
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Substrate specificity in decarboxylation of mixtures of acetate and propionate using oxidized Pt electrodes and galvanic square-wave pulsed electrolysis†
Margot
Olde Nordkamp
 a,
Talal
Ashraf
a,
Guido
Mul
*a and
Bastian Timo
Mei
*ab
a,
Talal
Ashraf
a,
Guido
Mul
*a and
Bastian Timo
Mei
*ab
aPhotocatalytic Synthesis Group, Science and Technology Faculty, University of Twente, Drienerlolaan 5, Enschede, 7522 NB, The Netherlands. E-mail: g.mul@utwente.nl
bLaboratory of Industrial Chemistry, Ruhr University Bochum, Universitätsstr. 150, Bochum, 44780, Germany. E-mail: bastian.mei@rub.de; g.mul@utwente.nl
First published on 12th December 2024
Abstract
The decarboxylation (of mixtures) of short-chain carboxylic acids (C2 and C3) on oxidized platinum anodes was investigated using constant current and galvanic square-wave pulse electrolysis. At constant current, a high ethylene to ethane product ratio indicates that propionate is the substrate of preferential decarboxylation in propionate/acetate mixtures, depending on the feed ratio. The specificity of (oxidized) Pt electrodes towards C3 decarboxylation can be further enhanced by the application of cathodic and anodic pulses. The application of relatively long cathodic pulses and very short anodic pulses has been demonstrated to facilitate the formation of high ethylene to ethane ratio product mixtures, which are higher than those obtained under constant current conditions. In particular, extended cathodic pulses have been observed to enhance the faradaic efficiency towards oxygen and to reduce carboxylate conversion. Based on isotherm and RRDE data, we propose that the selectivity for propionate is attributable to a higher affinity for the oxidized Pt electrode, which is further enhanced by cathodic and anodic pulses. The use of galvanic square wave-pulse electrolysis thus offers a promising pathway for the efficient conversion of bio-derived acids into fuels and chemicals.
Introduction
Replacement of fossil resources by biomass for the synthesis of fuels and chemicals can significantly contribute to reaching the climate targets set by the European Commission by 2050.1 Industrial efforts to optimize the use of biomass-derived carbon are rapidly maturing, resulting in an increased availability of bio-based liquids.2,3 Although research and development have led to advancement in biomass liquefaction technologies,4 the quality of these liquids is still fairly poor and requires upgrading before processing in a refinery is feasible. As large scale traditional upgrading of bio-based liquids is energy intensive, alternatively electrochemical processes are developed to improve bioliquid quality and hence establish a circular and bio-based economy.5 In particular Kolbe electrolysis using Pt electrodes receives increased attention to convert bio-derived acids and to upgrade pyrolysis oil and make it suitable for the production of fuels and chemicals.6The effect of reaction conditions on performance of (Pt) electrodes has been frequently studied and a broad substrate scope has been evaluated. Nevertheless, a vast majority of studies examines electrolyte solutions containing single (complex) carboxylic acids in often non-aqueous solvents such as acetonitrile, dimethylformamide or methanol, containing supporting electrolytes like sodium sulphate to increase electrical conductivity.7–11 Utilization of bio-based liquids – such as the water-soluble fraction of pyrolysis oil – in refineries likely requires treatment of concentrated aqueous solutions of mixtures with a high content of short-chain carboxylic acids, with acetic acid (C2 acid) and propionic acid (C3 acid) representing up to 30% of the acid content.12,13
Recent work from Angulo et al. studied the effect of electrolyte composition and operating current density on the product distribution during the electrochemical oxidation of propionic acid in aqueous media using Pt electrodes.14 They showed that high faradaic efficiency (>50%) towards ethylene was obtained, while butane (the Kolbe product) was not detected being in agreement with earlier work of Levy and Sanderson investigating the electrochemical oxidation of (mixtures of) short-chain n-alkanoic acids (C3–C6) in aqueous media.15,16 Interestingly, the earlier work of Levy and Sanderson indicated that the decarboxylation of C3 acid results in butane formation exclusively in the presence of hexanoic acid (C6 acid).15,16 Klocke et al.,17 studied the influence of carboxylic acid chain length during anodic decarboxylation of 3-oxanonanoic acid and 3-oxapentadecanoic acid in methanol confirming favourable formation of (cross-) coupling products in the presence of a carboxylic acid with a longer chain length. They attributed this phenomenon to enhanced interaction of longer chain acids with the electrode surface. A recent work by Neubert et al.18 investigated the (hetero-) coupling (of mixtures) of C4, C6 and C8 acids for bio-fuel production purposes. Overall, the results of the extensive analysis of product distributions and acid conversion suggest a high substrate specificity for C6 acid when non-equimolar mixtures of C4, C6 and C8 are present in the feed (approximately 60% of the total conversion is assigned to C6 acid decarboxylation). These three studies mainly discuss the product selectivity in mixtures of long chain carboxylic acids, while decarboxylation of mixtures of small molecules (C2 or C3) was not examined in detail. Moreover, constant-current or constant potential conditions were used to evaluate acid decarboxylation.
Recently pulsed electrolysis emerged as an interesting alternative to improve reactivity and selectivity for many synthetic organic electrochemical reactions.19–24 In fact, pulsed electrolysis was already used by Hickling and Wilkins to investigate dimer formation during anodic decarboxylation of C2 acid.25 Negative current pulses were found to interfere with dimer production, and instead permit oxygen evolution to the expense of an overall lower reactant conversion.25 Hioki et al.26 used rapid alternating polarity instead of classical direct current measurements highlighting that both product selectivity and reactant conversion for a wide range of carboxylic acids, including biomass-derived acids, are positively affected. The favourable performance was explained by a lower local acidification at the electrode surface resulting in a higher concentration of deprotonated acids (required for the decarboxylation process). Clearly pulsed electrolysis has recently been revealed to provide means to enable electrolysis with unprecedented conversion and selectivity.
In this study, we discuss decarboxylation of mixtures of short-chain carboxylic acids (C2 and C3) using constant current and galvanic square-wave pulse electrolysis. It is shown that C3 is the preferred substrate for conversion using (oxidized) Pt electrodes, and that using galvanic square-wave pulsed electrolysis is a means to enhance this substrate specificity of C3 over C2. We will explain the observations by considering adsorption isotherms of (deprotonated) acids on Pt (oxide) surfaces and rotating-ring disk measurements. Methods to enhance the efficient and selective conversion of bio-derived acids via mixed Kolbe electrolysis are discussed in relation to its economic viability for the production of fuels and chemicals.
Experimental
Materials and chemicals
Platinum foil (0.025 mm thick, 99.9% pure, Alfa Aesar), platinized titanium mesh (Magneto Special Anodes B.V.), and a Pt ring-disk electrode (Metrohm) were used. Nitric acid (ACS reagent, 70%), acetic acid (glacial, ReagentPlus®, >99%), sodium acetate (ACS reagent, >99.0%), propionic acid (ACS reagent, ≥99.5%), sodium propionate (≥99.0%), hexanoic acid (≥99%) and sodium hydroxide (reagent grade, >97%) were used as purchased from Sigma Aldrich without purification. Ethanol (technical grade, BOOM BV) was used for cleaning purposes. Ultrapure water (18.2 MΩ cm−1, from a Millipore, Milli-Q Advantage A10) was used as solvent.Electrochemical measurements and product analysis
All measurements were performed in a single compartment three electrode glass cell (100 mL) equipped with a platinum foil working electrode (0.8 cm2 geometric area), a platinized titanium mesh counter electrode (6 cm2 geometric area) and a Ag/AgCl (3 M NaCl, ProSense) reference electrode connected to a Biologic VMP3 Potentiostat. A custom-made Teflon electrode holder was used to cover the backside of the working electrode and to establish a defined exposed electrode area. Two needle ports used as gas inlet and outlet, respectively enabled continuous purging of the glass cell and were used for the detection of gaseous products by gas chromatography. Prior to each experiment the glass cell, working-, and counter electrode were cleaned with 10% nitric acid and Milli-Q water to remove organic residues. The electrolyte in mixed Kolbe electrolysis experiments always consisted of a total acid concentration of 1 M. The pH of the electrolyte was adjusted to pH 5. The acid ratio of acetic and propionic acid was varied by adjusting the concentrations of the acids. The freshly prepared electrolyte (70 mL) was deoxygenated using 30 mL min−1 Helium (>5.0). Electrochemical cleaning of the Pt working electrode was performed using cyclic voltammetry at a scan rate of 200 mV s−1 after which cyclic voltammograms at 100 and 50 mV s−1 were recorded. Unless mentioned otherwise, potentials were not corrected for ohmic drop, preventing artefacts caused by overcompensation. If not stated otherwise, results are presented against the RHE scale, calculated according to the following equation:| ERHE = EAg/AgCl + 0.059 pH + E0Ag/AgCl | (1) |
Faradaic efficiencies were determined using galvanostatic conditions at a current density of 250 mA cm−2 to ensure Kolbe electrolysis occurs.27 Throughout the measurements the solution was continuously stirred by a magnetic stirring bar at a stirring rate of 900 rpm. Liquid analysis to determine the amount of liquid products and the C2/C3 conversion was performed after a constant charge of 5000 C had passed.
Pulsed electrolysis experiments using fast galvanic square-wave pulses were performed in the three-electrode electrochemical cell described above using a VersaSTAT 3 potentiostat (Princeton Applied Research). Anodic pulses were performed similarly to the constant current experiments at 250 mA cm−2, while negative galvanic pulses were performed at −1 mA cm−2. Experiments were performed with an anodic pulse length of 10 or 200 ms, while the duration of the cathodic pulse was varied from 50 to 500 ms. Galvanic square-wave pulses were performed for a total duration of 2 hours irrespectively of the anodic and cathodic pulse duration.
Product analysis was performed by gas chromatography (GC, Interscience CompactGC, the Netherlands) and high-performance liquid chromatography (HPLC, Agilent technology 1200 series, Agilent). Online detection of gaseous products by GC was performed at 10 minutes intervals with a He (>5.0) purge at a constant flow rate of 30 mL min−1. Light gases (H2, O2, CO2) were detected with a ShinCarbon micropacked column (ST 80/100 2 m, 0.53 mm at 90 °C) connected to a thermal conductivity detector (TCD) operating at 110 °C. Hydrocarbons (C1–C4) were detected using a Rt Q BOND PLOT (0.32 mm ID, 10 μm, 15 m, at 60 °C) column connected to a flame ionization detector (FID) operating at 150 °C. Liquid products were measured by injecting liquid aliquots of the electrolyte into a HPLC equipped with a refractive index detector. Acetic acid, propionic acid and ethanol were detected on a Hi-Plex H column which was heated to 65 °C. The mobile phase consists of 5 mM H2SO4 (flow rate of 0.6 mL min−1). H2SO4 converted the acetate or propionate present in the sample to acetic or propionic acid, which was also detected as such.
Temkin isotherms were constructed from the underpotential deposition of hydrogen (Hupd) determined by cyclic voltammetry measurements performed at a scan rate of 100 mV s−1 unless mentioned otherwise. As supporting electrolyte, a 100 mM acetic acid/sodium acetate buffer with a pH of 5 was used and different concentrations of propionic acid (or hexanoic acid for comparison) were added to the electrolyte, while maintaining a pH of 5. The charge (the integral of current over time) related to Hupd was determined form both adsorption and desorption after subtracting the capacitance current. The plotted values represent the charge associated with Hupd desorption, which generally matches the Hupd charge from adsorption. The adsorption isotherms were determined by fitting the coverages at various concentrations in OriginPro using the Temkin adsorption model, as this model resulted in the best fit.28
Rotating ring disc electrode (RRDE) experiments were performed using a Metrohm Autolab RRDE with a Pt disk used as working electrode and a Pt ring electrode for in situ detection of products. The counter electrode (CE) was a 2 cm2 Pt flag electrode. The collection efficiency of the Pt ring electrodes was determined to be 24%.
Results and discussion
Chronopotentiometry for faradaic efficiency analysis
Product selectivity and substrate specificity in (mixtures of) C2 and C3 acid were determined using chronopotentiometry, online-GC measurements to detect gaseous products, and analysis of liquid products at the end of the experiments using HPLC. Using electrolyte mixtures with different C2 to C3 acid ratios, the faradaic efficiency (%) towards the different reaction products and the conversion (mmoles) of C2 and C3 acid were determined during electrolysis at 250 mA cm−2 on a Pt electrode after a charge of 5000 C passed the cell. Electrolysis of pure C2 acid electrolyte exclusively yields carbon dioxide and ethane (the Kolbe product), in agreement with earlier work.27,29,30 Carbon dioxide is not included in the product distribution in Fig. 1 as it is considered a byproduct resulting from the decarboxylation of carboxylic acid. The electron transfer that occurs prior to decarboxylation is already accounted for in the calculation of the faradaic efficiency of the final products (Kolbe, Hofer-Moest, non-Kolbe, disproportionation, and ester products, see Fig. S1† in ESI for the reaction mechanism). A conversion of approximately 47 mmoles of C2 acid was determined which results in a selectivity of 92% for C2 acid decarboxylation (the maximum possible conversion considering the charged passed was 51 mmoles, calculated by dividing the transferred charge by the Faraday constant and number of electrons required for acid decarboxylation). This is also in agreement with previous literature on Kolbe electrolysis of C2 acid.27,29–31 | ||
| Fig. 1 Kolbe electrolysis of mixtures of acetic acid (C2) and propionic acid (C3) with different ratios (left y-axis). Faradaic efficiency (%) to ethylene, ethane, propane, butane and ethanol (right y-axis). Conversion (in mmoles) of C2 acid (circle) and C3 acid (square). Conversion was determined after a charge of 5000 C passed through the cell. The electrolyte pH was approximately the pKa of both acids (pH ∼5), the total acid concentration was 1 M and measurements were performed with an applied current density of 250 mA cm−2. The error margins are based on N = 2. | ||
Electrolysis of the C3 acid resulted in ethylene and ethanol formation with a FEethylene of ∼60% and a FEethanol ∼27%, respectively. These values are in excellent agreement with data of earlier work on the electrochemical decarboxylation of C3 acid, which reported product yields of 61% and 31% towards ethylene and ethanol, respectively, in a 2 M C3 acid electrolyte with similar pH.15 We also identified formation of ethane, butane and ethyl propionate, in minor quantities. In total, the sum of the individual FE remains short of 100%, which is likely explained by alternative oxidation processes such as the oxidation of ethanol to carbon dioxide, the formation of hydrogen peroxide via water oxidation, or Pt dissolution during electrolysis.32 Interestingly, the conversion of only 28 mmoles of C3 in comparison to 47 mmoles of C2 acid, suggests significant differences in reaction mechanism of decarboxylation of C3 acids compared to C2 acid. C3 acids yield primarily products generated via a two-electron pathway, whereas for C2 acid oxidation, product formation usually requires only one electron transfer step.
Electrolysis of mixtures of C2 and C3 acids was performed either using equimolar amounts of the acids or excess of either C2 or C3 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio. Electrolysis of a 1:1 mixture of C2 and C3 acid revealed a clear substrate specificity towards conversion of the C3 acid with the main products being ethylene and ethanol (with a summed FE of ∼64%), while ethane formation only accounted for a FE of 16%. Additionally, the conversion of C3 acid of 21 mmoles closely resembled the conversion of a pure C3 acid electrolyte. Decarboxylation of the C2 acid amounted to 11 mmoles, in agreement with the lower electron demand for the Kolbe dimer formation, ethane. Even for electrolytes containing an excess of C2 acid (ratio C2:C3 = 2:1), C3 acid was still favourably oxidized. This is evident from a high contribution of ethylene and ethanol (total FE: 56%) in the product composition and a slightly higher conversion of C3 compared to C2 acid (17 mmoles vs. 15 mmoles). The preferred conversion of C3 acid is likely associated with preferred (stronger) adsorption characteristics.
:2 ratio. Electrolysis of a 1:1 mixture of C2 and C3 acid revealed a clear substrate specificity towards conversion of the C3 acid with the main products being ethylene and ethanol (with a summed FE of ∼64%), while ethane formation only accounted for a FE of 16%. Additionally, the conversion of C3 acid of 21 mmoles closely resembled the conversion of a pure C3 acid electrolyte. Decarboxylation of the C2 acid amounted to 11 mmoles, in agreement with the lower electron demand for the Kolbe dimer formation, ethane. Even for electrolytes containing an excess of C2 acid (ratio C2:C3 = 2:1), C3 acid was still favourably oxidized. This is evident from a high contribution of ethylene and ethanol (total FE: 56%) in the product composition and a slightly higher conversion of C3 compared to C2 acid (17 mmoles vs. 15 mmoles). The preferred conversion of C3 acid is likely associated with preferred (stronger) adsorption characteristics.
To verify this hypothesis, mixtures of C3 and C6 acid (having an even higher adsorption strength than C3 acid) were electrolysed (Fig. S2†). Contrasting electrolyte mixtures of C2 and C3 with a ratio of 2:1, the presence of C6 acid significantly decreased the conversion of C3 acid by a factor of four from 28 mmol to 7 mmoles, most likely at the expense of C6 acid conversion as oxygen (formed via competitive water oxidation) was not detected. Even in a 6:1 excess of C3 acid, the conversion of C3 acid appeared to be significantly reduced (18 mmoles) in comparison to electrolytes containing only C3 acid. Furthermore in the presence of C6 acid, the product distribution of C3 acid oxidation products was altered and a significant contribution of butane of 29% (2:1) and 13% (6:1) was observed in agreement with the work of Levy and Sanderson.15 Though a quantitative analysis of the different (cross coupled) oxidation products of C6 acid was not feasible,15,33 these results support the conclusion that substrate specificity of oxidized Pt electrodes is determined by differences in surface affinity of adsorbates.
Pulsed electrolysis
Galvanic square-wave pulsed electrolysis was performed in a 1:1 mixture of acetic acid and propionic acid using positive current pulses (Ja = 250 mA cm−2) with a fixed duration (ta of 10 or 200 ms), while negative current pulses (Jc = −1 mA cm−2) with varying durations (tc = 50–500) (see Fig. S3† in the ESI† for a schematic representation) were used. In the following primarily the ratio of the major products C2H4 and C2H6 (the major products of Kolbe oxidation of C3 and C2 acid respectively) is considered to analyse the impact of the perturbation profile (see Fig. 2).
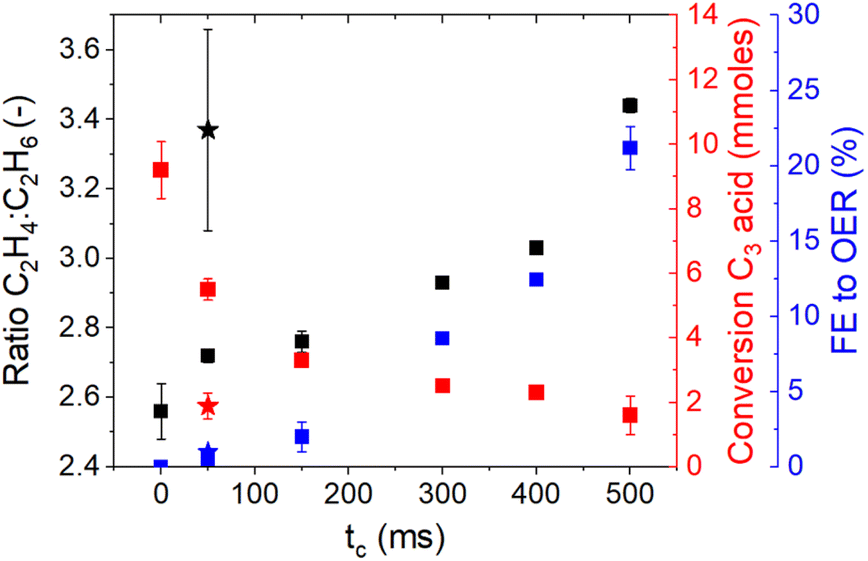 | ||
| Fig. 2 Kolbe electrolysis performed with a 1:1 mixture of C2 and C3 acid (1 M, pH 5) using pulsed electrolysis. The ratio of ethylene and ethane, the C3 acid conversion and the faradaic efficiency towards oxygen were determined during alternating anodic (pulse length of 10 ms and 200 ms at 250 mA cm−2) and cathodic (at −1 mA cm2; pulse length indicated by the x-axis). Experiments performed at an anodic pulse length of 10 ms are shown using star symbols and anodic pulse was fixed with a duration of 200 ms are shown by squares. | ||
Under constant current conditions (tc = 0), a C2H4:C2H6 ratio of 2.5 was obtained (compare also Fig. 1). Using alternating pulses (using ta = 200 ms) the ratio is clearly increasing, with the duration of the cathodic pulse. At the longest cathodic pulse duration tc = 500 ms, a maximum ratio of C2H4:C2H6 of ∼3.5 is obtained. The favourable formation of ethylene, i.e. the product of C3 acid decarboxylation is accompanied by a significant decrease in the total conversion of C3 acid and a significant increase in oxygen evolution as revealed by a FEoxygen of approximately 22% at the longest cathodic pulse durations. It is noteworthy that the use of shorter anodic pulses (ta = 10 ms at Jc = −1 mA cm−2, as indicated by star symbols in Fig. 2) enables the decarboxylation process to be conducted at a favourable C2H4:C2H6 ratio. The ratio of the products was approximately 3.4, while the oxygen evolution reaction (OER) was suppressed. However, variations in the cathodic current density (Jc = −5 mA cm−2 or Jc = 0.1 mA cm−2) did not result in any favourable product composition.
Voltammetry and adsorption isotherms
Rotating-ring disk measurements (Pt disc and Pt ring electrode) were used to further evaluate the electrochemical behaviour of C2 and C3 carboxylic acids (Fig. 3). While linear sweep voltammetry (LSV) was performed at the Pt disk (solid lines, Jdisc), chronoamperometry was performed at the Pt ring at 0.1 V vs. RHE allowing for oxygen detection by the oxygen reduction reaction (dashed lines, Jring). | ||
| Fig. 3 Rotating-ring disk measurements (3000 rpm) of 0.5 M C2 (black curves) acid and 0.5 M C3 (red curve) acid (both at pH 5) using a Pt disk (solid lines) and Pt ring (dashed lines) electrode. Linear sweep voltammetry (10 mV s−1) was performed at the disk while the ring potential was Ering = +0.1 V vs. RHE. The ring currents have been corrected for the collection efficiency and inverted to allow for comparison to the Jdisc. | ||
Independent of the carboxylic acid used, below an applied disk potential of 1.7 V vs. RHE, no significant disc or ring current was detected and only from ∼1.7 V vs. RHE an anodic current was observed at the disk independent of the acid used.
Considering that Jring is simultaneously increasing, oxygen evolution occurs at the disk.27,30 In acetic acid (black traces in Fig. 3), Jdisc reaches a plateau in current density of 17 mA cm−2 at 2.4–2.5 V vs. RHE, which is commonly known as the inflection zone, and according to literature coincides with a transition between OER at lower potentials to hydrocarbon production (Kolbe reaction) at higher potentials.30 Although less pronounced, an inflection zone is also visible at approximately 2.5 V when using C3 acids as electrolyte. In line with Jdisc also the Jring increases from ∼1.7 V to more positive potentials, confirming the formation of oxygen. At potentials above the inflection zone, Jring drops to (almost) zero, indicating that oxygen evolution was (largely) suppressed at these high anodic potentials. In addition to the slight shift in applied Pt disk potential, a clear difference in Jring between C2 (black traces in Fig. 3) and C3 (red traces in Fig. 3) acid was observed. Despite similar disk currents obtained in the plateau region, a maximum Jring,C2 of ∼3.2 mA cm−2 at 2.4 V was obtained for C2 acids, while for Jring,C3 only a maximum current density of ∼1.6 mA cm−2 was obtained. The difference in Jring is assigned to suppression of the OER with increasing carbon chain length of the carboxylic acid used.
The interaction and adsorption strength of the different acids with the Pt surface were obtained from measurements of the underpotential deposition of hydrogen (Hupd) in the presence of different concentrations of the acids using cyclic voltammetry (CV)28 (Fig. S4†) and adsorption isotherms constructed (Fig. S5†) thereof. A buffer of C2 acid was used to exclude the introduction of foreign anions and to maintain a stable concentration of deprotonated carboxylic acid. Examples of the cyclic voltammetry (CV) measurements performed with a Pt electrode in a C2 acid buffer (100 mM) with 50 mM of C3 or C6 acid are shown in Fig. S4.†
In the C2 acid buffer, hydrogen adsorption and desorption on the different facets of Pt is observed at ∼0.25 and ∼0.1 V and ∼0.05 and 0.2 V when sweeping towards negative and positive potentials, respectively.34,35 The current associated with hydrogen underpotential deposition (Hupd) is proportional to the amount of hydrogen ad- or desorbed from the Pt surface and can therefore be used to reveal the fraction of H sites that are occupied by adsorbed (foreign) molecules.28 Thus, the decrease in Hupd peak current density upon addition of C3 acid to the C2 acid buffer (red trace in Fig. S4a†), is directly related to the adsorption of C3 acid with the Pt surface and agrees well with reports on adsorption of other organic molecules such as phenol in acidic media.28,36 Integrating the Hupd current obtained for different concentrations of C3 allows to determine the total charge, i.e., the concentration of adsorbed hydrogen. Comparison to the Hupd charge determined for the Pt surface in the C2 acid buffer only allows to estimate the fraction of Hupd that was inhibited by the presence of the C3 acid. The fraction of Hupd inhibited vs. the logarithmic concentration of C3, reflecting the Temkin isotherm, results in a linear correlation (Fig. S5b†) that allows to estimate the heat of adsorption or Temkin constant of the C3 carboxylic acid from the slope of a linear fit to be α = 0.365.37
The Temkin constant for the adsorption of hexanoic acid was determined for comparison. As shown in Fig. S5b,† a higher α (α = 0.403) for C6 acid, and hence a higher heat of adsorption indicates a stronger interaction of C6 acid with the platinum electrode surface compared to C3 acid. The adsorption data of C2 acid on a Pt electrode surface suggest an α value of only 0.08.38 Overall the RRDE measurements and adsorption isotherms support the hypothesis of a stronger adsorption with increasing carboxylic acid chain length and thus the product distribution of Kolbe electrolysis of mixtures of carboxylic acids will likely depend on the adsorption strength of the individual components and their concentrations.
Discussion
To explain the effect of a longer cathodic duration in square-wave pulse experiments (Fig. 2), we assume the following: during a negative current pulse, the established carboxylate layer will likely desorb and the double layer structure will thus rearrange as a function of the cathodic pulse duration (tc) and hence on the amount of passed cathodic charge (Qc). With increasing Qc, adsorbed carboxylate anions will desorb from the Pt surface, yet it is reasonable to assume that desorption of C2 occurs preferentially due to the significantly lower adsorption strength, as demonstrated by the Temkin isotherms. During the positive pulse, the stronger adsorption of C3 acid will lead to a successive change in the ratio of adsorbed C3 to C2 anions at the Pt surface and thus to a preferred conversion of C3 acid compared to C2 acid, consequently increasing the share of C2H4 in the product distribution.At a high Qc, the anodic pulse charge (Qa) is likely not sufficient to fully restore Kolbe conditions. As a result, oxygen evolution is no longer suppressed by the formation of a (dense) carboxylate layer resulting in the exponential increase in O2 formation at tc > 150 ms (see Fig. 3). Despite variations in electrolyte composition and pulse conditions, an exponential increase in oxygen evolution was also observed in the work of Hickling et al. when cathodic pulses were applied.25 They investigated the effect of cathodic pulses using aqueous acetic acid solutions and found that when Qa was roughly 75 times higher than Qc, oxygen was formed with a CE of ∼20%, which closely resembles the OER FE obtained here. It is important to note that a shift in product selectivity and substrate specificity as a result of the galvanic pulses as well as the implications of changes in pulse duration were not revealed previously.25
Based on the presented results it is expected that by applying galvanic square-wave pulses in mixtures with other carboxylic acids, substrate specificity will also shift to the stronger adsorbing acid. Thus, utilization of galvanic square-wave pulses to influence product selectivity and substrate selectivity in mixed Kolbe electrolysis poses opportunities in the field of electrosynthesis using bio-based liquids and potentially allows for the formation of high-value products. For example, selective production of ethylene from propionic acid can contribute to the production of bio-based fuels or plastics. Furthermore, besides manipulation of acid specificity of the oxidized Pt surface, using galvanic square-wave pulses might also prevent undesired oxidation of other compounds naturally present in bio-based liquids, e.g. alcohols. Nevertheless, further research is necessary to gain a deeper understanding of these potential opportunities and likely additional means to suppress the undesired occurrence of the OER might be essential. Examples of additives which have been used previously to suppress the OER, include sodium dodecyl sulphate, which forms a hydrophobic layer on the Pt anode and improves selectivity to conversion of hydrocarbons.39 Alternatively, the platinum anode could be modified by a PTFE-layer to increase the hydrophobic nature of the electrode to similarly suppress OER.40
Despite the opportunities that pulsed electrolysis in mixed Kolbe electrolysis can offer, it is important that the results obtained in this work are approached with caution. Firstly, the results do not account for the differences in diffusion constants of the carboxylic acids. Changes in the thickness of the diffusion layer can lead to variations in acid conversion and product selectivity. To provide more insight in this regard, measurements using forced transport such as rotating disk electrode (RDE) measurements or measurements in flow cell configurations could be employed. Secondly, the precise composition of the adsorbed layer on the platinum surface is still not known and detailed understanding of the structure of the adsorbed layer during pulsed electrolysis is required. In situ techniques such as surface-enhanced infrared spectroscopy (SEIRAS) or surface-enhanced Raman spectroscopy (SERS) could reveal the composition of the adsorbed layer in (mixed) Kolbe electrolysis. Nevertheless, this work clearly emphasizes the potential of pulsed electrolysis as a strategic tool to modulate product distributions and substrate specificities beyond mixed Kolbe electrolysis of C2 and C3 acid, i.e., for electrolysis of more complex technical feeds.
Conclusions
Kolbe electrolysis of C2 and C3 acid as well as mixtures thereof was investigated using Pt electrodes. The reactant specificity and product selectivity were analyzed and it is highlighted that C3 acid is preferably oxidized over C2 acid. Conversion of C3 was further improved using a galvanic square-wave pulse electrolysis, but at the expense of lower conversion and formation of oxygen at extended cathodic pulse durations which was efficiently suppressed by modulation of the anodic pulse duration. The preferential conversion of C3 acids is explained by adsorption properties of the acids on the (oxidized) Pt surfaces, confirmed by Temkin adsorption isotherms measurements of C3 and C6 acid. Galvanic square-wave electrolysis is a means to manipulate the substrate specificity towards the acid with the strongest affinity for the electrode surface and can be used to advance the production of fuels and chemicals from bio-based resources.Data availability
Data for this article, including gas chromatography, cyclic voltammetry and chronopotentiometry data will be made available at Zenodo at https://doi.org/10.5281/zenodo.14515195.Author contributions
M. Olde Nordkamp: conceptualization, data curation, formal analysis, investigation, methodology, visualization, writing – original draft. T. Ashraf: conceptualization, data curation, formal analysis, methodology. G. Mul: conceptualization, funding acquisition, resources, supervision, writing – review & editing. B. Mei: formal analysis, methodology, conceptualization, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank and acknowledge support from the Topconsortia voor Kennis en Innovatie – Bio-Based Economy (TKI-BBE) who provided valuable financial support for the EC2fuels project. This work was also supported by the European Union's Horizon 2020 Research and Innovation Program under grant agreement no. 101006612. The authors extend their thanks to Dr Robbie Venderbosch from Biomass Technology Group (BTG) for his help with conceptualizing this work.Notes and references
- European Commission, 2050 long-term strategy, https://climate.ec.europa.eu/eu-action/climate-strategies-targets/2050-long-term-strategy_en Search PubMed.
- X. Hu and M. Gholizadeh, J. Energy Chem., 2019, 39(x), 109–143, DOI:10.1016/j.jechem.2019.01.024.
- R. H. A. Venderbosch, ChemSusChem, 2015, 8(8), 1306–1316, DOI:10.1002/cssc.201500115.
- G. Perkins, N. Batalha, A. Kumar, T. Bhaskar and M. Konarova, Renew. Sustainable Energy Rev., 2019, 115, 109400, DOI:10.1016/j.rser.2019.109400.
- J. R. Page, Z. Manfredi, S. Bliznakov and J. Valla, Materials, 2023, 16(394), 1–32, DOI:10.3390/ma16010394.
- F. J. Holzhäuser, J. B. Mensah and R. Palkovits, Green Chem., 2020, 22(2), 286–301, 10.1039/c9gc03264a.
- J. Ranninger, P. Nikolaienko, K. J. J. Mayrhofer and B. B. Berkes, ChemSusChem, 2022, 15(5), 1–7, DOI:10.1002/cssc.202102228.
- H. J. Schäfer, Chem. Phys. Lipids, 1979, 24(4), 321–333, DOI:10.1016/0009-3084(79)90117-8.
- J. Coleman, R. Lines, J. Utley and B. Weedon, J.C.S. Perkin II, 1974, 1064–1069, 10.1039/P29740001064.
- T. Ashraf, A. Paradelo Rodriguez, B. T. Mei and G. Mul, Faraday Discuss., 2023, 247, 252–267, 10.1039/d3fd00066d.
- T. Ashraf, T. Mei and G. Mul, ChemElectroChem, 2024, 11, 252–267, DOI:10.1002/celc.202400274.
- S. V. Vassilev, D. Baxter, L. K. Andersen and C. G. Vassileva, Fuel, 2010, 89(5), 913–933, DOI:10.1016/j.fuel.2009.10.022.
- S. Arnold, T. Tews, M. Kiefer, M. Henkel and R. Hausmann, GCB Bioenergy, 2019, 11(10), 1159–1172, DOI:10.1111/gcbb.12623.
- A. Angulo, C. Elizarraras, J. H. Shin, A. van Riel, T. Akashige and M. Modestino, RSC Sustainable, 2023, 1(9), 2270–2276, 10.1039/d3su00347g.
- P. F. Levy, J. E. Sanderson and L. K. Cheng, J. Electrochem. Soc., 1984, 131(4), 773–777, DOI:10.1149/1.2115697.
- J. E. Sanderson, P. F. Levy, L. K. Cheng and G. W. Barnard, J. Electrochem. Soc., 1983, 130(9), 1844–1848, DOI:10.1149/1.2120109.
- E. Klocke, A. Matzeit, M. Gockeln and H. Schäfer, J. Chem. Ber., 1993, 126(7), 1623–1630, DOI:10.1002/cber.19931260720.
- K. Neubert, M. Hell, M. Chávez Morejón and F. Harnisch, ChemSusChem, 2022, 15(21), 1–10, DOI:10.1002/cssc.202201426.
- J. Zhong, C. Ding, H. Kim, T. McCallum and K. Ye, Green Synth. Catal., 2022, 3(1), 4–10, DOI:10.1016/j.gresc.2022.01.003.
- D. Gunasekera, J. P. Mahajan, Y. Wanzi and S. Rodrigo, J. Am. Chem. Soc., 2022, 144(22), 9874–9882, DOI:10.1021/jacs.2c02605.
- S. Rodrigo, D. Gunasekera, J. P. Mahajan and L. Luo, Curr. Opin. Electrochem., 2021, 28, 100712, DOI:10.1016/j.coelec.2021.100712.
- S. Rodrigo, C. Um, J. C. Mixdorf and D. Gunasekera, Org. Lett., 2020, 22(17), 6719–6723, DOI:10.1021/acs.orglett.0c01906.
- L. Zeng, J. Wang, D. Wang, H. Yi and A. Lei, Angew. Chem., Int. Ed., 2023, 62, e202309620, DOI:10.1002/anie.202309620.
- Y. Kawamata, K. Hayashi, E. Carlson and S. Shaji, J. Am. Chem. Soc., 2021, 143(40), 16580–16588, DOI:10.1021/jacs.1c06572.
- A. Hickling and R. Wilkins, Discuss. Faraday Soc., 1968, 45, 261–268, 10.1039/DF9684500261.
- Y. Hioki, M. Costantini, J. Griffin and K. C. Harper, Science, 2023, 380(6640), 81–87, DOI:10.1126/science.adf4762.
- M. O. Nordkamp, B. Mei, R. Venderbosch and G. Mul, ChemCatChem, 2022, 14(16) DOI:10.1002/cctc.202200438.
- N. Singh, U. Sanyal, J. L. Fulton and O. Y. Gutiérrez, ACS Catal., 2019, 9(8), 6869–6881, DOI:10.1021/acscatal.9b01415.
- T. Dickinson and W. F. K. Wynne-Jones, Trans. Faraday Soc., 1961, 58, 382–387, 10.1039/TF9625800382.
- S. Liu, N. Govindarajan, H. Prats and K. Chan, Chem. Catal., 2022, 2(5), 1100–1113, DOI:10.1016/j.checat.2022.02.014.
- X. Peng, E. Faegh, M. Elam, I. Street and E. Mustain, Chem Catal., 2025, 101190, DOI:10.1016/j.checat.2024.101190.
- M. Olde Nordkamp, T. Ashraf, M. Altomare and A. C. Borca, Surf. Interfaces, 2024, 44(103684), 1–7, DOI:10.1016/j.surfin.2023.103684.
- K. Neubert, M. Schmidt and F. Harnisch, ChemSusChem, 2021, 14(15), 3097–3109, DOI:10.1002/cssc.202100854.
- G. Jerkiewicz, Electrocatalysis, 2010, 1(4), 179–199, DOI:10.1007/s12678-010-0022-1.
- N. M. Marković, T. J. Schmidt, B. N. Grgur and H. A. Gasteiger, J. Phys. Chem. B, 1999, 103(40), 8568–8577, DOI:10.1021/jp991826u.
- K. Sasaki, A. Kunai, J. Harada and S. Nakabori, Electrochim. Acta, 1983, 28(5), 671–674, DOI:10.1016/0013-4686(83)85062-2.
- N. Ayawei, A. N. Ebelegi and D. Wankasi, J. Chem., 2017, 1–11, DOI:10.1155/2017/3039817.
- S. Gilman, Electrochim. Acta, 2012, 65, 141–148, DOI:10.1016/j.electacta.2012.01.016.
- L. Lang, Y. Li, J. C. H. Lam and Y. Ding, Sustainable Energy Fuels, 2022, 6(11), 2797–2804, 10.1039/d1se02067f.
- Y. Ono, S. H. Kim, M. Yasuda and T. Nonaka, Electrochemistry, 1999, 67(11), 1042–1045, DOI:10.5796/electrochemistry.67.1042.
Footnote |
| † Electronic supplementary information (ESI) available: Cyclic voltammograms of Pt electrodes, faradaic Efficiencies, Temkin adsorption isotherm measurements, calibration curves. See DOI: https://doi.org/10.1039/d4se01274g |
| This journal is © The Royal Society of Chemistry 2025 |