
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unusual temperature-promoted carbon dioxide capture in deep-eutectic solvents: the synergistic interactions†
Shashi Kant
Shukla
 *a and
Jyri-Pekka
Mikkola
*ab
*a and
Jyri-Pekka
Mikkola
*ab
aTechnical Chemistry, Department of Chemistry, Chemical-Biological Centre, Umeå University, SE-90187 Umeå, Sweden. E-mail: shashi.kant.shukla@umu.se; jyri-pekka.mikkola@umu.se
bIndustrial Chemistry & Reaction Engineering, Department of Chemical, Engineering, Johan Gadolin Process Chemistry Centre, Åbo Akademi University, FI-20500 Åbo-Turku, Finland
First published on 1st March 2019
Abstract
A series of novel ethylenediamine (EDA)-based deep-eutectic solvents (DESs) gave rise to unexpectedly large carbon dioxide (CO2) capture capacities at higher temperatures owing to the “synergistic interaction” between the donor and acceptor moieties.
Reversible fixation–release of carbon dioxide (CO2) has attracted much attention during the last few decades due to an excessive increase in the consumption of fossil fuels as the main source of energy, thus causing an upsurge in CO2 emissions, leading to global warming. The current state-of-the-art industrial technology uses amine-based solvents in reversible CO2 capture.1 This methodology has critical issues such as high regeneration energy, amine instability, high volatility, corrosion and high maintenance costs.2,3 In order to optimize the CO2 uptake, the flue gas is cooled to room temperature before absorption, whereas high temperatures are required in the desorption–regeneration step. Both these steps increase the cost of the setup and hence developing a solvent system that can absorb CO2 directly from hot flue gas and undergo rapid desorption is the most attractive solution but have not been introduced until now.
The last century witnessed a surge in the development of a number of “green and sustainable” alternatives to volatile solvents. Among them, ionic liquids (ILs) and deep-eutectic solvents (DESs) have been the most explored ones for various applications because of their advantageous properties such as their negligible vapour pressure, wide liquidus range, low flammability, high thermal stability and recyclability.4,5 Blanchard et al. reported for the first time the potential of ILs in CO2 capture.6 Later, various ILs differing in terms of cations, anions and alkyl chain length were applied to optimize CO2 uptake and to gain understanding about the functioning of these neoteric media.7–10 The basicity of the anion was identified as the key factor in achieving high CO2 uptake and has been most explored to date.11–14 For example, in carbonyl-containing anion-functionalized ILs, high CO2 intake was favoured by H-bonding interactions.15 Pan et al. reported an equimolar CO2 uptake in anion-functionalized hydroxypyridine-based ILs owing to intramolecular proton transfer.16 However, the high molar uptake in ILs is attributed to their large molar mass and thus is less impressive when converted to the gravimetric values.10,17 Similarly, task-specific ILs for CO2 capture are expensive and require a long time for synthesis and are therefore rendered unsuitable for industrial applications.18
In contrast, DESs, a new generation of solvent analogous to ILs, provide a more promising alternative in CO2 capture and involve cost-effective, easy preparation, exhibiting properties similar to ILs, and resulting in higher gravimetric CO2 uptake.19,20 In a short time-span, DESs have found application in organic synthesis, polymerization, stabilization of biomolecules, nanotechnology, separation, extraction of target compounds and so on.21–26 The performance of DESs as sorbents in CO2 capture relies on pairing an efficient hydrogen bond donor (HBD) with hydrogen bond acceptor (HBA) with an optimal molar ratio.27–29 Later developments have shown moderate to high CO2 uptakes for different classes of DESs.30–33 We recently presented an in-depth discussion about the impact of intermolecular interactions on the CO2 uptake in ethylenediamine ([EDA])-, 3-amino-1-propanol ([AP])-, and polyamine-based DESs.33 Unlike ILs and amine-based solvent systems where basicity is a prerequisite for high CO2 capture, the interplay between the hydrogen bond donor acidity (α) and hydrogen bond acceptor basicity (β) governs the CO2 absorption in DESs.33 Additionally, the reaction conditions also influence the CO2 solubility and maximum uptake takes place at low temperatures and high pressures.34 In line with the current technology, flue gas passes through a cooler before traveling to the absorption tank.35 Therefore, employing a medium that has a low enthalpy of absorption (ΔHabs) and can reversibly absorb CO2 in the post-combustion process, at typical flue gas temperatures, will eliminate the necessity of a cooler unit and extra steam (for desorption), thus bringing down the cost of the overall setup and making the whole process more lucrative.
In this study, we synthesized DESs containing [EDA], [AP], diethylenetriamine ([DETA]), tetraethylenepentamine ([TEPA]) and pentaethylenehexamine ([PEHA]) as HBDs and monoethanolammonium chloride ([MEA·Cl]) and 1-methylimidazolium chloride ([HMIM·Cl]) as their HBA counterparts (see the ESI† for details) and measured the CO2 uptakes at temperatures 298 K–328 K, at ambient pressure (Table 1). The structures of the different DESs are shown in Fig. 1. As depicted in Fig. 2 and 3, respectively, the [EDA]- and [AP]-based DESs have opposite CO2 uptake behaviours within the same temperature range. The [EDA]-based DESs show a positive trend in terms of temperature, while a negative temperature effect is evident for the [AP]-class of DESs. So far, only a negative impact of increasing temperatures on CO2 capture has been perceived in previous investigations.32,35,36
| DESs | E a,η | ΔH° | ΔS° | E T30 | α | β |
|---|---|---|---|---|---|---|
| E a,η = kJ mol−1, ΔH° = kJ mol−1, ΔS° = J K−1 mol−1, ET(30) = kcal mol−1; the error in Ea,η, ΔH°, ΔS° and ET(30) are ±0.003, ±0.002, ±0.001 and 0.004, respectively. | ||||||
[MEA·Cl][EDA] = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
42.7 | 11.9 | 4.3 | 59.2 | 0.948 | 0.688 |
| [MEA·Cl][EDA] = 1:2 |
42.4 | 10.8 | 4.0 | 56.5 | 0.785 | 0.681 |
| [MEA·Cl][EDA] = 1:3 |
42.2 | 8.0 | 3.1 | 54.6 | 0.673 | 0.658 |
| [MEA·Cl][EDA] = 1:4 |
41.8 | 7.3 | 2.9 | 53.2 | 0.599 | 0.791 |
| [HMIM·Cl][EDA] = 1:1 |
48.0 | 17.0 | 5.7 | 54.7 | — | — |
| [HMIM·Cl][EDA] = 1:2 |
42.1 | 7.9 | 3.0 | 54.7 | 0.653 | 0.615 |
| [HMIM·Cl][EDA] = 1:3 |
38.9 | 7.2 | 2.9 | 53.9 | 0.646 | 0.861 |
| [HMIM·Cl][EDA] = 1:4 |
38.8 | 7.1 | 2.8 | 52.7 | 0.613 | 0.901 |
| [MEA·Cl][AP] = 1:1 |
48.3 | −10.0 | −4.5 | 60.2 | 1.013 | 0.526 |
| [MEA·Cl][AP] = 1:2 |
47.3 | −8.6 | −3.7 | 59 | 1.045 | 0.628 |
| [MEA·Cl][AP] = 1:3 |
43.0 | −7.5 | −3.1 | 58 | 0.932 | 0.696 |
| [MEA·Cl][AP] = 1:4 |
35.6 | −6.6 | −2.8 | 57.2 | 0.895 | 0.720 |
| [HMIM·Cl][AP] = 1:1 |
49.3 | −25.2 | −12.5 | 59.2 | 1.000 | 0.631 |
| [HMIM·Cl][AP] = 1:2 |
43.2 | −9.1 | −4.5 | 58.1 | 0.941 | 0.629 |
| [HMIM·Cl][AP] = 1:3 |
42.7 | −8.0 | −3.6 | 57.4 | 0.910 | 0.720 |
| [HMIM·Cl][AP] = 1:4 |
39.8 | −5.8 | −2.6 | 56.6 | 0.869 | 0.713 |
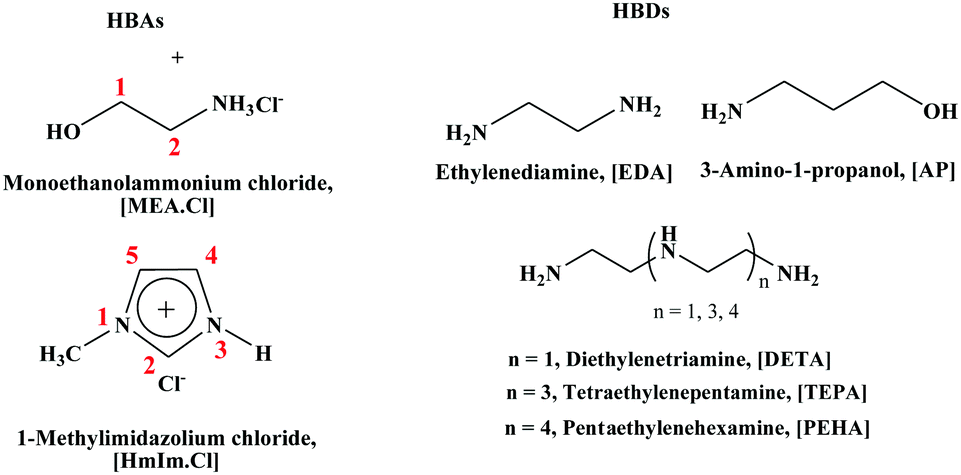 | ||
| Fig. 1 Structures of DESs. | ||
 | ||
Fig. 2 CO2 uptake kinetics in 1:1 (a) [MEA·Cl][EDA]- and (b) [HMIM·Cl][EDA]-based DESs at 298 K (■), 308 K ( ), 318 K ( ), 318 K ( ) and 328 K ( ) and 328 K ( ). ). | ||
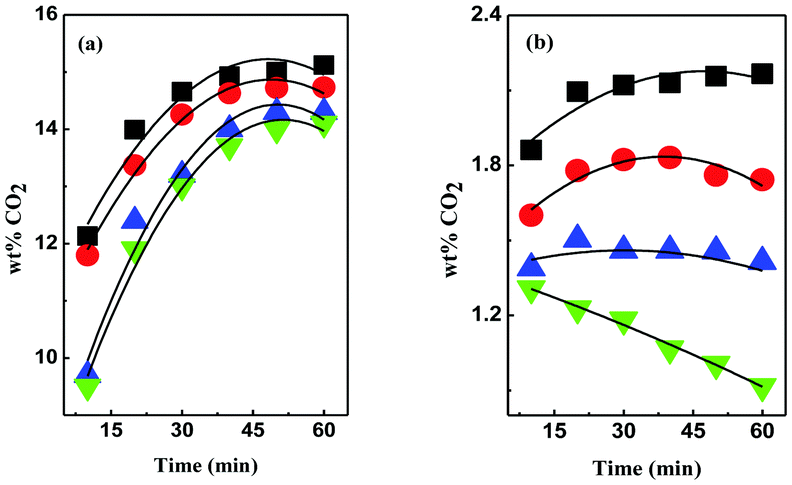 | ||
Fig. 3 CO2 uptake kinetics in 1:1 (a) [MEA·Cl][AP]- and (b) [HMIM·Cl][AP]-based DESs at 298 K (■), 308 K ( ), 318 K ( ), 318 K ( ) and 328 K ( ) and 328 K ( ). ). | ||
As shown in Fig. 2, the gravimetric CO2 uptake increases from 11.4% to 24.8% in 1:1 [MEA·Cl][EDA] and 3.6% to 12.1% in 1:1 [HMIM·Cl][EDA], respectively, in the temperature range 298 K–328 K in 1 h. The CO2 capture capacity increases further upon increasing the HBA:HBD ratio from 1:1 to 1:4. In 1:4 [MEA·Cl][EDA], the CO2 uptake reaches ∼36% in 1 h at 328 K, whereas in 1:4 [HMIM·Cl][EDA] ∼34% of CO2 is sorbed at 328 K (ESI,† Fig. S1–S4). Contrary to our reports, Trivedi et al. reported lower CO2 uptake in 1:3 [MEA·Cl][EDA] at a higher temperature, which is surprising to us.32 The temperature-promoted CO2 uptake was not specific to the [EDA]-based DESs noted in [DETA]-, [TEPA]- and [PEHA]-based DESs; though, the efficiency was lower than in the former case (ESI,† Fig. S5 and S6).
In general, an increasing temperature results in a lower viscosity but increases the activation energy of the gas molecule at the same time. The resultant of these opposing effects directs the course of CO2 capture, at higher temperatures. Therefore, the temperature-favoured CO2 uptake in the [EDA]-based DESs cannot be attributed to the reduction in viscosity only. Additionally, [MEA·Cl][EDA] and [HMIM·Cl][EDA] possess low viscosity, which further decreases upon increasing the HBA:HBD ratio from 1:1 to 1:4. In the latter case, the viscosity is already low so the influence of temperature is hard to detect. To discern the impact of viscosity on CO2 uptake more clearly, we calculated the activation energy of viscous flow (Ea,η) from the temperature-dependent viscosity data using the Arrhenius-type equation (ESI,† Fig. S7) as shown in Table 1. Ea,η accounts for the hindrance in the diffusion of CO2 by the DES. At HBA:HBD = 1:1, the [EDA]-based DESs possess lower Ea,η than the [AP]-class of DESs but, at higher HBA:HBD ratios, the trend in Ea,η reverses (Table 1). The Ea,η values of the [DETA], [TEPA] and [PEHA]-based DESs vary between 43.2 and 60.6 kJ mol−1 despite the positive effect of temperature on CO2 capture (ESI,† Table S1). Thus, the positive and negative temperature effects on the CO2 solubility in the [EDA]- and [AP]-based DESs, respectively, might originate from the different intermolecular interactions between the DESs and CO2.
To account for the impact of intermolecular interactions on the course of CO2 capture, at elevated temperatures, the thermodynamics (standard enthalpy change (ΔH°) and standard entropy change (ΔS°)) of capture along with the solvent polarity parameters (ET(30), α and β) are discussed further to unravel the underlying mechanisms33,37 (see the ESI,† Fig. S8 and S9 and Table S2).
As shown in Table 1, a positive ΔH° for the [EDA]-based DESs indicates an endothermic nature of CO2 uptake, while a negative ΔH° for the [AP]-based DESs signals that the process is exothermic. The standard entropy change (ΔS°) is positive for the [EDA]-class and negative for the [AP]-class of DESs. Following the spontaneity criteria (ΔG° = ΔH° − TΔS°), it is evident that the CO2 uptake becomes more spontaneous (−TΔS° > ΔH°) in [MEA·Cl][EDA] and [HMIM·Cl][EDA] than [MEA·Cl][AP] and [HMIm·Cl][AP] as the temperature increases. A shift in HBA:HBD from 1:1 to 1:4 decreases ΔH° and ΔS° in the [EDA]-based DESs and thus renders CO2 uptake more spontaneous. In the [AP]-class of DESs with an increase of the HBA:HBD ratio, ΔH° and ΔS° become less negative and thus spontaneous and result in higher CO2 uptake. The [DETA]-, [TEPA]- and [PEHA]-based DESs have positive ΔH° and ΔS° akin to the [EDA]-class of DESs and hence dissolve CO2 spontaneously as the temperature increases (ESI,† Table S1). More importantly, a very small value of ΔH° for the [EDA]-based DESs makes regeneration of the absorbent energy-saving, which is a key factor in practical applications.38
The nature of DES–CO2 interactions was then investigated in terms of the Kamlet–Taft parameters α and β, which measure donor and acceptor strengths, respectively (ESI,† Table S2 and Fig. S10). The positive effect of CO2 uptake indicates “synergistic interaction” of the donor and acceptor sites in the [EDA]-based DESs. The synergistic action is widely acknowledged in catalysis and polarity measurements of binary systems.39,40 “Hyperpolarity” was observed in a binary mixture of ILs when the donor (α) and acceptor (β) strengths become similar and favour synergistic interaction.41 In other words, for synergistic interaction, the relative difference between the donor and acceptor (α – β) should be zero. If α – β ≠ 0, the system will have acidic/basic characteristics depending on the relative differences in the values of α and β. This will cause an energy difference in the HBA and HBD in a DES and lower the probability for synergistic interactions (Scheme 1). The synergy in the DES results in weak interactions, which is reflected in the low polarity (ET(30)) and ΔH° and ΔS° values.
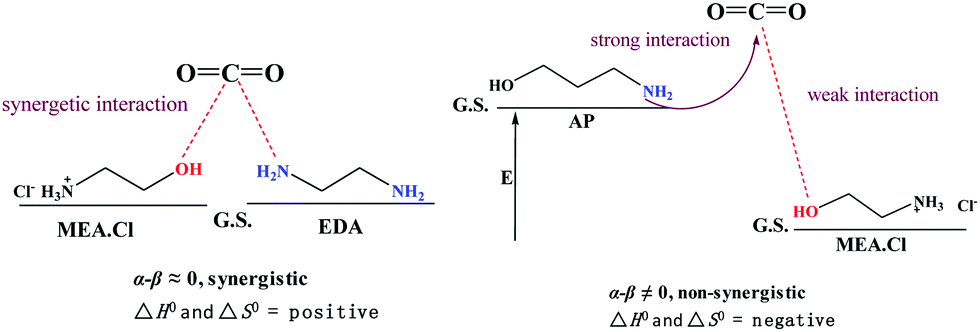 | ||
| Scheme 1 Representation of synergistic and non-synergistic interactions and thermodynamic criteria in DESs. | ||
As shown in Table 1, the relative α – β for [MEA·Cl][EDA] is lower than those of [MEA·Cl][AP] and [HMIM·Cl][AP] at 1:1. The small α – β suggests alignment of the donor and acceptor energy levels for synergistic interactions. At higher HBA:HBD ratios, in the [EDA]-based DESs, relative α – β nears zero and renders the synergistic interactions more feasible. Decreases in the ΔH° and ΔS° further support this trend. Similarly, the α – β trend in the [HMIm·Cl][EDA]-class of DESs also indicates synergistic interactions at different molar ratios. The relative α – β for the [AP]-class of DESs is comparatively large and hence results in negative ΔH° and ΔS° (Table 1). The large α – β gives rise to differences in the ground state energy of the donor and acceptor and forbids synergistic interactions (Scheme 1). Thus, the greater CO2 uptake in the [AP]-class of DESs, at higher HBA:HBD ratios, is due to their reduced viscosity.
The [DETA]-, [TEPA]- and [PEHA]-based DESs possess large α – β but have positive ΔH° and ΔS°, which suggests the possibility of synergistic interactions. The large in α – β difference is caused by the multiple amine groups in the HBDs.
In summary, we, for the first time, have reported temperature-promoted CO2 capture in novel [EDA]-based DESs. The positive effect of temperature on CO2 capture arises from the synergistic interactions between the donor and acceptor moieties. The synergistic interactions in DESs are caused by small α – β and ET(30) and positive ΔH° and ΔS°. Lower CO2 uptake in the [AP]-class of DESs, at elevated temperatures, is due to the large α – β and negative ΔH° and ΔS°. Very low ΔH° indicates quick regeneration of the [EDA]-based DESs and hence these DESs evolve as excellent substitutes for the amine-based technology in a CO2 capture plant.
We are thankful to the Wallenberg Wood Science Center (WWSC), Kempe Foundations, and the Bio4Energy programme. This work is also part of the activities of the Johan Gadolin Process Chemistry Centre at Åbo Akademi University.
Conflicts of interest
There are no conflicts to declare.References
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- R. López-Rendón, M. A. Mora, J. Alejandre and M. E. Tuckerman, J. Phys. Chem. B, 2006, 110, 14652–14658 CrossRef PubMed.
- S. Zeng, X. Zhang, L. Bai, X. Zhang, H. Wang, J. Wang, D. Bao, M. Li, X. Liu and S. Zhang, Chem. Rev., 2017, 117, 9625–9673 CrossRef CAS PubMed.
- X. P. Zhang, X. C. Zhang, H. F. Dong, Z. J. Zhao, S. J. Zhang and Y. Huang, Energy Environ. Sci., 2012, 5, 6668–6681 RSC.
- Q. R. Sheridan, W. F. Schneider and E. J. Maginn, Chem. Rev., 2018, 118, 5242–5260 CrossRef CAS PubMed.
- L. A. Blanchard, D. Hancu, E. J. Beckman and J. F. Brennecke, Nature, 1999, 399, 28–29 CrossRef.
- B. E. Gurkan, J. C. de la Fuente, E. M. Mindrup, L. E. Ficke, B. F. Goodrich, E. A. Price, W. F. Schneider and J. F. Brennecke, J. Am. Chem. Soc., 2010, 132, 2116–2117 CrossRef CAS PubMed.
- Y. Q. Zhang, S. J. Zhang, X. M. Lu, Q. Zhou, W. Fan and X. P. Zhang, Chem. – Eur. J., 2009, 15, 3003–3011 CrossRef CAS PubMed.
- C. M. Wang, X. Y. Luo, H. M. Luo, D. E. Jiang, H. R. Li and S. Dai, Angew. Chem., Int. Ed., 2011, 50, 4918–4922 CrossRef CAS PubMed.
- C. M. Wang, H. M. Luo, D. E. Jiang, H. R. Li and S. Dai, Angew. Chem., Int. Ed., 2010, 49, 5978–5981 CrossRef CAS PubMed.
- I. Niedermaier, M. Bahlmann, C. Papp, C. Kolbeck, W. Wei, S. K. Calderon, M. Grabau, P. S. Schulz, P. Wasserscheid, H. P. Steinruck and F. Maier, J. Am. Chem. Soc., 2014, 136, 436–441 CrossRef CAS PubMed.
- B. F. Goodrich, J. C. de la Fuente, B. E. Gurkan, D. J. Zadigian, E. A. Price, Y. Huang and J. F. Brennecke, Ind. Eng. Chem. Res., 2011, 50, 111–118 CrossRef CAS.
- C. M. Wang, H. M. Luo, X. Y. Luo, H. R. Li and S. Dai, Green Chem., 2010, 12, 2019–2023 RSC.
- T. Oncsik, R. Vijayaraghavan and D. R. MacFarlane, Chem. Commun., 2018, 54, 2106–2109 RSC.
- F. Ding, X. He, X. Luo, W. Lin, K. Chen, H. Lia and C. Wang, Chem. Commun., 2014, 50, 15041–15044 RSC.
- M. Pan, R. Vijayaraghavan, F. Zhou, M. Kar, H. Li, C. Wang and D. R. MacFarlane, Chem. Commun., 2017, 53, 5950–5953 RSC.
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 926–927 CrossRef CAS PubMed.
- B. Gurkan, B. F. Goodrich, E. M. Mindrup, L. E. Ficke, M. Massel, S. Seo, T. P. Senftle, H. Wu, M. F. Glaser, J. K. Shah, E. J. Maginn, J. F. Brennecke and W. F. Schneider, J. Phys. Chem. Lett., 2010, 1, 3494–3499 CrossRef CAS.
- A. P. Abbott, G. Capper, D. L. Davies, H. L. Munro, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2001, 2010–2011 RSC.
- A. Zhu, T. Jiang, B. Han, J. Zhang, Y. Xie and X. Ma, Green Chem., 2007, 9, 169–172 RSC.
- A. Abo-Hamad, M. Hayyan, M. A. H. AlSaadi and M. A. Hashim, Chem. Eng. J., 2015, 273, 551–567 CrossRef CAS.
- B. Tang, H. Zhang and K. H. Row, J. Sep. Sci., 2015, 38, 1053–1064 CrossRef CAS PubMed.
- J. García-Álvarez, Eur. J. Inorg. Chem., 2015, 5147–5157 CrossRef.
- F. del Monte, D. Carriazo, M. C. Serrano, M. C. Gutierrez and M. L. Ferrer, ChemSusChem, 2014, 7, 999–1009 CrossRef CAS PubMed.
- H. Zhao, J. Chem. Technol. Biotechnol., 2015, 90, 19–25 CrossRef CAS.
- C. Vidal, J. García-Álvarez, A. Hernán-Gómez, A. R. Kennedy and E. Hevia, Angew. Chem., Int. Ed., 2016, 55, 16145–16148 CrossRef CAS PubMed.
- Q. H. Zhang, K. D. Vigier, S. Royer and F. Jerome, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC.
- M. Francisco, A. van den Bruinhorst and M. C. Kroon, Angew. Chem., Int. Ed., 2013, 52, 3074–3085 CrossRef CAS PubMed.
- Y. Dai, J. van Spronsen, G.-J. Witkamp, R. Verpoorte and Y. H. Choi, J. Nat. Prod., 2013, 76, 2162–2173 CrossRef CAS PubMed.
- X. Li, M. Hou, B. Han, X. Wang and L. Zou, J. Chem. Eng. Data, 2008, 53, 548–550 CrossRef CAS.
- M. Francisco, A. van den Bruinhorst, L. F. Zubeir, C. J. Peters and M. C. Kroon, Fluid Phase Equilib., 2013, 340, 77–84 CrossRef CAS.
- T. J. Trivedi, J. H. Lee, H. J. Lee, Y. K. Jeong and J. W. Choi, Green Chem., 2016, 18, 2834–2842 RSC.
- S. K. Shukla and J.-P. Mikkola, Phys. Chem. Chem. Phys., 2018, 20, 24591–24601 RSC.
- S. Sarmad, Y. Xie, J.-P. Mikkola and X. Jia, New J. Chem., 2017, 41, 290–301 RSC.
- I. Kim and H. F. Svendsen, Ind. Eng. Chem. Res., 2007, 46, 5803–5809 CrossRef CAS.
- A. Finotello, J. E. Bara, D. Camper and R. D. Noble, Ind. Eng. Chem. Res., 2008, 47, 3453–3459 CrossRef CAS.
- S. K. Shukla and A. Kumar, J. Phys. Chem. B, 2015, 119, 5537–5545 CrossRef CAS PubMed.
- J. Ren, L. Wu and B.-G. Li, Ind. Eng. Chem. Res., 2013, 52, 8565–8570 CrossRef CAS.
- K. Herodes, I. Leito, I. Koppel and M. Rosés, J. Phys. Org. Chem., 1999, 12, 109–115 CrossRef CAS.
- L. Hongliang, W. Liangbing, D. Yizhou, P. Zhengtian, L. Zhuhan, C. Yawei, W. Menglin, Z. Xusheng, Z. Junfa, Z. Wenhua, S. Rui, M. Chao and Z. Jie, Nat. Nanotechnol., 2018, 13, 11–417 CrossRef PubMed.
- A. Sarkar, S. Trivedi, G. A. Baker and S. Pandey, J. Phys. Chem. B, 2008, 112, 14927–14936 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables and figures, synthesis and characterization of DESs, polarity, viscosity, activation energy of viscous flow and thermodynamic characterization, and temperature-dependent CO2 capture profiles. See DOI: 10.1039/c9cc00831d |
| This journal is © The Royal Society of Chemistry 2019 |