
Recent advances in the development of phenothiazine and its fluorescent derivatives for optoelectronic applications
Faizal
Khan
and
Rajneesh
Misra
 *
*
Department of Chemistry, Indian Institute of Technology Indore, Indore 453552, India. E-mail: rajneeshmisra@iiti.ac.in
First published on 17th January 2023
Abstract
Phenothiazine is one of the most extensively investigated aromatic compounds owing to its unique optical and electronic properties, low cost, and easy functionalization. In recent years, phenothiazine and its derivatives have provided a versatile platform for developing materials with wide range of applications. The electron-rich sulfur and nitrogen atoms make phenothiazine a stronger electron donor and its non-planar butterfly shaped bent structure sufficiently suppresses the molecules to form aggregates. In this review, we discuss the recent advances in the development of phenothiazine-based fluorescent materials for applications in various fields. We highlight prominent examples of phenothiazine-based materials that have been effectively exploited in applications such as mechanochromism, aggregation-induced emission, phosphorescence, and sensor probes. This review will pave the way for researchers to design and develop new phenothiazine functionalized materials and use them for various optoelectronic applications.
 Faizal Khan | Faizal Khan is currently a PhD student at the Indian Institute of Technology (IIT) Indore, working under the supervision of Prof. Rajneesh Misra. He has completed his Bachelor of Science (BSc) in 2013 and Master of Science (MSc) in 2015, from St. Andrew*s College, Gorakhpur. His research at IIT Indore focuses on the design and synthesis of organic mechanochromic luminogens. |
 Rajneesh Misra | Dr Rajneesh Misra is currently a full professor in the Department of Chemistry, Indian Institute of Technology (IIT) Indore. He received his master*s degree from the University of Gorakhpur, in 2001. After that, he moved to the Indian Institute of Technology (IIT) Kanpur for his PhD in chemical sciences, in 2007. After two successive postdoctoral stays at the Georgia Institute of Technology, Atlanta, USA (2008), and Kyoto University, Japan (2009), he joined IIT Indore, India, as an assistant professor. His research interests lie in the areas of organic photonics and organic electronics. |
1. Background
Photoluminescence (PL) is the phenomenon of light emission from a substance, after the absorption of photons, which occurs from electronically excited states.1 Materials that exhibit strong PL characteristics have attracted a lot of research interest owing to their potential uses in a variety of optoelectronic devices such as light emitting diodes (LEDs),2 in non-linear optics (NLO),3 bioimaging and biomedical applications,4–6 sensors,7 deformation detectors,8 data storage devices9 and fluorescent tags.10 The electronically excited molecules either decay or relax back to the ground state by dispersing their excited state energy via radiative or non-radiative routes. Many studies have been carried out to fully understand the radiative and non-radiative mechanisms used to dissipate the excited state energy.11–15 It should be noted that the radiative relaxation of the excited state leads to light emission principally via three processes: fluorescence, phosphorescence, and delayed fluorescence.16 Researchers have investigated the various luminescence phenomena and have developed an understanding of the mechanics involved in producing effective luminescence.17Organic luminescent materials have several advantages over their inorganic counterparts, such as their compatibility with various substrates, definite chemical structures, low fabrication costs and their favourable ease of processing.18 Devices based on organic materials can be fabricated using either a thermal evaporation method or a solution processing method, which enables simple fabrication steps such as drop-casting19 and spin-coating,20 even at room temperatures. The luminescence properties of an organic compound can be tuned easily via precise alterations in the π-conjugation length or by inserting various electron donating or electron withdrawing units into their molecular structure.21,22 Owing to these advantages, organic luminescent materials are frequently used in various optoelectronic applications such as memory devices, solid-state lasers, sensors, security ink, and bioimaging.23,24 Most of the practical applications of organic luminescent materials require them to be in the solid state. For example, the development of organic light emitting diodes (OLEDs) is accomplished through vacuum deposition of the emissive material as a solid film.25 In addition, various biomedical applications such as fluorescence imaging use luminophores in the aggregated form to trap various biocomponents and bioprocesses in living cells.26 A plethora of organic luminophores have been reported in the scientific literature for use in advanced optoelectronic technologies, although only a handful show strong emission in the solid state. Traditional organic luminophores emit no or very little light when aggregated and exhibit aggregation caused quenching (ACQ) due to the significant π–π stacking interactions in the solid state. In 1954, Förster identified quenching of the emission with increasing concentration or upon aggregation,27 and since then a large amount of research has been devoted to studying and mitigating this phenomenon. In 1970, Birks reported that the majority of well-known organic aromatic molecules exhibit ACQ.28 In the solution state, the aromatic molecules are well-separated and do not interact with each other, resulting in strong emission. However, when the molecules aggregate, they are in close proximity and the aromatic rings of the luminophores interact with each other and form excessive π–π stacking interactions. For example, perylene molecules are highly emissive in their isolated form or in the solution state, but upon aggregation the molecules interact with each other like stacked discs (Fig. 1(a)). This creates non-radiative pathways for the relaxation of excited state molecules, resulting in suppressed emission in the solid state.29,30
 | ||
| Fig. 1 (a) Schematic representation showing the general ACQ phenomenon in perylene molecules; and (b) the restriction of intramolecular rotation (RIR) mechanism of phenyl rotors against the silole stator in HPS aggregates. | ||
The emission of a fluorophore in the solid state can be improved via the aggregation-induced emission (AIE) mechanism in which the use of several non-planar moieties containing rotatable aromatic rings enables non-emissive materials to become emissive when aggregated. Tang et al. established the AIE mechanism in which the twisted molecular structure of AIE luminogens prevents the molecules from stacking on top of each other and reduces the π–π stacking interactions in the solid state.31 This decreases the rate of non-radiative pathways for the dissipation of excited state energy and increases the emission in the solid state.32–36 The AIE phenomenon is the reverse of the ACQ effect, and was first observed in hexaphenylsilole (HPS) derivatives. Hexaphenylsilole is non-emissive in the solution state due to free rotation of the phenyl rings, which dissipates the excited state energy non-radiatively. In the aggregated state, the intramolecular rotations of the HPS phenyl rings are restricted, which reduces the rate of non-radiative decay of the excited state energy and results in intense emission in the aggregated state (Fig. 1(b)).37 Over the past few years, a wide range of AIE luminogens that have the potential to produce enhanced emission in the solid state via restricting the intramolecular motion have been synthesized through careful manipulation of the chemical structure of various organic compounds.38,39 The chemical structures of commonly employed AIEgens are shown in Chart 1.
 | ||
| Chart 1 Chemical structures of various aggregation induced emission (AIE) type luminogens. | ||
2. Mechanochromism
The phenomenon in which a luminescent material changes its luminescence colour in response to an external mechanical force such as grinding, rubbing, shearing, or scratching, is called mechanochromism.40,41 Mechanochromic materials are frequently used in data storage devices, rewritable inks, information encryption, and sensors due to their simple operation and rapid responsiveness.42,43 The terms mechanochromic (MC), mechanofluorochromic (MFC) and mechanochromic luminescent (MCL) materials can be used for fluorescent materials that can show changes in their colour by changing their emission wavelength when subjected to a mechanical stimulus. By contrast, mechanoresponsive luminescent (MRL) materials are materials that generally cover any changes in their emission wavelength, emission intensity, or emission lifetime when subjected to a mechanical stimulus. In general, MC, MFC and MCL materials are types of MRL material.44 The emission colour of mechanochromic materials has a broad luminous range, including the whole visible spectrum and extending into the near-infrared region.45 Based on the different phenomena exhibited by the mechanochromic materials, mechanochromism can be classified into various groups, such as two-color and multicolor mechanochromism,46 reversible and irreversible mechanochromism,47 fluorescence–phosphorescence mechanochromism,48 turn-on and turn-off type mechanochromism,49 and bathochromically and hypsochromically shifted mechanochromism.50The stimuli responsive changes in the emission of a mechanochromic material are a result of perturbations in the molecular packing patterns and the intermolecular interactions. The application of a mechanical force does not alter the chemical structure of a mechanochromic luminogen; however, it alters the various intermolecular and intramolecular interactions. A mechanical stimulus leads in either perturbation to the close association of the molecules or the disruption of certain intermolecular interactions, which ultimately affects the emission characteristics.51 A mechanical force can change the crystalline nature of a compound to an amorphous phase,52,53 but sometimes the mechanical force causes a transition between different crystalline states.54 The various interactions, such as hydrogen bonding or intramolecular and intermolecular charge transfer, become more noticeable when external forces are applied, causing different colours to be emitted. The mechanical force occasionally induces a change in the molecular structure; for example, the lactam rhodamine-B molecule 1 changes into its ring-open isomer when subjected to mechanical stimulation (Fig. 2).55 This change is accompanied by the appearance of red fluorescence.
 | ||
| Fig. 2 Mechanical stimulus induced spectral changes in the rhodamine-B derivative 1. Reproduced from ref. 55 with permission from © 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
The mechanical stimulus induced conformational change is one of the most significant factors that contribute to the shift in the emission colour. Mechanochromic materials usually have non-planar molecules in their crystalline state, but, upon the application of an external force, these compounds become planar and increase in their extent of conjugation, resulting in a color change with a bathochromic shift.56 A reverse phenomenon was reported by Wu et al. in which the mechanical force perturbed the planar structures of the molecules in the crystalline state, and thus a hypsochromic shift in the emission was observed.57 Several mechanochromic materials exhibit excimer (i.e., excited-state dimer) formation upon the application of a mechanical force, in which the emission spectrum shifts bathochromically because the excimer emission often occurs at a lower energy than the corresponding monomer emission.58 Various mechanisms have been proposed to explain mechanochromic behavior, such as the transformation of dimeric or excimer emission to monomer emission,59 single crystals to microcrystalline states,60 phosphorescence emission in the crystalline state and prompt fluorescence in the ground state,61 the transformation of a large molecular assembly into smaller molecular assemblies or into isolated molecules,62 and many more. Recent research has demonstrated that adjustable emissions can be obtained by controlling the intramolecular torsional angles of a luminogen in the solid state. Yang et al. developed three crystalline polymorphs of a phenothiazine based luminogen by changing the crystallization conditions.63 These polymorphs display very different photoluminescence properties owing to the differences in their packing modes.
In recent years, organic compounds that contain electron donor (D) and acceptor (A) moieties have surfaced as potential candidates for the development of mechanochromic materials.64 These D–A framework based compounds exhibit intramolecular charge-transfer (ICT) transitions from the donor to the acceptor moiety. The D–A systems display mechanochromic behaviour by changing the dihedral angles between the donor and acceptor moieties.65 Alterations in the dihedral angles have an effect on the ICT emission and, thus, multicolor emission can be achieved in a D–A system either by exploiting the multiple ICT states or by switching from the localized excited (LE) state to the charge-transfer (CT) state. Tian et al. synthesized a non-planar D–A molecule based on the tetraphenylethylene-acridonyl moiety (2) which shows blue emission.66 On mechanical grinding, the blue emission of 2 turns to a bright cyan emission (Fig. 3). This change in emission colour was associated with the grinding induced transformation of the LE state to the CT state. Ma et al. synthesized the D–A derivative 3 that is based on the triphenylamine and diphenylacrylonitrile moieties.67 The triphenylamine derivative 3 displayed a colour shift from green to yellow upon mechanical grinding, which was associated with the partial conversion of the LE state to the CT excited state and the coexistence of LE–CT states. Additional grinding altered the colour from yellow to red, which is associated with the dominance of the CT excited state (Fig. 3).
 | ||
| Fig. 3 Chemical structures and mechanical stimuli induced emission colour changes in 2 and 3. Reproduced from ref. 66 and ref. 67, respectively, with permission from © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
In general, a compound must have the following characteristics in order to exhibit mechanochromism: (i) a non-planar or twisted molecular structure; (ii) effective emission in the solid state; (iii) a heteroatom containing a lone pair of electrons, such as N, O and S; and (iv) effective intramolecular charge transfer (ICT) characteristics.68 In this context, various N, O and S atom based mechanochromic compounds have been reported so far. Herein, we focus primarily on the contribution of the phenothiazine core for the reasons stated in the next section.
3. Phenothiazine
Phenothiazine is an electron-rich tricyclic heteroarene, synthesized in 1883 by Bernthsen via the reaction of diphenylamine with sulfur.69 It has recently attracted a lot of research interest owing to its versatility of functionalization and its unique optoelectronic properties. The phenothiazine moiety exhibits a butterfly-shaped non-planar structure with a dihedral angle of 158.51° between the planes of the two benzene rings. Owing to the presence of electron rich S and N atoms, the phenothiazine has a strong electron donating character that facilitates the formation of enhanced intramolecular charge transfer (ICT) complexes. In addition, its butterfly non-planar structure inhibits π–π stacking upon aggregation, resulting in enhanced fluorescence in the solid state.70–73Multiple functional sites are present on the phenothiazine core, including C-2, C-3, C-7, C-8, N-10, and the S-atom of the thiazine ring (Fig. 4(a)). In most cases, the C-3 and C-7 positions of the phenothiazine core are functionalized with a wide range of electron-donating or electron-withdrawing groups. Furthermore, to acquire the appropriate solubility in common organic solvents, the N-10 position of phenothiazine can be functionalized with suitable solubilizing groups such as alkyl or aryl substituents.74 The sulphur atom in the phenothiazine unit can be easily oxidized to sulfoxide (4+ oxidation) and sulfone (6+ oxidation) via reaction with an appropriate oxidizing agent such as hydrogen peroxide or 3-chloroperbenzoic acid (m-CPBA).75–77 The bowl-shaped molecular structure of phenothiazine enables its derivatives to exist as two different conformers with significantly different optical and electronic properties. These are the H-intra (quasi-axial) conformer and H-extra (quasi-equatorial) conformer, depending on the S–N axis and plane of the aromatic rings (Fig. 4(b)). Among them, the quasi-axial conformation exhibits a much larger energy gap than the quasi-equatorial conformer. Studies on these two heterogenous conformers have produced some exciting findings.78 For example, Chi et al. investigated the conformation dependent delayed fluorescence in a phenothiazine derivative in which mechanical pressure changes the molecular conformation from the quasi-axial to the quasi-equatorial state;79 Minakata et al. reported a mechanochromically active fluorophore by utilizing two phenothiazine units, where the fluorophore exhibited distinct emission properties depending on the conformation of the phenothiazine moiety.80 Li et al. successfully demonstrated dynamic mechanoluminescence from blue to yellow in a phenothiazine derivative which was associated with the mechanical pressure induced conformation transition from the quasi-axial to the quasi-equatorial state.81 The development of “donor–acceptor approach” based molecular engineering in π-conjugated organic materials accelerated the synthesis of a vast variety of phenothiazine derivatives with customized optical and electronic properties.
 | ||
| Fig. 4 (a) Chemical structure of 10H-phenothiazine showing various functionalization positions; and (b) quasi-equatorial and quasi-axial conformations of phenothiazine. | ||
A number of phenothiazine based organic small molecules and polymers have been synthesized and used effectively as photoactive materials in various organic electronic devices such as organic light-emitting diodes (OLEDs), dye-sensitized solar cells (DSSCs), perovskite solar cells (PSCs), organic field effect transistors (OFETs), sensors, batteries, and many more.82,83 Some reports have previously explored the major optoelectronic applications of phenothiazines, particularly in solar cells.84–87 The use of the phenothiazine core in the development of organic fluorescent materials has not yet been fully investigated. In this review, we intend to bridge this knowledge gap by providing a summary of the recent advances in the development of phenothiazine-based fluorescent materials for applications in various fields. We cover multiple methods for the synthesis of the phenothiazine core, as well as the complexity of structural modifications, and then highlight prominent examples of phenothiazine-based materials that have been effectively exploited in applications such as mechanochromism, aggregation induced emission, phosphorescence, and sensor probes.
3.1. Synthesis of the phenothiazine core
The phenothiazine core can be synthesized using the following principal methods: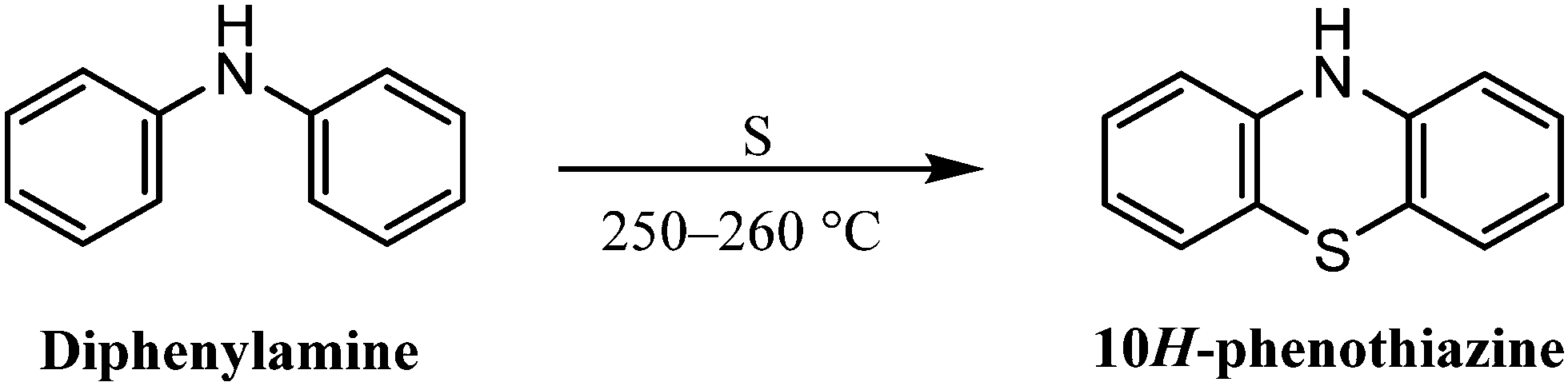 | ||
| Scheme 1 Synthesis of 10H-phenothiazine from diphenylamine. | ||
 | ||
| Scheme 2 Synthesis of 1,3-dinitro-10H-phenothiazine 4via Smiles rearrangement. | ||
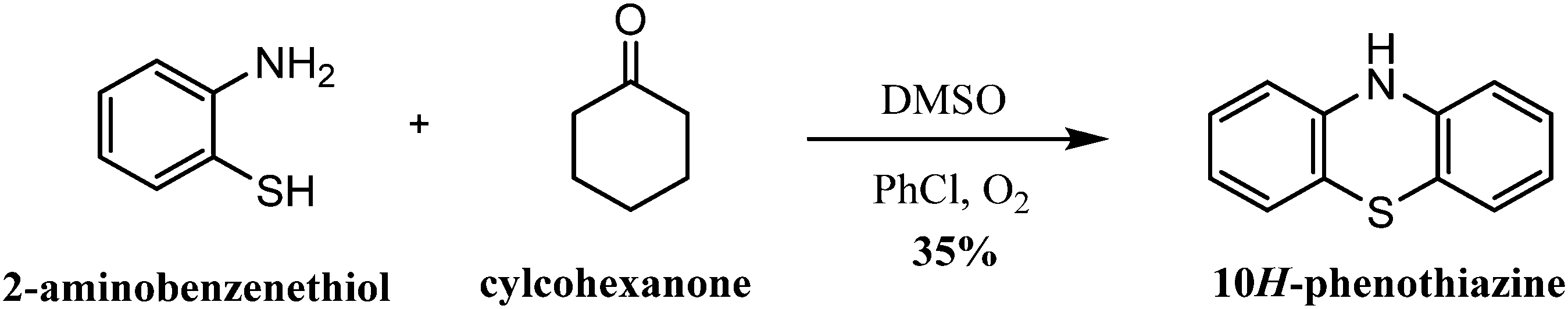 | ||
| Scheme 3 Synthesis of 10H-phenothiazine from 2-aminobenzenethiol and cylcohexanone. | ||
(ii) The metal (Cu or Fe) catalyzed coupling reaction of 2-aminobenzenethiol with 1-bromo-2-chlorobenzene results in the formation of 10H-phenothiazine in 67% yield (Scheme 4).91
 | ||
| Scheme 4 Synthesis of 10H-phenothiazine from 2-aminobenzenethiol and 1-bromo-2-chlorobenzene. | ||
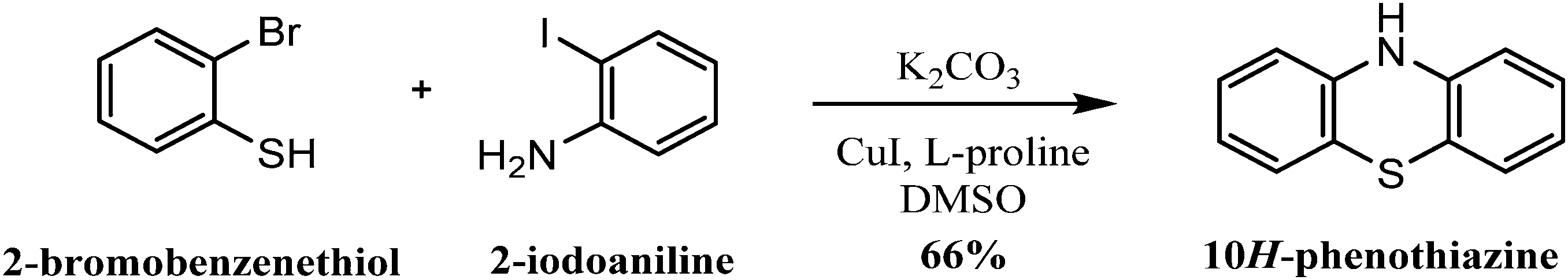 | ||
| Scheme 5 Synthesis of 10H-phenothiazine from 2-bromobenzenethiol and 2-iodoaniline. | ||
3.2. Functionalization of the phenothiazine core
Phenothiazine core can be functionalized in the following ways: amino-H functionalization, nuclear functionalization, and sulfide linkage functionalization.3.2.1.1. N-Alkylation. Longer alkyl chains can be introduced to the phenothiazine core in order to improve its solubility in common organic solvents. The reaction of 10H-phenothiazine with NaOH and n-butyl iodide in DMSO at room temperature results in the formation of 10-butyl-10H-phenothiazine 5 in 92% yield (Scheme 6).93
 | ||
| Scheme 6 Synthesis of 10-butyl-10H-phenothiazine 5. | ||
3.2.1.2. N-Arylation. Wang et al. reported some phenothiazine derivatives for applications in room temperature phosphorescence (RTP). The 10H-phenothiazine was reacted with iodobenzene in the presence of potassium tert-butoxide, palladium acetate and tri-tert-butylphosphine at 100 °C to give 10-phenyl-10H-phenothiazine 6 in 85% yield (Scheme 7).94
 | ||
| Scheme 7 Synthesis of 10-phenyl-10H-phenothiazine 6. | ||
3.2.2.1. Halogenation (bromination). 3-Bromo-10-butyl-10H-phenothiazine (7) and 3,7-dibromo-10-butyl-10H-phenothiazine (8) were synthesized in two different pathways:
(i) The reaction of 10-butyl-10H-phenothiazine with liquid bromine (1 eq. and 2 eq.) in glacial acetic acid, resulting in the formation of monobrominated and dibrominated phenothiazines 7 and 8 in 64% and 70% yields, respectively.95
(ii) The reaction of 10-butyl-10H-phenothiazine with N-bromosuccinimide (NBS) (1 eq. and 2 eq.) in the presence of a catalytic amount of benzoyl peroxide in CCl4, resulting in the formation of monobrominated and dibrominated phenothiazines 7 and 8 in 60% and 85% yields, respectively (Scheme 8).96
 | ||
| Scheme 8 Synthesis of 3-bromo-10-butyl-10H-phenothiazine 7 and 3,7-dibromo-10-butyl-10H-phenothiazine 8. | ||
3.2.2.2. Formylation. The mono- and di-formylated phenothiazines 9 and 10, respectively, were synthesized via the dropwise addition of POCl3 to a mixture of DMF and 1,2-dichloroethane at 0 °C, followed by the addition of N-alkylated phenothiazine (Scheme 9). The reaction proceeds at 80 °C and requires aqueous work-up for isolation of the formylated products.97
 | ||
| Scheme 9 Synthesis of mono- and di-formylated phenothiazines 9 and 10. | ||
3.2.2.3. Nitration. A solution of the N-alkylated phenothiazine in 1,2-dichloroethane was allowed to react with concentrated nitric acid at 10 °C for two hours, which resulted in the formation of 11 in 90% yield (Scheme 10).98
 | ||
| Scheme 10 Synthesis of compound 11. | ||
Hosseinnezhad et al. synthesized several fluorescent phenothiazine materials and investigated their photophysical properties. The nitro group on the phenothiazine was converted to the corresponding amine by reacting the 3-nitrophenothiazine derivative with SnCl2 and HCl in ethanol (Scheme 11).99
 | ||
| Scheme 11 Synthesis of compound 12. | ||
 | ||
| Scheme 12 Synthesis of compounds 13 and 14. | ||
3.2.4.1. Sonogashira cross-coupling. Zhang et al. reported the preparation of mono- and di-ferrocene substituted phenothiazine derivatives and studied their optoelectronic properties. The monobromo and dibromo derivatives of phenothiazine were reacted with ethynyl ferrocene in the presence of bis(triphenylphosphine)palladium(II) dichloride and copper(I) iodide in triethylamine, which gives compounds 15 and 16 in 52% and 42% yields, respectively (Scheme 13).101
 | ||
| Scheme 13 Synthesis of compounds 15 and 16. | ||
3.2.4.2. Suzuki cross-coupling. Zhang et al. synthesized a phenothiazine-based luminogen via the Suzuki cross-coupling reaction and investigated its photophysical properties for mechanochromic applications. Reaction of the boronate ester of phenothiazine with 2-(4-bromophenyl)-3,3-diphenylacrylonitrile in the presence of the Pd catalyst and K2CO3 gives 17 in 62% yield (Scheme 14).102
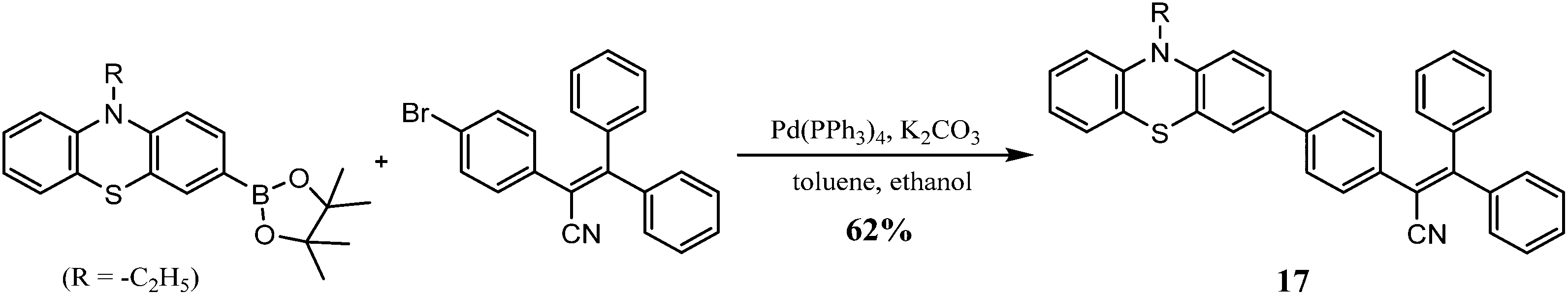 | ||
| Scheme 14 Synthesis of compound 17. | ||
3.2.4.3. Stille cross-coupling. Sang et al. synthesized a phenothiazine and thiophene based polymer via the Stille coupling reaction and investigated the photophysical and electrochemical properties for photovoltaic applications. Reaction of the dibromo phenothiazine with stannyl thiophene in the presence of the Pd-catalyst and toluene resulted in the formation of 18 in 54% yield (Scheme 15).103
 | ||
| Scheme 15 Synthesis of compound 18. | ||
3.2.4.4. Heck cross-coupling. Ravivarma et al. reported a series of phenothiazine-based derivatives for DSSC applications. The derivatives were synthesized via the Heck cross-coupling reaction. The dibromo phenothiazine was reacted with the vinyl functionalized phenothiazine in the presence of the Pd-catalyst, K2CO3 and tetrabutylammonium bromide (TBAB) to give derivative 19 in 62% yield (Scheme 16).104
 | ||
| Scheme 16 Synthesis of compound 19. | ||
3.3. Phenothiazine based mechanochromic materials
Phenothiazine has been used as an efficient building block to construct various mechanochromic materials. The non-planar structure of phenothiazine resulted in stronger emission in the solid state which is an essential criterion for a molecule to be mechanochromically active.70 In the solid state, the central ring of the phenothiazine moiety causes conformational changes, and the N and S atoms potentially trigger supramolecular interactions, which can impact the photophysical properties.71,72 The stimuli responsive luminescence behaviour of various phenothiazine derivatives is discussed below.Wan et al. reported fluorinated phenothiazine derivatives 20, 21 and 22 (Chart 2).105 The crystalline sample of monofluoro derivative 20, when crushed, showed a hypsochromic shift in its emission (Δλ = −35 nm) due to the loose molecular packing that could be easily disturbed by the weak external stimulus. with strong grinding, a bathochromic shift (Δλ = +41 nm) in its emission was observed (Fig. 5(a)). Two polymorphs of the difluoro derivative 21 were reported, in which the emission was bathochromically shifted (Δλ1 = +76 nm, Δλ2 = +35 nm; Table 1) when subjected to mechanical grinding. The trifluoro derivative 22 shows a small bathochromic shift of +10 nm in its emission in response to mechanical grinding.
 | ||
| Chart 2 Chemical structures of phenothiazine based mechanochromic luminogens 20–63. | ||
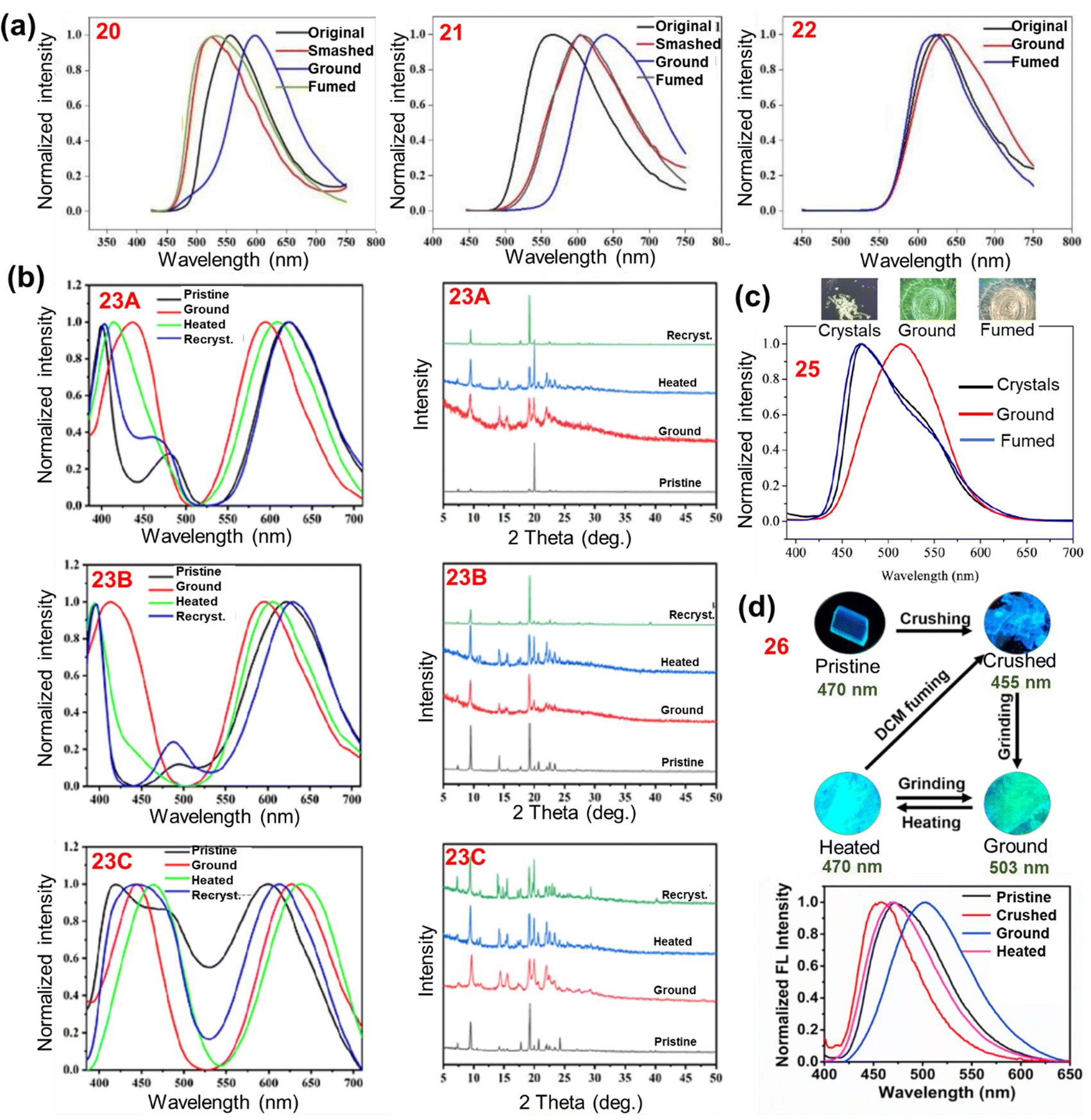 | ||
| Fig. 5 (a) Emission spectra of 20, 21 and 22 in various solid states. Reproduced from ref. 105 with permission from © 2022 Elsevier Ltd., all rights reserved. (b) Emission spectra (left) and PXRD patterns (right) of the three polymorphs of 23 (23A, 23B and 23C) in various solid states. Reproduced from ref. 106 with permission from © 2022 Elsevier Ltd., all rights reserved. (c) Fluorescence images and spectra of 25 in the crystalline, ground and fumed states. Reproduced from ref. 108 with permission from © 2021 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (d) Fluorescence images (top) and emission spectra (bottom) of 26 in different solid states. Reproduced from ref. 109 with permission from © 2021 Elsevier Ltd., all rights reserved. | ||
| Compound | λ 1 (nm) | λ 2 (nm) | λ 3 (nm) | Δλa (nm) |
|---|---|---|---|---|
λ
1 = emission maximum in the pristine or as-synthesized state; λ2 = emission maximum in the slightly ground state; λ3 = emission maximum in the completely ground state.a Grinding induced spectral shift; Δλ = λ2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (or3) − λ1.b Emission maxima of different polymorphs. (or3) − λ1.b Emission maxima of different polymorphs. |
||||
| 20 | 557 | 522 | 598 | −35, +41 |
| 21 | 566b | — | 642 | +76 |
| 607b | — | 642 | +35 | |
| 22 | 623 | — | 633 | +10 |
| 23 | 622b | — | 595 | −27 |
| 623b | — | 596 | −27 | |
| 599b | — | 627 | +28 | |
| 24 | 570 | — | 600 | +30 |
| 25 | 478 | — | 525 | +47 |
| 26 | 470 | 455 | 503 | −15, +33 |
| 27 | 519 | — | 569 | +50 |
| 28 | 506 | — | 564 | +58 |
| 29 | 478 | — | 538 | +60 |
| 30 | 565 | — | 604 | +39 |
| 31 | 591 | — | 619 | +28 |
| 32 | 602 | — | 622 | +20 |
| 33 | 597 | — | 622 | +25 |
| 34 | 597 | — | 628 | +31 |
| 35 | 467 | — | 512 | +45 |
| 36 | 517 | — | 544 | +27 |
| 37 | 545 | — | 591 | +46 |
| 38 | 579 | — | 616 | +37 |
| 39 | 543 | — | 567 | +24 |
| 40 | 567 | — | 635 | +68 |
| 41 | 560 | — | 596 | +36 |
| 42 | 534 | — | 523 | −11 |
| 43 | 567 | — | 554 | −13 |
| 44 | 495 | — | 535 | +40 |
| 45 | 452 | — | 462 | +10 |
| 46 | 491 | — | 520 | +29 |
| 47 | 528 | — | 559 | +31 |
| 48 | 536 | — | 608 | +72 |
| 49 | 572 | — | 598 | +26 |
| 50 | 514 | — | 568 | +54 |
| 51 | 537 | — | 567 | +30 |
| 52 | 529 | — | 557 | +28 |
| 53 | 560 | — | 580 | +20 |
| 54 | 513 | — | 568 | +55 |
| 55 | 519 | — | 563 | +44 |
| 56 | 517 | — | 559 | +42 |
| 57 | 568b | — | 673 | +105 |
| 640b | — | 673 | +33 | |
| 58 | 547 | — | 663 | +116 |
| 59 | 449 | — | 570 | +121 |
| 60 | 467, 492 | — | 467 | — |
| 61 | 550 | — | 563 | +13 |
| 62 | 440, 552 | — | 570 | +18 |
| 63 | 440, 535 | — | 570 | +35 |
Wang et al. reported the N-aryl substituted phenothiazine derivative 23 (Chart 2)106 and successfully developed its three crystalline polymorphs 23A, 23B and 23C. The mechanochromic response of crystals 23A and 23B were the same, whereas 23C shows different mechanochromic behaviour. The 23B crystals exhibit a significant hypsochromic shift (from 623 nm to 596 nm) in the emission in response to mechanical grinding due to grinding induced conformational distortion. Upon heating the ground powder of 23B, its emission was restored to 606 nm, and when recrystallised using dichloromethane, the emission maximum was recovered to 631 nm. For crystal 23A, a shift in its emission was seen from 622 nm (yellow fluorescence) to 595 nm (yellowish green fluorescence) after grinding. In response to heating or recrystallization of the ground form of 23A, the yellow emission could be partially restored. The emission of crystal 23C (599 nm) was similar to the emission of the ground forms of crystals 23A and 23B, which shows that the crystals of 23C and the ground powders of 23A and 23B have similar molecular arrangements. Upon mechanical grinding, a red shift in the emission (from 599 to 627 nm) was observed, and upon heating the ground sample of 23C this further shifted its emission bathochromically to 638 nm. The PXRD pattern of crystals of 23C was similar to the ground samples of 23A and 23B (Fig. 5(b)). For 23A, 23B and 23C the PXRD study revealed that the ground samples retained their crystalline state, and their crystallinity was not completely destroyed after being ground.
Qian et al. reported the dihydroazulene based phenothiazine derivative 24 (Chart 2).107 Compound 24 exhibited a red-shift in its emission from 570 nm to 600 nm upon mechanical grinding due to the conformational planarization of the phenothiazine and dihydroazulene moieties. Hasan et al. reported the carbazole and phenothiazine based derivative 25 (Chart 2).108 The pristine sample of 25 exhibited two emission bands at 478 nm and 538 nm, which corresponded to the charge-transfer emission of the carbazole moiety and the fluorescence and TADF of the phenothiazine moiety (quasi-equatorial conformation). Upon mechanical grinding, the peak at 538 nm disappeared and the ICT peak of the carbazole was bathochromically shifted to 525 nm. A colour change from khaki to green was observed (Fig. 5(c)). The bathochromic shift in the emission was due to the grinding induced-planarization between the carbazole donor and the benzonitrile acceptor unit, whereas the elimination of the peak at 538 nm was due to the breaking of the dimers in the crystalline sample. The emission of the crystalline sample was restored on treating the ground sample with dichloromethane vapour. The mechanochromic behaviour of 25 was associated with the crystalline-to-amorphous phase transition, as revealed via PXRD studies before and after grinding.
Fan et al. reported a D–π–A design based benzothiazole–phenothiazine derivative 26 (Chart 2).109 Upon crushing the crystalline sample of 26 with a pestle, its emission maximum hypsochromically shifted from 470 nm to 455 nm and a colour change from cyan to blue was observed (Fig. 5(d)). Upon strong grinding, the emission bathochromically shifted to 503 nm and the colour changed to green. The crystalline fluorescence can be restored without the influence of any secondary stimulus. The time of the self-recovery process was about 14 h at room temperature, and heating the ground powder was found to accelerate the recovery process to the level of seconds. The blue shift in the emission was assigned to the transformation of one crystalline state (which has a dimeric structure) to another crystalline state (which has a monomeric structure) with a relatively loose packing mode. Strong grinding leads to planarity in the molecular conformation, and the dihedral angle between the phenothiazine center and the phenyl ring decreases, which results the bathochromic shift in the emission.
Sych et al. reported the quinoline based isomeric derivatives of phenothiazine 27, 28 and 29 (Chart 2).110 Upon grinding the crystalline samples of 27–29, their emission spectra show a bathochromic shift of 50–60 nm (Table 1). The mechanochromism of all the derivatives was found to be reversible i.e., when their ground powder was treated with dichloromethane vapour for about 2 minutes, the original emission was restored. In all the derivatives, the di-tert-butyl phenothiazine fragment of the crystalline and fumed samples adopted the quasi-equatorial conformation. The compounds with phenyl spacers (28 and 29) experienced TADF when their crystalline samples were ground mechanically (Fig. 6(a)). Reversible switching between the prompt fluorescence (in the crystalline/fumed state) and TADF (in the ground state) was observed for up to four grinding/fuming cycles. The compound without the phenyl spacer (27) was found to exhibit TADF in both the crystalline/fumed and ground states. This study shows that 28 and 29 with the phenyl spacer between the quinoline and phenothiazine moieties exhibited grinding induced TADF (Fig. 6(a)).
 | ||
| Fig. 6 (a) Fluorescence images of 27, 28 and 29 under UV light before and after grinding; (b) daylight (left) and UV (right) images of 32, 33 and 34 before and after grinding; (c) fluorescence images of 35, 36, 37 and 38 films on pieces of weighing paper in response to grinding and fuming under UV light; (d) grinding and fuming induced changes of the π-stacking model for 35, 36, 37 (top) and 38 (bottom); and (e) emission spectra and (f) PXRD patterns of 39 in the as-synthesized, ground and fumed forms. Reproduced from ref. 110 with permission from © 2021 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim; ref. 113 and 114 with permission from © 2020 and © 2019 Elsevier Ltd., all rights reserved; and ref. 115 with permission from the Royal Society of Chemistry. | ||
Yang et al. reported the dibenzo[a,c]phenazine–phenothiazine derivative 30 (Chart 2).111 Upon grinding, the pristine powder of 30 showed a bathochromic shift in its emission from 565 nm (yellow fluorescence) to 604 nm (red fluorescence). When the ground powder was exposed to dichloromethane vapour, the emission returned to 560 nm. PXRD studies revealed that the pristine form and the fumed forms have different molecular packing arrangements. The ground form was found to be amorphous in nature. The emission of the pristine and ground forms was assigned to the ICT transition. The molecules adopted a twisted conformation in the pristine state. Mechanical grinding causes conformational planarization and the dihedral angle between the donor and acceptor units decreases, which increases the effective conjugation and results in the bathochromic shift of the emission.
Zhang et al. reported mono substituted phenothiazine derivative 31 (Chart 2).112 The pristine powder of 31, when ground, showed a bathochromic shift in its emission from 591 nm to 619 nm. PXRD studies revealed the crystalline nature of the pristine form, which contrasted with the amorphous nature of the ground form. When the ground form was exposed to solvent fumes, the pristine emission was restored. Zhang et al. reported three phenothiazine derivatives 32, 33 and 34 (Chart 2), which differ in the nature of their alkyl chain length.113 All the derivatives exhibited reversible mechanochromism in which the emission maximum of each derivative was bathochromically shifted in response to mechanical grinding. Compound 32, with a two-carbon chain, exhibited an emission maximum at 602 nm, whereas 33 and 34, with four- and eight-carbon chains, respectively, exhibited similar emission maximum at 597 nm. The emission maximum of 32, 33 and 34 bathochromically shifted to 622 nm (Δλ = +20 nm), 622 nm (Δλ = +25 nm), and 628 nm (Δλ = +31 nm) (Table 1), respectively, upon mechanical grinding. Fig. 6(b) shows the colour change of compounds 32–34 from orange to red. As revealed from the PXRD studies, the ordered crystalline-to-unordered amorphous phase transition was responsible for the mechanochromic behaviour of 32, 33 and 34.
Xue et al. reported four phenothiazine derivatives 35–38 (Chart 2) that contain different electron donating and electron withdrawing groups.114 Compounds 35–38 exhibited reversible mechanochromism, and their emission maxima were bathochromically shifted (Δλ for 35 = +45 nm, Δλ for 36 = +27 nm, Δλ for 37 = +46 nm, Δλ for 38 = +37 nm; Table 1) in response to mechanical grinding. Fig. 6(c) shows the fluorescence images of thin films of 35, 36, 37 and 38 coated on a piece of paper in response to grinding and fuming. For all the derivatives, their mechanochromism is associated with the phase transformation from the crystalline state to the amorphous state. The application of mechanical force to the crystalline samples of 35–37 promotes a reduction in the sliding angle, resulting in the formation of metastable J-aggregates, and in 38, the grinding force causes the molecules to come into close proximity (Fig. 6(d)).
Yang et al. reported a phenothiazine derivative 39 (Chart 2).115 Crystals of 39 exhibited an emission maximum at 543 nm, which bathochromically shifted to 567 nm upon mechanical grinding and the emission colour changed from yellow to orange (Fig. 6(e)). In addition, crystals of 39 exhibited a wide absorption band with an absorption maximum at 372 nm, which was blue-shifted compared with the absorption maximum of 39 in cyclohexane (400 nm), revealing face-to-face H-aggregate formation in the crystalline state. The absorption maximum of the ground form of 39 (415 nm) was red-shifted compared with the absorption maximum of 39 in cyclohexane solution. This revealed that the grinding of the crystalline form of 39 significantly destroyed the H-aggregate. PXRD studies revealed the crystalline-to-amorphous transition of 39 when its crystals were mechanically ground (Fig. 6(f)).
Wang et al. reported the dumbbell shaped D–π–A–π–D design based phenothiazine derivative 40 (Chart 2).116 In response to mechanical grinding, the emission colour of 40 was changed from orange (567 nm) to red (635 nm) (Fig. 7(a)). The orange emission was restored upon fuming the ground sample of 40 with dichloromethane vapour for about 10 seconds. PXRD studies revealed the crystalline nature of 40 in the pristine state but showed amorphous characteristics in the ground state (Fig. 7(b)). Compound 40 in its pristine form, ground form and in the solution state (cyclohexane) exhibited absorption maxima at 432 nm, 486 nm and 448 nm, respectively. The absorption peak of the pristine sample of 40 was blue shifted compared with the absorption peak of 40 in cyclohexane, which indicated the formation of head-to-head H-aggregates in the pristine state because the H-aggregate always results in a hypsochromically shifted absorption band relative to isolated molecules. The absorption peak of the ground sample of 40 was red shifted compared with the absorption peak of 40 in cyclohexane. This shows that the formation of J-aggregates might be taking place upon mechanical grinding because J-aggregates always result in a red-shifted absorption band relative to the isolated molecules. Hence, the mechanochromic response of 40 was attributed to the grinding force-induced transformation of H-aggregates to J-aggregates.
 | ||
| Fig. 7 Fluorescence spectra of (a) 40 and (c) 41 in various solid states (insets: fluorescence images of pristine and ground samples of (a) 40 and (c) 41 and the types of H- and J-aggregate); and PXRD spectra of (b) 40 and (d) 41 in various solid states. Reproduced from ref. 116 with permission from the Royal Society of Chemistry; and ref. 117 with permission from © 2018 Elsevier Ltd., all rights reserved. | ||
Shen et al. reported phenothiazine derivative 41 (Chart 2),117 which in its pristine form exhibited yellow fluorescence with an emission maximum at 560 nm. Upon mechanical grinding, the emission of 41 changed to 596 nm (orange fluorescence) (Fig. 7(c)). When the ground powder of 41 was exposed to solvent vapour, its pristine emission was restored. The mechanochromism of 41 is reversible, i.e., its yellow fumed form could be again changed to orange upon further grinding. PXRD studies indicated that the mechanochromism of 41 was associated with the reversible phase transformation from the crystalline state to the amorphous state (Fig. 7(d)). H-aggregates existed in the crystals of 41. Grinding induces molecular sliding along the long axis, resulting in complete transformation of the stacking modes from the H-aggregation state to the J-aggregation state.
Xi et al. reported the N-aryl substituted phenothiazine derivatives 42 and 43 (Chart 2),118 which exhibited a hypsochromic shift of 11 nm and 13 nm, respectively, in their emission when subjected to mechanical grinding. The results of PXRD studies revealed that the mechanochromism of 42 and 43 was associated with the transition between the crystalline and amorphous states. Single-crystal XRD analysis revealed the existence of enhanced π-conjugation and a relatively planar molecular conformation of 42 and 43 in the crystalline state. Upon grinding, the torsion angle may be increased, and the relatively planar conformation of 42 and 43 may be transformed to a more distorted conformation, thus exhibiting the hypsochromic shift upon mechanical grinding.
Arivazhagan et al. reported the four phenothiazine derivatives 44–47 (Chart 2).119 These derivatives exhibited bathochromically shifted (Δλ = 10–40 nm; Table 1) reversible mechanochromism (Fig. 8(a)). PXRD studies of 44–47 revealed that grinding causes the loss of crystallinity of their pristine samples. Zhang et al. reported the three phenothiazine based mechanochromic luminogens 48–50 (Chart 2).120 Compounds 48, 49 and 50 in the as-synthesized forms exhibited an intense yellow (536 nm), yellow-orange (572 nm) and yellow-green (514 nm) emission that changed to red (608 nm), orange-red (598 nm) and orange (568 nm) emission, respectively, after grinding. The mechanochromism of 48, 49 and 50 was found to be reversible, and the pristine emission could be restored when the ground samples were exposed to solvent vapour. For 48, 49 and 50 the mechanochromic behaviour was associated with the phase transformation from the crystalline to the amorphous state.
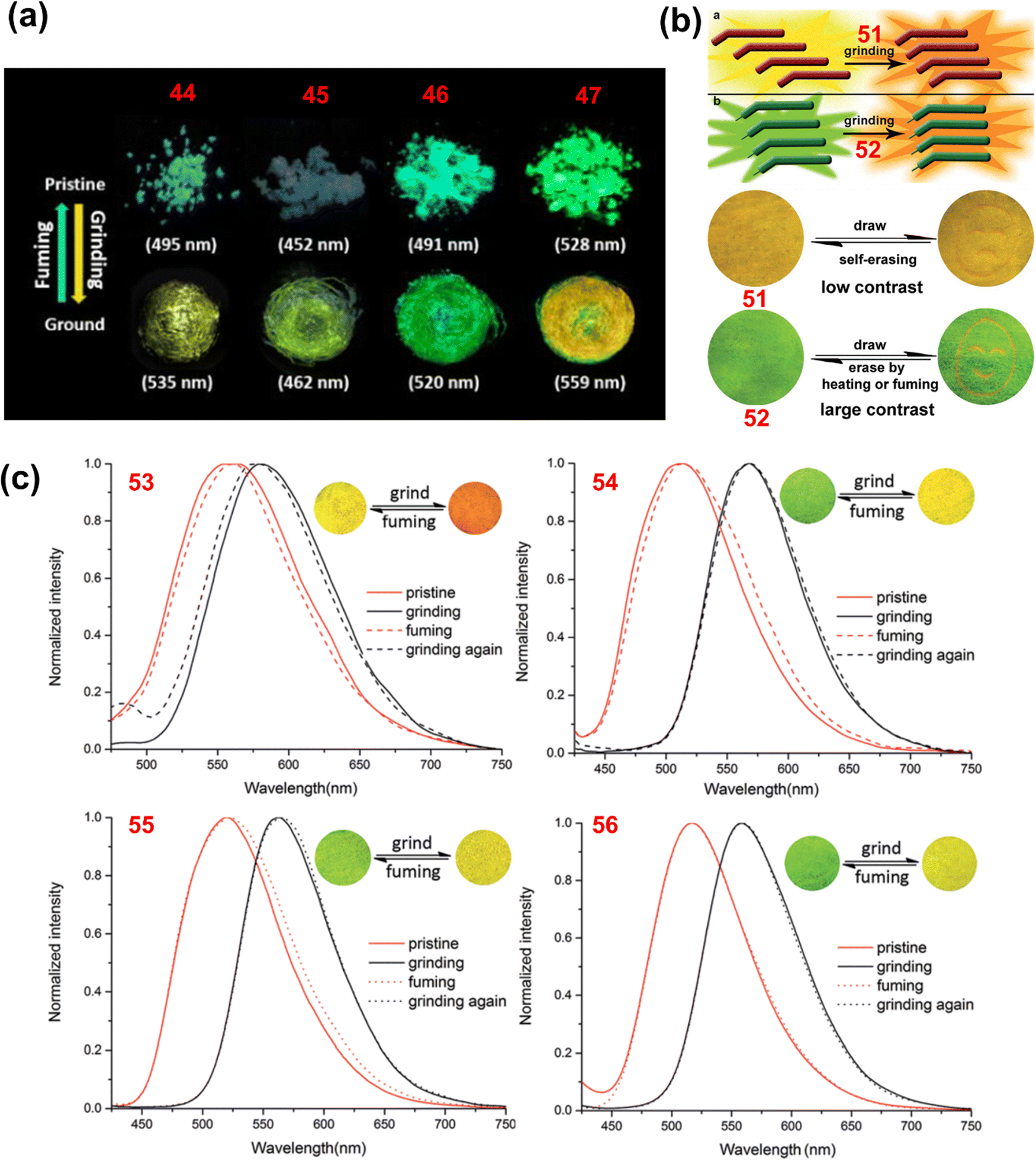 | ||
| Fig. 8 (a) Fluorescence images of pristine and ground forms of 44, 45, 46 and 47 under UV light; (b) schematic diagram of the mechanism of mechanochromism for 51 and 52 (top) and fluorescence images of films of 51 and 52 on pieces of weighing paper in response to grinding, fuming and heating under UV light; and (c) emission spectra of 53, 54, 55 and 56 in their pristine, ground and fumed states (insets: fluorescence images of ground and fumed forms). Reproduced from ref. 119 with permission from © 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim; and from ref. 121 and 122 with permission from the Royal Society of Chemistry. | ||
Xue et al. reported the two phenothiazine derivatives 51 and 52 (Chart 2).121 Crystals of 51 showed yellow fluorescence with an emission maximum at 537 nm and a shoulder band at 560 nm. Upon mechanical grinding, the emission of 51 was bathochromically shifted to 567 nm and the ground powder emits orange fluorescence (Fig. 8(b)). Compound 51 in toluene solution exhibited an absorption maximum at 400 nm, whereas in the crystalline state it shows an absorption maximum at 475 nm. The bathochromically shifted absorption maximum of 51 in the crystalline state suggested that the molecules are stacked in the J-aggregate form. A blue-shifted absorption band (422 nm) relative to the crystalline form of 51 was observed when it was ground. The large J-aggregates were split into smaller aggregates. The colour of crystals of 52 changed from green (529 nm) to orange (557 nm) when ground (Fig. 8(b)). The mechanochromism of 51 and 52 was reversible and the crystalline emission could be restored when their ground samples were fumed with dichloromethane. The colour change of 51 and 52 in response to mechanical grinding was associated with the phase transformation from the crystalline to the amorphous state.
Xue et al. reported the phenothiazine and anthracene based derivatives 53–56 (Chart 2)122 in which the number of C-atoms in the alkyl chain on the phenothiazine core was different. Compound 53, with the smallest alkyl chain (–C2H5), shows a red shifted absorption and emission compared with 54, 55 and 56. The smaller alkyl chain causes excessive π–π stacking in 53. Upon mechanical grinding, the crystals of 53, 54, 55 and 56 shifted their emission from 560, 513, 519 and 517 nm to 580, 568, 563 and 559 nm, respectively (Fig. 8(c)). The crystalline emission of 53–56 could be restored when their ground samples were fumed with dichloromethane vapour. Moreover, in response to heating, the crystallinity of the ground samples of 53–56 was restored. At 110 °C, the ground form of 56 was restored to its original state after 2 seconds. The recovery time was 3 seconds for 55 and 360 seconds for 54; however, 53 could not be restored, even after heating for 2 hours. The self-reversible mechanochromism was observed only for compound 56. The yellow fluorescence color of the ground sample of 56 gradually reverted to the original green emission after five days when the sample was left to stand at room temperature.
Okazaki et al. reported the D–A–D based multi-functional molecules 57 and 58, comprising phenazine as the acceptor and phenothiazine as the donor.123,124 These derivatives exhibited conformation dependent multi-colour mechanochromism. Two polymorphs of 57 were developed, from which needle-like crystals of 57 (57-Y) exhibited bright yellow emission at 568 nm, whereas block-like crystals of 57 (57-O) exhibited orange emission at 640 nm. Upon mechanical grinding, the colour changed drastically to red in both cases, and the resulting red powder of 57 exhibited an emission maximum at 673 nm. Compound 58 exhibited yellow-green emission at 547 nm which, upon mechanical grinding, was bathochromically shifted to 663 nm. Single-crystal X-ray analysis of 57-O revealed the “quasi-equatorial” conformation of one phenothiazine unit and the “quasi-axial” conformation of another phenothiazine unit (Fig. 9(a)). The red emission from the ground powder of 57 was attributed to the molecular state where both the phenothiazine moieties adopted the equatorial conformation. The orange emission of the 57-O polymorph was attributed to emission from the weaker ICT state in which one phenothiazine moiety adopted the equatorial conformation and the other phenothiazine moiety adopted the axial conformation. However, the most blue-shifted emission of 57-Y was attributed to emission from the axial–axial conformer (Fig. 9(b)).
 | ||
| Fig. 9 (a) ORTEP drawings of the single crystal of 57O showing the quasi-equatorial conformation of one phenothiazine unit and the quasi-axial conformation of another phenothiazine unit; and (b) schematic correlation between the emission colour and the molecular conformations of the phenothiazine units in 57. Reproduced from ref. 123 and 124 with permission from the Royal Society of Chemistry. | ||
Yang et al. reported the phenothiazine and 4-fluorobenzophenone based derivative 59.125 Upon slight grinding of the crystals of 59, bright blue emission at 449 nm was observed, which upon continuous grinding changed to white and then to yellow (570 nm). The white emission appeared due to the rational mixture ratio of blue and yellow emissions. The molecular conformation transition from quasi-axial to quasi-equatorial was responsible for the interesting dynamic mechanochromism in 59. Tian et al. reported the phenothiazine derivative 60, which contains the non-conjugated chiral cholesterol, group for mechanochromic applications.126 The crystal of 60 exhibited fluorescence–phosphorescence dual emission peaks at 467 and 492 nm. Upon mechanical grinding, the emission band at 492 nm was eliminated and the fluorescence peak at 467 nm was retained. Deng et al. reported the three phenothiazine and benzophenone derivatives 61, 62 and 63.127 Compound 61 exhibited a single emission band at 550 nm, whereas 62 and 63 exhibited two emission bands. They both possessed a higher energy emission band at 440 nm and a shoulder band in the lower energy region at 552 and 535 nm for 62 and 63, respectively. Upon mechanical grinding, the emission of 61 was bathochromically shifted to 563 nm, whereas compounds 62 and 63 exhibited an obvious enhancement in the emission intensity of the lower energy emission band, which was bathochromically shifted to 570 nm for both derivatives. For 62 and 63, the higher energy emission band was a fluorescent emission with a typical nano-second lifetime; however, the lower energy emission band belonged to a TADF emission with a longer lifetime in the region of micro-seconds.
3.4. Other applications of phenothiazine based luminescent materials
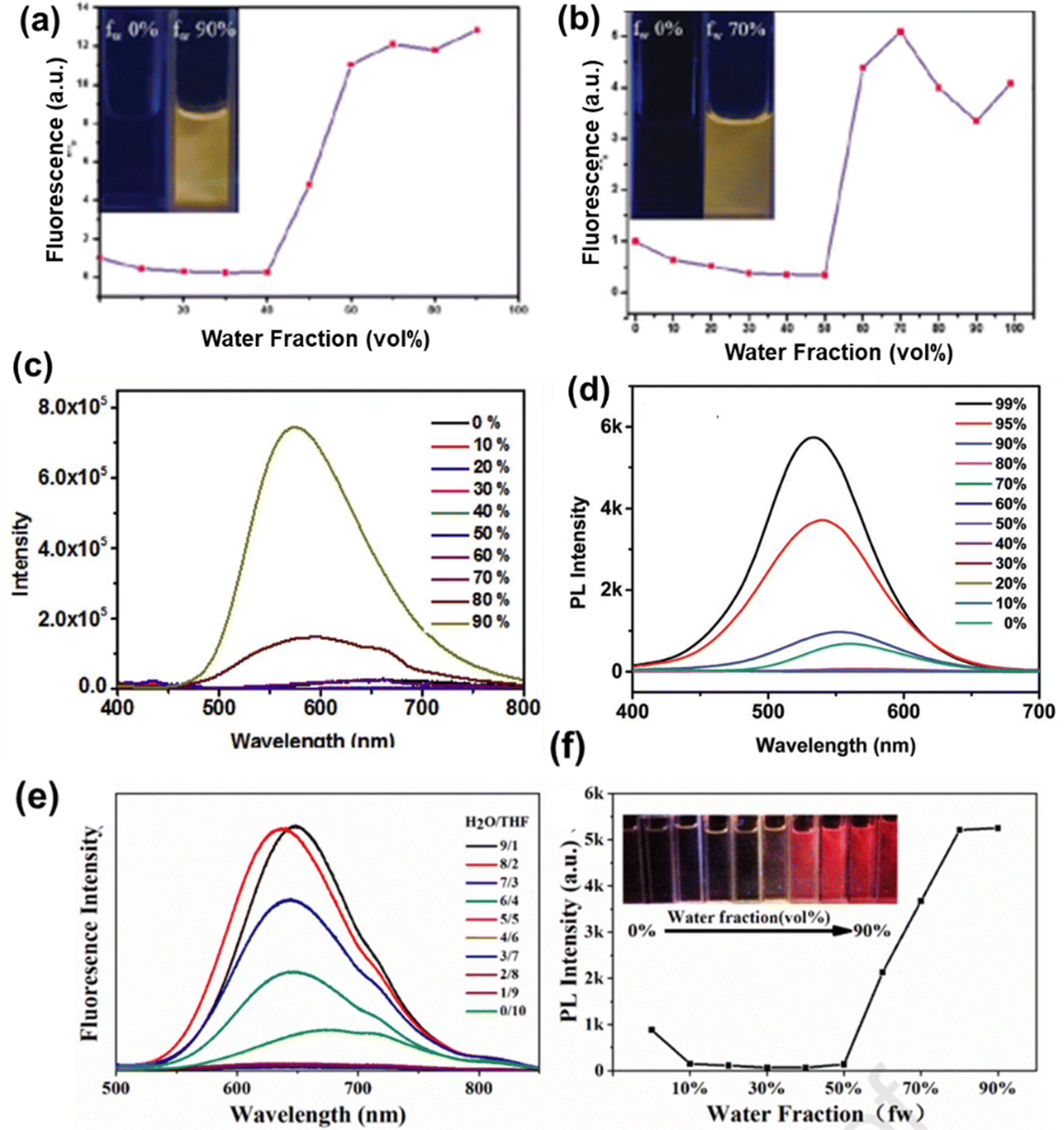
Shivaji et al. reported the perylene diimide-fused [7]helicene derivative of phenothiazine 64 (Chart 3) and investigated its AIE behaviour in THF–water binary mixtures.128 The emission intensity of 64 decreases as the content of water was increased from 0 to 20%. A significant enhancement in the emission intensity was observed at the 70% water fraction (fw) (Fig. 10(a)). At a higher water percentage, the aggregation of 64 strengthens the emission intensity by stabilizing the intramolecular charge transfer (ICT) states. Li et al. reported the phenothiazine derivative 65 (Chart 3) and investigated its AIE behaviour in DMSO–water binary mixtures.129 A reduction in the emission intensity of 65 was observed when the content of water in the DMSO solution was increased from 0% to 70%. The formation of aggregates takes place at 70–90% water fractions and the emission intensity is significantly improved (Fig. 10(b)).
 | ||
| Chart 3 Chemical structures of phenothiazine based AIEgens 64–90. | ||
 | ||
| Fig. 10 Fluorescence spectra of (a) 64, (c) 68 and (d) 69 in different water–THF mixtures; and plot of fluorescence intensity vs. water fraction (%) for compounds (b) 65 (inset: fluorescence images of 65 in different water–DMSO mixtures), (e) 68 (black line) and (e) 69 (red line). (f) Photo of a suspension of 68 with a 90% water fraction in THF with the Tyndall effect (right) and in pure THF without the Tyndall effect (left). Reproduced from ref. 128 with permission from the Royal Society of Chemistry; from ref. 129 with permission from © 2022 Elsevier Ltd., all rights reserved; and from ref. 131 with permission, copyright 2021 American Chemical Society. | ||
Song et al. reported tetra-coordinated boron based derivatives of phenothiazine 66 and 67 (Chart 3), which are weakly emissive in pure THF.130 The successive addition of water to the THF solution of 66 and 67 results in the formation of aggregates and an increase in the emission intensity. A strong AIE effect was observed in compounds 68 and 69 reported by Huang et al.131 Compounds 68 and 69 are completely non-emissive in THF solution, and when water was added to their THF solution, the emission spectra did not change much until the water content reached 60%. After that, the luminescence of 68 and 69 started to get very bright (Fig. 10(c)–(e)) because the molecules could not move as freely inside the aggregates, which stopped the non-radiative transition processes. The Fig. 10(f) shows a suspension of 68 with a 90% water fraction in THF solution exhibiting the Tyndall effect.
Chakraborty et al. reported the two anthracene based phenothiazine derivatives 70 and 71 (Chart 3).132 The AIE behaviour of 70 and 71 was examined in acetonitrile–water binary mixtures. Upon the slow addition of water to the acetonitrile solution of 70 and 71, a reduction in the fluorescence intensity up to a 40–50% water fraction was observed initially due to the polarity effect. At the 40% water fraction, there was a sharp increase in the fluorescence intensity of 70, and the emission intensity was found to be highest at the 90% water fraction (Fig. 11(a)). For 71, the fluorescence enhancement started at the 50% water fraction and reached a maximum at the 70% water fraction (Fig. 11(b)). Zhang et al. reported the quinoline based phenothiazine derivative 72 (Chart 3), which was not emissive in pure THF.133 The addition of an 80% water content to the THF solution of 72 significantly enhanced the emission and at 90% water fraction, and the fluorescence intensity of 72 was much higher than that in pure THF (Fig. 11(c)). The quinoline based phenothiazine derivative 73 (Chart 3) reported by Shen et al. is weakly emissive in pure THF.134 The successive addition of water to the THF solution of 73 led to the formation of nano-aggregates and the emission became substantially stronger as the water percentage was increased to 90–99% (Fig. 11(d)).
 | ||
| Fig. 11 Plot of fluorescence intensity vs. water fraction (%) for compounds (a) 70 (inset: fluorescence images of 70 in 0 and 90% water–acetonitrile mixtures), (b) 71 (inset: fluorescence images of 71 in 0 and 90% water–acetonitrile mixtures) and (f) 74 (inset: fluorescence images of 74 in different water–THF mixtures; and fluorescence spectra of (c) 72, (d) 73 and (e) 74 in different water–THF mixtures. Reproduced from ref. 132 and 134 with permission from the Royal Society of Chemistry; and from ref. 133 and 135 with permission from © 2021 Elsevier Ltd., all rights reserved. | ||
Yu et al. reported the triphenylamine and benzothiadiazole based phenothiazine derivative 74 (Chart 3).135 In pure THF, 74 shows an emission band at 674 nm. After the content of water reached a value of 50% in its THF solution, the PL intensity increased (Fig. 11(e) and (f)). The D–A type architecture of 74 may be ascribed to the low PL intensity at a water content below 50%, which may prompt a twisted intramolecular charge transfer transition, resulting in diminished emission. The major reasons for the AIE phenomenon in 74 are the impedance to free rotation of the benzene ring in the propeller-like configuration of triphenylamine and the suppression of the conformation change of phenothiazine.
Lin et al. reported the phenothiazine derivative 75 (Chart 3).136 The emission intensity of 75 in pure THF solution is quite low, and it reduced steadily when the content of water in the THF solution was increased from 10% to 50%. At the 80% water fraction, the highest PL intensity was seen to be centred at 429 nm, which is 187 times more than in pure THF solution. The further addition of water to the 90–95% water fraction reduced the emission intensity. The PXRD and SEM patterns of two types of aggregates of 75 produced at the water fractions 80% and 95%, respectively, were determined. In the PXRD pattern, the aggregates formed at the 80% water fraction showed several sharp and strong diffraction peaks, but the aggregates formed at the 90% water fraction did not exhibit any sharp diffraction peaks (Fig. 12(a) inset). In the SEM studies, the aggregates formed at the 80% water contents displayed rod-like microstructures, whereas the aggregates formed at the 95% water content exhibited irregular particles. The results from these XRD and SEM studies indicate that the aggregates formed at the 80% water fraction exhibit a crystalline morphology, while the aggregates formed at the 95% water fraction exhibit an amorphous morphology.
 | ||
| Fig. 12 (a) Fluorescence images (top) and fluorescence spectra (bottom) of 75 in different THF/water mixtures (inset: PXRD spectra of aggregates of 75 formed at 80% and 95% water fractions in THF); (b) selected DLS profiles of 76 as a function of fw; emission spectra of (c) 76 (inset: fluorescence images of 76 in pure THF and at the 90% water fraction in THF), (e) 78 and (f) 79 in different THF/water mixtures; (d) emission intensity changes in the two emission bands of 76 (λem = 390 and 550 nm) at various fw; plot of fluorescence intensity vs. water fraction (%) for compounds (g) 78 and (h) 79. Reproduced from ref. 136 with permission from © 2020 Elsevier Ltd., all rights reserved; from ref. 137 with permission, copyright 2020 American Chemical Society; and from ref. 139 with permission from the Royal Society of Chemistry. | ||
Lei et al. reported the triptycene based phenothiazine derivative 76 (Chart 3),137 which emits poorly in THF, and the solution stays almost dark until the water content reaches 50%. A rapid increase in the emission was then seen at 390 nm, and loss of the solution transparency was accompanied by an apparent turn on of the fluorescence. Above a water content of 70%, a new emission band appeared at 550 nm (Fig. 12(c) and (d)). At the 90% water fraction, a highly emissive solution with a considerably improved fluorescence quantum yield was obtained. Dynamic light scattering (DLS) profiles revealed that the average particle size of the aggregates increased rapidly, with the hydrodynamic diameter (D) increasing from 6 nm for particles in pure THF (fw = 0%) to 460 nm for aggregates formed at the 90% water fraction (Fig. 12(b)). The rigid molecular geometry of the triptycene moiety and the twisted molecular structure of the phenothiazine moiety have cooperative impacts on the AIE features of 76.
Li et al. reported the phenothiazine derivative 77 (Chart 3)138 which exhibited an AIE effect when the content of water in its THF solution was more than 50%. At water percentages greater than 50%, the formation of nano-aggregates takes place and thus the intramolecular motions are inhibited, which increased the emission intensity. Liu et al. reported the two phenothiazine based derivatives 78 and 79 (Chart 3).139 Compound 78 exhibited orange-red emission with an emission maximum at 580 nm in pure THF. When the content of water in the THF solution of 78 was increased from 0% to 50%, the emission intensity decreased, and the emission maximum was shifted bathochromically. At the 70% water fraction, the emission intensity of 78 increased (Fig. 12(e) and (g)). The emission of 78 at the lower water fractions (0–50%) was attributed to intramolecular charge transfer transitions, whereas at the higher water fractions (>50%) the emission of 78 originated mainly due to the formation of nano-aggregates. Compound 79 also exhibited similar AIE behaviour (Fig. 12(f) and (h)).
Li et al. reported the two phenothiazine derivatives 80 and 81 (Chart 3)140 for which the AIE effect was observed above the 50% and 30% water fraction, respectively. The emission intensity of 80 decreases as the content of water is increased from 0% to 50%, whereas for 81 the emission intensity decreases up to the 30% water fraction. This was due to the rise in the solvent polarity upon the addition of water, and the fluorophores remain soluble at this point. After that, the emission intensities of 80 and 81 rise dramatically, showing that molecular aggregation occurs at this step, which restricts the intramolecular motions and boosts their emission intensities. The sulphone-based phenothiazine derivative 82 (Chart 3) was reported by Zheng et al., which exhibited a weak emission band at 550 nm in pure THF.141 Upon increasing the water content in the THF solution of 82 to 90%, its emission intensity increased by 37 times due to the formation of nano-aggregates.
Ekbote et al. reported the benzothiadiazole and phenothiazine based AIEgens 83, 84, and 85 (Chart 3).142 In pure DMSO, 83 exhibits a strong ICT emission band at 639 nm. The strength of the ICT emission of 83 drops as the water fraction increases up to 30% owing to the stability of the ICT peak in a polar medium. The formation of aggregates occurs at the 40% water fraction, which increases the emission intensity and the emission maximum is hypsochromically shifted to 485 nm. At the 60% water fraction, there is a modest decrease in the emission intensity of 83, which is due to the formation of bigger aggregates. The emission intensity of 83 again increases at the 70% water fraction with the appearance of a new emission band at 511 nm. In pure DMSO solution, the isomers 84 and 85 show weak emission. The AIE effect was observed at the 50% and 60% water fractions for 84 and 85, respectively.
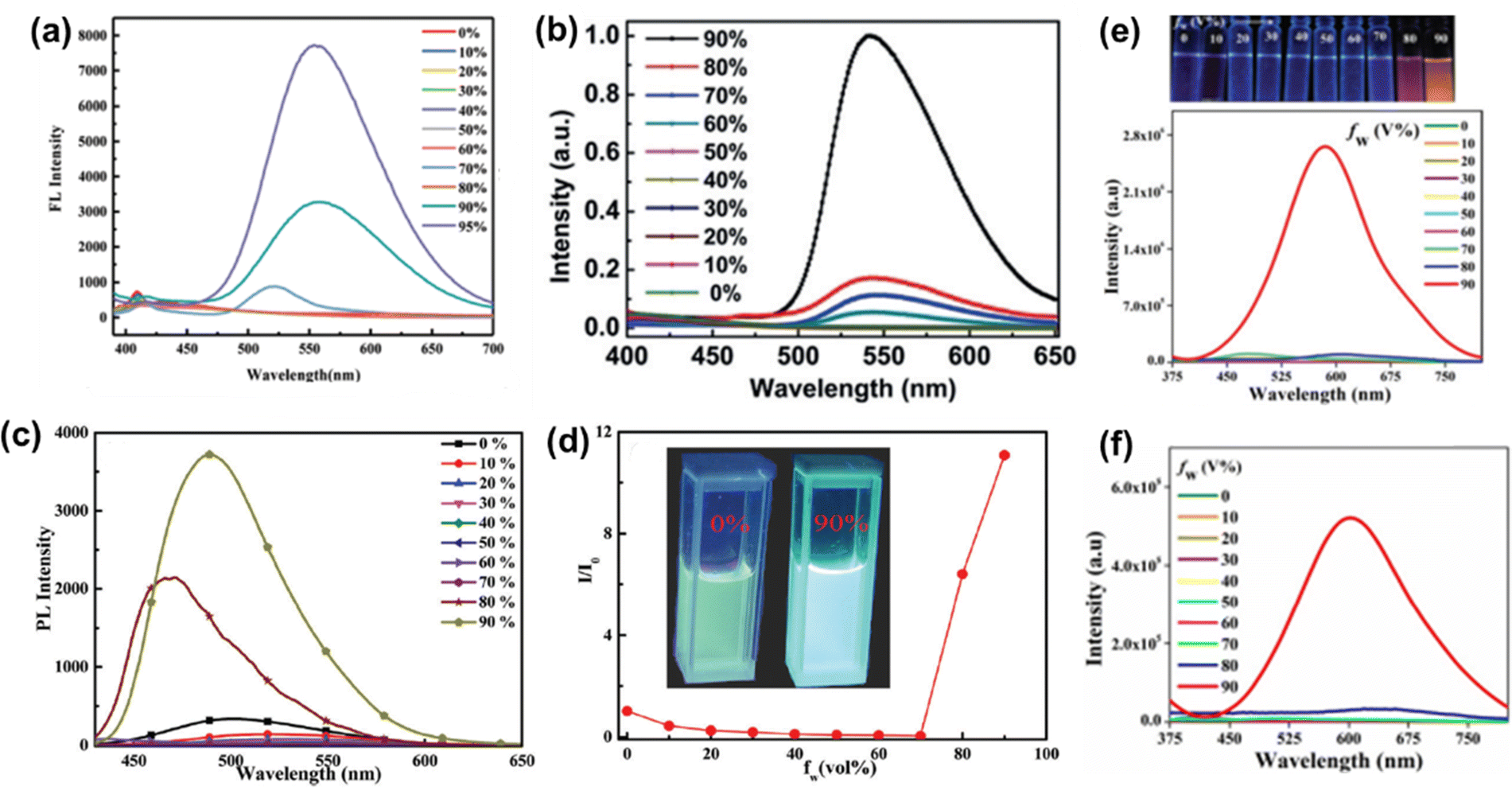
Huang et al. reported the phenothiazine derivative 86 (Chart 3).143 The THF solution of 86 has a faint emission band, but as the water content reaches 70% in the THF solution, the emission intensity increases. When the fw reaches 95%, the aggregation states changed and a new emission peak at 550 nm with increased yellow fluorescence is observed (Fig. 13(a)). Lee et al. reported the phenothiazine derivative 87 (Chart 3), which is weakly emissive in pure THF.144 The fluorescence intensity increases progressively as the water content in the THF solution is increased. When the fw reaches 90%, the fluorescence intensity of 87 is 540 times greater than in pure THF solution (Fig. 13(b)). Xiang et al. reported the phenoxazine-based phenothiazine derivative 88 (Chart 3), which shows a weak emission band at 502 nm in pure THF.145 The emission intensity falls and the emission peak is bathochromically shifted from 502 nm to 550 nm when the content of water is gradually increased from 0% to 70%, which might be attributed to the stabilization of the ICT state. The emission intensity of 88 increases dramatically when the water percentage is increased to 80%, and then to 90% (Fig. 13(c) and (d)).
 | ||
| Fig. 13 Fluorescence spectra of (a) 86, (b) 87, (c) 88, (e) 89 (inset: fluorescence images of 89 in 0–90% water/THF mixture) and (f) 90 in different water/THF mixtures; and (d) plot of fluorescence intensity vs. water fraction (%) for compound 88 (inset: fluorescence images of 88 in 0 and 90% water fractions in THF). Reproduced from ref. 143, 145 and 146 with permission from the Royal Society of Chemistry; and from ref. 144 with permission from © 2018 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Neena et al. reported the two phenothiazine derivatives 89 and 90 (Chart 3)146 for which the emission intensity remains constant until the water content in the THF solution reaches 80% of the total volume. At fw = 90%, a significant increase in the emission intensity is observed, and the emission maximum is located at 585 nm and 600 nm for 89 and 90 (Fig. 13(e) and (f)), respectively. In the aggregated state (fw = 90%), the emission intensity of 89 and 90 is enhanced by 190 and 180 times, respectively, compared with the emission intensity of 89 and 90 in pure THF.
The mechanochromically active derivatives 44, 45, 48 and 49 (Chart 2)119,120 also exhibit AIE behaviour. Compounds 44 and 45 show a maximum emission intensity at 90% water fractions in THF solution. Compound 48 in pure THF exhibits weak emission, but as the fw is increased to 90%, the emission at 610 nm grows rapidly and is 14.0-fold stronger than in pure THF. The emission of 49 is also quite moderate in pure THF, but it increased dramatically in a THF–water mixed solvent, with a 13.8-fold enhancement in the emission intensity when the fw reaches 90% in THF solution.
Keruckiene et al. reported the biphenyl bridged phenothiazine derivative 91 (Chart 4).147 At 77 K, solid molecular dispersions of 91 in Zeonex demonstrate dual-state emission (Fig. 14(a)). This emission is caused by the radiative decay of both the singlet and triplet excited states. This radiative route dominates as a result of the constrained vibrational movements of molecules of 91 in Zeonex, which inhibit charge transfer (CT) from the donor to acceptor moieties. The major radiative channels for the excited states were found to show prompt fluorescence and phosphorescence. The emission peaks of the dual-state were shown to occur at 456 nm and 533 nm, and their lifetimes were measured to be 11.81 ns and 179 ns, respectively. In this case, the phosphorescence was broad and overlapped with the fluorescence spectrum. It is well known that phenothiazine derivatives are capable of forming various conformers depending on the environment, and either TADF or RTP can be seen in them. In this instance, Zeonex seems to have constrained rotation around the C–N bond between the donor and acceptor units and the C–C bond in the biphenyl moiety. This results not only in delayed charge transfer (prompt fluorescence) but also in radiative decay of the excited triplet states at ambient temperature. Ma et al. reported the three benzo[5,6][1,4]thiazino[2,3,4-kl]phenothiazine 5,5,9,9-tetraoxide derivatives 92, 93 and 94 (Chart 4).148 All of these compounds show dual emission of varying intensities. The phosphorescence peaks with vibrational features can be seen clearly at around 450–650 nm for both 92 and 93 (Fig. 14(b) and (c), respectively), and their intensity proportions are more than twice as high as those of the fluorescence peaks. The triphenylethylene substituted derivative 94 exhibits substantial phosphorescence signals at 501 and 531 nm (Fig. 14(d)) with the fluorescence peak being one-third this proportion.
 | ||
| Chart 4 Chemical structures of phenothiazine based phosphors 91–122. | ||
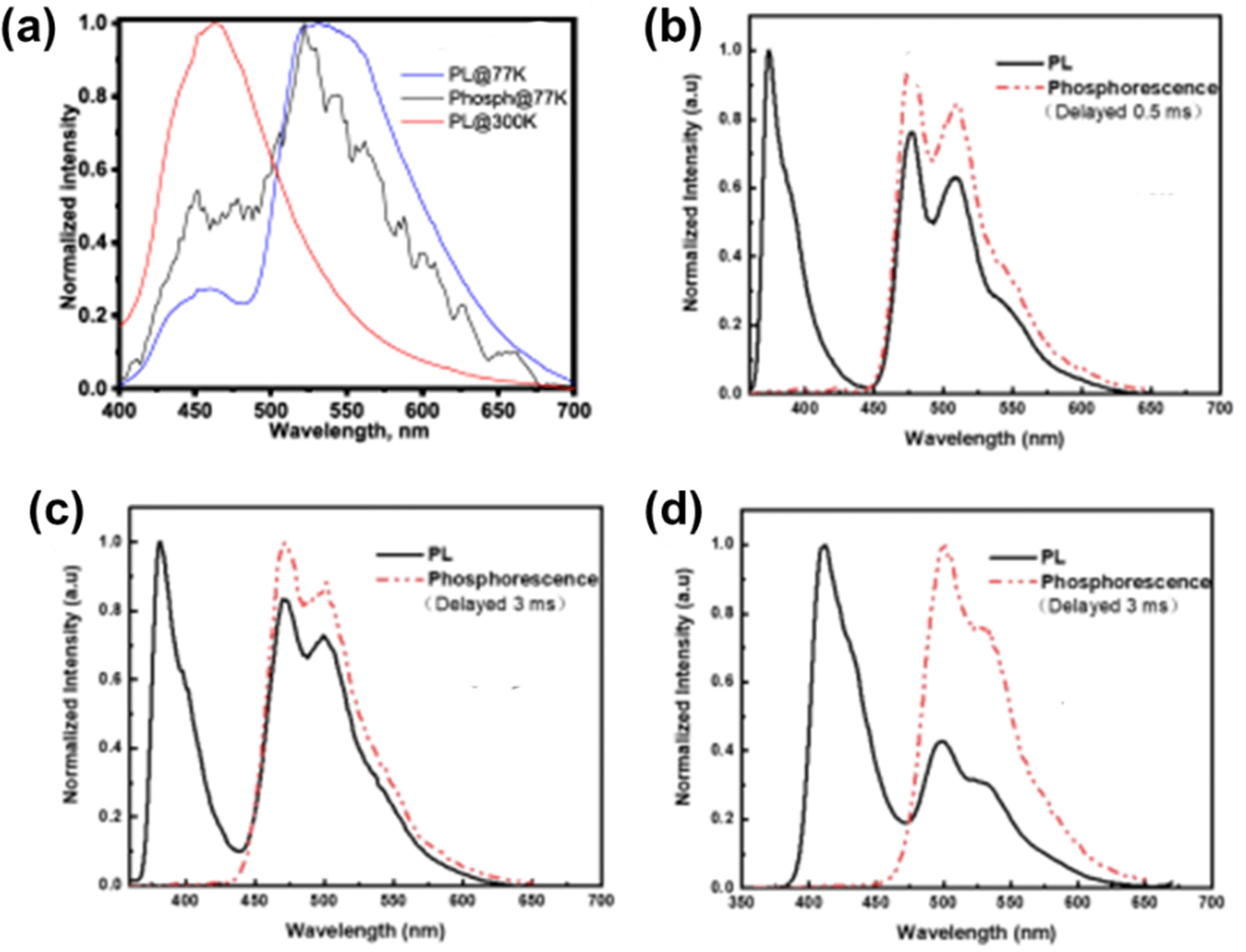 | ||
| Fig. 14 (a) Photoluminescence spectra of a 10% solid solution of 91 in Zeonex recorded at room temperature and at 77 K; and steady PL and delayed PL spectra of compounds (b) 92, (c) 93 and (d) 94 measured in 10−5 M toluene at 77 K. Reproduced from ref. 147 with permission from © 2022 Elsevier Ltd., all rights reserved; and from ref. 148 with permission from the Royal Society of Chemistry. | ||
Gao et al. reported the phenothiazine derivative 95 (Chart 4) with three chlorine atoms.149 Derivative 95 displayed conformation-dependent RTP emission, with the quasi-axial conformation exhibiting a clear RTP effect whereas the quasi-equatorial conformation did not (Fig. 15(a) and (b), respectively). The quasi-equatorial and quasi-axial conformations of 95 exhibited blue and green emission, respectively, when seen under UV light. When the UV lamp was turned off, the quasi-axial conformation of 95 showed a green afterglow (Fig. 15(b)) that lasted for about 0.3 s; by contrast, the quasi-equatorial conformation did not show any afterglow (Fig. 15(a)). In the steady-state PL spectrum of the quasi-equatorial conformation, there was only one emission peak at 444 nm with a lifetime of 2.07 ns, whereas no emission was found in the delayed mode (Fig. 15(a)), showing the RTP inactivity of the quasi-equatorial conformation. The quasi-axial conformation produced dual fluorescence–phosphorescence emissions, with the emission at 382 nm being fluorescence with a lifetime of 1.11 ns and the emission at 517 nm being phosphorescence (Fig. 15(b)) with a lifetime of 32.07 ms.
 | ||
| Fig. 15 (a) Delayed PL (blue line) and steady-state PL (green line) spectra of the 95 quasi-equatorial crystal (insets: photographs of the 95-equatorial crystals taken before and after turning off the UV light). (b) Delayed PL (red line) and steady-state PL (blue line) spectra of the 95 quasi-axial crystal (insets: photographs of 95-axial crystals taken before and after turning off the UV light). PL spectra of solid samples of (c) 96 and (d) 97 in vacuum and air environments; PL spectra of 1 wt% solid solutions of (e) 96, (f) 97, (g) 98 and (h) 99 in PMMA recorded in vacuum and in air. Reproduced from ref. 149 with permission from the Royal Society of Chemistry; and from ref. 150 with permission from © 2021 Elsevier Ltd., all rights reserved. | ||
Leitonas et al. reported the four thianthrene based phenothiazine derivatives 96–99 (Chart 4).150 In the presence of air, the solid sample of 96 was mostly phosphorescent, with just a minor influence of oxygen on the emission (Fig. 15(c)). The PL spectrum of the solid sample of 96 has an emission maximum at 555 nm and a weak shoulder at 430 nm. The solid sample of 97 displays blue fluorescence with faint RTP when it is exposed to air. By contrast, when it is deoxygenated, it displays strong RTP with an emission maximum at 558 nm and fluorescence with an emission maximum at 438 nm (Fig. 15(d)). Thus, the solid powder of 97 clearly emits both fluorescence and RTP under vacuum conditions. Compound 96 showed the highest ratio of RTP to fluorescence intensity among all these derivatives. For 98 and 99, there was no difference in the RTP intensity, regardless of whether it was measured in air or under vacuum conditions. For the molecular dispersions of 96–99 in PMMA, only fluorescence was seen in air. However, the solid films of molecular dispersions of 96–99 in PMMA showed robust RTP in a vacuum, with emission maximum values at 512–533 nm and emission durations in the millisecond range (Fig. 15(e)–(h)).
Wang et al. reported the three phenothiazine based phosphors 100, 101 and 102 (Chart 4).151 Amorphous films were developed by doping compounds 100, 101 and 102 into a stiff PMMA matrix with a mass fraction of 0.1%. After around 30 seconds of UV irradiation, a distinct photoinduced RTP emission was seen for all of them, ranging from almost non-RTP emission to the strong RTP emission. Among these derivatives, 102 performed the best (Fig. 16(a)), with an RTP efficiency reaching 22% after photoinduction. Tian et al. reported the three phenothiazine based phosphors 103, 104 and 105 (Chart 4).152 A co-crystal sample of 104, obtained by removing the solvent from a solution of 104 and triphenylphosphine oxide (OPPh3) (1:100 mass ratio) in ethanol, produces a bright afterglow lasting about 9 seconds. However, only mild RTP can be found in co-crystal samples of 103 and 105 (Fig. 16(b)). The PL spectrum of 104 includes emission bands at 295 nm and 399 nm, which are ascribed to the distinctive emission peaks of OPPh3 and the compound 104, respectively. In addition, the PL spectrum reveals a novel emission band extending from 460 to 620 nm, which is assumed to represent the spectrum of the green afterglow.
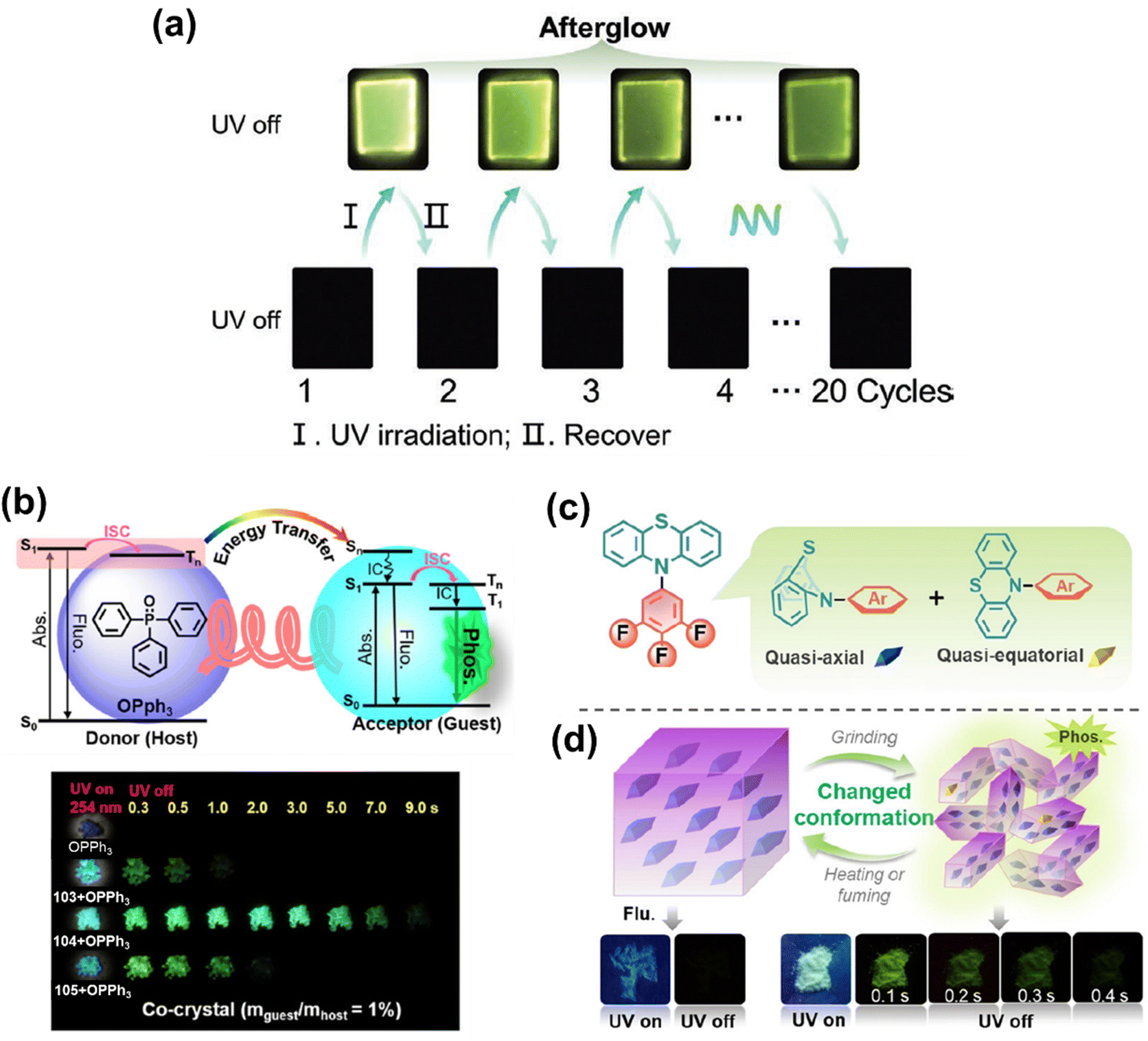 | ||
| Fig. 16 (a) Phosphorescence images of 102 doped into a PMMA film taken after turning the UV light on and off (I = UV light on for 30 s, and II = standing for 30 min after turning off the UV light); (b) (top) energy transfer between the donor (host) and acceptor (guest), and (bottom) photographs of co-crystals of 103–105 with OPPh3 under and after excitation; (c) quasi-axial and quasi-equatorial conformations of 106; and (d) schematic representation for the RTP mechanism in 106 and phosphorescence images of 106 taken after turning the UV light on–off. Reproduced from ref. 151–153 with permission from © 2021 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Ren et al. reported the phenothiazine based phosphor 106 (Chart 4).153 There are two possible conformations for 106: quasi-axial and quasi-equatorial (Fig. 16(c)). When the crystal of 106 was excited, it gave off only blue fluorescence. However, when 106 was ground, it demonstrated phosphorescence with a clear green afterglow, showing the unique force-induced RTP effect (Fig. 16(d)). Following stimulation via heating or fuming, the ground powder reverted to its non-RTP phase. The change in molecular conformation from quasi-axial to quasi-equatorial is expected to be the primary cause of these findings.
Qiu et al. reported the four phenothiazine derivatives 107–110 (Chart 4), which have a planar conformation in the ground state.154 Upon excitation, substantial conformational relaxation occurs in these derivatives, resulting in tunable fluorescence and phosphorescence emissions, depending on the degree of ICT. Multiple emissions, including fluorescence (from dual conformation) and phosphorescence, were seen in the compounds that have a decreased amount of ICT character (107 and 108). However, in the compounds with a greater ICT character (109 and 110), the singlet excitons favour ISC and undergo triplet state, thus the emission is dominated by the phosphorescence.
Ward et al. reported the phenothiazine based phosphors 111 and 112 (Chart 4).155 These compounds are highly phosphorescent at ambient temperature. Herein, the sulphur atom of the phenothiazine moiety is a key part of the RTP emission. The authors synthesized several other compounds in which the sulphur atom of phenothiazine was replaced with an oxygen atom, and no phosphorescence characteristics were observed. Yang et al. reported the six phenothiazine-S,S-dioxide based derivatives 113–118 (Chart 4).156 The electron withdrawing character of the substituents improves the RTP lifespan in the crystals of these luminogens. The RTP lifetime values were found to be 88 ms, 96 ms, 188 ms, 268 ms, 256 ms and 410 ms for 113 (–OCH3 group), 114 (–CH3 group), 115 (–H atom), 116 (–Br atom), 117 (–Cl atom) and 118 (–F atom), respectively (Fig. 17). The electron-withdrawing substituents seem to maintain the excited triplet state associated with the ultralong RTP effect in this system.
 | ||
| Fig. 17 Room temperature phosphorescence (RTP) behaviour of 113–118. Reproduced from ref. 156 with permission from © 2018 Springer Nature, Creative Commons CC BY license. | ||
Huang et al. reported the dibenzothiophene based phenothiazine phosphors 119–122 (Chart 4).157 Compound 119 exhibited phosphorescence with an emission maximum at 540 nm, which is similar to the phosphorescence of phenothiazine. This indicates that the lowest energy triplet state in 119 is mainly concentrated on the phenothiazine unit. The phosphorescence of 121 and 122 matches completely with the phosphorescence of the dibenzothiophene unit, indicating the presence of lowest energy triplet states on the dibenzothiophene unit. By contrast, the phosphorescence in 120 occurs from both the dibenzothiophene acceptor and the phenothiazine donor units. The phenothiazine derivative 86 (Chart 3) shows phosphorescence characteristics.143 Under a nitrogen atmosphere, a significant green afterglow was seen with the naked eye for 86 after the UV radiation had been removed. It should be noted that, at low temperatures, it is difficult for the excited triplet excitons to come back into the excited singlet states through the RISC process. As a consequence, T1 excitons immediately return to the ground state, resulting in phosphorescence.
Wang et al. reported the bifunctional fluorescent probe 123 (Chart 5) that is based on phenothiazine for the selective sensing of Fe3+ and ClO− ions.158 An Fe3+-promoted spirolactone ring opening mechanism in 123 causes a unique fluorescence response for Fe3+ ions (Fig. 18(a)). By contrast, 123 can interact with the ClO− ion through the ClO− induced oxidation of the phenothiazine moiety, which results in selective and sensitive changes in the fluorescence of 123. Probe 123 was employed for tracing Fe3+ ions in living HeLa cells. It was found that the cells loaded with the probe do not exhibit any emission signal; however, a significant emission band was observed when the probed cells were treated with Fe3+ (Fig. 22(a)). These results demonstrated that probe 123 can be effectively used to trace Fe3+ in living cells.
 | ||
| Chart 5 Chemical structures of phenothiazine based chemosensor probes 123–140. | ||
 | ||
| Fig. 18 Sensing mechanism of (a) 123, (b) 124 and (c, top) 125; and (c, bottom) fluorescence response of 125 (left) to Hg2+ and [125 + Hg2+] (right) to I−. Reproduced from ref. 160 with permission from © 2015 Elsevier Ltd., all rights reserved. | ||
Sun et al. reported the phenothiazine-based fluorescence sensor 124 (Chart 5) for the imaging of Hg2+ ions in living cells.159 In comparison to other metal ions such as Cu2+, Mg2+, Ag+, Hg2+, Cr3+, Co2+, Ni2+, Al3+, Ca2+, Zn2+, Fe2+, Fe3+, Na+, Cd2+, and K+, probe 124 detected Hg2+. Compound 124 was used to detect and visualize Hg2+ in actual water samples and live cells, indicating that it has a lot of potential in environmental and biological systems. Hg2+ triggers the deprotection process of thioaldehyde to form the corresponding aldehyde (Fig. 18(b)). This results in the formation of a donor–acceptor system and initiates the process of intramolecular charge transfer. Probe 124 was found to have low toxicity to the living cells, even at high concentrations. Hence, this probe was employed for tracing Hg2+ in HeLa cells. Probe 124 was first incubated into the HeLa cells and then Hg2+ was added. The cells showed stronger fluorescence as the concentration of Hg2+ ions was increased (Fig. 22(b)). Yang et al. reported the phenothiazine based sensor 125 (Chart 5), which exhibited high selectivity toward Hg2+ by forming a 1:1 complex.160 The Hg2+ ensemble of 125 is described as a very sensitive and selective sensor for detecting iodide in aqueous solutions (Fig. 18(c)). Later, the fluorescence quenching detection mechanism of 125 was comprehensively investigated by Sun et al.161 Sensor 125 exhibited strong orbital coupling with Hg2+ which opens up two non-emissive routes, namely intramolecular electron transfer and intersystem crossing. The interplay between the two channels resulted in a substantial degree of fluorescence quenching, making 125 a viable candidate for Hg2+ sensor fabrication. In addition, probe 125 was used to detect the presence of mercury and iodine in living HUVEC cells. The cells were incubated with probe 125 and then treated with Hg(ClO4)2. After the intracellular uptake of Hg2+ ions, a non-fluorescent ensemble of probe 125 with the Hg2+ ions was formed, and when this ensemble was treated with various amounts of I−, the green fluorescence was restored (Fig. 22(c)).
Karmegam et al. reported the phenothiazine–rhodamine based sensor 126 (Chart 5) for Zn2+ detection.162 In the presence of Zn2+ ions, the absorption spectrum of 126 indicated the emergence of a new peak in a higher wavelength region (Fig. 19(a)). The emission band at 528 nm for probe 126 showed its ring closed form. As for Zn2+ ion binding to 126, the λem is shifted to 608 nm and is followed by green to pinkish red fluorescence emission owing to the ring opening of the spirolactam moiety in 126 (Fig. 19(b) and (c)). The spectral overlap that occurs between the emission band of the donor and the absorption band of the ring-opened form of the acceptor moiety leads to the fluorescence resonance energy transfer-ON process for the detection of Zn2+ ions. The cytotoxicity of probe 126 was evaluated by adding it in various concentrations to HeLa cells, and it was found that more than 98% of the cells remained viable. The HeLa cells incubated with probe 126 showed clear green fluorescence, but after the intracellular uptake of Zn2+, the green fluorescence changed to bright red (Fig. 22(d)). Suganya et al. reported the phenothiazine based fluorescence probe 127 (Chart 5)163 for the selective detection of CN− (Fig. 19(d)). After the CN− ion was introduced, intramolecular charge transfer from phenothiazine to the cyanovinyl unit was totally inhibited (Fig. 19(e)). This resulted in the fluorescence being completely quenched.
 | ||
| Fig. 19 (a) UV-visible spectrum of 126 (10−5 M) in the presence of various metal ions; (b) concentration-dependent fluorescence enhancement of 126 (10−5 M) upon the addition of Zn2+ (0–1.1 eq.); sensing mechanism for (c) 126 and (e) 127; and (d) daylight (top) and UV light (bottom) images of 127 (20 μM) in the presence of various anions. Reproduced from ref. 162 with permission from © 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim; and from ref. 163 with permission from © 2019 Elsevier Ltd., all rights reserved. | ||
Hou et al. reported the fluorescent probe 128 (Chart 5) based on phenothiazine for the detection of thiophenols in living cells and real water samples.164 Probe 128 has the ability to detect thiophenols with a fast response time of 3 min and a large Stokes shift of 126 nm. Compound 128 is not emissive in solution, but upon the addition of thiophenol an emission band appears at 527 nm. Upon increasing the concentration of thiophenol to 20.0 μM, the intensity of the emission band at 527 nm was enhanced up to 176-fold. This enhancement in the emission intensity is due to the formation of an excited state intramolecular proton transfer (ESIPT) type fluorophore (Fig. 20(a)).
 | ||
| Fig. 20 Sensing mechanism of (a) 128 and (b) 129; and (c) portions of the 1H NMR spectrum of probe 130 exhibiting spectral changes upon the addition of an increasing concentration of cyanide ions. Reproduced from ref. 165 and 166 with permission from © 2018 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Feng et al. reported fluorescence probe 129 (Chart 5) for the monitoring of hypochlorous acid (HOCl) levels in biological samples.165 When hypochlorous acid was added, the colour of the solution changed. This made it easier to spot the presence of hypochlorous acid with the naked eye. HRMS titration analysis demonstrated that the sensor worked via oxidation of the thioether by HOCl (Fig. 20(b)). Probe 129 was employed to trace the presence of HOCl in living cells. The cells were stained with probe 129 and then treated with HOCl. After the intracellular uptake of HOCl, the fluorescence was clearly enhanced (Fig. 22(e)). This demonstrated that probe 129 easily enters into cells and can be used for the visualization of HOCl in living cells. Dhamodharan et al. reported the fluorophores 130 and 131 (Chart 5) based on ethylenedioxythiophene and phenothiazine for the selective detection of cyanide ions.166 The addition of cyanide ions to the DMSO solution of the probe resulted in a significant increase in the emission. The weak yellowish and yellow-orange emissions of probes 130 and 131 changed to bright green and bright yellow emissions, respectively, upon the addition of cyanide. This shows that the compounds 130 and 131 worked as “turn-on” type fluorescent probes for the detection of cyanide ions. The addition of cyanide ions caused a change in the chemical shift of the vinylic protons, which indicated the nucleophilic addition of cyanide ion to the vinyl carbon (Fig. 20(c)).
Anand et al. reported derivative 132 based on a Schiff base (Chart 5) for the selective detection of Cu2+ ions.167 Schiff base 132 is highly selective for Cu2+ ions in the presence of the other important cations (Fig. 21(b)). Quenching of the emission of probe 132 was observed with a limit of detection (LOD) of 4.88 ppb Cu2+ ions, and the emission intensity of probe 132 was reduced by 13-fold (Fig. 21(a)). Vengaian et al. reported the phenothiazine–diaminomaleonitrile based colorimetric and fluorescence sensor 133 (Chart 5) for the detection of Hg2+ and S2−.168 With the addition of Hg2+, the absorption peak of probe 133 shifted from 425 nm (black line) to 448 nm (pink line) (Fig. 21(c)), indicating the existence of ground state interactions between the probe and the Hg2+ ion. Probe 133 emitted strong fluorescence at 550 nm, but the addition of Hg2+ decreased its emission intensity (Fig. 21(d)). The addition of S2− ions to the [133 + Hg2+] ensemble caused the quenched fluorescence to return at 550 nm (Fig. 21(e)).
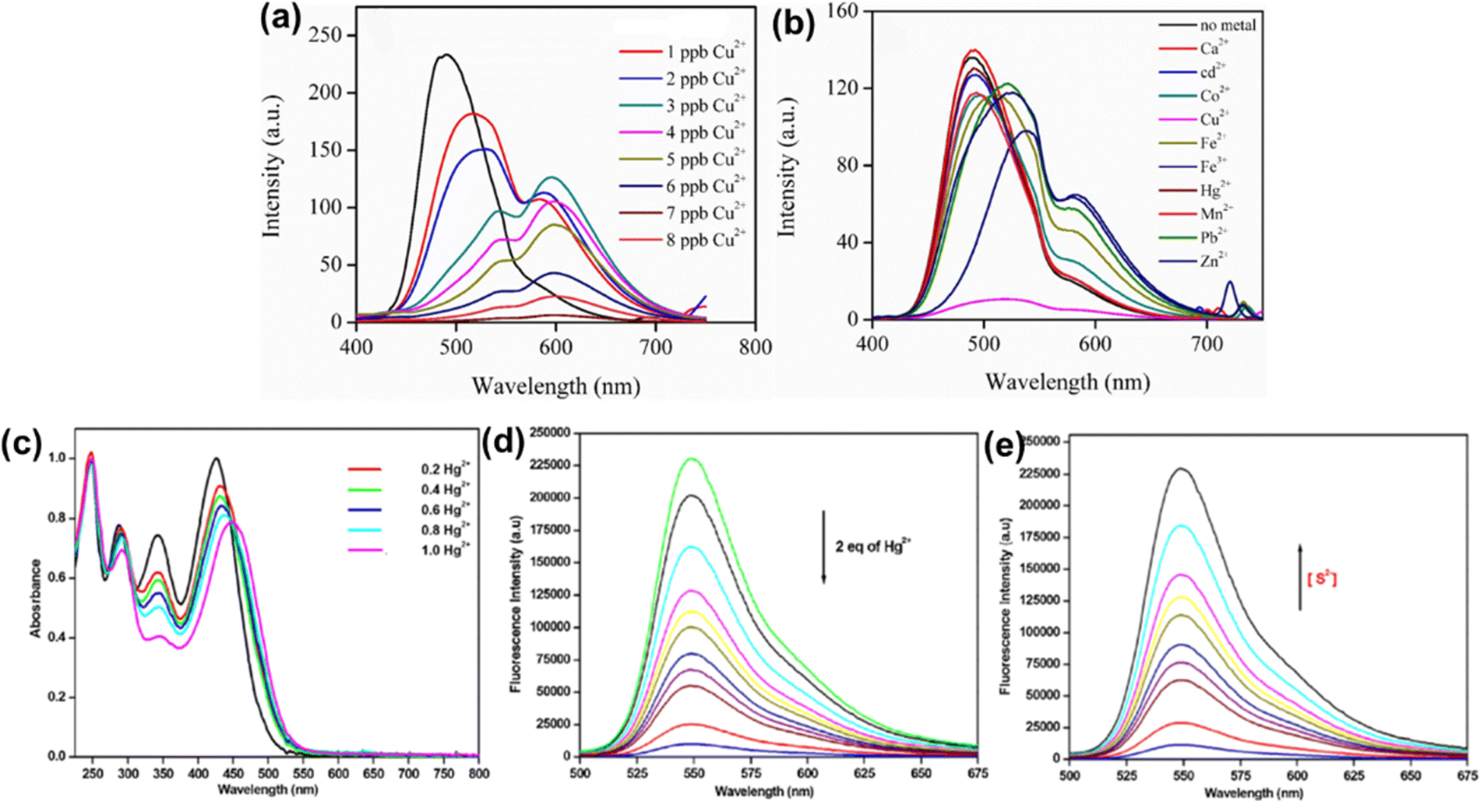 | ||
| Fig. 21 Fluorescence spectra of 132 (a) with the addition of different concentrations of Cu2+ (black line = emission spectrum of probe 132) and (b) in presence of different cations; (c) absorption and (d) emission spectra of probe 133 upon the addition of Hg2+ in ethanol–water (6:4) mixture; and € fluorescence response of [133 + Hg2+]. Reproduced from ref. 167 with permission from the Royal Society of Chemistry; and from ref. 168 with permission from © 2016 Elsevier Ltd., all rights reserved. | ||
Probe 133 was employed to detect the presence of Hg2+ and S2− in HeLa cells. The cells loaded with probe 133 exhibited green fluorescence under UV light. After the intracellular uptake of Hg2+, the green fluorescence was quenched due to the formation of the non-fluorescent [133−Hg2+] ensemble (Fig. 22(f)). The green fluorescence was restored when this ensemble was treated with S2−, indicating that probe 133 could be used for on–off fluorescence imaging in HeLa cells.
 | ||
| Fig. 22 Fluorescence images of cells incubated with probe (a) 123, (b) 124, (c) 125, (d) 126, (e) 129 and (f) 133 before and after the addition of various analytes. Reproduced from ref. 158, 160 and 168 with permission from © 2021, 2015 and 2016 Elsevier Ltd., all rights reserved; and from ref. 159, 162 and 165 with permission from © 2020 and 2018 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Zou et al. reported the dicyanovinyl-functionalized phenothiazine based “on–off” fluorescent chemodosimeter 134 (Chart 5).169 Compound 134 exhibited two major absorption bands at 321 and 488 nm. The addition of CN− ions reduced the strength of the CT absorption band at 488 nm, and a new absorption band appeared in the shorter wavelength region (Fig. 23(a)). The color of the solution changed from pink to colorless due to the considerable decrease in absorption. The sensing mechanism of probe 134 is shown in Fig. 23(b). Wie et al. reported the Schiff base of phenothiazine 135 (Chart 5) for the ratiometric sensing of Cd2+ ions.170 Compound 135 was weakly emissive, but as the Cd2+ was added to its solution the fluorescence intensity increased linearly, and the highest intensity fluorescence peak showed a blue shift from 575 to 525 nm (Fig. 23(c)). The absorption maximum of probe 135 was 356 nm. When Cd2+ ions were added, a new absorption band appeared at 471 nm, and the strength of the absorption band at 356 nm dropped (Fig. 23(d)).
 | ||
| Fig. 23 (a) Absorption spectrum of probe 134 (10 mM) in the presence of increasing concentrations of CN− ions; sensing mechanism of (b) 134 and of (e) and (f) 136; and (c) emission and (d) absorption spectra of probe 135 in the presence of increased concentrations of Cd2+. Reproduced from ref. 169 with permission from the Royal Society of Chemistry; from ref. 170 with permission from © 2013 Springer Nature; and from ref. 171 with permission, copyright 2022 American Chemical Society. | ||
Huang et al. reported the dual-responsive ratiometric fluorescent probe 136 (Chart 5) for the separate and simultaneous detection of hypochlorite (ClO−) and peroxynitrite (ONOO−).171 In this probe, the phenothiazine–coumarin moiety was selected as the ClO− responsive fragment, and the precursor of 2-(benzo[d]thiazol-2-yl)aniline was used as the ONOO− sensor. Compound 136 initially emitted red fluorescence at 640 nm. This fluorescence changed to green fluorescence (520 nm) and blue fluorescence (450 nm) in the presence of ClO− and ONOO− ions, respectively. Fig. 23(e) and (f) depict the sensing mechanism of 136.
Hu et al. reported the phenothiazine-based turn-on fluorescent probe 137 (Chart 5) for the selective detection of hydrogen sulfide (H2S) in food, live cells and animals.172 Upon the addition of NaHS (a source of H2S), the absorption maximum of probe 137 was hypsochromically shifted from 480 nm to 403 nm (Fig. 24(a)) and the brown coloured solution was decolourized. Probe 137 exhibited weak emission at 596 nm. The addition of NaHS increases the intensity of emission band at 596 nm (Fig. 24(b)) and the fluorescence colour of the probe 137 solution changes from blue to pink. The nucleophilic attack of H2S at the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of 137via a Michael addition reaction prevented the ICT process, resulting in a 34-fold increase in the fluorescence emission. Bao et al. reported water-soluble fluorescence probe 138 (Chart 5) for the imaging of mitochondrial hypochlorous acid.173 In a pure aqueous solution, the probe showed an ultrafast response to HOCl (<5 s) – Fig. 24(c) depicts the sensing mechanism of 138. Probe 138 exhibits an absorption maximum at 604 nm, which is hypsochromically shifted to 556 nm in the presence of HOCl. The presence of HOCl also causes the non-emissive solution of probe 138 to show strong emission.
C double bond of 137via a Michael addition reaction prevented the ICT process, resulting in a 34-fold increase in the fluorescence emission. Bao et al. reported water-soluble fluorescence probe 138 (Chart 5) for the imaging of mitochondrial hypochlorous acid.173 In a pure aqueous solution, the probe showed an ultrafast response to HOCl (<5 s) – Fig. 24(c) depicts the sensing mechanism of 138. Probe 138 exhibits an absorption maximum at 604 nm, which is hypsochromically shifted to 556 nm in the presence of HOCl. The presence of HOCl also causes the non-emissive solution of probe 138 to show strong emission.
 | ||
| Fig. 24 (a) Absorption and (b) emission spectra of probe 137 in the presence of various analytes and NaHS; and (c) the sensing mechanism of 138. Reproduced from ref. 172 with permission from the Royal Society of Chemistry; and from ref. 173 with permission from © 2021 Elsevier Ltd., all rights reserved. | ||
Liang et al. reported the phenothiazine-based turn-on fluorescent probe 139 (Chart 5) for the selective quantification of HOCl in living cells.174 An increase in the concentration of ClO− causes the oxidation of probe 139 to the sulfoxide product. A further increase in the concentration of hypochlorite ions causes the breaking of one or two CC bonds (Fig. 25(a)). This is followed by a change in colour from light pink to light yellow with a shift in the absorption spectrum maximum from 580 to 493 nm. There was a significant increase in the fluorescence emission at 595 nm in the presence of ClO− ions.
 | ||
| Fig. 25 (a) Sensing mechanism of 139; and (b) binding of Hg2+ ion with probe 140 (left) and fluorescence responses of probe 140 with the addition of incremental concentrations of Hg2+ ions (right). Reproduced from ref. 175 with permission from © 2021 Springer Nature. | ||
Govindasamy et al. reported the phenothiazine–thiophene hydrazone based sensor 140 (Chart 5) for the detection of Hg2+ ion.175 After the addition of Hg2+ ions, the absorption peak at 304 nm was diminished, and the peak at 397 nm was bathochromically shifted to 422 nm. When Hg2+ ions were introduced to the probe, the color of the probe changed from pale green to bright yellow. Sensor 140 exhibited an intense emission at 537 nm. The emission intensity at 537 nm was completely quenched with the addition of Hg2+ ions (Fig. 25(b)).
The phenothiazine-based derivative 65 (Chart 3)129 was used as a probe for the detection of hydrazine. The recognition group of probe 65 was methyl cyanoacetate, while the fluorescent chromophore was phenothiazine. Probe 65 exhibited a strong emission band at 657 nm, which shifted to 475 nm in the presence of hydrazine. The presence of hydrazine also affected the absorption spectrum of probe 65 and shifted its absorption maximum from 470 nm to 375 nm. In the presence of hydrazine, the detection limit of probe 65 was determined to be 5.2 ppb.
Marghad et al. reported phenothiazine derivative 141, which exhibits dual emission characteristics (Chart 6).176 The calculated ΔEST value for 141 is substantially lower (0.01 eV), making it a suitable candidate to be used for an optimal TADF performance. Xiang et al. reported the TADF active fluorophore 142, which comprises phenoxazine and phenothiazine S,S-dioxide units.177 TD-DFT calculations revealed effective reverse intersystem crossing from T1 to S1, and ΔEST between S1 and T1 was found to be 0.03 eV. After achieving positive findings in derivative 142 for TADF applications, the same group reported three more derivatives of phenothiazine S,S-dioxide, 143, 144 and 145 (Chart 6).178 TD-DFT calculations revealed that the singlet–triplet energy splitting (ΔEST) was 0.69, 0.73 and 0.13 eV for 143, 144 and 145, respectively. Compound 145, with a smaller ΔEST value compared with 143 and 144, exhibited improved the reverse intersystem crossing (RISC) process.
 | ||
| Chart 6 Chemical structures of phenothiazine based TADF emitters 141–145. | ||
4. Summary and outlook
In this review, we have provided the significant advancements in the development of fluorescent phenothiazine derivatives for applications in mechanochromism, aggregation-induced emission, phosphorescence, and sensor probes. The careful molecular engineering of the phenothiazine core is an effective technique for developing conjugated materials with adjustable optoelectronic properties. The phenothiazine core has primary reactive sites at the 3-, 7-, and N10-positions (Fig. 4(a)), which enables many synthetic alterations to produce new phenothiazine-based conjugated materials with tunable optoelectronic properties. Phenothiazine derivatives serve as excellent candidates for a variety of optoelectronic applications owing to their favourable optical properties, such as wide absorption with high molar extinction coefficients, high phosphorescence quantum yields with extended lifetimes of up to milliseconds, and improved photochemical and photothermal stabilities. The achievements of phenothiazine derivatives in various applications are as follows:• The presence of electron rich S and N atoms make phenothiazine a strong electron donor. The electron donating ability of phenothiazine facilitates the formation of intramolecular charge transfer (ICT) complexes. Compounds with ICT characteristics exhibit mechanochromic phenomena by exploiting the various ICT states in response to external mechanical stimuli.
• The bent shaped phenothiazine in combination with substituents present at the 3-, 7- and N10-positions provides twisting in the target structures, which not only helps in suppressing excessive π–π stacking interactions in the solid state but also enables the molecule to undergo conformational changes in response to external mechanical forces, endowing the materials with mechanochromic properties.
• The mechanochromic properties of phenothiazine derivatives are largely attributed to their D–A structure. The effect of mechanochromism is realized through the conversion of crystalline materials into amorphous materials and, in some cases, via the conversion that is observed between two crystal forms.
• Conformation dependent prompt fluorescence and TADF characteristics were observed in the phenothiazine derivatives.
• The phenothiazine derivatives exhibit alkyl chain length-dependent mechanochromic behavior.
• The phenothiazine derivatives exhibit high quantum yields in solution and this solid state, as well as long excited-state lifetimes, which make them suitable candidates for producing efficient phosphorescence emission.
• Conformation dependent mechanochromism and phosphorescence characteristics were realized in the phenothiazine derivatives. Mechanical stimulus-induced transformation between the two conformations of phenothiazine (quasi-axial and quasi-equatorial) changes the emission colour and can turn-on/off the phosphorescence characteristics.
• It was found that the quasi-equatorial conformation of the phenothiazine derivative is more likely to give RTP emission, and the mechanical stimulus-induced transition between the two conformations leads to stimulus responsive RTP effects.
• The phenothiazine-S,S-dioxide based derivative was found to exhibit a long-lived phosphorescence lifetime of up to 822 ms with a high quantum yield of 11.6%.
• The unique non-planar shape of the phenothiazine moiety can lock the intramolecular motions, thereby suppressing the non-radiative decay and endow the materials with AIE properties.
• The electron rich S-atom of phenothiazine is susceptible to undergoing oxidation in the presence of ClO− ions, and thus can be used for the selective detection of hypochlorous acid.
• Fluorescent phenothiazine derivatives have been successively employed as probe sensors for the detection of a variety of chemical species such as Fe3+, Hg2+, Zn2+, Cu2+, CN−, Cd2+, ClO−, NO2−, S2− and many more.
We believe that this review will stimulate further investigations towards the rational design and synthesis of innovative phenothiazine-based materials with the potential to be used in a variety of applications such as sensors, fluorescent tags, organic light-emitting diodes (OLEDs), non-linear optics (NLO) and optical data storage devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by SERB CRG/2018/000032 and CSIR 01(2934)/18/EMR-II. F. K. thanks CSIR-New Delhi for a fellowship.References
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, US, 3rd edn, 2006 Search PubMed.
- (a) X. Chen, Z. Yang, W. Li, Z. Mao, J. Zhao, Y. Zhang, Y.-C. Wu, S. Jiao, Y. Liu and Z. Chi, ACS Appl. Mater. Interfaces, 2019, 11, 39026–39034 CrossRef CAS PubMed; (b) X. Cai and S.-J. Su, Adv. Funct. Mater., 2018, 28, 1802558 CrossRef; (c) T. Miwa, S. Kubo, K. Shizu, T. Komino, C. Adachi and H. Kaji, Sci. Rep., 2017, 7, 284 CrossRef PubMed.
- (a) Y. Jiang, Y.-Y. Liu, X. Liu, H. Lin, K. Gao, W.-Y. Lai and W. Huang, Chem. Soc. Rev., 2020, 49, 5885–5944 RSC; (b) H.-H. Fang, J. Yang, J. Feng, T. Yamao, S. Hotta and H.-B. Sun, Laser Photonics Rev., 2014, 8, 687–715 CrossRef; (c) J. Gierschner, S. Varghese and S. Y. Park, Adv. Opt. Mater., 2016, 4, 348–364 CrossRef CAS.
- (a) X. Wu and W. Zhu, Chem. Soc. Rev., 2015, 44, 4179–4184 RSC; (b) S. Zhu, R. Tian, A. L. Antaris, X. Chen and H. Dai, Adv. Mater., 2019, 1900321 CrossRef PubMed.
- (a) P. Gao, W. Pan, N. Li and B. Tang, Chem. Sci., 2019, 10, 6035–6071 RSC; (b) B. A. D. Neto, J. R. Correa and J. Spencer, Chem. – Eur. J., 2022, 28, e202103262 CrossRef CAS PubMed; (c) D. Wu, L. Chen, W. Lee, G. Ko, J. Yin and J. Yoon, Coord. Chem. Rev., 2018, 354, 74–97 CrossRef CAS.
- (a) H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640 CrossRef CAS PubMed; (b) W. Qin, D. Ding, J. Liu, W. Z. Yuan, Y. Hu, B. Liu and B. Z. Tang, Adv. Funct. Mater., 2012, 22, 771–779 CrossRef CAS.
- (a) S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339–1386 CrossRef CAS PubMed; (b) Y.-P. Li, X.-H. Zhu, S.-N. Li, Y.-C. Jiang, M.-C. Hu and Q.-G. Zhai, ACS Appl. Mater. Interfaces, 2019, 11, 11338–11348 CrossRef CAS PubMed.
- (a) Y. Gong, Y. Tan, J. Liu, P. Lu, C. Feng, W. Z. Yuan, Y. Lu, J. Z. Sun, G. He and Y. Zhang, Chem. Commun., 2013, 49, 4009 RSC; (b) E. Ubba, Y. Tao, Z. Yang, J. Zhao, L. Wang and Z. Chi, Chem. – Asian J., 2018, 13, 3106–3121 CrossRef CAS PubMed.
- (a) Y. Zhuang, L. Wang, Y. Lv, T.-L. Zhou and R.-J. Xie, Adv. Funct. Mater., 2018, 28, 1705769 CrossRef; (b) J. Yu, M. Luo, Z. Lv, S. Huang, H.-H. Hsu, C.-C. Kuo, S.-T. Han and Y. Zhou, Nanoscale, 2020, 12, 23391–23423 RSC.
- N. Kumar, Y. Hori, M. Nishiura and K. Kikuchi, Chem. Sci., 2020, 11, 3694–3701 RSC.
- S. Suzuki, S. Sasaki, A. S. Sairi, R. Iwai, B. Z. Tang and G. Konishi, Angew. Chem., Int. Ed., 2020, 132, 9940–9951 CrossRef.
- F. Bernardi, M. Olivucci and M. A. Robb, Chem. Soc. Rev., 1996, 25, 321 RSC.
- A. L. Sobolewski, W. Domcke, C. Dedonder-Lardeux and C. Jouvet, Phys. Chem. Chem. Phys., 2002, 4, 1093–1100 RSC.
- B. G. Levine and T. J. Martínez, Annu. Rev. Phys. Chem., 2007, 58, 613–634 CrossRef CAS PubMed.
- B. Valeur and M. N. Berberan-Santos, J. Chem. Educ., 2011, 88, 731–738 CrossRef CAS.
- R. Gao, M. S. Kodaimati and D. Yan, Chem. Soc. Rev., 2021, 50, 5564–5589 RSC.
- (a) E. M. S. Stennett, M. A. Ciuba and M. Levitus, Chem. Soc. Rev., 2014, 43, 1057–1075 RSC; (b) M. Shimizu and T. Hiyama, Chem. – Asian J., 2010, 5, 1516–1531 CrossRef CAS PubMed; (c) A. N. Woodward, J. M. Kolesar, S. R. Hall, N.-A. Saleh, D. S. Jones and M. G. Walter, J. Am. Chem. Soc., 2017, 139, 8467–8473 CrossRef CAS PubMed.
- (a) G. Zhang, J. Sun, P. Xue, Z. Zhang, P. Gong, J. Peng and R. Lu, J. Mater. Chem. C, 2015, 3, 2925–2932 RSC; (b) Y. Sagara, T. Mutai, I. Yoshikawa and K. Araki, J. Am. Chem. Soc., 2007, 129, 1520–1521 CrossRef CAS PubMed; (c) M. Sase, S. Yamaguchi, Y. Sagara, I. Yoshikawa, T. Mutai and K. Araki, J. Mater. Chem., 2011, 21, 8347 RSC.
- D. Arnaud, R. K. Pandey, S. Miyajima, S. Nagamatsu, R. Prakash, W. Takashima, S. Hayase and K. Kaneto, Trans. Mater. Res. Soc. Jpn., 2013, 38, 305–308 CrossRef CAS.
- K. Wei Chou, H. Ullah Khan, M. R. Niazi, B. Yan, R. Li, M. M. Payne, J. E. Anthony, D.-M. Smilgies and A. Amassian, J. Mater. Chem. C, 2014, 2, 5681–5689 RSC.
- (a) S. Cai, H. Shi, J. Li, L. Gu, Y. Ni, Z. Cheng, S. Wang, W. Xiong, L. Li, Z. An and W. Huang, Adv. Mater., 2017, 29, 1701244 CrossRef PubMed; (b) Y. Yu, C. Wang, Y. Wei, Y. Fan, J. Yang, J. Wang, M. Han, Q. Li and Z. Li, Adv. Opt. Mater., 2019, 7, 1900505 CrossRef.
- Q. Li and Z. Li, Acc. Chem. Res., 2020, 53, 962–973 CrossRef CAS PubMed.
- C. H. Kim and T. Joo, Phys. Chem. Chem. Phys., 2009, 11, 10266 RSC.
- H. Tahara, K. Uranaka, M. Hirano, T. Ikeda, T. Sagara and H. Murakami, ACS Appl. Mater. Interfaces, 2019, 11, 1–6 CrossRef CAS PubMed.
- C.-T. Chen, Chem. Mater., 2004, 16, 4389–4400 CrossRef CAS.
- S. Liu, X. Zhou, H. Zhang, H. Ou, J. W. Y. Lam, Y. Liu, L. Shi, D. Ding and B. Z. Tang, J. Am. Chem. Soc., 2019, 141, 5359–5368 CrossRef CAS PubMed.
- G. v Bünau, Ber. Bunsenges. Phys. Chem., 1970, 74, 1294–1295 Search PubMed.
- J. B. Birks, Photophysics of aromatic molecules, Wiley-Interscience, London, New York, 1970 Search PubMed.
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS.
- (a) E. A. Jares-Erijman and T. M. Jovin, Nat. Biotechnol., 2003, 21, 1387–1395 CrossRef CAS PubMed; (b) H. Saigusa and E. C. Lim, J. Phys. Chem., 1995, 99, 15738–15747 CrossRef CAS.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, B. Z. Tang, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu and D. Zhu, Chem. Commun., 2001, 1740–1741 RSC.
- J. Liang, B. Z. Tang and B. Liu, Chem. Soc. Rev., 2015, 44, 2798–2811 RSC.
- R. Hu, N. L. C. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494–4562 RSC.
- W. Z. Yuan, Y. Gong, S. Chen, X. Y. Shen, J. W. Y. Lam, P. Lu, Y. Lu, Z. Wang, R. Hu, N. Xie, H. S. Kwok, Y. Zhang, J. Z. Sun and B. Z. Tang, Chem. Mater., 2012, 24, 1518–1528 CrossRef CAS.
- C. Chen, R.-H. Li, B.-S. Zhu, K.-H. Wang, J.-S. Yao, Y.-C. Yin, M.-M. Yao, H.-B. Yao and S.-H. Yu, Angew. Chem., Int. Ed., 2018, 57, 7106–7110 CrossRef CAS PubMed.
- Y.-X. Zhu, Z.-W. Wei, M. Pan, H.-P. Wang, J.-Y. Zhang and C.-Y. Su, Dalton Trans., 2016, 45, 943–950 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- H. Tong, Y. Dong, Y. Hong, M. Häussler, J. W. Y. Lam, H. H.-Y. Sung, X. Yu, J. Sun, I. D. Williams, H. S. Kwok and B. Z. Tang, J. Phys. Chem. C, 2007, 111, 2287–2294 CrossRef CAS.
- Y. Dong, J. W. Y. Lam, A. Qin, J. Liu, Z. Li, B. Z. Tang, J. Sun and H. S. Kwok, Appl. Phys. Lett., 2007, 91, 011111 CrossRef.
- (a) F. Ciardelli, G. Ruggeri and A. Pucci, Chem. Soc. Rev., 2013, 42, 857–870 RSC; (b) Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878 RSC.
- (a) B. R. Crenshaw, M. Burnworth, D. Khariwala, A. Hiltner, P. T. Mather, R. Simha and C. Weder, Macromolecules, 2007, 40, 2400–2408 CrossRef CAS; (b) X. Wu, J. Guo, Y. Cao, J. Zhao, W. Jia, Y. Chen and D. Jia, Chem. Sci., 2018, 9, 5270–5277 RSC; (c) A. Kishimura, T. Yamashita, K. Yamaguchi and T. Aida, Nat. Mater, 2005, 4, 546–549 CrossRef CAS PubMed.
- S. Yagai, S. Okamura, Y. Nakano, M. Yamauchi, K. Kishikawa, T. Karatsu, A. Kitamura, A. Ueno, D. Kuzuhara, H. Yamada, T. Seki and H. Ito, Nat. Commun., 2014, 5, 4013 CrossRef CAS PubMed.
- H. Ito, M. Muromoto, S. Kurenuma, S. Ishizaka, N. Kitamura, H. Sato and T. Seki, Nat. Commun., 2013, 4, 2009 CrossRef PubMed.
- Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073–1095 CrossRef CAS PubMed.
- X.-Y. Wang, L. Lv, L. Sun, Y. Hou, Z. Hou and Z. Chen, Front. Chem., 2022, 10, 865198 CrossRef CAS PubMed.
- W. Yang, C. Liu, S. Lu, J. Du, Q. Gao, R. Zhang, Y. Liu and C. Yang, J. Mater. Chem. C, 2018, 6, 290–298 RSC.
- H. Qian, N. S. Purwanto, D. G. Ivanoff, A. J. Halmes, N. R. Sottos and J. S. Moore, Chem, 2021, 7, 1080–1091 CAS.
- G. He, L. Du, Y. Gong, Y. Liu, C. Yu, C. Wei and W. Z. Yuan, ACS Omega, 2019, 4, 344–351 CrossRef CAS PubMed.
- (a) P. S. Hariharan, V. K. Prasad, S. Nandi, A. Anoop, D. Moon and S. P. Anthony, Cryst. Growth Des., 2017, 17, 146–155 CrossRef CAS; (b) P. S. Hariharan, D. Moon and S. P. Anthony, J. Mater. Chem. C, 2015, 3, 8381–8388 RSC.
- F. Khan, A. Ekbote, G. Singh and R. Misra, J. Mater. Chem. C, 2022, 10, 5024–5064 RSC.
- (a) S. Sasaki, S. Suzuki, W. M. C. Sameera, K. Igawa, K. Morokuma and G. Konishi, J. Am. Chem. Soc., 2016, 138, 8194–8206 CrossRef CAS PubMed; (b) S. Sekiguchi, K. Kondo, Y. Sei, M. Akita and M. Yoshizawa, Angew. Chem., Int. Ed., 2016, 55, 6906–6910 CrossRef CAS PubMed.
- A. Li, N. Chu, J. Liu, H. Liu, J. Wang, S. Xu, H. Cui, H. Zhang, W. Xu and Z. Ma, Mater. Chem. Front., 2019, 3, 2768–2774 RSC.
- Y. Qi, Y. Wang, G. Ge, Z. Liu, Y. Yu and M. Xue, J. Mater. Chem. C, 2017, 5, 11030–11038 RSC.
- C. Duan, Y. Zhou, G.-G. Shan, Y. Chen, W. Zhao, D. Yuan, L. Zeng, X. Huang and G. Niu, J. Mater. Chem. C, 2019, 7, 3471–3478 RSC.
- M.-J. Teng, X.-R. Jia, X.-F. Chen and Y. Wei, Angew. Chem., Int. Ed., 2012, 51, 6398–6401 CrossRef CAS PubMed.
- G. Li, Y. Xu, W. Zhuang and Y. Wang, RSC Adv., 2016, 6, 84787–84793 RSC.
- J. Wu, Y. Cheng, J. Lan, D. Wu, S. Qian, L. Yan, Z. He, X. Li, K. Wang, B. Zou and J. You, J. Am. Chem. Soc., 2016, 138, 12803–12812 CrossRef CAS PubMed.
- A. Pucci, Sensors, 2019, 19, 4969 CrossRef CAS PubMed.
- K. Nagura, S. Saito, H. Yusa, H. Yamawaki, H. Fujihisa, H. Sato, Y. Shimoikeda and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 10322–10325 CrossRef CAS PubMed.
- X. Feng, J. Zhang, Z. Hu, Q. Wang, Md. M. Islam, J.-S. Ni, M. R. J. Elsegood, J. W. Y. Lam, E. Zhou and B. Z. Tang, J. Mater. Chem. C, 2019, 7, 6932–6940 RSC.
- P. Xue, J. Ding, P. Wang and R. Lu, J. Mater. Chem. C, 2016, 4, 6688–6706 RSC.
- W. Liu, Y. Wang, G. Ge, L. Ma, L. Ren and Y. Zhang, Dyes Pigm., 2019, 171, 107704 CrossRef CAS.
- J. Yang, Z. Ren, B. Chen, M. Fang, Z. Zhao, B. Z. Tang, Q. Peng and Z. Li, J. Mater. Chem. C, 2017, 5, 9242–9246 RSC.
- J. Wu, Y. Cheng, J. Lan, D. Wu, S. Qian, L. Yan, Z. He, X. Li, K. Wang, B. Zou and J. You, J. Am. Chem. Soc., 2016, 138, 12803–12812 CrossRef CAS PubMed.
- B. Roy, M. C. Reddy and P. Hazra, Chem. Sci., 2018, 9, 3592–3606 RSC.
- Q. Qi, J. Qian, X. Tan, J. Zhang, L. Wang, B. Xu, B. Zou and W. Tian, Adv. Funct. Mater., 2015, 25, 4005–4010 CrossRef CAS.
- Y. Zhang, K. Wang, G. Zhuang, Z. Xie, C. Zhang, F. Cao, G. Pan, H. Chen, B. Zou and Y. Ma, Chem. – Eur. J., 2015, 21, 2474–2479 CrossRef CAS PubMed.
- X. Ma, J. Li, C. Lin, G. Chai, Y. Xie, W. Huang, D. Wu and W.-Y. Wong, Phys. Chem. Chem. Phys., 2019, 21, 14728–14733 RSC.
- A. Bernthsen, Ber. Dtsch. Chem. Ges., 1883, 16, 2896–2904 CrossRef.
- P. Siva Gangadhar, G. Reddy, S. Prasanthkumar and L. Giribabu, Phys. Chem. Chem. Phys., 2021, 23, 14969–14996 RSC.
- S. Revoju, A. Matuhina, L. Canil, H. Salonen, A. Hiltunen, A. Abate and P. Vivo, J. Mater. Chem. C, 2020, 8, 15486–15506 RSC.
- G. Kim, H. R. Yeom, S. Cho, J. H. Seo, J. Y. Kim and C. Yang, Macromolecules, 2012, 45, 1847–1857 CrossRef CAS.
- I. J. Al-Busaidi, A. Haque, N. K. Al Rasbi and M. S. Khan, Synth. Met., 2019, 257, 116189 CrossRef CAS.
- (a) C. S. Krämer and T. J. J. Müller, Chem. – Eur. J., 2003, 3534–3548 CrossRef; (b) P. Acker, J. S. Wössner, G. Desmaizieres and B. Esser, ACS Sustainable Chem. Eng., 2022, 10, 3236–3244 CrossRef CAS; (c) G. Taurand, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2000, p. a19_387 Search PubMed.
- P. Huang, Manju, S. Kazim, L. Lezama, R. Misra and S. Ahmad, ACS Appl. Mater. Interfaces, 2021, 13, 33311–33320 CrossRef CAS PubMed.
- Y. Rout, C. Montanari, E. Pasciucco, R. Misra and B. Carlotti, J. Am. Chem. Soc., 2021, 143, 9933–9943 CrossRef CAS PubMed.
- Y. Rout, A. Ekbote and R. Misra, J. Mater. Chem. C, 2021, 9, 7508–7531 RSC.
- J. Daub, R. Engl, J. Kurzawa, S. Miller, S. Schneider, A. Stockmann and M. Wasielewski, J. Phys. Chem. A, 2001, 105, 5655–5665 CrossRef CAS.
- Y. Chen, C. Xu, B. Xu, Z. Mao, J. Li, Z. Yang, N. Peethani, C. Liu, G. Shi, F. Gu, Y. Zhang and Z. Chi, Mater. Chem. Front., 2019, 3, 1800–1806 RSC.
- M. Okazaki, Y. Takeda, P. Data, P. Pander, H. Higginbotham, A. Monkman and S. Minakata, Chem. Sci., 2017, 8, 2677–2686 RSC.
- J. Yang, J. Qin, P. Geng, J. Wang, M. Fang and Z. Li, Angew. Chem., Int. Ed., 2018, 57, 14174–14178 CrossRef CAS PubMed.
- N. Duvva, Y. K. Eom, G. Reddy, K. S. Schanze and L. Giribabu, ACS Appl. Energy Mater., 2020, 3, 6758–6767 CrossRef CAS.
- G. Tang, A. A. Sukhanov, J. Zhao, W. Yang, Z. Wang, Q. Liu, V. K. Voronkova, M. Di Donato, D. Escudero and D. Jacquemin, J. Phys. Chem. C, 2019, 123, 30171–30186 CrossRef CAS.
- S. Thokala and S. P. Singh, ACS Omega, 2020, 5, 5608–5619 CrossRef CAS PubMed.
- J.-S. Luo, Z.-Q. Wan and C.-Y. Jia, Chin. Chem. Lett., 2016, 27, 1304–1318 CrossRef CAS.
- A. F. Buene and D. M. Almenningen, J. Mater. Chem. C, 2021, 9, 11974–11994 RSC.
- Z.-S. Huang, H. Meier and D. Cao, J. Mater. Chem. C, 2016, 4, 2404–2426 RSC.
- E. Knovenagel, J. Prakt. Chem., 1914, 89(2), 1 CrossRef.
- F. Kehrmann and J. Steinberg, Ber. Dtsch. Chem. Ges., 1911, 44, 3011–3017 CrossRef.
- Y. Liao, P. Jiang, S. Chen, F. Xiao and G.-J. Deng, RSC Adv., 2013, 3, 18605 RSC.
- C. Dai, X. Sun, X. Tu, L. Wu, D. Zhan and Q. Zeng, Chem. Commun., 2012, 48, 5367 RSC.
- D. Ma, Q. Geng, H. Zhang and Y. Jiang, Angew. Chem., Int. Ed., 2010, 49, 1291–1294 CrossRef CAS PubMed.
- D. H. Yun, H. S. Yoo, Y. S. Park and J. W. Woo, Adv. Mater. Res., 2011, 418–420, 153–158 Search PubMed.
- Y. Wang, J. Yang, M. Fang, Y. Gong, J. Ren, L. Tu, B. Z. Tang and Z. Li, Adv. Funct. Mater., 2021, 31, 2101719 CrossRef CAS.
- (a) M. Sailer, A. W. Franz and T. J. J. Müller, Chem. – Eur. J., 2008, 14, 2602–2614 CrossRef CAS PubMed; (b) J. Doskocz, J. Sołoducho, J. Cabaj, M. Łapkowski, S. Golba and K. Palewska, Electroanalysis, 2007, 19, 1394–1401 CrossRef CAS.
- A. Bieliauskas, V. Martynaitis, V. Getautis, T. Malinauskas, V. Jankauskas, E. Kamarauskas, W. Holzer and A. Šačkus, Tetrahedron, 2012, 68, 3552–3559 CrossRef CAS.
- (a) T.-Y. Wu, M.-H. Tsao, F.-L. Chen, S.-G. Su, C.-W. Chang, H.-P. Wang, Y.-C. Lin, W.-C. Ou-Yang and I.-W. Sun, Int. J. Mater. Sci., 2010, 11, 329–353 CAS; (b) V. Zilinskaite, D. Gudeika, J. V. Grazulevicius and J. Sidaravicius, Mol. Cryst. Liq. Cryst., 2014, 590, 80–89 CrossRef CAS.
- A. C. Schmalz and A. Burger, J. Am. Chem. Soc., 1954, 76, 5455–5459 CrossRef CAS.
- M. Hosseinnezhad, K. Gharanjig, S. Moradian and S. Tafaghodi, Arabian J. Chem., 2019, 12, 2069–2076 CrossRef CAS.
- M. Toşa, C. Paiz, C. Majdik, L. Poppe, P. Kolonits, L. A. Silberg, L. Novák and F.-D. Irimie, Heterocycl. Commun., 2001, 7, 277–282 Search PubMed.
- W.-W. Zhang, Y.-G. Yu, Z.-D. Lu, W.-L. Mao, Y.-Z. Li and Q.-J. Meng, Organometallics, 2007, 26, 865–873 CrossRef CAS.
- G. Zhang, J. Sun, P. Xue, Z. Zhang, P. Gong, J. Peng and R. Lu, J. Mater. Chem. C, 2015, 3, 2925–2932 RSC.
- G. Sang, Y. Zou and Y. Li, J. Phys. Chem. C, 2008, 112, 12058–12064 CrossRef CAS.
- M. Ravivarma, C. Satheeshkumar, S. Ganesan and P. Rajakumar, Mater. Chem. Front., 2017, 1, 2117–2124 RSC.
- Z. Wan, Y. Zan, B. Wang, W. Fang, S. Cui, Y. Teng, F. Zhang, Y. Li, L. Chen and G. Bai, J. Lumin., 2022, 242, 118555 CrossRef CAS.
- Z. Wang, Y. Li, Y. Yang, Y. Chen and H. Wu, Opt. Mater., 2022, 123, 111886 CrossRef CAS.
- C. Qian, Z. Ma, J. Liu, X. Zhang, S. Wang and Z. Ma, Chem. – Asian J., 2021, 16, 3713–3718 CrossRef CAS PubMed.
- N. Hasan, Z. Ma, J. Liu, Z. Li, C. Qian, Y. Liu, M. Chen, H. Jiang, X. Jia and Z. Ma, ChemPhysChem, 2021, 22, 2093–2098 CrossRef CAS PubMed.
- M. Fan, Y. Cheng, B. Fang, L. Lai and M. Yin, Dyes Pigm., 2021, 190, 109311 CrossRef CAS.
- G. Sych, R. Pashazadeh, Y. Danyliv, O. Bezvikonnyi, D. Volyniuk, A. Lazauskas and J. V. Grazulevicius, Chem. – Eur. J., 2021, 27, 2826–2836 CrossRef CAS PubMed.
- Y. Yang, A. Li, Z. Ma, J. Liu, W. Xu, Z. Ma and X. Jia, Dyes Pigm., 2020, 181, 108575 CrossRef CAS.
- T. Zhang, Y. Han, J. Huo and P. Xue, CrystEngComm, 2020, 22, 5137–5144 RSC.
- T. Zhang, Y. Han, K. Wang, M. Liang, W. Bian, Y. Zhang, X. Li, C. Zhang and P. Xue, Dyes Pigm., 2020, 172, 107835 CrossRef CAS.
- T. Zhang, Y. Han, M. Liang, W. Bian, Y. Zhang, X. Li, C. Zhang and P. Xue, Dyes Pigm., 2019, 171, 107692 CrossRef CAS.
- Z. Yang, P. Xue, L. Zhang and P. Chen, New J. Chem., 2019, 43, 14603–14608 RSC.
- S. Wang, L. Li, K. Li, T. Zhang, Z. Zhao and P. Xue, New J. Chem., 2019, 43, 12957–12962 RSC.
- Y. Shen, P. Chen, J. Liu, J. Ding and P. Xue, Dyes Pigm., 2018, 150, 354–362 CrossRef CAS.
- Y. Xi, Y. Cao, Y. Zhu, H. Guo, X. Wu, Y. Li and B. Wang, Chem. Lett., 2018, 47, 650–653 CrossRef CAS.
- C. Arivazhagan, A. Maity, K. Bakthavachalam, A. Jana, S. K. Panigrahi, E. Suresh, A. Das and S. Ghosh, Chem. – Eur. J., 2017, 23, 7046–7051 CrossRef CAS PubMed.
- G. Zhang, J. Sun, P. Xue, Z. Zhang, P. Gong, J. Peng and R. Lu, J. Mater. Chem. C, 2015, 3, 2925–2932 RSC.
- P. Xue, B. Yao, J. Sun, Q. Xu, P. Chen, Z. Zhang and R. Lu, J. Mater. Chem. C, 2014, 2, 3942–3950 RSC.
- P. Xue, B. Yao, X. Liu, J. Sun, P. Gong, Z. Zhang, C. Qian, Y. Zhang and R. Lu, J. Mater. Chem. C, 2015, 3, 1018–1025 RSC.
- M. Okazaki, Y. Takeda, P. Data, P. Pander, H. Higginbotham, A. P. Monkman and S. Minakata, Chem. Sci., 2017, 8, 2677–2686 RSC.
- P. Data, M. Okazaki, S. Minakata and Y. Takeda, J. Mater. Chem. C, 2019, 7, 6616–6621 RSC.
- J. Yang, J. Qin, P. Geng, J. Wang, M. Fang and Z. Li, Angew. Chem., Int. Ed., 2018, 57, 14174–14178 CrossRef CAS PubMed.
- Y. Tian, X. Yang, Y. Gong, Y. Wang, M. Fang, J. Yang, Z. Tang and Z. Li, Sci. China: Chem., 2021, 64, 445–451 CrossRef CAS.
- H. Deng, Z. Yang, G. Li, D. Ma, Z. Xie, W. Li, Z. Mao, J. Zhao, Z. Yang, Y. Zhang and Z. Chi, Chem. Eng. J., 2022, 438, 135519 CrossRef CAS.
- B. S. Shivaji, R. Boddula, A. Saeki and S. P. Singh, J. Mater. Chem. C, 2022, 10, 5173–5182 RSC.
- L. Li, R. Wang, L. Wang and L. Huang, J. Mol. Struct., 2022, 1249, 131596 CrossRef CAS.
- S. Song, P. Zhang, H. Liu, X. Zhu, X. Feng, Z. Zhao and B. Z. Tang, Dyes Pigm., 2021, 196, 109776 CrossRef CAS.
- M. Huang, H. Lu, M. Wang, B. Liu, Z. Ma, Z. Wang and J. Yang, J. Phys. Chem. B, 2021, 125, 11548–11556 CrossRef CAS PubMed.
- M. Chakraborty and M. Chakravarty, Mater. Adv., 2021, 2, 6418–6427 RSC.
- L. Zhang, Y.-F. Wang, M. Li, Q.-Y. Gao and C.-F. Chen, Chin. Chem. Lett., 2021, 32, 740–744 CrossRef CAS.
- Y.-F. Shen, M. Li, W.-L. Zhao, Y.-F. Wang, H.-Y. Lu and C.-F. Chen, Mater. Chem. Front., 2021, 5, 834–842 RSC.
- Y. Yu, M. Cang, W. Cui, L. Xu, R. Wang, M. Sun, H. Zhou, W. Yang and S. Xue, Dyes Pigm., 2021, 184, 108770 CrossRef CAS.
- Y. Lin, Y. Song, Y. Jin, B. Wang and C. Fan, Dyes Pigm., 2020, 183, 108711 CrossRef CAS.
- P. Lei, S. Zhang, N. Zhang, X. Yin, N. Wang and P. Chen, ACS Omega, 2020, 5, 28606–28614 CrossRef CAS PubMed.
- S. Li, T. Cheng, C. Yin, S. Zhou, Q. Fan, W. Wu and X. Jiang, ACS Appl. Mater. Interfaces, 2020, 12, 43466–43473 CrossRef CAS PubMed.
- F. Liu, Y. Tan, H. Liu, X. Tang, L. Gao, C. Du, J. Min, H. Jin and P. Lu, J. Mater. Chem. C, 2020, 8, 6883 RSC.
- S. Li, C. Yin, R. Wang, Q. Fan, W. Wu and X. Jiang, ACS Appl. Mater. Interfaces, 2020, 12, 20281–20286 CrossRef CAS PubMed.
- S. Zheng, T. Liu, Z. Song, Z. He, Z. Yang, H. Wang and Z. Zeng, Dyes Pigm., 2020, 176, 108204 CrossRef CAS.
- A. Ekbote, S. M. Mobin and R. Misra, J. Mater. Chem. C, 2020, 8, 3589–3602 RSC.
- L. Huang, X. Wen, J. Liu, M. Chen, Z. Ma and X. Jia, Mater. Chem. Front., 2019, 3, 2151–2156 RSC.
- B. Huang, W.-C. Chen, Z. Li, J. Zhang, W. Zhao, Y. Feng, B. Z. Tang and C.-S. Lee, Angew. Chem., Int. Ed., 2018, 57, 12473–12477 CrossRef CAS PubMed.
- S. Xiang, Z. Huang, S. Sun, X. Lv, L. Fan, S. Ye, H. Chen, R. Guo and L. Wang, J. Mater. Chem. C, 2018, 6, 11436–11443 RSC.
- K. K. Neena, P. Sudhakar, K. Dipak and P. Thilagar, Chem. Commun., 2017, 53, 3641–3644 RSC.
- R. Keruckiene, N. Kusas, L. Dvylys, E. Skuodis, V. E. Matulis, E. G. Ragoyja, D. A. Lyakhov, I. Klymenko and J. V. Grazulevicius, J. Lumin., 2022, 241, 118502 CrossRef CAS.
- Z. Ma, Z. Yang, L. Mu, L. Deng, L. Chen, B. Wang, X. Qiao, D. Hu, B. Yang, D. Ma, J. Peng and Y. Ma, Chem. Sci., 2021, 12, 14808–14814 RSC.
- M. Gao, Y. Tian, J. Yang, X. Li, M. Fang and Z. Li, J. Mater. Chem. C, 2021, 9, 15375–15380 RSC.
- K. Leitonas, A. Tomkeviciene, G. Baratte, A. Dabuliene, S. M. Punniyakoti, D. Volyniuk and J. V. Grazulevicius, Sens. Actuators, B, 2021, 345, 130369 CrossRef CAS.
- Y. Wang, J. Yang, M. Fang, Y. Gong, J. Ren, L. Tu, B. Z. Tang and Z. Li, Adv. Funct. Mater., 2021, 31, 2101719 CrossRef CAS.
- Y. Tian, J. Yang, Z. Liu, M. Gao, X. Li, W. Che, M. Fang and Z. Li, Angew. Chem., Int. Ed., 2021, 60, 20259–20263 CrossRef CAS PubMed.
- J. Ren, Y. Wang, Y. Tian, Z. Liu, X. Xiao, J. Yang, M. Fang and Z. Li, Angew. Chem., Int. Ed., 2021, 60, 12335–12340 CrossRef CAS PubMed.
- W. Qiu, X. Cai, M. Li, L. Wang, Y. He, W. Xie, Z. Chen, M. Liu and S.-J. Su, J. Mater. Chem. C, 2021, 9, 1378–1386 RSC.
- J. S. Ward, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, J. Santos, F. B. Dias and M. R. Bryce, J. Org. Chem., 2018, 83, 14431–14442 CrossRef CAS PubMed.
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef PubMed.
- R. Huang, J. S. Ward, N. A. Kukhta, J. Avó, J. Gibson, T. Penfold, J. C. Lima, A. S. Batsanov, M. N. Berberan-Santos, M. R. Bryce and F. B. Dias, J. Mater. Chem. C, 2018, 6, 9238–9247 RSC.
- Q. Wang, D. Zheng, Q. Cao, K. Huang and D. Qin, Spectrochim. Acta, Part A, 2021, 261, 120061 CrossRef CAS PubMed.
- Y. Sun, L. Wang, J. Zhou, D. Qin and H. Duan, Appl. Organomet. Chem., 2020, 34, e5945 CAS.
- W. Yang, S. Yang, Q. Guo, T. Zhang, K. Wu and Y. Hu, Sens. Actuators, B, 2015, 213, 404–408 CrossRef CAS.
- B. Sun and L. Liu, Spectrochim. Acta, Part A, 2020, 229, 117939 CrossRef CAS PubMed.
- M. V. Karmegam, S. Karuppannan, D. B. Christopher Leslee, S. Subramanian and S. Gandhi, Luminescence, 2020, 35, 90–97 CrossRef CAS PubMed.
- S. Suganya, E. Ravindran, M. K. Mahato and E. Prasad, Sens. Actuators, B, 2019, 291, 426–432 CrossRef CAS.
- P. Hou, J. Wang, S. Fu, L. Liu and S. Chen, Anal. Bioanal. Chem., 2019, 411, 935–942 CrossRef CAS PubMed.
- H. Feng, Q. Meng, Y. Wang, C. Duan, C. Wang, H. Jia, Z. Zhang and R. Zhang, Chem. – Asian J., 2018, 13, 2611–2618 CrossRef CAS PubMed.
- E. Ramachandran, S. A. A. Vandarkuzhali, G. Sivaraman and R. Dhamodharan, Chem. – Eur. J., 2018, 24, 11042–11050 CrossRef CAS PubMed.
- V. Anand, B. Sadhasivam and R. Dhamodharan, New J. Chem., 2018, 42, 18979–18990 RSC.
- K. M. Vengaian, C. D. Britto, K. Sekar, G. Sivaraman and S. Singaravadivel, Sens. Actuators, B, 2016, 235, 232–240 CrossRef CAS.
- Q. Zou, X. Li, Q. Xu, H. Ågren, W. Zhao and Y. Qu, RSC Adv., 2014, 4, 59809–59816 RSC.
- W. Wang, Y. Zhang, Y. Li and Q. Zhao, Chem. Res. Chin. Univ., 2013, 29, 632–637 CrossRef CAS.
- T. Huang, S. Yan, Y. Yu, Y. Xue, Y. Yu and C. Han, Anal. Chem., 2022, 94, 1415–1424 CrossRef CAS PubMed.
- Y. Hu, Z. Shang, J. Wang, M. Hong, R. Zhang, Q. Meng and Z. Zhang, Analyst, 2021, 146, 7528–7536 RSC.
- X. Bao, X. Cao, Y. Yuan, B. Zhou and C. Huo, Sens. Actuators, B, 2021, 344, 130210 CrossRef CAS.
- L. Liang, Y. Sun, C. Liu, X. Zeng and J. Zhao, Dyes Pigm., 2021, 190, 109344 CrossRef CAS.
- V. Govindasamy, S. Perumal, I. Sekar, B. Madheswaran, S. Karuppannan and S. B. Kuppannan, J. Fluoresc., 2021, 31, 667–674 CrossRef CAS PubMed.
- I. Marghad, F. Bencheikh, C. Wang, S. Manolikakes, A. R érat, C. Gosmini, D. H. Kim, J. C. Ribierre and C. Adachi, RSC Adv., 2019, 9, 4336–4343 RSC.
- S. Xiang, Z. Huang, S. Sun, X. Lv, L. Fan, S. Ye, H. Chen, R. Guo and L. Wang, J. Mater. Chem. C, 2018, 6, 11436–11443 RSC.
- R. Guo, Y. Wang, Z. Huang, Q. Zhang, S. Xiang, S. Ye, W. Liu and L. Wang, J. Mater. Chem. C, 2020, 8, 3705–3714 RSC.
| This journal is © The Royal Society of Chemistry 2023 |