
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure–function relationship for CO2 methanation over ceria supported Rh and Ni catalysts under atmospheric pressure conditions
Natalia M.
Martin
 *a,
Felix
Hemmingsson
a,
Andreas
Schaefer
a,
Martin
Ek
b,
Lindsay R.
Merte
c,
Uta
Hejral
d,
Johan
Gustafson
d,
Magnus
Skoglundh
a,
Ann-Christin
Dippel
e,
Olof
Gutowski
e,
Matthias
Bauer
f and
Per-Anders
Carlsson
a
*a,
Felix
Hemmingsson
a,
Andreas
Schaefer
a,
Martin
Ek
b,
Lindsay R.
Merte
c,
Uta
Hejral
d,
Johan
Gustafson
d,
Magnus
Skoglundh
a,
Ann-Christin
Dippel
e,
Olof
Gutowski
e,
Matthias
Bauer
f and
Per-Anders
Carlsson
a
aDepartment of Chemistry and Chemical Engineering and Competence Centre for Catalysis, Chalmers University of Technology, Göteborg, 412 96, Sweden. E-mail: Natalia.Martin@chalmers.se; Fax: +46 31160062; Tel: +46 31 772 29 04
bCentre for Analysis and Synthesis, Lund University, 22100 Lund, Sweden
cDepartment of Materials Science and Applied Mathematics, Malmö University, 204 06 Malmö, Sweden
dDivision of Synchrotron Radiation Research, Lund University, 22100 Lund, Sweden
eDeutsches Elektronen-Synchrotron DESY, 22607 Hamburg, Germany
fDepartment of Chemistry, Paderborn University, 33098 Paderborn, Germany
First published on 5th March 2019
Abstract
In situ structural and chemical state characterization of Rh/CeO2 and Ni/CeO2 catalysts during atmospheric pressure CO2 methanation has been performed by a combined array of time-resolved analytical techniques including ambient-pressure X-ray photoelectron spectroscopy, high-energy X-ray diffraction and diffuse reflectance infrared Fourier transform spectroscopy. The ceria phase is partially reduced during the CO2 methanation and in particular Ce3+ species seem to facilitate activation of CO2 molecules. The activated CO2 molecules then react with atomic hydrogen provided from H2 dissociation on Rh and Ni sites to form formate species. For the most active catalyst (Rh/CeO2), transmission electron microscopy measurements show that the Rh nanoparticles are small (average 4 nm, but with a long tail towards smaller particles) due to a strong interaction between Rh particles and the ceria phase. In contrast, larger nanoparticles were observed for the Ni/CeO2 catalyst (average 6 nm, with no crystallites below 5 nm found), suggesting a weaker interaction with the ceria phase. The higher selectivity towards methane of Rh/CeO2 is proposed to be due to the stronger metal–support interaction.
1 Introduction
The conversion of CO2 to methane (methanation) has received wide interest during the last decade because of societal challenges related to climate change and finiteness of fossil fuels, which drive the change of the energy economy towards a system with a higher share of renewables.1–4 The methanation reaction from CO2 and H2 has the potential to (re-)use CO2 not only as an environmentally friendly carbon source but also as an alternative to H2 storage provided that the process can be made sufficiently efficient. In this context, CO2 conversion promoted by heterogeneous catalysis arouses interest.Methanation of CO2 has previously been investigated for a number of catalytic systems based on transition metals supported on various oxides.5–10 Nickel is one of the most studied metal catalysts for the methanation reaction, but some previous results have shown deactivation of Ni catalysts even at low temperatures due to formation of mobile Ni carbonyls or carbon deposition.11,12 Ruthenium and rhodium supported on various metal oxides have been shown to be highly active for the hydrogenation of CO2 and also the most selective catalysts towards formation of methane.10,13,14 Among the support materials, alumina, ceria and titania have shown superior catalytic activity. CeO2 is one of the most interesting support materials in catalysis, mainly because of its high reducibility and high oxygen storage capacity, which are key to the functionality of ceria in many applications.15–17 Although many ceria supported metal catalysts, with high stability and activity, have been reported,18 the origin of their good performance is not unambiguously understood but attributed to various factors, which need to be clarified by confirming the active sites and the corresponding mechanism for the reaction. Operando characterization techniques are superior tools to obtain detailed structural information so as to understand the structure of active sites and reaction mechanisms under practical conditions. All these factors are necessary for a knowledge-based development of high-performance catalysts.
We have recently shown that supported Rh and Ni catalysts are active for methane production from CO2 hydrogenation under atmospheric pressure conditions and at relatively low temperatures (≤625 K).19 The selectivity towards methane formation was found to be higher for Rh as compared to Ni when supported on ceria. Thus a detailed comparison of these two systems during CO2 methanation can reveal information about the active sites and reaction intermediates responsible for the enhanced activity towards methane production over Rh/Ce2. Several other studies, including our previous work, have shown a support-dependent reaction mechanism toward CO2 methanation via either a CO route (for catalysts supported on non-reducible oxides) or a formate route (for catalysts supported on reducible oxides).5–7,20 In related work we have shown that, for Rh catalysts supported on non-reducible oxides, the linearly adsorbed CO species is a more active precursor during the hydrogenation of CO2 as compared to the bridge-bonded CO species,20 which is attributed to the smaller particle sizes of the Rh-based catalysts. Even though the previous work suggested a different reaction path for the catalysts supported on reducible oxides, the correlation between activity/selectivity and structural properties for ceria-based catalysts was still not fully understood and the oxygen vacancies on the surface of CeO2 have been shown to play an important role in a number of catalytic reactions.
In this work we investigate how the choice of the active metal may influence the catalytic activity and selectivity for ceria supported catalysts. For this, Ni and Rh supported on CeO2 have been chosen and compared and the results show that the metal particle sizes are different for the two catalysts. It is suggested that for the highly dispersed catalyst (Rh/CeO2), the stronger interaction of Rh with the ceria support leads to its enhanced activity during the methanation of CO2. Special attention is paid to the identification of surface species and the influence of the ceria support during the CO2 methanation reaction. The scope of this work is to relate catalytic activity with observed chemical or structural changes of the active metal (Rh or Ni) and the support phase. To this end, we have employed a combined array of high-energy X-ray diffraction (HE-XRD), ambient-pressure X-ray photoelectron spectroscopy (AP-XPS) and diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) under time-resolved, in situ conditions. This unique combination of methods allows for addressing both the chemical and structural dynamics important for the CO2 conversion specifically targeting the site requirement. The results provide evidence that an active state of the ceria-based catalysts is rich in Ce3+ sites which can be associated with oxygen vacancies on reduced ceria activating CO2 molecules forming formate, an important species for the reaction mechanism. Further, the results show the reversible oxidation of Ni during the transient CO2 hydrogenation and the lack of adsorbed CO species for the Ni/CeO2 sample. This may be related to the increased particle sizes, as determined from transmission electron microscopy (TEM) measurements on the as-prepared catalyst, indicating that Ni interacts more weakly with the ceria support. It is suggested that the linearly adsorbed CO species can be later hydrogenated to methane over the Rh/CeO2 catalyst, while the weaker interaction between the CO and the Ni/CeO2 catalyst leads to its fast desorption and thus lower methane selectivity.
2 Experimental section
2.1 Catalyst preparation and ex situ characterization
The catalysts were prepared by incipient wetness impregnation using ceria powder samples (99.5 H.S.A. 514, Rhone-Pôulenc). The ceria samples were calcined in air at 875 K for 2 h starting from room temperature with a heating rate of 5 K min−1 to remove carbonaceous impurities and stabilise the structure of the support materials. Precursor solutions of nickel and rhodium were prepared by dissolving Ni(NO3)2·6H2O (Alfa Aesar) and Rh(NO3)2·6H2O (Alfa Aesar) salts in Milli-Q water (18 MΩ cm). To increase the solubility of the rhodium salt, 25 droplets of 70% HNO3 were added. The specific amount of precursor solution to obtain 3 wt% of the metal was added to 3 g of each support. The theoretical metal loading is expected to be close to the actual loading as previously reported for incipient wetness impregnation.21 The impregnated ceria samples were instantly frozen with liquid nitrogen, freeze-dried for 24 h and finally calcined in air at 825 K for 1 h. The specific surface area of the catalysts was determined by nitrogen sorption at 77 K (Micromeritics Tristar 3000) using the Brunauer–Emmett–Teller (BET) method.22 The samples were dried in a N2 flow at 500 K for 3 h prior to the measurements.Structural characterization by transmission electron microscopy (TEM) imaging was performed using a JEOL 3000F microscope (300 kV, Cs = 0.6 mm). The catalysts were ground and dry dispersed onto plasma cleaned (10 s) lacy carbon copper grids. Rh and Ni/NiO nanoparticles were identified through their (111) plane spacings (ca. 2.1 Å for metallic Rh and 2.0/2.4 Å for Ni/NiO, respectively), which falls between the (200) and (220) plane spacings of the CeO2 support (2.7 and 1.9 Å, respectively).
2.2 In situ characterization
![[S with combining breve]](https://www.rsc.org/images/entities/char_0053_0306.gif) unijć26 line shape convoluted with a Gaussian was used. The spectra were normalized to the background at the low binding energy side and a Shirley-type background was subtracted from all the spectra.27 Prior to the measurements the samples were exposed to 1.33 × 10−5 hPa O2 and the temperature was increased from room temperature to about 600 K to remove any C impurities. The recorded Ce 4d spectrum was considered to correspond to CeO2 and was later used to determine the stoichiometry of the ceria. Oxidation and reduction measurements were performed in situ at 595 K by exposing the catalyst to either O2 or H2. The C 1s level was measured continuously during the exposure and core-level spectra were recorded after a stable signal was observed. The methanation reaction was performed under a gas mixture of CO2 and H2, with a total pressure of 0.2 hPa and a partial pressure ratio of 1
unijć26 line shape convoluted with a Gaussian was used. The spectra were normalized to the background at the low binding energy side and a Shirley-type background was subtracted from all the spectra.27 Prior to the measurements the samples were exposed to 1.33 × 10−5 hPa O2 and the temperature was increased from room temperature to about 600 K to remove any C impurities. The recorded Ce 4d spectrum was considered to correspond to CeO2 and was later used to determine the stoichiometry of the ceria. Oxidation and reduction measurements were performed in situ at 595 K by exposing the catalyst to either O2 or H2. The C 1s level was measured continuously during the exposure and core-level spectra were recorded after a stable signal was observed. The methanation reaction was performed under a gas mixture of CO2 and H2, with a total pressure of 0.2 hPa and a partial pressure ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :14 (CO2:H2) at temperatures from 515 to 625 K.
:14 (CO2:H2) at temperatures from 515 to 625 K.
3 Results and discussion
3.1 Ex situ characterization of the as-prepared catalysts
The surface area and microstructure of the calcined catalysts were characterized prior to the in situ investigations. A summary of the specific surface area measurements is provided in Table 1. The high resolution TEM micrographs of the two catalysts are shown in Fig. 1. The diameter of the CeO2 nanoparticles of around 7 nm found by TEM is consistent with the specific surface area found by the BET method for both catalysts. The Rh crystallites are on average smaller than the Ni crystallites (average diameters of 4 and 6 nm for N = 11 and 10, respectively) as illustrated in Fig. 1. Particles 2–3 nm in diameter constitute nearly half of the found Rh crystallites, while no Ni crystallites smaller than 5 nm in diameter are found. The identified metallic crystallites however cannot account for the full loading of the catalysts, particularly in the case of Rh, indicating that there is an additional material that does not produce fringes in the electron micrographs with the characteristic spacings of the respective metals. Such a material could be present as a highly dispersed, possibly non-crystalline phase. For Ni, the presence of an oxidised material, formed during the calcination and maintained during storage, is indicated by the presence of d = 2.4 Å lattice planes.| Sample | Metal loading (wt%) | Calc. temperature (K) | Specific surface area (m2 g−1) | CO2 conversion | CH4 production | CO production |
|---|---|---|---|---|---|---|
| Rh/CeO2 | 3.0 | 825 | 125 | ∼46% | ∼41% | ∼0.05% |
| Ni/CeO2 | 3.0 | 825 | 128 | ∼44% | ∼30% | ∼0.14% |
 | ||
| Fig. 1 High-resolution TEM micrographs of the (a) Rh/CeO2 and (b) Ni/CeO2 catalysts. The inset in (a) shows a 2 nm Rh crystallite with a plane spacing of 2.2 Å. For Ni/CeO2 in (b), spacing consistent with both metallic and oxidised Ni was found as indicated by the outlined areas. The inset diffractogram shows the corresponding crystalline reflections used for identifying the two phases. The white dashed lines indicate the (111), (200) and (220) reflections from the CeO2 support. | ||
3.2 In situ characterization of the redox behavior
Similar to our recent publication,20 an oxidation/reduction experiment was performed over the Rh/CeO2 catalyst to obtain information on the characteristics of the catalyst under oxidation or reduction conditions. Fig. 2 shows the XPS spectra of the Rh 3d and Ce 4d levels upon exposure of the Rh/CeO2 catalyst to 1.33 × 10−5 hPa O2 or 1.33 × 10−6 hPa H2 at 595 K. The exposure to either O2 or H2 has been monitored by recording the C 1s or Rh 3d levels continuously. When constant signals were obtained, the relevant core levels were still recorded during the gas treatment. Due to the superposition of several components in the O 1s core level arising not only from the sample but also from the silicon wafer used as the substrate, we will exclude the O 1s core level in the XPS analysis. | ||
| Fig. 2 XPS spectra of the Rh 3d (a) and Ce 4d (b) regions recorded in situ from the reduced (bottom spectra) and oxidised (top spectra) Rh/CeO2 catalyst at 595 K. For comparison, the spectrum of the reduced sample (dotted red) is superimposed on the spectrum of the oxidised sample in Ce 4d (b). | ||
For the reduced sample, the Rh 3d5/2 XPS data in Fig. 2(a) show the presence of a main peak at 307.3 eV typical for bulk metal Rh (Rh0) and a minor peak at 308.2 eV, associated with oxidised rhodium,28 suggesting partial oxidation of Rh. The sustained oxidation state of Rh suggests the existence of a strong metal–support interaction between Rh and ceria, thus rhodium oxide is not easily reduced by H2 exposure. For the oxidised sample, the bulk Rh component decreases in intensity and the Rh oxide peak increases. In addition, a new weak component at 309.1 eV forms, which has previously been assigned to Rh species (Rh3+) located on the ceria lattice.29 However, the metallic Rh component is still present after the sample has been exposed to 1.33 × 10−5 hPa O2 at 595 K for a long time period (∼1 h). Thus, the Rh 3d XPS results suggest that Rh is not fully oxidised or reduced during the oxidation/reduction treatment, which is likely due to small Rh nanoparticles interacting strongly with the ceria support as previously reported for reducible oxides30–32 and also supported by the high degree of dispersion observed by TEM for the as-prepared catalysts and the AP-XPS measurements during CO2 methanation at higher pressures (see section 3.3.2 below). The strong metal–support interaction between Rh and the ceria support facilitates good anchoring of the Rh particles and favors the catalytic activity for CO2 methanation as will be discussed in more detail below.
In addition, XPS was used to determine the oxidation state of ceria during the oxidation/reduction treatment. The synthesized Rh/CeO2 was reduced to Rh/CeOx when exposed to 1.33 × 10−6 hPa H2 at 595 K. The Ce 4d spectra exhibit an intrinsically broad spectral shape with several contributions due to different electron coupling mechanisms.33 The curve fitting of these spectra is difficult. However, the spectra allow for differentiation between Ce4+ and Ce3+ and changes in the oxide's oxidation state can be monitored. Particularly useful to this end are the signals designated as W′′′ and X′′′ in Fig. 2(b). These signals are interpreted to originate from the spin orbit split 4d9O2p64f0 final state configuration.34 Thus their intensity should be proportional to the Ce4+ concentration in the sampling volume of the XPS measurement. We used the W′′′ and X′′′ signals, located at binding energies of 126.1 and 122.8 eV, respectively, to estimate the stoichiometry (x in CeOx) of the oxide.35,36 The “degree of oxidation” refers to the amount of Ce4+ and Ce3+ in the oxide: “100% oxidised” refers to all Ce present as Ce4+ (CeO2) and “0% oxidised” refers to all Ce present as Ce3+ (CeO1.5), i.e., reduced ceria. The presence of Ce3+ is usually associated with the formation of oxygen vacancies on the ceria surface. Comparison with the sample exposed to O2 during a heating ramp to 600 K, for which we assumed a CeO2 stoichiometry, indicates that the reduced sample contains about 23% (±5%) Ce3+, designated as Rh/CeO1.89±0.03. The redox behaviour of the Rh/CeO2 catalyst has been found to be reversible when switching between reduction/oxidation periods as previously reported.16 Thus, the results indicate that the Ce3+ species formed by the reduction in H2 can be regenerated to Ce4+ by oxygen.
3.3 In situ studies of the CO2 methanation reaction
 | ||
| Fig. 3 X-ray diffractograms during transient hydrogenation of 0.5 vol% CO2 over the as-prepared Rh/CeO2 catalyst during periodic variation of the feed gas composition between 2 vol% H2 + 0.5 vol% CO2 and 0.5 vol% CO2 at 625, 575 and 525 K for 10 min recorded using HE-XRD. The top panel shows the color coded intensities of the XRD patterns in the 0–8 Å−1 region versus time. The bottom panel shows selected XRD patterns at the end of selected CO2 (red) or CO2 + H2 (black) pulses together with the pattern recorded from the ceria reference sample (green). The inset indicates a zoom in between 2.7 and 4.1 Å−1. | ||
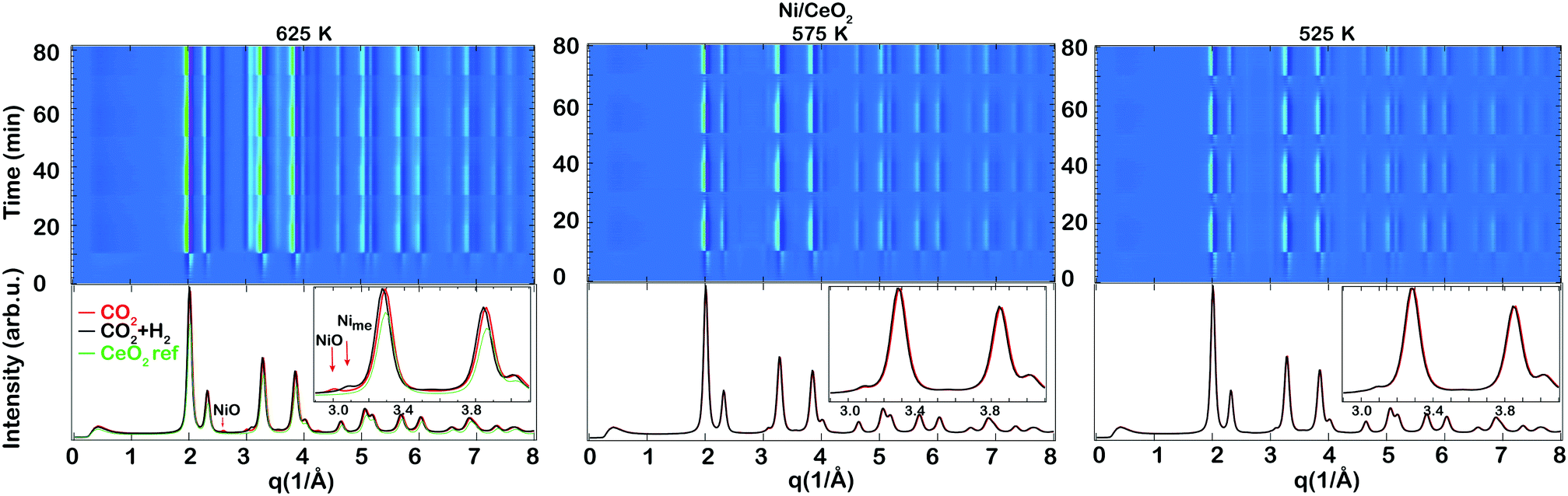 | ||
| Fig. 4 X-ray diffractograms during transient hydrogenation of 0.5 vol% CO2 over the as-prepared Ni/CeO2 catalyst during periodic variation of the feed gas composition between 2 vol% H2 + 0.5 vol% CO2 and 0.5 vol% CO2 at 625, 575 and 525 K for 10 min recorded using HE-XRD. The top panel shows the color coded intensities of the XRD patterns in the 0–8 Å−1 region versus time. The bottom panel shows selected XRD patterns at the end of selected CO2 (red) or CO2 + H2 (black) pulses together with the pattern recorded from the ceria reference sample (green). The inset indicates a zoom in between 2.9 and 4.1 Å−1. | ||
Some clear changes could be observed in the XRD patterns between the CO2 and CO2 + H2 pulses during the time-resolved measurements. Selected XRD patterns are shown at the bottom of Fig. 3 and 4 together with the pattern recorded from the ceria reference sample (green). For simplicity, the ceria reference pattern is only included in the panel presenting the measurements conducted at 625 K. The main reflections are due to scattering from the ceria lattice except the highlighted reflections around 3 Å−1 which will be discussed more below. For both samples the ceria phase undergoes changes during the pulsed CO2 hydrogenation experiment. During the CO2 pulse, Ce seems to be in an oxidised state similar to the ceria reference, while the shift toward lower q (i.e., larger d spacing) during the CO2 hydrogenation suggests a reduction of the ceria support due to formation of more Ce3+ species which have a larger ionic radius than Ce4+.
In addition, there are some differences observed between the ceria supported Rh and Ni catalysts. For the Rh/CeO2 catalyst, a weak broad reflection is observed at q = 2.85 Å−1 during the CO2 methanation (see the inset), similar to the observations in our recent work of CO2 hydrogenation over Rh/Al2O3.20 As already mentioned in our previous work, this corresponds to a d spacing of the reflecting planes of 2.2 Å which is close to the d spacing of metallic Rh (∼2.1 Å)37 (ref no. #171677). The small difference in the cell parameter from metallic bulk Rh here is attributed to the strong interaction between the small Rh nanoparticles and oxygen in the ceria support. We therefore denote the reflection at q = 2.85 Å−1 as Rhsupp. When in the presence of CO2 only, the ceria is fully oxidised and the formation of (amorphous) Rh oxides cannot be excluded (cf. XPS results in Fig. 2), which may explain the lower intensity of Rhsupp. During CO2 methanation, however, the presence of Rh oxides is unlikely and the ceria is somewhat reduced, which results in an increased intensity of the Rhsupp. For the Ni/CeO2 catalyst, a new peak appears at q = 3.1 Å−1 as the intensity of the peak at q = 3.0 Å−1 decreases during the CO2 + H2 pulse. The corresponding d spacing of the peak at q = 3.1 Å−1 is d = 2.02 Å, which is similar to the tabulated values for Ni available in the ICSD data collection37 (ref no. #260172). Thus, the results obtained for the Ni supported catalyst suggest that Ni is reduced to a metallic state during the CO2 methanation reaction.
Based on the XRD results it can be concluded that under CO2 methanation conditions the ceria is slightly reduced for both samples; crystalline Rh is influenced by the ceria support and does not resemble the bulk Rh structure, whereas the Ni phase exists as metallic Ni with a structure close to that of bulk Ni.
 | ||
| Fig. 5 AP-XPS spectra of the C 1s region of the Rh/CeO2 catalyst measured in situ during a temperature ramp from 515 to 625 K and under a gas mixture of CO2 (0.0133 hPa) and H2 (0.1867 hPa) (a). The corresponding C 1s (b), Rh 3d (c) and Ce 4d (d) levels recorded in situ for the Rh/CeO2 catalyst after exposure to a gas mixture of CO2 (0.0133 hPa) and H2 (0.1867 hPa) at 515 K. For comparison, the Ce 4d spectrum recorded after H2 treatment as presented in Fig. 2(b) (dotted red) of ceria supported Rh is superimposed on the shown Ce 4d spectra (d). | ||
Fig. 5(b–d) show a series of XPS spectra collected for the Rh/CeO2 catalyst under CO2 (0.0133 hPa) and H2 (0.1867 hPa) at 515 K. In the C 1s region, there are several contributions observed in addition to the weak component at about 293.5 eV from gaseous CO2. The strong peak at 285 eV with a shoulder at higher binding energy can be attributed to Cx/CO species on Rh. The component at about 285 eV with a slightly lower intensity can be observed before the catalyst is exposed to the CO2 + H2 mixture (not shown) and therefore it is suggested that some C is already present on the catalyst prior to CO2 hydrogenation. However, we cannot exclude that some CO2 is fully dissociated on the Rh/CeO2 catalyst during the methanation reaction since previous studies have reported facile decomposition of CO over Rh deposited on reduced ceria.35 Thus, reduced ceria facilitates the (complete) decomposition of CO2 or CO when CO2 or CO2 + H2 interact with the Rh/CeO2 catalyst. Further, another strong component is observed at around 290 eV with a shoulder on the low binding energy side (289.5 eV) which resembles the formate/carboxylate peaks on CeOx/Cu(111) previously observed by Graciani et al.38 This component appears to be slightly shifted to a higher binding energy as compared to the study by Graciani et al., and therefore, it is suggested that the carboxylate species are likely formed at the interface between ceria and Rh. It is interesting to note that no signals could be detected for CHx species at around 285–286 eV, which implies that any other surface intermediates produced by the hydrogenation of CO2 or CO are short-lived.
In the corresponding spectra for the Rh 3d and Ce 4d regions shown in Fig. 5(c and d), a clear change is seen in the line shape of the Ce 4d spectrum indicating the reduction of ceria, with the formation of Ce3+ (i.e., oxygen vacancies), and a change in the Rh 3d spectrum is observed indicating the reduction of RhOx, similar to the results of the reduction treatment discussed above. The Rh 3d spectrum shows a similar behavior to when the catalyst was exposed to H2 indicating still the presence of small amounts of Rh–O species during the methanation reaction. In the corresponding Ce 4d spectrum some signs of a change in the oxidation state to reduced ceria during CO2 methanation are evident. Similar to the H2 exposure, the synthesized Rh/CeO2 is rapidly reduced to Rh/CeOx when exposed to the CO2 + H2 reactant mixture. The recorded spectrum after the H2 exposure is included for comparison (dotted red). Further reduction of the ceria is observed during the methanation reaction as compared to the H2 exposure, which is likely due to the higher H2 pressure used during the methanation reaction (0.1867 hPa) as compared to the H2 treatment (1.33 × 10−6 hPa). The net amount of Ce3+ formed during exposure to the CO2 + H2 mixture is found to be about 40% (±5%) (Rh/CeO1.80±0.03).
The DRIFTS results from a steady-state measurement obtained after the Rh/CeO2 and Ni/CeO2 catalysts have been exposed to a flow of 0.2 vol% CO2 and 0.8 vol% H2 at 625 K for 20 min are presented in Fig. 6. The interaction between the Rh- and Ni-based catalysts with 0.2 vol% CO2 and 0.8 vol% H2 at 625 K results in the development of intense absorption bands between 1200–1600 cm−1, indicating the formation of formate and carbonate species. Although it is difficult to assign these peaks to specific structures on the surface, it is clear that formate and carbonate species are present on the surface of both catalysts. Some differences are observed between the investigated catalysts. The intensity of the IR bands is much higher for the Ni/CeO2 sample indicating that some higher amounts of formates and carbonates are present on this sample during the CO2 methanation reaction. Also the Ni/CeO2 sample shows a sharp peak around 1000 cm−1 typical for C–O symmetric stretching expected when formates/carbonates/carbonyls are present. However, its origin is not fully understood at this moment and the formation of some other species over the Ni/CeO2 catalyst during CO2 hydrogenation reaction is not excluded.
 | ||
| Fig. 6 In situ DRIFTS results in the wavenumber region of 800–3800 cm−1 for the Rh/CeO2 (red) and Ni/CeO2 (black) catalysts exposed to 0.2 vol% CO2 and 0.8 vol% H2 at 625 K for 20 min. Note that the overall intensity of the FTIR absorption bands for the Ni/CeO2 catalyst is higher and the intensity of the spectrum has therefore been divided by a factor of 5. | ||
For the Rh/CeO2 sample, some additional peaks at 2020 and 1740 cm−1 appear. These peaks can be attributed to CO species linearly bound on reduced Rh sites (Rh–CO, 2020 cm−1) and bridge-bonded CO at the Rh/ceria interface (1740 cm−1) (see Table 2 for reference). This indicates that the Rh/ceria interface plays a role in the CO2 hydrogenation reaction over Rh/CeO2. The broad peak at 1800 cm−1 may indicate the presence of some bridge-bonded CO species on Rh. No vibrations from CO species adsorbed on Rh+ sites (2800–3000 cm−1) could be seen for the Rh/CeO2 sample indicating that the formed CO during the methanation reaction adsorbs only on metallic Rh. The adsorbed CO species may arise from either the direct dissociation of CO2 over metallic Rh or from formate dissociation. The presence of adsorbed CO species on metallic Rh for the Rh/CeO2 catalyst together with the formation of OH species (3500–3800 cm−1) suggests that the adsorbed formate species may dissociate forming CO and OH on the surface. In contrast, no adsorbed CO species are observed for the Ni/CeO2 catalyst, suggesting its fast desorption since gas phase CO has previously been observed for this sample.19 Thus, the fact that CO is more strongly bound to the Rh/CeO2 catalyst could explain its higher selectivity towards methane compared to the Ni/CeO2 catalyst. Additional DRIFTS measurements of CO2 hydrogenation over ceria19 indicated the formation of a much smaller amount of formate/carbonate species than over Rh/CeO2 thus, strengthening the assumption that the metal–ceria interface is important for the reaction in agreement with previous work and related reactions.38–42
As summarised in Table 1, even though the Rh/CeO2 and Ni/CeO2 catalysts show similar CO2 conversion, the methane selectivity is slightly lower for the Ni/CeO2 catalyst. The TEM measurements on the as-prepared samples show the presence of larger nanoparticles (∼6 nm, likely oxidised) for the Ni/CeO2 sample, while smaller nanoparticles are observed for the Rh/CeO2 sample (≤4 nm, but with a long tail towards smaller particles). This supports the assumption that there is a stronger interaction with the support for the Rh/CeO2 catalyst. The stronger interaction between the metal and the support for the Rh/CeO2 catalyst leads to a higher CH4 selectivity during the CO2 hydrogenation.
The AP-XPS experiments in the present work indicate the activation of CO2 on the Rh/CeO2 catalyst in the presence of H2 at T = 515 K, generating formate, carboxylate and CO/Cx species on the surface, in addition to the methane observed in the gas phase. Further, photoelectron spectroscopy provides evidence showing that the surface formate is located at the Rh–ceria interface rather than on the metal. These results are supported by the DRIFTS measurements that also reveal that linearly bound CO (carbonyl hydrides) and formate species are present on the surface of the Rh/CeO2 catalyst during the reaction. Since with IR spectroscopy we are not able to observe C species on the surface, the results suggest that the C species observed by AP-XPS during the methanation reaction are formed as a consequence of the complete decomposition of CO2. Regarding the chemical state of the catalyst during the CO2 methanation reaction, the results provide evidence of small Rh particles (partially oxidised even under reducing conditions due to the strong interaction with the ceria support) on partially reduced ceria denoted as CeOx. The results strongly indicate that the presence of Ce3+ formed during the reduction of ceria is highly connected to the high activity of the Rh/CeO2 catalyst. For the comparative study on Ni/CeO2, the results show a similar behaviour to Rh/CeO2 with reduction of ceria during the methanation of CO2. Thus, Ce3+ is linked to the increased activity for CO2 hydrogenation over ceria-based catalysts while the metal surface is likely to act as the supplier of hydrogen for the subsequent hydrogenation of activated CO2. The main differences between the two catalysts are the reversible oxidation of Ni under the transient operation conditions and the lack of adsorbed CO species for the Ni/CeO2 sample, which may be related to the increased particle sizes. It is suggested that the linearly adsorbed CO species can be hydrogenated to methane subsequently over the Rh/CeO2 catalyst, while the weaker interaction between CO and the Ni/CeO2 surface leads to fast desorption of CO. Rh seems to be more active and selective towards CH4 than Ni likely due to its stronger interaction with the ceria support which can facilitate further steps during CO/Cx hydrogenation to methane.
The promoting effect of ceria is mainly related to its ability to undergo changes in its oxidation state (Ce3+–Ce4+ redox couple) with the consequent formation/annihilation of surface defects (O vacancies).15,49 Some previous reports have suggested Ce3+ as the active site for CO2 adsorption and conversion along with a valence change from Ce3+ to Ce4+.38,50–52 A recent study by Wang et al.53 on Ru supported on ceria showed also that the surface oxygen vacancies on ceria rather than on Ru are more likely to be the active sites in CO2 methanation. This supports our interpretation that the Ce3+ species, associated with oxygen vacancies, are the active sites for CO2 hydrogenation.
Although CO2 methanation is a comparatively simple reaction, its mechanism appears to be difficult to establish. Still, there are discussions on the nature of the intermediate compounds involved in the reaction process and on the methane formation scheme.54–56 Traditionally, transient studies are used to distinguish between intermediate or spectator species, but our recent transient DRIFTS measurements where the catalysts are subjected to alternating pulses of CO2 and CO2 + H2 indicate that both formates/carbonates and some CO species form under transient operation conditions.19 It is difficult to study the stability of such species from a temperature ramped AP-XPS study as the selectivity of the catalyst changes at higher temperatures. Further, from the DRIFTS measurements we are not able to separate the different formate/carbonate/carboxylate species and thus we cannot confirm or deny that any of these species is the reaction intermediate. In related work, Graciani et al.38 suggested the less stable carboxylate species to be the reaction intermediate for CO2 hydrogenation to methanol over CeOx/Cu(111), ruling out the formate reaction pathway due to the high stability of these species. Even though we see that the formate species are efficient as transient species, further measurements are needed to confirm if formates are indeed a reaction intermediate in the CO2 methanation over Rh/CeO2. Still, from the results we have achieved so far, we consider the most likely reaction pathway to involve the formation of formates and propose that the hydrogen molecules can dissociate on the metallic sites and react with the activated CO2 to form formates (HCOO) at the metal–support interface (see eqn (1) below). The formates can then dissociate into CO + OH and this step needs available metallic sites from where CO can be later hydrogenated to methane.
 | (1) |
From the transient experiments where the catalysts are subjected to alternating pulses of CO2 and CO2 + H2, we can conclude that the Ce3+ species needed to activate the CO2 molecules are re-generated during the methanation reaction since we observe a reversible change in the oxidation state of ceria under these conditions. Since hydrogen is the only reductant introduced into the feed during the reaction, we think that hydrogen, after being dissociated on Rh, may spill over to the ceria leading to the reduction of the ceria support.
By comparison with our recent study of CO2 methanation over Rh/Al2O3, which also showed a comparable activity to Rh/CeO2, we cannot exclude that formates are also important in the reaction mechanism for CO2 methanation over Rh/Al2O3 even though a smaller amount of formates is formed when alumina is used as the support. The results of our studies do not provide a conclusive assignment of a reaction path for the CO2 to CH4 conversion over the highly active Rh/CeO2 catalyst. However, the results reveal some of the important initial steps in the CO2 methanation reaction over Rh/CeO2, which may be used as a starting point for further investigations. In order to reach a final conclusion concerning the reaction mechanism, it will be helpful to investigate the H2 + CO reaction, the CO dissociation and the reactivity of surface C formed on the Rh catalyst.
4 Conclusions
Ceria supported Rh and Ni catalysts were structurally characterized during CO2 hydrogenation to methane. In situ measurements employing AP-XPS, HE-XRD and DRIFT spectroscopy revealed that the Ce3+ species are likely the active sites in CO2 methanation for ceria-based catalysts. The mechanism of the methanation reaction is complex and even though we are not able to reach a final conclusion, our results suggest that the formates created from the activated CO2 molecules by Ce3+ play an important part in the reaction mechanism and are dissociated to CO and OH species. A stronger interaction between the metal and the support for the Rh/CeO2 catalyst, as evident from its high degree of dispersion and as supported by the TEM measurements, leads to more strongly bound CO from formate dissociation, and thus a higher methane selectivity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank ALS for providing the beamtime. This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231. We also acknowledge the support of Dr. Ethan Crumlin at the beamline 9.3.2 at ALS. Parts of this research were carried out at PETRA III at DESY, a member of the Helmholtz Association (HGF). P. Velin and E. C. Adams are acknowledged for their contribution to sample preparation. This work was financially supported by the Swedish Research Council through the project “Synergistic development of X-ray techniques and applicable thin oxides for sustainable chemistry” (Dnr. 2017-06709) and the Röntgen-Ångström collaboration “Time-resolved in situ methods for design of catalytic sites within sustainable chemistry” (No. 349-2013-567), the Swedish Foundation for Strategic Research through the project “Novel two-dimensional systems obtained on SiC as a template, for electronics, sensing and catalysis” (RMA15-0024), the Knut and Alice Wallenberg Foundation through the project “Atomistic design of catalysts” (KAW 2015.0058), and the Competence Centre for Catalysis, which is financially supported by the Chalmers University of Technology, the Swedish Energy Agency and the member companies AB Volvo, ECAPS AB, Johnson Matthey AB, Preem AB, Scania CV AB, Umicore Denmark ApS and Volvo Car Corporation AB.References
- S. Rönsch, J. Schneider, S. Matthischke, M. Schluter, M. Götz, J. Lefebre, P. Prabhakaran and S. Bajohr, Fuel, 2016, 166, 276–296 CrossRef.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- P. Frontera, A. Macario, M. Ferraro and P. L. Antonucci, Catalysts, 2017, 7(59), 1–28 Search PubMed.
- X. Su, J. Xu, B. Liang, H. Duan, B. Hou and Y. Huang, J. Energy Chem., 2016, 25, 553–565 CrossRef.
- F. Wang, S. We, H. Chen, B. Wang, L. Zheng, M. Wei, D. G. Evans and X. Duan, J. Am. Chem. Soc., 2016, 138, 6298–6305 CrossRef CAS PubMed.
- Q. Pan, J. Peng, S. wang and S. Wang, Catal. Sci. Technol., 2014, 4, 502–509 RSC.
- P. A. Ussa Aldana, F. Ocampo, K. Kobl, B. Louis, F. Thibault-Starzyk, M. Daturi, P. Bazin, S. Thomas and A. C. Roger, Catal. Today, 2013, 215, 201–207 CrossRef.
- D. C. Upham, A. R. Derk, S. Sharma, H. Metiu and E. W. McFarland, Catal. Sci. Technol., 2015, 5, 1783–1791 RSC.
- A. Karelovic and P. Ruiz, J. Catal., 2013, 301, 141–153 CrossRef CAS.
- F. Solymosi, A. Erdöhelyi and T. Bánsági, J. Catal., 1981, 68, 371–382 CrossRef CAS.
- M. Agneli, M. Kolb and C. Mirodatos, J. Catal., 1994, 148, 9–21 CrossRef.
- M. Agneli, H. Swaan, C. Marquez-Alvarez, G. Martin and C. Mirodatos, J. Catal., 1998, 175, 117–128 CrossRef.
- F. Solymosi, I. Tombácz and M. Kocsis, J. Catal., 1982, 75, 78–93 CrossRef CAS.
- T. Iizuka, Y. Tanaka and K. Tanabe, J. Mol. Catal., 1982, 17, 381–389 CrossRef CAS.
- C. T. Campbell and C. H. F. Peden, Science, 2005, 309, 713–714 CrossRef CAS PubMed.
- A. Trovarelli and P. Fornasiero, Catalysis by Ceria and related materials. Catalytic Science Series, Imperial College Press, 2013 Search PubMed.
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116(10), 5987–6041 CrossRef CAS PubMed.
- F. Wang, M. Wei, D. G. Evans and X. Duan, J. Mater. Chem. A, 2016, 4, 5773–5783 RSC.
- N. M. Martin, P. Velin, M. Skoglundh, M. Bauer and P.-A. Carlsson, Catal. Sci. Technol., 2017, 7, 1086–1094 RSC.
- N. M. Martin, et al. , Catal. Sci. Technol., 2018, 8, 2686–2696 RSC.
- E. C. Adams, M. Skoglundh, M. Folic, E. C. Bendixen, P. Gabrielsson and P.-A. Carlsson, Appl. Catal., B, 2015, 165, 10–19 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- N. Schell, A. King, F. Beckmann, H.-U. Ruhnau, R. Kirchhof, R. Kiehn, M. Müller, A. Schreyer, R. Garrett and I. Gentle, et al., The high energy materials science beamline (hems) at petra iii, AIP Conf. Proc., 2010, 1234(1), 391 CrossRef CAS.
- M. E. Grass, et al. , Rev. Sci. Instrum., 2010, 81, 053106 CrossRef PubMed.
- D. R. Mullins, S. H. Overbury and D. R. Huntley, Surf. Sci., 1998, 409, 307–319 CrossRef CAS.
- S. Doniach and M. Sunijć, J. Phys. C: Solid State Phys., 1970, 3, 285–291 CrossRef CAS.
- D. A. Shirley, Phys. Rev. B: Solid State, 1972, 5(12), 4709–4714 CrossRef.
- C. Force, E. Román, J. M. Guil and J. Sanz, Langmuir, 2007, 23(8), 4569–4574 CrossRef CAS PubMed.
- L. S. Kibis, T. Yu. Kardash, E. A. Derevyannikova, O. A. Stonkus, E. M. Slavinskaya, V. A. Svetlichnyi and A. I. Boronin, J. Phys. Chem. C, 2017, 121, 26925–26938 CrossRef CAS.
- Z. Zhang, A. Kladi and X. E. Verykios, J. Catal., 1994, 148, 737–747 CrossRef CAS.
- C. de Leitenburg and A. Trovarelli, J. Catal., 1995, 156, 171–174 CrossRef CAS.
- D. Bounechada, S. Fouladvand, L. Kylhammar, T. Pingel, E. Olsson, M. Skoglundh, J. Gustafson, M. Di Michiel, M. A. Newton and P.-A. Carlsson, Phys. Chem. Chem. Phys., 2013, 15, 8648–8661 RSC.
- A. Kotani and H. Ogasawara, J. Electron Spectrosc. Relat. Phenom., 1992, 60, 257–299 CrossRef CAS.
- P. Burroughs, A. Hamnett, A. F. Orchard and G. Thornton, J. Chem. Soc., Dalton Trans., 1976, 1686–1698 RSC.
- D. R. Mullins and S. H. Overbury, J. Catal., 1999, 188, 340–345 CrossRef CAS.
- A. Schaefer, B. Hagman, J. Höcker, U. Hejral, J. Ingo Flege and J. Gustafson, Phys. Chem. Chem. Phys., 2018, 20, 19447–19457 RSC.
- https://icsd.fiz-karlsruhe.de (accessed May2018).
- J. Graciani, et al. , Science, 2014, 345, 546–550 CrossRef CAS PubMed.
- Z. Liu, et al. , Angew. Chem., Int. Ed., 2017, 56(42), 13041–13046 CrossRef CAS PubMed.
- S. D. Senanayake, et al. , J. Phys. Chem. C, 2016, 120(3), 1778–1784 CrossRef CAS.
- J. A. Rodriguez, P. Liu, D. J. Stacchiola, S. D. Senanayake, M. G. White and J. G. Chen, ACS Catal., 2015, 5(11), 6696–6706 CrossRef CAS.
- S. D. Senanayake, K. Mudiyanselage, A. Bruix, S. Agnoli, J. Hrbek, D. Stacchiola and J. A. Rodriguez, J. Phys. Chem. C, 2014, 118(43), 25057–25064 CrossRef CAS.
- F. Solymosi, A. Erdohelyi and T. Bansagi, J. Chem. Soc., Faraday Trans. 1, 1981, 77, 2645–2657 RSC.
- R. J. Behm, S. Eckle and Y. Denkwitz, J. Catal., 2010, 269, 255–268 CrossRef.
- A. Kiennemann, R. Breault, J.-P. Hindermann and M. Laurin, J. Chem. Soc., Faraday Trans. 1, 1987, 83, 2119–2128 RSC.
- I. A. Fisher and A. T. Bell, J. Catal., 1996, 162, 54–65 CrossRef CAS.
- H. Y. Luo, H. W. Zhou, L. W. Lin, D. B. Liang, C. Li, D. Fu and Q. Xin, J. Catal., 1994, 145, 232–234 CrossRef CAS.
- G. Busca, Phys. Chem. Chem. Phys., 1999, 1, 723–736 RSC.
- A. B. Kroner, M. A. Newton, M. Tromp, O. M. Roscioni, A. E. Russell, A. J. Dent, C. Prestipino and J. Evans, ChemPhysChem, 2014, 15, 3049–3059 CrossRef CAS PubMed.
- C. Leitenburgh, A. Trovarelli and J. Kaspar, J. Catal., 1997, 166, 98–107 CrossRef.
- D. R. Mullins, Surf. Sci. Rep., 2015, 70, 42–85 CrossRef CAS.
- M. V. Konishcheva, D. I. Potemkin, P. V. Snytnikov, O. A. Stonkus, V. D. Belyaev and V. A. Sobyanin, Appl. Catal., B, 2018, 221, 413–421 CrossRef CAS.
- F. Wang, C. Li, X. Zhang, M. Wei, D. G. Evans and X. Duan, J. Catal., 2015, 329, 177–186 CrossRef CAS.
- P. Panagiotopoulou, D. I. Kondarides and X. E. Verykios, Catal. Today, 2012, 181, 138–147 CrossRef CAS.
- A. Beuls, C. Swalus, M. Jacquemin, G. Heyen, A. Karelovic and P. Ruiz, Appl. Catal., B, 2012, 113–114, 2–10 CrossRef CAS.
- A. Karelovic and P. Ruiz, Appl. Catal., B, 2012, 113–114, 237–249 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |