
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoisomers and functional groups in oxidorhenium(V) complexes: effects on catalytic activity†
J. A.
Schachner
 *,
B.
Berner
,
F.
Belaj
and
N. C.
Mösch-Zanetti
*
*,
B.
Berner
,
F.
Belaj
and
N. C.
Mösch-Zanetti
*
Institute of Chemistry, University of Graz, Schubertstr. 1, 8010 Graz, Austria. E-mail: joerg.schachner@uni-graz.at
First published on 24th April 2019
Abstract
The syntheses of oxidorhenium(V) complexes [ReOCl(L1a–c)2] (3a–c), equipped with the bidentate, mono-anionic phenol–dimethyloxazoline ligands HL1a–c are described. Ligands HL1b–c contain functional groups on the phenol ring, compared to parent ligand 2-(4,4-dimethyl-4,5-dihydro-1,3-oxazol-2-yl)-phenol H1a; namely a methoxy group ortho to the hydroxyl position (2-(4,4-dimethyl-4,5-dihydro-1,3-oxazol-2-yl)-6-methoxyphenol, H1b), or a nitro group para to the hydroxyl position (2-(4,4-dimethyl-4,5-dihydro-1,3-oxazol-2-yl)-4-nitrophenol, H1c). Furthermore, oxidorhenate(V) complexes (NBu4)[ReOCl3(L1a–b)] (2a–b) were synthesized for solid state structural comparisons to 3a–b. All novel complexes are fully characterized including NMR, IR and UV-Vis spectroscopy, MS spectrometry, X-ray crystallography, elemental analysis as well as cyclic voltammetry. The influence of functional groups (R = –H, –OMe and –NO2) on the catalytic activity of 3a–c was investigated in two benchmark catalytic reactions, namely cyclooctene epoxidation and perchlorate reduction. In addition, the previously described oxidorhenium(V) complex [ReOCl(oz)2] (4), employing the phenol–oxazoline ligand 2-(4,5-dihydro-2-oxazolyl)phenol Hoz, was included in these catalysis studies. Complex 4 is a rare case in oxidorhenium(V) chemistry where two stereoisomers could be separated and fully characterized. With respect to the position of the oxazoline nitrogen atoms on the rhenium atom, these two stereoisomers are referred to as N,N-cis and N,N-trans isomer. A potential correlation between spectroscopic and structural data to catalytic activity was evaluated.
Introduction
The interest in the chemistry of high oxidation state rhenium oxides was certainly triggered by the two seminal publication on methyltrioxorhenium(VII) (MTO) by Herrmann in 1991.1 After finding an elegant synthesis of MTO, the group of Herrmann and others demonstrated the impressive catalytic activities in oxidation catalysis by MTO, including catalytic epoxidation of a wide variety of olefins.2,3–5 To this day, MTO remains the most active rhenium-based epoxidation catalyst, reaching turnover numbers (TONs) of >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000.3,6,7 However, MTO suffered from epoxide hydrolysis, when acid-sensitive epoxides are formed.8 This observed drawback, as well as decomposition reactions of MTO, prompted the investigation of oxidorhenium(V) complexes as potential epoxidation catalysts. The first two examples of such rhenium(V) complexes again came from the group of Herrmann, using tetra- and bidentate Schiff-base ligands.9,10 Over the last few years the chemistry of oxidorhenium(V) complexes and their application in homogeneous catalytic epoxidation of cyclooctene further developed.6,11–18 Compared to the impressive activities of MTO however, oxidorhenium(V) complexes had been inferior epoxidation catalysts, with the published complexes until 2014 only reaching turnovers of TON < 75.17 Our initial research efforts concentrated on phenol–pyrazole (HpyzR) ligands, equipped with various substituents R (R = H, Me, OMe, Br, NO2) on the phenol moiety (Fig. 1).14,16
000.3,6,7 However, MTO suffered from epoxide hydrolysis, when acid-sensitive epoxides are formed.8 This observed drawback, as well as decomposition reactions of MTO, prompted the investigation of oxidorhenium(V) complexes as potential epoxidation catalysts. The first two examples of such rhenium(V) complexes again came from the group of Herrmann, using tetra- and bidentate Schiff-base ligands.9,10 Over the last few years the chemistry of oxidorhenium(V) complexes and their application in homogeneous catalytic epoxidation of cyclooctene further developed.6,11–18 Compared to the impressive activities of MTO however, oxidorhenium(V) complexes had been inferior epoxidation catalysts, with the published complexes until 2014 only reaching turnovers of TON < 75.17 Our initial research efforts concentrated on phenol–pyrazole (HpyzR) ligands, equipped with various substituents R (R = H, Me, OMe, Br, NO2) on the phenol moiety (Fig. 1).14,16
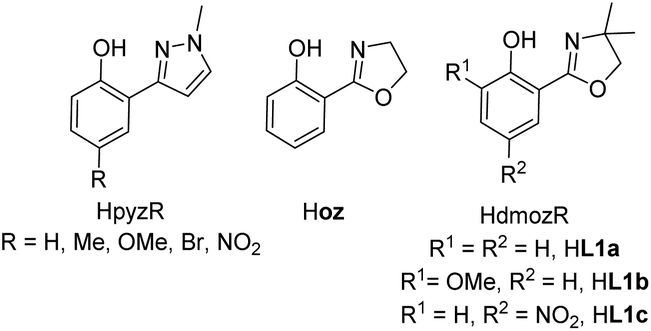 | ||
| Fig. 1 Phenol–pyrazole and phenol–oxazoline ON-bidentate ligands used in oxidorhenium(V) chemistry, shown with their commonly used abbreviations. | ||
The introduction of functional groups on the phenol moiety was found to have a beneficial effect on epoxidation activity of the resulting oxidorhenium(V) complexes [ReOCl(pyzR)2].14 Especially the set of complexes with R = OMe, Br and NO2 showed enhanced activities (TON > 95) in the epoxidation of cyclooctene (1 mol% cat. loading, 3 equiv. TBHP) as well as activity with the green oxidant H2O2 (TONs between 55 to 61).14 In contrast, complexes [ReOCl(pyzR)2] with R = H and Me showed either reduced epoxidation activity (TON < 60, with TBHP) or none at all (TON = 0, with H2O2) under the same conditions.14 Whereas MTO and related oxidorhenium(VII) complexes were shown to form side-on coordinated mono-peroxo and bis-peroxo complexes as the active catalyst,4,5,18,19 no such structures have been observed or isolated for oxidorhenium(V) complexes like 3a–c. A peroxide activation mechanism via deprotonation of the incoming peroxide similar to dioxidomolybdenum(VI) complexes20 seems less likely for oxidorhenium(V) complexes. Here, the oxido ligand is much less nucleophilic due to strong π-bonding.21
A second type of catalytic reaction effected by an oxidorhenium(V) complex was first described in 2000. The group of Abu-Omar reported the capability of complex [ReOCl(oz)2] (4), equipped with the phenol–oxazoline ligand Hoz (Fig. 1), to catalytically reduce perchlorate anions to chloride.22–24 We also became interested in this remarkable chemistry of the Hoz ligand, and began to investigate the oxazoline–dimethyl version of the Hoz ligand, the Hdmoz ligand (HL1a, Fig. 1), in perchlorate reduction chemistry.12 Based on the ground laying work of the Abu-Omar group, a dissociative oxygen atom transfer (OAT) mechanism of redox-catalysis was postulated, with the Re atom cycling between Re(V) and Re(VII) (Scheme 1).22–25
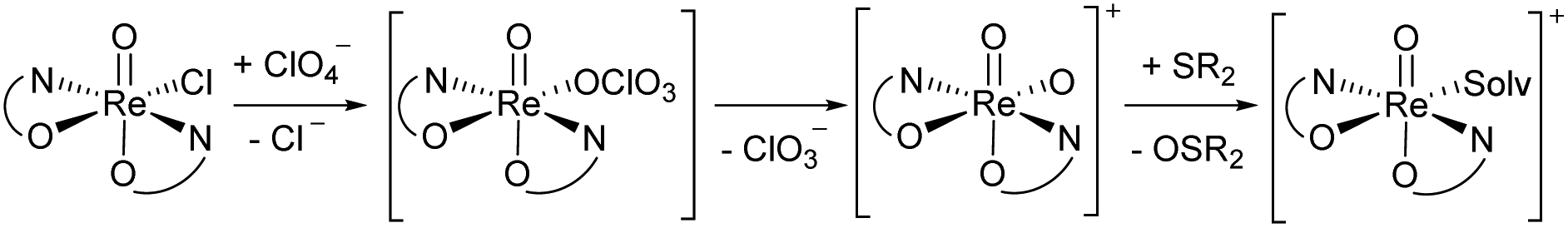 | ||
| Scheme 1 Proposed dissociative oxygen atom transfer mechanism of perchlorate reduction via Re(V)/(VII) redox catalysis; solv = solvent. | ||
In perchlorate reduction catalysis, we could show that isomers play an important role for catalyst activity.11,12 For an octahedral complex with two ON-bidentate and two monodentate ligands like 3a–c, six stereoisomers could form in principle (Scheme 2). Isomers A and B have a trans arrangement of the oxido and X ligand, with the bidentate ON-ligand in the equatorial plane. Both isomers contain an element of symmetry (σ or C2) and are therefore also referred to as “symmetric isomers”. Depending on the relative position of the nitrogen donor atom, isomer A is referred to as an N,N-cis isomer, B as an N,N-trans isomer. In isomers C to F, ligands O and X adopt a mutual cis orientation to each other, resulting in total loss of symmetry. Most of the isolated oxidorhenium(V) complexes belong to asymmetric isomers C and D.17 Again C constitutes an N,N-cis and D an N,N-trans isomer. Isomers E and F have not been observed yet in rhenium chemistry. This might be due to the strong trans influence of the oxido ligand, making the coordination of the neutral nitrogen donor in this position unlikely.
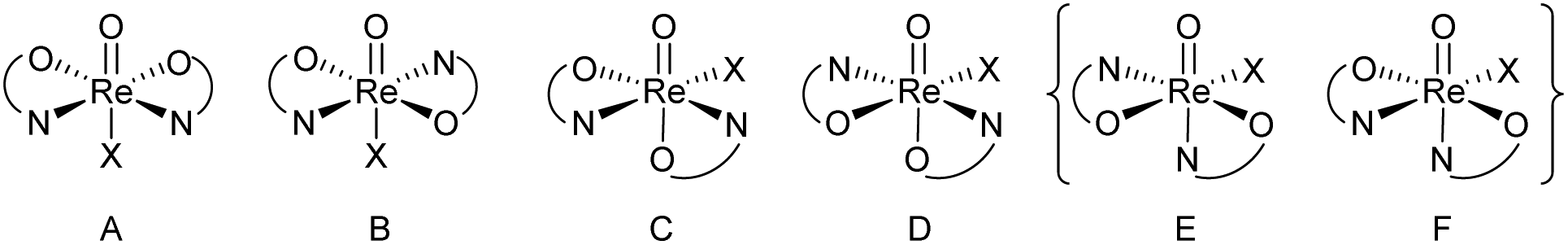 | ||
| Scheme 2 Possible stereoisomers A–F for octahedral [ReOX(ON)2] complexes (E and F have not been observed yet). | ||
For parent complex [ReOCl(oz)2] 4, both the N,N-cis (C, cis-4) and N,N-trans (D, trans-4) isomer are formed in the synthesis (Scheme 4). It could be shown that trans-4 is more active in perchlorate reduction compared to cis-4.12 The initial step of the catalytic cycle is loss of the chlorido ligand, creating a vacant site on the rhenium atom. In cis-4, the neutral nitrogen atom of the oxazoline moiety is trans to the chlorido ligand, exerting a weaker trans-influence compared to trans-4, where the phenolate oxygen atom is trans to the chlorido ligand (Scheme 2). This situation elongates the Re–Cl bond in trans-4 (2.4093(10) Å), compared to cis-4 (2.383(3) Å).12 Hence the loss of chlorido ligand requires less energy for trans-4. The same is true for the subsequent steps in the catalytic cycle. Those are OAT from the substrate to the rhenium atom and subsequent OAT from rhenium to the sulfide acceptor. For all these steps, the energy barriers are higher for cis-4 compared to trans-4.12 In the synthesis of complexes 3a–c, special attention was paid on the potential formation of N,N-cis or N,N-trans isomers (Scheme 4), because of their significant influence on catalytic activity.12,22,26,27 Stereocontrol in the synthesis of oxidorhenium(V) complexes to obtain the desired N,N-trans isomer is therefore highly desired. In literature, it was shown that a single methyl group on the oxazoline moiety is sufficient for the isomerically pure formation of an N,N-trans isomer.26,27 The resulting steric hindrance in an hypothetical N,N-cis complex was identified as one source of stereo-control.27 The observation that complexes cis/trans-4, without a substituent on the oxazoline moiety, are obtained as a mixture of N,N-cis and N,N-trans isomers supports this conclusion. Within this manuscript we would like to present an additional element of stereocontrol, based on the coordination chemistry of the Hdmoz ligands HL1a–c.
With this manuscript, we would like to present results obtained for the set of oxidorhenium(V) complexes [ReOCl(L1a–c)2] in epoxidation and perchlorate reduction catalysis. By systematic introduction of electron-donating (–OMe, HL1b) and an electron-withdrawing (–NO2, HL1c) groups (Fig. 1) we are relating structural and spectroscopic factors to catalytic activity. In addition factors that control the stereoselectivity of the synthesis of these complexes were investigated with the help of model complexes 2a–b. Finally the influence of stereoisomerism on catalytic epoxidation activity was tested with complexes cis- and trans-4.
Results and discussion
Synthetic procedures
Ligands H1a–b were synthesized according to literature,12,28,29 HL1c based on established literature procedures30 with modifications (see ESI†). Anionic oxidorhenate(V) complexes (NBu4)[ReOCl3(L1a–b)] 2a–b were obtained by reaction of precursor complex (NBu4)[ReOCl4] with one equivalent of ligand HL1a or HL1b at room temperature in EtOH (Scheme 3). Only this precursor, together with short reaction times (0.5–1 h) at room temperature, allows for isolation of mono-ligated complexes 2a–b. Under the same reaction conditions, precursor [ReOCl3(OPPh3)(SMe2)] gives mixtures with bis-ligated complexes 3a and 3b, respectively.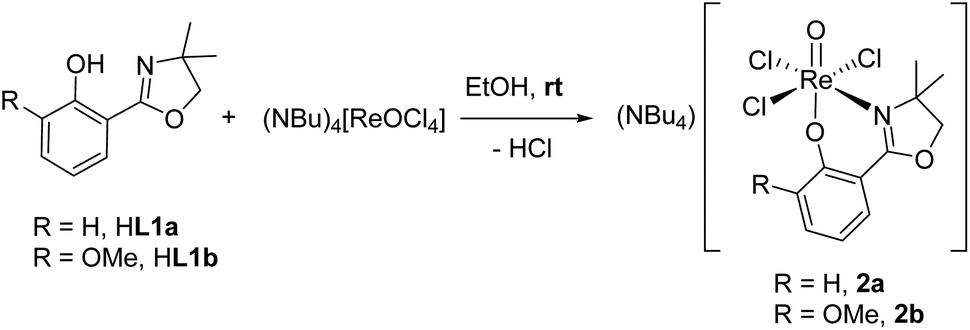 | ||
| Scheme 3 Synthesis of (NBu4)[ReOCl3(L1a–b)] 2a–b. | ||
Complexes [ReOCl(L1a–c)2] 3a–c were obtained by reaction of precursor complex [ReOCl3(OPPh3)(SMe2)] with two equivalent of ligands HL1a–c respectively, under refluxing conditions in CH3CN (Scheme 4). Complete dissolution of the poorly soluble precursor complex as well as a typical color change of the reaction mixtures to green (3a and 3c) or greenish-brown (3b) indicate successful synthesis of the respective complexes. The isolated complexes are stable to air and moisture, and are in general insoluble in non-polar solvents like pentane or heptane. Based on their substituents on the phenol moiety, complex 3b (–OMe) shows the highest solubility in polar solvents like CH2Cl2, CHCl3, or CH3CN, followed by 3a (–H) and 3c (–NO2) with the lowest solubility. Also alcohols like CH3OH and EtOH or CH3CN/H2O mixtures are possible solvents. When using the HdmozR ligands HL1a–c in the synthesis of complexes 3a–c, 1H NMR spectroscopy and solid state structural data obtained by single-crystal X-ray analyses (Fig. 3) confirmed the exclusive formation of N,N-trans isomers.
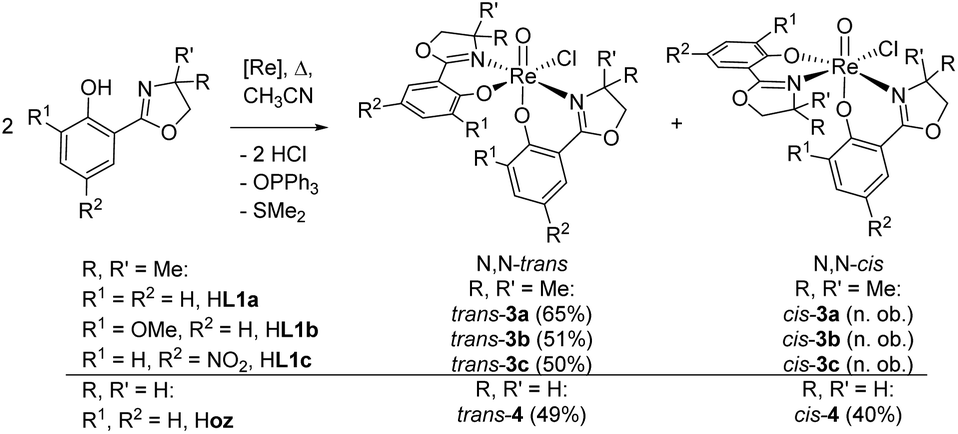 | ||
| Scheme 4 Synthesis (isolated yield) of single isomer of trans-3a–c and isomeric mixture of complexes cis/trans-4 ([Re] = [ReOCl3(OPPh3)(SMe2)]; n. ob. = not observed). | ||
Crystallography
Single crystals for all five novel complexes 2a–b and 3a–c were obtained, and their solid state structure could be solved by X-ray crystallography. The molecular structures of 2a and 2b revealed the expected anionic oxidorhenate(V) complexes with (NBu4)+ as counterion (Fig. 2). Both complexes adopt a slightly distorted octahedral conformation with similar bond lengths and angles (Table 1). The presence of the methoxy group in 2b does not result in a significantly different coordination around the rhenium atom, compared to 2a. Several other anionic tri-halo oxidorhenate(V) complexes have been structurally characterized and show the same coordination pattern.9,26,31–33 In all cases, the alkoxide oxygen of the ligand moiety coordinates trans to the oxido ligand, resulting in an axial–equatorial coordination of the bidentate ligand, with the three chlorido ligands in a meridional coordination. This preferred coordination might also explain the absence of isomers E and F (Scheme 2) in rhenium chemistry.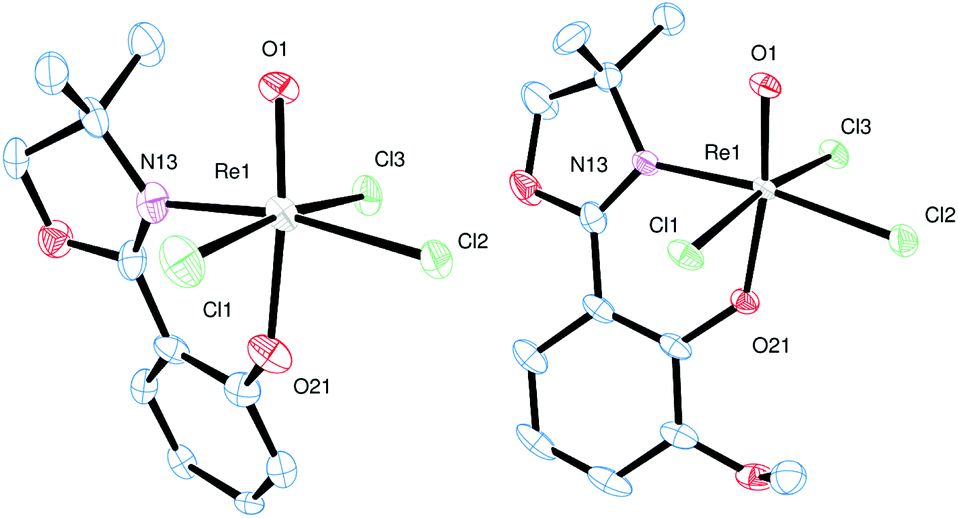 | ||
| Fig. 2 Molecular views (50% level) of 2a and 2b (H atoms, solvent and (NBu4)+ molecules omitted for clarity). | ||
| [Å] | 2a | 2b |
|---|---|---|
Re1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O1 O1 |
1.690(7) | 1.6801(16) |
| Re1–N13 | 2.172(8) | 2.1386(16) |
| Re1–O21 | 2.011(8) | 1.9800(16) |
| Re1–Cl1 | 2.381(3) | 2.3909(4) |
| Re1–Cl2 | 2.377(2) | 2.3717(4) |
| Re1–Cl3 | 2.420(3) | 2.3891(4) |
| [°] | ||
|---|---|---|
| O1–Re1–O21 | 171.4(3) | 171.18(9) |
| Cl1–Re1–Cl3 | 171.51(10) | 169.71(3) |
| N13–Re1–Cl2 | 169.7(2) | 171.40(6) |
Single crystals of complex 3a were obtained from an EtOH solution at 8 °C, of complex 3b from an EtOH/EtOAc mixture at room temperature, and of complex 3c from a saturated EtOH solution by slow evaporation at 8 °C. X-ray diffraction analysis revealed quite similar solid state structures, with the rhenium centers in a distorted octahedral coordination. In all cases the chlorido ligand is located cis to the oxido ligand. Furthermore, the N,N-trans orientation of the two bidendate ligands was confirmed for all three complexes (Fig. 3).
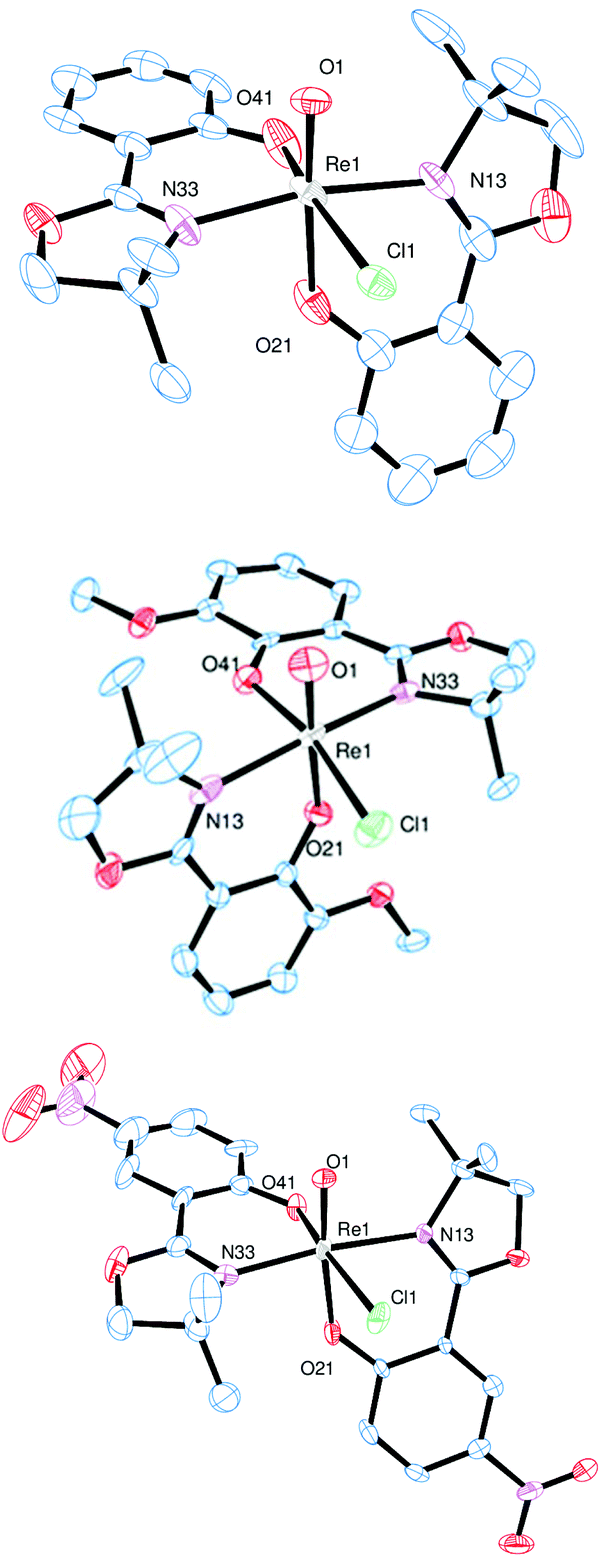 | ||
| Fig. 3 Molecular views (50% probability level) of complex 3a (top), 3b (middle) and 3c (bottom). Hydrogen atoms were omitted for clarity. | ||
A summary of selected bond angles and distances for 3a–c can be found in Table 2. The observed bond lengths in complexes 3a–c show that the solid state structures are sensitive to the substituents on the ligand moieties L1a–c. However, a clear trend is not easily extracted from the data at hand. For example, 3a displays the longest Re–Cl1 distance, indicating a stronger trans influence of the L1a ligand moiety, compared to L1b–c. Also, complex 3a displays the shortest ReO bond, compared to complexes 3b–c, which show comparable ReO bond lengths, regardless of the electron-donating or -withdrawing nature of the phenol substituents. The rhenium–oxido and rhenium–chlorido bond lengths in 3c are actually very similar to 3b, equipped with the electron-donating ligand L1b.
| [Å] | 3a | 3b | 3c |
|---|---|---|---|
| Re1O1 |
1.682(6) | 1.757(4) | 1.719(4) |
| Re1–Cl1 | 2.440(2) | 2.400(2) | 2.4087(18) |
| Re1–N13 | 2.203(5) | 2.118(5) | 2.205(3) |
| Re1–N33 | 2.058(5) | 2.096(4) | 2.037(3) |
| Re1–O21 | 2.056(7) | 1.987(4) | 2.033(3) |
| Re1–O41 | 1.944(7) | 1.999(4) | 1.962(3) |
| [°] | |||
|---|---|---|---|
| O1–Re1–O21 | 173.2(3) | 177.3(3) | 172.91(17) |
| Cl1–Re1–O41 | 163.8(6) | 168.07(18) | 166.62(10) |
| N13–Re1–N33 | 165.9(2) | 165.4(3) | 168.63(13) |
Stereo-control of isomers
Switching from the Hoz to the Hdmoz ligand class, with two methyl groups on the oxazoline moiety, only leads to the respective N,N-trans complexes (Scheme 4). In order to study this stereoselective formation of N,N-trans isomers of 3a–c in more detail, the two mono-ligated, anionic oxidorhenate(V) complexes (NBu4)[ReOCl3(L1a)] (2a) and (NBu4)[ReOCl3(L1b)] (2b) were synthesized (Scheme 3). The formation of mono-ligated complexes 2a–b provides evidence that the synthesis of bis-ligated complexes [ReOCl(L1a–c)2] is a step-wise process. Thus, whether the N,N-trans or N,N-cis isomer is formed in the next step depends on the respective trans influence of the initially coordinated ligand moiety (Scheme 5).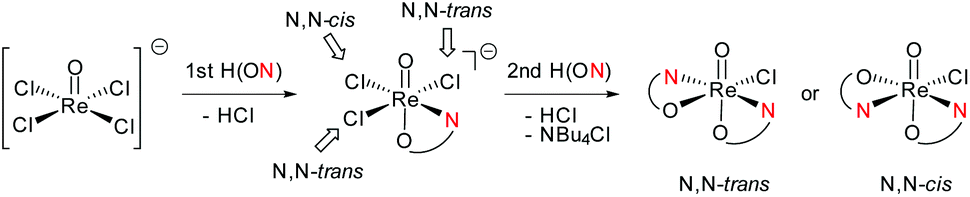 | ||
| Scheme 5 Schematic representation of step-wise coordination of a bidentate ligand H(ON) (e.g. HL1a–c), with formation of N,N-trans or N,N-cis isomers. Arrows indicate point of substitution of incoming phenolate oxygen of the second H(ON) ligand and resulting stereoisomer. | ||
Complex 2a can be compared to previously published complex (H2oz)[ReOCl3(oz)] (2-oz).26 As mentioned above, the respective bis-ligated complex 3a is isomerically pure, whereas 4 is isolated as an isomeric N,N-trans and N,N-cis mixture. As summarized in Table 3, for complex 2a, the two trans coordinated chlorido ligands Cl1 and Cl3 exhibit longer bond distances to the Re center (2.381(3) and 2.420(3) Å, respectively) compared to chlorido ligand Cl2, which is trans to the oxazoline nitrogen N13. Here the Re–Cl bond distance is at 2.377(2) Å, as expected based on the weaker trans influence of N13. The same observations are made in case of 2b, where bis-ligated complex 3b also only shows the N,N-trans isomer. Complex 2-oz displays an unusually long Re–Cl1 bond (2.4701(6) Å), which is caused by two H-bonds to neighboring (H2oz)+ cations in the unit cell.26 Therefore this bond has to be disregarded for the structural discussion. The remaining two bonds Re–Cl2 (2.3690(6) Å) and Re–Cl3 (2.3621(7) Å) are of very similar distances. Such a similar bonding situation could explain the observed mixtures of N,N-cis and N,N-trans isomers for 4, as there is no preference for the point of coordination of the second incoming Hoz ligand (Scheme 5). The shorter and therefore stronger Re–Cl2 bonds observed in 2a and 2b would explain the stereoselective synthesis of N,N-trans complexes 3a–b.
Cyclic voltammetry
In order to evaluate the electronic situation on the Re center with respect to the ligand substituents, cyclic voltammetry experiments on bis-ligated complexes 3a–c were performed. In addition, also the two isomers of N,N-cis/trans4 were investigated. Analyte solutions for cyclic voltammetry were near 1 mM in acetonitrile, with (NBu4)PF6 used as supporting electrolyte (0.1 M). The recorded currents Ip [μA] were divided by the actual concentration of analyte to normalize the voltammograms for better comparability. All five complexes displayed quasi-reversible Re(V)/(VI) redox couples at a positive potential,14,34 which also proved to be scan-rate independent. At negative potentials only irreversible redox events occurred. Half-wave potentials E1/2 (E1/2 = (Ep,c + Ep,a)/2) are given in Table 4 (referenced to ferrocene/ferrocenium).| [V] | 3a | 3b | 3c | cis-4 | trans-4 |
|---|---|---|---|---|---|
| E 1/2 | 0.642 | 0.613 | 0.922 | 0.576 | 0.582 |
For complexes 3a–c (Table 4 und Fig. 4), the half-wave potential of 3c is shifted by 280 mV to higher potential, compared to unsubstituted 3a, as expected for the electron-withdrawing nature of the nitro groups on the ligand moiety of L1c. Methoxy substituted complex 3b on the other hand is only shifted by 29 mV to lower potential, compared to 3a, indicating a small electron-donating effect of the methoxy substituents in 3b. The influence of the methyl groups on the oxazoline moiety can be compared between 3a and trans-4. Here, the Re(V)/(VI) redox couple for trans-4 is 60 mV lower than for 3a, indicating an electron-richer metal center in trans-4. The data in Table 4 and Fig. 4 show that the two stereoisomer of 4 have virtually identical redox potentials. Obviously, stereoisomerism does not have an effect on the redox potential of the Re center.
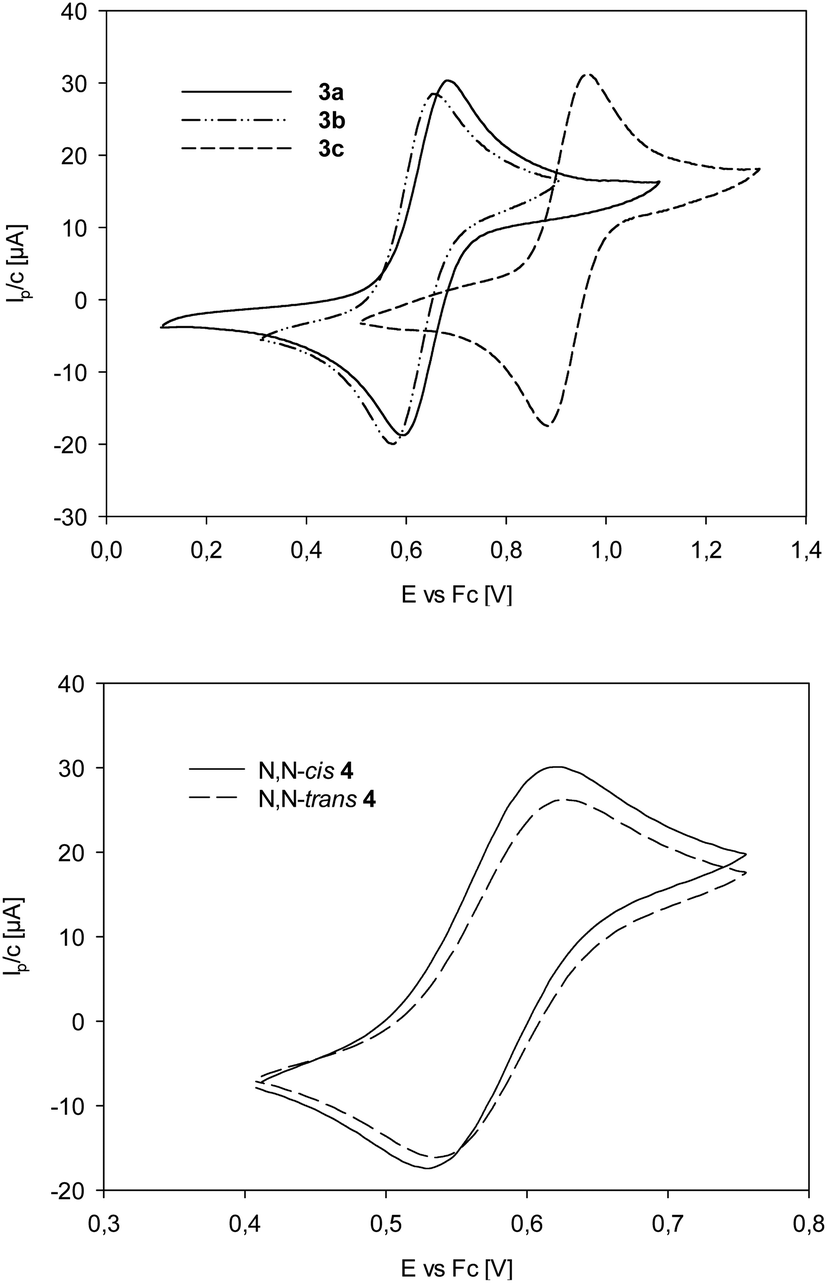 | ||
| Fig. 4 Top: Comparison of Re(V)/Re(VI) redox couples of complexes 3a–c; bottom: comparison of Re(V)/Re(VI) redox couples of complexes cis/trans-4. | ||
Epoxidation of cyclooctene
Complexes 2a–b, 3a–c and cis/trans-4 were evaluated as catalysts in the epoxidation of standard substrate cyclooctene. The effect of the different isomers cis/trans-4 and the introduction of electron-donating (–OMe in 3b) and withdrawing (–NO2 in 3c) groups on catalytic activity was of particular interest. Complexes 2a and 2b were also tested for potential catalytic activity, as other mono-ligated complexes have proven to be active epoxidation catalysts.14,15 In typical catalytic experiments 3 equiv. of tert-butylhydroperoxide (TBHP) as oxidant with a 1 mol% catalyst loading in CHCl3 at 50 °C and the substrate cyclooctene were used (Scheme 6). Reaction progress was monitored by withdrawing aliquots during the reaction and analysis with GC-MS. In general, all six tested complexes are catalytically active without induction period. A summary of turnover numbers (TON) with the respective turnover frequency (TOF [h−1]) is given in Table 5. The lowest activity was displayed by complex 2a, the highest by 3c. Complexes 3a and 3b showed activities in a similar range as cis/trans-4. All complexes tested showed a significant brightening to complete discoloration from the initially green solutions during catalytic experiments, hinting at the formation of catalytically inactive Re(VII) complexes.31 | ||
| Scheme 6 Epoxidation of cyclooctene using tert-butylhydroperoxide (TBHP). | ||
| TON (TOF) | |
|---|---|
| TOFs [h−1] were calculated at time of maximum yield of epoxide, not maximum activity of catalyst. If not mentioned otherwise, this was after 24 h.a Maximum yield of epoxide reached after 8.5 h. | |
| 2a | <10 |
| 2b | 23 (0.96) |
| 3a | 37 (1.5) |
| 3b | 34 (1.4) |
| 3c | 80 (9.4) |
| cis-4 | 40 (1.7) |
| trans-4 | 30 (1.3) |
Time-conversion plots of cyclooctene epoxidation are given in Fig. 5.
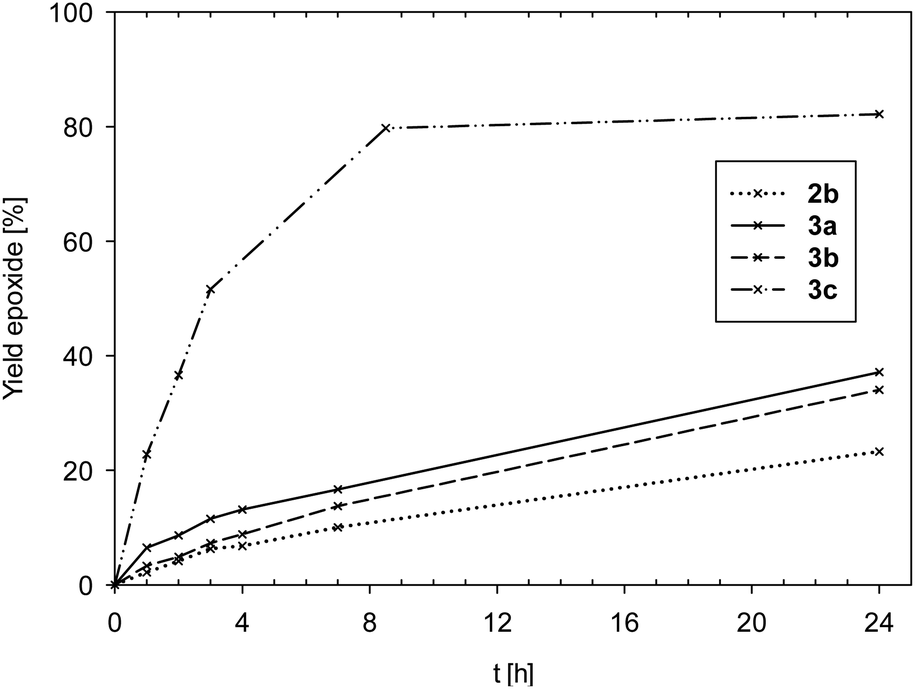 | ||
| Fig. 5 Comparison of yield of epoxide for complexes 2b and 3a–c (2a is not shown due to low activity). | ||
Data in Fig. 5 and Table 5 show complexes 2a and 2b to exhibit the lowest activities (2a: TON < 10; 2b: TON = 23) of all seven investigated complexes. The few examples of oxidorhenate(V) complexes, that were also tested in epoxidation, were of similar low activity.31,33 Also the bis-ligated complexes 3a (TON = 37) and 3b (TON = 34) showed low activities in the epoxidation of cyclooctene. In the cases of complexes 2b and 3b, significant discoloration after addition of oxidant TBHP could be observed, indicating a fast oxidation to unproductive perrhenate(VII) salts.31 Hence, ligand L1b seems to result in unstable complexes under strong oxidative conditions. This is quite in contrast to the OMe-substituted pyrazole–phenol complex [ReOCl(pyzOMe)2], which showed excellent activities in cyclooctene epoxidation for both TBHP and H2O2.14 A potential reason for the lack of activity for 2b and 3b could be ring-opening or decomposition reactions of the non-aromatic oxazoline moiety in L1b, a known problem in oxazoline chemistry.24 In contrast to 3b, complex 3c, coordinated by the electron-withdrawing ligand moiety L1c, showed the highest activity (TON = 80) of all seven complexes (Fig. 5). In this case, the same positive effect on catalytic activity was observed as with [ReOCl(pyzNO2)2].14 Obviously, the electron-withdrawing property of a nitro group results in enhanced epoxidation activity.
Data in Fig. 6 reveals a slight difference in catalytic performance of isomeric complexes cis- and trans-4, with cis-4 (TON = 40) performing slightly better compared to trans-4 (TON = 30).
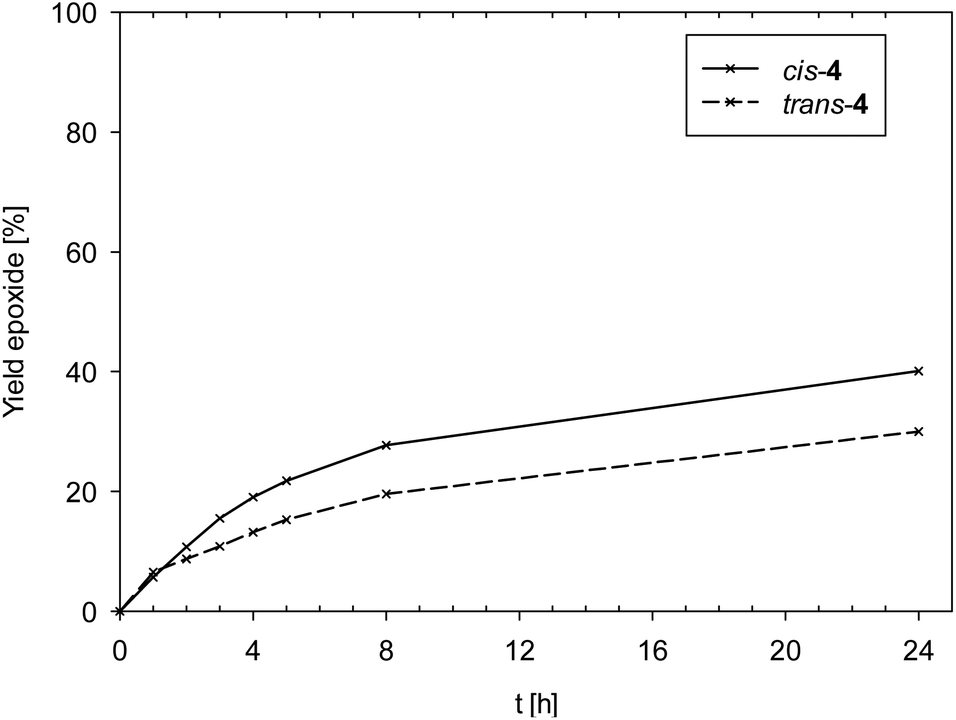 | ||
| Fig. 6 Yield of epoxide for isomeric complexes cis/trans-4. | ||
With the results from epoxidation catalysis in hand, a potential correlation to structural and spectroscopic data can be probed. Table 6 shows a summary of such data together with TONs in cyclooctene epoxidation. By comparing spectroscopic data of complexes 3a–c and cis/trans-4 no obvious correlation however between electronic properties and catalyst activity can be drawn.12,22 Neither the ReO IR absorption frequency nor the ReO bond lengths show a clear trend correlating to epoxidation activity. For example, cis-4 and 3c show similar ReO IR absorptions (957 vs. 963 cm−1), but very different activities in epoxidation (TON 40 vs. 80). Similarly, the ReO bond distance of 3c does not vary considerably from the other six ReO bond distances. Nevertheless, 3c is the most active epoxidation catalyst tested in this series. The only property of 3c that is significantly different compared to the other six complexes is the measured redox potential E1/2 of 0.922 V. This seems to indicate a more active catalyst if an electron poor rhenium center is present. The similarly active catalyst [ReOCl(pyzNO2)2] also showed a similar high redox potential E1/2 of 0.992 mV.14
|
ν(CN) [cm−1] |
ν(ReO) [cm−1] |
λ max (ε)a [] |
d(ReO) [Å] |
E
1/2b [V] |
TONb [] | |
|---|---|---|---|---|---|---|
| a In CH2Cl2, ε = molar extinction coefficient [dm−3 mol−1 cm−1]. b This work. c Only irreversible redox waves observed. | ||||||
| 2a | 1611 | 962 | 710 (73) | 1.690(7) | Irr.c | <10 |
| 2b | 1609 | 969 | 580 (56) | 1.6801(16) | Irr.c | 23 |
| 3a | 1604 | 955 | 635 (69)b | 1.682(6) | 0.642 | 37 |
| 3b | 1582 | 955 | 660 (65) | 1.757(4) | 0.613 | 34 |
| 3c | 1588 | 963 | 670 (85) | 1.719(4) | 0.922 | 80 |
| trans-422 | 1630 | 972 | 665 (87) | 1.693(2) | 0.582 | 30 |
| cis-412 | 1620 | 957 | 650 (69) | 1.689(8) | 0.576 | 40 |
To summarize, stereoisomers as in cis/trans-4 do not play a significant role in epoxidation activity. The observed differences in electronic and steric properties are most likely too small to cause a significant difference in catalytic activity. Electron-withdrawing substituents seem to have a beneficial effect on epoxidation, as both [ReOCl(dmozNO2)2] 3c as well as the previously published complex [ReOCl(pyzNO2)2] showed enhanced catalytic activity.14 These results indicate that an electron-deficient rhenium center results in higher catalyst activity and must therefore facilitate the oxygen atom transfer from TBHP to the olefin. The exact mode of interaction between the rhenium center and TBHP remains unclear at this point. The redox potential E1/2 might have predictive power concerning catalyst activity.
Perchlorate reduction
Complexes 2a–b, 3a–c and cis/trans-4 were tested in the challenging reduction of perchlorate to chloride, with a 3.2 mol% catalyst loading in the presence of four equivalents of the sulfide acceptor SMe2 (DMS) (Scheme 7). Reactions were conducted in CD3CN/D2O mixtures of 95/5 or 50/50 vol%, and the conversion of SMe2 to OSMe2 (DMSO) was monitored by 1H NMR spectroscopy over 24 h. Whereas the 95/5 vol% mixture is necessary to fully dissolve the LiClO4 substrate, we were interested if higher water contents in the reaction affect catalyst activity. This could either occur by potential hydrolytic decomposition of the catalyst or coordination of the H2O molecule to the vacant site on the rhenium thereby inhibiting catalysis. From reaction of 4 with AgOTf (OTf = SO2CF3−) it is known that H2O will coordinate to the resulting cationic oxidorhenium(V) triflate complex. The solid state structure of complex [ReO(oz)2(H2O)]OTf revealed the formation of a symmetric N,N-trans stereoisomer (isomer B, Scheme 7), with the water oxygen atom coordinated trans to the oxido ligand and the oz ligands in equatorial position.23 From DFT calculations it was shown that a symmetric cation B is not catalytically active in perchlorate reduction, as the coordination site trans to the oxido ligand is deactivated for perchlorate coordination.12 The catalytically active species therefore should remain an isomer D (Scheme 7), with the vacant coordination site cis to the oxido ligand.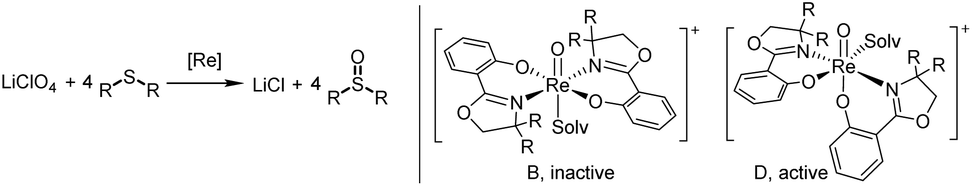 | ||
| Scheme 7 Left: catalytic perchlorate reduction by oxidorhenium(V) complexes; right: catalytically inactive and active stereoisomers in perchlorate reduction; solv = solvent. | ||
Results are summarized in Table 7. While mono-ligated rhenate complexes 2a–b are not active, complexes 3a–c and cis/trans-4 were capable to reduce perchlorate at both water contents (5 and 50 vol%), with the exception of nitro-substituted complex 3c. Complex 3c showed complete loss of catalytic activity at 50 vol% water. In this case however, we attribute this to the low solubility of 3c in aqueous media. In both solvent mixtures, solid 3c can be observed throughout the 24 h experiment time. Similar to our previous findings in 5 vol% water,12trans-4 remains the more active complex also at 50 vol% water (91%), compared to cis-4 (58%). It is interesting to observe though that only for cis-4 the catalytic activity increases with increasing water content (33% at 5 vol% vs. 58% at 50 vol%). At 5 vol% water, 3a and 3b show essentially the same catalytic activity (75 and 78% respectively, Table 7). At 50 vol% water however, 3a shows a significantly decreased activity (75 vs. 32%). With 3b, the difference is not as pronounced (78 vs. 64%).
For all active complexes, catalyst decomposition can be observed by 1H NMR spectroscopy, as more and more free ligand is visible over the 24 h reaction time. A discoloration of the initially green solutions points to decomposition to perrhenate.31 The inactivity of complexes 2a–b is consistent to previously tested, neutral mono-ligated complexes.12 The observed differences in catalytic activity in regard to water content are subject to current investigations in our lab.
Conclusions
Oxidorhenium(V) complexes equipped with phenol–dimethloxazoline ligands remain as a unique class of complexes capable to both catalytically epoxidize cyclooctene and reduce the challenging substrate perchlorate to harmless chloride under mild, aqueous conditions. In olefin epoxidation, stereoisomers do not play a significant role in catalyst activity, as demonstrated by the very similar TONs of cis/trans-4 (40/30). Electron withdrawing substituents rather seem to result in more active catalysts. Nitro-substituted complex 3c showed the highest activity with a TON of 80 for cyclooctene. This is line with the nitro-substituted complex [ReOCl(pyzNO2)2], which also proved to be a similarly active epoxidation catalyst.14 From comparison of spectral and structural data, it seems the redox potential E1/2 obtained from cyclic voltammetry measurements correlates with enhanced activity in epoxidation, in contrast to IR absorptions or bond lengths.In contrast to epoxidation catalysis, stereoisomers play a vital role for catalyst activity in perchlorate reduction. Here, only N,N-trans isomers are active. The formation of such N,N-trans isomers is a unique feature of the oxazoline ligand moiety. All related pyrazole based complexes [ReOCl(pyzR)2] adopted the N,N-cis conformation in the solid state, which also explains their inactivity in perchlorate reduction catalysis.16 For the observed stereoselective synthesis of complexes 3a–c, a novel mechanism is proposed, based on the two-step coordination of the respective HL1a–c ligands. Coordination of the first ligand gives intermediate oxidorhenate(V) complexes [ReOCl3(L1a–c)]−, with a strengthened Re–Cl2 bond thereby directing the second incoming ligand to form an N,N-trans complex. Finally, H2O seems to play a far more important role in perchlorate catalysis than simply dissolving the perchlorate salt. It potentially can also act as an inhibitor by competing with substrate for the vacant coordination site on rhenium and favoring the formation of inactive cationic stereoisomers. Complexes 3a and 3b showed a diminished activity at 50 vol% H2O compared to 5 vol%, whereas conversion to DMSO actually increased for less active stereoisomer cis-4. The exact role of H2O however is subject to ongoing investigations in our lab.
Experimental section
General
The rhenium precursors [ReOCl3(OPPh3)(SMe2)]35 and (NBu4)[ReOCl4]36 were prepared according to previously published methods. The ligands HL1a,27,29 HL1b28 as well as complexes 3a12 and cis/trans-412 were prepared via published routes. Synthesis of HL1c can be found in the ESI.† Chemicals were purchased from commercial sources and used without further purification. NMR spectra were recorded with a Bruker Avance (300 MHz) instrument. Chemical shifts are given in ppm and are referenced to residual protons in the solvent. Coupling constants (J) are given in Hertz (Hz). Mass spectra were recorded with an Agilent 5973 MSD – Direct Probe using the EI ionization technique. Samples for infrared spectroscopy were measured on a Bruker Optics ALPHA FT-IR Spectrometer equipped with an ATR diamond probe head. GC-MS measurements were performed on an Agilent 7890 A with an Agilent 19091J–433 column coupled to a mass spectrometer type Agilent 5975 C. Electrochemical measurements were performed under an inert N2 atmosphere in a glove box in dry acetonitrile (stored over molecular sieve) with a Gamry Instruments Reference 600 Potentiostat using a three electrode setup. Platin was used as working electrode, Pt wire (99.99%) as supporting electrode; the reference electrode was a Ag wire immersed in a solution of 0.01 M AgNO3 and 0.1 M (NBu4)PF6 in CH3CN separated from the solution by a Vycor® tip. Supporting electrolyte used was (NBu4)PF6 (0.1 M). Elemental analyses were carried out using a Heraeus Vario Elementar automatic analyzer at the University of Technology Graz.X-ray structure determinations
Crystallographic data (excluding structure factors) for 2a, 2b, 3a, 3b, and 3c were deposited with the Cambridge Crystallographic Data Center as supplementary publication no. 1895410 (2a), 1850856 (2b), 1562677 (3a), 1562678 (3b) and 1562679 (3c).†Epoxidation of cyclooctene
A Heidolph Parallel Synthesizer 1 was used for all epoxidation experiments. In a typical experiment, 2–3 mg of catalyst (1 mol%) were dissolved in 0.5 mL CHCl3 and mixed with cyclooctene (1 equiv.) and 50 μL of mesitylene (internal standard) and heated to the respective reaction temperature (50 °C). Then the oxidant (3 equiv.) was added. Aliquots for GC–MS (20 μL) were withdrawn at given time intervals, quenched with MnO2 and diluted with HPLC grade ethyl acetate. The reaction products were analysed by GC–MS (Agilent 7890 A with an Agilent 19091J–433 column coupled to a mass spectrometer type Agilent 5975 C), and the epoxide produced from each reaction mixture was quantified vs. mesitylene as the internal standard. Experimental error of GC-MS measurements is ±5%.Synthesis of complex (NBu4)[ReOCl3(L1a)] 2a
Ligand HL1a (0.030 g, 0.16 mmol, 1 equiv.) and (NBu4)[ReOCl4] (0.10 g, 0.16 mmol, 1 equiv.) were dissolved in 20 ml EtOH and stirred at rt for 1 h. The mixture turns green within 5 minutes. After 1 h, a small amount of white precipitate (probably NBu4Cl) is filtered off and the solvent removed completely. The crude product is a mixture of 2a and bis-ligated 3a, which can be separated by dissolving the better soluble 3a in small amounts of EtOAc, leaving behind analytical pure yellowish–greenish 2a (62 mg, 52%). Recrystallization from EtOH/EtOAc (1/1 vol%) yielded 2a as single crystals. 1H NMR (300 MHz, chloroform-d) δ 7.61 (dd, J = 7.9, 1.8 Hz, 1H), 7.12 (ddd, J = 8.7, 7.1, 1.8 Hz, 1H), 7.00–6.90 (m, 1H), 6.90 (ddd, J = 8.2, 7.1, 1.2 Hz, 1H), 4.51 (s, 2H), 3.35–3.23 (m, 8H), 1.74 (s, 6H), 1.72–1.61 (m, 8H), 1.53–1.38 (m, 8H), 1.00 (t, J = 7.3 Hz, 12H). 13C NMR (75 MHz, chloroform-d) δ 136.33, 131.32, 118.80, 116.96, 108.59, 78.98 (CH2), 73.54, 59.41 (NBu4+), 26.78 (Me2), 24.39 (NBu4+), 19.95 (NBu4+), 13.90 (NBu4+), (two C obscured); ATR-IR νmax/cm−1: 2960, 2930, 2871, 1612 (CN), 1482, 1378, 1323, 1271, 1082, 962 and 955 (ReO), 879, 860, 756, 616; EI-MS (m/z): 497.9 (M+ − NBu4); UV λmax(CH2Cl2)/nm: 710 (ε/dm3 mol−1 cm−1 73); elemental analysis calculated for C27H48Cl3N2O4Re (741.3 g mol−1): C 43.75, H 6.53, N 3.78; found C 42.83, 6.51 H, 3.30 N.
Synthesis of complex (NBu4)[ReOCl3(L1b)] 2b
Ligand HL1b (0.083 g, 0.375 mmol, 1.1 equiv.) and (NBu4)[ReOCl4] (0.20 g, 0.34 mmol, 1 equiv.) were dissolved in EtOH and stirred at rt for 0.5 h. An intense green color formed moments after dissolution. Upon concentration unreacted (NBu4)[ReOCl4] precipitated and was removed by filtration. Removal of solvent yielded crude 2b. Recrystallization from EtOH/EtOAc (1/1 vol%) yielded 2b as single crystals (105 mg, 42%). 1H NMR (300 MHz, CDCl3) δ 7.30 (dd, J = 5.6, 4.1 Hz, 1H), 6.81–6.77 (m, 2H), 4.51 (s, 2H), 3.83 (s, 3H), 3.26–3.15 (m, 8H), 1.74 (s, 6H), 1.65–1.52 (m, 8H), 1.37 (m, 8H), 0.95 (t, J = 7.3 Hz, 12H); 13C NMR (75 MHz, CDCl3) δ 166.53, 157.48, 148.69, 124.86, 123.98, 116.77, 109.78, 100.11, 78.91 (CH2), 73.66, 59.54 (OCH3), 58.83 (NBu4+), 26.81 (Me2), 24.25 (NBu4+), 19.83 (NBu4+), 13.89 (NBu4+); ATR-IR νmax/cm−1: 2959, 2933, 1609 (CN), 1582, 1458, 1379, 1254, 1055, 969 and 957 (ReO), 845, 737, 670, 567, 435; EI-MS (m/z): 528.1 (M+ − NBu4); UV λmax(CH2Cl2)/nm: 580 (ε/dm3 mol−1 cm−1 56); elemental analysis calculated for C28H50Cl3N2O4Re (771.3 g mol−1): C 43.60, H 6.53, N 3.63; found C 43.78, H 6.48, N 3.66.
UV-Vis data of complex 3a. UV λmax(CH2Cl2)/nm: 650 (ε/dm3 mol−1 cm−1 70); other analytical data has been previously published.12
Synthesis of complex [ReOCl(L1b)2] 3b
Ligand HL1b (0.63 g, 2.85 mmol, 2 equiv.) and precursor [ReOCl3(OPPh3)(SMe2)] (0.93 g, 1.43 mmol, 1 equiv.) was heated under reflux in 30 ml CH3CN for 3 h. Analytically pure 3b precipitated upon cooling and was isolated by filtration (495 mg, 51%). Single crystals suitable for X-ray diffraction analysis were obtained from EtOH/EtOAc. 1H NMR (300 MHz, chloroform-d) δ 7.53 (dd, J = 8.3, 1.6 Hz, 1H), 7.34 (dd, J = 8.0, 1.6 Hz, 1H), 6.92–6.82 (m, 2H), 6.76 (dd, J = 8.0, 1.6 Hz, 1H), 6.67 (t, J = 8.1 Hz, 1H), 4.67–4.51 (m, 4H), 3.73 (s, 3H), 3.67 (s, 3H), 2.00 (s, 3H), 1.90 (s, 6H), 1.64 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 150.22, 121.98, 121.33, 118.65, 117.54, 116.48, 115.70, 110.92, 81.89, 79.47, 78.75, 72.92, 55.86 (OCH3), 55.43 (OCH3), 27.61, 27.10, 27.07, 25.97 (some C are obscured); ATR-IR νmax/cm−1: 1582 (s, CN), 1379 (s), 1052 (m), 955 (s, ReO); EI-MS (m/z): 678.2 (M+); UV λmax(CH2Cl2)/nm: 660 (ε/dm3 mol−1 cm−1 65); elemental analysis calculated for C24H28ClN2O7Re (678.15 g mol−1): C 42.51, H 4.16, N 4.13; found C 42.74, H 4.06, N 4.08.
Synthesis of complex [ReOCl(L1c)2] 3c
Ligand HL1c (0.50 g, 2.12 mmol, 2 equiv.) and precursor [ReOCl3(OPPh3)(SMe2)] (0.69 g, 1.06 mmol, 1 equiv.) was heated under reflux in 30 ml CH3CN for 4 h. Analytically pure 3c precipitated upon cooling and was isolated by filtration (375 mg, 50%). Single crystals suitable for X-ray diffraction analysis were obtained from DCM/heptane. 1H NMR (300 MHz, CDCl3) δ 8.98 (d, J = 2.8 Hz, 1H), 8.72 (d, J = 2.9 Hz, 1H), 8.24 (dd, J = 9.3, 2.9 Hz, 1H), 8.07 (dd, J = 9.2, 2.9 Hz, 1H), 6.90 (d, J = 9.4 Hz, 2H), 6.74 (d, J = 9.2 Hz, 1H), 4.79 (dd, J = 8.5, 4.1 Hz, 2H), 4.70 (d, J = 8.5 Hz, 1H), 4.60 (d, J = 8.5 Hz, 1H), 1.96 (s, 3H), 1.95 (s, 3H), 1.84 (s, 3H), 1.67 (s, 3H); because of the low solubility of 3c no meaningful 13C NMR spectra could be acquired; HSQC (CDCl3) δ 131.05, 130.26, 128.19, 128.01, 122.44, 119.35, 82.40, 82.24, 79.67, 79.62, 27.15, 27.13, 27.10, 25.86; ATR-IR νmax/cm−1: 1611, 1588 (s, CN), 1312, 1269 (s, NO2), 963 (s, ReO); EI-MS (m/z): 708.3 (M+); UV λmax(CH2Cl2)/nm: 670 (ε/dm3 mol−1 cm−1 85); elemental analysis calculated for C22H22ClN4O9Re·0.5CH2Cl2 (708.06 g mol−1): C 36.00, H 3.09; N 7.46; found C 36.01; H 2.98; N 7.00.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The organizers of the 7th EuCheMS conference on N-ligands in September 2018, Lisbon, where parts of this research was presented, are thankfully acknowledged. Financial support from NAWI Graz is gratefully acknowledged.Notes and references
- (a) W. A. Herrmann, R. W. Fischer and D. W. Marz, Angew. Chem., Int. Ed. Engl., 1991, 30, 1638–1641 CrossRef; (b) W. A. Herrmann and M. Wang, Angew. Chem., Int. Ed. Engl., 1991, 30, 1641–1643 CrossRef.
- (a) R. G. Harms, W. A. Herrmann and F. E. Kühn, Coord. Chem. Rev., 2015, 296, 1–23 CrossRef CAS; (b) J. Rudolph, K. L. Reddy, J. P. Chiang and K. B. Sharpless, J. Am. Chem. Soc., 1997, 119, 6189–6190 CrossRef CAS; (c) C. Copéret, H. Adolfsson and K. Barry Sharpless, Chem. Commun., 1997, 1565–1566 RSC; (d) C. C. Romão, F. E. Kühn and W. A. Herrmann, Chem. Rev., 1997, 97, 3197–3246 CrossRef.
- S. Huber, M. Cokoja and F. E. Kühn, J. Organomet. Chem., 2014, 751, 25–32 CrossRef CAS.
- F. E. Kühn, A. Scherbaum and W. A. Herrmann, J. Organomet. Chem., 2004, 689, 4149–4164 CrossRef.
- W. A. Herrmann, J. Organomet. Chem., 1995, 500, 149–173 CrossRef CAS.
- J. W. Kück, R. M. Reich and F. E. Kühn, Chem. Rec., 2016, 16, 349–364 CrossRef PubMed.
- S. Yamazaki, Org. Biomol. Chem., 2007, 5, 2109–2113 RSC.
- (a) W. Adam and C. M. Mitchell, Angew. Chem., Int. Ed. Engl., 1996, 35, 533–535 CrossRef CAS; (b) T. R. Boehlow and C. D. Spilling, Tetrahedron Lett., 1996, 37, 2717–2720 CrossRef CAS; (c) P. Altmann, M. Cokoja and F. E. Kühn, Eur. J. Inorg. Chem., 2012, 2012, 3235–3239 CrossRef CAS.
- W. A. Herrmann, M. U. Rauch and G. R. J. Artus, Inorg. Chem., 1996, 35, 1988–1991 CrossRef CAS.
- F. E. Kühn, M. U. Rauch, G. M. Lobmaier, G. R. J. Artus and W. A. Herrmann, Chem. Ber., 1997, 130, 1427–1431 CrossRef.
- N. Zwettler, J. A. Schachner, F. Belaj and N. C. Mösch-Zanetti, Inorg. Chem., 2016, 55, 5973–5982 CrossRef CAS PubMed.
- J. A. Schachner, B. Terfassa, L. M. Peschel, N. Zwettler, F. Belaj, P. Cias, G. Gescheidt and N. C. Mösch-Zanetti, Inorg. Chem., 2014, 53, 12918–12928 CrossRef CAS.
- (a) B. Terfassa, J. A. Schachner, P. Traar, F. Belaj and N. C. Mösch-Zanetti, Polyhedron, 2014, 75, 141–145 CrossRef CAS; (b) B. Machura, M. Wolff, D. Tabak, J. A. Schachner and N. C. Mösch-Zanetti, Eur. J. Inorg. Chem., 2012, 3764–3773 CrossRef CAS; (c) S. Dinda, M. G. B. Drew and R. Bhattacharyya, Catal. Commun., 2009, 10, 720–724 CrossRef CAS.
- N. Zwettler, J. A. Schachner, F. Belaj and N. C. Mösch-Zanetti, Inorg. Chem., 2014, 53, 12832–12840 CrossRef CAS PubMed.
- B. Machura, M. Wolff, E. Benoist, J. A. Schachner and N. C. Mösch-Zanetti, Dalton Trans., 2013, 42, 8827–8837 RSC.
- P. Traar, J. A. Schachner, L. Steiner, A. Sachse, M. Volpe and N. C. Mösch-Zanetti, Inorg. Chem., 2011, 50, 1983–1990 CrossRef CAS PubMed.
- B. Machura, M. Wolff and I. Gryca, Coord. Chem. Rev., 2014, 275, 154–164 CrossRef CAS.
- F. E. Kühn, A. M. Santos and W. A. Herrmann, Dalton Trans., 2005, 34, 2483–2491 RSC.
- W. A. Herrmann, R. W. Fischer, M. U. Rauch and W. Scherer, J. Mol. Catal., 1994, 86, 243–266 CrossRef CAS.
- (a) L. F. Veiros, A. Prazeres, P. J. Costa, C. C. Romão, F. E. Kühn and M. J. Calhorda, Dalton Trans., 2006, 1383–1389 RSC; (b) J. Morlot, N. Uyttebroeck, D. Agustin and R. Poli, ChemCatChem, 2013, 5, 601–611 CrossRef CAS; (c) M. E. Judmaier, C. Holzer, M. Volpe and N. C. Mösch-Zanetti, Inorg. Chem., 2012, 51, 9956–9966 CrossRef CAS PubMed.
- J. R. Winkler and H. B. Gray, in Molecular electronic structures of transition metal complexes, ed. D. M. P. Mingos, J. P. Dahl and M. Atanasov, Springer, Berlin, 2012, pp. 17–28 Search PubMed.
- M. M. Abu-Omar, L. D. McPherson, J. Arias and V. M. Béreau, Angew. Chem., Int. Ed., 2000, 39, 4310–4313 CrossRef CAS.
- J. Arias, C. R. Newlands and M. M. Abu-Omar, Inorg. Chem., 2001, 40, 2185–2192 CrossRef CAS PubMed.
- L. D. McPherson, M. Drees, S. I. Khan, T. Strassner and M. M. Abu-Omar, Inorg. Chem., 2004, 43, 4036–4050 CrossRef CAS.
- M. M. Abu-Omar, Comments Inorg. Chem., 2003, 24, 15–37 CrossRef CAS.
- J. Liu, D. Wu, X. Su, M. Han, S. Y. Kimura, D. L. Gray, J. R. Shapley, M. M. Abu-Omar, C. J. Werth and T. J. Strathmann, Inorg. Chem., 2016, 55, 2597–2611 CrossRef CAS.
- J. Liu, X. Su, M. Han, D. Wu, D. L. Gray, J. R. Shapley, C. J. Werth and T. J. Strathmann, Inorg. Chem., 2017, 56, 1757–1769 CrossRef CAS PubMed.
- H. C. Aspinall, O. Beckingham, M. D. Farrar, N. Greeves and C. D. Thomas, Tetrahedron Lett., 2011, 52, 5120–5123 CrossRef CAS.
- P. G. Cozzi, C. Floriani, A. Chiesi-Villa and C. Rizzoli, Inorg. Chem., 1995, 34, 2921–2930 CrossRef CAS.
- M. Hoogenraad, K. Ramkisoensing, S. Gorter, W. L. Driessen, E. Bouwman, J. G. Haasnoot, J. Reedijk, T. Mahabiersing and F. Hartl, Eur. J. Inorg. Chem., 2002, 377–387 CrossRef CAS.
- G. M. Lobmaier, G. D. Frey, R. D. Dewhurst, E. Herdtweck and W. A. Herrmann, Organometallics, 2007, 26, 6290–6299 CrossRef CAS.
- (a) S. Gatto, T. I. A. Gerber, G. Bandoli, J. Perils and J. G. H. Du Preez, Inorg. Chim. Acta, 1998, 269, 235–240 CrossRef CAS; (b) C. Bolzati, F. Tisato, F. Refosco, G. Bandoli and A. Dolmella, Inorg. Chem., 1996, 35, 6221–6229 CrossRef CAS; (c) S. M. Harben, P. D. Smith, R. L. Beddoes, D. Collison and C. D. Garner, J. Chem. Soc., Dalton Trans., 1997, 2777–2784 RSC; (d) H. Luo, S. J. Rettig and C. Orvig, Inorg. Chem., 1993, 32, 4491–4497 CrossRef CAS; (e) M. Shivakumar, S. Banerjee, M. Menon and A. Chakravorty, Inorg. Chim. Acta, 1998, 275–276, 546–551 CrossRef CAS; (f) J. D. G. Correia, Â. Domingos, I. Santos, C. Bolzati, F. Refosco and F. Tisato, Inorg. Chim. Acta, 2001, 315, 213–219 CrossRef CAS; (g) E. J. de Souza, V. M. Deflon, A. G. de, A. Fernandes, S. S. Lemos, A. Hagenbach and U. Abram, Inorg. Chim. Acta, 2006, 359, 1513–1518 CrossRef CAS.
- A. Sachse, N. C. Mösch-Zanetti, G. Lyashenko, J. W. Wielandt, K. Most, J. Magull, F. Dall'Antonia, A. Pal and R. Herbst-Irmer, Inorg. Chem., 2007, 46, 7129–7135 CrossRef CAS PubMed.
- C. C. van Kirk, V. M. Béreau, M. M. Abu-Omar and D. H. Evans, J. Electroanal. Chem., 2003, 541, 31–38 CrossRef CAS.
- B. D. Sherry, R. N. Loy and F. D. Toste, J. Am. Chem. Soc., 2004, 126, 4510–4511 CrossRef CAS PubMed.
- R. Alberto, R. Schibli, A. Egli, P. August Schubiger, W. A. Herrmann, G. R. J. Artus, U. Abram and T. A. Kaden, J. Organomet. Chem., 1995, 493, 119–127 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details on synthesis of ligand H1c as well as crystallographic data of complexes 2a–b and 3a–c. CCDC 1895410, 1850856 and 1562677–1562679. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt01352k |
| This journal is © The Royal Society of Chemistry 2019 |