
Novel pyrazolo[3,4-d]pyrimidine derivatives: design, synthesis, anticancer evaluation, VEGFR-2 inhibition, and antiangiogenic activity†
Ahmed M.
Abdelhamed
a,
Rasha A.
Hassan
 *b,
Hanan H.
Kadry
b and
Amira A.
Helwa
*a
*b,
Hanan H.
Kadry
b and
Amira A.
Helwa
*a
aPharmaceutical Organic Chemistry Department, College of Pharmaceutical Sciences and Drug Manufacturing, Misr University for Science and Technology (MUST), 6th of October City, Egypt. E-mail: amira.helwa@must.edu.eg
bPharmaceutical Organic Chemistry Department, Faculty of Pharmacy, Cairo University, Kasr El-Aini Street, Cairo 11562, Egypt. E-mail: rasha.hasan@pharma.cu.edu.eg
First published on 7th November 2023
Abstract
A novel series of 12 pyrazolo[3,4-d]pyrimidine derivatives were created and evaluated in vitro for their antiproliferative activity against the NCI 60 human tumor cell line panel. Compounds 12a–d displayed significant antitumor activity against MDA-MB-468 and T-47D (breast cancer cell lines), especially compound 12b, which exhibited the highest anticancer activity against MDA-MB-468 and T-47D cell lines with IC50 values of 3.343 ± 0.13 and 4.792 ± 0.21 μM, respectively compared to staurosporine with IC50 values of 6.358 ± 0.24 and 4.849 ± 0.22 μM. The most potent cytotoxic derivatives 12a–d were studied for their VEGFR-2 inhibitory activity to explore the mechanism of action of these substances. Compound 12b had potent activity against VEGFR-2 with an IC50 value of 0.063 ± 0.003 μM, compared to sunitinib with IC50 = 0.035 ± 0.012 μM. Moreover, there was an excellent reduction in HUVEC migratory potential that resulted in a significant disruption of wound healing patterns by 23% after 72 h of treatment with compound 12b. Cell cycle and apoptosis investigations showed that compound 12b could stop the cell cycle at the S phase and significantly increase total apoptosis in the MDA-MB-468 cell line by 18.98-fold compared to the control. Moreover, compound 12b increased the caspase-3 level in the MDA-MB-468 cell line by 7.32-fold as compared to the control.
Introduction
Cancer, a global health concern, accounts for millions of deaths annually.1 In 2020, the number of reported cancer cases reached 19.3 million, with nearly 10 million cancer-related deaths recorded.2,3 The development of novel therapies for cancer poses a significant challenge because of its complex and heterogeneous nature. Cancer often exhibits common traits, such as self-proliferation induction, resistance to apoptosis, unlimited proliferative potential, and high invasiveness, which can arise from the hyperactivation of oncogenic pathways and/or disruption of tumor suppressor mechanisms.4–7Protein tyrosine kinases (PTKs) are crucial enzymes that mediate their function by transferring the phosphoryl group from the gamma position of ATP, resulting in the phosphorylation of tyrosine residues in proteins.8 These enzymes can be categorized based on their location as membrane-bound receptor tyrosine kinases (RTKs) or cytoplasmic non-receptor tyrosine kinases (NRTKs).8 PTKs play pivotal roles in regulating various essential cellular processes, including cell proliferation, growth, metabolism, motility, and apoptosis. Dysregulated PTK catalytic activity, often caused by mutations or overexpression, contributes significantly to several clinical diseases, including cancer.9–11 Substantial efforts have been dedicated to elucidating the physiological and pathological functions of receptor protein kinase signal transduction pathways over the past 35 years.8
The receptor tyrosine kinase superfamily includes three subtypes of vascular endothelial growth factor (VEGF) receptors, namely VEGFR-1–3. The extracellular domain of VEGFRs consists of approximately 750 amino acid residues and is organized into seven immunoglobulin (Ig)-like folds. VEGFR-1 and VEGFR-2 play critical roles in physiological and pathological angiogenesis, including tumor angiogenesis.11
The VEGFR signaling pathway holds substantial importance as a therapeutic target for various human malignancies because it exerts significant control over tumor angiogenesis. VEGFR-2 is a crucial target for angiogenesis. Inhibition of the VEGF/VEGFR signaling pathway has shown promising potential for impeding tumor angiogenesis and tumor growth.12 VEGFR-2 resembles a typical tyrosine kinase receptor and comprises an extracellular ligand-binding domain, a transmembrane domain, and a tyrosine kinase domain.13
VEGFR-2 inhibitors can be classified into three groups based on their binding positions to the receptor. Sunitinib (Fig. 1) belongs to the category of type I inhibitors, which competitively bind to the ATP-binding pocket in the active “DFG-in” conformation. It uses hydrogen bonds in the hinge region and hydrophobic interactions in the adenine region to establish binding interactions.14 Sorafenib (Fig. 1) is a type II inhibitor that occupies the inactive “DFG-out” conformation of the ATP-binding site and accesses the hydrophobic back pocket.14–17 Additionally, type III inhibitors bind to an inactive “DFG-out” conformation outside the ATP pocket. Recently, numerous VEGFR-2 inhibitors have been developed and used in cancer treatment. Notably, sunitinib and sorafenib have been approved for various types of cancer.18–20 Sunitinib, an indolinone-category kinase inhibitor, was first introduced on the market. It is prescribed to treat imatinib-resistant gastrointestinal stromal tumors and renal cell carcinoma.21 Sunitinib inhibits VEGFR-2 by binding to its kinase domain.22 Sunitinib and various VEGFR-2 inhibitors have been studied regarding their structure–activity relationship (SAR) and binding patterns. Studies have revealed four key characteristics of these inhibitors.23–25 First, most inhibitors exhibit an aromatic flat ring structure, occupying the ATP-binding domain and engaging in hydrogen bond interactions (shown in blue) with the Cys919 residues in the hinge region. In particular, sunitinib forms additional hydrogen bonds with Glu917.21 Second, an aryl ring acts as a hydrophobic spacer, occupying the region between the ATP-binding and DFG domains (shown in green). Third, VEGFR-2 inhibitors possess a linker that accepts and donates hydrogen bonds (HBA–HBD), establishing interactions (shown in red) with the DFG moiety residues (Glu885 and Asp1046). Finally, these inhibitors possess a terminal hydrophobic group that interacts with the allosteric hydrophobic pocket through multiple hydrophobic interactions (shown in brown).
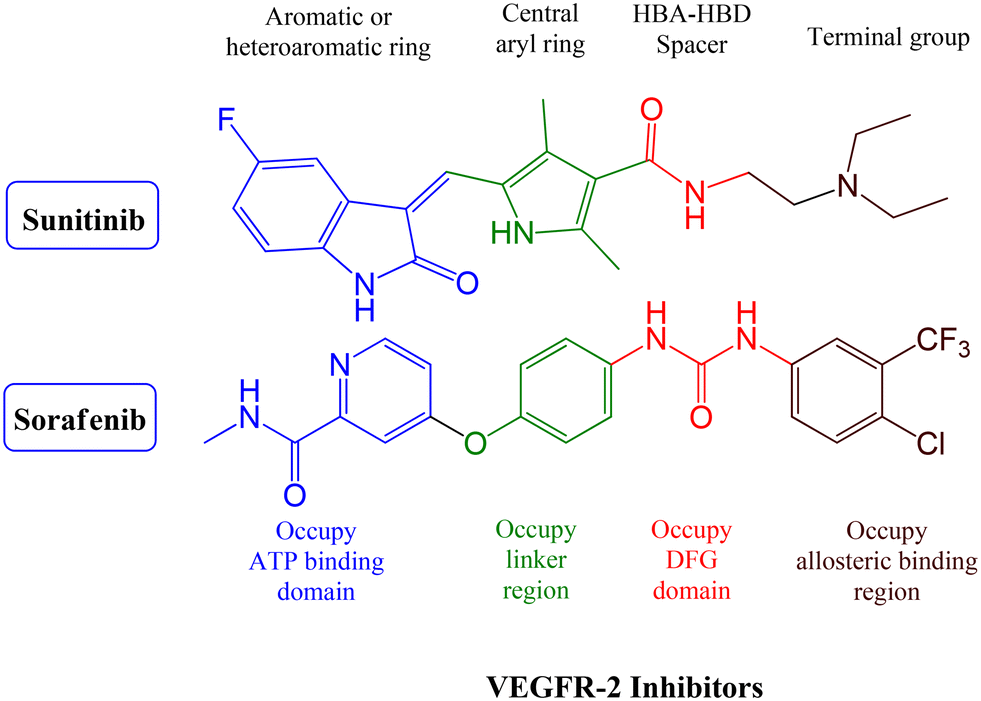 | ||
| Fig. 1 Inhibitors of VEGFR-2 and their pharmacophoric properties. | ||
Developing novel anticancer medications, including kinase inhibitors, with improved tumor selectivity, efficacy, and safety remains a crucial objective in research. Among the isosteres of the purine nuclei, the pyrazolo[3,4-d]pyrimidine scaffold is widely recognized as one of the most desirable heterocycles for drug discovery, particularly in cancer therapy. Extensive investigations have revealed diverse pharmacological properties associated with this scaffold,26,27 with notable attributes of potent anticancer activity.28–30 Previous reports suggest that this scaffold exerts its anticancer effects through various distinct mechanisms, such as inhibition of cyclin-dependent kinases (CDK)31–33 and the epidermal growth factor receptor (EGFR).34
The synthesized pyrazolo[3,4-d]pyrimidine compounds demonstrated significant potential as anticancer agents and have been identified as promising lead compounds for future cancer chemotherapy drugs. Fig. 2 illustrates a selection of both marketed VEGFR-2 inhibitors (sunitinib and sorafenib) and previously published pyrazolo[3,4-d]pyrimidine derivatives (I–VI), which have exhibited promising anticancer activity and VEGFR-2 inhibitory effects.29,35–39
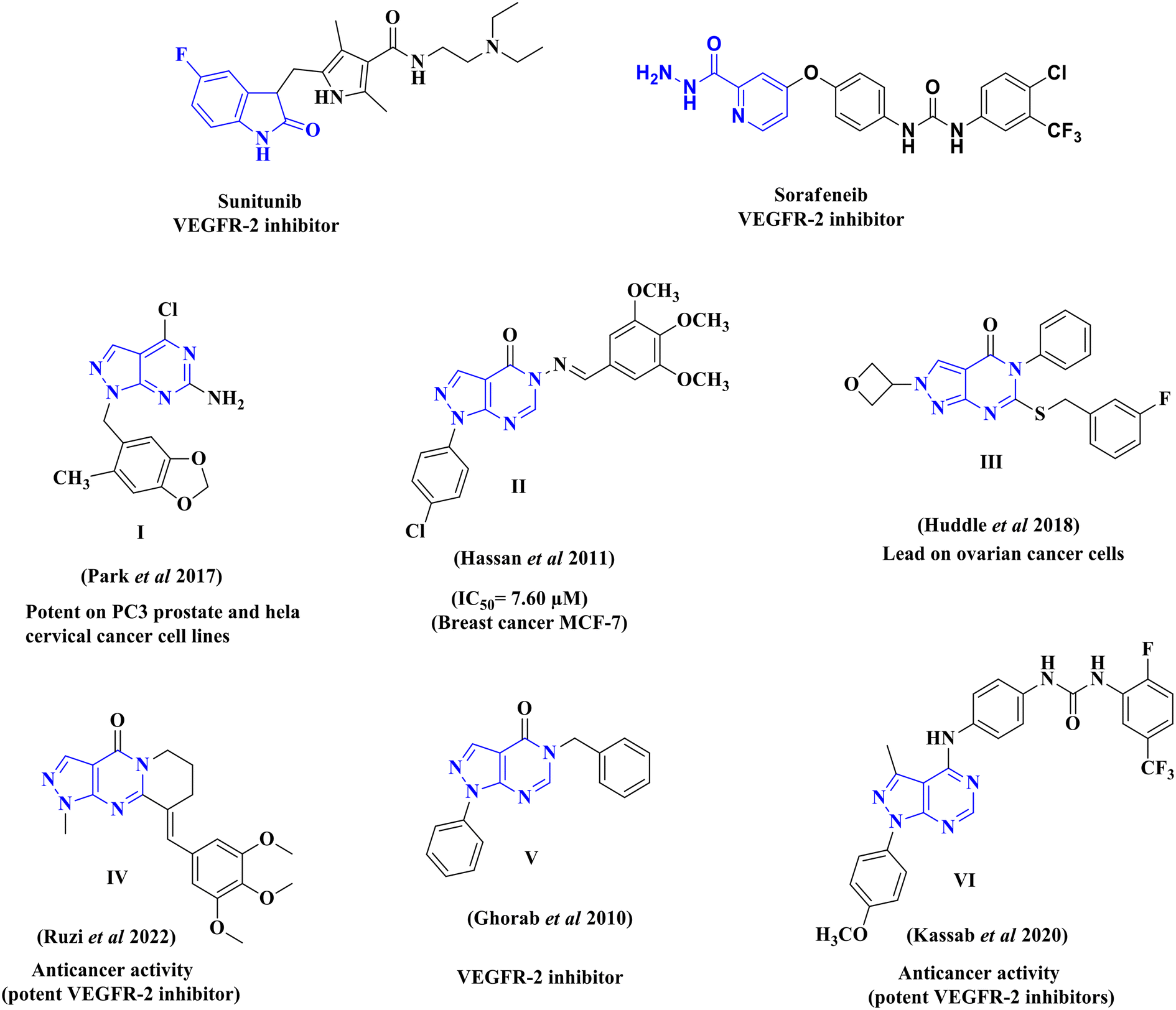 | ||
| Fig. 2 Structures of reported pyrazolo[3,4-d]pyrimidine derivatives, and some VEGFR-2 inhibitors with cytotoxic activity. | ||
Building upon these findings and building on our previous research on anticancer derivatives,40–47 specifically focusing on VEGFR-2 inhibitors,48–53 the main objective of this study was to synthesize novel derivatives of pyrazolo[3,4-d]pyrimidine with anticancer activity, while possessing the fundamental pharmacophoric properties found in first-generation VEGFR-2 inhibitors such as sunitinib, through ring variation and bio isosteric replacement (Fig. 3). Subsequently, the anticancer activities of all the synthesized derivatives were evaluated using the NCI (USA) 60-panel cell line. The most potent derivatives were further assessed using the MTT assay against two breast cancer cell lines, MDA-MB-468 and T47D, and a normal breast cell line, MCF-10a. Furthermore, this study evaluated the in vitro VEGFR-2 inhibition of the most promising derivatives. Additionally, a wound healing test was conducted to assess the antiangiogenic properties of the most potent derivatives. Furthermore, the impact on the normal cell cycle profile, caspase-3 activity, and induction of apoptosis in the breast cancer cell line MDA-MB-468 was investigated. Finally, a molecular docking study was performed to validate the binding mode of the most active derivatives within the VEGFR-2 binding site.
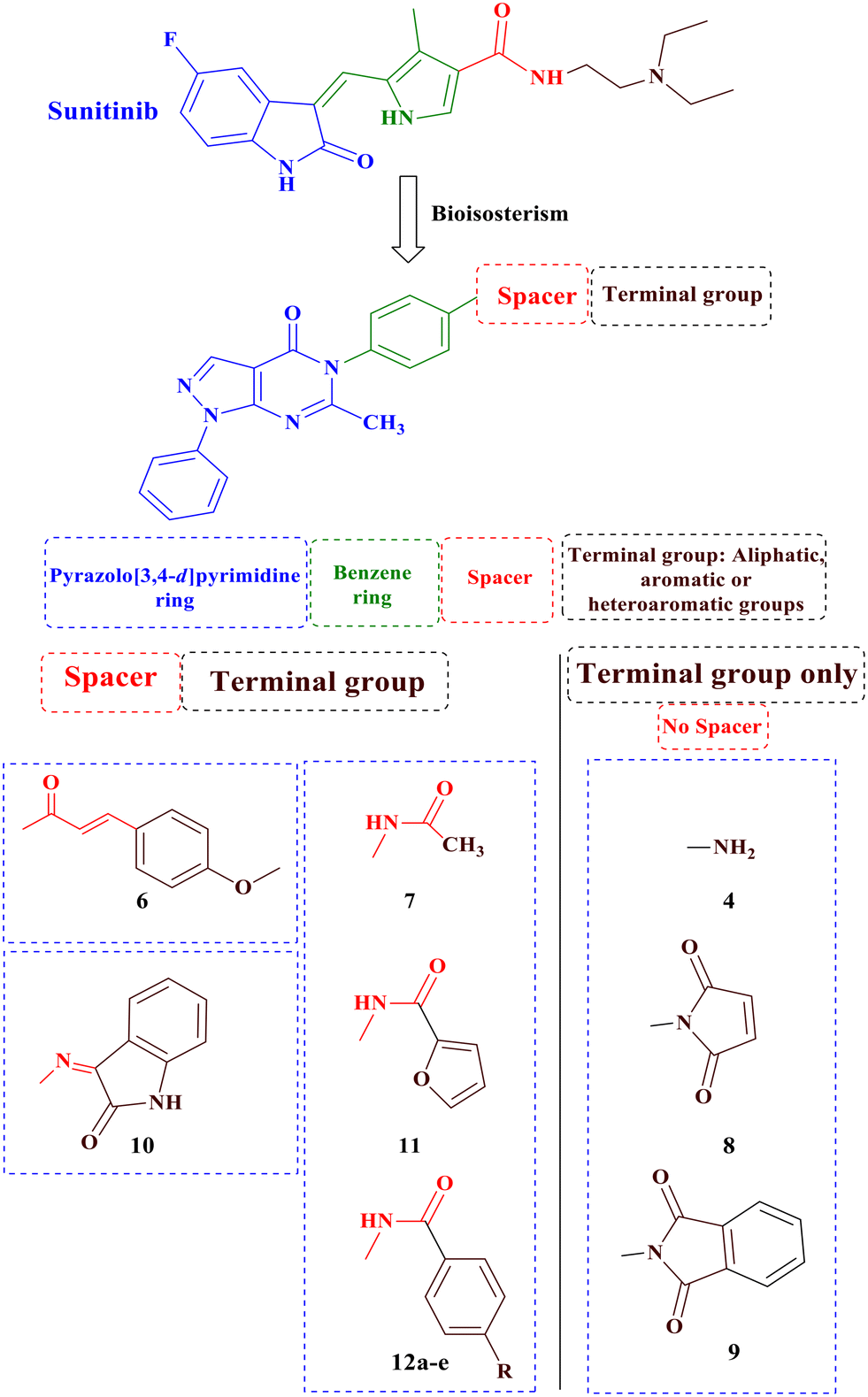 | ||
| Fig. 3 Rationale design of the novel pyrazolo[3,4-d]pyrimidine as VEGFR-2 inhibitors comparative to sunitinib's pharmacophoric properties. | ||
In the designed compounds, the indolinone scaffold of sunitinib was substituted for a pyrazolo[3,4-d]pyrimidine core. Furthermore, the terminal N,N,N-triethylamine group of sunitinib was replaced with aliphatic, aromatic, and heterocyclic moieties. Additionally, some compounds, such as compounds 7, 11, and 12a–e, retained the amide spacer present in sunitinib, whereas others had the amide spacer removed, as seen in compounds 4, 8, and 9. Alternative spacers, including an azomethine group in derivative 10 and an α, β-unsaturated carbonyl chain in derivative 6, were introduced (Fig. 3).
Experimental section
Chemistry
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1600 (CN), 1560 (CC). 1H NMR (400 MHz, DMSO-d6) δ; 8.31 (s, 1H, CH pyrazole), 8.09 (d, J = 7.6 Hz, 2H, ArH), 7.58 (t, J = 7.6 Hz, 2H, ArH), 7.41 (t, J = 7.6 Hz, 1H, ArH), 6.97 (d, J = 8.4 Hz, 2H, ArH), 6.68 (d, J = 8.4 Hz, 2H, ArH), 5.44 (s, 2H, NH2, D2O exchangeable), 2.20 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ; 160.6, 158.3, 151.1, 149.7, 138.8, 136.7, 129.7 (2C), 129.0 (2C), 127.4, 125.6, 122.0 (2C), 114.6 (2C), 106.0, 25.2; EI-MS: m/z = 317 [M+] (100%); Anal. Calcd. for C18H15N5O: C, 68.13; H, 4.76; N, 22.07; found C, 67.91; H, 4.89; N, 21.98.
O), 1593 (CN), 1508 (CC), 1H NMR (400 MHz, DMSO-d6) δ; 8.32 (s, 1H, CH pyrazole), 8.19 (d, J = 8.8 Hz, 2H, ArH), 7.93 (d, J = 8.8 Hz, 2H, ArH), 7.88–7.82 (m, 2H, ArH), 7.72 (d, J = 15.6 Hz, 2H, CHCH), 7.59–7.51 (m, 4H, ArH), 7.46–7.42 (m, 1H, ArH), 7.03 (d, J = 8.8 Hz, 2H, ArH), 3.83 (s, 3H, OCH3), 1.97 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ; 187.9, 170.6, 161.7, 161.0, 144.0, 143.8, 140.1, 138.8, 133.0, 132.3, 131.2 (2C), 130.1 (2C), 129.6 (2C), 128.4, 127.9 (2C), 124.0 (2C), 120.0, 119.5, 115.0 (2C), 114.9, 55.8, 23.4; EI-MS: m/z = 462 [M+] (1.51%); Anal. Calcd. for C28H22N4O3: C, 72.71; H, 4.79; N, 12.11; found C, 72.86; H, 4.96; N, 12.32.
O), 1600 (CN), 1543 (CC). 1H NMR (400 MHz, DMSO-d6) δ; 10.18 (s, 1H, NH, D2O exchangeable), 8.35 (s, 1H, CH pyrazole), 8.10 (d, J = 8.0 Hz, 2H, ArH), 7.75 (d, J = 8.8 Hz, 2H, ArH), 7.59 (t, J = 8.0 Hz, 2H, ArH), 7.42 (t, J = 7.4 Hz, 1H, ArH), 7.34 (d, J = 8.8 Hz, 2H, ArH), 2.19 (s, 3H, CH3), 2.10 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ; 169.3, 159.8, 158.1, 151.1, 140.2, 138.7, 136.7, 132.5, 129.7 (2C), 129.2 (2C), 127.6, 122.2 (2C), 120.2 (2C), 105.9, 25.1, 24.4; EI-MS: m/z = 359 [M+] (46.62%); Anal. Calcd. for C20H17N5O2: C, 66.84; H, 4.77; N, 19.49; found C, 67.12; H, 4.89; N, 19.72.
O), 1566 (CN), 1508 (CC). 1H NMR (400 MHz, DMSO-d6) δ; 8.39 (s, 1H, CH pyrazole), 8.11 (d, J = 7.4 Hz, 2H, ArH), 7.62–7.55 (m, 6H, ArH), 7.43 (t, J = 7.4 Hz, 1H, ArH), 7.26 (s, 2H, ArH), 2.22 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 170.2 (2C), 159.4, 158.0, 151.2, 138.7, 137.0, 136.8, 135.3 (2C), 132.6, 129.8 (2C), 129.6 (2C), 128.1, 127.6 (2C), 122.1 (2C), 105.9, 25.3; EI-MS: m/z = 397 [M+] (100%); Anal. Calcd. for C22H15N5O3: C, 66.49; H, 3.80; N, 17.62; found C, 66.72; H, 4.01; N, 17.85.
O), 1600 (CN), 1508 (CC). 1H NMR (400 MHz, DMSO-d6) δ; 8.36 (s, 1H, CH pyrazole), 8.12–8.10 (m, 2H, ArH), 8.03–7.94 (m, 2H, ArH), 7.92–7.87 (m, 2H, ArH), 7.71–7.68 (m, 1H, ArH), 7.64–7.58 (m, 4H, ArH), 7.45–7.42 (m, 1H, ArH), 7.39 (d, J = 8.8 Hz, 1H, ArH), 2.24 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ; 168.1, 167.8, 167.3, 159.8, 158.1, 151.1, 138.8, 136.8, 132.0, 129.8 (4C), 129.2, 128.8, 127.6, 124.0 (2C), 122.1 (4C), 120.7 (2C), 105.9, 25.3. EI-MS: m/z = 447 [M+] (5.46%); Anal. Calcd. for C26H17N5O3: C, 69.79; H, 3.83; N, 15.65; found C, 69.57; H, 3.98; N, 15.87.
O), 1600 (CN), 1500 (CC). 1H NMR (400 MHz, DMSO-d6) δ; 11.14 (s, 1H, NH, D2O exchangeable), 8.39 (s, 1H, CH pyrazole), 8.12 (d, J = 8.0 Hz, 2H, ArH), 7.61 (t, J = 8.0 Hz, 2H, ArH), 7.54 (d, J = 8.4 Hz, 1H, ArH), 7.46–7.36 (m, 3H, ArH), 7.19 (d, J = 8.4 Hz, 2H, ArH), 6.96 (d, J = 7.6 Hz, 1H, ArH), 6.75 (t, J = 7.6 Hz, 1H, ArH), 6.44 (d, J = 7.6 Hz, 1H, ArH), 2.23 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 163.8, 159.7, 158.1, 156.1, 151.6, 151.2, 147.7, 138.7, 136.8, 135.2, 134.6, 130.4 (2C), 129.8 (2C), 127.6, 126.0, 122.2 (2C), 118.9 (2C), 116.2, 112.2, 106.0, 96.8, 25.2; EI-MS: m/z = 446 [M+] (11.97%); Anal. Calcd. for C26H18N6O2: C, 69.95; H, 4.06; N, 18.82; found C, 70.18; H, 4.24; N, 19.08.
O), 1566 (CN), 1508 (CC). 1H NMR (400 MHz, DMSO-d6) δ; 10.57 (s, 1H, NH, D2O exchangeable), 8.36 (s, 1H, CH pyrazole), 8.11 (d, J = 7.6 Hz, 2H, ArH), 8.03 (d, 2H, ArH), 7.97 (d, J = 8.8 Hz, 2H, ArH), 7.65 (d, J = 8.8 Hz, 2H, ArH), 7.59 (t, J = 8.8 Hz, 2H, ArH), 7.42 (d, J = 8.8 Hz, 2H, ArH), 2.23 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ; 165.2, 159.7, 158.1, 151.1, 140.0, 138.7, 137.1, 136.8, 133.9, 133.2, 130.2, 130.0, 129.8, 129.2, 129.0, 128.9, 128.8, 127.6, 122.1, 121.6, 121.2, 105.9, 25.2; EI-MS: m/z = 412 [M+ + H] (0.73%); Anal. Calcd. for C23H17N5O: C, 67.15; H, 4.17; N, 17.02; found C, 67.43; H, 4.25; N, 17.31.
O), 1600 (CN), 1560 (CC). 1H NMR (400 MHz, DMSO-d6) δ; 8.31 (s, 1H, CH pyrazole), 8.09 (d, J = 7.6 Hz, 2H, ArH), 7.58 (t, J = 7.6 Hz, 2H, ArH), 7.41 (t, J = 7.6 Hz, 1H, ArH), 6.97 (d, J = 8.4 Hz, 2H, ArH), 6.68 (d, J = 8.4 Hz, 2H, ArH), 5.44 (s, 2H, NH2, D2O exchangeable), 2.20 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ; 160.6, 158.3, 151.1, 149.7, 138.8, 136.7, 129.7 (2C), 129.0 (2C), 127.4, 125.6, 122.0 (2C), 114.6 (2C), 106.0, 25.2; EI-MS: m/z = 317 [M+] (100%); Anal. Calcd. for C18H15N5O: C, 68.13; H, 4.76; N, 22.07; found C, 67.91; H, 4.89; N, 21.98.
O), 1593 (CN), 1508 (CC), 1H NMR (400 MHz, DMSO-d6) δ; 8.32 (s, 1H, CH pyrazole), 8.19 (d, J = 8.8 Hz, 2H, ArH), 7.93 (d, J = 8.8 Hz, 2H, ArH), 7.88–7.82 (m, 2H, ArH), 7.72 (d, J = 15.6 Hz, 2H, CHCH), 7.59–7.51 (m, 4H, ArH), 7.46–7.42 (m, 1H, ArH), 7.03 (d, J = 8.8 Hz, 2H, ArH), 3.83 (s, 3H, OCH3), 1.97 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ; 187.9, 170.6, 161.7, 161.0, 144.0, 143.8, 140.1, 138.8, 133.0, 132.3, 131.2 (2C), 130.1 (2C), 129.6 (2C), 128.4, 127.9 (2C), 124.0 (2C), 120.0, 119.5, 115.0 (2C), 114.9, 55.8, 23.4; EI-MS: m/z = 462 [M+] (1.51%); Anal. Calcd. for C28H22N4O3: C, 72.71; H, 4.79; N, 12.11; found C, 72.86; H, 4.96; N, 12.32.
O), 1600 (CN), 1543 (CC). 1H NMR (400 MHz, DMSO-d6) δ; 10.18 (s, 1H, NH, D2O exchangeable), 8.35 (s, 1H, CH pyrazole), 8.10 (d, J = 8.0 Hz, 2H, ArH), 7.75 (d, J = 8.8 Hz, 2H, ArH), 7.59 (t, J = 8.0 Hz, 2H, ArH), 7.42 (t, J = 7.4 Hz, 1H, ArH), 7.34 (d, J = 8.8 Hz, 2H, ArH), 2.19 (s, 3H, CH3), 2.10 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ; 169.3, 159.8, 158.1, 151.1, 140.2, 138.7, 136.7, 132.5, 129.7 (2C), 129.2 (2C), 127.6, 122.2 (2C), 120.2 (2C), 105.9, 25.1, 24.4; EI-MS: m/z = 359 [M+] (46.62%); Anal. Calcd. for C20H17N5O2: C, 66.84; H, 4.77; N, 19.49; found C, 67.12; H, 4.89; N, 19.72.
O), 1566 (CN), 1508 (CC). 1H NMR (400 MHz, DMSO-d6) δ; 8.39 (s, 1H, CH pyrazole), 8.11 (d, J = 7.4 Hz, 2H, ArH), 7.62–7.55 (m, 6H, ArH), 7.43 (t, J = 7.4 Hz, 1H, ArH), 7.26 (s, 2H, ArH), 2.22 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 170.2 (2C), 159.4, 158.0, 151.2, 138.7, 137.0, 136.8, 135.3 (2C), 132.6, 129.8 (2C), 129.6 (2C), 128.1, 127.6 (2C), 122.1 (2C), 105.9, 25.3; EI-MS: m/z = 397 [M+] (100%); Anal. Calcd. for C22H15N5O3: C, 66.49; H, 3.80; N, 17.62; found C, 66.72; H, 4.01; N, 17.85.
O), 1600 (CN), 1508 (CC). 1H NMR (400 MHz, DMSO-d6) δ; 8.36 (s, 1H, CH pyrazole), 8.12–8.10 (m, 2H, ArH), 8.03–7.94 (m, 2H, ArH), 7.92–7.87 (m, 2H, ArH), 7.71–7.68 (m, 1H, ArH), 7.64–7.58 (m, 4H, ArH), 7.45–7.42 (m, 1H, ArH), 7.39 (d, J = 8.8 Hz, 1H, ArH), 2.24 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ; 168.1, 167.8, 167.3, 159.8, 158.1, 151.1, 138.8, 136.8, 132.0, 129.8 (4C), 129.2, 128.8, 127.6, 124.0 (2C), 122.1 (4C), 120.7 (2C), 105.9, 25.3. EI-MS: m/z = 447 [M+] (5.46%); Anal. Calcd. for C26H17N5O3: C, 69.79; H, 3.83; N, 15.65; found C, 69.57; H, 3.98; N, 15.87.
O), 1600 (CN), 1500 (CC). 1H NMR (400 MHz, DMSO-d6) δ; 11.14 (s, 1H, NH, D2O exchangeable), 8.39 (s, 1H, CH pyrazole), 8.12 (d, J = 8.0 Hz, 2H, ArH), 7.61 (t, J = 8.0 Hz, 2H, ArH), 7.54 (d, J = 8.4 Hz, 1H, ArH), 7.46–7.36 (m, 3H, ArH), 7.19 (d, J = 8.4 Hz, 2H, ArH), 6.96 (d, J = 7.6 Hz, 1H, ArH), 6.75 (t, J = 7.6 Hz, 1H, ArH), 6.44 (d, J = 7.6 Hz, 1H, ArH), 2.23 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 163.8, 159.7, 158.1, 156.1, 151.6, 151.2, 147.7, 138.7, 136.8, 135.2, 134.6, 130.4 (2C), 129.8 (2C), 127.6, 126.0, 122.2 (2C), 118.9 (2C), 116.2, 112.2, 106.0, 96.8, 25.2; EI-MS: m/z = 446 [M+] (11.97%); Anal. Calcd. for C26H18N6O2: C, 69.95; H, 4.06; N, 18.82; found C, 70.18; H, 4.24; N, 19.08.
O), 1566 (CN), 1508 (CC). 1H NMR (400 MHz, DMSO-d6) δ; 10.57 (s, 1H, NH, D2O exchangeable), 8.36 (s, 1H, CH pyrazole), 8.11 (d, J = 7.6 Hz, 2H, ArH), 8.03 (d, 2H, ArH), 7.97 (d, J = 8.8 Hz, 2H, ArH), 7.65 (d, J = 8.8 Hz, 2H, ArH), 7.59 (t, J = 8.8 Hz, 2H, ArH), 7.42 (d, J = 8.8 Hz, 2H, ArH), 2.23 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ; 165.2, 159.7, 158.1, 151.1, 140.0, 138.7, 137.1, 136.8, 133.9, 133.2, 130.2, 130.0, 129.8, 129.2, 129.0, 128.9, 128.8, 127.6, 122.1, 121.6, 121.2, 105.9, 25.2; EI-MS: m/z = 412 [M+ + H] (0.73%); Anal. Calcd. for C23H17N5O: C, 67.15; H, 4.17; N, 17.02; found C, 67.43; H, 4.25; N, 17.31.
N-(4-(6-Methyl-4-oxo-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-5(4H)-yl)phenyl)benzamide (12a). Gray solid: 90% yield; m.p. 308–310 °C; IR (KBr, cm−1) 3356 (NH), 3062, 3005 (CH aromatic), 2970, 2916 (CH aliphatic), 1705, 1659 (2 C
O), 1550 (CN), 1508 (CC). 1H NMR (400 MHz, DMSO-d6) δ; 10.50 (s, 1H, NH, D2O exchangeable), 8.37 (s, 1H, CH pyrazole), 8.11 (d, J = 8.0 Hz, 2H, ArH), 7.99 (t, J = 7.6 Hz, 4H, ArH), 7.65–7.55 (m, 5H, ArH), 7.45–7.41 (m, 3H, ArH), 2.23 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 166.4, 159.8, 158.1, 151.2, 140.2, 138.7, 136.8, 135.2, 133.1, 132.2, 129.8 (2C), 129.2 (2C), 128.9 (2C), 128.2 (2C), 127.6, 122.2 (2C), 121.5 (2C), 105.9, 25.2; EI-MS: m/z = 421 [M+] (1.55%); Anal. Calcd. for C25H19N5O2: C, 71.25; H, 4.54; N, 16.62; found C, 70.98; H, 4.63; N, 16.89.
4-Fluoro-N-(4-(6-methyl-4-oxo-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-5(4H)-yl)phenyl)benzamide (12b). Gray solid: 50% yield; m.p. 310–312 °C; IR (KBr, cm−1) 3298 (NH), 3051, 3001 (CH aromatic), 2932, 2900 (CH aliphatic), 1694, 1670 (2 C
O), 1566 (CN), 1504 (CC). 1H NMR (400 MHz, DMSO-d6) δ; 10.50 (s, 1H, NH, D2O exchangeable), 8.36 (s, 1H, CH pyrazole), 8.12–8.06 (m, 4H, ArH), 7.96 (d, J = 8.4 Hz, 2H, ArH), 7.60 (t, J = 8.0 Hz, 2H, ArH), 7.45–7.37 (m, 5H, ArH), 2.23 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ; 165.3, 163.4, 159.7, 158.2, 151.1, 140.0, 138.6, 136.7, 133.1, 131.6, 131.0, 130.9, 129.7 (2C), 129.1(2C), 127.6, 122.2 (2C), 121.7 (2C), 116.0, 115.8, 105.8, 25.1; EI-MS: m/z = 439 [M+] (73.26%); Anal. Calcd. for C25H18FN5O2: C, 68.33; H, 4.13; N, 15.94; found C, 68.56; H, 4.29; N, 16.12.
4-Chloro-N-(4-(6-methyl-4-oxo-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-5(4H)-yl)phenyl)benzamide (12c). Gray solid: 46% yield; m.p. 303–305 °C; IR (KBr, cm−1) 3294 (NH), 3043, 3012 (CH aromatic), 2978, 2924 (CH aliphatic), 1690, 1651 (2 C
O), 1550 (CN), 1508 (CC). 1H NMR (400 MHz, DMSO-d6) δ; 10.58 (s, 1H, NH, D2O exchangeable), 8.36 (s, 1H, CH pyrazole), 8.10 (d, J = 7.6 Hz, 2H, ArH), 7.98 (d, J = 8.8 Hz, 4H, ArH), 7.60 (t, J = 8.0 Hz, 2H, ArH), 7.51 (d, J = 3.2 Hz, 1H, ArH), 7.44–7.37 (m, 3H, ArH), 6.74–6.73 (m, 1H, ArH), 2.22 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) 159.8, 158.1, 156.9, 151.1, 147.7, 146.4, 139.6, 138.7, 136.8, 133.1, 129.8 (4C), 129.1 (2C), 127.6, 122.2 (2C), 121.6 (2C), 115.8, 112.7, 105.9, 25.2; EI-MS: m/z = 455 [M+] (5.62%); Anal. Calcd. for: C25H18ClN5O2: C, 65.86; H, 3.98; N, 15.36; found C, 66.04; H, 4.13; N, 15.62.
4-Methyl-N-(4-(6-methyl-4-oxo-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-5(4H)-yl)phenyl)benzamide (12d). Gray solid: 55% yield; m.p. 306–308 °C; IR (KBr, cm−1) 3337 (NH), 3060, 3032 (CH aromatic), 2974, 2947 (CH aliphatic), 1686, 1647 (2 C
O), 1570 (CN), 1519 (CC). 1H NMR (400 MHz, DMSO-d6) δ; 10.41 (s, 1H, NH, D2O exchangeable), 8.36 (s, 1H, CH pyrazole), 8.11 (d, J = 7.2 Hz, 2H, ArH), 7.98 (d, J = 7.6 Hz, 2H, ArH), 7.90 (t, J = 8.8 Hz, 2H, ArH), 7.60 (t, J = 8.0 Hz, 2H, ArH), 7.41–7.33 (m, 5H, ArH), 2.40 (s, 3H, CH3), 2.23 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ; 165.6, 160.0, 151.5, 141.9, 140.0, 138.5, 136.8, 135.4, 132.5, 129.7, 129.43, 129.35, 129.1, 128.2, 128.1, 127.5, 122.1, 121.6, 121.1 (4C), 105.0, 25.2, 21.5; EI-MS: m/z = 435 [M+] (9.77%); Anal. Calcd. for C26H21N5O2: C, 71.71; H, 4.86; N, 16.08; found C, 71.53; H, 4.97; N, 16.29.
N-(4-(6-Methyl-4-oxo-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-5(4H)-yl)phenyl)-4-nitrobenzamide (12e). Gray solid: 46% yield; m.p. 298–300 °C; IR (KBr, cm−1) 3306 (NH), 3101, 3062 (CH aromatic), 2924, 2858 (CH aliphatic), 1674 broad peak (2 C
O), 1597 (CN), 1508 (CC). 1H NMR (400 MHz, DMSO-d6) δ; 10.80 (s, 1H, NH, D2O exchangeable), 8.41 (d, J = 8.8 Hz, 2H, ArH), 8.36 (s, 1H, CH pyrazole), 8.23 (d, J = 8.8 Hz, 2H, ArH), 8.11 (d, J = 7.6 Hz, 2H, ArH), 7.98 (d, J = 8.4 Hz, 2H, ArH), 7.60 (t, J = 7.6 Hz, 2H, ArH), 7.46–7.41 (m, 3H, ArH), 2.23 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 164.8, 159.7, 158.2, 151.1, 149.7, 140.8, 139.6, 138.6, 136.7, 133.6, 129.75, 129.69, 129.6, 129.3, 127.6, 124.1 (2C), 122.2 (2C), 121.8 (2C), 121.4 (2C), 105.8, 25.1; EI-MS: m/z = 466 [M+] (8.85%); Anal. Calcd. for: C25H18N6O4: C, 64.37; H, 3.89; N, 18.02; found C, 64.59; H, 4.02; N, 18.19.
Biological evaluation
The biological assays were performed according to the documented protocols, which can be found in the ESI.† The assays included antiproliferative activity screening by NCI,57–61 MTT assay,62in vitro VEGFR-2 inhibitory evaluation,51 wound healing test,49 cell cycle analysis,63 apoptosis evaluation,64 and caspase-3 enzyme evaluation.30Molecular modeling studies
The X-ray crystallographic structure of VEGFR-2 co-crystallized with sunitinib (PDB ID: 4AGD)65 was obtained from the Protein Data Bank (PDB) (https://www.Rcsb.Org/Structure/4AGD). Molecular docking was conducted using the Molecular Operating Environment software (MOE, version 2015.10). The receptor was prepared for docking using the Protonate 3D protocol in MOE with default settings. Sunitinib was initially docked to the VEGFR-2 active site to validate the docking protocol. Once the protocol was validated, the newly synthesized compounds 12a–d were examined for their interactions with the VEGFR-2 active site, and their binding patterns were predicted. The compounds were built using the MOE builder, and their structures were energy-minimized using the MMFF94x force field. The Triangle Matcher placement method and London dG scoring function were employed for the docking process.Results and discussion
Chemistry
Twelve pyrazolo[3,4-d]pyrimidine derivatives were synthesized in several stages, as illustrated in Schemes 1 and 2. The newly prepared compounds were characterized by IR, 1H NMR, 13C NMR, mass spectroscopy and elemental analyses. The synthesis began with the reaction of ethyl 2-cyano-3-ethoxypropanoate and phenylhydrazine in absolute ethanol, resulting in the formation of ethyl 5-amino-1-phenyl-1H-pyrazole-4-carboxylate (1).54 Subsequent basic hydrolysis of compound 1 yielded 5-amino-1-phenyl-1H-pyrazole-4-carboxylic acid (2),55 which was further refluxed in acetic anhydride to obtain the key intermediate, 6-methyl-1-phenylpyrazolo[3,4-d][1,3]oxazin-4(1H)-one (3).56 Compound 3 was then used in the reaction with p-phenylenediamine and p-aminoacetophenone, leading to the formation of the corresponding 5-(4-substituted phenyl)-1H-pyrazolo[3,4-d]pyrimidine-4-one derivatives, namely compounds 4 and 5, respectively.56 The presence of NH2 groups in compound 4 was confirmed by the peaks observed at 3447 and 3420 cm−1 in the IR spectrum. In the 1H NMR spectrum of compound 4, a singlet signal at δ 5.44 ppm was observed, which could be attributed to the presence of an NH2 group that underwent exchange with D2O, indicating its presence. Furthermore, the mass spectrum of compound 4 showed its molecular ion peak [M+], which also corresponds to its base peak at m/z 317.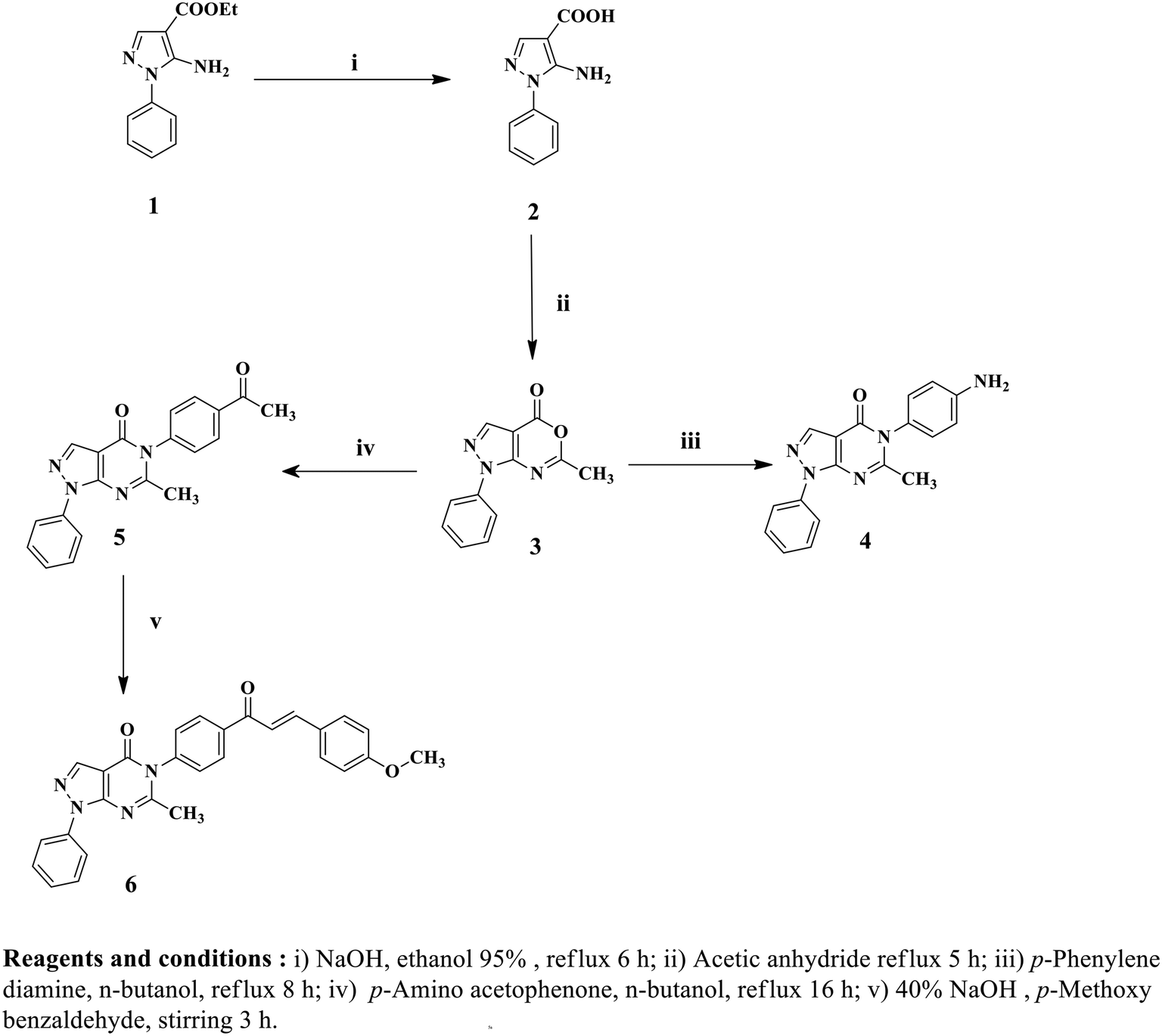 | ||
| Scheme 1 Synthetic pathways of pyrazolopyrimidine derivatives 4–6. | ||
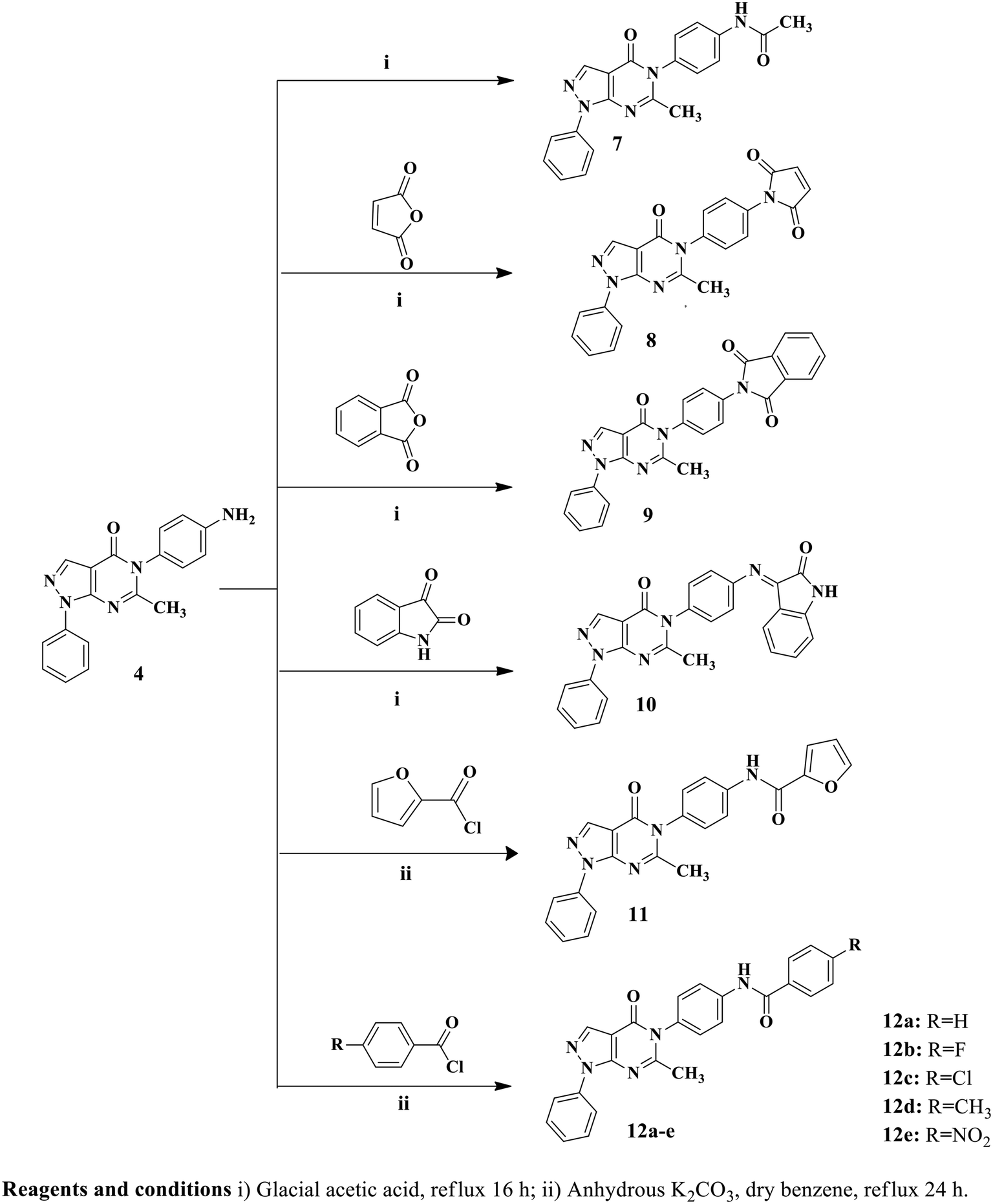 | ||
| Scheme 2 Synthetic pathways of pyrazolopyrimidine derivatives 7–11, and 12a–e. | ||
Furthermore, 5-(4-(3-(4-methoxyphenyl)acryloyl)phenyl)-6-methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (6) was synthesized by stirring compound 5 with p-methoxybenzaldehyde in the presence of NaOH. The 1H NMR spectrum of compound 6 displayed a doublet signal at δ 7.72, indicating the presence of a (CHCH) group with coupling constant 15.6 ppm. The coupling constant value of the enone protons confirmed the trans (E) configuration of the chalcone. Additionally, a singlet signal at δ 3.83 ppm was observed, which could be attributed to the OCH3 group. Moreover, the mass spectrum of compound 6 displayed its molecular ion peak at m/z 462. Upon refluxing compound 4 with glacial acetic acid, N-(4-(6-methyl-4-oxo-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-5(4H)-yl)phenyl)acetamide (7) was obtained. The IR spectrum of compound 7 exhibited a peak at 3414 cm−1, indicating the presence of NH. In the 1H NMR spectrum, a D2O exchangeable singlet signal at δ 10.18 ppm corresponding to the NH proton was observed, along with a singlet signal at δ 2.19 ppm representing CH3. Additionally, the mass spectrum of compound 7 showed its molecular ion peak at 359 m/z. In contrast, compounds 8 and 9 were synthesized through the reaction of compound 4 with the corresponding acid anhydride in the presence of glacial acetic acid following a documented procedure.66 The 1H NMR spectra of compounds 8 and 9 were confirmed by the disappearance of the NH2 signal present in starting compound 4 at δ 5.44 ppm. Additionally, an increase in the number of aromatic protons further supported the successful synthesis of compound 8. Another significant piece of evidence for the synthesis of compound 8 was the emergence of signals at δ 170.2 ppm in the 13C NMR spectrum, indicating the presence of CO groups. Furthermore, the mass spectrum of compound 8 exhibited its molecular ion peak, which corresponds to its base peak at m/z 397. Compound 9 exhibited three peaks in the IR spectrum at 1716, 1681, and 1643 cm−1, along with signals at δ 168.1, 167.8, and 167.3 ppm in the 13C NMR spectrum, providing further confirmation of the presence of three CO groups. Additionally, the mass spectrum of compound 9 showed its molecular ion peak at m/z 447. As depicted in Scheme 2, the reaction of compound 4 with isatin in the presence of glacial acetic acid resulted in the formation of hydrazone derivative 10. The 1H NMR spectrum of derivative 10 exhibited a singlet signal at δ 11.14 ppm, which could be attributed to the NH proton, and showed exchangeability with D2O. Moreover, the integration of aromatic protons indicated an increase in their abundance. Analysis of the IR spectrum of derivative 10 revealed the presence of an NH group, as evidenced by a peak at 3252 cm−1, and three CO groups, indicated by peaks at 1732, 1693, and 1659 cm−1. Moreover, the mass spectrum of compound 10 exhibited its molecular ion peak at m/z 446. Conversely, the reaction of key intermediate 4 with furoyl chloride yielded the desired compound 11. The 1H NMR spectrum of compound 11 confirmed the presence of a D2O exchangeable singlet signal at δ 10.57 ppm, which corresponded to the NH proton. Additionally, evidence of the formation of compound 11 was observed in the 13C NMR spectrum, where the two signals at δ 165.2 and 159.7 ppm indicated the presence of two CO groups. The IR spectrum further supports the formation of a new compound, as evidenced by the broad peak at 1685 cm−1, which corresponded to the presence of two CO groups. Additionally, the mass spectrum of compound 11 displayed [M+ + H] at m/z 412.
Compounds 12a–e were synthesized by refluxing compound 4 with substituted benzoyl chloride, in the presence of anhydrous K2CO3 and dry benzene. The 1H NMR spectra of compounds 12a–e displayed D2O exchangeable singlet signals in the range of δ 10.41–10.80 ppm, corresponding to the NH group. Another confirmation of the formation of compounds 12a–e is the increase in aromatic protons observed in their respective 1H NMR spectra. Furthermore, the structures of compounds 12a–e were confirmed by their 13C NMR spectra, which displayed signals in the range of δ 158.1–166.4 ppm, corresponding to the CO group. Finally, the mass spectra of compounds 12a–e exhibited their molecular ion peaks at m/z 421, 439, 455, 435 and 466, respectively.
Biological evaluation
| Panel/cell lines | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compounds | 4 | 6 | 7 | 8 | 9 | 10 | 11 | 12a | 12b | 12c | 12d | 12e |
| Blank entries (—) indicate weak growth inhibition of less than 15%; entries in bold indicate strong growth inhibition (GI% > 70%). | ||||||||||||
| Leukemia | ||||||||||||
| CCRF-CEM | 21.04 | 53.62 | 30.83 | 32.21 | — | 37.81 | 23.57 | 49.51 | 65.18 | 25.29 | 53.73 | 52.54 |
| HL-60(TB) | — | 55.3 | 28.32 | — | — | — | 19.80 | 18.49 | 57.49 | 48.07 | 48.64 | 54.04 |
| K-562 | 52.08 | 87.56 | 55.38 | — | — | 34.44 | 30.15 | 66.83 | 72.79 | 61.27 | 68.79 | 68.85 |
| MOLT-4 | — | 55.26 | 43.58 | — | — | 34.13 | — | 68.09 | 94.62 | 30.70 | 65.69 | 63.4 |
| RPMI-8226 | 48.17 | >100 | 47.47 | — | — | 36.46 | 26.08 | 43.56 | 64.75 | 52.90 | 41.57 | 47.70 |
| Non-small cell lung cancer | ||||||||||||
| A549/ATCC | — | — | — | — | — | — | — | 28.89 | 45.01 | 26.15 | 34.23 | 33.03 |
| EKVX | 63.32 | 57.12 | 66.76 | — | — | 52.13 | 26.92 | 65.66 | 69.78 | 68.54 | 69.52 | 69.34 |
| HOP-62 | 16.18 | 19.48 | — | — | — | — | — | 37.86 | 35.43 | 41.13 | 21.19 | — |
| HOP-92 | — | — | 16.97 | — | — | 17.59 | — | 29.21 | — | 37.84 | — | — |
| NCI-H226 | 18.04 | 29.42 | 33.45 | — | — | 24.73 | — | 44.67 | 43.77 | 49.07 | 45.20 | 50.76 |
| NCI-H23 | 40.11 | 27.59 | 31.69 | — | — | 30.66 | — | 45.55 | 49.78 | 47.22 | 46.45 | 50.64 |
| NCI-H322M | — | — | — | — | — | — | — | — | — | — | — | 17.22 |
| NCI-H460 | 16.76 | — | — | — | — | — | — | 27.93 | 45.65 | 28.81 | 24.63 | 26.41 |
| NCI-H522 | — | 42.64 | 25.04 | — | — | 16.75 | — | 20.71 | 36.06 | 32.28 | 28.77 | 38.94 |
| Colon Cancer | ||||||||||||
| COLO 205 | — | — | — | — | — | — | — | 20.17 | 37.69 | 18.80 | 22.64 | 25.92 |
| HCC-2998 | — | — | — | — | — | — | — | — | 20.11 | 15.77 | — | — |
| HCT-116 | 25.88 | 90.29 | 28.85 | — | — | 17.88 | — | 41.89 | 51.66 | 44.53 | 48.49 | 55.63 |
| HCT-15 | — | 46.48 | 15.14 | — | — | 15.65 | — | 20.24 | 33.94 | 23.21 | 24.65 | 28.57 |
| HT29 | — | 28.51 | — | — | — | — | — | 25.19 | 36.52 | 28.04 | 28.83 | 21.55 |
| KM12 | — | 68.43 | — | — | — | — | — | 24.72 | 35.89 | 19.25 | 22.48 | 20.44 |
| SW-620 | — | 24.86 | — | — | — | — | — | — | 23.50 | 17.33 | — | — |
| CNS Cancer | ||||||||||||
| SF-268 | 17.43 | — | — | — | — | — | — | 21.49 | — | — | — | — |
| SF-295 | 27.07 | 34.45 | 36.45 | — | — | 24.74 | 15.35 | 46.55 | 60.44 | 45.85 | 49.36 | 56.31 |
| SF-539 | 19.39 | 22.44 | 18.95 | — | — | 20.95 | — | 19.79 | — | — | 18.65 | 18.68 |
| SNB-19 | — | 18.23 | 16.43 | — | — | 36.04 | — | 23.64 | 19.63 | 16.28 | 17.93 | 25.41 |
| SNB-75 | 28.10 | — | — | — | — | — | — | 43.20 | 35.47 | 27.47 | 17.25 | 32.88 |
| U251 | — | 25.89 | — | — | — | 39.63 | — | — | 18.89 | — | — | — |
| Melanoma | ||||||||||||
| LOX IMVI | 18.48 | 56.05 | 21.15 | — | — | 18.26 | — | 26.77 | 38.21 | 33.44 | 32.84 | 34.09 |
| MALME-3M | 17.83 | 39.45 | 22.27 | — | — | 23.07 | 16.63 | 29.80 | 31.56 | 33.30 | 32.32 | 33.98 |
| M14 | — | 46.93 | — | — | — | 18.76 | — | 23.60 | 37.10 | 24.55 | 26.77 | 24.65 |
| MDA-MB-435 | 22.95 | 88.84 | 21.14 | — | — | — | — | 27.23 | 46.24 | 30.21 | 29.45 | 33.64 |
| SK-MEL-2 | 17.82 | 29.37 | 24.32 | — | — | — | — | — | 22.12 | 26.05 | 21.66 | 29.33 |
| SK-MEL-28 | — | — | — | — | — | — | — | — | 16.22 | 17.05 | 19.33 | 18.2 |
| SK-MEL-5 | 66.85 | 72.33 | 77.39 | — | — | 42.92 | 23.07 | 76.15 | >100 | 80.35 | 92.95 | 82.37 |
| UACC-257 | 34.27 | 25.36 | 41.66 | — | — | 29.34 | — | 46.94 | 68.16 | 53.87 | 57.98 | 50.77 |
| UACC-62 | 34.55 | 49.98 | 38.28 | — | — | 36.16 | 16.55 | 36.84 | 36.89 | 32.25 | 39.24 | 39.64 |
| Ovarian Cancer | ||||||||||||
| IGROV1 | 15.00 | — | — | — | — | 34.67 | — | — | 26.50 | 21.34 | 28.41 | 31.06 |
| OVCAR-3 | — | 38.96 | — | — | — | 24.31 | — | 16.81 | — | 17.45 | — | — |
| OVCAR-4 | 48.74 | 34.28 | 44.83 | — | — | 43.94 | 21.16 | 53.18 | 59.89 | 53.9 | 50.21 | 48.22 |
| OVCAR-5 | — | — | — | — | — | — | — | — | — | — | — | — |
| OVCAR-8 | — | 16.55 | 20.03 | — | — | 24.77 | — | 38.40 | 36.29 | 43.44 | 23.2 | 34.15 |
| NCI/ADR-RES | — | 37.65 | — | — | — | — | — | 30.38 | 57.22 | 44.50 | 47.31 | 47.21 |
| SK-OV-3 | — | — | — | — | — | 19.63 | — | 19.01 | — | — | — | — |
| Renal Cancer | ||||||||||||
| 786-0 | — | 15.44 | — | — | — | — | — | 20.37 | 17.49 | 20.30 | — | 16.29 |
| A498 | 16.77 | — | 16.13 | — | — | — | — | — | — | — | — | 19.56 |
| ACHN | 27.07 | 29.04 | 29.80 | — | — | 27.71 | — | 36.66 | 39.02 | 32.64 | 38.25 | 39.61 |
| CAKI-1 | 31.71 | 25.01 | 31.09 | — | — | 31.69 | — | 33.41 | 34.29 | 29.71 | 27.56 | 31.49 |
| RXF 393 | — | 15.63 | — | — | — | 28.96 | — | 18.94 | 20.95 | — | — | 26.37 |
| SN12C | 21.06 | 18.43 | 26.06 | — | — | 37.33 | — | 24.32 | 25.59 | 17.01 | 26.66 | 28.96 |
| TK-10 | 21.82 | — | 20.38 | — | — | — | — | 18.56 | 28.96 | 19.85 | 17.71 | 31.47 |
| Prostate Cancer | ||||||||||||
| PC-3 | 17.99 | 34.93 | 30.34 | — | — | 24.01 | — | 29.52 | 46.89 | 36.72 | 28.35 | 33.7 |
| DU-145 | 17.28 | 16.81 | 17.82 | — | — | — | — | 18.59 | 19.29 | 17.78 | 15.14 | 21.02 |
| Breast Cancer | ||||||||||||
| MCF7 | 37.65 | 91.70 | 44.36 | — | — | 33.68 | 23.40 | 47.09 | 64.15 | 46.75 | 52.21 | 56.70 |
| HS 578T | 21.44 | — | — | — | — | 17.60 | — | 28.95 | 21.28 | 20.05 | 15.17 | 23.14 |
| BT-549 | 31.16 | 55.01 | 48.01 | — | — | 22.15 | — | 39.07 | 54.03 | 35.98 | 40.84 | 48.8 |
| T-47D | 64.88 | 65.3 | 70.86 | — | — | 38.28 | 34.05 | 72.41 | 81.97 | 72.47 | 65.39 | 66.48 |
| MDA-MB-468 | 62.38 | 88.68 | 64.22 | 18.65 | — | 51.42 | 37.27 | 71.01 | 85.50 | 84.33 | 78.94 | 68.64 |
| Mean inhibition | 20.71 | 33.65 | 23.39 | 0.44 | 1.05 | 20.94 | 9.46 | 31.57 | 39.63 | 32.42 | 31.99 | 34.14 |
Screening conducted by the National Cancer Institute (NCI) revealed that compound 4 exhibited moderate antitumor activity against 13 cell lines, with an overall mean inhibition of 20.71%. Compound 5 reacted with p-methoxybenzaldehyde, leading to the formation of chalcone 6. Chalcone 6 demonstrated a lethal effect on the leukemia cell line RPMI-8226, along with strong antiproliferative activity against leukemia K-562, colon cancer HCT-116, melanoma (MDA-MB-435 and SK-MEL-5), and breast cancer (MCF7 and MDA-MB-468) cell lines, resulting in an overall mean inhibition of 33.65%.
The acetylated derivative, compound 7, demonstrated potent antiproliferative activity against the melanoma SK-MEL-5 and breast cancer T-47D cell lines, resulting in an overall mean inhibition of 23.39%. Conversely, compounds 8 and 9 exhibited no significant inhibitory activity, with mean inhibitions of 0.44% and 1.05%, respectively.
The isatin derivative (compound 10) displayed a mean inhibition of 20.94% and moderate activity against 17 cancer cell lines, with inhibition ranging from 30.66% to 52.13%.
Furthermore, the furoyl derivative, compound 11, exhibited decreased antiproliferative activity and only demonstrated moderate anticancer activity against three cell lines, leukemia K-562, breast cancer cell lines T-47D and MDA-MB-468, with a mean inhibition of 9.46%.
In contrast to compound 11, benzoyl derivatives 12a–e displayed increased activity, with a mean inhibition range of 31.57–39.63%. Compound 12a exhibited strong inhibitory activity against the melanoma SK-MEL-5 cell line, as well as the breast cancer cell lines T-47D and MDA-MB-468, with growth inhibition percentages (GI%) of 76.15%, 72.41%, and 71.01%, respectively. Additionally, it demonstrated moderate anticancer activity against 20 cancer cell lines with a mean inhibition of 31.57%.
The p-fluorobenzoyl derivative compound 12b showed a lethal effect on the melanoma cell line SK-MEL-5, along with strong anticancer activity against the leukemia cell lines K-562 and MOLT-4, with GI% values of 72.79% and 94.62%, respectively. It also exhibited strong anticancer activity against breast cancer cell lines T-47D and MDA-MB-468, with GI% values of 81.97% and 85.50%, respectively. Furthermore, compound 12b exhibited moderate anticancer activity against 31 cancer cell lines and recorded the highest mean inhibition among all the tested compounds, with a score of 39.63%.
When the p-fluoro group was substituted with a p-chloro group, as in compound 12c, strong antitumor activity was observed against three cell lines: melanoma SK-MEL-5 with a GI% of 80.35% and breast cancer cell lines T-47D and MDA-MB-468 with GI% values of 72.47% and 84.33%, respectively. Additionally, it displayed moderate anticancer activity against 24 cell lines, with GI% ranging from 30.70% to 68.54%. The p-methyl benzoyl derivative 12d demonstrated a decrease in antiproliferative activity compared to 12c, exhibiting only strong antitumor activity against two cell lines namely melanoma SK-MEL-5 and breast cancer MDA-MB-468, with growth inhibition of 92.95 and 78.94%, respectively. In addition, compound 12d demonstrated moderate anticancer activity against 21 cell lines, with a GI range of 32.32–69.52%. Altering the p-methyl group with a p-nitro group led to the formation of compound 12e, which showed strong antitumor activity only against the melanoma SK-MEL-5 cell line with a growth inhibition of 82.37%, together with moderate activity against several cell lines of different types of cancer with growth inhibition ranging from 31.06–69.34%.
Among the newly synthesized derivatives with varying structures and antiproliferative activities, compounds containing amide spacers (12a–e) exhibited higher anticancer activities than those with an azomethine spacer (10) or compounds directly attached without spacers (8 and 9). Notably, the presence of a 4-substituted phenyl group (12a–e) in the terminal hydrophobic region demonstrated the most potent anticancer activity, surpassing compounds with an amino group (4), methyl group (7), or various heterocyclic rings, such as 1-H-pyrrole (8), isoindoline (9), indoline (10), and furoyl (11).
Among the most active series (12a–e), compound 12b, featuring a 4-fluorophenyl group, exhibited the highest potency, followed by the 4-chlorophenyl derivative 12c. Conversely, compound 12e, which was substituted with a strongly electron-withdrawing nitro group, displayed the lowest activity. The SAR of the synthesized compounds is summarized in Fig. 4.
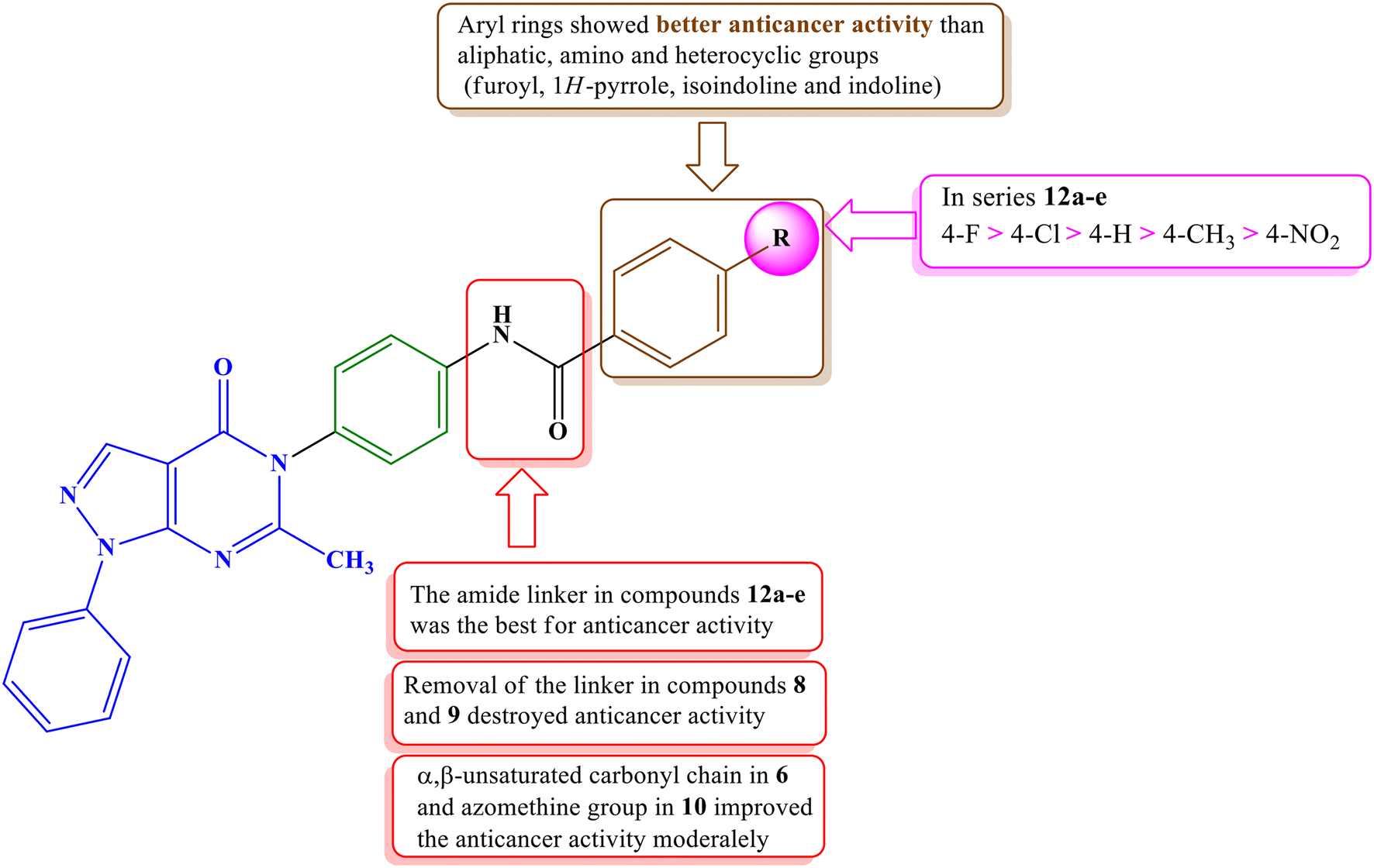 | ||
| Fig. 4 Structure–activity relationship of the pyrazolo[3,4-d]pyrimidine synthesized compounds. | ||
| Cytotoxicity IC50 μM | Selectivity index | ||||
|---|---|---|---|---|---|
| Codes | MCF-10a | MDA-MB-468 | T47D | MDA-MB-468 | T47D |
| a Not tested. | |||||
| 12a | 55.48 ± 2.37 | 27.02 ± 1.02 | 35.511 ± 1.58 | 2.05 | 1.56 |
| 12b | 22.77 ± 0.97 | 3.343 ± 0.13 | 4.792 ± 0.21 | 6.81 | 4.75 |
| 12c | 34.57 ± 1.48 | 10.661 ± 0.4 | 20.11 ± 0.9 | 3.24 | 1.72 |
| 12d | 58.18 ± 2.49 | 16.030 ± 0.6 | NTa | 3.63 | NTa |
| Staurosporine | 18.01 ± 0.77 | 6.358 ± 0.24 | 4.849 ± 0.22 | 2.83 | 3.71 |
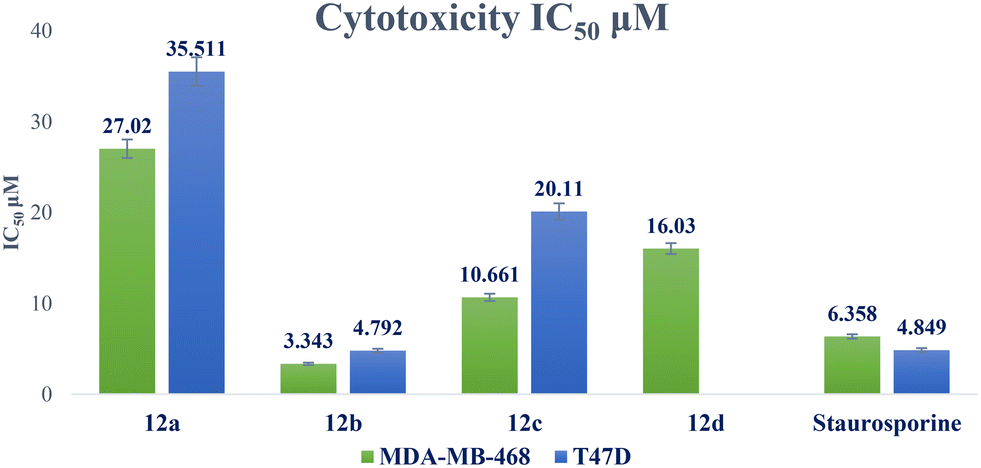 | ||
| Fig. 5 Graphical illustration of IC50 of compounds 12a–d against breast cancer cell lines compared to staurosporine. | ||
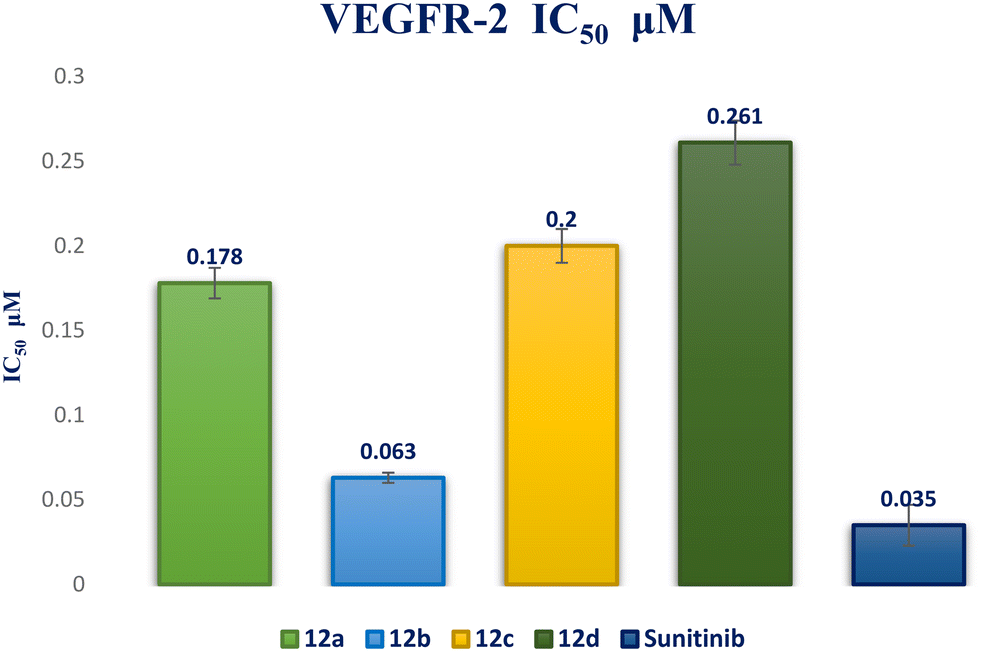 | ||
| Fig. 6 IC50 bar graph for compounds 12a–d against VEGFR-2 in comparison to sunitinib. | ||
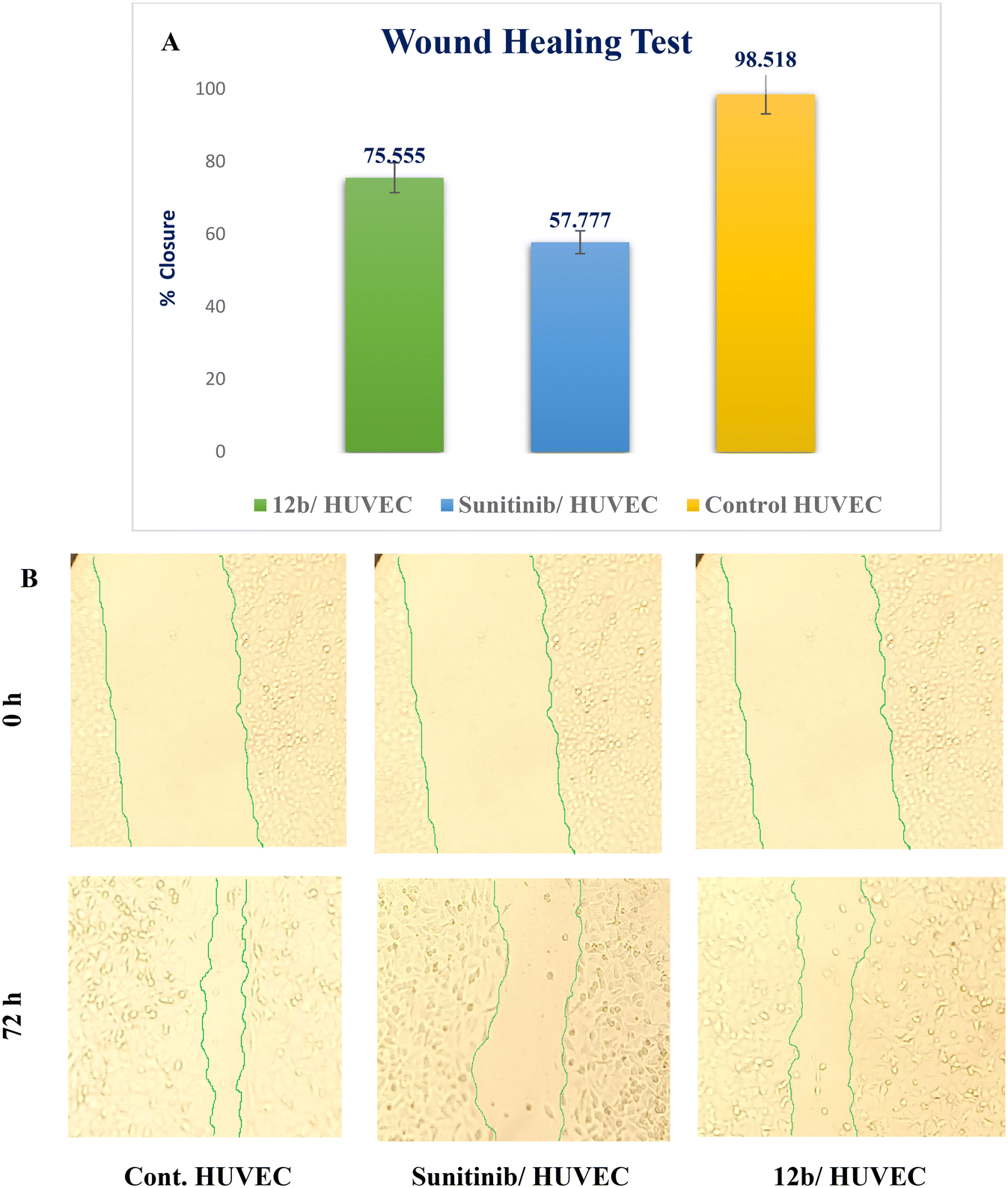 | ||
| Fig. 7 Effect of sunitinib and compound 12b on endothelial cell migration in HUVEC cells. A) The following graph illustrates the percentage of wound closure for control and treated cells; B) a 72 h study using compound 12b and sunitinib was conducted on HUVECs. Values are expressed as mean ± standard deviation. | ||
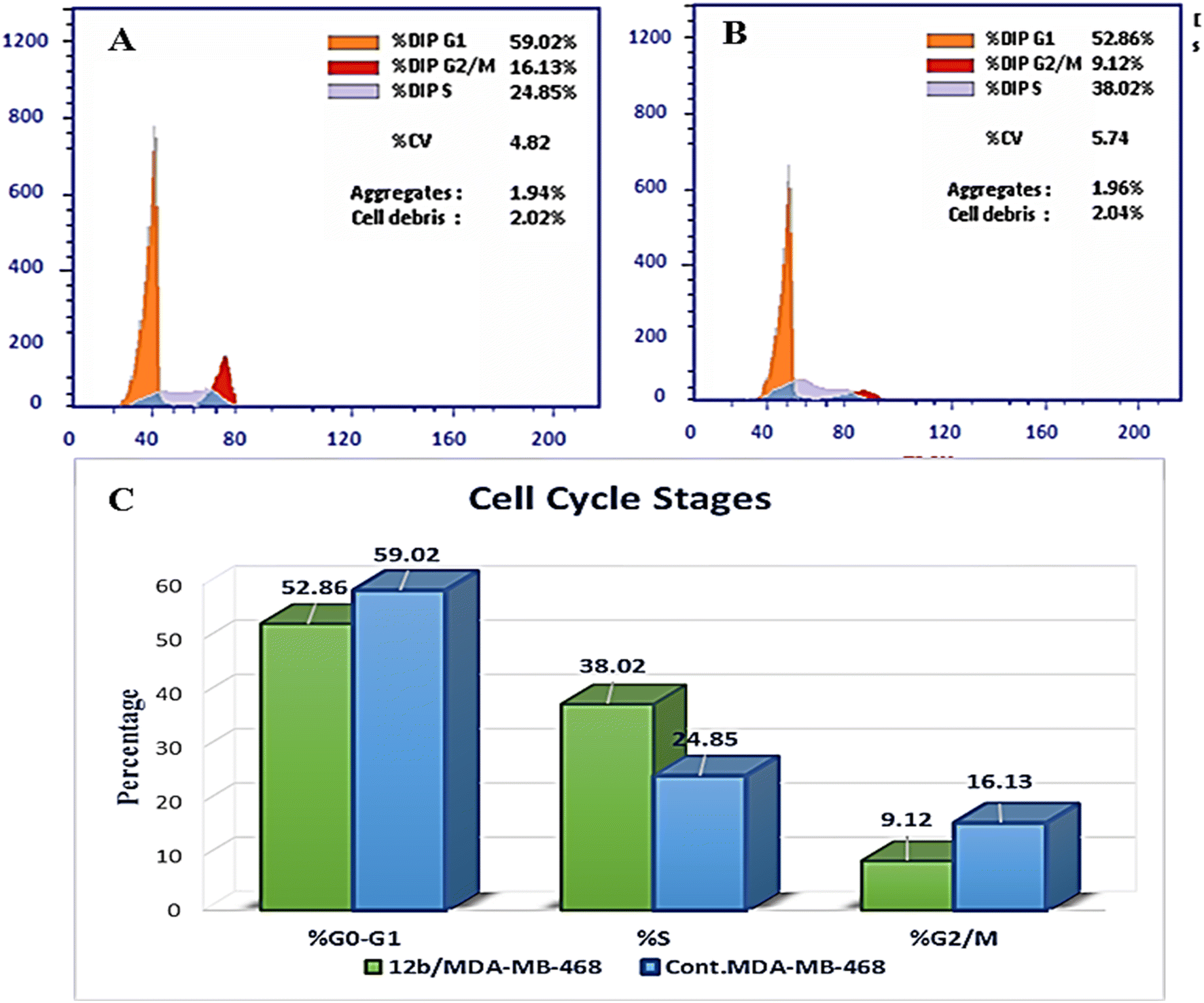 | ||
| Fig. 8 Cell cycle arrest is caused by compound 12b in MDA-MB-468 cells. A) Control MDA-MB-468; B) cells treated with 12b; C) the diagram illustrates the distribution of cell cycle phases in control and treated cells. | ||
| Compound | Apoptosis | Necrosis | ||
|---|---|---|---|---|
| Total | Early | Late | ||
| 12b/MDA-MB-468 | 43.28 | 15.85 | 22.49 | 4.94 |
| Cont. MDA-MB-468 | 2.28 | 0.52 | 0.17 | 1.59 |
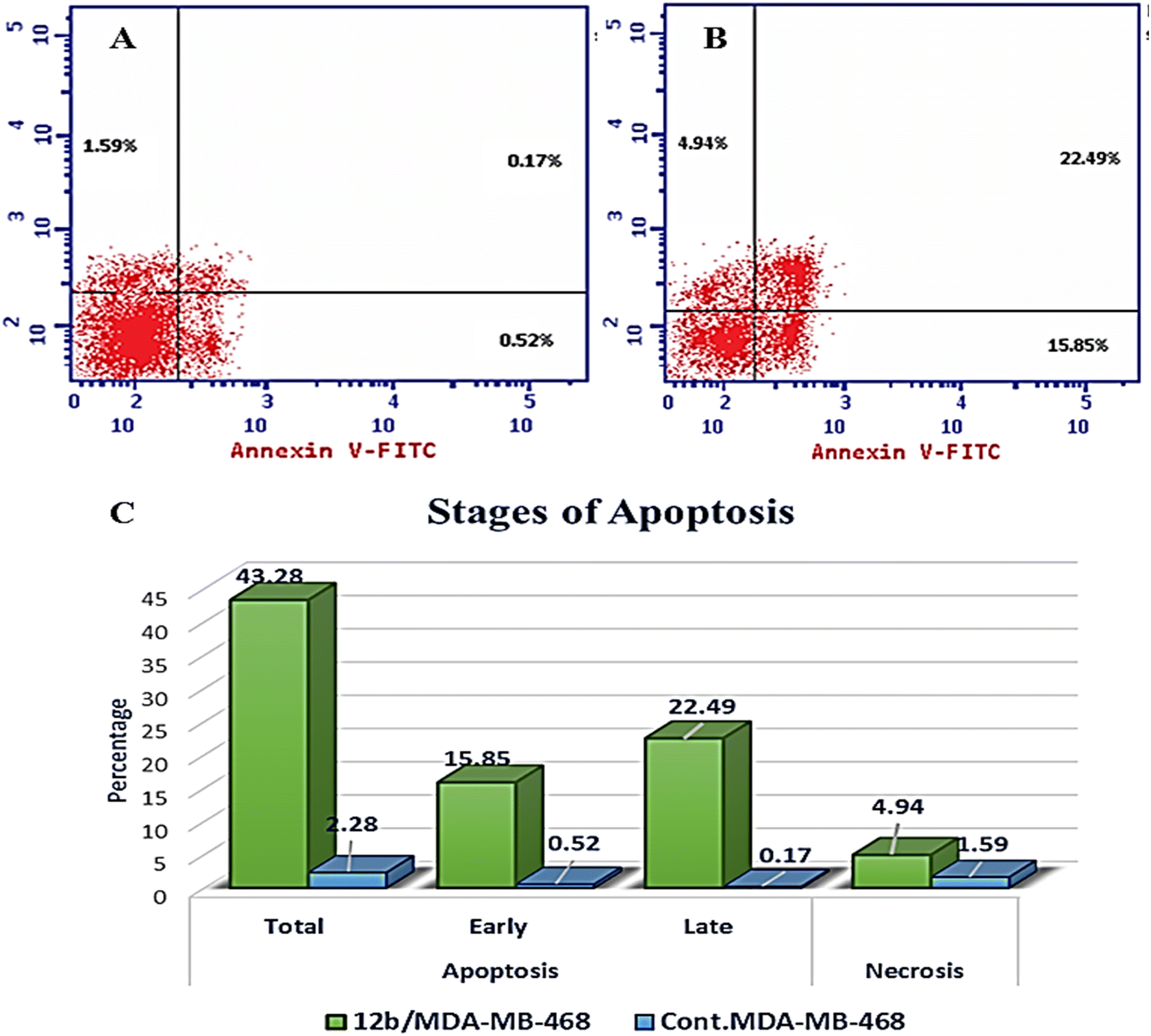 | ||
| Fig. 9 The effect of compound 12b on annexin V staining in MDA-MB-468 cells. A) Control MDA-MB-468; B) cells treated with 12b; C) graph displaying the percentage of apoptotic and necrotic cells in control and 12b-treated cells. | ||
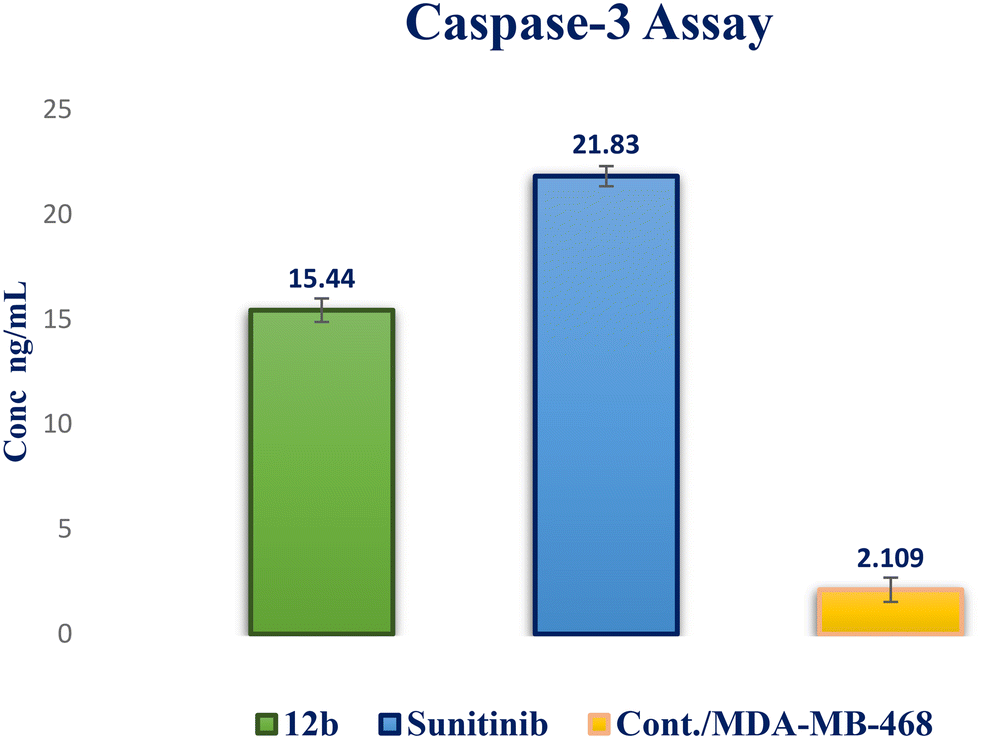 | ||
| Fig. 10 Caspase-3 activity in MDA-MB-468 cells of compound 12b compared to sunitinib. | ||
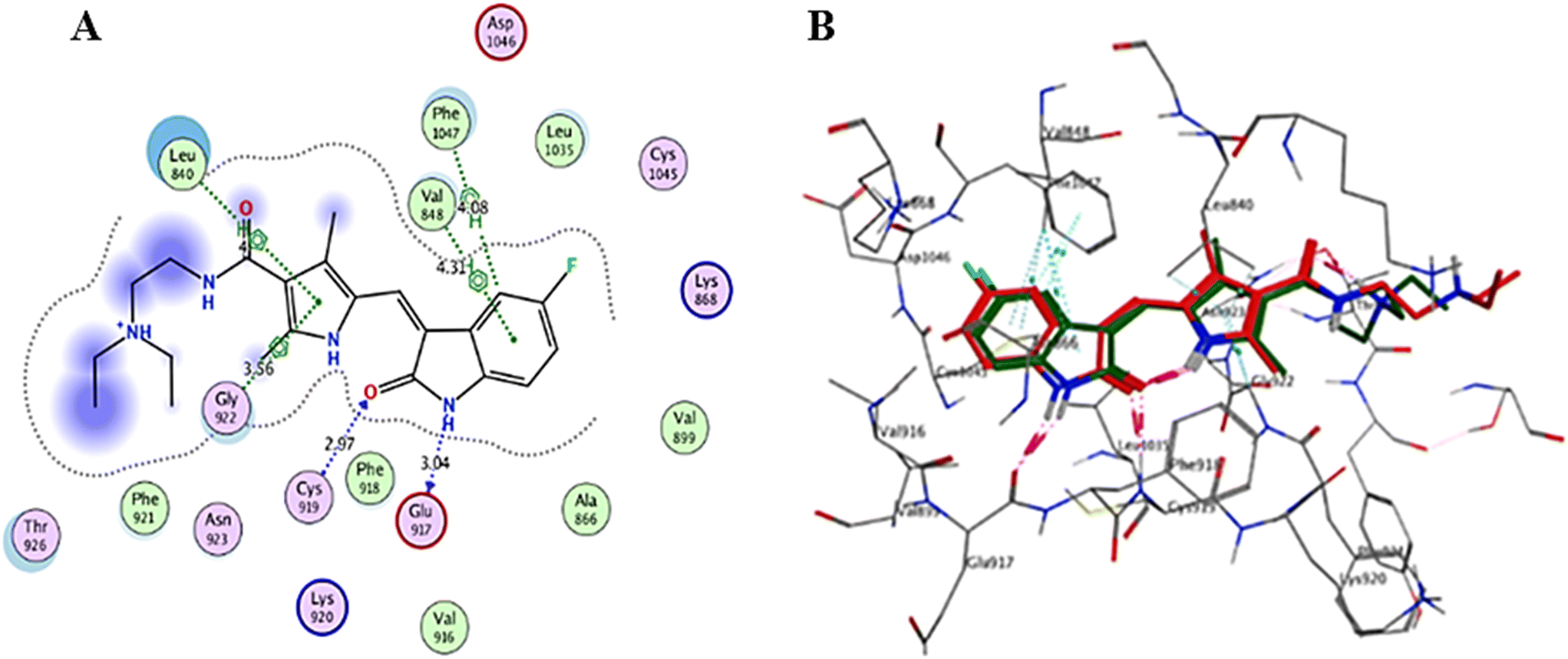 | ||
| Fig. 11 (A) Sunitinib in the VEGFR-2 active site (2D interaction diagram); (B) the superimposition of the co-crystallized sunitinib (red) and its docking poses (green) in the VEGFR-2 active site (3D representation). | ||
| Compound | S (kcal mol−1) | Amino acids | Types of interactions | Distance (Å) |
|---|---|---|---|---|
| 12a | −6.4953 | Asp1046 | HB donor | 3.15 |
| Cys919 | HB acceptor | 2.96 | ||
| Lys868 | HB acceptor | 3.10 | ||
| Phe1047 | H–π | 4.06 | ||
| Leu840 | π–H | 4.21 | ||
| Leu840 | π–H | 3.98 | ||
| Leu840 | π–H | 3.84 | ||
| Val848 | π–H | 4.36 | ||
| 12b | −7.6497 | Cys919 | HB acceptor | 3.46 |
| Leu840 | π–H | 3.71 | ||
| Leu840 | π–H | 3.88 | ||
| Val848 | π–H | 4.12 | ||
| Val848 | π–H | 4.42 | ||
| 12c | −5.8195 | Cys919 | HB acceptor | 3.03 |
| Leu840 | π–H | 4.28 | ||
| Leu840 | π–H | 4.29 | ||
| Leu840 | π–H | 3.89 | ||
| 12d | −5.8969 | Glu885 | HB donor | 3.15 |
| Cys919 | HB acceptor | 3.03 | ||
| Leu840 | π–H | 4.29 | ||
| Leu840 | π–H | 4.30 | ||
| Leu840 | π–H | 3.89 | ||
| Asp1046 | π–H | 4.52 | ||
| Sunitinib | −8.4031 | Glu917 | HB donor | 3.04 |
| Cys919 | HB acceptor | 2.97 | ||
| Phe1047 | H–π | 4.08 | ||
| Leu840 | π–H | 4.00 | ||
| Val848 | π–H | 4.31 | ||
| Gly922 | π–H | 3.56 |
Interaction analysis of the highly active designed compounds 12a–d with crucial amino acids in the VEGFR-2 binding site indicated favorable binding outcomes overall. These compounds exhibited a good fit within the binding region of the enzyme and formed strong binding interactions, as evidenced by the 2D and 3D representations of the docked poses. Specifically, compound 12b demonstrated a binding pattern in the VEGFR-2 binding site that closely resembled that of sunitinib, along with a predicted docking energy of −7.6497 kcal mol−1 (Fig. 12). In the active “DFG-in” conformation, the N2 of the pyrazolopyrimidinone ring within compound 12b formed a hydrogen bond with Cys919 in the ATP-binding pocket of VEGFR-2. Additionally, the pyrazole and pyrimidinone rings establish π–H bonds with Leu840. Furthermore, the N1-phenyl ring of the compound interacts with Val848 through two π–H bonds. Conversely, the hydrophobic 4-fluorophenyl ring of the compound fitted into an allosteric hydrophobic pocket. The docking poses of compounds 12a, 12c, and 12d are shown in Fig. S1–S3 of the ESI.†
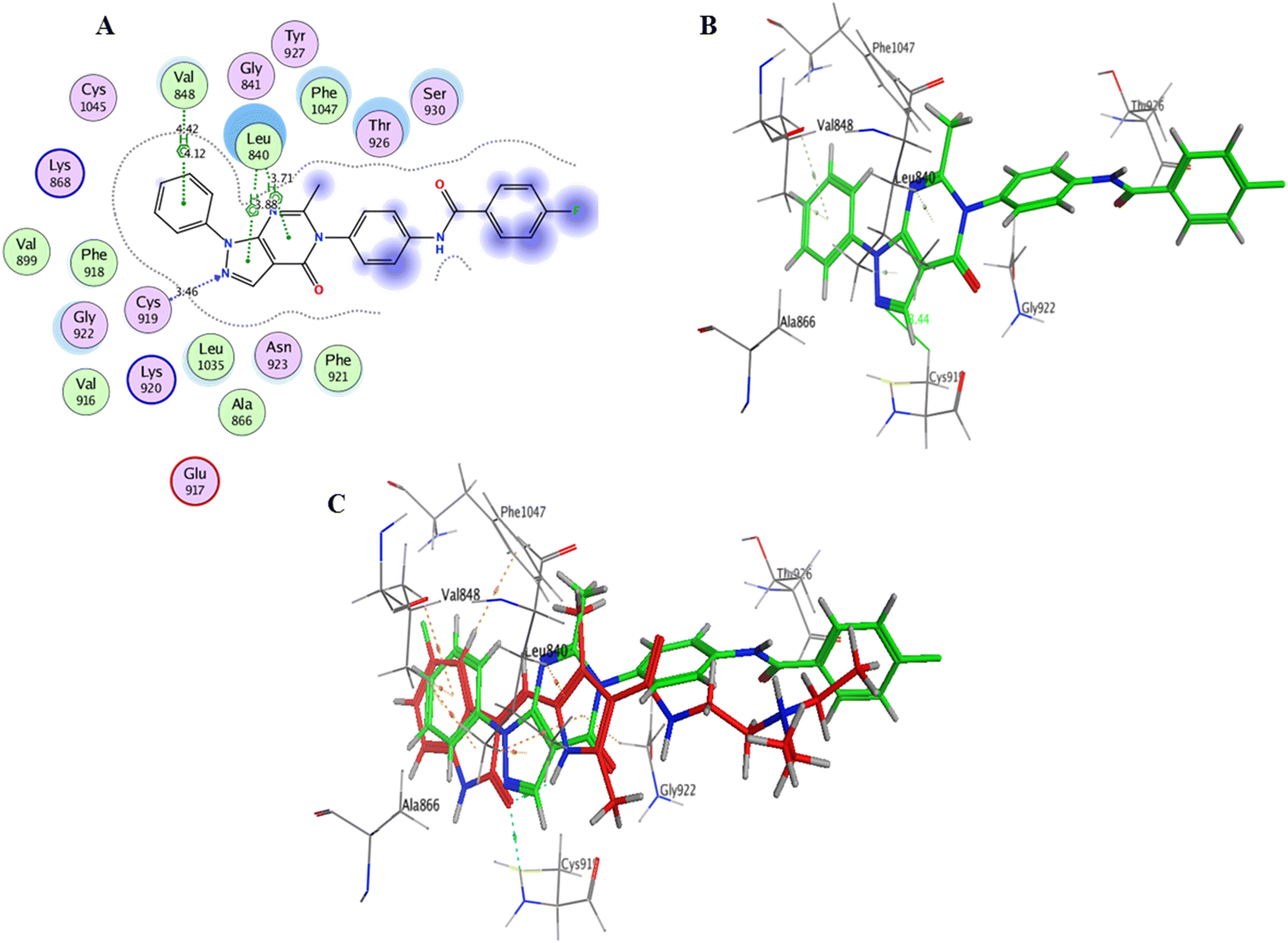 | ||
| Fig. 12 A) Compound 12b within the VEGFR-2 binding site (2D interactions); B) compound 12b within the VEGFR-2 binding site (3D representation); C) superimposition of compound 12b (green) with sunitinib (red) interactions within the VEGFR-2 binding site (3D representation). | ||
Conclusion
In this study, a novel series of pyrazolo[3,4-d]pyrimidine compounds, specifically 4, 6–11, and 12a–e, was synthesized and evaluated for their cytotoxic effects. Among these compounds, the most active, namely 12a–d, demonstrated significant anticancer activity against breast cancer cell lines MDA-MB-468 and T-47D. Notably, compound 12b exhibited the highest antitumor potency against both MDA-MB-468 and T-47D cell lines, with IC50 values of 3.343 ± 0.13 μM and 4.792 ± 0.21 μM, respectively. Furthermore, compound 12b emerged as the most potent VEGFR-2 inhibitor, with an IC50 value of 0.063 ± 0.003 μM, surpassing that of sunitinib (IC50 = 0.035 ± 0.012 μM). These findings provide additional support for molecular docking results. Compound 12b significantly reduced wound healing by 23%. Further analysis of the cell cycle and apoptosis demonstrated that compound 12b induced a substantial increase in total apoptosis and arrested the cell cycle in the S phase in the MDA-MB-468 cell line. Moreover, compound 12b induced a 7.32-fold increase in the expression of apoptotic caspase-3. Based on these findings, compound 12b showed significant promise as a candidate for further research to develop more potent anticancer drugs.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors would like to thank the National Institutes of Health, USA, for their support in conducting the antiproliferative screening in this study. Additionally, the authors acknowledge the Confirmatory Diagnostic Unit of VACSERA-EGYPT for their contribution to performing the biological assays.References
- S. C. Parija, Pondicherry J. Nurs., 2022, 15, 1–2 CrossRef.
- J. Ferlay, M. Colombet, I. Soerjomataram, D. M. Parkin, M. Piñeros, A. Znaor and F. Bray, Int. J. Cancer, 2021, 149, 778–789 CrossRef CAS PubMed.
- B. S. Chhikara and K. Parang, Chem. Biol. Lett., 2023, 10, 451 Search PubMed.
- D. J. Baillache and A. Unciti-Broceta, RSC Med. Chem., 2020, 11, 1112–1135 RSC.
- N. M. Saleh, M. G. El-Gazzar, H. M. Aly and R. A. Othman, Front. Chem., 2020, 7, 917 CrossRef PubMed.
- A. A. Gaber, A. H. Bayoumi, A. M. El-morsy, F. F. Sherbiny, A. B. M. Mehany and I. H. Eissa, Bioorg. Chem., 2018, 80, 375–395 CrossRef CAS PubMed.
- N. S. Abdou, R. A. T. Serya, A. Esmat, M. F. Tolba, N. S. M. Ismail and K. A. M. Abouzid, MedChemComm, 2015, 6, 1518–1534 RSC.
- M. Kim, M. Baek and D. J. Kim, Curr. Pharm. Des., 2017, 23, 4226–4246 CAS.
- M. A. Abdelgawad, R. B. Bakr, O. A. Alkhoja and W. R. Mohamed, Bioorg. Chem., 2016, 66, 88–96 CrossRef CAS PubMed.
- C. Wang, H. Liu, Z. Song, Y. Ji, L. Xing, X. Peng, X. Wang, J. Ai, M. Geng and A. Zhang, Bioorg. Med. Chem. Lett., 2017, 27, 2544–2548 CrossRef CAS PubMed.
- S. J. Modi and V. M. Kulkarni, Med. Drug Discovery, 2019, 2, 100009 CrossRef.
- K. M. Cook and W. D. Figg, Ca-Cancer J. Clin., 2010, 60, 222–243 CrossRef PubMed.
- N. Ferrara, in Tumor Angiogenes, Springer International Publishing, Cham, 2019, pp. 211–226 Search PubMed.
- Z. Zhao, H. Wu, L. Wang, Y. Liu, S. Knapp, Q. Liu and N. S. Gray, ACS Chem. Biol., 2014, 9, 1230–1241 CrossRef CAS PubMed.
- H. Zhang, F. Gong, H. Fei, C. Li, C. Zhang, Y. Wang, Y. Xu, R. Hu and L. Sun, Eur. J. Med. Chem., 2016, 109, 371–379 CrossRef CAS PubMed.
- S. Sun, J. Zhang, N. Wang, X. Kong, F. Fu, H. Wang and J. Yao, Molecules, 2018, 23, 24–37 CrossRef PubMed.
- W. M. Eldehna, S. M. Abou-Seri, A. M. El Kerdawy, R. R. Ayyad, A. M. Hamdy, H. A. Ghabbour, M. M. Ali and D. A. A. El Ella, Eur. J. Med. Chem., 2016, 113, 50–62 CrossRef CAS PubMed.
- Z. Hao and I. Sadek, OncoTargets Ther., 2016, 9, 5495–5505 CrossRef CAS PubMed.
- G. Gadaleta-Caldarola, S. Infusino, R. Divella, E. Ferraro, A. Mazzocca, F. De Rose, G. Filippelli, I. Abbate and M. Brandi, Future Oncol., 2015, 11, 1863–1880 CrossRef CAS PubMed.
- P. Wu, T. E. Nielsen and M. H. Clausen, Trends Pharmacol. Sci., 2015, 36, 422–439 CrossRef CAS PubMed.
- H. K. Mahmoud, T. A. Farghaly, H. G. Abdulwahab, N. T. Al-Qurashi and M. R. Shaaban, Eur. J. Med. Chem., 2020, 208, 112752 CrossRef CAS PubMed.
- R. Roskoski, Biochem. Biophys. Res. Commun., 2007, 356, 323–328 CrossRef CAS PubMed.
- E. B. Elkaeed, R. G. Yousef, H. Elkady, I. M. M. Gobaara, B. A. Alsfouk, D. Z. Husein, I. M. Ibrahim, A. M. Metwaly and I. H. Eissa, Molecules, 2022, 27, 4606 CrossRef CAS PubMed.
- A. E. Abdallah, R. R. Mabrouk, M. M. S. Al Ward, S. I. Eissa, E. B. Elkaeed, A. B. M. Mehany, M. A. Abo-Saif, O. A. El-Feky, M. S. Alesawy and M. A. El-Zahabi, J. Enzyme Inhib. Med. Chem., 2022, 37, 573–591 CrossRef CAS PubMed.
- H. Elkady, A. Elwan, H. A. El-Mahdy, A. S. Doghish, A. Ismail, M. S. Taghour, E. B. Elkaeed, I. H. Eissa, M. A. Dahab, H. A. Mahdy and M. M. Khalifa, J. Enzyme Inhib. Med. Chem., 2022, 37, 397–410 Search PubMed.
- Y. Y. Wang, F. Z. Xu, Y. Y. Zhu, B. Song, D. Luo, G. Yu, S. Chen, W. Xue and J. Wu, Bioorg. Med. Chem. Lett., 2018, 28, 2979–2984 CrossRef CAS PubMed.
- M. Abdel-Megid, Synth. Commun., 2020, 50, 3563–3591 CrossRef CAS.
- A. A. Gaber, A. M. El-Morsy, F. F. Sherbiny, A. H. Bayoumi, K. M. El-Gamal, K. El-Adl, A. A. Al-Karmalawy, R. R. Ezz Eldin, M. A. Saleh and H. S. Abulkhair, Arch. Pharm., 2021, e2100258 CrossRef PubMed.
- Z. Ruzi, K. Bozorov, L. Nie, J. Zhao and H. A. Aisa, Biomed. Pharmacother., 2022, 156, 113948 CrossRef CAS PubMed.
- M. Maher, A. E. Kassab, A. F. Zaher and Z. Mahmoud, J. Enzyme Inhib. Med. Chem., 2019, 34, 532–546 CrossRef CAS PubMed.
- D. C. Kim, Y. R. Lee, B. S. Yang, K. J. Shin, D. J. Kim, B. Y. Chung and K. H. Yoo, Eur. J. Med. Chem., 2003, 38, 525–532 CrossRef CAS PubMed.
- S. Cherukupalli, B. Chandrasekaran, R. R. Aleti, N. Sayyad, G. A. Hampannavar, S. R. Merugu, H. R. Rachamalla, R. Banerjee and R. Karpoormath, J. Mol. Struct., 2019, 1176, 538–551 CrossRef CAS.
- A. A. Mandour, I. F. Nassar, M. T. A. Aal, M. A. E. Shahin, W. A. El-Sayed, M. Hegazy, A. M. Yehia, A. Ismail, M. Hagras, E. B. Elkaeed, H. M. Refaat and N. S. M. Ismail, J. Enzyme Inhib. Med. Chem., 2022, 37, 1957–1973 CrossRef CAS PubMed.
- F. F. Sherbiny, A. H. Bayoumi, A. M. El-Morsy, M. Sobhy and M. Hagras, Bioorg. Chem., 2021, 116, 105325 CrossRef CAS PubMed.
- H. K. Park, H. Jeong, E. Ko, G. Lee, J. E. Lee, S. K. Lee, A. J. Lee, J. Y. Im, S. Hu, S. H. Kim, J. H. Lee, C. Lee, S. Kang and B. H. Kang, J. Med. Chem., 2017, 60, 7569–7578 CrossRef CAS PubMed.
- G. S. Hassan, H. H. Kadry, S. M. Abou-Seri, M. M. Ali and A. E. E. D. Mahmoud, Bioorg. Med. Chem., 2011, 19, 6808–6817 CrossRef CAS PubMed.
- B. C. Huddle, E. Grimley, C. D. Buchman, M. Chtcherbinine, B. Debnath, P. Mehta, K. Yang, C. A. Morgan, S. Li, J. Felton, D. Sun, G. Mehta, N. Neamati, R. J. Buckanovich, T. D. Hurley and S. D. Larsen, J. Med. Chem., 2018, 61, 8754–8773 CrossRef CAS PubMed.
- M. M. Ghorab, F. A. Ragab, S. I. Alqasoumi, A. M. Alafeefy and S. A. Aboulmagd, Eur. J. Med. Chem., 2010, 45, 171–178 CrossRef CAS PubMed.
- A. E. Kassab, Y. El-Dash and E. M. Gedawy, Arch. Pharm., 2020, 353, e1900319 CrossRef PubMed.
- A. Sonousi, R. A. Hassan, E. O. Osman, A. M. Abdou and S. H. Emam, J. Enzyme Inhib. Med. Chem., 2022, 37, 2644–2659 CrossRef CAS PubMed.
- P. A. Halim, R. A. Hassan, K. O. Mohamed, S. O. Hassanin, M. G. Khalil, A. M. Abdou and E. O. Osman, J. Enzyme Inhib. Med. Chem., 2022, 37, 189–201 CrossRef CAS PubMed.
- A. El-Malah, Z. Mahmoud, H. Hamed Salem, A. M. Abdou, M. M. H. Soliman and R. A. Hassan, Green Chem. Lett. Rev., 2021, 14, 220–232 CAS.
- R. A. Hassan, S. H. Emam, D. Hwang, G.-D. Kim, S. O. Hassanin, M. G. Khalil, A. M. Abdou and A. Sonousi, Bioorg. Chem., 2022, 118, 105487 CrossRef CAS PubMed.
- A. A. Helwa, N. M. El-Dydamony, R. A. Radwan, S. M. Abdelraouf and R. M. Abdelnaby, Bioorg. Chem., 2020, 102, 104051 CrossRef CAS PubMed.
- N. M. El-Dydamony, R. M. Abdelnaby, R. Abdelhady, O. Ali, M. I. Fahmy, R. R. F. Eldeen and A. A. Helwa, J. Enzyme Inhib. Med. Chem., 2022, 37, 895–911 CrossRef CAS PubMed.
- E. O. Osman, S. H. Emam, A. Sonousi, M. M. Kandil, A. M. Abdou and R. A. Hassan, Drug Dev. Res., 2023, 84, 888–906 CrossRef CAS PubMed.
- M. T. M. Sayed, P. A. Halim, A. K. El Ansary and R. A. Hassan, Drug Dev. Res., 2023, 84, 1299–1319 CrossRef CAS PubMed.
- L. M. A. Abdel Ghany, N. M. El-Dydamony, A. A. Helwa, S. M. Abdelraouf and R. M. Abdelnaby, New J. Chem., 2022, 46, 17394–17409 RSC.
- A. E. Kassab, E. M. Gedawy, M. I. A. Hamed, A. S. Doghish and R. A. Hassan, J. Enzyme Inhib. Med. Chem., 2021, 36, 922–939 CrossRef CAS PubMed.
- R. A. Hassan, M. I. A. Hamed, A. M. Abdou and Y. El-Dash, Bioorg. Chem., 2022, 125, 105861 CrossRef CAS PubMed.
- Y. El-Dash, E. Elzayat, A. M. Abdou and R. A. Hassan, Bioorg. Chem., 2021, 114, 105137 CrossRef CAS PubMed.
- S. H. Emam, R. A. Hassan, E. O. Osman, M. I. A. Hamed, A. M. Abdou, M. M. Kandil, E. M. Elbaz and D. S. Mikhail, Drug Dev. Res., 2023, 84, 475–499 CrossRef PubMed.
- S. E. Seif, Z. Mahmoud, W. W. Wardakhan, A. M. Abdou and R. A. Hassan, Drug Dev. Res., 2023, 84, 839–860 CrossRef CAS PubMed.
- I. M. Salem, S. M. Mostafa, I. Salama, O. I. El-Sabbagh, W. A. H. Hegazy and T. S. Ibrahim, J. Enzyme Inhib. Med. Chem., 2023, 38, 203–215 CrossRef CAS PubMed.
- B. S. Ferreira, R. C. Silva, B. A. Souto and M. S. dos Santos, Lett. Org. Chem., 2020, 18, 335–343 CrossRef.
- G. N. Tageldin, S. M. Fahmy, H. M. Ashour, M. A. Khalil, R. A. Nassra and I. M. Labouta, Bioorg. Chem., 2018, 78, 358–371 CrossRef CAS PubMed.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS PubMed.
- R. H. Shoemaker, Nat. Rev. Cancer, 2006, 6, 813–823 CrossRef CAS PubMed.
- M. R. Boyd and K. D. Paull, Drug Dev. Res., 1995, 34, 91–109 CrossRef CAS.
- M. C. Alley, D. A. Scudiere, A. Monks, M. L. Hursey, M. J. Czerwinski, D. L. Fine, B. J. Abbott, J. G. Mayo, R. H. Shoemaker and M. R. Boyd, Cancer Res., 1988, 48, 589–601 CAS.
- M. R. Grever, S. A. Schepartz and B. A. Chabner, Semin. Oncol., 1992, 19, 622–638 CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- M. F. Tolba, A. Esmat, A. M. Al-Abd, S. S. Azab, A. E. Khalifa, H. A. Mosli, S. Z. Abdel-Rahman and A. B. Abdel-Naim, IUBMB Life, 2013, 65, 716–729 CrossRef CAS PubMed.
- A. M. Rieger, K. L. Nelson, J. D. Konowalchuk and D. R. Barreda, J. Visualized Exp., 2011, 50, e2597 Search PubMed.
- M. McTigue, B. W. Murray, J. H. Chen, Y. L. Deng, J. Solowiej and R. S. Kania, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18281–18289 CrossRef CAS PubMed.
- K. E. Anwer and G. H. Sayed, J. Heterocycl. Chem., 2020, 57, 2339–2353 CrossRef CAS.
- J. A. Pietenpol and Z. A. Stewart, Toxicology, 2002, 181–182, 475–481 CrossRef CAS PubMed.
- W. Gorczyca, Endocr.-Relat. Cancer, 1999, 6, 17–19 CAS.
- I. Vermes, C. Haanen, H. Steffens-Nakken and C. Reutellingsperger, J. Immunol. Methods, 1995, 184, 39–51 CrossRef CAS PubMed.
- C. L. J. Teng, C. T. R. Yu, W. L. Hwang, J. R. Tsai, H. C. Liu, G. Y. Hwang and S. L. Hsu, Ann. Hematol., 2013, 92, 301–313 CrossRef CAS PubMed.
- M. S. Taghour, H. A. Mahdy, M. H. Gomaa, A. Aglan, M. G. Eldeib, A. Elwan, M. A. Dahab, E. B. Elkaeed, A. A. Alsfouk, M. M. Khalifa, I. H. Eissa and H. Elkady, J. Enzyme Inhib. Med. Chem., 2022, 37, 2063–2077 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3md00476g |
| This journal is © The Royal Society of Chemistry 2023 |