
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSequence-defined peptoids via iterative exponential growth†
Abigail M.
Clapperton
 a,
Christine
Hood
a and
Helen
Tran
*abc
a,
Christine
Hood
a and
Helen
Tran
*abc
aDepartment of Chemistry, University of Toronto, Toronto, Ontario M5S 3H6, Canada. E-mail: tran@utoronto.ca
bDepartment of Chemical Engineering and Applied Chemistry, University of Toronto, Toronto, Ontario M5S 3E5, Canada
cAcceleration Consortium, University of Toronto, Toronto, Ontario M5G 1Z5, Canada
First published on 28th April 2025
Abstract
Synthetic control over polymer sequence, composition, and stereochemistry is critical to understanding their influence on the intramolecular and intermolecular interactions of polymers. We report an iterative exponential growth (IEG) strategy for peptoids, a class of sequence-defined peptidomimetics, relying on orthogonally protected monomers. The IEG technique enables the synthesis of monodisperse peptoids with varied sequences, side chains, and stereoconfigurations on a scale that is useful for material science applications. The method allows for direct monitoring of the reaction progress without the need for cleavage from a solid-support. This IEG strategy offers higher molecular weights than other solution-phase sequence-defined synthetic strategies for peptoids and seeks to mimic the precise structural organization of sequence-defined biopolymers for a synthetic polymer system, which we anticipate will enable the rational design of functional polymer materials.
Introduction
Sequence-definition in synthetic polymers allows for precise control over the arrangement of monomers in a polymer chain and is a powerful tool to give access to defined conformations, controlled intermolecular interactions, and information storage.1–4 Peptoids are a chemically diverse class of sequence-defined polymers that are a versatile platform to investigate the functional effects of monomer sequence. Polypeptoids are analogous to polypeptides with the side chain appended to the nitrogen, resulting in an achiral backbone with no hydrogen bond donors (Fig. 1A).5 This imparts unique properties to peptoids including improved processibility, thermal and protease stability, maintained biocompatibility, and access to self-assembled nanostructures through sequence design.6–9 Control over the sequence, composition, and stereoconfigurations of peptoid polymers has been transformative in expanding the understanding of structure–function relationships in these materials. The importance of molecular design has been demonstrated in the influence of peptoid sequence on conformations,10–13 nanostructure morphologies,14–17 molecular interactions,18–21 and targeted RNA delivery.22 For example, changes to peptoid chain length, sequence, and monomer substitution have been shown to strongly influence the self-assembly of peptoids into nanosheets. Zuckermann and coworkers have demonstrated that for a specific peptoid sequence, a difference of 4 units in the chain length (i.e., 12-mer vs. 16-mer) can determine whether the peptoid will successfully assemble into stable nanosheets.23 However, the advancement of these sequence-defined polypeptoids hinges on the development of scalable synthetic methods that can maintain control over the sequence without sacrificing efficiency or synthetic versatility.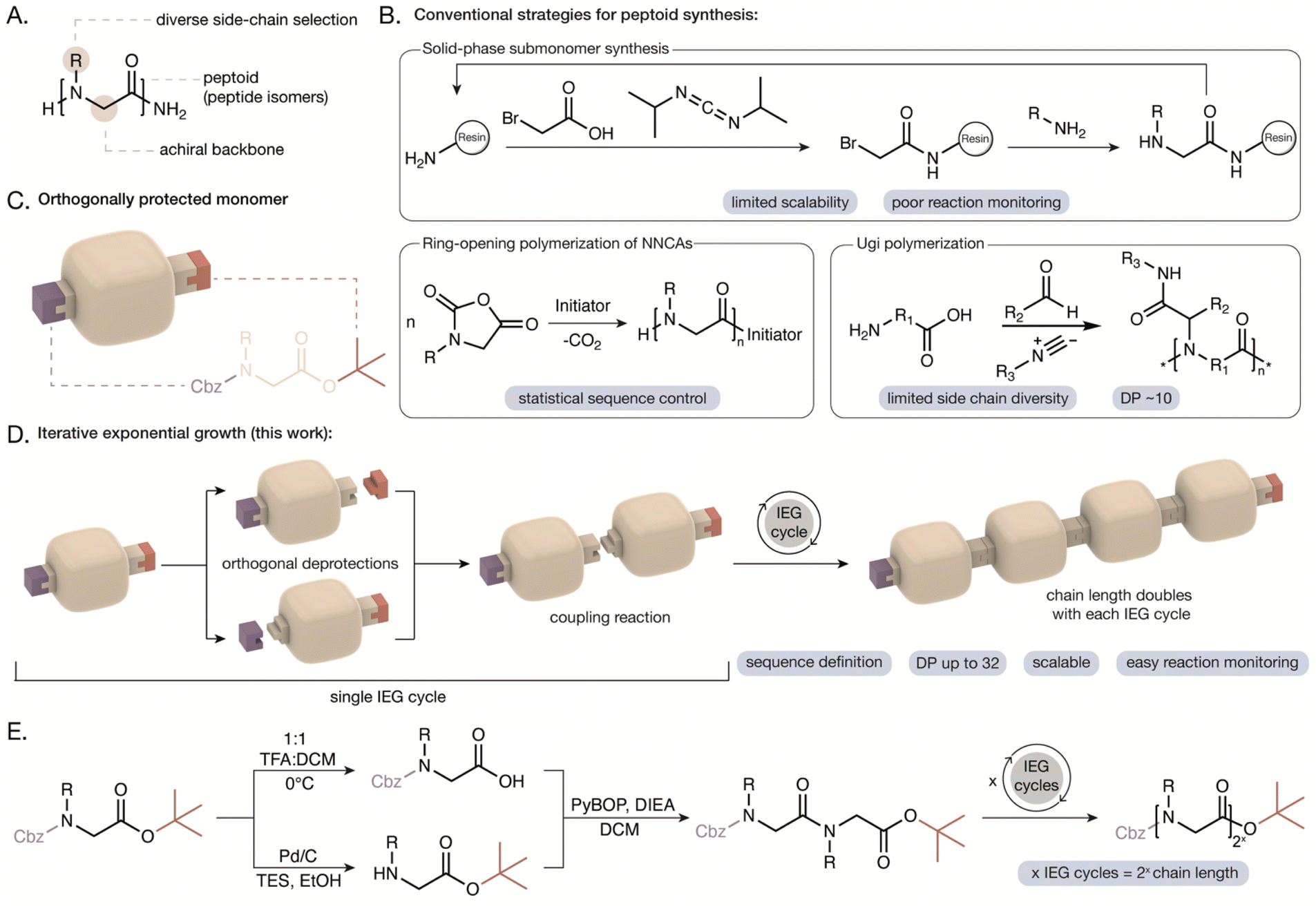 | ||
| Fig. 1 (A) Chemical structure of a peptoid with the achiral backbone and side chain highlighted. (B) Conventional strategies for the synthesis of peptoids include solid-phase submonomer synthesis, ring opening polymerization of N-substituted N-carboxyanhydrides (NNCAs) and the Ugi multicomponent reaction. The current limitations of each strategy are emphasized. (C) Illustration of an orthogonally protected peptoid monomer with the secondary amine protected with the carboxybenzyl (Cbz) group and the carboxylic acid protected as a tert-butyl ester. (D) Illustration of the IEG cycle involving orthogonal deprotections to reveal reactive functional groups, followed by a coupling reaction. The chain length will double with each IEG cycle. (E) The IEG cycle developed in this work for peptoid synthesis involves orthogonal deprotections using acid and hydrogenation, followed by amide coupling. For x IEG cycles, the chain length is 2x (i.e., 8 monomer units after 3 IEG cycles). | ||
Significant progress in synthetic strategies for polypeptoids has given access to a large diversity of sequences and chain lengths, yet achieving both sequence definition and scalability remains a challenge. The solid-phase submonomer method offers absolute sequence control through a two-step cycle involving acylation with bromoacetic acid followed by displacement with a primary amine (Fig. 1B).5,24 However, this strategy requires reagents in 10–25× excess to achieve high conversions, which is undesirable for costly or synthetically complex amines.25 Peptoids with chain lengths of up to 60 units have been synthesized using the submonomer approach.24,26,27 However, the synthesis is still limited to milligram scales in typical research lab environments and reaction progress cannot be monitored without cleavage from the resin. The ring-opening polymerization of N-substituted N-carboxyanhydrides (NNCAs) can generate peptoids on larger scales and has been optimized for improved control over molecular weights and dispersity. However, this technique can only provide statistical sequence-control through the ratio of monomers added (Fig. 1B).28–31 Scalable strategies for peptoid synthesis like the Ugi multicomponent reaction and iterative solution-phase approaches offer sequence-definition for low degrees of polymerization (i.e., up to 10-mer alpha-peptoids) with limited side chain diversity.32–35
Iterative exponential growth (IEG) approaches provide access to sequence-defined polymers but have not yet been translated to peptoid synthesis. Typical IEG methods involve an asymmetric difunctionalized monomer that is subjected to orthogonal reactions to generate two different functional groups that undergo a coupling reaction.36–43 One set of reactions in an IEG cycle doubles the polymer chain length while maintaining precise control over the primary sequence. IEG strategies are attractive as they are scalable, can access higher degrees of polymerization in fewer reactions than multistep growth methods, and don't require reagents in excess.44 However, many IEG approaches have low sequence complexity, intensive purification steps, lower yields with increasing molecular weight, and longer reaction times than solid-phase approaches.36–43 The extension of IEG strategies to diverse polymer backbones and side chains will help to address the classic trade-off between scalability and structural control in polymer synthesis, increasing the relevance of sequence-defined polymers beyond research labs.
Here we report an IEG strategy to prepare sequence-defined peptoids on multi-gram scales (e.g., an octamer on 8-gram scale), relying on orthogonal protecting groups and amide coupling reactions. In this approach, the C-terminus of the peptoid was protected as an acid labile tert-butyl ester (Fig. 1C). Recent work by Faure and coworkers demonstrated that increased steric hindrance at the C-terminus prevents undesirable diketopiperazine formation during the stepwise synthesis of peptoids in the C to N direction.33 The carboxybenzyl (Cbz) group was selected as the N-terminus protecting group as it is orthogonal to the tert-butyl ester and is removed via hydrogenation (Fig. 1C).45 In an IEG cycle, the doubly protected peptoid monomer is divided to undergo orthogonal deprotections, revealing a secondary amine and a carboxylic acid which can undergo amide coupling to double the chain length (Fig. 1D and E). The nature of this IEG strategy also enables direct reaction monitoring since cleavage from a resin is not required. We report the compatibility of this IEG strategy with a variety of hydrophobic, hydrophilic, and chiral monomers to generate peptoids with varied chain lengths (i.e., up to a 32-mer demonstrated), controlled stereochemistry, and unique sequences (i.e., homopolymer, block, alternating, periodic, and aperiodic). The monodisperse peptoids were characterized by nuclear magnetic resonance (NMR) spectroscopy, gel permeation chromatography (GPC), ultra-high-performance liquid chromatography (UHPLC), and mass spectrometry (matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF MS) or direct analysis in real time (DART-MS)). The self-assembly of peptoids into nanosheets and helical secondary structures is maintained after synthesis via IEG and characterized using transmission electron microscopy (TEM) and circular dichroism (CD) spectroscopy. Furthermore, post-IEG modification of peptoids is shown using thiol–ene and copper-catalyzed azide–alkyne cycloaddition (CuAAC) reactions. This allowed us to incorporate functional groups that were incompatible with the deprotection reactions (i.e., alcohols and thiols), thus expanding functional group diversity. The versatile IEG approach described herein will enable the design and synthesis of functional peptoid-based materials with highly specific properties controlled through their primary structure.
Results and discussion
Peptoid homopolymers
We evaluated the performance of the proposed IEG strategy using monomers that have been previously investigated in a variety of peptoid applications (i.e., N-decylglycine (Ndc), N-phenethylglycine (Npe), N-2-(2-(2-methoxyethoxy)ethoxy)ethylglycine (Nte), and N-(2-methoxyethyl)glycine (Nme)).46 The orthogonally protected monomers for IEG were prepared in one to three steps from each primary amine. Substitution of the amine with tert-butyl bromoacetate generated the tert-butyl ester protected monomer with a free secondary amine (ESI Scheme 1†). A portion of this product underwent amine protection with benzyl chloroformate followed by deprotection of the tert-butyl ester with trifluoroacetic acid (TFA) to prepare the Cbz-protected monomer with a free carboxylic acid (ESI Schemes 2 and 3†). These singly protected monomers were prepared on multigram scales in 65–80% yield over the three steps. Preliminary work explored fluorenylmethoxycarbonyl (Fmoc) as the amine protecting group, but unwanted deprotection occurred during amide coupling by the amine monomer, causing uncontrolled polymerization (Fig. S2†). The selected Cbz group is unreactive to the tert-butyl ester cleavage conditions (i.e., TFA at 0 °C) and the amide coupling, making it compatible with IEG for peptoid synthesis.45Since chain length increases exponentially with every IEG cycle, each of the prepared monomers was subjected to three IEG cycles to access octamers (Fig. 2A). One IEG cycle is comprised of three reactions corresponding to the tert-butyl ester deprotection, Cbz deprotection, and amide coupling (Fig. 1E). The prepared amine and carboxylic acid monomers were coupled using PyBOP ((benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate) as the amide coupling agent with N,N-diisopropylethylamine (DIEA) to achieve dimers up to a 16 g scale in 80–95% isolated yields (ESI Scheme 4†). PyBOP is a greener alternative to BOP as it doesn't generate the carcinogenic byproduct, hexamethylphosphoramide. Phosphonium coupling reagents are known to have good solubility at high concentrations and can be used in excess without side reactions and chain terminations.47 PyBOP has been previously explored in the preparation of cyclic peptoids with high efficiency and was therefore hypothesized to be successful in coupling long peptoid chains together in this IEG approach.48,49 We performed preliminary investigations with several carbodiimide coupling reagents but found PyBOP provided the highest reaction efficiency. After coupling, a portion of the dimers was then subjected to the tert-butyl ester deprotection using 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 TFA/dichloromethane (DCM) at 0 °C to provide the singly protected carboxylic acid dimer after aqueous work-up in 70–95% isolated yields (ESI Scheme 5†). In a separate reaction, the Cbz protecting group of the dimer was removed via catalytic transfer hydrogenation with triethylsilane, generating the amine dimer after a silica plug in 90–95% isolated yields (ESI Scheme 6†).50 An amide coupling reaction provided the tetramers and subjecting these to another three-step IEG cycle generated the octamer peptoids on a multigram scale in 45–90% isolated yields (i.e., Ndc octamer was obtained on an 8-gram scale, ESI Schemes 7–10†).
:1 TFA/dichloromethane (DCM) at 0 °C to provide the singly protected carboxylic acid dimer after aqueous work-up in 70–95% isolated yields (ESI Scheme 5†). In a separate reaction, the Cbz protecting group of the dimer was removed via catalytic transfer hydrogenation with triethylsilane, generating the amine dimer after a silica plug in 90–95% isolated yields (ESI Scheme 6†).50 An amide coupling reaction provided the tetramers and subjecting these to another three-step IEG cycle generated the octamer peptoids on a multigram scale in 45–90% isolated yields (i.e., Ndc octamer was obtained on an 8-gram scale, ESI Schemes 7–10†).
 | ||
| Fig. 2 Peptoid homopolymers synthesized via IEG. (A) Illustration of the synthesis of an 8-mer peptoid after an orthogonally protected monomer undergoes three IEG cycles. (B) Chemical structure of orthogonally protected Ndc peptoid and GPC traces for chain lengths of 2–8. (C) Chemical structure of orthogonally protected Npe peptoid and UHPLC traces for chain lengths of 2–8. (D) Chemical structure of orthogonally protected Nte peptoid and UHPLC traces for chain lengths of 2–8. (E) Chemical structure of orthogonally protected Nme peptoid and UHPLC traces for chain lengths of 8–32. (F) GPC traces for Nme peptoid series with chain lengths of 4, 8, 16, and 32. (G) 1H NMR spectrum of Nme32 with some peak splitting due to the presence of rotamers from the Cbz and tert-butyl ester protecting groups. | ||
The peptoid dimers and tetramers were purified by silica gel chromatography while the octamers were purified using a preparative recycling GPC. The preparative recycling GPC separates molecules based on size, rather than polarity. We found that purification is more efficient with the IEG method rather than solid-phase or other iterative solution-phase methods since the product and starting materials in IEG have significantly different sizes (i.e., 8-mer and 4-mer). Purification after solid-phase peptoid synthesis requires an 8-mer to be purified from a 7-mer or 6-mer, which have very small differences in size and polarity, thus making separation more challenging. The amide coupling gave decreased yields with each cycle, as expected from other IEG protocols.44 For example, the cumulative yield of the Nme8 peptoid was 56% after 7 steps when starting from the singly protected monomers. A summary of the yields for each step of the IEG cycle as well as cumulative yields is included in the ESI as Table S1† for all peptoids synthesized in this study. 1H NMR, 13C NMR, UHPLC, GPC, MALDI-TOF MS, and DART-MS demonstrated the successful preparation of monodisperse peptoids with a variety of side chains (Fig. 2B–D and see ESI† for detailed characterization data). We would also like to note that the hydrophobicity of peptoids can be challenging to predict due to the differing contributions of the side chains and the peptoid backbone. For example, Nte4 and Nme4 had shorter UHPLC retention times than Nte2 and Nme2, respectively (Fig. 2D and 3B). For all other less polar side chains, UHPLC retention time increased with peptoid chain length.
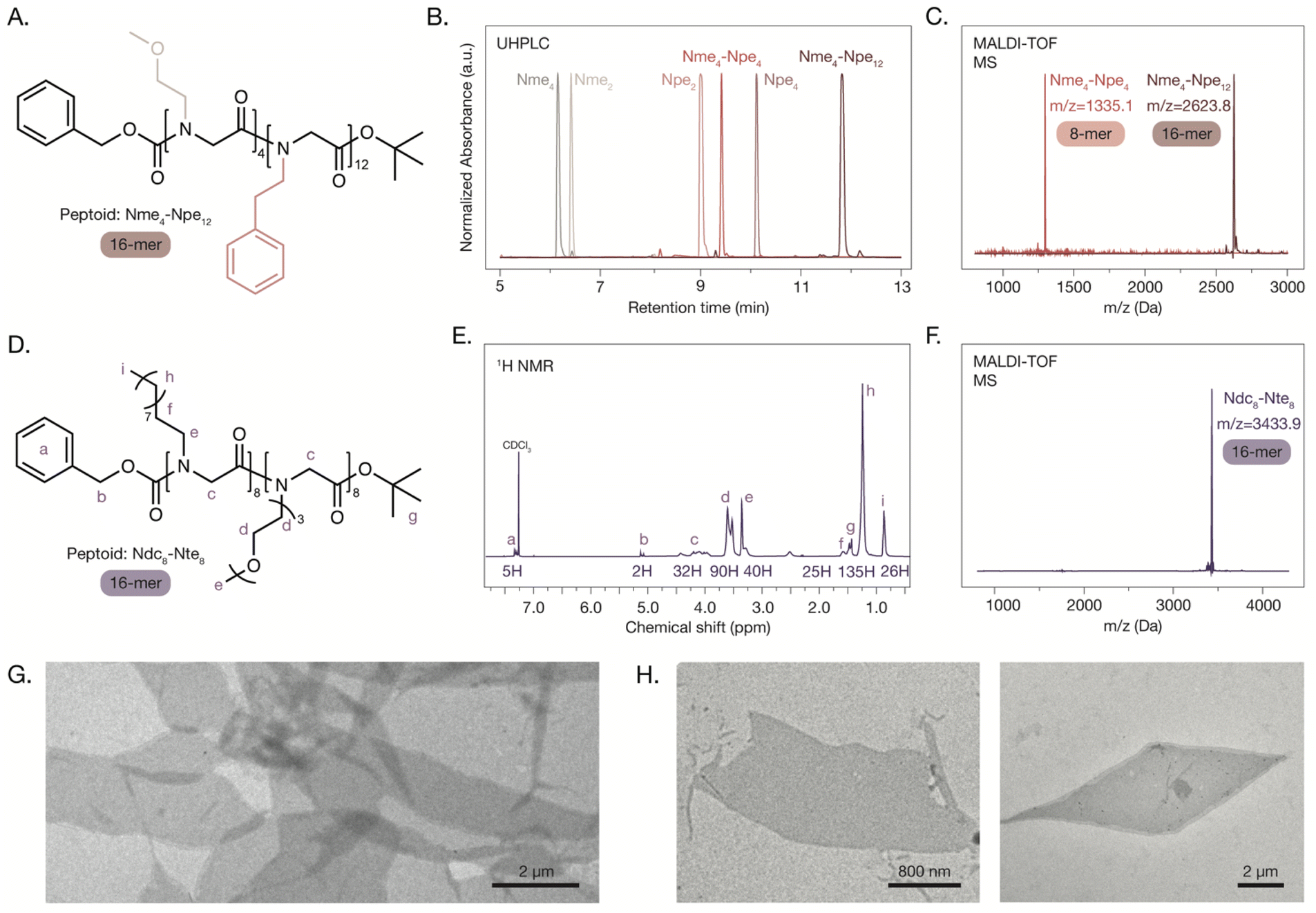 | ||
| Fig. 3 Peptoids with diblock sequences synthesized via IEG and their self-assembly. (A) Chemical structure of orthogonally protected Nme4–Npe12 block peptoid. (B) UHPLC traces from each IEG cycle in the synthesis of the block 16-mer. (C) MALDI-TOF MS traces for the Nme4–Npe4 and Nme4–Npe12 peptoids synthesized using the IEG method. (D) Chemical structure of orthogonally protected Ndc8–Nte8 block peptoid. (E) 1H NMR spectrum of the Ndc8–Nte8 peptoid with peaks labelled. (F) MALDI-TOF MS trace for the Ndc8–Nte8 peptoid. (G and H) TEM micrographs of overlapping (G) and individual (H) orthogonally protected Ndc8–Nte8 nanosheets. | ||
Beyond the synthesis of peptoid octamers, we expect that the IEG strategy can give access to higher molecular weight peptoids compared to other solution-phase sequence-defined peptoid synthesis strategies (i.e., beyond 10-mer peptoids). The Nme8 peptoid was subjected to two additional IEG cycles to extend the chain length to 16-mer and 32-mer peptoids (ESI Schemes 11–16†). The UHPLC, MALDI, and GPC traces highlighted that precise chain length control was achieved with IEG for the series of Nme peptoids (Fig. 2E–G). The peptoids were also characterized using NMR spectroscopy as the selected protecting groups allow for end-group analysis (Fig. 2G). Some peaks in the 1H NMR spectrum of the orthogonally protected peptoids are split due to the presence of rotamers from the two protecting groups, as has been observed previously.51 We hypothesize that further extension of the chain lengths is possible with appropriate reaction scaling to account for decreased amide coupling yields. Additional optimization of the solvents for the three reactions of this IEG strategy may be required for peptoids with more hydrophobic side chains (i.e., Ndc or Npe) as we observed decreased solubility when attempting to access longer chain lengths (>16).
Diblock peptoids and self-assembly
Peptoid block copolymers are of significant interest as they can form discrete morphologies upon self-assembly.52 The synthesis of diblock peptoids with defined block sizes was achieved by combining peptoid blocks with different side chains in later IEG cycles. A block of Nme4 and a block of Npe4 were obtained after two IEG cycles from each monomer, as previously described. The tetramers were coupled after a tert-butyl ester deprotection of Nme4 and a Cbz deprotection of Npe4, generating the block Nme4-Npe4 octamer (ESI Scheme 17†). The Npe block was successfully extended after undergoing another IEG cycle with Npe8, generating the Nme4–Npe12 asymmetric block peptoid (Fig. 3A and ESI Schemes 18–20). The precise control over the composition of each block and their order are indicated by the monodisperse UHPLC and MALDI-TOF MS traces and confirmed with 1H NMR end group analysis, where signals for both the Nme and the Npe blocks were observed with expected integrations (Fig. 3B and C). Absolute control over the block size and end groups of diblock peptoids can allow for improved investigation of structure–function relationships, as small changes in the peptoid sequence have been shown to have significant effects on self-assembly.53–56Another diblock peptoid was prepared from Ndc and Nte monomers and its self-assembly into two-dimensional nanosheets was observed using TEM. The self-assembly of this peptoid diblock has been the focus of recent investigations, where interactions at the N-terminus have a significant impact on chain packing.56 Using the Ndc8 and Nte8 peptoids, we performed the tert-butyl ester deprotection of Ndc8 and the Cbz deprotection of Nte8 to generate singly protected 8-mer blocks (ESI Schemes 21 and 22†). These blocks underwent an amide coupling reaction to generate the 16-mer peptoid, Ndc8-Nte8 (Fig. 3D and ESI Scheme 23†). UHPLC, MALDI-TOF MS and NMR were used to characterize the resulting peptoid, confirming its defined sequence (Fig. 3E and F). The peptoid was subjected to self-assembly conditions and nanosheets with a range of sizes (1–12 μm) were observed via TEM. The protecting groups used for this IEG strategy did not negatively affect the self-assembly of diblock peptoids and contributed to the unique nanoscale morphologies observed (Fig. 3G and H). After Cbz deprotection of the Ndc8–Nte8 peptoid followed by self-assembly, only peptoid aggregates were observed by TEM, highlighting the importance of the hydrophobic N-terminal end group on nanosheet stability (ESI Fig. S1†). An investigation of the influence of the end-groups on the self-assembly of these diblock peptoids will require further systematic study.
Sequence-defined periodic and aperiodic peptoids
While IEG sequences are inherently repetitive, peptoid sequences of increasing complexity could be achieved by combining different monomers during early IEG cycles. This sequence definition is useful for applications that require the precise positioning of specific side chains along the peptoid backbone, like biosensing. The synthesis of an octamer peptoid comprised of four different side chains with an ABXY–ABXY sequence is depicted in Fig. 4A. The singly protected N-hexylglycine (Nhx), Npe, Ndc, and Nme monomers were prepared and coupled to produce dimers Nhx–Npe (AB) and Ndc–Nme (XY) on multigram scales in 89% yields (ESI Schemes 24–28†). Two subsequent IEG cycles generated 1.7 g of the ABXY tetramer in 88% yield and 0.8 g of the ABXY–ABXY octamer in 88% yield (ESI Schemes 29–34†). The sequence-definition was confirmed with UHPLC, MALDI-TOF MS, and NMR with no evidence of other chain lengths or sequences present (Fig. 4B–D). The ABXY–ABXY octamer underwent another IEG cycle to produce the monodisperse 16-mer (ABXY)4 (Fig. 4D and ESI Schemes 35–37†). We were also successful in combining the ABXY–ABXY octamer with the block peptoid Npe4–Nme4, producing an aperiodic 16-mer peptoid with a precisely controlled sequence (Fig. 4E and ESI Scheme 38†). All characterization methods are consistent with the resulting peptoid structures (Fig. 4F and G).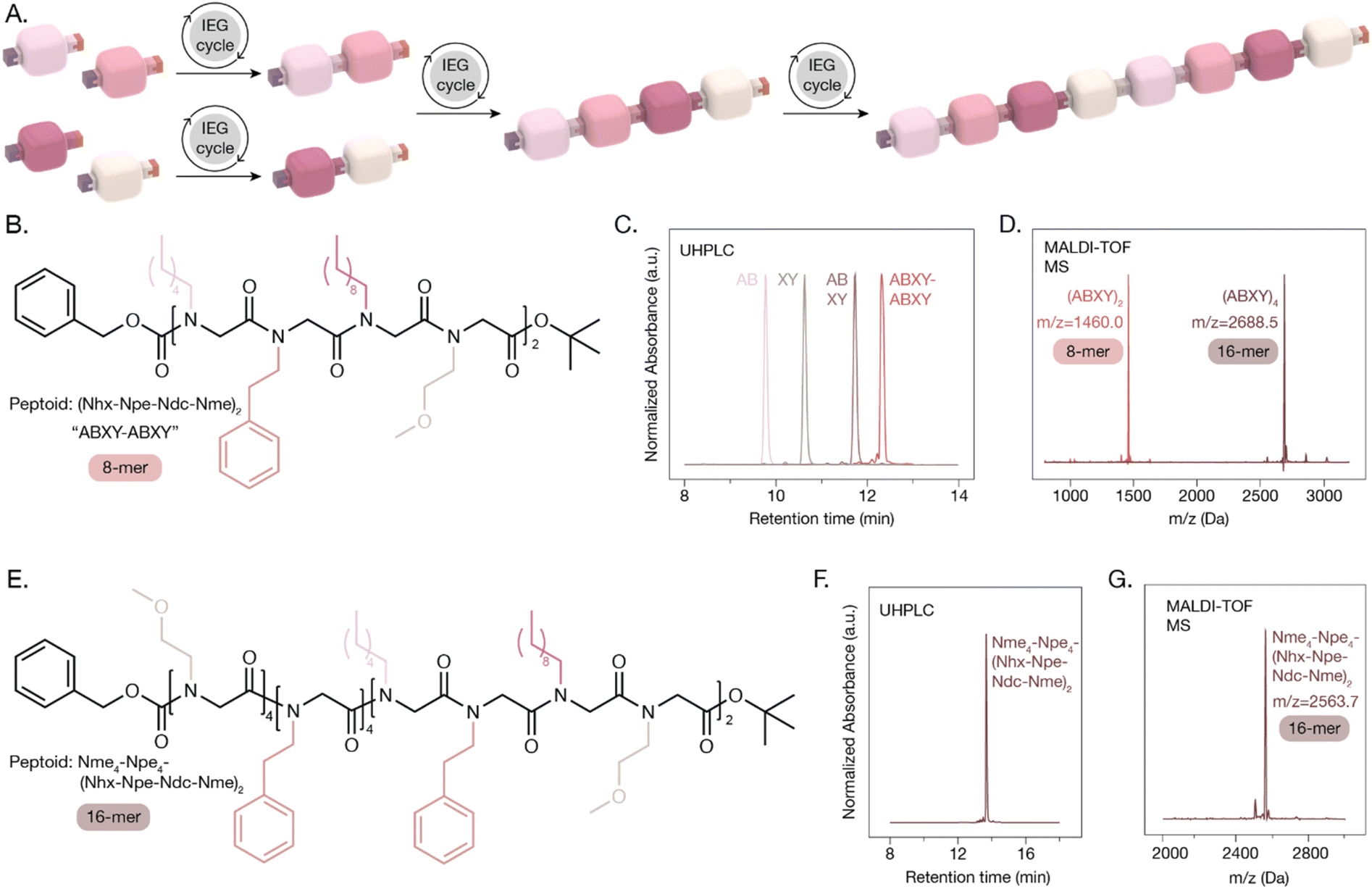 | ||
| Fig. 4 Sequence-defined periodic and aperiodic peptoids synthesized via IEG. (A) Illustration of the synthesis of an ABXY–ABXY octamer from four different monomers. (B) Chemical structure of the ABXY–ABXY peptoid with the (Nhx–Npe–Ndc–Nme)2 sequence. (C) UHPLC traces of the 2-mers, 4-mer, and 8-mer after the three IEG cycles. (d) MALDI-TOF MS traces of the 8-mer and 16-mer peptoids synthesized with (ABXY)2 and (ABXY)4 sequences. (E) Chemical structure of the aperiodic sequence-defined peptoid with Nme4–Npe4–(Nhx–Npe–Ndc–Nme)2 sequence. (F) UHPLC trace and (G) MALDI-TOF MS trace of the aperiodic sequence-defined 16-mer. | ||
Stereo-defined alternating peptoids
The peptoid backbone is inherently achiral but chiral monomers like (S)–N-(1-phenylethyl)glycine (Nspe) and (R)-N-(1-phenylethyl)glycine (Nrpe) can be incorporated using the IEG strategy (Fig. 5A and D). Nspe and Nrpe are more sterically hindered than the previous monomers explored, making the addition of the Cbz protecting group and the amide coupling steps low yielding. To address these challenges, Nspe and Nrpe were coupled to less sterically hindered Cbz-protected Nme monomers (ESI Schemes 39–40†). Furthermore, the amide coupling conditions were optimized by changing the solvent from DCM to N,N-dimethylformamide (DMF) and increasing the reaction temperature to 45 °C, increasing coupling yields from 4% to 74%. Increased temperatures and reaction times have also been used in solid-phase submonomer methods to improve reaction efficiencies for longer chain lengths and bulky side chains.27 | ||
| Fig. 5 Stereo-defined alternating peptoids synthesized via IEG. (A) Chemical structure of alternating 8-mer peptoid (Nme–Nrpe)4. (B) UHPLC traces of the 2-mer, 4-mer, and 8-mer and (C) MALDI-TOF MS traces for the 4-mer and 8-mer containing the Nrpe monomer. (D) Chemical structure of alternating 8-mer peptoid (Nme–Nspe)4. (E) UHPLC traces of the 2-mer, 4-mer, and 8-mer and (F) MALDI-TOF MS traces for the 4-mer and 8-mer containing the Nspe monomer. (G) Chemical structure of alternating 16-mer peptoid (Nme–Nrpe)4–(Nme–Nspe)4. (H) UHPLC trace and (I) MALDI-TOF MS trace for the stereo-defined 16-mer. (J) CD spectra for the series of stereo-defined peptoids measured at 60 μmol L−1 in acetonitrile at 25 °C. | ||
Alternating Nme–Nrpe and Nme–Nspe peptoids were synthesized with the optimized reaction conditions and studied using CD spectroscopy to characterize their backbone conformations in solution. Monodisperse 8-mers (Nme–Nrpe)4 and (Nme–Nspe)4 were successfully produced after three IEG cycles and the NMR, UHPLC, and MALDI-TOF MS spectra are identical for each set of enantiomers, as expected (Fig. 5A–F and ESI Schemes 41–46†). The uniform 16-mer peptoid (Nme–Nrpe)4–(Nme–Nspe)4 was synthesized after subjecting both alternating 8-mers to another IEG cycle (Fig. 5G–I and ESI Schemes 47–49†). Peptoids with sequences comprised of 50% or more of α-chiral, aromatic side chains (i.e., Nspe or Nrpe), with one placed at the C-terminus, have been previously shown to form highly robust chiral helical structures in organic solvents with a handedness that is determined by the chirality of the side-chains.13,57,58 CD spectroscopy is an effective method to characterize the structure of chiral peptoids since the amide bond absorbs in the 240–180 nm region and CD signals can be correlated to specific aspects of peptoid secondary structures. The 8-mers exhibit a strong and well-defined peak at 220 nm, corresponding to the n → π* transition of the amide bond and indicating the presence of a regular repeating backbone conformation, as has been observed previously for peptoids with similar sequences (Fig. 5J).58 This peak is reduced in magnitude and less defined for the dimers and tetramers, indicating that these chain lengths are not sufficient to give rise to a defined and regular repeating peptoid secondary structure and instead likely form protohelical structures. This is consistent with previous observations for Nrpe3 and Nrpe4.13 The terminal protecting groups used in this study prevent hydrogen bonding interactions between the N- and C-terminus, which have been previously shown to contribute to helical heterogeneity in peptoids.58–61 As expected, the 16-mer containing equal amounts of Nrpe and Nspe side chains exhibited no peaks associated with peptoid secondary structure between 200-220 nm.
Post-IEG modifications with thiol–ene and CuAAC click reactions
Post-functionalization of peptoids prepared with the IEG method allows for the incorporation of functional groups that are incompatible with the three reactions of an IEG cycle. For example, side chains containing primary or secondary amines, carboxylic acids, alcohols, and thiols could not be incorporated directly with this IEG protocol. Click reactions such as the thiol–ene and CuAAC reactions offer efficient and mild methods to introduce new functionalities without the need for additional orthogonal protecting groups. We added the alkene (Nall) and alkyne (Nalk) monomers to the peptoids through an amide coupling with the tert-butyl ester deprotected Npe4 (ESI Schemes 50–51, 53 and 54†). The irradiation of the Npe4-Nall peptoid at 370 nm with 1.2 equivalents of benzyl mercaptan gave the thioether functionalized peptoid, Npe4-Nthio, in 90% conversion by UHPLC using 2,2-dimethoxy-2-phenylacetophenone (DMPA) as the radical initiator (Fig. 6A–C, Supplementary Scheme 52). We subjected the Npe4–Nalk peptoid to modified CuAAC reaction conditions which used a CuSO4 catalyst, sodium ascorbate as a reducing agent, tert-butanol/H2O 2:1 (v/v) as the solvent system, and an elevated temperature (65 °C) (Fig. 6D and ESI Scheme 55†). Following reaction with 3-azidopropan-1-ol using these conditions, the triazole peptoid, Npe4–Ntriazo, was obtained in quantitative yield (Fig. 6E and F). While these two examples show modification to the end of the peptoid sequence, we also synthesized a 7-mer peptoid with an Npe4–Nalk–Npe2 sequence (ESI Scheme 56†). The CuAAC reaction with 3-azidopropan-1-ol was successful with the alkyne group in the middle of the peptoid sequence (Fig. 6G–I and ESI Scheme 57†), demonstrating that additional functional groups can be incorporated at various positions in the peptoid sequence.
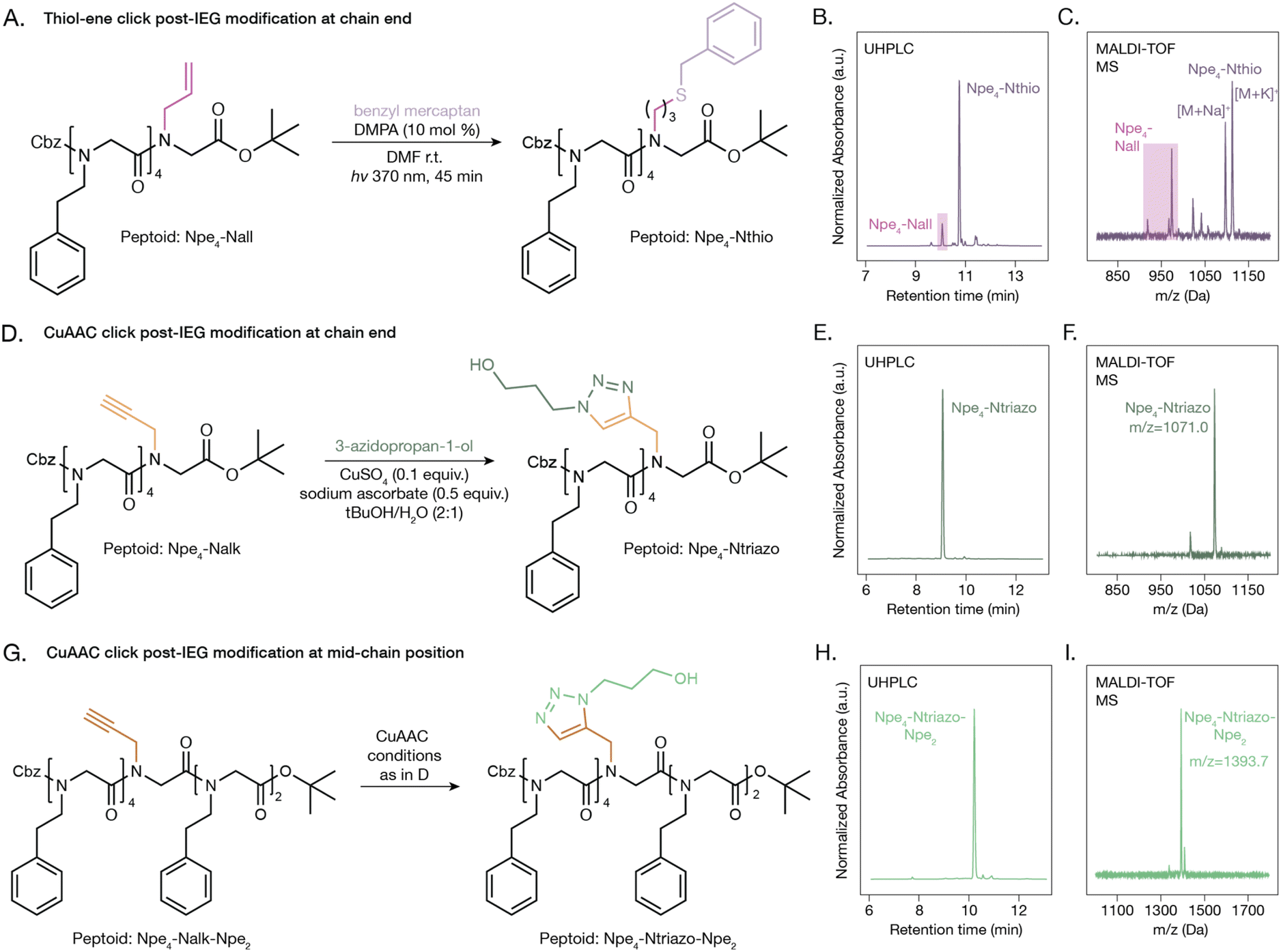 | ||
| Fig. 6 Post-IEG modifications of peptoids. (A) Synthesis of thioether functionalized peptoid from photo-initiated thiol–ene click reaction. (B) UHPLC and (C) MALDI-TOF MS traces of the resulting thioether peptoid, Npe4–Nthio, with 90% conversion. (D) Synthesis of the triazole functionalized peptoid from copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction. (E) UHPLC and (F) MALDI-TOF MS traces of the resulting triazole peptoid, Npe4–Ntriazo. (G) Synthesis of the triazole functionalized peptoid from copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction at the mid-chain position. (H) UHPLC and (I) MALDI-TOF MS traces of the resulting triazole peptoid, Npe4–Ntriazo–Npe2. | ||
Conclusions
We have demonstrated an IEG synthetic method for the preparation of peptoids that enables precise control over their sequence, chain length, and stereoconfiguration. The developed IEG approach is compatible with a variety of commonly explored side chains and can access peptoids with increased molecular weights compared to other sequence-defined solution-phase methods (i.e., up to a 32-mer is demonstrated). Peptoids can be synthesized on a multi-gram scale with this IEG method, going beyond what can typically be achieved with solid-phase syntheses (i.e., accessing an octamer on an 8-gram scale). Furthermore, peptoids with unique sequence arrangements (i.e., block, alternating, aperiodic) were generated using this orthogonal protecting group strategy. The self-assembly of peptoids synthesized via IEG was not disrupted by the presence of the protecting groups, which is promising for the deliberate design of future peptoid materials. While we note there are limitations with the compatibility of certain side chains with this technique, post-modification of the peptoids with thiol–ene and CuAAC click reactions enabled the incorporation of additional functionalities. Post-functionalization at precise locations along the peptoid backbone has important implications for applications in biosensing and targeted delivery. There is potential to further optimize this IEG approach to enhance the translation of peptoids from academic labs to industrial synthesis. This can be done through the elimination of intermediate isolation and purification by leveraging continuous flow chemistry and automated workflows. The reported IEG method generates sequence- and stereo-defined peptoids without the need for excess reagents and with easy reaction monitoring, which will facilitate the intentional design and synthesis of peptoids for functional materials.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
A. M. C. and H. T. conceptualized the work. A. M. C. and H. T. developed the molecular designs and synthetic protocols. A. M. C. carried out the syntheses, characterizations, and self-assemblies. C. H. carried out self-assemblies and collected TEM images of the peptoid nanosheets. A. M. C. and H. T. analyzed all results and A. M. C. wrote the manuscript. All authors reviewed and edited the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council (NSERC) of Canada (H.T., RGPIN2021-03554; A.M.C., CGS-D and CGS-M scholarship) and the University of Toronto's Acceleration Consortium from the Canada First Research Excellence Fund (H.T., CDG13-2023). The authors also thank Yuran Shi (DMF GPC) and Ernest Tse (illustration assistance). GPC characterization was performed at the Stanford Nano Shared Facilities (SNSF), supported by the National Science Foundation under Award ECCS-1542152.References
- C. Yang, K. B. Wu, Y. Deng, J. Yuan and J. Niu, ACS Macro Lett., 2021, 10, 243–257 CrossRef CAS PubMed.
- J. Babi, L. Zhu, A. Lin, A. Uva, H. El-Haddad, A. Peloewetse and H. Tran, J. Polym. Sci., 2021, 59, 2378–2404 CrossRef CAS.
- A. J. DeStefano, R. A. Segalman and E. C. Davidson, JACS Au, 2021, 1, 1556–1571 CrossRef CAS.
- R. Aksakal, C. Mertens, M. Soete, N. Badi and F. Du Prez, Advanced Science, 2021, 8, 2004038 CrossRef CAS PubMed.
- R. N. Zuckermann, J. M. Kerr, S. B. H. Kent and W. H. Moos, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef CAS.
- S. M. Miller, R. J. Simon, S. Ng, R. N. Zuckermann, J. M. Kerr and W. H. Moos, Bioorg. Med. Chem. Lett., 1994, 4, 2657–2662 CrossRef CAS.
- B. Sanii, R. Kudirka, A. Cho, N. Venkateswaran, G. K. Olivier, A. M. Olson, H. Tran, R. M. Harada, L. Tan and R. N. Zuckermann, J. Am. Chem. Soc., 2011, 133, 20808–20815 CrossRef.
- J. M. Astle, D. G. Udugamasooriya, J. E. Smallshaw and T. Kodadek, Int. J. Pept. Res. Ther., 2008, 14, 223–227 CrossRef.
- J. Schwochert, R. Turner, M. Thang, R. F. Berkeley, A. R. Ponkey, K. M. Rodriguez, S. S. F. Leung, B. Khunte, G. Goetz, C. Limberakis, A. S. Kalgutkar, H. Eng, M. J. Shapiro, A. M. Mathiowetz, D. A. Price, S. Liras, M. P. Jacobson and R. S. Lokey, Org. Lett., 2015, 17, 2928–2931 CrossRef PubMed.
- X. Luo, T. Yu, N. K. Li, R. N. Zuckermann, X. Jiang, N. P. Balsara and D. Prendergast, ACS Nano, 2024, 18, 14917–14924 CrossRef.
- A. L. Patterson, S. P. O. Danielsen, B. Yu, E. C. Davidson, G. H. Fredrickson and R. A. Segalman, Macromolecules, 2019, 52, 1277–1286 CrossRef.
- X. Jiang, M. Seidler, G. L. Butterfoss, X. Luo, T. Yu, S. Xuan, D. Prendergast, R. N. Zuckermann and N. P. Balsara, ACS Macro Lett., 2023, 12, 632–638 CrossRef PubMed.
- C. W. Wu, T. J. Sanborn, R. N. Zuckermann and A. E. Barron, J. Am. Chem. Soc., 2001, 123, 2958–2963 CrossRef PubMed.
- Y. Luo, Y. Song, M. Wang, T. Jian, S. Ding, P. Mu, Z. Liao, Q. Shi, X. Cai, H. Jin, D. Du, W. Dong, C. Chen and Y. Lin, Small, 2019, 15(43), 1902485 CrossRef PubMed.
- R. Kudirka, H. Tran, B. Sanii, K. T. Nam, P. H. Choi, N. Venkateswaran, R. Chen, S. Whitelam and R. N. Zuckermann, Pept. Sci., 2011, 96, 586–595 CrossRef PubMed.
- K. T. Nam, S. A. Shelby, P. H. Choi, A. B. Marciel, R. Chen, L. Tan, T. K. Chu, R. A. Mesch, B.-C. Lee, M. D. Connolly, C. Kisielowski and R. N. Zuckermann, Nat. Mater., 2010, 9, 454–460 CrossRef.
- H. Jin, F. Jiao, M. D. Daily, Y. Chen, F. Yan, Y.-H. Ding, X. Zhang, E. J. Robertson, M. D. Baer and C.-L. Chen, Nat. Commun., 2016, 7, 12252 CrossRef.
- B. Yu, B. S. Chang, W. S. Loo, S. Dhuey, P. O'Reilly, P. D. Ashby, M. D. Connolly, G. Tikhomirov, R. N. Zuckermann and R. Ruiz, ACS Nano, 2024, 18, 7411–7423 CrossRef PubMed.
- S. T. Wang, M. A. Gray, S. Xuan, Y. Lin, J. Byrnes, A. I. Nguyen, N. Todorova, M. M. Stevens, C. R. Bertozzi, R. N. Zuckermann and O. Gang, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 6339–6348 CrossRef CAS.
- M. Wang, Y. Song, P. Mu, X. Cai, Y. Lin and C.-L. Chen, ACS Appl. Bio Mater., 2020, 3, 6039–6048 CrossRef CAS.
- A. Battigelli, J. H. Kim, D. C. Dehigaspitiya, C. Proulx, E. J. Robertson, D. J. Murray, B. Rad, K. Kirshenbaum and R. N. Zuckermann, ACS Nano, 2018, 12, 2455–2465 CrossRef CAS PubMed.
- E. R. Webster, N. E. Peck, J. D. Echeverri, S. Gholizadeh, W.-L. Tang, R. Woo, A. Sharma, W. Liu, C. S. Rae, A. Sallets, G. Adusumilli, K. Gunasekaran, O. A. W. Haabeth, M. Leong, R. N. Zuckermann, S. Deutsch and C. J. McKinlay, ACS Nano, 2024, 18, 22181–22193 CrossRef CAS PubMed.
- E. J. Robertson, C. Proulx, J. K. Su, R. L. Garcia, S. Yoo, E. M. Nehls, M. D. Connolly, L. Taravati and R. N. Zuckermann, Langmuir, 2016, 32, 11946–11957 CrossRef CAS.
- A. M. Clapperton, J. Babi and H. Tran, ACS Polym. Au, 2022, 2, 417–429 CrossRef CAS PubMed.
- R. N. Zuckermann, J. M. Kerr, S. B. H. H. Kent, W. H. Moosf and S. B. H. H. Kent, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef CAS.
- B.-C. Lee, R. N. Zuckermann and K. A. Dill, J. Am. Chem. Soc., 2005, 127, 10999–11009 CrossRef CAS PubMed.
- M. D. Connolly, S. Xuan, N. Molchanova and R. N. Zuckermann, Methods Enzymol., 2021, 656, 241–270 CAS.
- B. A. Chan, S. Xuan, A. Li, J. M. Simpson, G. L. Sternhagen, T. Yu, O. A. Darvish, N. Jiang and D. Zhang, Biopolymers, 2018, 109(1), e23070 CrossRef.
- P. Zhou, T. Shen and J. Ling, J. Polym. Sci., 2021, 59, 2946–2958 CrossRef CAS.
- M. Liao, Y. Yao, K. Gan, X. Su, N. Zhao, R. N. Zuckermann, S. Xuan and Z. Zhang, Angew. Chem., 2025, 64(6), e202417990 CrossRef CAS PubMed.
- S. Wang, M.-Y. Lu, S.-K. Wan, C.-Y. Lyu, Z.-Y. Tian, K. Liu and H. Lu, J. Am. Chem. Soc., 2024, 146, 5678–5692 CrossRef CAS PubMed.
- C. Caumes, T. Hjelmgaard, R. Remuson, S. Faure and C. Taillefumier, Synthesis, 2011, 2011, 257–264 CrossRef.
- N. Soumanou, D. Lybye, T. Hjelmgaard and S. Faure, Green Chem., 2023, 25, 3615–3623 RSC.
- S. Wang, Y. Tao, J. Wang, Y. Tao and X. Wang, Chem. Sci., 2019, 10, 1531–1538 RSC.
- Y. Tao and Y. Tao, Macromol. Rapid Commun., 2021, 42, 2000515 CrossRef CAS PubMed.
- F. A. Leibfarth, J. A. Johnson and T. F. Jamison, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10617–10622 CrossRef.
- Z. Li, X. Ren, P. Sun, H. Ding, S. Li, Y. Zhao and K. Zhang, ACS Macro Lett., 2021, 10, 223–230 CrossRef.
- K. Takizawa, C. Tang and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 1718–1726 CrossRef PubMed.
- Z. Huang, J. Zhao, Z. Wang, F. Meng, K. Ding, X. Pan, N. Zhou, X. Li, Z. Zhang and X. Zhu, Angew. Chem., Int. Ed., 2017, 56, 13612–13617 CrossRef.
- M. B. Koo, S. W. Lee, J. M. Lee and K. T. Kim, J. Am. Chem. Soc., 2020, 142, 14028–14032 CrossRef.
- O. I. Paynter, D. J. Simmonds and M. C. Whiting, J. Chem. Soc. Chem. Commun., 1982, 1165–1166 RSC.
- J. C. Barnes, D. J. C. Ehrlich, A. X. Gao, F. A. Leibfarth, Y. Jiang, E. Zhou, T. F. Jamison and J. A. Johnson, Nat. Chem., 2015, 7, 810–815 CrossRef PubMed.
- W. He, S. Wang, M. Li, X. Wang and Y. Tao, Angew. Chem., Int. Ed., 2022, 61, e202112439 CrossRef.
- S. Binauld, D. Damiron, L. A. Connal, C. J. Hawker and E. Drockenmuller, Macromol. Rapid Commun., 2011, 32, 147–168 CrossRef.
- A. Isidro-Llobet, M. Álvarez and F. Albericio, Chem. Rev., 2009, 109, 2455–2504 CrossRef.
- A. S. Culf and R. J. Ouellette, Molecules, 2010, 15, 5282–5335 CrossRef PubMed.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC.
- S. B. Y. Shin, B. Yoo, L. J. Todaro and K. Kirshenbaum, J. Am. Chem. Soc., 2007, 129, 3218–3225 CrossRef PubMed.
- J. R. B. Eastwood, L. Jiang, R. Bonneau, K. Kirshenbaum and P. D. Renfrew, J. Phys. Chem. B, 2022, 126, 5161–5174 CrossRef PubMed.
- P. K. Mandal and J. S. McMurray, J. Org. Chem., 2007, 72, 6599–6601 CrossRef PubMed.
- D. X. Hu, P. Grice and S. V. Ley, J. Org. Chem., 2012, 77, 5198–5202 CrossRef.
- S. Xuan and R. N. Zuckermann, J. Mater. Chem. B, 2020, 8, 5380–5394 RSC.
- J. Sun, X. Jiang, R. Lund, K. H. Downing, N. P. Balsara and R. N. Zuckermann, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3954–3959 CrossRef.
- X. Jiang, S. Xuan, J. Kundu, D. Prendergast, R. N. Zuckermann and N. P. Balsara, Soft Matter, 2019, 15, 4723–4736 RSC.
- D. R. Greer, M. A. Stolberg, J. Kundu, R. K. Spencer, T. Pascal, D. Prendergast, N. P. Balsara and R. N. Zuckermann, J. Am. Chem. Soc., 2018, 140, 827–833 CrossRef PubMed.
- T. Yu, X. Luo, D. Prendergast, G. L. Butterfoss, B. Rad, N. P. Balsara, R. N. Zuckermann and X. Jiang, ACS Nano, 2023, 17, 4958–4970 CrossRef.
- K. Kirshenbaum, A. E. Barron, R. A. Goldsmith, P. Armand, E. K. Bradley, K. T. V. Truong, K. A. Dill, F. E. Cohen and R. N. Zuckermann, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4303–4308 CrossRef.
- B. Yu, S. P. O. Danielsen, K.-C. Yang, R.-M. Ho, L. M. Walker and R. A. Segalman, ACS Macro Lett., 2020, 9, 849–854 CrossRef PubMed.
- J. R. Stringer, J. A. Crapster, I. A. Guzei and H. E. Blackwell, J. Am. Chem. Soc., 2011, 133, 15559–15567 CrossRef.
- S. Alamdari and J. Pfaendtner, J. Phys. Chem. B, 2023, 127, 6163–6170 CrossRef.
- P. Armand, K. Kirshenbaum, R. A. Goldsmith, S. Farr-Jones, A. E. Barron, K. T. V. Truong, K. A. Dill, D. F. Mierke, F. E. Cohen, R. N. Zuckermann and E. K. Bradley, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4309–4314 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, materials and methods, NMR, GPC, UHPLC, and MS data. See DOI: https://doi.org/10.1039/d5sc01296a |
| This journal is © The Royal Society of Chemistry 2025 |