
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA self-assembled Pd2L4 cage that selectively encapsulates nitrate†
Li-Peng
Zhou
and
Qing-Fu
Sun
*
State Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, P. R. China. E-mail: qfsun@fjirsm.ac.cn; Fax: +86 591 63173527; Tel: +86 591 63173527
First published on 29th September 2015
Abstract
A M2L4 cage with D4 symmetry was self-assembled from four anthracene-bridged benzimidazole ligands and two PdII ions. The cage features a concise hydrophobic pocket wrapped up by the anthracene walls with eight hydrogen-bond donors pointing inward, which provide a specific binding site for nitrate, with a binding affinity at least two orders of magnitude higher than all the other anions screened including halide anions, which have a very similar ionic radius and charge density.
Nitrate anion is ubiquitous and important in biology, the environment, and the food industry.1 Although nitrate is probably benign, it can be reduced to nitrite or other nitric oxides, which react with thiols, amines and amides to form carcinogenic compounds. Other health concerns associated with NO3− metabolism include diabetes, thyroid disorders, respiratory infections and congenital malformations.2,3 Therefore, the design of a synthetic host with the capability of selective encapsulation of nitrate is an important task.
Many artificial hosts that can bind nitrate have been documented, including pyrrole-,4,5 amide-,6–8 ammonium-,9,10 urea-,11 guanidinium-12 based tripod-,13 macrocycle-,14 rotaxane-,15 catenane-16 or cage-17 like receptors. Because of the intrinsic properties of nitrate, the reported receptors have several common problems. First of all, large hydration energy and weak basicity of the nitrate anion result in that NO3− is weakly coordinative and it is difficult to form robust hydrogen bonds with the host,2,17,18 though hydrogen-bonding interaction plays a crucial role in the anion recognition process.19,20 As a result, nitrate anion recognition has been mostly studied in less-polar solvents to favour the hydrogen bonding interactions, and in general poor binding affinity has been reported in polar solvents. Secondly, NO3− has a D3h symmetry with equivalent N–O bonds. Based on the principle of geometrical matching,20 the hydrogen bond donors were limited to a complementary trigonal arrangement in the reported systems.15,21 Thirdly, these receptors usually show limited selectivity for NO3−.17 Particularly, due to the negligible difference in ionic radii and charge densities between nitrate and halide anions, it is difficult to selectively recognize nitrate from halide anions.13,15 Fourth, the most reported organic hosts generally require tedious multistep synthesis and in most cases give low yield. This means the design of a specific nitrate receptor is still very challenging.
The coordination cages,22–28 readily self-assembled from simple organic ligand and metal components, have distinct advantages for the design of new ion receptors. Supramolecular organometallic cages avoid tedious synthesis; still they can be regulated easily via a rational symmetry consideration regarding the shape of the ligand, the coordination geometry of the metal, and the relative spatial orientation of the ligand and metal components. Although numerous coordination cages have been designed and synthesized for the encapsulation of anions in the literature,29–36 to the best of our knowledge, an example where the differentiation between nitrate and halides by the host has never been reported due to the difficulties raised above.
Herein, we succeeded in designing a cationic M2L4 cage by a quantitative self-assembly of four anthracene-bridged benzimidazole ligands and two PdII ions (Fig. 1A). The cage showed a D4 symmetry, but exhibited excellent capability in the selective encapsulation of nitrate. The binding constant (Kanion) for the inclusion of NO3− was at least two orders of magnitude higher than all the other anions screened.
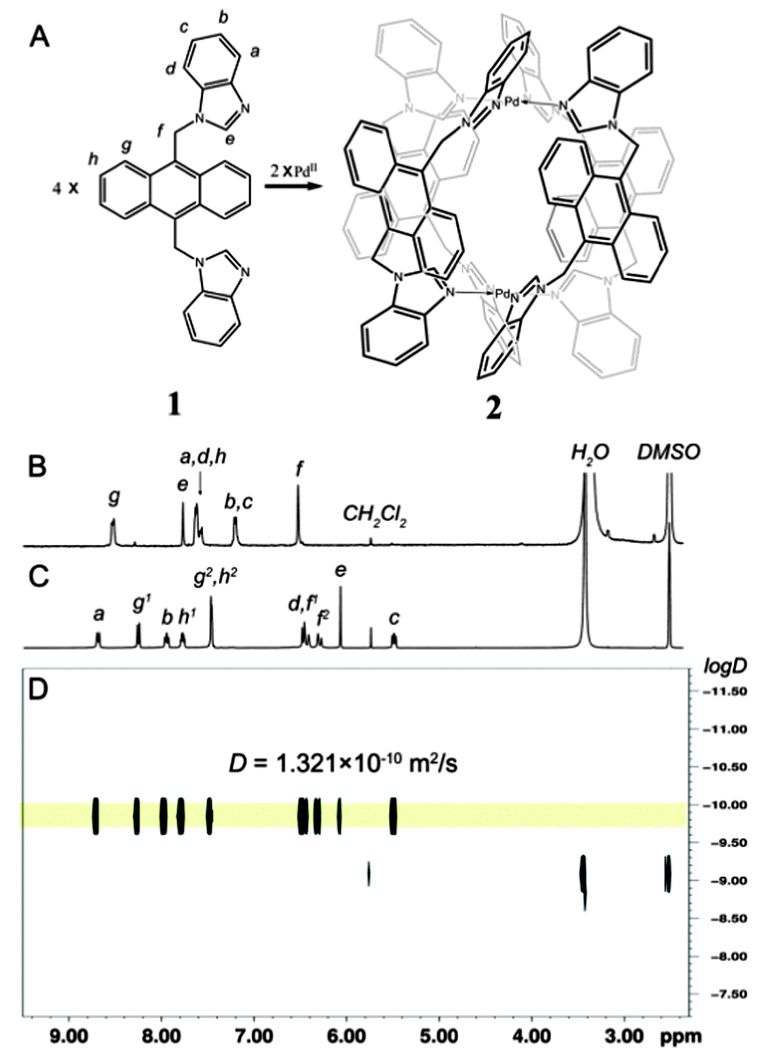 | ||
| Fig. 1 (A) Self-assembly of complex 2; the 1H NMR (400 MHz, d6-DMSO, 298 K) spectrum of (B) ligand 1 and (C) complex 2a; (D) 1H DOSY spectrum of complex 2a. | ||
Bidentate benzimidazole ligand 1, with an anthracene spacer, was synthesized in two steps according to an established method.37 After treating ligand 1 (18 µmol) with a half equivalent of Pd(NO3)2 (9 µmol) in 700 µL d6-DMSO (dimethyl sulfoxide) for 2 h at 70 °C, the turbid solution turned limpid. The signals of the protons on the complex (2a) strongly split and shifted in comparison with those of the free ligand in the 1H NMR spectrum (Fig. 1B and C). All the signals were assigned carefully based on coupling constants, integrals along with the corelationships obtained from the 1H–1H COSY spectrum (Fig. S5, ESI†). Ha and Hb of benzimidazole were significantly shifted downfield (from 7.62 ppm and 7.21 ppm to 8.70 ppm and 7.95 ppm, respectively), which is diagnostic for the metal coordination. Diffusion-ordered NMR spectroscopy (DOSY) showed a single product with a single band at the diffusion coefficient D = 1.321 × 10−10 m2 s−1 (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D = −9.879) (Fig. 1D). The radius of the complex calculated from the D value was 8.42 Å, in accordance with the crystal structures of the complex (see discussion below).
D = −9.879) (Fig. 1D). The radius of the complex calculated from the D value was 8.42 Å, in accordance with the crystal structures of the complex (see discussion below).
Solid structural confirmation of 2a was provided by X-ray crystallographic analysis.‡ Suitable single crystals were obtained by slow diffusion of 1,4-dioxane vapour into a solution of 2a in DMSO after about one week. Crystallographic data showed that four ligands in 2a arranged in a quadruple helicate conformation due to the steric repulsion between the anthracene panels, resulting in a D4 symmetry of the host framework with inherent P or M helicity (Fig. 2A). Such helical chirality of the host must be maintained in solution to account for the observed diastereomeric splitting for proton Hf,g,h signals on the complex (Fig. 1C).
 | ||
| Fig. 2 (A) A perspective view of the crystal structure of 2a with one encapsulated NO3− anion. (Note that the disorder of the central NO3− anion was not shown and four ligands were coloured differently to show the quadruple helicate conformation.) (B) Hydrogen bonding interactions (highlighted with green dashed lines) found between the NO3− and the benzimidazole ligands. All the other residual molecules were omitted for clarity. | ||
More interestingly, the four anthracene walls of the ligand wrap up a very concise hydrophobic cavity where all the benzimidazole protons are pointing inward, forming a perfect bind pocket that is occupied by a nitrate anion. Though the nitrate anion is in-plane disordered into four different orientations due to a mismatch of the symmetry, at each orientation its oxygen atoms are involved in at least six hydrogen bonding interactions with the benzimidazole He (Fig. 2B), with bonding distances of around 2.124–2.637 Å.
We happened to note that only three out of the four NO3− in 2a could be replaced by BF4− after anion exchange by addition of an excess amount of NaBF4 in a typical anion exchange procedure, as revealed by ESI-Q-TOF mass spectroscopy (Fig. 3), which showed prominent peaks observed at m/z 2202.5594, 1057.7774, 676.1837, corresponding to the [(NO3⊂2)·(BF4)3−n]n+, (n = 1–3) respectively. The finely resolved isotopic distribution at each MS signal was also in perfect agreement with the simulated pattern. The IR spectrum also confirmed that the nitrate occupied in the cavity of helicate (Fig. S8, ESI†). This finding inspired us to estimate that cage 2 has a much stronger binding affinity for NO3− than BF4−.
 | ||
| Fig. 3 ESI-Q-TOF mass spectrum of complex (NO3⊂2)·(BF4)3. | ||
When Pd(CH3CN)4(BF4)2 was used as the metal source, 1H and DOSY NMR spectra also suggested the quantitative formation of a similar metal-coordination cage (Fig. S9 and S12, ESI†). However, the 1H NMR spectrum (complex 2b) changed dramatically in comparison with that of 2a (Fig. S23, ESI†). This was the result of the encapsulation of BF4−, which was clearly confirmed by the 19F NMR spectrum (Fig. S11, ESI†) and the ESI-Q-TOF mass spectrum (Fig. S14, ESI†). In the 19F NMR spectrum, the signals corresponding to the encapsulated (−145.18 ppm) and the free (−148.24 ppm) BF4− anions were both observed.30 When the bulkier guest BF4−, with a radius of 2.27 Å,38 which is larger than NO3− (1.79 Å), was encapsulated in situ during the complexation, the cage had to adopt a more twisted configuration. So the difference of δHf1 with δHf2 increased, and Hb and Hc are more downfield shifted. The difference in distortions was also suggested by the coordination conditions. It was necessary to prolong the reaction time or increase the temperature to 110 °C for the formation of 2b, meaning that it had to overcome a higher energy barrier when BF4− was trapped into the cavity.
Though both NO3− and BF4− could be encapsulated, the cage showed distinct binding affinities between them. After treating 2b with one equivalent of KNO3 at 110 °C, the 1H and 19F NMR spectrum revealed that 2b converted to 2a almost quantitatively with the encapsulated BF4− released (Fig. S24 and S25, ESI†), which confirmed that cage 2 bound NO3− more strongly than BF4−.
These observations urged us to make the empty cage 2 for further studies of its specific anion binding properties. We chose Pd(PF6)2 (0.10 M solution in d6-DMSO, prepared by reacting PdCl2 with AgPF6 in a 1:2 ratio at room temperature for 10 h under dark conditions, the AgCl precipitate was then removed by filtration) as the metal source. In contrast to NO3− and BF4−, PF6− with an ionic radius of 2.54 Å38 is significantly larger, and thus should not be encapsulated in the cavity. After treating ligand 1 with Pd(PF6)2 for 2 h at 70 °C, 1H and DOSY NMR spectra all suggested the quantitative formation of a similar metal-coordination cage 2c (Fig. S16 and S19, ESI†). The 19F NMR spectrum also confirmed that the PF6− was not encapsulated by the cage (Fig. S18, ESI†). However, the ESI-Q-TOF mass spectrum revealed that a chloride ion was trapped into the cavity (Fig. S21, ESI†). The contamination of Cl− possibly comes from the preparation process of the Pd(PF6)2 solution. This was also confirmed by the anion exchange reactions (Fig. S26 and S27, ESI†) by treating 2b with 1.1 equivalent of N(C4H9)4Cl at 110 °C, where new emerging signals attributed to 2c were observed. The equilibrium constant, calculated by integrating the 1H NMR spectra, was around 40. The 19F NMR spectrum, showing that BF4− was replaced from the cavity by Cl−, further proved the presence of Cl− in 2c. Cl−, an ionic radius of 1.65 Å, is smaller than NO3− and BF4−, so 2c twisted less. This was also supported by the 1H NMR spectrum, where a smaller diastereomeric splitting between δHf1 with δHf2 was observed in comparison to 2a (Fig. S23, ESI†).
Nevertheless, the presence of Cl− in the cavity of 2c had no influence on the study of anion binding properties. Though the binding constants (Kanion) could not be obtained, the relative binding ability (versus Cl−) could be exhibited. A series of anions available in the lab were screened for binding ability measurement by treating with cage 2c (Fig. 4 and Fig. S28, ESI†), from which the equilibrium constants (K, i.e., Kanion/KCl) (Table 1) for the inclusion of anions, determined by the integration of the characteristic encapsulation signals in the 1H NMR experiments, were calculated.
 | ||
| Fig. 4 1H NMR (400 MHz, d6-DMSO, 298K) spectra showing the encapsulation of different anions by cage 2c. (1.1 equivalent of anions were added to the solution of 2c in d6-DMSO, * represents the new encapsulation signals.) | ||
|
|
|||
|---|---|---|---|
| Anion | K | Anion | K |
| a Determined by the integration of the He signals except for the case of BF4− where the integration of Hc takes place because of signal overlapping. b Determined by the exchange of 2b with N(C4H9)4Cl. c No distinct exchange peaks appeared. | |||
| NO3− | 2.56 × 102 | NO2− | 4.12 × 10−3 |
| Br− | 2.86 × 10−1 | F− | —c |
| I− | 2.33 × 10−2 | Ac− | —c |
| BF4−b |
2.5 × 10−2 | CO32− | —c |

For bulky anions, such as CF3SO3−, H2PO4−, and HSO4−, no signals referenced to the anion-cage complex were observed. This probably was because they are too bulky to enter the cavity. For Br−, the equilibrium constant was less than 1, meaning that the binding affinity is weaker than Cl−. Presumably because the larger Br− needs host 2 to keep a more high-energy twisted configuration, internal anions were exchanged. Similarly, but more obviously, less exchange for BF4− and I− was observed because of their larger size. As for F−, conversely, the smaller size made its spatial orientation inappropriate, so no distinct peaks appeared. The binding of cage 2 towards Ac− and CO32− was much weaker than Cl− though they have similar size and geometry compared to nitrate anions.
Notably, for NO3−, the equilibrium constant was up to 256, in other words, cage 2 shows two orders of magnitude higher binding affinity toward NO3− than Cl−. Moreover, in the kinetic experiments, the encapsulation of NO3− was faster than Br− (Fig. S29–S31, ESI†). We attributed such a big difference to the presence of maximal hydrogen bonding interactions between the nitrate and the host cage in spite of the mismatching on symmetry. In contrast, the lack of hydrogen bonding weakens this binding between anions and the host even if their symmetries are better matching when single-atom halide anions were placed in the D4 symmetrical host. Similarly, little exchange for NO2− was observed.
In conclusion, a chiral M2L4 cage was constructed from the coordination-driven self-assembly of the bidentate ligand 1 and PdII and showed distinct inclusion behaviour for different anions where unprecedented selective binding toward nitrate was observed.
This work was supported by the National Natural Science Foundation of China (Grant no. 21402201, 21471150, and 21221001), start-up foundation from FJIRSM-CAS, and award of “The Recruitment Program of Global Youth Experts”. We thank Prof. Da-Qiang Yuan (FJIRSM) for his kind assistance with the X-ray data collection.
Notes and references
- B. Hay, D. Dixon, G. Lumetta, R. Vargas and J. Garza, in Fundamentals and Applications of Anion Separations, ed. B. Moyer and R. Singh, Springer, US 2004, pp. 43–57 Search PubMed.
- M. Strianese, S. Milione, V. Bertolasi and C. Pellecchia, Inorg. Chem., 2013, 52, 11778–11786 CrossRef CAS PubMed.
- O. A. Okunola, P. V. Santacroce and J. T. Davis, Supramol. Chem., 2008, 20, 169–190 CrossRef CAS PubMed.
- J. L. Sessler, D. An, W.-S. Cho, V. Lynch and M. Marquez, Chem. – Eur. J., 2005, 11, 2001–2011 CrossRef CAS PubMed.
- J. L. Sessler, V. Roznyatovskiy, G. D. Pantos, N. E. Borisova, M. D. Reshetova, V. M. Lynch, V. N. Khrustalev and Y. A. Ustynyuk, Org. Lett., 2005, 7, 5277–5280 CrossRef CAS PubMed.
- A. P. Bisson, V. M. Lynch, M.-K. C. Monahan and E. V. Anslyn, Angew. Chem., Int. Ed., 1997, 36, 2340–2342 CrossRef CAS PubMed.
- J. M. Mahoney, K. A. Stucker, H. Jiang, I. Carmichael, N. R. Brinkmann, A. M. Beatty, B. C. Noll and B. D. Smith, J. Am. Chem. Soc., 2005, 127, 2922–2928 CrossRef CAS PubMed.
- B. Wu, J. Yang, X. Huang, S. Li, C. Jia, X.-J. Yang, N. Tang and C. Janiak, Dalton Trans., 2011, 40, 5687–5696 RSC.
- S. Mason, T. Clifford, L. Seib, K. Kuczera and K. Bowman-James, J. Am. Chem. Soc., 1998, 120, 8899–8900 CrossRef CAS.
- L. Cronin, P. A. McGregor, S. Parsons, S. Teat, R. O. Gould, V. A. White, N. J. Long and N. Robertson, Inorg. Chem., 2004, 43, 8023–8029 CrossRef CAS PubMed.
- P. Byrne, G. O. Lloyd, N. Clarke and J. W. Steed, Angew. Chem., Int. Ed., 2008, 47, 5761–5764 CrossRef CAS PubMed.
- P. Blondeau, J. Benet-Buchholz and J. de Mendoza, New J. Chem., 2007, 31, 736–740 RSC.
- M. Işıklan, M. A. Saeed, A. Pramanik, B. M. Wong, F. R. Fronczek and M. A. Hossain, Cryst. Growth Des., 2011, 11, 959–963 Search PubMed.
- R. Herges, A. Dikmans, U. Jana, F. Köhler, P. G. Jones, I. Dix, T. Fricke and B. König, Eur. J. Org. Chem., 2002, 3004–3014 CrossRef CAS.
- M. J. Langton, L. C. Duckworth and P. D. Beer, Chem. Commun., 2013, 49, 8608–8610 RSC.
- M. J. Langton and P. D. Beer, Chem. Commun., 2014, 50, 8124–8127 RSC.
- J. Romański and P. Pitek, J. Org. Chem., 2013, 78, 4341–4347 CrossRef PubMed.
- A. S. Singh and S.-S. Sun, RSC Adv., 2012, 2, 9502–9510 RSC.
- K. Kavallieratos, C. M. Bertao and R. H. Crabtree, J. Org. Chem., 1999, 64, 1675–1683 CrossRef CAS PubMed.
- P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486–516 CrossRef CAS.
- A. S. Singh and S.-S. Sun, J. Org. Chem., 2012, 77, 1880–1890 CrossRef CAS PubMed.
- S. M. Biros, R. M. Yeh and K. N. Raymond, Angew. Chem., Int. Ed., 2008, 47, 6062–6064 CrossRef CAS PubMed.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed.
- G. H. Clever, S. Tashiro and M. Shionoya, J. Am. Chem. Soc., 2010, 132, 9973–9975 CrossRef CAS PubMed.
- M. Han, J. Hey, W. Kawamura, D. Stalke, M. Shionoya and G. H. Clever, Inorg. Chem., 2012, 51, 9574–9576 CrossRef CAS PubMed.
- J. J. Henkelis and M. J. Hardie, Chem. Commun., 2015, 51, 11929–11943 RSC.
- N. J. Cookson, J. J. Henkelis, R. J. Ansell, C. W. G. Fishwick, M. J. Hardie and J. Fisher, Dalton Trans., 2014, 43, 5657–5661 RSC.
- J. J. Henkelis, J. Fisher, S. L. Warriner and M. J. Hardie, Chem. – Eur. J., 2014, 20, 4117–4125 CrossRef CAS PubMed.
- R. Custelcean, Chem. Soc. Rev., 2014, 43, 1813–1824 RSC.
- C. Klein, C. Gütz, M. Bogner, F. Topić, K. Rissanen and A. Lützen, Angew. Chem., Int. Ed., 2014, 53, 3739–3742 CrossRef CAS PubMed.
- H. Amouri, L. Mimassi, M. N. Rager, B. E. Mann, C. Guyard-Duhayon and L. Raehm, Angew. Chem., Int. Ed., 2005, 44, 4543–4546 CrossRef CAS PubMed.
- C. Desmarets, F. Poli, X. F. Le Goff, K. Muller and H. Amouri, Dalton Trans., 2009, 10429–10432 RSC.
- H. Amouri, C. Desmarets, A. Bettoschi, M. N. Rager, K. Boubekeur, P. Rabu and M. Drillon, Chem. – Eur. J., 2007, 13, 5401–5407 CrossRef CAS PubMed.
- S. Freye, R. Michel, D. Stalke, M. Pawliczek, H. Frauendorf and G. H. Clever, J. Am. Chem. Soc., 2013, 135, 8476–8479 CrossRef CAS PubMed.
- J. K. Clegg, J. Cremers, A. J. Hogben, B. Breiner, M. M. J. Smulders, J. D. Thoburn and J. R. Nitschke, Chem. Sci., 2013, 4, 68–76 RSC.
- I. A. Riddell, M. M. J. Smulders, J. K. Clegg and J. R. Nitschke, Chem. Commun., 2011, 47, 457–459 RSC.
- J.-L. Du, T.-L. Hu, S.-M. Zhang, Y.-F. Zeng and X.-H. Bu, CrystEngComm, 2008, 10, 1866 RSC.
- H. Tokuda, K. Hayamizu, K. Ishii, M. A. B. H. Susan and M. Watanabe, J. Phys. Chem. B, 2004, 108, 16593–16600 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, supporting figures and tables. CCDC 1048711. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc07306e |
| ‡ Crystal data for 2a: space group C2, a = 13.7980 (14) Å, b = 32.546 (4) Å, c = 13.7267 (16) Å, α = 90°, β = 100.330 (10)°, γ = 90°. V = 6064.3 (12) Å3, Z = 2, T = 102 K. R1 = 0.0971, wR2 = 0.2481, and goodness of fit = 1.075. CCDC 1048711. |
| This journal is © The Royal Society of Chemistry 2015 |