
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synergistic reinforcement of Diels–Alder cycloadducts with hydrogen bonding interactions in recyclable dual-dynamic polyurethane networks
Jennifer C.
Hughes
a,
James A.
Wilson
 b,
Devanshi
Singh
a,
Nick
Hawkins
c,
Yi.
Zhang
d,
Chris
Holland
e and
Andrew T.
Slark
*a
b,
Devanshi
Singh
a,
Nick
Hawkins
c,
Yi.
Zhang
d,
Chris
Holland
e and
Andrew T.
Slark
*a
aDepartment of Chemistry, School of Mathematical and Physical Sciences, The University of Sheffield, Sheffield, S3 7HF, UK. E-mail: a.slark@sheffield.ac.uk
bChemical Engineering and Biotechnologies, Aston University, Birmingham, B4 7ET, UK
cDepartment of Engineering Science, Oxford University, Oxford, OX1 3PJ, UK
dISIS Neutron and Muon Source, Rutherford Appleton Laboratory, Didcot, OX11 0QX, UK
eSchool of Chemical Materials and Biological Engineering, The University of Sheffield, Sheffield, S1 3JD, UK
First published on 30th January 2026
Abstract
Here we report a facile, efficient strategy to prepare dual-dynamic networks (DDNs) comprising both thermally reversible Diels–Alder (DA) covalent bonds and non-covalent hydrogen bonds which combine excellent mechanical properties and creep resistance with facile processability at mild temperatures. A series of DDNs was synthesised via the copolymerisation of maleimide-terminated poly(ε-caprolactone urethane) or poly(1,4-butadiene urethane) prepolymers with multifunctional furan crosslinkers containing ester, urethane or urea functional groups. The mechanical properties of the resulting DDNs are enhanced by increasing the strength of crosslinker hydrogen bonding or reducing the polarity of the bismaleimide backbone, achieving a broad range of tensile strength (11.7–26.5 MPa), elongation (210–690%) and toughness (14.4–75.7 MJ m−3) values. DDNs comprising crosslinkers with stronger hydrogen bonding groups produced higher gel transition temperatures (Tgel), creep-resistance and tensile strength, implying synergistic network reinforcement. Furthermore, DDNs comprising the non-polar poly(1,4-butadiene) also presented improved creep resistance. For these materials, rubbery plateaus extended over broader temperature ranges resulting in higher Tgel up to 150 °C. Poly(ε-caprolactone) conferred networks with superior Young's modulus, tensile strength, toughness and flexibility. We have shown that materials can be thermally reprocessed multiple times whilst maintaining high stress recovery efficiencies and display rapid healing abilities under mild temperatures. This work highlights the crucial role of crosslinked network reinforcement via hydrogen bonding interactions to design high-performance yet recyclable polymer networks with tailored properties.
1. Introduction
Covalent adaptable networks (CANs) are crosslinked polymers which incorporate dynamic covalent bonds. These chemical bonds can exchange upon the application of external stimuli such as light, heat or pH, enabling the network to rearrange its topology on a molecular level.1 CANs have become increasingly popular in recent years as they potentially offer a means of bridging the gap between traditional thermoplastics and thermosets, in addition to providing networks with reprocessing, recycling, self-healing and reshaping characteristics.2–4 CANs can be categorised as associative or dissociative, depending on the underlying bond exchange mechanism and their resulting temperature dependence.5 In associative CANs, bond exchange proceeds via an addition–elimination pathway without a temporary net loss of cross-link density, leading to flow at high temperatures but network integrity is maintained.6 This is a characteristic of “vitrimers” which were previously pioneered by Leibler et al., via transesterification in epoxies using ester/hydroxyl groups.7 Other types of chemistry platform have also been developed.8–10 In dissociative CANs, the exchange mechanism proceeds via an elimination–addition pathway, with a temperature dependant equilibrium between covalent bond formation and dissociation. At sufficiently high temperatures, there is a sol–gel transition leading to a loss in network integrity. Notable examples of dissociative dynamic covalent chemistries include Diels–Alder (DA) cycloaddition,9,10 imines,11,12 acylhydrazone bonds,13,14 hindered-ureas,15,16 and disulphide bonds.17,18 The thermally reversible DA cycloaddition between furan and maleimide remains popular due to high conversion at ambient temperature, in addition to its retro DA (rDA) reaction which occurs at relatively low temperatures (typically 100–110 °C).19 Different DA-based polymer architectures have been investigated. In the seminal work by Wudl, DA networks were made from low molecular weight polyfunctional maleimides and polyfunctional furans.20 A common methodology is to synthesise linear oligomers/prepolymers with pendant furan groups in the backbone which are crosslinked with low molar mass bismaleimide crosslinkers (typically BMI).21,22 In our approach, we synthesise and subsequently copolymerise higher molecular weight maleimide terminated-prepolymers with lower molar mass trifunctional furan crosslinkers. Furan-maleimide DA chemistry has been applied to several polymer backbones, including polyurethane,23–26 epoxy,27–29 polyacrylate,30–32 and polysiloxane,33–35 to create self-healing and thermally reprocessable CANs for diverse applications such as elastomers, adhesives, coatings and composites. The autonomous self-healing of polyether based DA networks occurs at room temperature if the surfaces are fresh, otherwise thermal healing (at least 80 °C) is required for repair.36 Autonomous self-healing at room temperature can also be promoted using an off-stoichiometric maleimide-to-furan ratio in the polymer network.37The preparation of CANs which combine both excellent reprocessability with high mechanical properties and creep resistance remains challenging due to conflicting requirements. To address such issues scientists have designed dual-dynamic networks (DDNs) comprising both dynamic covalent and non-covalent crosslinks. Dynamic non-covalent bonds are those formed by reversible supramolecular interactions such as hydrogen bonding, π–π stacking, dipole–dipole interactions and van der Waals forces. Dynamic non-covalent bonds typically exhibit low bond energies, meaning they can provide dynamic networks with fast thermal processing and healing abilities, but they tend to creep, limiting the materials dimensional stability.38 Dynamic covalent bonds possess higher bond dissociation energies than non-covalent bonds, meaning that they can impart stronger mechanical properties and creep-resistance to dynamically cross-linked materials. DDNs have the ability to combine dimensional stability, bond dynamicity and multi-responsiveness. The choice of chemistries can also provide easy access to diversified material properties and synergistic effects. An excellent review by Nicolaÿ et al. provides an overview of the advances in the emerging field of DDNs, with a special emphasis on their design, structure–property relationships and applications.39 Yan and co-workers have also thoroughly described the challenges and perspectives of synergistic dual-dynamic bonds in CANs in their recent mini-review.40
The use of hydrogen bonding to provide synergy with dynamic covalent bonds has proven an effective strategy. Several studies have demonstrated that strong hydrogen bonds are capable of catalysing the DA cycloaddition reaction and may even alter the stereochemistry.41,42 Sacrificial hydrogen bonds can also be introduced into crosslinked networks to increase toughness and tensile strength.43–46 In 2015, Konkolewicz and co-workers synthesised poly(2-hydroxyethyl acrylate)-based networks combining non-covalent crosslinks from reversible 2-ureido-4[1H]-pyrimidinone (UPy) with DA covalent cross-links.47 The materials combined excellent self-healing with creep-resistance behaviour whilst retaining malleability. Konkolewicz later studied dual-dynamic interpenetrating networks (IPNs) comprising thermoresponsive hydrogen bonds and DA bonds based on RAFT chemistry. Properties were tuned by varying crosslinker content and chain length to achieve a material with 100% self-healing recovery and a maximum stress of 6 MPa.48 In 2023, Gao et al. designed a self-healing polyurethane (PU) elastomer with a high fracture strength, elongation and ultra-high toughness for electronic applications exhibiting synergistic enhancement effects from a dual-dynamic DA/hydrogen bond crosslinked network.49 Recently Wang and coworkers studied stereochemistry-tuned hydrogen-bonding synergistic CANs to produce recyclable PU elastomers with excellent creep resistance.50
Inspired by this work, herein we report a facile approach to prepare DDNs from prepolymers and crosslinkers made with remarkably high atom efficiency and conversion from commercially available raw materials. This builds on our previous studies involving the copolymerisation of telechelic bismaleimide prepolymers with furan crosslinkers to produce robust, thermally reversible adhesives.51 In this paper, we investigate the copolymerisation of multifunctional furan crosslinkers comprising additional ester, urethane or urea functional groups with maleimide functional poly(ester urethane) or poly(1,4-butadiene urethane) prepolymers of different polarity. We demonstrate that the morphology and thermomechanical properties of the resulting DDNs vary systematically with the prepolymer and crosslinker structures. We show that increasing the crosslinker hydrogen bonding strength provides synergistic reinforcement of the DA covalent network and enhancement of thermomechanical properties. In particular, the gel transition temperature can be increased to 150 °C while maintaining very high creep resistance. The resulting elastomeric materials are also readily reprocessable and healable.
2. Results and discussion
2.1. Synthesis of trifunctional furan crosslinkers
Three trifunctional (TF) furan crosslinkers comprising isocyanurate cores and either multifunctional ester, urethane or urea functional groups were synthesised: TF-ester, TF-urethane, and TF-urea, respectively (Scheme 1). TF-ester was synthesised via the reaction of tris[2-acryloyloxyethyl] isocyanurate with furfuryl mercaptan. 1H NMR spectroscopy of TF-ester shows successful incorporation of the furan functional groups via facile, quantitative thia-Michael addition in high yield (Fig. S1). The multifunctional crosslinkers TF-urethane and TF-urea were synthesised from the reaction between hexamethylene diisocyanate (HDI) trimer and furfuryl alcohol (FA) or furfuryl amine (FAM), respectively. 1H NMR spectroscopy of TF-urethane and TF-urea also indicates successful incorporation of furan-urethane or furan-urea groups via reaction of the isocyanate functional groups with FA and FAM (Fig. S2 and S3). | ||
| Scheme 1 One step synthesis of (A) TF-ester, (B) TF-urethane, and (C) TF-urea trifunctional furan crosslinkers. | ||
ATR-FTIR spectroscopy reveals that TF-urethane and TF-urea display bands at νmax = 3328 and 3320 cm−1, representing N–H stretching from urethane and urea functional groups, respectively (Fig. 1(A)). All multifunctional crosslinkers show FTIR bands at νmax = 1685 cm−1 in the carbonyl region, corresponding to stretching from the isocyanurate ring carbonyl (Fig. 1(B)). The TF-urethane presents a broad band at νmax = 1685 cm−1, due to the overlap of isocyanurate ring carbonyl with hydrogen-bonded urethane carbonyl stretching. TF-urethane also exhibits a shoulder at νmax = 1720 cm−1 corresponding to free urethane carbonyl stretching. The TF-ester presents a prominent band at νmax = 1730 cm−1, representing ester carbonyl stretching from the crosslinker backbone whilst TF-urea displays a further band at νmax = 1625 cm−1, indicating hydrogen-bonded urea carbonyl stretching. Differential scanning calorimetry (DSC) analysis reveals that TF-ester is amorphous, whereas TF-urethane and TF-urea are semi-crystalline (Fig. 1(C)). The glass transition temperature (Tg) of the crosslinkers increases significantly with hydrogen bonding strength; TF-ester displays a low Tg of −34 °C, whilst TF-urethane and TF-urea exhibit Tg values of −1.4 and 71 °C, respectively. The TF-urethane and TF-urea also exhibit melting endotherms at 44 and 129–140 °C (Tm), respectively. This increase in Tg and Tm reflect the increasing strength of intermolecular forces from ester < urethane < urea groups in the furan crosslinkers.
 | ||
| Fig. 1 (A) ATR-FTIR spectra of TF crosslinkers, (B) ATR-FTIR spectra of TF-crosslinkers zoomed into the carbonyl region and (C) DSC thermograms (1st heating cycles) of TF crosslinkers. | ||
2.2 Synthesis of poly(ε-caprolactone urethane) and poly(1,4-butadiene urethane) bismaleimides
Maleimide-terminated poly(ε-caprolactone urethane) (PCL-M) and poly(1,4-butadiene urethane) (PBD-M) prepolymers were synthesised from the corresponding poly(ε-caprolactone) (PCL) and poly(1,4-butadiene) (PBD) polyols via consecutive addition reactions in bulk. Polyol was first copolymerised with excess 4,4′-methylene diphenyl diisocyanate (MDI) in step 1 to form an isocyanate (NCO) terminated intermediate, followed by reaction of terminal NCO groups with 2-hydroxyethyl maleimide (HEMI) in step 2 (Scheme 2).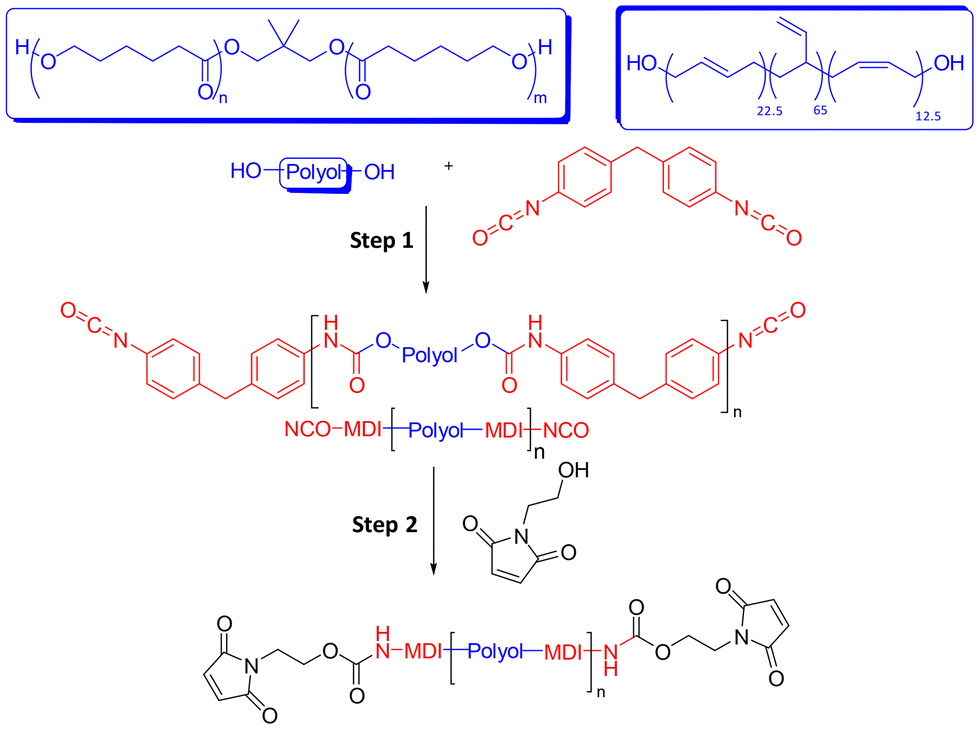 | ||
| Scheme 2 Synthesis of bismaleimide PU prepolymers. Step (1) bulk, 110 °C for 1 hour. Step (2) bulk, 120 °C for 1.5–2 hours. | ||
1H NMR spectroscopy of the prepolymers illustrates chemical shifts associated with the aromatic urethane and maleimide end-groups (Fig. S4 and S5). ATR-FTIR spectroscopy indicates complete disappearance of the free –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO stretching band at vmax = 2260 cm−1 and simultaneous appearance of a fingerprint region band at vmax = 696 cm−1, representing the maleimide B1 antisymmetric ring deformation mode, after step 2 (Fig. S6).52 Size exclusion chromatography (SEC) analysis shows a modest increase in apparent molecular weight compared to the starting polyols, as a result of the reagent stoichiometry (2
CO stretching band at vmax = 2260 cm−1 and simultaneous appearance of a fingerprint region band at vmax = 696 cm−1, representing the maleimide B1 antisymmetric ring deformation mode, after step 2 (Fig. S6).52 Size exclusion chromatography (SEC) analysis shows a modest increase in apparent molecular weight compared to the starting polyols, as a result of the reagent stoichiometry (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mol.eq. NCO:OH, in step 1 and 1.1:1 mol.eq. OH:NCO in step 2) (Table S1). DSC analysis reveals that both bismaleimides display sub-ambient Tg values at −30 °C (Table S1). PCL-M also presents a large endotherm between 32 and 45 °C, resulting from the melting of semi-crystalline PCL segments. In comparison, PBD-M exhibits endotherms at 66 and 146 °C which can be attributed to the melting of hard segments (HS) phase separated from a non-polar matrix (Fig. S7).
:1 mol.eq. NCO:OH, in step 1 and 1.1:1 mol.eq. OH:NCO in step 2) (Table S1). DSC analysis reveals that both bismaleimides display sub-ambient Tg values at −30 °C (Table S1). PCL-M also presents a large endotherm between 32 and 45 °C, resulting from the melting of semi-crystalline PCL segments. In comparison, PBD-M exhibits endotherms at 66 and 146 °C which can be attributed to the melting of hard segments (HS) phase separated from a non-polar matrix (Fig. S7).
2.3 Synthesis of dual dynamic networks
A series of six DDNs was synthesised with systematic changes in the type of furan crosslinker or bismaleimide backbone via the separate copolymerisation of PCL-M or PBD-M prepolymers separately with the TF-ester, TF-urethane, or TF-urea crosslinkers (Table 1). All networks exhibited similar fg values (0.59–0.61) enabling direct comparison. The correct masses of the corresponding furan and maleimide components (to achieve a 1:1 furan:maleimide stoichiometric ratio) were first dissolved in an appropriate solvent and mixed to form a homogeneous solution which was cast into a Teflon-lined Petri dish. Solvent was then removed, and the bulk networks were subsequently processed via compression moulding to give 1 mm thick elastomeric sheets. The resulting networks were conditioned at 25 °C for 7 days to enable DA-cycloaddition prior to testing. Soxhlet analysis of the DDNs reveals high gel fractions (90–97%) indicating high degrees of cross linking. Moreover, ATIR-FTIR spectroscopy of the networks reveals low absorption at νmax = 696 cm−1, implying that most free maleimide groups have successfully copolymerised with furan groups to form DA-cycloadducts (Fig. S8).
| DDN | Bismaleimide | TF-crosslinker | f g (mol kg−1) | Gel fractionb (%) |
|---|---|---|---|---|
| a f g = equivalent concentration of maleimide and furan functional groups. b Determined via Soxhlet extraction in THF after 24 hours. | ||||
| DA(PCL-ester) | PCL-M | TF-ester | 0.61 | 95 |
| DA(PCL-urethane) | PCL-M | TF-urethane | 0.61 | 95 |
| DA(PCL-urea) | PCL-M | TF-urea | 0.61 | 92 |
| DA(PBD-ester) | PBD-M | TF-ester | 0.59 | 90 |
| DA(PBD-urethane) | PBD-M | TF-urethane | 0.59 | 96 |
| DA(PBD-urea) | PBD-M | TF-urea | 0.59 | 97 |
2.4 Thermal properties
The presence and dissociation of DA-cycloadducts in the networks was confirmed using DSC analysis via the appearance of broad endotherms between 107–146 °C, with retro-DA enthalpies (ΔHrDA) between 10–12 J g−1 (Fig. 2(A) and (B) and Table 2). DDNs comprising PCL-M present additional endotherms between 36 and 44 °C for the melting of semi-crystalline PCL soft segments (SS). DA(PCL-ester) and DA(PCL-urethane) present similar ΔHmPCL values (17–20 J g−1), whilst DA(PCL-urea) exhibits a significantly smaller ΔHmPCL of 6.1 J g−1, implying that bidentate urea hydrogen bonding suppresses PCL crystallisation. DA(PCL-urea) exhibits a low Tg of −43 °C but the Tg values of DA(PCL-ester) and DA(PCL-urethane) cannot be detected, likely due to higher PCL crystallinity. The DDNs containing PBD-M display lower TgSS values at −29 °C and upper TgHS values between 41 and 55 °C, indicating microphase separation. These networks also exhibit noticeable exothermic inclines above 160 °C, which can be attributed to maleimide homopolymerisation and the alder–ene reaction.53 The influence of such side reactions can be minimised by incorporating free-radical scavengers.54 | ||
| Fig. 2 DSC thermograms (1st heating cycles) of (A) DDNs based on PCL and (B) DDNs based on PBD with different crosslinkers. TGA thermograms of (C) DDNs based on PCL and (D) DDNs based on PBD with different crosslinkers. | ||
| DDN | T gSS/HS (°C) | T mPCL (°C) | ΔHmPCLa (J g−1) | T rDA (°C) | ΔHrDAa (J g−1) | T deg5% (°C) |
|---|---|---|---|---|---|---|
| a Determined via DSC analysis. b Determined via TGA. | ||||||
| DA(PCL-ester) | — | 44.0 | 17.8 | 107 & 134 | 11.0 | 419 |
| DA(PCL-urethane) | — | 44.0 | 20.0 | 115 & 146 | 10.8 | 384 |
| DA(PCL-urea) | −43/— | 36/43 | 6.1 | 115 & 143 | 10.4 | 364 |
| DA(PBD-ester) | −29/41 | — | — | 129 | 12.4 | 359 |
| DA(PBD-urethane) | −29/54 | — | — | 137 | 11.2 | 333 |
| DA(PBD-urea) | −29/55 | — | — | 136 | 9.6 | 333 |
Thermogravimetric analysis (TGA) reveals that the decomposition temperature at 5% weight loss (Tdeg5%) for all networks exceeds 330 °C, indicating excellent thermal stability. The Tdeg5% values of the DDNs decrease with increasing strength of furan cross-linker hydrogen bonding (Fig. 2(C) and (D) and Table 2). The energies of the major chemical bonds in the DDNs decrease in the order CN (615 kJ mol−1) > CC (602 kJ mol−1) > C–O (358 kJ mol−1) > C–C (346 kJ mol−1) > C–N (305 kJ mol−1). This comparison indicates that the urethane bonds in the HS are likely to be weaker than the other bonds, with the presence of multiple C–N bonds further reducing the thermal stability.55
Dynamic Mechanical Analysis (DMA) was employed to investigate storage modulus (E′) and loss factor (tanδ) of the DDNs as a function of temperature (Fig. 3 and Table S2).
 | ||
| Fig. 3 Variation of storage modulus with temperature for (A) DDNs based on PCL and (B) DDNs based on PBD with different crosslinkers. Variation of tanδ with temperature for (C) DDNs based on PCL and (D) DDNs based on PBD with different crosslinkers. | ||
The PCL based networks initially display a gradual decrease in E′ with temperature. These materials also exhibit clear step changes in E′ and diffuse tanδ peaks between 40–45 °C corresponding to the melting of semi-crystalline PCL segments. On heating above the TmPCL, the modulus gradually reduces before the large reduction in stiffness due to the presence of thermoresponsive DA crosslinks. In comparison, the PBD based networks exhibit a clear initial reduction in E′ accompanied by narrow, strong tanδ peaks between −14 and −16 °C, corresponding to the PBD TgSS values. These networks also present small, diffuse tanδ peaks between 40 and 90 °C, indicating some microphase separation. A steep reduction in E′ occurs in all networks at higher temperatures (>90 °C) where network integrity is lost at Tgel when the equilibrium favours DA bond dissociation. Interestingly, for both PCL and PBD networks the onset temperature of Tgel increases by more than 40 °C depending on the type of crosslinker (TF-urea > TF-urethane > TF-ester). This implies that the hydrogen bonding in the multifunctional crosslinker provides synergistic network reinforcement. The viscoelastic properties can also be tuned by altering the type of backbone. For example, the PBD networks display broader and more stable rubbery plateaus in addition to higher Tgel values than the corresponding PCL networks, likely due to enhanced microphase separation and consequently stronger hydrogen bonding. It is noteworthy that the gel transition temperature for DA(PBD-urea) is 150 °C, typically 50 °C higher than the dissociation of cycloadducts from conventional DA networks based on maleimides and furans often reported.
2.5 Morphology
The morphology of the DDNs was investigated via small-angle X-ray scattering (SAXS) and wide-angle X-ray scattering (WAXS) experiments conducted at room temperature. WAXS shows that all DDNs display broad scattering features between 2θ = 5–30°, centred around 2θ = 20°, due to amorphous regions (Fig. 4(A) and (B)). The WAXS spectra of the networks based on PCL also contain superimposed sharp diffraction peaks at 2θ = 21.2°, 21.8° and 23.5°, characteristic of crystalline PCL segments.56 SAXS analysis implies that all networks comprise phase separated nanodomains. The Lorentz-corrected (Iq2) one-dimensional SAXS plots of the networks based on PCL display broad scattering features between qmax = 0.05–0.08 Å−1 with average interdomain d-spacings between 7.9–12.5 nm according to the Bragg equation (d = 2π/qmax) (Fig. 4(C)). These periodic scattering features indicate the presence of phase-separated nanodomains, likely resulting from the incompatibility between the copolymerised polar TF-crosslinkers and the less polar PCL rubber matrix. Similar SAXS patterns were observed by Wang and coworkers when they incorporated quadruple hydrogen bonds and boronic ester bonds into styrene–butadiene vitrimers.53 DA(PCL-urea) presents an additional scattering peak at qmax = 0.02 Å−1 which can be attributed to the aggregation of TF-urea crosslinker molecules, likely via bidentate hydrogen bonding and π–π stacking. This confines these bonding motifs to larger nanoscale hard domains (d-spacing = 31.4 nm) within the soft PCL matrix. Sun et al. observed similar results by exploiting hydrogen-bonding induced aggregation using dynamic covalent pyrazole-urea motifs to create phase-separated elastomers.57 In comparison, the networks based on PBD exhibit pronounced scattering peaks between qmax = 0.054–0.066 Å−1 corresponding to d-spacings between 9.5–11.6 nm, also indicating nanoscale phase separation (Fig. 4(D)). These scattering peaks are more intense and narrower than those produced by the DDNs based on PCL due to a higher electron density difference between the soft and hard domains. DA(PBD-ester) presents an additional broad scattering feature at qmax = 0.018 Å−1 with a d-spacing of 35 nm, whilst DA(PBD-urethane) and DA(PBD-urea) reveal scattering shoulders at low qmax values, indicating further ordering and possible formation of larger HS aggregates. | ||
| Fig. 4 WAXS profiles of (A) DDNs based on PCL and (B) DDNs based on PBD with different crosslinkers. Lorentz-corrected SAXS plots of (C) DDNs based on PCL and (D) DDNs based on PBD with different crosslinkers. | ||
2.6 Mechanical properties
To further assess the viscoelastic properties of the DDNs, creep recovery measurements were performed under a constant load stress of 0.5 MPa at 70 °C for 30 minutes followed by relaxation for 60 minutes (Fig. 5). The creep behaviour of the DDNs can be significantly reduced via stronger crosslinker hydrogen bonding, with networks comprising TF-urea providing the best results. This is because the additional hydrogen bonds from the furan crosslinkers can act as physical crosslinks to hinder chain segment mobility. There is a large reduction in creep from TF-ester to TF-urethane for both networks based on PCL and PBD. The load stress for the TF-ester containing DDNs led to either rapid creep to the drive limit or sample failure. Similar creep reductions were observed by Wang et al. when they reinforced boronic ester crosslinked styrene–butadiene rubber with quadruple hydrogen bonding moieties leading to phase separation.58 The results also demonstrate that networks based on PBD display significantly reduced creep compared to the respective networks based on PCL. For example, DA(PBD-urethane) reaches 18% creep strain whilst DA(PCL-urethane) advances to the accelerated creep stage (83.7% creep strain). This substantial improvement in creep resistance could be attributed to the enhanced degree of microphase separation occurring in the non-polar PBD. DA(PBD-urea) displays the lowest overall creep strain of 7.8% and almost complete recovery, benefiting from a combination of strong bidentate urea bonding and microphase separation. | ||
| Fig. 5 Creep recovery curves for (A) DDNs based on PCL and (B) DDNs based on PBD with different crosslinkers at 70 °C, under a constant stress of 0.5 MPa for 30 minutes followed by sample recovery for 60 minutes. | ||
The mechanical properties of the DDNs were obtained via uniaxial tensile testing at 25 °C using an extension rate of 10 mm min−1 (Fig. 6(A) and (B)). From the resulting stress–strain curves, the Young's modulus (Ey) (linear regression in the 0–2% strain interval), stress at break (σb), strain at break (εb) and modulus of toughness (UT, the total area under the stress–strain curve) of the networks were calculated (Table S3). The tensile data show that the network strength and Young's modulus generally increase with stronger crosslinker hydrogen bonding, whilst flexibility and overall toughness decrease. The increase in network strength and stiffness can be attributed to the incorporation of stronger hydrogen bonds which act as additional physical crosslinks and resist deformation. This effect is similar to the augmentation of DA networks by hydrogen bonding reported elsewhere. For example, polyurethane networks comprising ureidopyrimidinones produced improvements in mechanical properties.59 Synergy between reversible hydrogen bonds and heat responsive DA bonds has also been observed in polyurethane urea materials where the dual network produced the best mechanical properties.60 The networks based on PCL display enhanced tensile strengths (16–27 versus 12–15 MPa), elongations (390–690 versus 180–350%) and toughness values (55–81 versus 14–26 MJ m−3) over the corresponding networks based on PBD, due to the superior strain-hardening ability of PCL. The stress–strain curves of the PCL networks display prominent yield points due to their semi-crystalline nature, leading to significantly higher Young's modulus values than the corresponding amorphous PBD-based networks (136–300 versus 8–15 MPa, respectively). The Young's modulus increases from 136 MPa in DA(PCL-ester) to over double (299 MPa) in DA(PCL-urea), indicating that stronger crosslinker hydrogen bonding enhances material resistance to deformation. This trend was also extended to the yield stress of the PCL-based systems, although was not as evident in the yield strain, which supports the hypothesis that the distance between crosslinks is comparable, but their strength differs.
 | ||
| Fig. 6 Typical stress–strain curves of (A) DDNs based on PCL and (B) DDNs based on PBD with different crosslinkers. (C) Thermal reprocessing process. (D) Mean stress values for pristine network materials and the same materials after reprocessing cycles ± standard deviation from measurements conducted independently on 5 specimens. | ||
2.7 Recyclability and healing
The networks DA(PBD-urethane), DA(PBD-urea), DA(PCL-urethane), and DA(PCL-urea) were subjected to thermal reprocessing via compression moulding, using mild conditions (100–130 °C, 3 MPa for 5–10 minutes) and subsequently conditioned at 25 °C for 7 days to allow crosslinking before remeasurement of tensile properties (R1). This process was repeated twice with the same materials (R2, R3) (Fig. 6(C)). Infra-red spectra of the reprocessed materials are identical to the originals, showing that there is no apparent change in chemical structure with reprocessing (Fig. S9). All DDNs maintained very good mechanical properties achieving high stress recovery efficiencies (>77%) after 3 recycles, demonstrating their potential sustainability advantage over conventional thermoset PU networks (Fig. 6(D), Table S4 and Fig. S10). The PBD-based networks display slightly higher stress recovery ratios after thermal recycling over the corresponding PCL-networks, possibly due to their amorphous nature.A very brief, preliminary investigation of the healing capability of the networks was performed via optical microscopy using DA(PCL-urethane) as an example (Fig. 7). This elastomer exhibited excellent thermal healing capability, achieving full healing of a 20 µm defect after just 90 seconds at 120 °C, due to the synergistic thermoreversibility of the covalent DA and physical hydrogen bonding crosslinks, enabling chain mobility. In comparison, DAPCL(ester) displays thermal healing of a similar defect within 5 minutes at 80 °C (Fig. S11). Our observations are similar to other polyurethane networks based on maleimides and furans, where healing temperatures are typically in the range of 80 °C–120 °C.61–63 However, a recent study indicates that autonomous self-healing at ambient temperature is possible via the combination of hydrogen bonds and Diels–Alder bonds.64 It is clear that a much more detailed study is required to fully understand the healing behavior of our materials reported here.
 | ||
| Fig. 7 Optical microscopy images of (A) DA(PCL-urethane) with a 20 µm defect and (B) the same sample after heating to 120 °C for 90 seconds with the defect fully healed. | ||
3. Conclusions
Maleimide functional prepolymers and multifunctional furan crosslinkers were made via facile methods from commercially available raw materials with high atom efficiency. Maleimide-telechelic poly(ε-caprolactone urethane) (PCL-M) or poly(1,4-butadiene urethane) (PBD-M) were copolymerised with the multifunctional furan crosslinkers TF-ester, TF-urethane and TF-urea, containing isocyanurate cores and either ester, urethane, or urea functional groups. Successful DA-network formation was confirmed by high gel fractions and low maleimide absorption. The DDNs showed melting endotherms and sharp decreases in E′ between 100–146 °C, confirming the retro DA (rDA) reaction and network dissociation.The thermomechanical properties of the materials increased with stronger hydrogen bonding in the multifunctional crosslinker and a reduction in the polarity of the maleimide prepolymer. Thermal analysis demonstrated that the gel transition temperature and creep resistance were significantly increased, whilst mechanical analysis demonstrated that Young's modulus and network strength increased by incorporating furan crosslinkers with stronger hydrogen bonding groups. Networks based on PBD displayed higher Tgel values, broader rubbery plateaus and improved creep resistance over the corresponding networks based on PCL, whilst the latter exhibited superior strength and flexibility. SAXS analysis revealed that all networks exhibited nanophase separated domains. The DDNs could be thermally reprocessed 3 times via compression moulding using mild conditions whilst retaining very good mechanical properties. Moreover, the networks demonstrated rapid thermal healing capability. This study provides a simple strategy to prepare recyclable and healable DDNs which combine high mechanical properties with excellent creep resistance by incorporating thermoreversible DA cycloadducts and synergistic hydrogen bonds.
Conflicts of interest
There are no conflicts to declare.Data availability
The authors declare that the data supporting the findings of this study are available within the article and its supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5py00347d.Acknowledgements
ATS thanks the EPSRC for a Manufacturing Fellowship (EP/R012121/1).References
- S. J. Rowan, S. J. Cantrill, G. R. Cousins, J. K. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed.
- S. Huang, et al. , Eur. Polym. J., 2020, 141, 110094 CrossRef CAS.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS PubMed.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- J. M. Winne, L. Leibler and F. E. Du Prez, Polym. Chem., 2019, 10, 6091–6108 RSC.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- B. J. Adzima, H. A. Aguirre, C. J. Kloxin, T. F. Scott and C. N. Bowman, Macromolecules, 2008, 41, 9112–9117 CrossRef CAS PubMed.
- J. Inglis, L. Nebhani, O. Altintas, F. G. Schmidt and C. Barner-Kowollik, Macromolecules, 2010, 43, 5515–5520 Search PubMed.
- S. K. Schoustra, J. A. Dijksman, H. Zuilhof and M. M. J. Smulders, Chem. Sci., 2021, 12, 293 RSC.
- S. K. Schoustra, T. Groenevelda and M. M. J. Smulders, Polym. Chem., 2021, 12, 1635 RSC.
- C. e. Yuan, M. Z. Rong and M. Q. Zhang, Polymer, 2014, 55, 1782–1791 CrossRef CAS.
- H. Otsuka, K. Aotani, Y. Higaki and A. Takahara, J. Am. Chem. Soc., 2003, 125, 4064 CrossRef CAS PubMed.
- H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218 CrossRef PubMed.
- L. Zhang and S. J. Rowan, Macromolecules, 2017, 50, 5051–5060 CrossRef CAS.
- B. Sieredzinska, Q. Zhang, K. J. v. d. Berg, J. Flapper and B. L. Feringa, Chem. Commun., 2021, 57, 9838–9841 RSC.
- B. Lewis, J. M. Dennis and K. R. Shull, ACS App. Polym. Mater., 2023, 5, 2583–2595 CrossRef CAS.
- A. Gandini, Prog. Polym. Sci., 2013, 38, 1–29 CrossRef CAS.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- G. Misiakos, L. Parmentier and S. Van Vlierberghe, React. Funct. Polym., 2024, 204, 106024 Search PubMed.
- Q. Zhou, Z. Sang, K. K. Rajagopalan, Y. Sliozberg, F. Gardea and S. A. Sukhishvili, Macromolecules, 2021, 54, 10510–10519 CrossRef CAS.
- G. Rivero, L.-T. T. Nguyen, X. K. D. Hillewaere and F. E. D. Prez, Macromolecules, 2014, 47, 2010–2018 CrossRef CAS.
- Y. Heo and H. A. Sodano, Adv. Funct. Mater., 2014, 24, 5261–5268 CrossRef CAS.
- G. Fu, L. Yuan, G. Liang and A. Gu, J. Mater. Chem. A, 2016, 4, 4232–4241 Search PubMed.
- B. Willocq, F. Khelifa, J. Brancart, G. Van Assche, P. Dubois and J. M. Raquez, RSC Adv., 2017, 7, 48047–48053 RSC.
- X. Kuang, G. Liu, X. Dong, X. Liu, J. Xu and D. Wang, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2094–2103 CrossRef CAS.
- T. S. Coope, D. H. Turkenburg, H. R. Fischer, R. Luterbacher, H. van Bracht and I. Bond, Smart Mater. Struct., 2016, 25, 010201 CrossRef.
- J. Chi, G. Zhang, Q. Xie, C. Ma and G. Zhang, Prog. Org. Coat., 2020, 139, 105473 CrossRef CAS.
- A. Kavitha and N. K. Singha, Macromolecules, 2010, 43, 3193–3205 CrossRef CAS.
- D. Li, Y. Zhang, L. Yuan, G. Liang and A. Gu, Polym. Int., 2020, 69, 110–120 CrossRef CAS.
- G. Fortunato, E. Tatsi, B. Rigatelli, S. Turri and G. Griffini, Macromol. Mater. Eng., 2020, 305, 1900652 CrossRef CAS.
- Q. Yan, L. Zhao, Q. Cheng, T. Zhang, B. Jiang, Y. Song and Y. Huang, Ind. Eng. Chem. Res., 2019, 58, 21504–21512 CrossRef CAS.
- Z. Gou, Y. Zuo and S. Feng, RSC Adv., 2016, 6, 73140–73147 RSC.
- J. Zhao, R. Xu, G. Luo, J. Wu and H. Xia, J. Mater. Chem. B, 2016, 4, 982–989 RSC.
- M. M. Diaz, J. Brancart, G. Van Assche and B. Van Mele, Polymer, 2018, 153, 453–463 CrossRef CAS.
- A. Safaei, J. Brancart, Z. Wang, S. Yazdani, B. Vanderborght, G. Van Assche and S. Terryn, Polymers, 2023, 15(17), 3527 CrossRef CAS PubMed.
- Y. Liu, S. Wang, J. Dong, P. Huo, D. Zhang, S. Han, J. Yang and Z. Jiang, Polymers, 2024, 16, 621 CrossRef CAS PubMed.
- L. Hammer, N. J. Van Zee and R. Nicolaÿ, Polymers, 2021, 13, 396 CrossRef CAS PubMed.
- J. Zhao, Z. Zhang, C. Wang and X. Yan, CCS Chem., 2024, 6, 41–56 CrossRef CAS.
- A. Wittkopp and P. R. Schreiner, Chem. – Eur. J., 2003, 9, 407–414 CrossRef CAS PubMed.
- C. X. Yan, F. Yang, X. Yang, D. G. Zhou and P. P. Zhou, J. Org. Chem., 2017, 82, 3046–3061 CrossRef CAS PubMed.
- Y. Shen, B. Wang, D. Li, X. Xu, Y. Liu, Y. Huang and Z. Hu, Polym. Chem., 2022, 13, 1130–1139 Search PubMed.
- R. Tian, Y. Zhao, Y. Fu, S. Yang, L. Jiang and X. Sui, Food Chem., 2024, 456, 140055 CrossRef CAS PubMed.
- Z. Zhang, D. Lei, C. Zhang, Z. Wang, Y. Jin, W. Zhang, X. Liu and J. Sun, Adv. Mater., 2023, 35, 2208619 Search PubMed.
- D. Lei, Z. Zhang, W. Zhang and X. Liu, Macromolecules, 2024, 57, 1362 CrossRef CAS.
- B. Zhang, Z. A. Digby, J. A. Flum, E. M. Foster, J. L. Sparks and D. Konkolewicz, Polym. Chem., 2015, 6, 7368–7372 RSC.
- B. Zhang, J. Ke, J. R. Vakil, S. C. Cummings, Z. A. Digby, J. L. Sparks, Z. Ye, M. B. Zanjani and D. Konkolewicz, Polym. Chem., 2019, 10, 6290–6304 Search PubMed.
- X. Duan, W. Cao, X. He, M. Wang, R. Cong, Z. Zhang, C. Ning, C. Wang, S. Zhao, Z. Li and W. Gao, Chem. Eng. J., 2023, 476, 146536 Search PubMed.
- Z. Feng, M. Xie, J. Lai, Z. Wang and H. Xia, Angew. Chem., Int. Ed., 2025, 64, e202423712 CrossRef CAS PubMed.
- L. M. Sridhar, M. O. Oster, D. E. Herr, J. B. D. Gregg, J. A. Wilson and A. T. Slark, Green Chem., 2020, 22, 8669–8679 RSC.
- T. Woldbæk, P. Klaboe and C. J. Nielsen, J. Mol. Struct., 1975, 27, 283–301 CrossRef.
- V. Tungare and G. C. Martin, J. App. Polym. Sci., 1992, 46, 1125–1135 Search PubMed.
- T. McReynolds, K. D. Mojtabai, N. H. G. Penners, G. Kim, S. J. Lindholm, Y. Lee, J. D. McCoy and S. Chowdhury, Polymers, 2023, 15, 1106 CrossRef PubMed.
- Y. Song, J. Li, G. Song, L. Zhang, Z. Liu, X. Jing, F. Luo, Y. Zhang, Y. Zhang and X. Li, ACS Appl. Polym. Mater., 2023, 5, 1302–1311 CrossRef CAS.
- Y. Chatani, Y. Okita, H. Tadokoro and Y. Yamashita, Polym. J., 1970, 1, 555–562 Search PubMed.
- S. Sun, G. Fei, X. Wang, M. Xie, Q. Guo, D. Fu, Z. Wang, H. Wang, G. Luo and H. Xia, Chem. Eng. J., 2021, 412, 128675 CrossRef CAS.
- L. Wang, Y. Liu, Y. Qiao, Y. Wang, Z. Cui, S. Zhu, F. Dong, S. Fang and A. Du, Polym. Chem., 2022, 13, 4144–4153 RSC.
- M. Li, R. Zhang, X. Li, Q. Wu, T. Chen and P. Sun, Polymer, 2018, 148, 127–137 CrossRef CAS.
- P. Wu, H. Cheng, X. Wang, R. Shi, C. Zhang, M. Arai and F. Zhao, Green Chem., 2021, 23, 552 RSC.
- J. Xi and N. Wang, Macromol. Chem. Phys., 2024, 225, 240019 CrossRef.
- X. Duan, W. Cao, X. He, M. Wang, R. Cong, Z. Zhang, C. Ning, C. Wang, S. Zhao, Z. Li and W. Gao, Chem. Eng. J., 2023, 476, 146356 CrossRef.
- Y. Li, M. Zhou, R. Wang, H. Han, Z. Huang and J. Wang, Eur. Polym. J., 2024, 214, 113159 CrossRef CAS.
- X. Du, Y. Tong, T. Wang, A. Zhang and Q. Xu, J. Mol. Struct., 2025, 1321, 139793 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |