetic rubber matrices has been witnessed. In such a study by Košíková et al., styrene-butadiene (SBR) rubber was filled with sulfur-free lignin fractions.69 A significant improvement in mechanical properties for lignin filled SBR composites was recorded over the baseline unfilled formulation. For instance, the 60 phr lignin loaded specimen accounted for a 94% increment in the tensile strength at break over a pristine SBR specimen. The homogeneous dispersion of lignin spherical particles (100 nm) in the composite matrix was observed via scanning electron micrographs.
Furthermore, the physiochemical properties of a compostable polymer matrix (PLA) with two different lignin specimens (alkali lignin and organosolv lignin) were studied.70 Employing different amounts of individual lignin samples and poly(lactic acid) (PLA), various blends were melt-blended employing a twin-screw extruder. Furthermore, each type of lignin underwent acetylation. As hypothesized, the acetylated lignin samples exhibited superior compatibility with the polymer matrix to their unmodified counterparts. Moreover, the acetylated lignin blends displayed enhanced resistance to hydrolytic degradation, thermal stability, retained tensile strength and increased elongation at break values.70 Blends of lignin with various polymeric matrices could potentially help achieve multiple UN SDGs, including SDG 9 (industry, innovation and infrastructure), SDG 12 (responsible consumption and production), SDG 13 (climate action), and SDG 14 (conserve and sustainably use the oceans, seas and marine resources for sustainable development).
A German manufacturer Tecnaro – Arboform® produced a commercial lignin containing thermoplastic products.40,71 Made from an amalgam of lignin and various natural fibers sourced from hemp, wood, flax, and other fibrous plants, it exhibits high tensile strength (transverse) and very low swelling with good fire resistance. However, its higher price (around 2.5 € per kg) in comparison to the cost of conventional thermoplastic materials (1 € per kg to 2 € per kg) limits its extensive utilization in the plastic market.
3.2. Lignin microspheres and nanospheres
In recent times, polymer-based micro- and nanospheres have garnered significant attention. For instance, the use of polymer microspheres and nanospheres in industrial applications, including the encapsulation and controlled release of fertilizers, pesticides, and other bioactives has been studied.72 The surge in sustainable practices has further fueled transition efforts from conventional plastics towards the employment of biobased and renewable polymer alternatives, such as lignin in agricultural, food, biomedical, and cosmetic controlled release applications.73,74 The sphericity of designed micro- and nanospheres enables a homogeneous release of active substances into the surrounding environment, and the facile manufacturing processes make them a favorable option for widescale applications.75,76Many routes for the fabrication of lignin micro- and nanoparticles have been reported in the literature, including emulsion inversion, antisolvent exchange, acid precipitation, chemical modification, ultrasonic irradiation, controlled drying, and a combination of multiple methods.77–83 For instance, Österberg et al. provided a review of various routes to fabricate spherical lignin particles and their applications.76 To fabricate lignin-based micro and/or nano particles, the employment of antisolvent exchange and acid precipitation techniques has been most prevalent.83 In the acid precipitation technique, lignin samples are initially dissolved in alkaline solution with a pH more than 11. This is known to deprotonate hydroxyl (phenolic) and carboxylic acid moieties. Further addition of an acidic medium (pH 3) protonates the hydroxyl and carboxylic acid groups, thus reducing the surface charge and in turn initiating particle precipitation.83 On a similar note, in the antisolvent precipitation technique, the lignin moiety is dissolved in an organic solvent (for instance THF), and with the slow addition of an antisolvent, the lignin fractions form spherical colloidal particles, as illustrated in Fig. 3f.
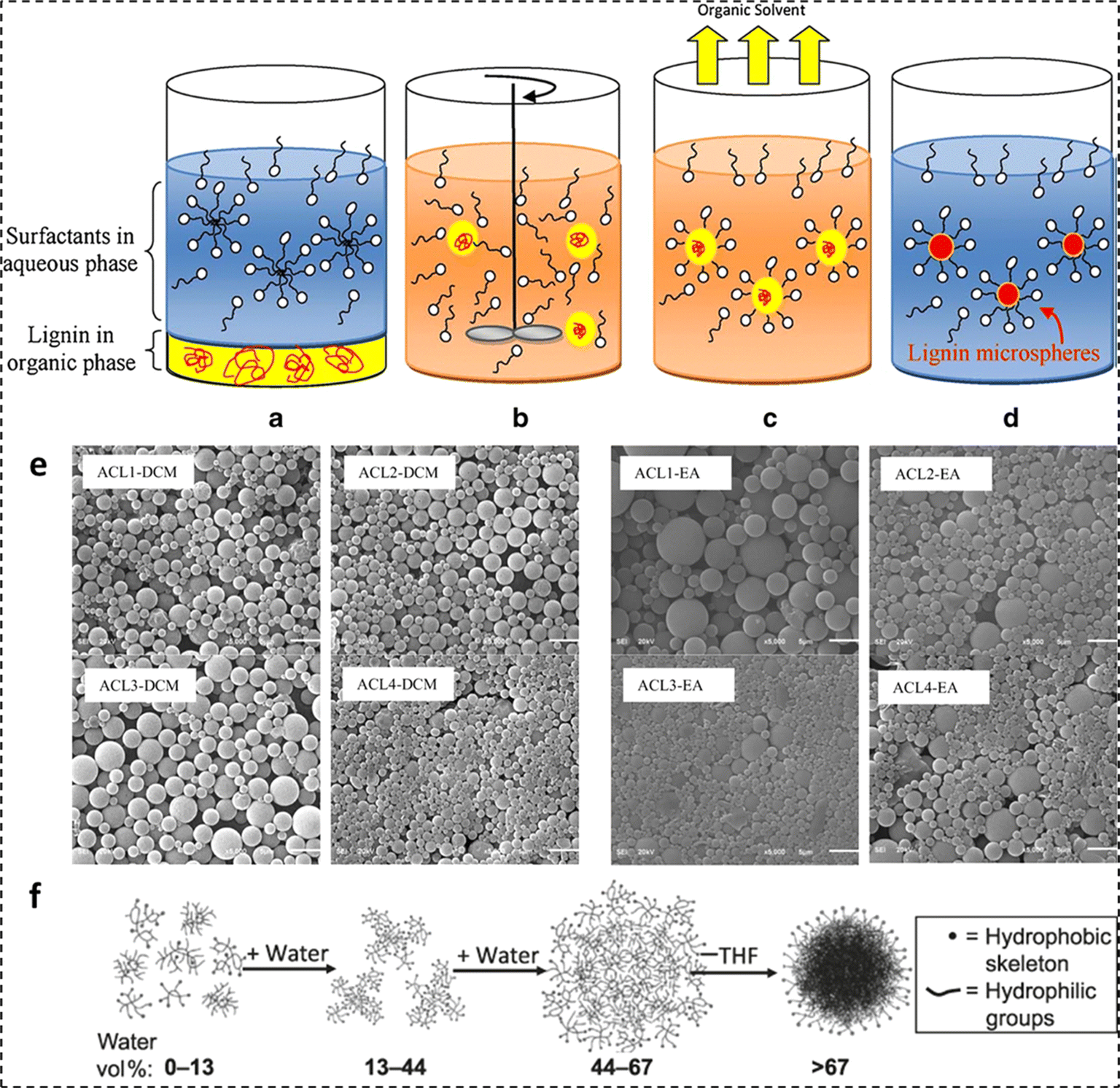 | ||
| Fig. 3 Four steps of ESE: (a) addition of two phases with opposite polarities, (b) emulsification, (c) evaporation of the solvent phase, and (d) microparticle formation, (e) lignin microsphere SEM micrographs (reproduced from ref. 72 with permission from Wiley, copyright 2012), and (f) lignin microparticle formation employing solvent exchange (reproduced from (ref. 84) with permission from RSC, copyright 2014). | ||
In a study by Sameni et al., microspheres of four types of lignin were fabricated employing a combination of the above-mentioned routes known as emulsion solvent evaporation (ESE).72 The four lignin samples underwent acetylation to improve lignin solubility in organic solvents. Then, the emulsion was prepared by homogenizing a lignin mixture of organic phase and aqueous phase (20 mL polyvinyl alcohol with a concentration of 0.2% w/v at 10000 rpm for 30 s. Next, 100 mL distilled water was added to the emulsion and the organic solvent was removed through magnetic stirring for 3 h in an open beaker to form lignin particles. The molecular weight of lignin is a critical parameter for the particle diameter when lignin acetate microspheres are prepared in an organic solvent. The reason for this is the fact that the diffusion and evaporation of the solvent take longer time and lignin with higher molecular weight generates more microspheres. After forming the particles and solvent removal, the colloid was centrifuged for 10 min at 9000 rpm and washed twice with deionized water. Finally, the micro-spheres were freeze-dried and stored. To emphasize the dependency of ESE produced microspheres and the type of lignin on the choice of the appropriate solvent, four different lignin samples including lignin isolated from industrial hardwood and non-wood residue (Bioethanol Plant, USA) (ACL1), lignin extracted from black liquor (Kraft pulp mill, Brazil) (ACL2), commercial softwood kraft lignin (Indulin AT, Westvaco Corporation, USA) (ACL3), and commercial non-wood soda lignin (Protobind 2000 ALM, India) (ACL4) were employed.72 The first step involved the addition of two phases with opposite polarities (lignin in organic solvent (dichloromethane (DCM) or ethyl acetate (EA)) and an emulsifier (polyvinyl alcohol (PVA)) in an aqueous phase) to a container, as illustrated in Fig. 3a. The next three steps were the emulsification of the two phases (Fig. 3b), evaporation of the solvent phase (Fig. 3c), and microparticle formation (Fig. 3d).
Scanning electron micrographs of the prepared microparticles were recorded. As evident from Fig. 3e, microparticle sizes ranging between 1280 nm and 1349 nm for DCM and 1313–2545 nm for EA were obtained. In another study to fabricate lignin nanoparticles, a solution of acetylated lignin (solubilized in THF) was precipitated with an anti-solvent (H2O). Using DLS, lignin nanoparticles with a hydrodynamic radius (Rh) of around 100 nm were recorded.
In a separate study by Saidane et al., lignin beads (micro-/nano-scale) were prepared employing epichlorohydrin as a crosslinking agent.85 Following the reaction (scheme outlined in Fig. 4a), smooth spherical lignin beads ranging in size between 100–800 μm were successfully obtained (Fig. 4b).85 The formulated beads underwent multiple functionalization reactions for final application in scavenging carbonyl compounds.85
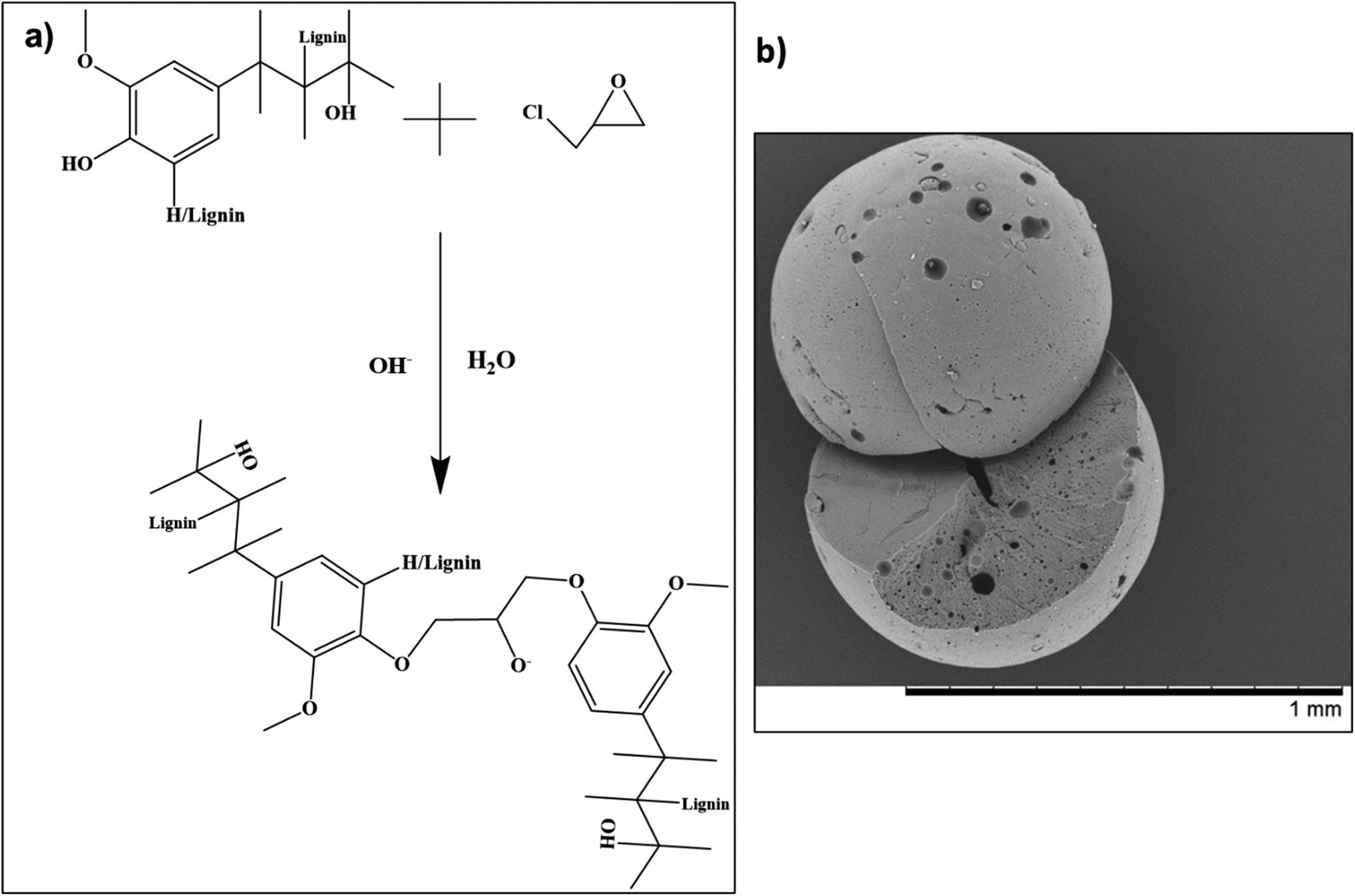 | ||
| Fig. 4 (a) Reaction mechanism for Kraft lignin with epichlorohydrin, and (b) scanning electron microscopy image of a fabricated lignin bead (reproduced from ref. 85 with permission from Wiley, copyright 2010). | ||
In another study, for the first time, lignin beads in conjunction with treated cellulose pulp were prepared and characterized for their antimicrobial activity against Escherichia coli and Staphylococcus aureus.86 The preparation procedure was initiated with a dispersion of lignin (up to 40%) and solvent treated (ethanol/HCl) cellulose pulp in an aqueous solution (constituting 12% urea/7% sodium hydroxide) with varying solid concentration ranges. This solution was dropped into an acidic medium to enable lignin–cellulose bead coagulation. The inherent fluorescence of lignin was vital in observing its distribution in the obtained beads. The distribution of lignin in the fabricated beads was homogeneous. Leaching of lignin fractions from the beads was recorded. While the lignin–cellulose beads did not display significant antibacterial activity against Escherichia coli, a significant reduction in the growth of Staphylococcus aureus was observed. With an increase in the lignin fraction, the growth of S. aureus plummeted.86
In another study, the impact of adding a commercial lignin extraction process to a kraft pulp mill on its environmental impact was studied. For this, a cradle-to-gate life cycle analysis was conducted.87,88 The results indicated a significant reduction in environmental impact, thus, signifying the benefits of lignin incorporating biomaterials as renewable alternatives to their conventional counterparts.87,88
In another study by Chen et al., the formulation of lignin beads from pine black liquor was investigated.89 Lignin beads ranging in size from 300–450 μm were prepared via reverse-phase suspension polymerization. The fabricated lignin beads displayed excellent L-lysine adsorption from aqueous media via electrostatic interaction.
To further enhance the uptake capacity of hollow spheres, a subclass of hollow spheres known as open-mouthed hollow capsules has gained significant attention.94–97 Various organic polymers and inorganic oxides have been employed in the preparation of open-mouthed hollow capsules, including silica (SiO2),98,99 polymethylsilsesquioxane (PMSQ),100 titanium dioxide (TiO2),101 polystyrene (PS),102 polystyrene/poly(3,4-ethylenedioxythiophene) (PS/PEDOT),103 chiral phenylacrylo-nitrile tartaric acids,104 polystyrene/poly(divinyl-benzene),105 and poly(acrylamide-ethylene glycol dimethacrylate).106
Lignin is a new entry material for the design and fabrication of hollow nanospheres. In a study by Xiong et al., a facile self-assembly method was employed by dissolving enzymatically hydrolysed lignin in THF and adding water to the solution to fabricate lignin hollow nanospheres (Fig. 5a).107 Due to π–π interactions, the layers were self-assembled from outside to inside, with the outside layer exhibiting hydrophobicity and the inside layer being hydrophilic. A positive correlation between the concentration of lignin, the thickness of the shell wall, and the diameter of the nanospheres was observed. On the flip side, with an increase in the concentration of lignin, the pore volume of nanospheres, the diameter of the single hole, and the surface area have been reduced. An average particle size of around 500 nm was observed. Furthermore, an increment in the agitation speed or speed at which water was added to the system resulted in a reduction in the lignin hollow nanosphere diameter, as evidenced by the SEM micrographs in Fig. 5.107
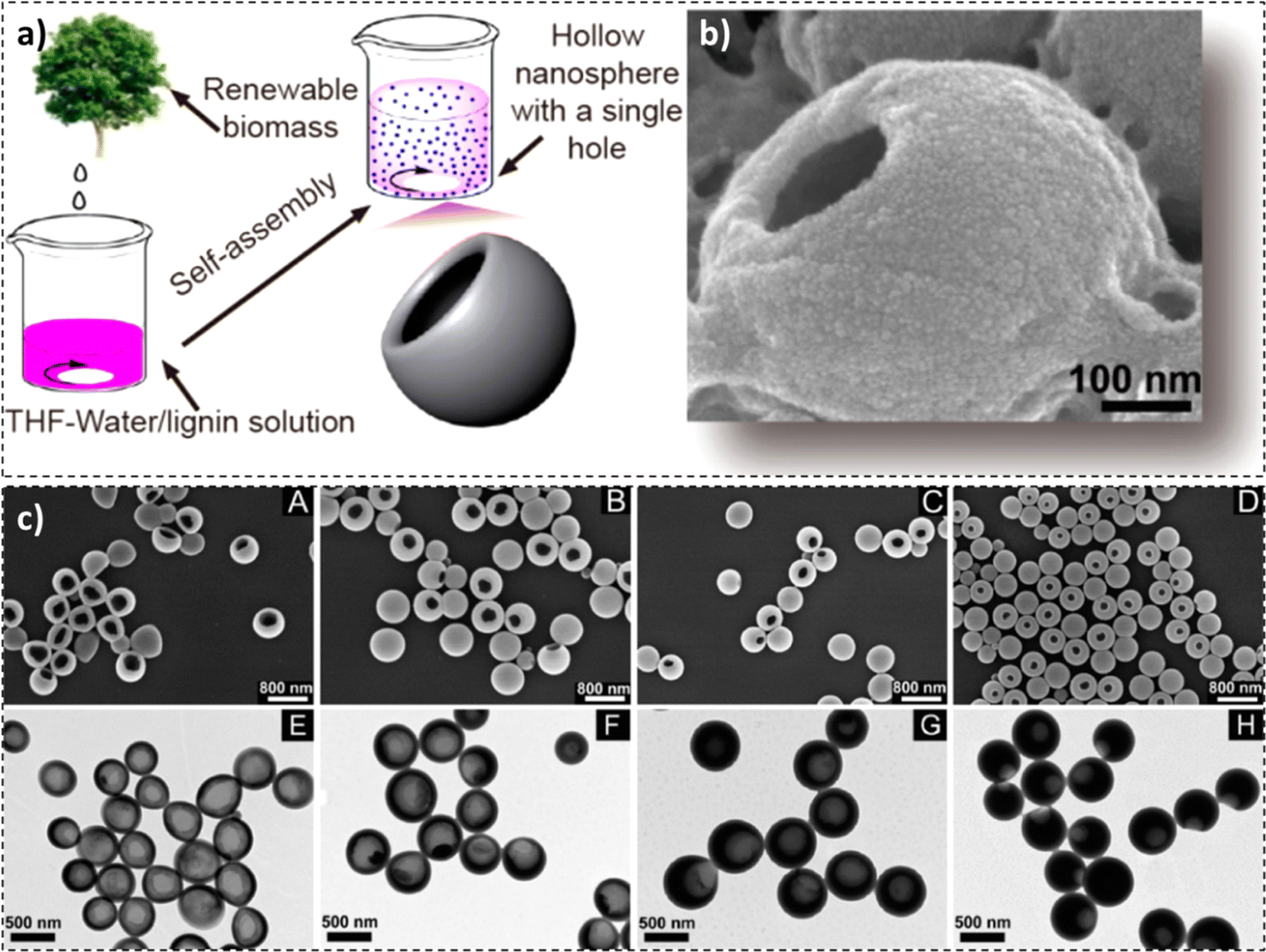 | ||
| Fig. 5 (a) Synthesis process, (b) SEM image, and (c) SEM images (A–D) and TEM images (E–H) at different predropping lignin concentrations: 0.5 mg mL−1 (A and E), 1 mg mL−1 (B and F), 1.5 mg mL−1 (C and G), and 2 mg mL−1 (D and H) (reproduced from ref. 107 with permission from ACS, copyright 2017). | ||
In a study by Sameni et al., the effect of process variables (mixing time, surfactant concentration, shear rate, and the organic solvent used) on the properties of lignin acetate microsphere fabrication employing ESE was studied. An average particle size between 20 and 100 μm was observed.108
3.3. Lignin nanotubes and nanofibers
In principle, cylindrical carbon nanotubes (buckminsterfullerene structure) can be utilized as universal delivery vehicles for therapeutic agents. However, their chemical inertness, sharp edges and toxicological concerns limit their versatility and suitability. Alternatively, carbon nanotubes can also be synthesized employing polymeric materials. In a study by Bianco et al., polymer based carbon nanotubes were fabricated employing polyelectrolytes, collagen, and poly(3,4-ethylenedioxythiophene) via template-mediated synthesis using nano-porous membranes containing arrays of aligned cylindrical pores.109 This approach could result in either polymer-functionalized nano-porous membranes or polymer-based nanotubes released from a sacrificial template. Although the development of polymer nanotubes appears to be a promising avenue for biomedical and biotechnological applications their commercial applicability is limited due to the inability of their components to maintain their structure indefinitely.110In a recent study by Ten et al., the use of lignin nanotubes as a gene delivery vehicle for human cells was studied.111 With the use of lignin nanotubes (LNTs), the cytotoxicity of carbon nanotubes and the immunogenicity of viral vectors could be avoided.111 Employing a sacrificial alumina template, aromatic plant cell wall (lignin) based nanotubes were synthesized. Five different lignin sources were selected for the isolation, including sorghum ((Sorghum bicolor (L.) Moench)), debarked stems of loblolly pine ((Pinus taeda L.)), debarked stems of poplar (Populus deltoides W. Bartram ex Humphry Marshall; POP), brown midrib6 (bmr6) and sugar cane bagasse from sugar cane (Saccharum spp.). As evident from the confocal micrographs (Fig. 6), the presence of lignin nanotubes (indicated by white arrows) in the immortalized human cell line (HeLa) cells and the nucleus of the cell without the presence of any auxiliary agents could be observed.
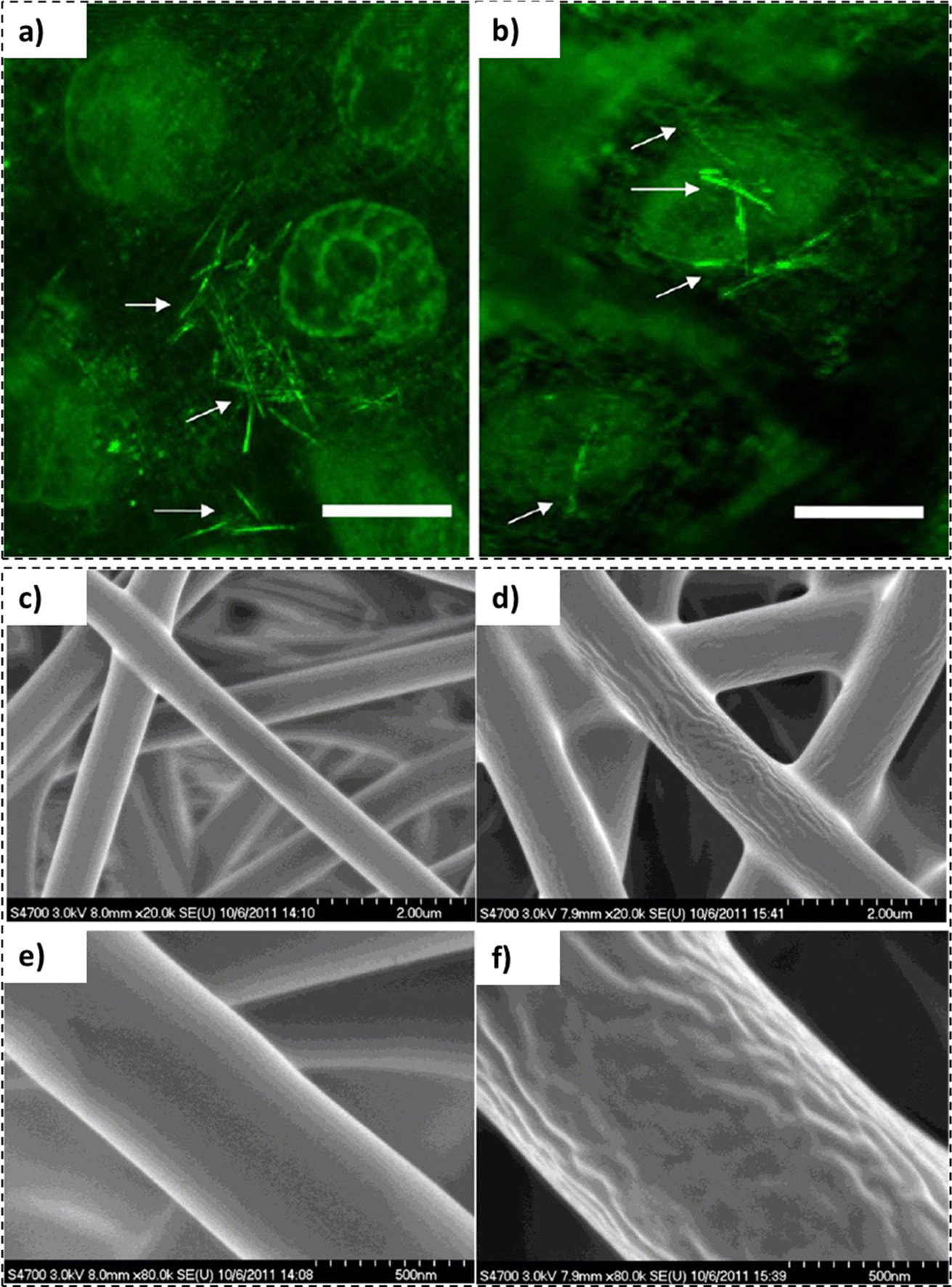 | ||
| Fig. 6 Confocal micrographs: (a) lignin nanotubes fabricated employing isolated pine lignin and thioglycolic acid and (b) isolated pine lignin nanotubes manufactured with NaOH (scale bar representing 15 μm) (reproduced from ref. 111 with permission from ACS, copyright 2012), SEM micrographs: lignin nanofibers (c and e) and PNIPAM grafted lignin nanofibers (d and f) (reproduced from ref. 112 with permission from ACS, copyright 2012). | ||
In recent times, increasing interest in the production and utilization of lignin nanofibers has been witnessed. In a study by Gao et al., surface-initiated atom-transfer radical polymerization (SI-ATRP) was employed to produce poly(N-isopropyl acrylamide)(PNIPAM) grafted softwood kraft lignin nanofiber mats.112 The nanofibers exhibited diameters between 790 and 1150 nm (Fig. 6c–f), thus, exhibiting potential applications in purification and separation devices.112,113
3.4. Lignin filled composites in additive manufacturing applications
In the recent decade, technological advances have made 3D printers more accessible to general consumers, thus, in turn fueling interest in additive manufacturing (AM) better known as 3D printing fabrication of materials. As illustrated in Fig. 7, 3D printing comprises three steps: development of a computer-aided design (CAD) model, preprocessing of the model, and additive manufacturing of the product.114 Although conventional fabrication materials (such as metals, conventional polymers, inorganic glasses, ceramics, and composites) are viable options for research and development at additive manufacturing's advent, their utilization at the consumer level raises sustainability concerns.34,115,116 The predicted 3D printing market size between the years 2022 and 2032 is presented in Fig. 7. In the year 2032, the global AM market size is projected to be 98.3 billion USD with a material sale potential of 900 million USD.117 Photopolymer sales are expected to contribute the biggest fraction (350 million USD) to this value, with its primary application being in the prototyping stage and molding application. Furthermore, AM metals and polymer powders could contribute to 14% and 25% of the total AM material sales, respectively. The remainder of sales are expected to be associated with polymer filaments employed primarily for fused deposition modeling (FDM).114,118 Not every polymeric material can be employed in additive manufacturing applications. For instance, elastomeric materials are unable to form 3D printable pellets due to their low Tg values. Certain semi-crystalline polymers (such as polylactic acid (PLA) and polyether ether ketone (PEEK)) are the most commonly used 3D printed materials. However, certain conventional polymers (for instance polypropylene) are difficult to 3D print without the employment of any additives. High Young's modulus and low viscosity of the printable materials facilitate smooth extrusion.34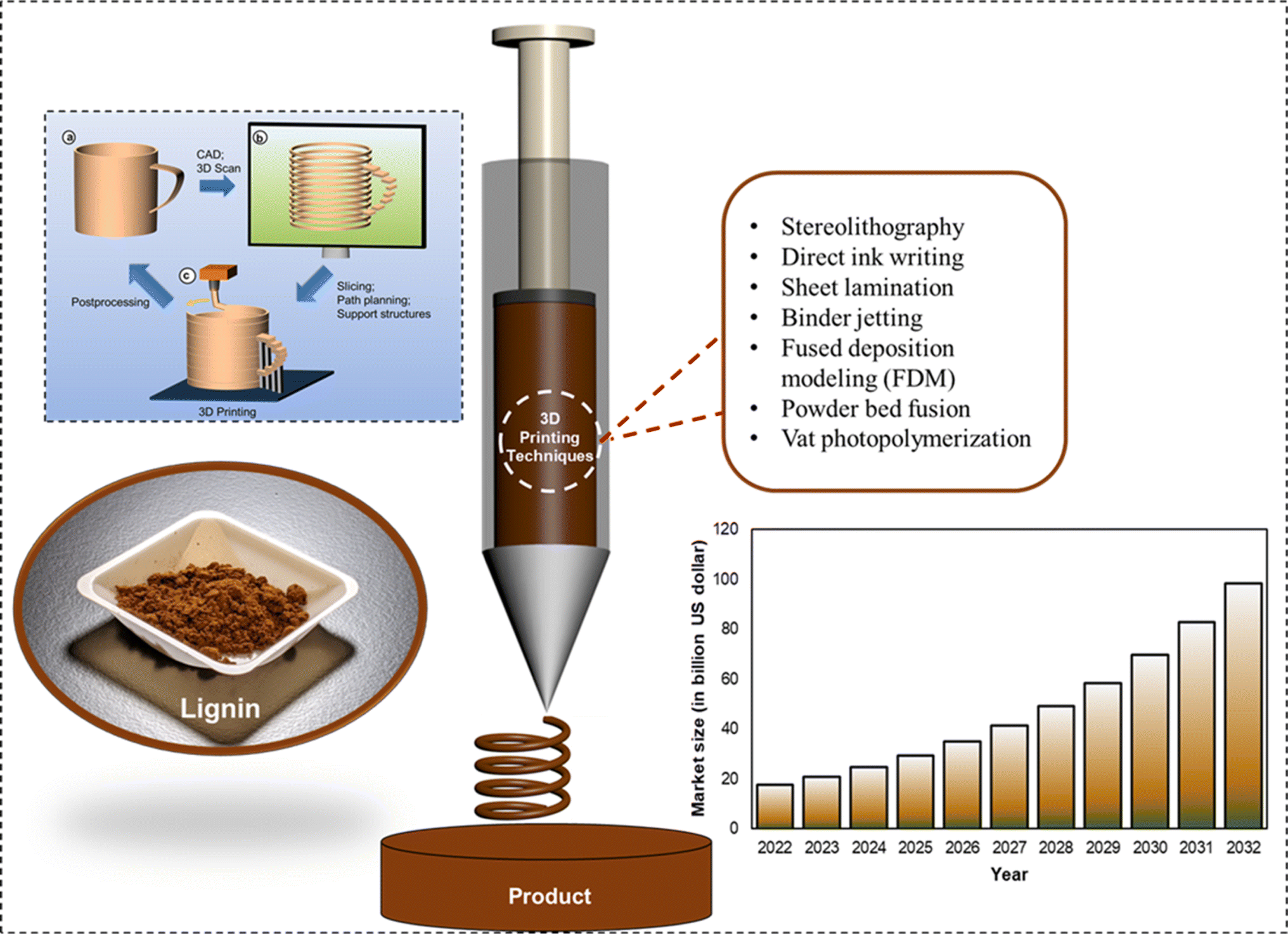 | ||
| Fig. 7 3D printing representation: fundamental steps of additive manufacturing: CAD model (development, preprocessing, and 3D printing of the product) (reproduced from ref. 114 with permission from ACS, copyright 2017) worldwide additive manufacturing market size (2022 to 2032) (reproduced from ref. 117 with permission from Elsevier, copyright 2020). | ||
An inclination towards the use of naturally abundant biopolymers such as lignocellulosic materials for AM feedstock has garnered significant attention.34,119–121 Since its advent in the later part of the previous centennial, several techniques for additive manufacturing have been developed, including stereolithography, direct ink writing, sheet lamination, binder jetting, fused deposition modeling (FDM), powder bed fusion, and vat photopolymerization.122 Among the various additive manufacturing techniques, stereolithography apparatus (SLA) is of particular interest.
Due to the inherent advantages of SLA such as good operator control, accuracy, and superior resolution, it can be a method of choice.123 However, due to the ability to solely process photoreactive resins, the choice of polymer feedstock materials is relatively narrow.123 To overcome this limitation, the development of modified photoreactive resins has been under intense investigation.124–129 The use of traditional fillers (such as graphene oxide, carbon nanotubes, SiO2, nanoclay, sepiolite nanofibers, aluminum oxide nanowires, and TiO2) in photoreactive resins for enhancing the tensile properties of stereolithography printed products has been recorded.125,130–134 Although conventional fillers enhance the mechanical properties of filled products, the associated concerns are fueling a shift towards more sustainable alternatives.135
The use of lignin as a filler in SLA-printed products is shown to enhance UV absorption,136 anti-aging properties137 and flame retardancy138 of 3D-printed lignin-based materials with applications in drug delivery materials and other biomedical applications.139,140 Due to the inherent stiffness that lignin imparts to the host polymer matrix, several studies have covered its application in 3D printing materials. In a study by Sutton et al., the use of methacrylic anhydride-modified lignin in conjunction with methacrylate resin to undergo stereolithography was explored.141 A 300% increment in elongation at break was recorded. This was attributed to the superior dispersibility of the modified lignin (15 wt%) in the filled matrix.
FDM is another additive manufacturing technique that can utilize lignin as ink. In a study by Mimini et al., FDM-printed samples of PLA composites with different types of lignin (kraft lignin, organosolv lignin, and lignosulfonate) were tested for their impact and flexural strength.142 A minimal reduction in both flexural and impact strength values of all the specimens was recorded. In a study by Nguyen et al., FDM was employed to incorporate hardwood lignin in a nylon 12 matrix.143 The resultant lignin (40 wt%) loaded specimens exhibited a tensile strength of 55 MPa with a 70% increment in Young's modulus values. This improvement in the mechanical properties was ascribed to the presence of extensive phenolic moieties in the lignin specimen and potential instances of hydrogen bonding between the lignin moieties and the amide bonds of nylon in the composite.
In another study, a similar increment in Young's modulus and a dip in tensile strength with higher loadings of pine kraft lignin in polylactide were observed.144,145 In a separate study, the use of lignin with PLA for additive manufacturing via the fused filament fabrication technique was explored.140 Polylactic acid pellets were coated with lignin (0–3 wt%) and castor oil prior to extrusion at 200 °C. The produced filament underwent fused filament fabrication. The filaments exhibited antioxidant capabilities and were further used to design and fabricate meshes. The 3D-printed meshes could be utilized in wound dressing and the healthcare sector.140 The above-mentioned studies were crucial in establishing the merits of employing lignin in additive manufacturing operations. The complexity of the systems arising due to the non-homogeneity of lignin, the lignin content of the formulations, the choice of the base polymer, and the employed additive manufacturing technique were established. Various lignin filled additive manufacturing composites could aid in fulfilling several Sustainable Development Goals (SDGs) including SDG 9, SDG 12, SDG 13, and SDG 14.
3.5. Utilization of lignin as a Pickering emulsifier
In recent years, the extensive use of Pickering emulsions in the cosmetic, food, and pharmaceutical industries has been witnessed.146 The use of organic and inorganic particles for stabilization of Pickering emulsions is widely practiced,147 ranging from latex,148 silica,149 and calcium carbonate,150 to LAPONITE®,151etc. However, some of these particles have safety and sustainability concerns. For instance, the inhalation of crystalline silica dust, which can be used as a Pickering emulsifier has been proven to be a leading cause of silicosis in human beings.152 Also, the mining operations for crystalline silica are known to be detrimental to the environment. Thus, there has been growing interest in finding sustainable alternatives such as lignin to conventional Pickering emulsifiers.153In a study by Shorey et al., native and esterified lignins for the stabilization of oil-in-water Pickering emulsions were studied. It was observed that due to the subpar dispersibility exhibited by the unmodified lignin in solvents, unstable Pickering emulsions were produced. In contrast, esterified lignins (LigHC, Lig DC, and LigPC) with hydrophobic properties displayed enhanced solvent dispersion and provided outstanding stability to oil-in-water Pickering emulsions. This was substantiated by a 47% reduction in average oil droplet size for he LigHC stabilized Pickering emulsion, as displayed in Fig. 8.44 The performance of lignin-stabilized Pickering emulsions was compared against an industrial grade emulsifying agent.44 A threefold higher viscosity value was recorded for the esterified lignin stabilized emulsion over its industrial surfactant counterpart. The significant increment in viscosity values was attributed to the superior shielding effect of lignin at the oil-water interface thus preventing their coalescence.44 These observations were in coherence with the optical micrographs recorded for the specimens.44
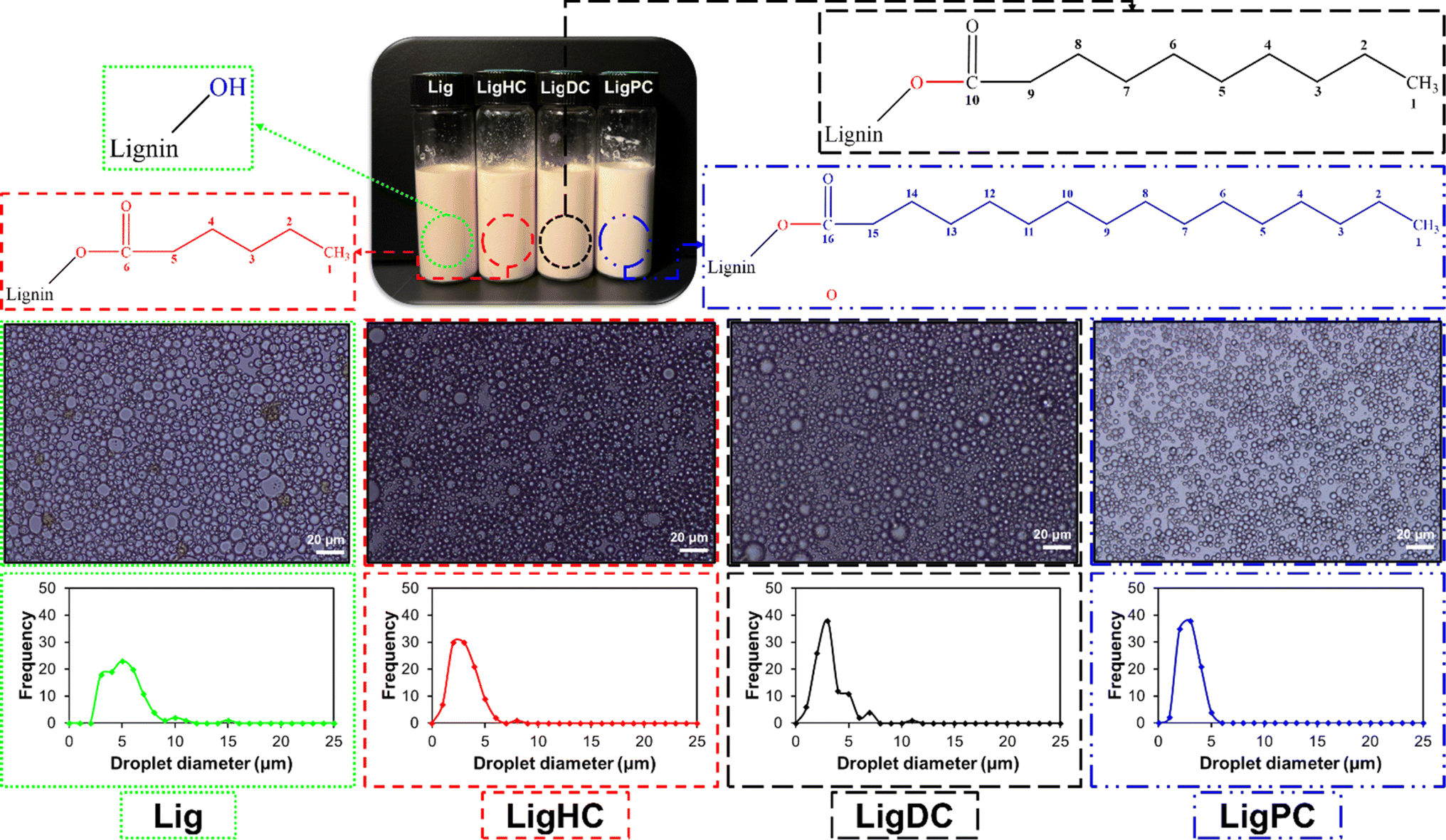 | ||
| Fig. 8 Optical micrographs of unmodified and esterified lignin stabilized oil-in-water Pickering emulsions (reproduced from ref. 44 with permission from Elsevier, copyright 2023). | ||
The use of novel Pickering emulsion formulations stabilized by employing lignin nanoparticles for controlled release of trans-resveratrol was explored.154 In this work, the stabilization effect of alkaline lignin grafted poly(N-isopropylacrylamide) (PNIPAM) via atom transfer radical polymerization (ATRP) was studied. The fabricated nanoparticles (with diameter <160 nm) absorbed at the interface of oil-in-water emulsions, thus, exhibiting stable Pickering emulsions with oil droplet diameter values in the vicinity of 200 μm. The stabilized Pickering emulsions exhibited stability improvement against UV radiation and exceptional controlled release performance. Other studies on utilization of lignin as a Pickering emulsifier are outlined in Table 2. An American firm, Ingevity Corporation, has been developing and commercializing proprietary lignin-based products. For instance, their lignin-based emulsifier (Indulin® SA-L) produces anionic asphalt emulsions.155
3.6. Lignin-based carbon fibers and their applications
Recently, exponential growth in the market size of carbon fibers has been witnessed.160 Evaluated at 2.99 billion USD in 2022, the carbon fiber market is forecast to be worth 5.83 billion USD by 2032, with a compound annual growth rate of 6.9%.160 Comprising at least 92 wt% carbon, the fibers exhibit high flexibility, low density, inertness, heat resistance, and low thermal expansion. In comparison to steel, a 10-fold increment in tensile strength at half the weight has been recorded for carbon fibers.161,162 Due to their exceptional properties, an influx of their use has been reported in various sectors, including – construction, automotive, aerospace, sports, oil and gas industries, etc. Amongst the various precursors available for the manufacture of carbon fibers, polyacrylonitrile (PAN) is the most widely employed. The expensive PAN precursor (approximating 50% of the manufacturing cost) renders the carbon fibers costly for widescale industrial and commodity composite applications.163–166 In addition, the environmental concerns posed by the use of petroleum-derived precursors have created interest in greener, renewable, and economical carbon fiber precursors.In recent times, the use of lignin as a carbon-fiber precursor has gained significant attention.167–171 This is primarily due to the vast availability, high carbon content, and high post-carbonization yield exhibited by lignin.168,170 However, the process of spinning carbon-fibers from lignin includes a series of processing steps – spinning (solution spinning, melt spinning, and electrospinning), pre-oxidation, and carbonization (low temperature or high temperature) (Fig. 9).172 In a critical review by Fang et al., an in-depth overview of manufacturing and applications of lignin-based carbon fibers and nanofibers was presented.171
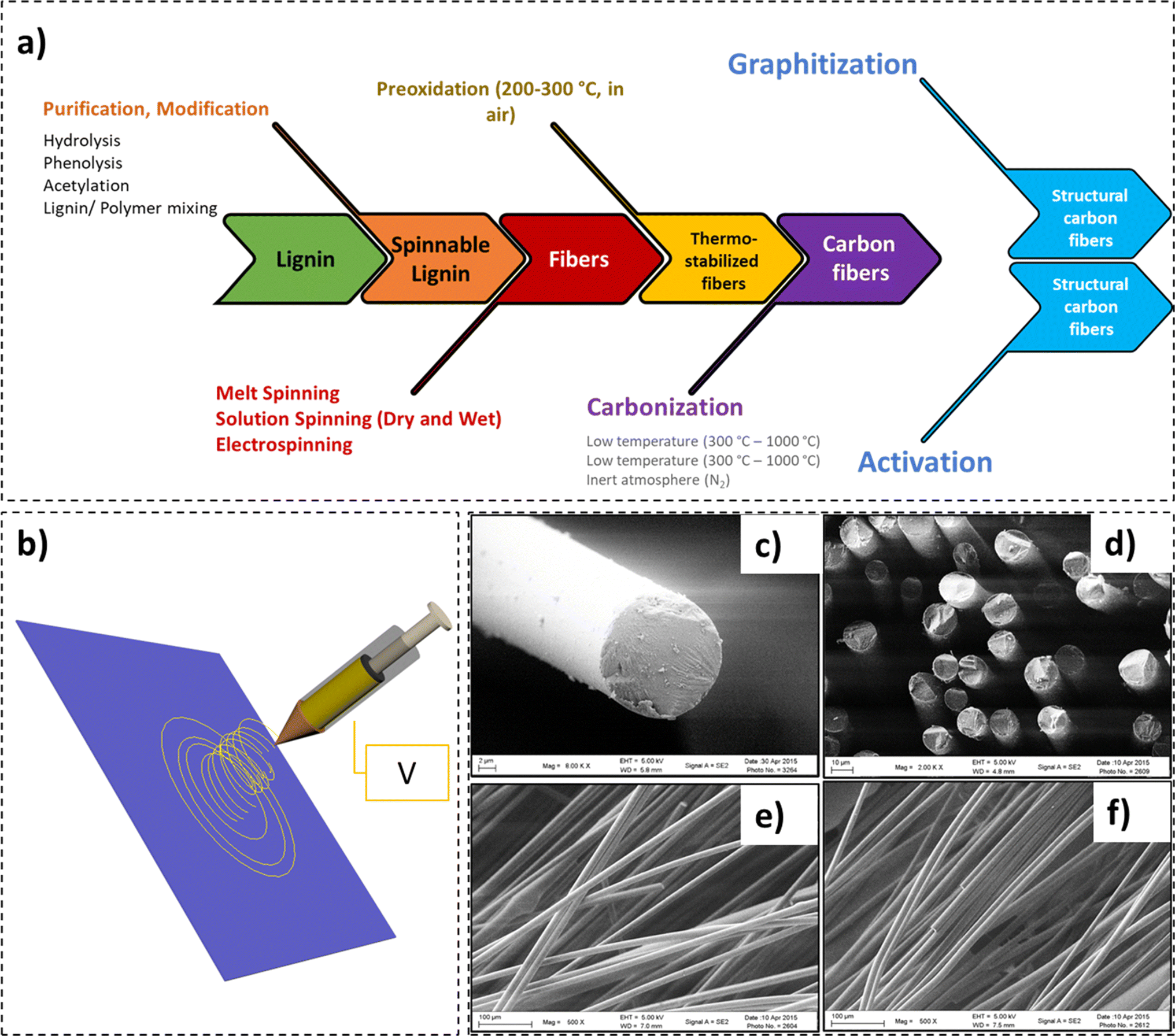 | ||
| Fig. 9 (a) Process of spinning carbon-fibers from lignin, (b) electrospinning apparatus, (c) SEM micrograph of a carbon fiber, (d) SEM micrograph of a cluster of carbon fibers (cross-section), and (e) and (f) SEM micrographs highlighting surface morphology of the carbon fibers (reproduced from ref. 179 with permission from Elsevier, copyright 2016). | ||
In a study by Jin et al., a tensile modulus of 98 GPa for unmodified lignin-based carbon fibers was obtained,173 which was perhaps one of the highest modulus values recorded for lignin. In this work, three fractionated-solvated lignin precursors (FSLPs) with increasing molecular weights (7200 g mol−1, 13800 g mol−1, and 28600 g mol−1) were employed. Lignin carbon fibers exhibited a uniform diameter (6–7 μm) with no surface deformations which defines their tensile values. These diameter values were noted to be much smaller than the values for other lignin-based carbon fibers previously reported.174,175 Moreover, they were near the diameter of PAN-based commercial carbon fibers.176 PAN based carbon fibers exhibit tensile modulus values more than 200 GPa with some manufacturers being able to achieve a tensile modulus as high as 517 GPa.177 Although the modulus values were the highest recorded for lignin-based carbon fibers, they were 52% lower than the tensile modulus values required for automotive industrial applications,113,173,178 indicating that there is a large gap that needs to be addressed before lignin-based carbon fibers find extensive industrial applications. Other studies on lignin-based carbon fibers are summarized in Table 3.
| Type of lignin | Polymer | Temperature | Tensile properties | Ref. | |
|---|---|---|---|---|---|
| Thermo-stabilization | Carbonization | ||||
| Softwood kraft lignin | — | 165 °C–250 °C@20 °C min−1, held 250 °C for 1 h | From room temperature to 1000 °C@7 °C min−1 | Tensile modulus: 98 ± 5 GPa | 173 |
| Softwood kraft lignin | Equicomponent blend with PAN | 300 °C@1 °C min−1, held for 1 h | Heated to 1200 °C@7 V min−1 and held for 1 h | Tensile modulus: 105.7 ± 3.1 GPa | 182 |
| Organosolv lignin, softwood kraft lignin, and z | — | Heated from 12 to 180 °C h−1 and held 250 °C for 1 h | 1000 °C@180 °C h−1 | Tensile modulus: 59 ± 8 GPa | 183 |
| Organosolv lignin | — | 250 °C@0.5 °C min−1 | Heated to 1000 °C | Modulus: 39.1 ± 13.3 GPa | 184 |
| Hardwood lignin | Copolymer with PAN | 105 °C for 1 h@1 °C min−1, then heated to 280 °C for 1 h | 800 °C@5 °C min−1 | Modulus: 22.92 gf/den | 185 |
| Softwood kraft lignin | 250 °C@4 °C min−1, held for 30 min | Heated to 600 °C@1 °C min−1 and to 1000 °C@3 °C min−1 | — | 186 | |
| Steam exploded lignin | Heated from 0.5 °C min−1 to 2.0 °C min−1 | Heated upto 1000 °C@5 °C min−1 | Modulus: 40.7 ± 6.3 GPa | 187 | |
On comparing the properties of lignin-based carbon fibers with those of their conventional counterparts, several differences and advantages of the former alternatives were recorded. The acceptable mechanical and interfacial properties of lignin-based carbon fibers for utilization in chopped carbon fiber composites render them advantageous over their PAN based counterparts, which require sizing.179 In another study by Li et al., lignin-based carbon fibers reported similar elastic modulus values as those of their commercial counterparts.180 These results will pave the way for eventual replacement of conventional carbon fibers by lignin fabricated variants.
In a study by Hermansson et al., the life cycle analysis (LCA) of lignin-based carbon fibers and recycled carbon fibers from discarded carbon fiber reinforced polymer (CFRP) products was conducted.181 Carbon fiber production accounted for more than 50% of the CFRP climate impact and energy consumption. With the replacement of the carbon fiber raw material (PAN) with lignin, reduction in energy consumption and climate impact was observed. In addition, recycled carbon fiber resulted in further energy conservation. These avenues require further research and future development.181
3.7. Lignin-incorporating sunscreen formulations
A relationship between the depleting ozone layer and increasing instances of skin cancer has been observed.188 Skin cancer instances in excess of 82000 (6500 cases of melanoma and 76100 cases of non-melanoma skin cancer (NMSC)) were reported in Canada (2014).189,190 With reduced exposure to artificial and natural UV radiation, most cases of skin cancer are preventable.191,192 The use of sunscreens as effective UV-blockers has been widely investigated. Since their advent in the United States cosmetic market in 1928, they have been a huge commercial success.193 Commercially available sunscreens incorporate various active ingredients (inorganic and organic) and hence vary in their respective UV-blocking mechanisms.188
Traditionally, the use of inorganic active ingredients (including zinc oxide (ZnO), talc, titanium oxide (TiO2), kaolin, etc.) in UV blockers is prominent.194 These active ingredients scatter and reflect UV rays off the surface of the skin, thus, in turn preventing sunlight from penetrating the skin. Although inorganic UV blockers mostly cover the broad UV spectrum, their inferior dispersion in cream bases and the photoreactive properties that could potentially harm cellular components make their organic counterparts an attractive and safe alternative.195–198
In contrast, organic UV blockers (including benzophenones, silatriazole, bisoctrizole, salicylates, ecamsule, bemotrizinol, etc.) work on the mechanism of absorbing the UV energy and converting it into heat energy.199 In numerous studies, the photostability and the synergistic effect of adding lignin to sunscreen formulations have been studied (up to 10 wt% loading), as covered in Table 4. There was an increment in both the sun protection factor and duration of protection against UV rays after the addition of lignin to creams and commercial sunscreen formulations.200 In a study by Zhang et al., with progressive loadings of acetylated lignin to the substrate sunscreen lotion, a three-time increase in the SPF value (75.2 SPF) was recorded (Fig. 10).201 This was attributed to the synergistic effect of the acetylated lignin in conjunction with synthetic actives.
| Sunscreen formulation base | Type of lignin | SPF value | Ref. | |
|---|---|---|---|---|
| Before | After | |||
| SPF 15 lotion | Eucalyptus kraft lignin | 15 | 75.2 | 201 |
| NIVEA moisturizing cream, sunscreen SPF 15, and SPF 50 | Enzymatic hydrolysis lignin and organosolv lignin | 47.71 ± 6.30 | 203 | |
| Pure cream | Soda lignin | 0.99 ± 0.01 | 6.81 ± 1.15 | 208 |
| NIVEA pure cream | Alkali lignin | 0.99 ± 0.01 | 5.72 ± 0.26 | 209 |
| SPF 15 sunscreen lotion | Kraft lignin | 18.22 ± 0.92 | 76.97 ± 6.38 | 208 |
| Glycerin hand cream | Alkali lignin | 1.06 ± 0.01 | 5.33 ± 0.47 | |
| Pure cream | Kraft lignin | 0.99 ± 0.01 | 6.33 ± 0.37 | 208 |
| SPF 15 sunscreen lotion | Soda lignin | 18.22 ± 0.92 | 91.61 ± 8.47 | 208 |
| Pure cream | Organosolv lignin | 0.99 ± 0.01 | 8.66 ± 0.25 | 208 |
| SPF 15 sunscreen lotion | Organosolv lignin | 18.22 ± 0.92 | 23.66 ± 1.28 | 208 |
| Pure cream | Enzymatic-hydrolyzed lignin | 0.99 ± 0.01 | 4.20 ± 0.50 | 208 |
| SPF 15 sunscreen lotion | Enzymatic-hydrolyzed lignin | 18.22 ± 0.92 | 20.13 ± 5.31 | 208 |
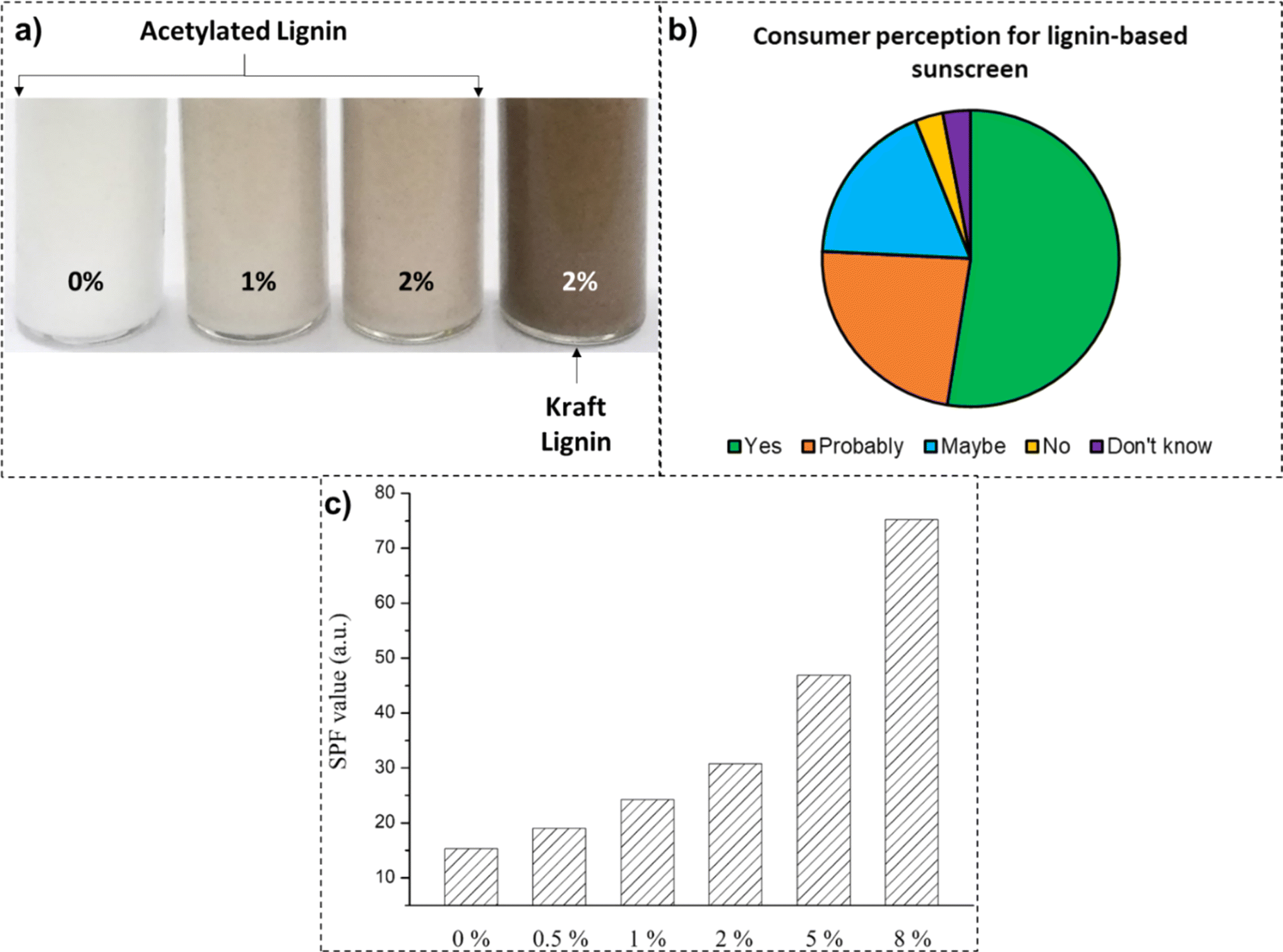 | ||
| Fig. 10 (a) Sunscreen formulations loaded with acetylated and kraft lignin samples (reproduced from ref. 201 with permission from Elsevier, copyright 2019), (b) consumer perception to the use of lignin-based sunscreen formulations (reproduced from ref. 202 with permission from MDPI, copyright 2021), and (c) recorded SPF values at various loading concentrations of acetylated lignin (reproduced from ref. 201 with permission from Elsevier, copyright 2019). | ||
The challenges for the mainstream application of lignin in sunscreen formulations are many, including heterogeneity of the chemical structure, dark color, broadening of the SPF value in the broad-spectrum UV blocking range, and consumer perception.203 Fractionation of lignin with organic solvents is usually employed to remove condensed structures, thus, increasing its dispersion in cream bases.198 In a study by Tran et al., the acetylation of the hydroxyl groups of the lignin specimen lightened the color for a better appeal.204 In a separate study by Wu et al., the color performance of unmodified and acetylated lignin specimens in sunscreen formulations at various loadings was compared. As is apparent from Fig. 10, sunscreen formulations loaded with 2 wt% of acetylated lignin demonstrated a much lighter shade than their unmodified counterpart, thus, being more acceptable to end-consumers. Another way to lighten the lignin color is through grafting other compounds onto the lignin structure, such as benzophenone,205 2,4-benzoyl-33-hydroxyphenyl acrylate (BHA),206 and poly(ethylene glycol) methacrylate (PEGMA).207 In a study by Sajinčič et al., the attitude and perception of consumers toward lignin-based sunscreen formulations were explored.202 While consumers in excess of 50% were willing to try lignin-based sunscreens, a mere 3% of the potential consumers refused trying them. While consumers tend to appreciate bio-sourced, healthy, and sustainable sunscreen alternatives, they are concerned about lower performance and protection. Additionally, because organic ingredients are perceived as luxurious, consumers are concerned about associated product cost.
3.8. Lignin-based smart materials
In a study by Shikinaka et al., the synthesis of lignin-based SMP exhibiting a dynamic shape-recovering behavior was reported.217 The synthesis procedure comprised mixing simultaneous enzymic saccharification and comminution (SESC) lignin and poly(ethylene carbonate) polymers (PEC), kneading the mixture (120 °C), and compression molding (120 °C, 3–5 MPa) into films. Thermoresponsive shape-memory behavior was noted in the molded films, as presented in Fig. 11a. The composite films were set into the original shape using heat (80 °C). Further cooling could straighten and fix the film shape (0–10 °C) and revert to their original shape with the reapplication of heat. The fabricated films could be remolded into different shapes many times and each shape recovery cycle took less than 60 seconds to reach the conclusion. Poly(ethylene carbonate) sheets exhibited thermosetting behavior. The shape memory characteristics of the baseline poly(ethylene carbonate) sheets were attributed to the formation of hydrogen bonds under cold conditions. The SESC lignin moieties tend to aggregate in the polymer matrix, thus, imparting dynamic shape recovery to the specimens. Thus, the synergistic movement of the polymer chains influenced by the presence of the lignin agglomeration resulted in the shape-recovery behavior of the composite films (Fig. 11b).217 In a study by Liu et al., the effective use of lignin as a crosslinking agent to develop crosslinked polycaprolactone (PCL) based thermal shape memory materials was explored.218 The process was initiated with the esterification of lignin and synthesis of PCL employing bulk ring-opening polymerization. Furthermore, the obtained PCL-OH moieties underwent a modification reaction to exhibit thiol terminal functional groups. The synthesis was concluded by crosslinking the lignin and PCL moieties via a photoredox thiolene reaction. The prepared samples exhibited an ability to transition between temporary and permanent shapes, and thus, a rapid thermal shape response was recorded. A reduction in the melting temperature of the crosslinked specimen with an increase in the lignin loading was observed. Thus, lignin could be employed as an effective crosslinker in polymeric matrices.218
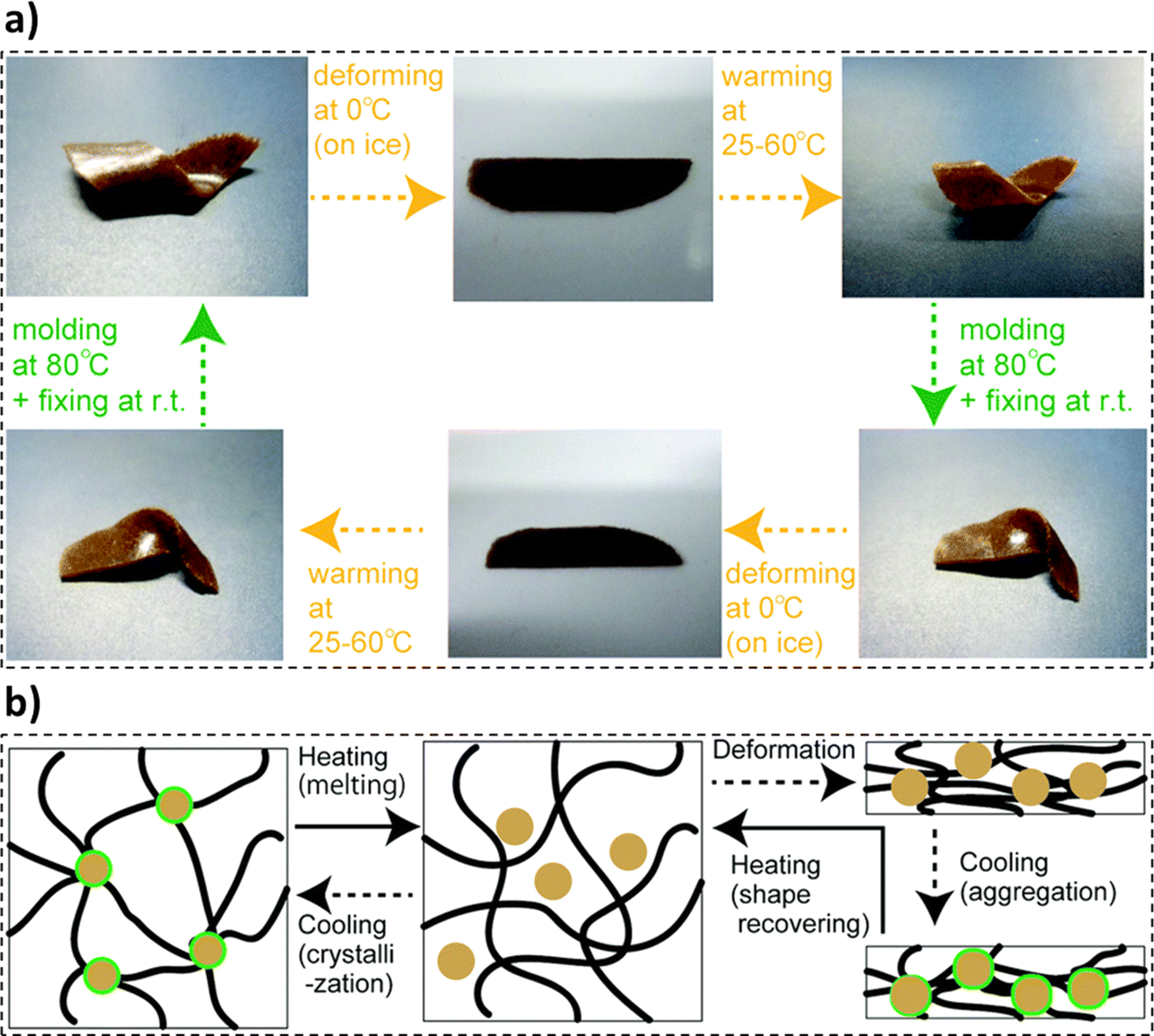 | ||
| Fig. 11 (a) Thermoresponsive shape memory behavior exhibited by the lignin composite film and (b) molecular transformations in one shape memory-recovery cycle of the lignin composite film (reproduced from ref. 217 with permission from RSC, copyright 2018). | ||
In a study by Larrañeta et al., lignin-based hydrogels employing an esterification reaction were fabricated.222 The synthesized lignin-poly(ethylene glycol)-poly(methyl vinyl ether-co-maleic acid) based hydrogels exhibited potential applications in medical material coatings. For lignin loaded hydrogel formulation (between 40% and 24%), a water uptake value of up to 500% was recorded. The formulated gels sustainably released curcumin fractions up to 4 days post loading.222
As illustrated in Fig. 12, lignin based photodynamic hydrogels exhibiting self-healing properties were synthesized in another study.223 These hydrogels were synthesized and functionalized with rose bengal (RB) lignin-based silver nanocomplexes (L-AgNCs) (Fig. 12a and b). The hydrogels constituted 2 to 7.5 mg mL−1 of lignin loadings with declining transparency for the higher loaded specimens, as displayed in Fig. 12c. In this case, 85% transmittance of the light through the hydrogels was recorded for the 2 mg mL−1 lignin loaded samples (Fig. 12d). In the SEM micrographs, an extensively cross-linked and porous structure was noted in the 2 mg mL−1 (Figure 12e) and 3 mg mL−1 (Fig. 12f) hydrogel formulations. In addition, the fabricated hydrogels exhibited self-healing properties. As shown in Fig. 12g, two dissected pieces of such hydrogel self-healed upon being placed together for 30 min. In another study by Ma et al., the degradability of lignin-poly(acrylic acid) based hydrogels was tested.224
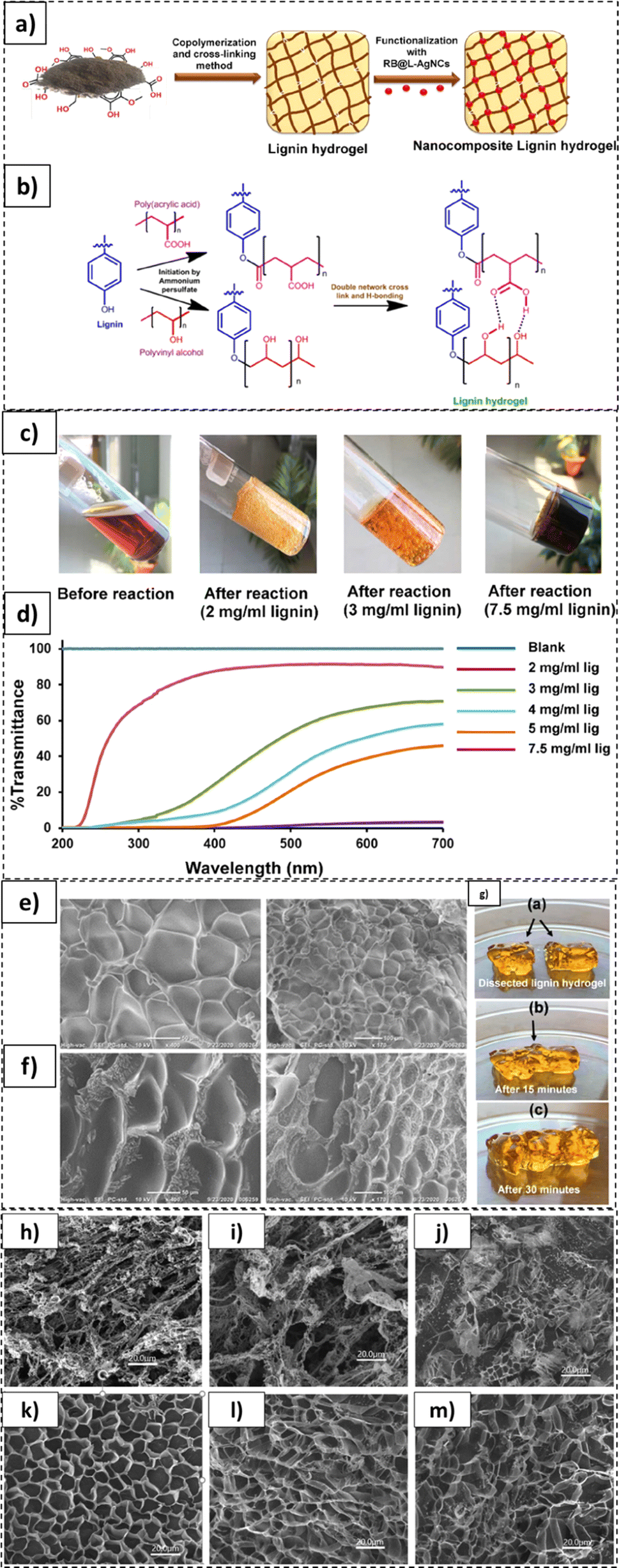 | ||
| Fig. 12 Lignin-based hydrogel formulations: (a) functionalization of lignin-based hydrogels, (b) lignin based hydrogel synthesis steps, and (c) various lignin hydrogel formulations with different lignin loadings. (d) hydrogel transmittance, (e) SEM images of 2 mg mL−1 lignin-based hydrogel, (f) SEM images of 3 mg mL−1 lignin based hydrogel, (g) self-healing properties of lignin based hydrogels (reproduced from ref. 223 with permission from ACS, copyright 2022), and SEM micrographs of hydrogel recorded post different days of degradation: 40% lignin loaded formulation (60 PAA/40 LM) (h) 0 day, (i) 35 days, and (j) 180 days; baseline PAA degradation: (k) 0 day, (l) 35 days, and (m) 180 days (reproduced from ref. 224 with permission from Elsevier, copyright 2016). | ||
As apparent from Fig. 12h and j, significant degradation of lignin loaded hydrogel was recorded. While the lignin loaded hydrogel exhibited a well-defined structure before the initiation of degradation, the porous structure underwent severe disintegration after 180 days. In contrast, for the baseline poly(acrylic acid) specimen no cracks or surface deformities were recorded. The deterioration of the lignin loaded sample was ascribed to the enzymatic and chemical degradation of the lignin-polymer chain covalent bonds. The 40% lignin loaded hydrogel sample lost approx. 5% weight in 35 days, varying minimally with the type of testing soil employed.224
In a study by Cerrutti et al., electrical impedance spectroscopy (EIS) based immunosensors were fabricated. These sensors constituted lignin and antigenic peptides on interdigitated gold electrodes and exhibited anti-p17 (HIV) antibody detection capabilities (Fig. 13a). The manufactured immunosensors could detect low concentrations of anti-bodies (0.1 ng mL−1) for up to 2 months.226 In another study, Jędrzak et al. prepared biosensors based on a silica (SiO2)/lignin material for glucose detection.227 The developed SiO2/lignin hybrid material underwent glucose oxidase immobilization, as presented in Fig. 13b. Incorporating lignin into the matrix resulted in twice (25.28 mg g−1) the amount of GOx immobilization compared to its baseline (SiO2) counterpart. Lignin-based biosensors demonstrated high sensitivity (0.78 μA m/M−1) and an ability to detect glucose in biological samples with great accuracy.
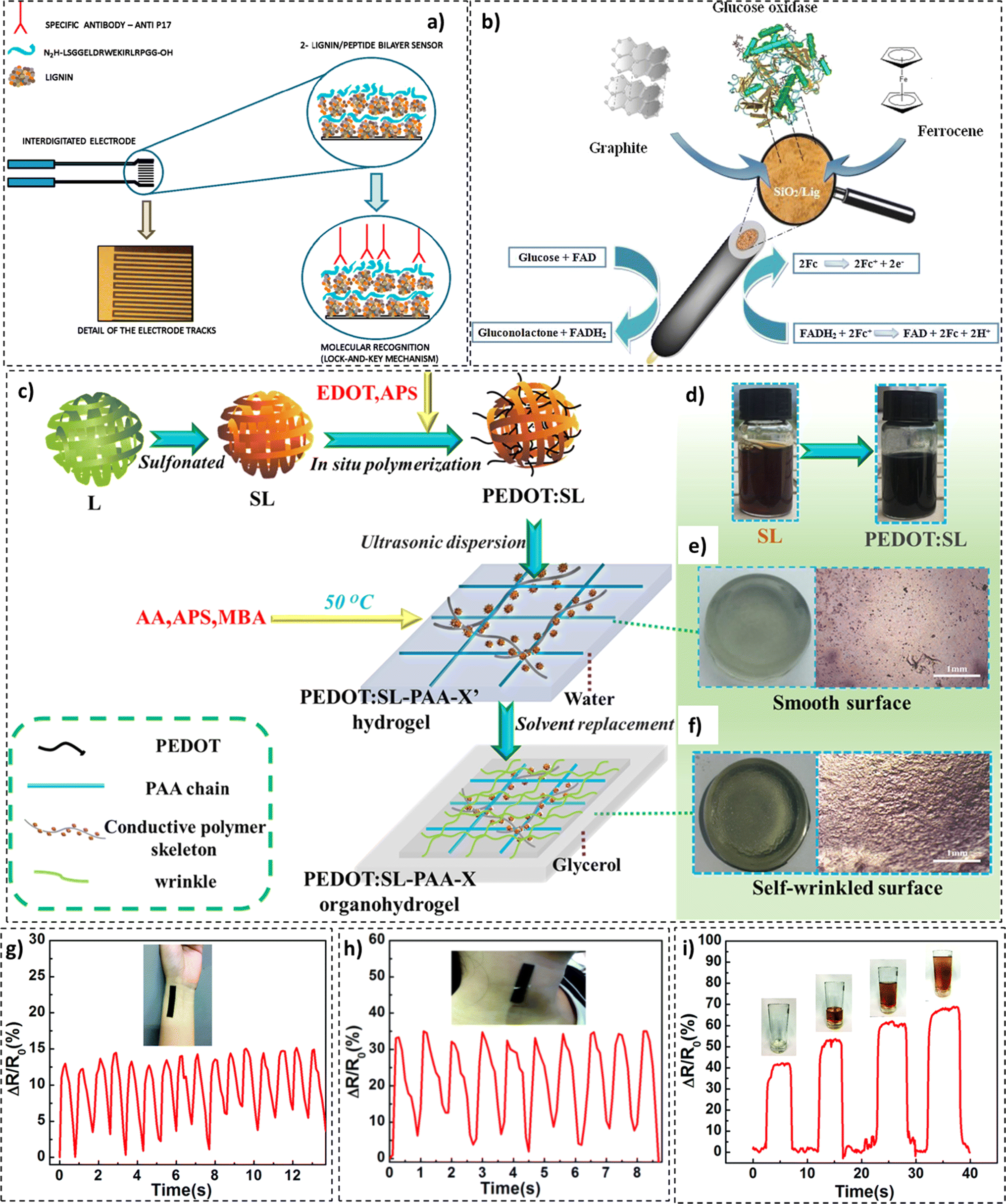 | ||
| Fig. 13 (a) Electrical impedance spectroscopy (EIS) based immunosensors (reproduced from ref. 226 with permission from Elsevier, copyright 2015), (b) functional GOx/silica/lignin system based carbon paste electrode (reproduced from ref. 229 with permission from Elsevier, copyright 2018), (c) synthesis of PEDOT:SL-PAA organohydrogel, (d) color change during synthesis, (e) morphology of PEDOT:SL-PAA hydrogel, and (f) morphology of a PEDOT:SL-PAA organohydrogel composite based wearable device: (reproduced from ref. 230 with permission from Elsevier, copyright 2019) CL/PDMS (g) for wrist pulse measurement, (h) for neck pulse measurement, and (i) power measurement in picking up a cup (reproduced from ref. 231 with permission from RSC, copyright 2018). | ||
Poly(3,4-ethylenedioxythiophene) (PEDOT) in conjunction with sulfonated lignin was incorporated as the conductive material in a poly(acrylic acid) (PAA) skeleton for the fabrication of hydrogel-based wearable sensors, as illustrated in Fig. 13c–f.228 The high sensitivity of the sensors produced was capable of detecting bodily motion, physiological signals, throat vibrations, and pulses. Potential applications in healthcare, sports science, and speech recognition were proposed. Other developments in the domain of lignin-based sensing materials have been reported.226,229–231 In one such stduy by Wang et al., lignin-based pressure sensors for wearable electronics were developed.228 For the prototype sensor, high sensitivity (57 kPa−1) and a wide working pressure range (0 to 130 kPa) were recorded, as illustrated in Fig. 13g–i.
3.9. Use of lignin in self-healing elastomers
The self-healing properties exhibited by many materials stem from the presence of supramolecular interactions,232 such as π–π stacking,233 ionic interactions,234,235 hydrogen bonding,236,237 metal–ligand interactions,238,239 and host–guest interactions.240 These materials exhibit instantaneous self-healing properties at the expense of mechanical strength. To counter this reduction in tensile properties, a dual phase material can be employed. While the hard phase would impart the material with stiffness, the soft phase would contribute to the material's self-healing properties.241 Lignin's high Young's modulus and mechanical strength, combined with its chemical versatility, offer a unique opportunity to address a standing challenge in self-healing materials.242In a study by Cui et al., a reaction of diglycidyl ether-terminated polyethylene glycol with lignin moieties was reported.241 The existence of covalent and hydrogen bonds in the resulting self-healing elastomers was observed, as illustrated in Fig. 14a and b. While the lignin moieties made the material stiff, the PEG soft phase fractions enabled chain movement.241 An elastic modulus value of 86 MPa with a tensile strength of 12 MPa was recorded for the 58% lignin-loaded sample. Furthermore, the 55 wt% lignin samples were observed to be extremely stretchable, reporting an elongation at break value exceeding 2200%. The unfolding and sliding of polymer chains due to breakage and rejuvenation of hydrogen bonds imparted the material with high stretchability.239,241 As represented in Fig. 15b, a recovery in elongation at break exceeding 50% was recorded after 1 min for the 50 wt% lignin-loaded samples.241 An inverse relationship between lignin loadings and self-healing time was also noted.
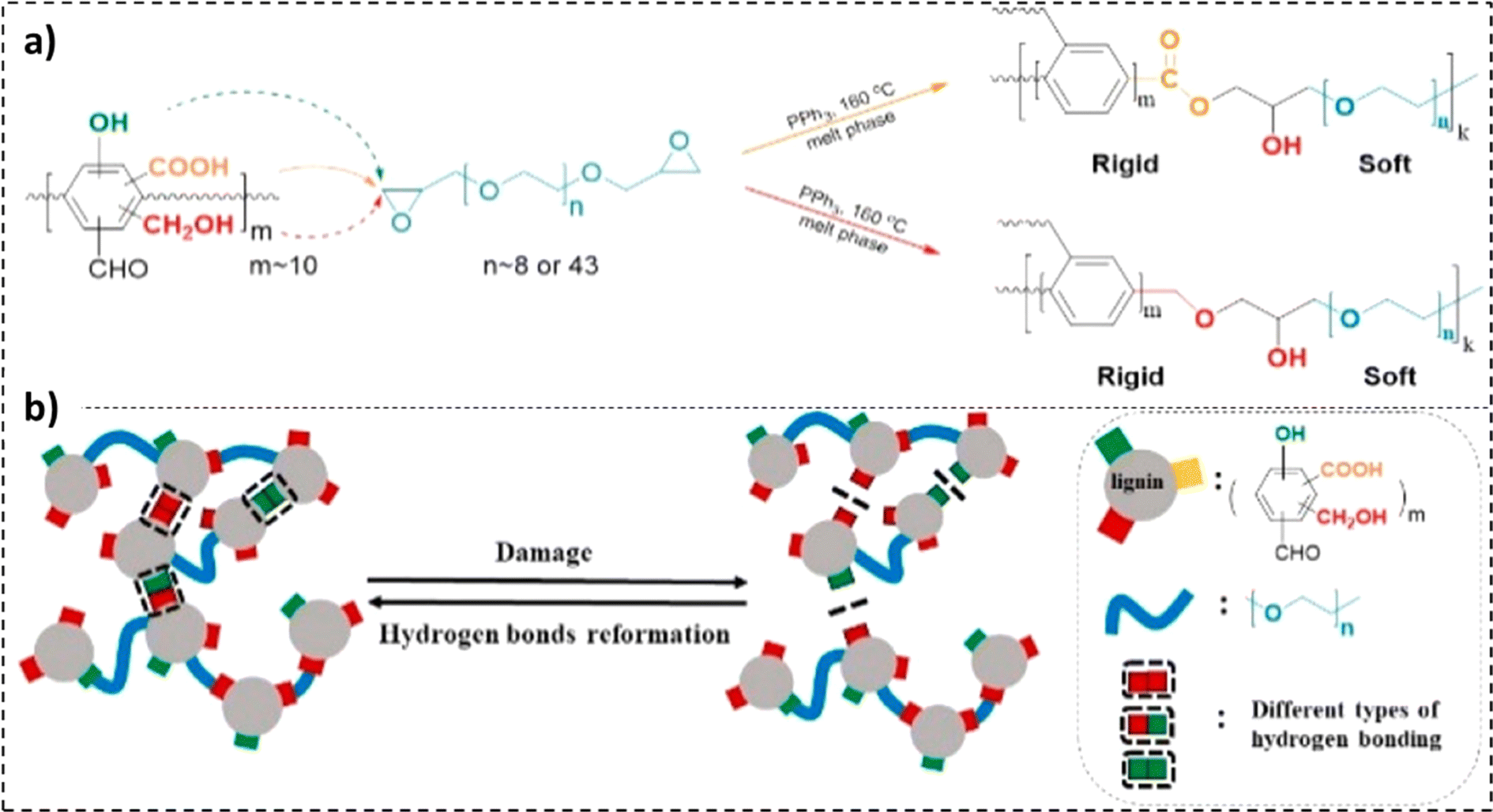 | ||
| Fig. 14 (a) Reaction mechanism of ACN-lignin and diglycidyl ether polyethylene glycol (PEG) (melt phase), and (b) illustration of the network formed by covalent and non-covalent bonds (reproduced from ref. 241 with permission from ACS, copyright 2018). | ||
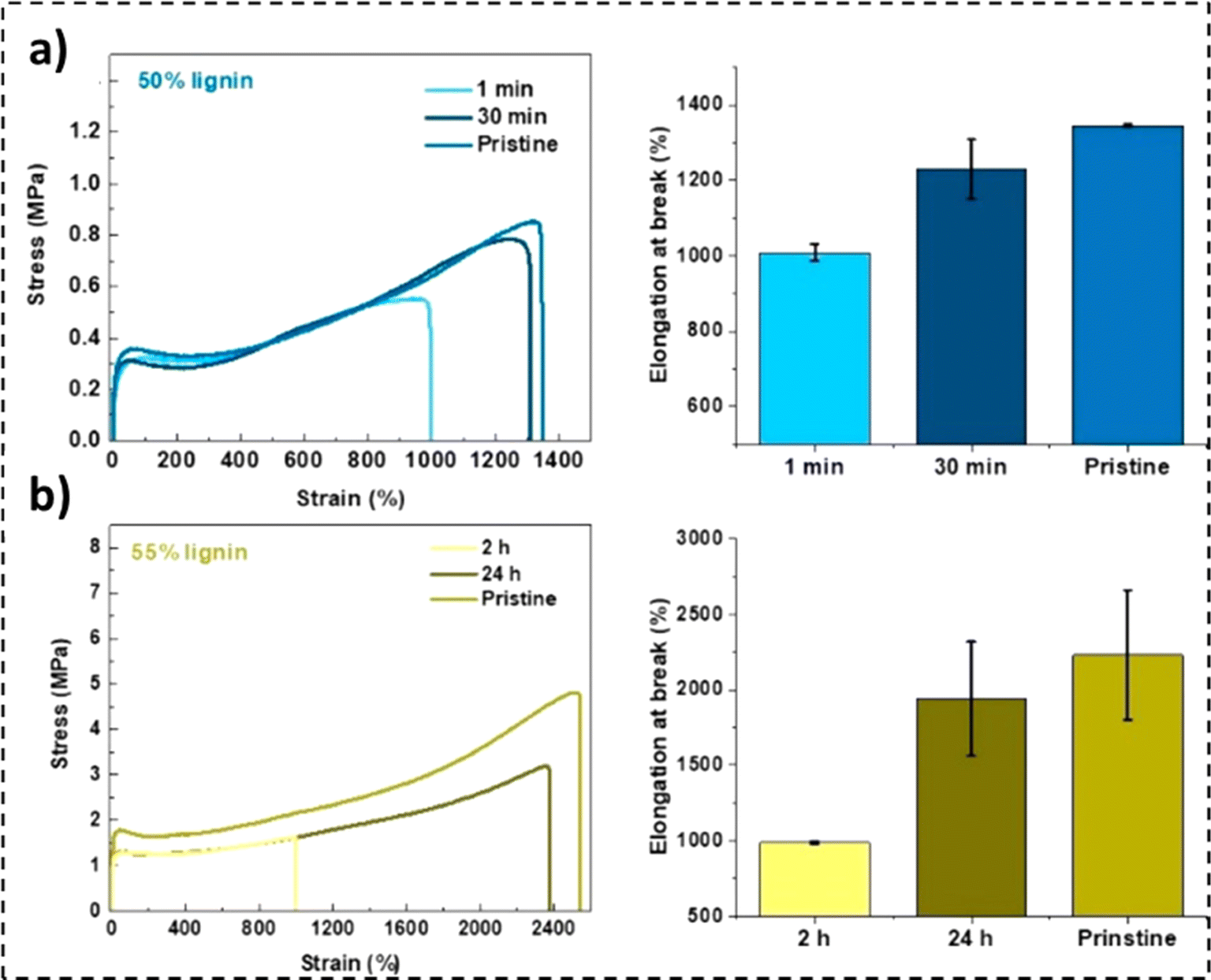 | ||
| Fig. 15 Tensile properties and recovery performance of (a) 50% ACN-lignin (1 min and 30 min) at ambient temperature and (b) 55% ACN-lignin (2 h and 24 h) exposed to 37 °C (reproduced from ref. 241 with permission from ACS, copyright 2018). | ||
In another study, the preparation of lignin constituting self-healing polyurethane elastomers was discussed.243 Good mechanical properties (tensile strength 10.77 MPa) in conjunction with self-healing properties (in excess of 93%) were observed. Moreover, the presence of dynamic hydrogen and disulfide bonds has been correlated with superior thermal stability, mechanical properties, resistance against fatigue, and self-healing capability.
3.10. Antimicrobial properties of lignin incorporating composites for active food packaging applications
Food packaging materials predominantly employ petroleum-sourced and non-biodegradable conventional plastics. A shift towards sustainable and biodegradable packaging materials is attracting substantial interest. However, their widescale application is primarily inhibited owing to their high cost and inferior barrier and other physical properties. Thus, the use of functional fillers to counter the inherent disadvantages of virgin biopolymers is gaining significant interest. One of the crucial roles of food packaging materials is to impart defense against microbial invaders. The choice of functional fillers for antimicrobial material is not only limited by the enhancement in physical properties but also by their antimicrobial activity. Of all the functional fillers available, the employment of lignin in synergy with other active agents to impart antimicrobial properties to the host packing material has been studied.244–249In a study by Shankar et al., the use of agar/lignin/silver nanoparticles for the fabrication of antibacterial composite sheets was studied.244 The use of lignin as a capping and reducing agent for the preparation of silver nanoparticles was observed. Furthermore, the incorporation of lignin-capped silver nanoparticles in an agar matrix via solution casting to produce composite sheets was witnessed, as evident from Fig. 16a. With the addition of lignin, an increment in the mechanical strength, UV barrier, and water vapor barrier properties of the films was recorded, which witnessed a further improvement with the addition of silver nanoparticles. The incorporation of silver nanoparticles up to 1 wt% in the polymer matrix exhibited clear benefits; a further increase in the concentration demonstrated detrimental effects. Antibacterial activity against E. coli and L. monocytogenes was noted in the cast films and thus, their potential application in active food packaging applications was proposed.244 In a similar study by Hu et al., lignin-capped silver nanoparticles were incorporated on the surface of cellulose nanofibers (up to 2.9 wt%).245 As evident from Fig. 16b, the alkali lignin-containing samples exhibited no bacterial growth in comparison to the original cellulose sample.
 | ||
| Fig. 16 (a) Agar/lignin/silver nanoparticle composite film synthesis procedure (reproduced from ref. 244 with permission from Elsevier, copyright 2017), (b) four diluted E. coli suspensions exposed (10 min) to: (1) cells and (2) AgNPs-AL-Cell 10 (reproduced from ref. 245 with permission from Elsevier, copyright 2015) and antibacterial activity against Escherichia coli (c and d), and Staphylococcus aureus (e and f) of CNF (c and e) composite films and CNF@CLNPs (d and f) composite films (reproduced from ref. 246 with permission from Elsevier, copyright 2019). | ||
A different approach was undertaken in a study by Yang et al., where the synergic effect of the incorporation of lignin nanoparticles (LNPs) and cellulose nanocrystals (CNCs) in a polylactic acid based (PLA and Glycidyl methacrylate (GMA) grafted PLA) matrix was investigated.247 The antibacterial activity of binary (PLA/3LNP) and tertiary (PLA-3LNP/1CNC) melt extruded nanocomposite films against Pseudomonas syringae pv. tomato (Pst) was recorded.247 With addition of 3 wt% LNP to the PLA matrix in binary nanocomposite films, the highest resistance to bacterial growth was observed. This was attributed to the antibacterial activity of lignin nanoparticles (LNPs).247
In another study, the incorporation of varying amounts of corncob lignin nanoparticles (0.5 wt% to 10 wt%) in cellulose nanofibrils for the production of cellulose composite films was studied.246 Superior antibacterial activity against Escherichia coli and Staphylococcus aureus was exhibited by the composite films. As evident from Fig. 16c, the bacterial growth (Escherichia coli and Staphylococcus aureus) in the lignin-incorporating sheets was inferior to that in the baseline cellulose counterparts. The superior antibacterial activity of lignin-loaded sheets against Escherichia coli was attributed to the presence of side chains and phenolic groups in the lignin molecular structure. Thus, the authors proposed potential application of the formulated films in antibacterial polymers.246 Lignin incorporating formulations shall conform to various Sustainable Development Goals (SDGs) including, SDG 9 and SDG 12.
4. Concluding remarks
Lignin is one of the most widely available biopolymers; thus, its effective derivation for advanced material applications is lucrative. Lignin can be distinguished on the basis of sourced-wood and extraction operation. The inherent limitation of lignin samples can be countered by their effective functionalization, with chemical modification being the most widely employed. Furthermore, the various venues for effective valorization of functionalized lignin moieties were discussed. Lignin-based materials and their applications in various domains were extensively covered. Furthermore, the influx in the use of additive manufacturing materials, properties of lignin-incorporating materials in AM, and their potential applications in the healthcare sector were explored. Other valorization pathways including lignin-based carbon fibers, Pickering emulsion stabilizers, and smart materials were discussed.Lignin-incorporating sunscreen formulations were discovered to exhibit dark color. 201However, consumer perception studies revealed that a vast majority of people favoring trying lignin-incorporating sunscreen formulations. Another medium for valorizing lignin is its use in self-healing elastomers, where the presence of supramolecular interactions is utilized for imparting the material with self-healing properties. Moreover, the antimicrobial properties and nontoxicity of lignin fractions in conjunction with other active agents in composites render it a suitable material for food packaging applications.244–247 Efforts to valorize lignin on a commercial scale have been made. Thus, the studies discussed in this review were paramount in establishing the need for and the significance of valorizing lignin for advanced material applications.
Abbreviations
| ACL1 | Lignin isolated from industrial hardwood and non-wood residue (Bioethanol plant, USA) |
| ACL2 | Lignin extracted from black liquor (Kraft pulp mill, Brazil) |
| ACL3 | Commercial softwood kraft lignin (Indulin AT, Westvaco Corporation, USA) |
| ACL4 | Commercial non wood soda lignin (Protobind 2000 ALM, India) |
| AM | Additive Manufacturing |
| ASTM | American Society for Testing Materials |
| atm | Standard Atmospheric Pressure |
| BHA | Benzoyl-33-Hydroxyphenyl Acrylate |
| DCM | Dichloromethane |
| DI | Deionized |
| DMSO | Dimethyl sulfoxide |
| DNA | Deoxyribonucleic acid |
| DLS | Dynamic Light Scattering |
| EA | Ethyl Acetate |
| ECH | Epichlorohydrin rubber |
| ESE | Emulsion Solvent Evaporation |
| FDM | Fused Deposition Modeling |
| FTIR | Fourier transformed infrared |
| G units | Guiaiacyl units |
| H units | Hydroxy-phenyl units |
| HW | Hardwood |
| IR | Infrared |
| KPS | Potassium per-sulfate |
| Lig | Unmodified Lignin |
| LigHC | Hexanoyl chloride modified Lignin |
| LNP | Lignin nanoparticles |
| LNT | Lignin nanotubes |
| Mn | Number average molecular weight |
| OSL | Organosolv lignin |
| PAN | Polyacrylonitrile |
| PDI | Polydispersity index |
| PEDOT | Poly(3,4-ethylenedioxythiophene) |
| PEGMA | Poly(ethylene glycol) methacrylate (PEGMA) |
| pH | Potential of hydrogen |
| PMSQ | Polymethylsilsesquioxane |
| PNIPAM | Poly(N-isopropyl acrylamide) |
| R h | Hydrodynamic radius |
| S units | Syringyl units |
| SBS | Styrene–Butadiene–Styrene |
| SLA | Stereolithography apparatus |
| SPF | Sun Protection Factor |
| SW | Softwood |
| T g | Glass transition temperature |
| THF | Tetrahydrofuran |
| TiO2 | Titanium(II) oxide |
| wt% | Percent weight |
| ZnO | Zinc oxide |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support of the Natural Sciences and Engineering Research Council of Canada (NSERC), the Institute of Polymer Research (IPR) at the University of Waterloo, the Canada Research Chair, and the Canada Foundation for Innovation is greatly appreciated.References
- J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, R. Narayan and K. L. Law, Science, 2015, 347, 768–771 CrossRef CAS PubMed
.
- S. A. Miller, ACS Macro Lett., 2013, 2, 550–554 CrossRef CAS PubMed
- O. Hauenstein, S. Agarwal and A. Greiner, Nat. Commun., 2016, 7, 1–7 Search PubMed
- T. Mekonnen, P. Mussone, H. Khalil and D. Bressler, J. Mater. Chem. A, 2013, 1, 13379–13398 RSC
- R. Muthuraj and T. Mekonnen, Polymer, 2018, 145, 348–373 CrossRef CAS
- E. A. B. da Silva, M. Zabkova, J. D. Araújo, C. A. Cateto, M. F. Barreiro, M. N. Belgacem and A. E. Rodrigues, Chem. Eng. Res. Des., 2009, 87, 1276–1292 CrossRef
- F. S. Chakar and A. J. Ragauskas, Ind. Crops Prod., 2004, 20, 131–141 CrossRef CAS
- R. Saad and J. Hawari, J. Porous Mater., 2013, 20, 227–233 CrossRef CAS
- R. C. Pettersen, Adv. Chem., 1984, 207, 57–126 CAS
- D. S. Bajwa, G. Pourhashem, A. H. Ullah and S. G. Bajwa, Ind. Crops Prod., 2019, 139, 111526 CrossRef CAS
-
H. Luo and M. M. Abu-Omar, Encyclopedia of Sustainable Technologies, 2017, pp. 573–585 Search PubMed
-
N. Mandlekar, A. Cayla, F. Rault, S. Giraud, F. Salaün, G. Malucelli and J.-P. Guan, Lignin – Trends and Applications, 2017, DOI:10.5772/INTECHOPEN.72963
-
N. Smolarski, High-value Opportunities for Lignin: Unlocking its Potential, 2012 Search PubMed
- L. Dessbesell, M. Paleologou, M. Leitch, R. Pulkki and C. (Charles) Xu, Renewable Sustainable Energy Rev., 2020, 123, 109768 CrossRef CAS
-
J. H. Lora, Chemical Modification, Properties, and Usage of Lignin, Springer, New York, US, 1st edn, 2012, DOI:10.1007/978-1-4615-0643-0
- Q. Li, R. Gao, Y. Li, B. Fan, C. Ma and Y. C. He, Bioresour. Technol., 2023, 384, 129292 CrossRef CAS PubMed
- W. Dessie, X. Luo, F. He, Y. Liao, G. J. Duns and Z. Qin, Biocatal. Agric. Biotechnol., 2023, 51, 102777 CrossRef CAS
- M. Lawoko and J. S. M. Samec, Curr. Opin. Green Sustainable Chem., 2023, 40, 100738 CrossRef CAS
-
A. Boston, H. London and N. York, Lignin in Polymer Composites, ed. O. Faruk, 2016 Search PubMed
- A. Eraghi Kazzaz and P. Fatehi, Ind. Crops Prod., 2020, 154, 112732 CrossRef CAS
- S. Sabaghi, N. Alipoormazandarani and P. Fatehi, ACS Omega, 2021, 6, 6393–6403 CrossRef CAS PubMed
- S. Sen, S. Patil and D. S. Argyropoulos, Green Chem., 2015, 17, 4862–4887 RSC
-
G. Rodsrud, Potential Applications for Different Lignin Sources Based on Experience from Borregaard and what about the Future?, 2015 Search PubMed
-
A. Maiorana, S. Venkata, S. Arbuckle and G. Viswanathan, Manufacture of novolacs and resoles using lignin, US Pat., US20210009745A1, 2021, pp. 1–7 Search PubMed
-
C. Christopher, P. Griet, T. Mathlide, L. Jorge, US Pat., WO/2017/067769, 2020, pp. 1–12 Search PubMed
- J. Pérez, J. Muñoz-Dorado, T. De La Rubia and J. Martínez, Int. Microbiol., 2002, 5, 53–63 CrossRef PubMed
- C. S. A. Antunes, M. Bietti, M. Salamone and N. Scione, J. Photochem. Photobiol., A, 2004, 163, 453–462 CrossRef CAS
- J. J. Liao, N. H. A. Latif, D. Trache, N. Brosse and M. H. Hussin, Int. J. Biol. Macromol., 2020, 162, 985–1024 CrossRef CAS PubMed
- M. Kaneko, J. Nemoto, H. Ueno, N. Gokan, K. Ohnuki, M. Horikawa, R. Saito and T. Shibata, Electrochem. Commun., 2006, 8, 336–340 CrossRef CAS
- G. Gellerstedt, A. Majtnerova and L. Zhang, C. R. Biol., 2004, 327, 817–826 CrossRef CAS PubMed
- R. B. González-González, H. M. N. Iqbal, M. Bilal and R. Parra-Saldívar, Curr. Opin. Green Sustainable Chem., 2022, 38, 100699 CrossRef
- J.-G. Rosenboom, R. Langer and G. Traverso, Nat. Rev. Mater., 2022, 7, 117–137 CrossRef PubMed
-
W. Fraser, AmallinTM Kraft Lignin Safety Data Sheet (SDS), 2020 Search PubMed
- L. S. Ebers, A. Arya, C. C. Bowland, W. G. Glasser, S. C. Chmely, A. K. Naskar and M. P. Laborie, Biopolymers, 2021, 112, e23431 CrossRef CAS PubMed
- S. Laurichesse and L. Avérous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef CAS
- A. Vishtal, P. Rousu, T. Hultholm, K. Turku, P. Paananen and J. Käyhkö, Bioresources, 2011, 6, 791–806 CAS
- J. M. Lang, U. M. Shrestha and M. Dadmun, Front. Energy. Res., 2018, 6(4), 1–14 CrossRef PubMed
- X. Pan and J. N. Saddler, Biotechnol. Biofuels, 2013, 6, 12 CrossRef CAS PubMed
- J. Domínguez-Robles, T. Tamminen, T. Liitiä, M. S. Peresin, A. Rodríguez and A. S. Jääskeläinen, Int. J. Biol. Macromol., 2018, 106, 979–987 CrossRef PubMed
- S. Laurichesse and L. Avérous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef CAS
- F. Verdini, E. C. Gaudino, E. Canova, S. Tabasso, P. J. Behbahani and G. Cravotto, Molecules, 2022, 27(11) DOI:10.3390/MOLECULES27113598
- C. Scarica, R. Suriano, M. Levi, S. Turri and G. Griffini, ACS Sustain. Chem. Eng., 2018, 6, 3392–3401 CrossRef CAS
- J. Ruwoldt, M. Tanase-Opedal and K. Syverud, ACS Omega, 2022, 7, 46371–46383 CrossRef CAS PubMed
- R. Shorey and T. H. Mekonnen, Int. J. Biol. Macromol., 2023, 230, 123143 CrossRef CAS PubMed
- R. Shorey and T. H. Mekonnen, Int. J. Biol. Macromol., 2022, 209, 472–484 CrossRef CAS PubMed
- R. Shorey, A. Gupta and T. H. Mekonnen, Ind. Crops Prod., 2021, 174, 114189 CrossRef CAS
-
I. A. Pearl, The Chemistry of Lignin, 1967 Search PubMed
- P. Buono, A. Duval, P. Verge, L. Averous and Y. Habibi, ACS Sustain. Chem. Eng., 2016, 4, 5212–5222 CrossRef CAS
- M. S. Karunarathna and R. C. Smith, Sustainability, 2020, 12, 734 CrossRef CAS
- Y. Y. Wang, X. Meng, Y. Pu and A. J. Ragauskas, Polymers, 2020, 2277 CrossRef CAS PubMed
- J. J. Meister, J. Macromol. Sci., Polym. Rev., 2002, 42, 235–289 CrossRef
- A. Boarino and H. A. Klok, Biomacromolecules, 2023, 24(3), 1065–1077 CrossRef CAS PubMed
- Y. Sun, Z. Ma, X. Xu, X. Liu, L. Liu, G. Huang, L. Liu, H. Wang and P. Song, ACS Sustain. Chem. Eng., 2020, 8, 2267–2276 CrossRef CAS
- D. Kun and B. Pukánszky, Eur. Polym. J., 2017, 93, 618–641 CrossRef CAS
- I. Fortelný, D. Kamenická and J. Kovář, Angew. Makromol. Chem., 1988, 164, 125–141 CrossRef
- G. Taylor, Proc. R. Soc. A, 1934, 146, 501–523 CAS
- H.-W. Kammer, Z. Phys. Chem., 1977, 258, 1149–1161 CrossRef CAS
- J. Sternberg, O. Sequerth and S. Pilla, Prog. Polym. Sci., 2021, 113, 101344 CrossRef CAS
- B. M. Upton and A. M. Kasko, Chem. Rev., 2016, 116, 2275–2306 CrossRef CAS PubMed
- D. Kai, M. J. Tan, P. L. Chee, Y. K. Chua, Y. L. Yap and X. J. Loh, Green Chem., 2016, 18, 1175–1200 RSC
- A. Sakakibara, Wood Sci. Technol., 1980, 14(2), 89–100 CrossRef CAS
- M. N. Collins, M. Nechifor, F. Tanasă, M. Zănoagă, A. McLoughlin, M. A. Stróżyk, M. Culebras and C. A. Teacă, Int. J. Biol. Macromol., 2019, 131, 828–849 CrossRef CAS PubMed
- S. Kubo and J. F. Kadla, Biomacromolecules, 2005, 6, 2815–2821 CrossRef CAS PubMed
- J. E. G. Van Dam, M. J. A. Van Den Oever, W. Teunissen, E. R. P. Keijsers and A. G. Peralta, Ind. Crops Prod., 2004, 19, 207–216 CrossRef CAS
- D. Kai, M. J. Tan, P. L. Chee, Y. K. Chua, Y. L. Yap and X. J. Loh, Green Chem., 2016, 18, 1175–1200 RSC
- D. Kun and B. Pukánszky, Eur. Polym. J., 2017, 93, 618–641 CrossRef CAS
- R. R. N. Sailaja and M. V. Deepthi, Mater. Des., 2010, 31, 4369–4379 CrossRef CAS
- F. Chen, H. Dai, X. Dong, J. Yang and M. Zhong, Polym. Compos., 2011, 32, 1019–1025 CrossRef CAS
- B. Košíková and A. Gregorová, J. Appl. Polym. Sci., 2005, 97, 924–929 CrossRef
- O. Gordobil, I. Egüés, R. Llano-Ponte and J. Labidi, Polym. Degrad. Stab., 2014, 108, 330–338 CrossRef CAS
- E. R. Inone-Kauffmann, Int. Sugar J., 2009, 111, 10–11 CAS
- J. Sameni, S. Krigstin, S. A. Jaffer and M. Sain, Ind. Crops Prod., 2018, 117, 58–65 CrossRef CAS
- R. M. Wilkins, Br. Polym. J., 1983, 15, 177–178 CrossRef CAS
- S. Dubey, V. Jhelum and P. K. Patanjali, J. Sci. Ind. Res., 2011, 70, 105–112 CAS
-
J. Asrar and Y. Ding, US Pat., 7771749B2, 2010, pp. 1–26 Search PubMed
- M. Österberg, M. H. Sipponen, B. D. Mattos and O. J. Rojas, Green Chem., 2020, 22, 2712–2733 RSC
- C. Frangville, M. Rutkevičius, A. P. Richter, O. D. Velev, S. D. Stoyanov and V. N. Paunov, ChemPhysChem, 2012, 13, 4235–4243 CrossRef CAS PubMed
- I. A. Gilca, R. E. Ghitescu, A. C. Puitel and V. I. Popa, Iran. Polym. J., 2014, 23, 355–363 CrossRef CAS
- I. A. Gilca, V. I. Popa and C. Crestini, Ultrason. Sonochem., 2015, 23, 369–375 CrossRef CAS PubMed
- R. del P. Castillo, J. Araya, E. Troncoso, S. Vinet and J. Freer, Anal. Chim. Acta, 2015, 866, 10–20 CrossRef CAS PubMed
- W. Yang, J. M. Kenny and D. Puglia, Ind. Crops Prod., 2015, 74, 348–356 CrossRef CAS
- A. Barakat, C. Gaillard, D. Lairez, L. Saulnier, B. Chabbert and B. Cathala, Biomacromolecules, 2008, 9, 487–493 CrossRef CAS PubMed
- M. Ago, B. L. Tardy, L. Wang, J. Guo, A. Khakalo and O. J. Rojas, MRS Bull., 2017, 42, 371–378 CrossRef CAS
- Y. Qian, Y. Deng, X. Qiu, H. Li and D. Yang, Green Chem., 2014, 16, 2156–2163 RSC
- D. Saidane, J. C. Barbe, M. Birot and H. Deleuze, J. Appl. Polym. Sci., 2010, 116, 1184–1189 CrossRef CAS
- K. Gabov, T. Oja, T. Deguchi, A. Fallarero and P. Fardim, Cellulose, 2017, 24, 641–658 CrossRef CAS
- M. Ago, B. L. Tardy, L. Wang, J. Guo, A. Khakalo and O. J. Rojas, MRS Bull., 2017, 42, 371–378 CrossRef CAS
- C. Culbertson, T. Treasure, R. Venditti, H. Jameel and R. Gonzalez, Nord. Pulp. Pap. Res. J., 2016, 31, 30–40 CrossRef CAS
- G. F. Chen and M. H. Liu, BioResources, 2012, 7, 298–314 CAS
- F. Xiong, Y. Han, S. Wang, G. Li, T. Qin, Y. Chen and F. Chu, ACS Sustain. Chem. Eng., 2017, 5, 2273–2281 CrossRef CAS
- H. Hah, J. Kim and B. Jeon, Chem. Commun., 2003, 1712, 1713 Search PubMed
- Q. Zhang, W. Wang, J. Goebl and Y. Yin, Nano Today, 2009, 4(6), 494–507 CrossRef CAS
- M. Chen, L. Wu, S. Zhou and B. You, Adv. Mater., 2006, 18, 801–806 CrossRef CAS
- S. Ding, T. Lin, Y. Wang, X. Lü and F. Huang, New J. Chem., 2013, 37, 784–789 RSC
- Q. Shi, P. Zhang, Y. Li, H. Xia, D. Wang and X. Tao, Chem. Sci., 2015, 6, 4350–4357 RSC
- X. Li, L. Zhou, Y. Wei, A. M. El-Toni, F. Zhang and D. Zhao, J. Am. Chem. Soc., 2015, 137, 5903–5906 CrossRef CAS PubMed
- J. Xu, A. Ma, Z. Xu, X. Liu, D. Chu and H. Xu, J. Phys. Chem. C, 2015, 119, 28055–28060 CrossRef CAS
- X. Li, L. Zhou, Y. Wei, A. M. El-Toni, F. Zhang and D. Zhao, J. Am. Chem. Soc., 2015, 137, 5903–5906 CrossRef CAS PubMed
- F. Xiong, Y. Han, S. Wang, G. Li, T. Qin, Y. Chen and F. Chu, ACS Sustainable Chem. Eng., 2017, 5, 2273–2281 CrossRef CAS
- M. W. Chang, E. Stride and M. Edirisinghe, Langmuir, 2010, 26, 5115–5121 CrossRef CAS PubMed
- S. Ding, T. Lin, Y. Wang, X. Lü and F. Huang, New J. Chem., 2013, 37, 784–789 RSC
- S. H. Im, U. Jeong and Y. Xia, Nat. Mater., 2005, 4(9) DOI:10.1038/nmat1448
- S. C. Luo, J. Jiang, S. S. Liour, S. Gao, J. Y. Ying and H. H. Yu, Chem. Commun., 2009, 2664–2666 RSC
- D.-M. Li and Y.-S. Zheng, J. Org. Chem, 2011, 76, 1100–1108 CrossRef CAS PubMed
- H. Minami, H. Kobayashi and M. Okubo, ACS Publications, 2005, 21, 5655–5658 CAS
- G. Guan, Z. Zhang, Z. Wang, B. Liu, D. Gao and C. Xie, Adv. Mater., 2007, 19, 2370–2374 CrossRef CAS
- F. Xiong, Y. Han, S. Wang, G. Li, T. Qin, Y. Chen and F. Chu, ACS Sustain. Chem. Eng., 2017, 5, 2273–2281 CrossRef CAS
- J. Sameni, K. Krigstin and M. Sain, International Journal of Engineering and Innovative Technology, 2015, 4, 2277–3754 Search PubMed
- A. Bianco, K. Kostarelos and M. Prato, Curr. Opin. Chem. Biol., 2005, 9, 674–679 CrossRef CAS PubMed
- E. Ten and W. Vermerris, Polymers, 2013, 5, 600–642 CrossRef
- E. Ten, C. Ling, Y. Wang, A. Srivastava, L. A. Dempere and W. Vermerris, Biomacromolecules, 2014, 15, 327–338 CrossRef CAS PubMed
- G. Gao, J. I. Dallmeyer and J. F. Kadla, Biomacromolecules, 2012, 13, 3602–3610 CrossRef CAS PubMed
- J. Sameni, S. A. Jaffer, J. Tjong and M. Sain, Curr. For. Rep., 2020, 6, 159–171 CrossRef
- S. C. Ligon, R. Liska, J. Stampfl, M. Gurr and R. Mülhaupt, Chem. Rev., 2017, 117, 10212–10290 CrossRef CAS PubMed
- D. Han and H. Lee, Curr. Opin. Chem. Eng., 2020, 28, 158–166 CrossRef
- E. Sanchez-Rexach, T. G. Johnston, C. Jehanno, H. Sardon and A. Nelson, Chem. Mater., 2020, 32, 7105–7119 CrossRef CAS
- 3D Printing Market Size To Hit USD 98.31 Billion By 2032, https://www.precedenceresearch.com/3d-printing-market, accessed 20 May 2023.
- Wohlers Report 2015 – Wohlers Associates, https://wohlersassociates.com/product/wohlers-report-2015/, accessed 12 April 2023.
- A. Ji, S. Zhang, S. Bhagia, C. G. Yoo and A. J. Ragauskas, RSC Adv., 2020, 10, 21698–21723 RSC
- M. E. Lamm, L. Wang, V. Kishore, H. Tekinalp, V. Kunc, J. Wang, D. J. Gardner and S. Ozcan, Polymers, 2020, 12(9), 2115 CrossRef CAS PubMed
- D. Mohan, Z. K. Teong, A. N. Bakir, M. S. Sajab and H. Kaco, Polymers, 12, 9, 1876 Search PubMed
- S. Zhang, M. Li, N. Hao and A. J. Ragauskas, ACS Omega, 2019, 4, 20197–20204 CrossRef CAS PubMed
- X. Wang, M. Jiang, Z. Zhou, J. Gou and D. Hui, Compos. B Eng., 2017, 110, 442–458 CrossRef CAS
- N. B. Palaganas, J. D. Mangadlao, A. C. C. De Leon, J. O. Palaganas, K. D. Pangilinan, Y. J. Lee and R. C. Advincula, ACS Appl. Mater. Interfaces, 2017, 9, 34314–34324 CrossRef CAS PubMed
- D. Lin, S. Jin, F. Zhang, C. Wang, Y. Wang, C. Zhou and G. J. Cheng, Nanotechnology, 2015, 26 DOI:10.1088/0957-4484/26/43/434003
- Y. Zhang, H. Li, X. Yang, T. Zhang, K. Zhu, W. Si, Z. Liu and H. Sun, Polym. Compos., 2018, 39, E671–E676 CAS
- Y. Y. C. Choong, S. Maleksaeedi, H. Eng, J. Wei and P. C. Su, Mater. Des., 2017, 126, 219–225 CrossRef CAS
- M. Gurr, D. Hofmann, M. Ehm, Y. Thomann, R. Kubier and R. Mülhaupt, Adv. Funct. Mater., 2008, 18, 2390–2397 CrossRef CAS
- R. J. Vora, I. A. Ashcroft and R. Hague, Plast., Rubber Compos., 2007, 36, 68–76 CrossRef CAS
- J. Z. Manapat, J. D. Mangadlao, B. D. B. Tiu, G. C. Tritchler and R. C. Advincula, ACS Appl. Mater. Interfaces, 2017, 9, 10085–10093 CrossRef CAS PubMed
- J. H. Sandoval, K. F. Soto, L. E. Murr and R. B. Wicker, J. Mater. Sci., 2007, 42, 156–165 CrossRef CAS
- H. Eng, S. Maleksaeedi, S. Yu, Y. Y. C. Choong, F. E. Wiria, C. L. C. Tan, P. C. Su and J. Wei, Procedia Eng., 2017, 216, 1–7 CrossRef
- D. E. Yunus, W. Shi, S. Sohrabi and Y. Liu, Nanotechnology, 2016, 27 DOI:10.1088/0957-4484/27/49/495302
- L. Wang and X. Ni, Polym. Bull., 2017, 74, 2063–2079 CrossRef CAS
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344(6185) DOI:10.1126/SCIENCE.1246843
- S. I. Falkehag, J. Marton and E. Adler, Sven. Papperstidn., 1966, 75–89 CAS
- P. Yu, H. He, Y. Jia, S. Tian, J. Chen, D. Jia and Y. Luo, Polym. Test., 2016, 54, 176–185 CrossRef CAS
- A. De Chirico, M. Armanini, P. Chini, G. Cioccolo, F. Provasoli and G. Audisio, Polym. Degrad. Stab., 2003, 79, 139–145 CrossRef CAS
- W. Xu, X. Wang, N. Sandler, S. Willför and C. Xu, ACS Sustain. Chem. Eng., 2018, 6, 5663–5680 CrossRef CAS
- J. Domínguez-Robles, N. K. Martin, M. L. Fong, S. A. Stewart, N. J. Irwin, M. I. Rial-Hermida, R. F. Donnelly and E. Larrañeta, Pharmaceutics, 2019, 11(4) DOI:10.3390/PHARMACEUTICS11040165
- J. T. Sutton, K. Rajan, D. P. Harper and S. C. Chmely, ACS Appl. Mater. Interfaces, 2018, 10, 36456–36463 CrossRef CAS PubMed
- V. Mimini, E. Sykacek, S. N. A. Hashim, J. Holzweber, H. Hettegger, K. Fackler, A. Potthast, N. Mundigler and T. Rosenau, J. Wood Chem. Technol., 2019, 39, 14–30 CrossRef CAS
- N. A. Nguyen, S. H. Barnes, C. C. Bowland, K. M. Meek, K. C. Littrell, J. K. Keum and A. K. Naskar, Sci. Adv., 2018, 4(12) DOI:10.1126/SCIADV.AAT4967
- N. Sallem-Idrissi, M. Sclavons, D. P. Debecker and J. Devaux, J. Appl. Polym. Sci., 2016 DOI:10.1002/APP.42963
- E. Gkartzou, E. P. Koumoulos and C. A. Charitidis, Manuf. Rev., 2017 DOI:10.1051/MFREVIEW/2016020
- D. J. McClements, L. Bai and C. Chung, Annu. Rev. Food Sci. Technol., 2017, 8, 205–236 CrossRef PubMed
- R. Zheng, B. P. Binks and Z. Cui, Langmuir, 2020, 36, 4619–4629 CrossRef CAS PubMed
- B. P. Binks and S. O. Lumsdon, Langmuir, 2001, 17, 4540–4547 CrossRef CAS
- B. P. Binks, J. Philip and J. A. Rodrigues, Langmuir, 2005, 21, 3296–3302 CrossRef CAS PubMed
- Z. G. Cui, K. Z. Shi, Y. Z. Cui and B. P. Binks, Colloids Surf., A, 2008, 329, 67–74 CrossRef CAS
- N. P. Ashby and B. P. Binks, Phys. Chem. Chem. Phys., 2000, 2, 5640–5646 RSC
- Our Endangered World, Is Silica Bad for the Environment?, https://www.ourendangeredworld.com/eco/is-silica-bad-for-the-environment/, accessed 15 August 2022.
- D. Klemm, F. Kramer, S. Moritz, T. Lindström, M. Ankerfors, D. Gray and A. Dorris, Angew. Chem., Int. Ed., 2011, 50, 5438–5466 CrossRef CAS PubMed
- L. Dai, Y. Li, F. Kong, K. Liu, C. Si and Y. Ni, ACS Sustain. Chem. Eng., 2019, 7, 13497–13504 CrossRef CAS
- P. Jędrzejczak, M. N. Collins, T. Jesionowski and Ł. Klapiszewski, Int. J. Biol. Macromol., 2021, 187, 624–650 CrossRef PubMed
- K. Lintinen, Y. Xiao, R. Bangalore Ashok, T. Leskinen, E. Sakarinen, M. Sipponen, F. Muhammad, P. Oinas, M. Österberg and M. Kostiainen, Green Chem., 2018, 20, 843–850 RSC
- T. Zou, M. H. Sipponen and M. Österberg, Front. Chem., 2019, 7, 449471 Search PubMed
- M. Ago, S. Huan, M. Borghei, J. Raula, E. I. Kauppinen and O. J. Rojas, ACS Appl. Mater. Interfaces, 2016, 8, 23302–23310 CrossRef CAS PubMed
- M. H. Sipponen, M. Smyth, T. Leskinen, L. S. Johansson and M. Österberg, Green Chem., 2017, 19, 5831–5840 RSC
- Carbon Fiber Market Size To Hit USD 5.83 Billion By 2032, https://www.precedenceresearch.com/carbon-fiber-market, accessed 22 May 2023.
- A. H. Wazir, L. Kakakhel and X. T. Cailiao, New Carbon Mater., 2009, 24, 83–88 CrossRef CAS
- K. Sudo and K. Shimizu, J. Appl. Polym. Sci, 1992, 44, 127–134 CrossRef CAS
- Y. Xu, Y. Liu, S. Chen and Y. Ni, Bioresources, 2020, 15, 7234–7259 Search PubMed
- D. A. Baker and T. G. Rials, J. Appl. Polym. Sci, 2013, 130, 713–728 CrossRef CAS
- M. Zhang, W. Liu, H. Niu and D. Wu, High Perform. Polym., 2019, 31, 168–177 CrossRef CAS
- L. Zhang, A. Aboagye, A. Kelkar, C. Lai and H. Fong, J. Mater. Sci., 2013, 49(2), 463–480 CrossRef
- D. A. Baker and T. G. Rials, J. Appl. Polym. Sci., 2013, 130, 713–728 CrossRef CAS
- E. Frank, L. M. Steudle, D. Ingildeev, J. M. Spörl and M. R. Buchmeiser, Angew. Chem., Int. Ed., 2014, 53, 5262–5298 CrossRef CAS PubMed
- E. Frank, F. Hermanutz and M. R. Buchmeiser, Macromol. Mater. Eng., 2012, 297, 493–501 CrossRef CAS
- M. Norgren and H. Edlund, Curr. Opin. Colloid Interface Sci., 2014, 19, 409–416 CrossRef CAS
- W. Fang, S. Yang, X.-L. Wang, T.-Q. Yuan and R.-C. Sun, Green Chem., 2017, 19, 1794–1827 RSC
- Y. Xu, Y. Liu, S. Chen and Y. Ni, Bioresources, 2020, 15, 7234–7259 Search PubMed
- J. Jin, J. Ding, A. Klett, M. C. Thies and A. A. Ogale, ACS Sustain. Chem. Eng., 2018, 6, 14135–14142 CrossRef CAS
- Y. Nordström, I. Norberg, E. Sjöholm and R. Drougge, J. Appl. Polym. Sci., 2013, 129, 1274–1279 CrossRef
- L. Salmén, E. Bergnor, A.-M. Olsson, M. Åkerström and A. Uhlin, Bioresources, 2015, 10, 7544–7554 CrossRef
- X. Huang, Materials, 2009, 2, 2369–2403 CrossRef CAS
- Y. Huang and R. J. Young, Carbon, 1995, 33, 97–107 CrossRef CAS
- D. A. Baker and T. G. Rials, J. Appl. Polym. Sci., 2013, 130, 713–728 CrossRef CAS
- N. Meek, D. Penumadu, O. Hosseinaei, D. Harper, S. Young and T. Rials, Compos. Sci. Technol., 2016, 137, 60–68 CrossRef CAS
- Q. Li, S. Xie, W. K. Serem, M. T. Naik, L. Liu and J. S. Yuan, Green Chem., 2017, 19, 1628–1634 RSC
- F. Hermansson, M. Janssen and M. Svanström, J. Clean. Prod., 2019, 223, 946–956 CrossRef CAS
- J. Jin and A. A. Ogale, J. Appl. Polym. Sci., 2017 DOI:10.1002/APP.45903
- J. F. Kadla, S. Kubo, R. A. Venditti, R. D. Gilbert, A. L. Compere and W. Griffith, Carbon, 2002, 40, 2913–2920 CrossRef CAS
- Y. Uraki, S. Kubo, N. Nigo, Y. Sano and T. Sasaya, Holzforschung, 1995, 49, 343–350 CrossRef CAS
- S. P. Maradur, C. H. Kim, S. Y. Kim, B. H. Kim, W. C. Kim and K. S. Yang, Synth. Met., 2012, 162, 453–459 CrossRef CAS
- I. Norberg, Y. Nordström, R. Drougge, G. Gellerstedt and E. Sjöholm, J. Appl. Polym. Sci., 2013, 128, 3824–3830 CrossRef CAS
- K. Sudo and K. Shimizu, J. Appl. Polym. Sci., 1992, 44, 127–134 CrossRef CAS
- H. A. Ariyanta, E. B. Santoso, L. Suryanegara, E. T. Arung, I. W. Kusuma, M. N. Azman Mohammad Taib, M. H. Hussin, Y. Yanuar, I. Batubara and W. Fatriasari, Sustain. Chem. Pharm., 2023, 32, 100966 CrossRef CAS
- H. Li, S. Colantonio, A. Dawson, X. Lin and J. Beecker, J. Cutan. Med. Surg., 2019, 23, 357–369 CrossRef PubMed
-
Canadian Cancer Society’s Advisory Committee on Cancer Statistics, Canadian Cancer Statistics, 2014, pp. 1–132 Search PubMed
- A. C. Green, G. M. Williams, V. Logan and G. M. Strutton, J. Clin. Oncol., 2011, 29, 257–263 CrossRef CAS PubMed
- B. K. Armstrong and A. Kricker, J. Photochem. Photobiol. B, 2001, 63, 8–18 CrossRef CAS PubMed
- P. Kullavanijaya and H. W. Lim, J. Am. Acad. Dermatol., 2005, 52, 937–958 CrossRef PubMed
- M. Latha, J. Martis and V. Shobha, J. Clin. Aesthet. Dermatol., 2013, 16–26 CAS
- M. Morsella, N. D'Alessandro, A. E. Lanterna and J. C. Scaiano, ACS Omega, 2016, 1, 464–469 CrossRef CAS PubMed
- M. Morsella, M. Giammatteo, L. Arrizza and N. d’Alessandro, RSC Adv., 2015, 5, 57453–57461 RSC
- A. Morlando, V. Sencadas, D. Cardillo and K. Konstantinov, Powder Technol., 2018, 329, 252–259 CrossRef CAS
- H. Ariyanta, E. Santoso, L. Suryanegara and E. Arung, Sustainable Chem. Pharm., 2023, 32, 100966 CrossRef CAS
- K. Geoffrey, A. Mwangi and S. Maru, Saudi Pharm. J., 2019, 27, 1009–1018 CrossRef CAS PubMed
- S. Lee, T. Tran, J. Choi and K. Wan, Int. J. Biol. Macromol., 2019, 122, 549–554 CrossRef CAS PubMed
- H. Zhang, X. Liu, S. Fu and Y. Chen, Int. J. Biol. Macromol., 2019, 133, 86–92 CrossRef CAS PubMed
- N. Sajinčič, O. Gordobil, A. Simmons and A. Sandak, Cosmetics, 2021, 8, 78 CrossRef
- Y. Qian, X. Zhong, Y. Li and X. Qiu, Ind. Crops Prod., 2017, 101, 54–60 CrossRef CAS
- M. Tran, D. Phan and E. Lee, Green Chem., 2021, 23, 4633–4646 RSC
- Y. Wu, Y. Qian, H. Lou, D. Yang and X. Qiu, ACS Sustainable Chem. Eng., 2019, 7, 15966–15973 CrossRef CAS
- Y. Wu, Y. Qian, A. Zhang, H. Lou, D. Yang and X. Qiu, Ind. Eng. Chem. Res., 2020, 59, 17057–17068 CrossRef CAS
- D. Kai, Y. Chua, L. Jiang, C. Owh, S. Chan and X. L., RSC Adv., 2016 Search PubMed
- Y. Qian, X. Qiu and S. Zhu, ACS Sustain. Chem. Eng., 2016, 4, 4029–4035 CrossRef CAS
- Y. Qian, X. Qiu and S. Zhu, ACS Sustainable Chem. Eng., 2016, 17, 320 Search PubMed
- N. Rapoport, Prog. Polym. Sci., 2007, 32, 962–990 CrossRef CAS
- H. Jiang, S. Kelch and A. Lendlein, Adv. Mater., 2006, 18, 1471–1475 CrossRef CAS
- H. Chen, H. Xia, Y. Qiu and Q. Q. Ni, Compos. Sci. Technol., 2018, 163, 105–115 CrossRef CAS
- J. Lu, A. Arsalan, Y. Dong, Y. Zhu, C. Qian, R. Wang, C. Cuilan, Y. Fu, Q. Q. Ni and K. N. Ali, Polym. Test, 2017, 59, 462–469 CrossRef CAS
- Y. Dong, H. Xia, Y. Zhu, Q. Q. Ni and Y. Fu, Compos. Sci. Technol., 2015, 120, 17–25 CrossRef CAS
- Y. Liu, H. Du, L. Liu and J. Leng, Smart Mater. Struct., 2014, 23 DOI:10.1088/0964-1726/23/2/023001
- J. Xu, J. Song, J. Xu and J. Song, Biomed. Eng.: Front. Challenges, 2011 DOI:10.5772/19256
- K. Shikinaka, Y. Funatsu, Y. Kubota, Y. Tominaga, M. Nakamura, R. R. Navarro and Y. Otsuka, Soft Matter, 2018, 14, 9227–9231 RSC
- H. Liu, N. Mohsin, S. Kim and H. Chung, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 2121–2130 CrossRef CAS
- E. S. Dragan, Chem. Eng. J., 2014, 243, 572–590 CrossRef CAS
- M. F. Akhtar, M. Hanif and N. M. Ranjha, Saudi Pharm. J., 2016, 24, 554–559 CrossRef PubMed
- D. Rico-García, L. Ruiz-Rubio, L. Pérez-Alvarez, S. L. Hernández-Olmos, G. L. Guerrero-Ramírez and J. L. Vilas-Vilela, Polymers, 2020, 12, 81 CrossRef PubMed
- E. Larrañeta, M. Imízcoz, J. X. Toh, N. J. Irwin, A. Ripolin, A. Perminova, J. Domínguez-Robles, A. Rodríguez and R. F. Donnelly, ACS Sustain. Chem. Eng., 2018, 6, 9037–9046 CrossRef PubMed
- S. Chandna, S. Paul, R. Kaur, K. Gogde and J. Bhaumik, ACS Appl. Polym. Mater., 2022, 4, 8962–8976 CrossRef CAS
- Y. Ma, Y. Sun, Y. Fu, G. Fang, X. Yan and Z. Guo, Chemosphere, 2016, 163, 610–619 CrossRef CAS PubMed
- A. Moreno and M. H. Sipponen, Mater. Horiz., 2020, 7, 2237–2257 RSC
- B. M. Cerrutti, M. L. Moraes, S. H. Pulcinelli and C. V. Santilli, Biosens. Bioelectron., 2015, 71, 420–426 CrossRef CAS PubMed
- A. Jędrzak, T. Rębiś, Ł. Klapiszewski, J. Zdarta, G. Milczarek and T. Jesionowski, Sens. Actuators, B, 2018, 256, 176–185 CrossRef
- Q. Wang, X. Pan, C. Lin, D. Lin, Y. Ni, L. Chen, L. Huang, S. Cao and X. Ma, Chem. Eng. J., 2019, 370, 1039–1047 CrossRef CAS
- A. Jędrzak, T. Rębiś, Ł. Klapiszewski, J. Zdarta, G. Milczarek and T. Jesionowski, Sens. Actuators, B, 2018, 256, 176–185 CrossRef
- Q. Wang, X. Pan, C. Lin, D. Lin, Y. Ni, L. Chen, L. Huang, S. Cao and X. Ma, Chem. Eng. J., 2019, 370, 1039–1047 CrossRef CAS
- B. Wang, T. Shi, Y. Zhang, C. Chen, Q. Li and Y. Fan, J. Mater. Chem. C, 2018, 6, 6423–6428 RSC
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446–7467 RSC
- S. Burattini, B. W. Greenland, D. H. Merino, W. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. MacKay, I. W. Hamley and S. J. Rowan, J. Am. Chem. Soc., 2010, 132, 12051–12058 CrossRef CAS
- S. J. Kalista, T. C. Ward and Z. Oyetunji, Mech. Adv. Mater. Struct., 2007, 14, 391–397 CrossRef CAS
- Y. Huang, M. Zhong, Y. Huang, M. Zhu, Z. Pei, Z. Wang, Q. Xue, X. Xie and C. Zhi, Nat. Commun., 2015, 6 DOI:10.1038/NCOMMS10310
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed
- B. Ghosh and M. W. Urban, Science, 2009, 323, 1458–1460 CrossRef CAS PubMed
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS PubMed
- C. H. Li, C. Wang, C. Keplinger, J. L. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel, C. Linder, X. Z. You and Z. Bao, Nat. Chem., 2016, 8, 618–624 CrossRef CAS PubMed
- K. Miyamae, M. Nakahata, Y. Takashima and A. Harada, Angew. Chem., Int. Ed., 2015, 54, 8984–8987 CrossRef CAS PubMed
- M. Cui, N. A. Nguyen, P. V. Bonnesen, D. Uhrig, J. K. Keum and A. K. Naskar, ACS Macro Lett., 2018, 7, 1328–1332 CrossRef CAS PubMed
- T. Elder, Biomacromolecules, 2007, 8, 3619–3627 CrossRef CAS PubMed
- N. Sun, Z. Wang, X. Ma, K. Zhang, Z. Wang, Z. Guo, Y. Chen, L. Sun, W. Lu, Y. Liu and M. Di, Ind. Crops Prod., 2021, 174, 114178 CrossRef CAS
- S. Shankar and J. W. Rhim, Food Hydrocoll., 2017, 71, 76–84 CrossRef CAS
- S. Hu and Y. Lo Hsieh, Carbohydr. Polym., 2015, 131, 134–141 CrossRef CAS PubMed
- X. Wang, S. Wang, W. Liu, S. Wang, L. Zhang, R. Sang, Q. Hou and J. Li, Carbohydr. Polym., 2019, 225, 115213 CrossRef PubMed
- W. Yang, E. Fortunati, F. Dominici, G. Giovanale, A. Mazzaglia, G. M. Balestra, J. M. Kenny and D. Puglia, Eur. Polym. J., 2016, 79, 1–12 CrossRef CAS
- C. C. O. Santos, F. V. Ferreira, I. F. Pinheiro and L. M. F. Lona, J. Environ. Chem. Eng., 2023, 11, 109691 CrossRef CAS
- A. Boarino and H. A. Klok, Biomacromolecules, 2023, 24, 1065–1077 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2024 |