
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comment on “An unexpected formation of the novel 7-oxa-2-azabicyclo[2.2.1]hept-5-ene skeleton during the reaction of furfurylamine with maleimides and their bioprospection using a zebrafish embryo model” by C. E. Puerto Galvis and V. V. Kouznetsov, Org. Biomol. Chem., 2013, 11, 407†
F. I.
Zubkov
 *a,
E. A.
Kvyatkovskaya
a,
E. V.
Nikitina
a,
P. N.-A.
Amoyaw
a,
V. V.
Kouznetsov
b,
V. A.
Lazarenko
c and
V. N.
Khrustalev
d
*a,
E. A.
Kvyatkovskaya
a,
E. V.
Nikitina
a,
P. N.-A.
Amoyaw
a,
V. V.
Kouznetsov
b,
V. A.
Lazarenko
c and
V. N.
Khrustalev
d
aDepartment of Organic Chemistry, Peoples Friendship University of Russia (RUDN University), 6 Miklukho-Maklaya St., Moscow, 117198, Russian Federation. E-mail: fzubkov@sci.pfu.edu.ru
bLaboratorio de Química Orgánica y Biomolecular, CMN, Universidad Industrial de Santander, Parque Tecnológico Guatiguara, Km 2 vía refugio, Piedecuesta, A.A. 681011, Colombia
cNRC Kurchatov Institute, 1 Acad. Kurchatov Sq., Moscow 123182, Russia
dDepartment of Inorganic Chemistry, Peoples Friendship University of Russia (RUDN University), 6 Miklukho-Maklaya St., Moscow 117198, Russia
First published on 10th July 2017
Abstract
It has been proved that the reaction between furfuryl amines and N-R-maleimides leads to the formation of aza-Michael addition products – 3-(furylmethylamino)-N-R-pyrrolidine-2,5-diones, instead of 7-oxa-2-azabicyclo[2.2.1]hept-5-enes, as this journal reported previously.
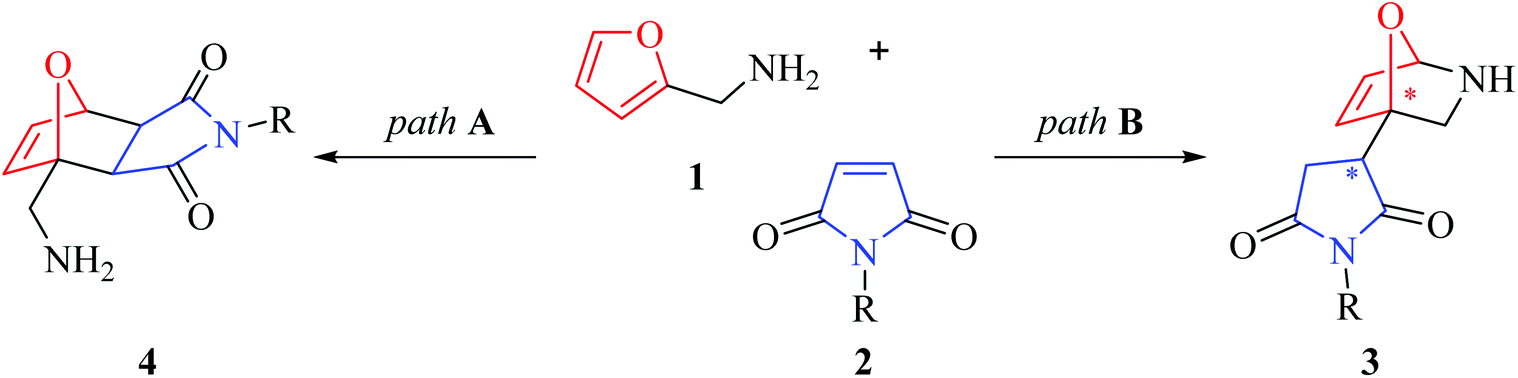
In 2013, one of us (V. V. Kouznetsov) hastily published in this journal the results of the work on the interaction of furfuryl amine 1 and N-substituted maleimides 2 in the presence of boric acid in PEG, in which the formation of 7-oxa-2-azabicyclo[2.2.1]hept-5-enes 3 was described (Scheme 1).1 The evaluation of catalyst and solvent effects on the yields of titled compounds, as well as a plausible reaction mechanism and some biomedical properties of the structures obtained were also discussed.
 | ||
| Scheme 1 | ||
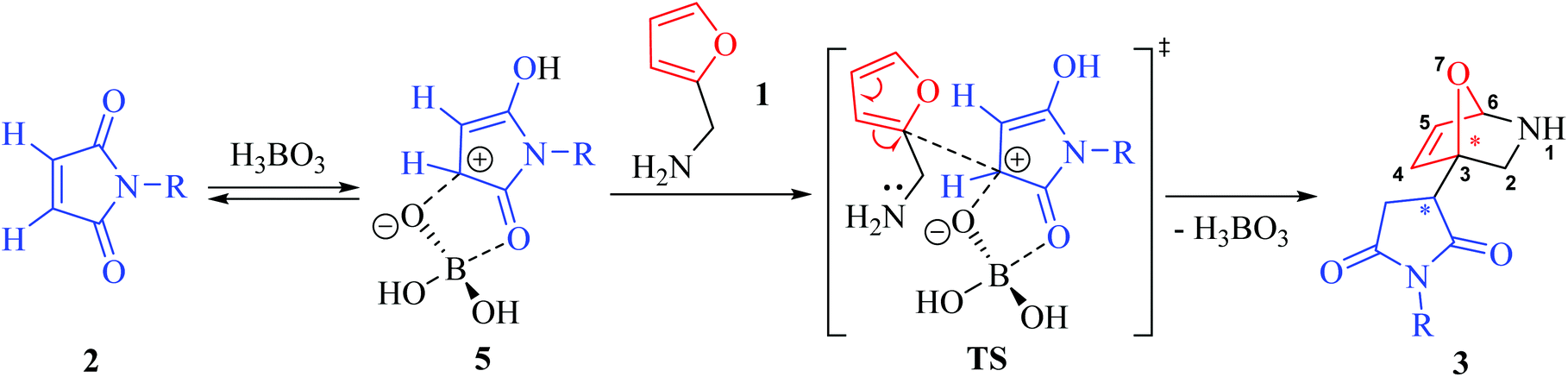
Based on the spectral data of the products obtained, it was concluded that path A, which would give Diels–Alder cycloadducts, 7-oxabicyclo[2.2.1]hept-5-enes (4), was not plausible, while a cyclisation (path B) through an electrophilic attack of the protonated maleimide (5, see Scheme 2) on the C-2 atom of the furan ring took place under acid catalysis. A sophisticated mechanism of this transformation, including a concerted rearrangement promoted by a molecule of H3BO3, was suggested in this work.1 The simplified version of this conversion is given in Scheme 2.
 | ||
| Scheme 2 | ||
Using the environmentally friendly conditions (PEG-400 as a solvent), the authors1 obtained the target compounds 3 in good yields and “without the observation of any collateral products or isomers”.
During an independent research into this synthesis, and after careful examination of the work done by Puerto and Kouznetsov, Professor Harald Stöver's research group respectfully cautioned us that the structure of the obtained derivatives 3 might be a different product.2 This suggestion by Professor Stöver, and our own observation that these compounds possess a unique heterocyclic core prompted us to begin a detailed study of its chemical properties. Primarily, it was expected that the Wagner–Meerwein rearrangement3 will occur to give the epoxy derivatives of 7-oxa-2-azabicyclo[2.2.1]hept-5-enes (3). However, in the first experiments we were surprised to find that the double bond in 7-oxa-2-azabicycloheptenes (3) remained intact under m-CPBA action. This strange observation prompted us to take a critical look at the data obtained earlier1 because we began to doubt the results of the study mentioned above in Schemes 1 and 2. To resolve the problem, we deem it necessary to find answers to the following questions.
1. Why the nucleophilic attack of the amino group of 1 proceeds on the C-2 atom of the furan ring instead of a much more accessible carbocation at C-2 of the maleimide ring?
2. 7-Oxa-2-azabicyclo[2.2.1]hept-5-enes (3) have two stereocenters, but for unexplained reasons, they are formed as a single diastereomer, without any structure confirmation of its spatial structure.
3. The NMR spectral data have a slight correlation with the proposed structures of 7-oxa-2-azabicyclo[2.2.1]heptanes, for example, the presence of a characteristic set of signals belonging to the furan ring in 1H NMR spectra cannot be overlooked.
Nevertheless, it should be noted that the chemical information on the interaction of furfurylamines and N-substituted maleimides is difficult to substantiate and ambiguous. Depending on the nature of the substituents on the maleimide core and the reaction conditions, the reaction could furnish different adducts.4–6 For example, Saxena et al.4 reported that the reaction of N-phenyl maleimides with one equivalent of furfuryl amine in benzene at 80 °C led to the formation of cycloadducts 4via path A (Scheme 1). Moreover, the synthesis of cycloadducts 4 through a reaction of maleimides and N-acetylfurfurylamine was described in the work being criticized.1 The same product formation was reported in the work,5 whereas Kondoli et al.6 described the formation of adducts 6 derived from the interaction between an activated alkene 2 and furfuryl amine 1 through the most obvious way of interaction, the aza-Michael addition, path C, a process not considered in the work1 (Scheme 3).
 | ||
| Scheme 3 | ||
Considering the comments of Prof. Stöver and the reasonable possibility that the Michael addition pathway may govern the reaction between furfurylamine 1 and maleimides 2, under the developed reaction conditions,1 we therefore reproduced the previous experiments and examined carefully the structure of the obtained products, and the same starting materials and the same experimental conditions were chosen for the first three reactions (Table 1). It was established that indeed the interaction of furfuryl amines 1 with maleimides 2 proceeds easily both in boiling acetonitrile without a catalyst (method A), and in polyethylene glycol (PEG-400) at 90 °C in the presence of boric acid (10 mol%) (method B) giving products 6a–c with yields of around 80%. Additionally, a new series of compounds 6d–i were also synthesized in boiling MeCN in good yields (Scheme 4, Table 1).
 | ||
| Scheme 4 | ||
| Compound 6 | R1 | R2 | Yield, % | |
|---|---|---|---|---|
Method A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
Method Bb |
|||
| a All yields are given for reactions in boiling MeCN after recrystallization or flash chromatography. b Data from the paper1 (90 °C, 1 h, PEG-400). | ||||
| a | H | H | 73 | 93 |
| b | H | Bn | 76 | 82 |
| c | H | Ph | 75 | 91 |
| d | H | Me | 86 | — |
| e | H | 4-NO2-C6H4 | 60 | — |
| f | Me | H | 61 | — |
| g | Me | Me | 64 | — |
| h | Me | Ph | 70 | — |
| i | Me | 4-NO2-C6H4 | 75 | — |
The confusion during the elucidation of the correct structure of the obtained compounds involves various aspects. First, the proposed 7-oxa-2-azabicyclo[2.2.1]hept-5-enes 3 and the respective Michael adducts 6 have the same molecular formula, so the spectrometric analysis did not contribute to the correct elucidation. Second, 1H NMR analysis was not enough; most of the signals present in the 1H NMR spectra are consistent with both structures. The careful analysis of both 1H and 13C NMR spectra, along with DEPT 135 spectra, might avoid this accident. Now that we realize that the correct structure corresponds to the Michael adduct, we evidenced that the two protons at the 3-position are not equivalents, they are diastereotopic rather than being an exo-/endo- pair and that the signals at 6.2–6.4 and 7.3–7.4 ppm are also consistent with a 2-substituted furan ring, characteristic of the Michael adduct.
For this correction, a revision and a detailed study on the NMR spectra of products 6a–c revealed their complete identity with the spectra given in the initial paper1 for the same sets of initial compounds (see the ESI† for the present study). All the 1H NMR spectra of adducts 6 were characterized by two repeated spin-isolated systems of signals in weak and strong fields, respectively. The first set is at δ ∼ 7.40 (dd, J = 0.8 and 1.8 Hz), 6.35 (dd, J = 1.8 and 3.2 Hz), and 6.25 (br. d or dd, J = 0.8 and 3.2 Hz) ppm, and the second is at δ ∼ 3.75 (dd, J = 5.0 and 8.3 Hz), 2.90 (dd, J = 8.3 and 18.0 Hz), and 2.50 (dd, J = 5.0 and 18.0 Hz) ppm. The first set represents the typical signals of the 2-substituted furan nucleus, while the second belongs to the AMX system of 3-substituted succinimide. The 13C NMR spectra of compounds 6a–c and 2D COSY (and/or HSQC) experiments revealed the same patterns. The signals of the furan nucleus are observed at δ ∼ 150, 140 (C-2 and C-5), 110 (C-3 and C-4) ppm and the signals of the C-3 and C-4 atoms of the imide part are located at 55 and 35 ppm. All these parameters are in good correlation with the structure of Michael adducts 6 (Scheme 4).
The resulting compounds 6 are mostly viscous oils, therefore, in order to further confirm their structures, some well-crystallizing N-aryl samples were obtained. Two of them (6c and h) turned out to be suitable for X-ray structural analysis (see ESI† and Fig. 1).
 | ||
| Fig. 1 Molecular structures of 6c (on the left) and 6h (on the right). Displacement ellipsoids are shown at the 50% probability level. The alternative position of the disordered fragment in 6c is depicted by dashed lines. | ||
The obtained data unequivocally confirmed the structure of the target products 6 and this suggests that here we are dealing with the simple nucleophilic aza-Michael addition, instead of the formation of a 7-oxa-2-azabicyclo[2.2.1]heptene system.
In the upshot of this study, we think that it is appropriate to cite the aphorism by Erich Maria Remarque: “Never look for complex ways where there is a simple road”.
Acknowledgements
Funding for this research was provided by the Ministry of Education and Science of the Russian Federation (award no. 4.1154.2017/4.6).Notes and references
- C. E. P. Galvis and V. V. Kouznetsov, Org. Biomol. Chem., 2013, 11, 407 Search PubMed.
- Prof. Harald Stöver and his students: S. Ros and N. Burke, Department of Chemistry and Chemical Biology, McMaster University, Hamilton, ON L8S 4 M1, Canada Search PubMed.
- E. W. Baxter, D. Labaree, S. Chao and P. S. Mariano, J. Org. Chem., 1989, 54, 2893 CrossRef CAS.
- N. Saxena, N. Singh, M. Mishra, C. B. Shiva Keshava, P. K. Shukla and R. P. Tripathi, J. Enzyme Inhib. Med. Chem., 2008, 23, 476 CrossRef CAS PubMed.
- S. Takano, Y. Oshima, F. Ito and K. Ogasawara, Yakugaku Zasshi, 1980, 100, 1194 ( Chem. Abstr. , 1981 , 95 , 24871x ) CrossRef CAS.
- J. C. Kondoli, D. Prajapati, J. S. Sandhu and B. J. Wakefield, J. Chem. Res., 1987, 851 ( J. Chem. Research (S) , 1987 , 76 ) Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray description for single-crystals 6c and 6h, detailed synthetic procedures and spectral data for compounds 6. CCDC 1547011 and 1547012. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ob01207a |
| This journal is © The Royal Society of Chemistry 2017 |