
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual-state emission of pyrazolyl-pyrrolo[3,4-b]pyridin-5-ones via excited-state intramolecular proton transfer (ESIPT): multicomponent synthesis and optical characterization†
Julio C.
Flores-Reyes‡
a,
Yoarhy A.
Amador-Sánchez‡
 a,
Alejandro
Valderrama-Celestino
a,
Bertha D.
Barrios-Campos
a,
Ricardo A.
Peralta
a,
Michael T.
Huxley
b,
Ilich A.
Ibarra
c,
Alejandro
Islas-Jácome
a,
Diego
Solis-Ibarra
*c and
Eduardo
González-Zamora
*a
a,
Alejandro
Valderrama-Celestino
a,
Bertha D.
Barrios-Campos
a,
Ricardo A.
Peralta
a,
Michael T.
Huxley
b,
Ilich A.
Ibarra
c,
Alejandro
Islas-Jácome
a,
Diego
Solis-Ibarra
*c and
Eduardo
González-Zamora
*a
aDepartamento de Química, Universidad Autónoma Metropolitana-Iztapalapa, Av. Ferrocarril San Rafael Atlixco 186, Col. Leyes de Reforma 1A Sección, Iztapalapa, 09310, Ciudad de México, Mexico. E-mail: egz@xanum.uam.mx
bSchool of Physics, Chemistry and Earth Sciences, Faculty of Sciences, Engineering and Technology, The University of Adelaide, Adelaide, SA 5005, Australia
cLaboratorio de Fisicoquímica y Reactividad de Superficies, (LaFReS), Instituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México, Circuito Exterior S/N, CU, Coyoacán, Ciudad de México, Mexico. E-mail: diego.solis@unam.mx
First published on 17th February 2025
Abstract
The dual-state emission (DSE) phenomenon has become crucial for developing dual-state emission luminogens (DSEgens) that exhibit efficient luminescence in both solution and solid states, addressing the limitations of conventional, phase-restricted fluorophores. Compounds exhibiting excited-state intramolecular proton transfer (ESIPT) mechanisms are especially valuable for enhancing emission stability across these states, offering significant potential in optoelectronics, bioimaging, and sensing applications. In this study, we report the synthesis of six new pyrazolyl-pyrrolo[3,4-b]pyridin-5-ones through an Ugi-Zhu-3CR coupled to an aza-Diels–Alder/N-acylation/decarboxylation/dehydration cascade sequence. An X-ray ORTEP confirms unequivocally the structure of one of the synthesized compounds. These ones demonstrate intriguing photophysical properties such as large Stokes shifts (>11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 cm−1) in solution and robust solid-state emission via ESIPT mechanism. Complementary DFT and TD-DFT calculations confirm weak but allowed transitions involving both pyrazole and pyrrolo[3,4-b]pyridin-5-one moieties, in agreement with experimental observations. This work represents the first application of an isocyanide-based multicomponent reaction for DSEgen synthesis, paving the way for innovative advances in the design of organic luminescent materials.
900 cm−1) in solution and robust solid-state emission via ESIPT mechanism. Complementary DFT and TD-DFT calculations confirm weak but allowed transitions involving both pyrazole and pyrrolo[3,4-b]pyridin-5-one moieties, in agreement with experimental observations. This work represents the first application of an isocyanide-based multicomponent reaction for DSEgen synthesis, paving the way for innovative advances in the design of organic luminescent materials.
Introduction
The DSE phenomenon has garnered significant attention within the scientific community due to its potential to enable DSEgens that emit effectively in both solution and solid states.1 Such materials bridge a critical gap left by traditional fluorophores, which are often restricted to single-phase emission. Achieving efficient DSE requires strategic incorporation of functional groups to sustain emission across varying states. Notably, the integration of triphenylamine (TPA)2 and tetraphenylethylene (TPE)3 units has proven effective for their strong emission properties and ability to minimize aggregation-caused quenching (ACQ)4 by reducing detrimental π–π stacking-type interactions in conjugated aromatic systems.5 Further, modifications with long-chain C–C organic species create “isolated” solid-state environments, enhancing emission with improved quantum yields. Incorporating aliphatic chains also enhances compound solubility, facilitating solution-processability for device applications and supporting robust DSE behavior.6 Compounds with ESIPT capabilities are also under active investigation, as this mechanism improves emission stability and intensity in both solution and solid phases. These advancements in DSE design show substantial promise for optoelectronics, bioimaging, and sensing technologies, where stable and versatile emission characteristics are essential.7Alongside, pyrazole-based heteroaromatic compounds have gained significant interest due to their presence in various biologically and industrially important molecules, including natural products,8 antimicrobial agents,9 and emissive chromophores.10 These aromatic systems serve as essential building blocks for the rapid development of optoelectronic devices, such as organic light-emitting diodes (OLEDs).11 To this end, synthetic strategies that provide high yields and excellent atom economy are highly desirable, enabling the efficient generation of diverse molecular architectures. Among these approaches, multicomponent reactions (MCRs) stand out for their ability to produce key emissive motifs with valuable biological12 and optical properties13 while circumventing the limitations of multi-step procedures, such as low yields and labor-intensive purification steps.14 Moreover, pyrazole-based compounds are particularly appealing for designing luminescent materials due to their neighboring nitrogen atoms, which can act as hydrogen bond acceptors, facilitating highly desirable emissive phenomena like the ESIPT effect.15 This process begins with the photoexcitation of the material, resulting in the formation of two tautomers (enol and keto forms) with distinct electronic structures. In the ground state these molecules adopt an enol (E) configuration since they are stabilized by an intramolecular hydrogen bond. Upon light absorption, the enol tautomer enters an excited state (E*), during which the electronic charge is redistributed resulting in an increased acidity of the H-donor and an increased basicity of the H-acceptor, causing a proton transfer to occur, generating a new excited keto tautomer (K*). As the system relaxes to its ground state, a recombination mechanism takes place, resulting in light emission (K) distinct than from absorbed in the first step of the ESIPT process. The K form tautomerizes back to the E form by a reverse proton transfer, completing a four-level photochemical cycle (Fig. 1).
 | ||
| Fig. 1 Representation of the ESIPT mechanism in emissive compounds. | ||
Our research group has previously developed novel methodologies based on the Ugi-Zhu reaction (UZ-3CR), utilizing various amines, aldehydes, and key α-isocyanoacetamides to assemble 5-aminooxazoles.16 These latter are subsequently subjected to a cascade sequence (intermolecular aza Diels–Alder cycloaddition/N-acylation/decarboxylation/dehydration) with maleic anhydride, yielding a series of pyrrolo[3,4-b]pyridin-5-ones with promising biological17 and optical18 properties (Scheme 1).
 | ||
| Scheme 1 Ugi-Zhu/aza Diels–Alder sequence for the multicomponent assembly of pyrrolo[3,4-b]pyridin-5-ones. | ||
As part of our ongoing efforts to synthesize novel and complex polyheterocyclic compounds, we have developed several methodologies that utilize the Ugi-Zhu three-component reaction as a key synthetic tool. This strategy has proven effective for preparing various pyrrolo[3,4-b]pyridin-5-one series, either fused, bound, or linked to heterocyclic frameworks of interest for their optical and medicinal properties. In this study, we successfully synthesized a series of pyrazole-derived pyrrolo[3,4-b]pyridin-5-one dual-state emitters through an Ugi-Zhu/cascade (aza Diels–Alder/N-acylation/decarboxylation/dehydration) sequence with emission in both solution and solid states. To the best of our knowledge, this is the first report in which an isocyanide-based multicomponent reaction has been employed for the creation of DSEgens. Additionally, optical characterization revealed intriguing fluorescence properties, underscoring the potential applications of these compounds in the field of photonics and as bioactive agents.
Results and discussion
Organic synthesis
To access the target pyrazolyl-pyrrolo[3,4-b]pyridin-5-one scaffold, we synthesized the corresponding O-protected pyrazole-derived aldehyde, which is crucial for the Ugi-Zhu reaction. First, a condensation was performed between phenylhydrazine and 2′-hydroxyacetophenone, followed by the incorporation of the aldehyde moiety using the classic Vilsmeier–Haack protocol, yielding the desired pyrazole 1′ with an isolated yield of 38% in a two-step process. After this, the hydroxy group was protected by introducing the methoxymethyl (MOM) protecting group by reacting the aldehyde with methoxymethyl chloride in the presence of sodium hydride. The MOM-protected aldehyde 1 was obtained in 64% yield (see ESI S1 and S2†).19 The protection of the hydroxyl group is essential to obtaining the desired compounds, as the reaction does not proceed when using the unprotected aldehyde. We attributed this to an increased resonance stabilization upon the loss of the acidic proton, rendering the aldehyde unreactive toward nucleophilic attack by the amine. With compound 1 in hand, the α-isocyanoacetamide 3 was synthesized following a slightly modified protocol established by Zhu and Bienaymé (see ESI S3†).16 It is noteworthy that the previously obtained synthetic modules are pivotal for the expedited access to the desired pyrazolyl-pyrrolo[3,4-b]pyridin-5-one scaffolds using the Ugi-Zhu/cascade (aza Diels–Alder/N-acylation/decarboxylation/dehydration) sequence. As shown in Scheme 2, the multicomponent methodology proceeded efficiently by incorporating the corresponding O-protected pyrazole-carbaldehyde 1, the α-isocyanoacetamide 3, various aliphatic amines (2a–f), and maleic anhydride (4) in dry toluene. The reaction was catalyzed by 10 mol% of Sc(OTf)3 under microwave heating (60 °C, 100 W) for 1.5 hours. After purification and isolation, the desired MOM-protected heterocycles 5a–f were obtained in 22% to 45% yields. At this stage, the MOM deprotection was necessary to obtain the desired ESIPT scaffolds. The MOM deprotection protocol was conducted under mild conditions (0.4 M HCl in THF), yielding the expected 6a–f products with yields ranging from 42% to 79% (see Scheme 2). Considering factors such as atom economy (up to 84.82%), structural complexity, and the number of steps composing the overall one-pot process, the isolated yields are deemed satisfactory. It is worth noting that all compounds were characterized by 1H, 13C, and 19F NMR, as well as high-resolution mass spectrometry (HRMS). Fortunately, high-quality crystals of compound 6e were successfully obtained for X-ray diffraction analysis, which unequivocally confirmed its polycyclic structure (see the ESI Table S1† for complete X-ray analysis). | ||
| Scheme 2 Pyrazolyl-pyrrolo[3,4-b]pyridin-5-ones and their respective deprotections synthetized by an Ugi-Zhu/cascade (aza Diels–Alder/N-acylation/decarboxylation/dehydration) sequence. (a) Thermal ellipsoids are drawn at 50% probability, with all hydrogen atoms omitted for clarity, except for the hydrogen atom of the pyrazole moiety (key for the ESIPT photochemical cycle). | ||
The process begins with a condensation between aldehyde 1 and the primary amines 2a–f, resulting in the formation of imines 7, which are activated by Sc[III]. This activation promotes α-addition by the isocyanide 3, leading to the formation of nitrilium ion intermediates 8. These crucial intermediates are then intramolecularly captured by the amide oxygen through a 5-endo-dig cyclization, forming 5-aminooxazoles 10.16 The 5-aminooxazoles 10 react with maleic anhydride (4) in a cascade sequence of reactions, beginning with an intermolecular aza Diels–Alder cycloaddition producing the oxa-bridged intermediates 11. These ones undergo an N-acylation followed by a decarboxylation, and a final dehydration process, ultimately yielding the target pyrazolyl-pyrrolo[3,4-b]pyridin-5-ones 5 (Scheme S5†).
Photophysical properties
To determine the optical properties of compounds 6a–f, we first measured the absorption spectra of the deprotected pyrazole aldehyde 1 at a concentration of 10−5 M. The molar absorptivity coefficients were found to be 1256 M−1 cm−1 in chloroform (CHCl3), 11637 M−1 cm−1 in acetonitrile (MeCN), and 13342 M−1 cm−1 in dimethyl sulfoxide (DMSO). Based on these results, we recorded the full absorption and emission spectra in these three solvents. The deprotected compound showed absorption maxima at 254 nm, 275 nm, and 288 nm in CHCl3, MeCN, and DMSO, respectively, indicating a bathochromic shift with increasing solvent polarity. The emission maxima of deprotected 1, corresponding to its keto form, were observed at 350 nm, 348 nm, and 354 nm, with Stokes shifts of 10799 cm−1, 7628 cm−1, and 6474 cm−1 in CHCl3, MeCN, and DMSO, respectively. Conversely, an additional emission maximum at 308 nm was observed, corresponding to enol emission in both MeCN and DMSO. Fig. 2 presents the normalized absorption and emission spectra in these solvents, while Table 1 summarizes the optical characterization data.
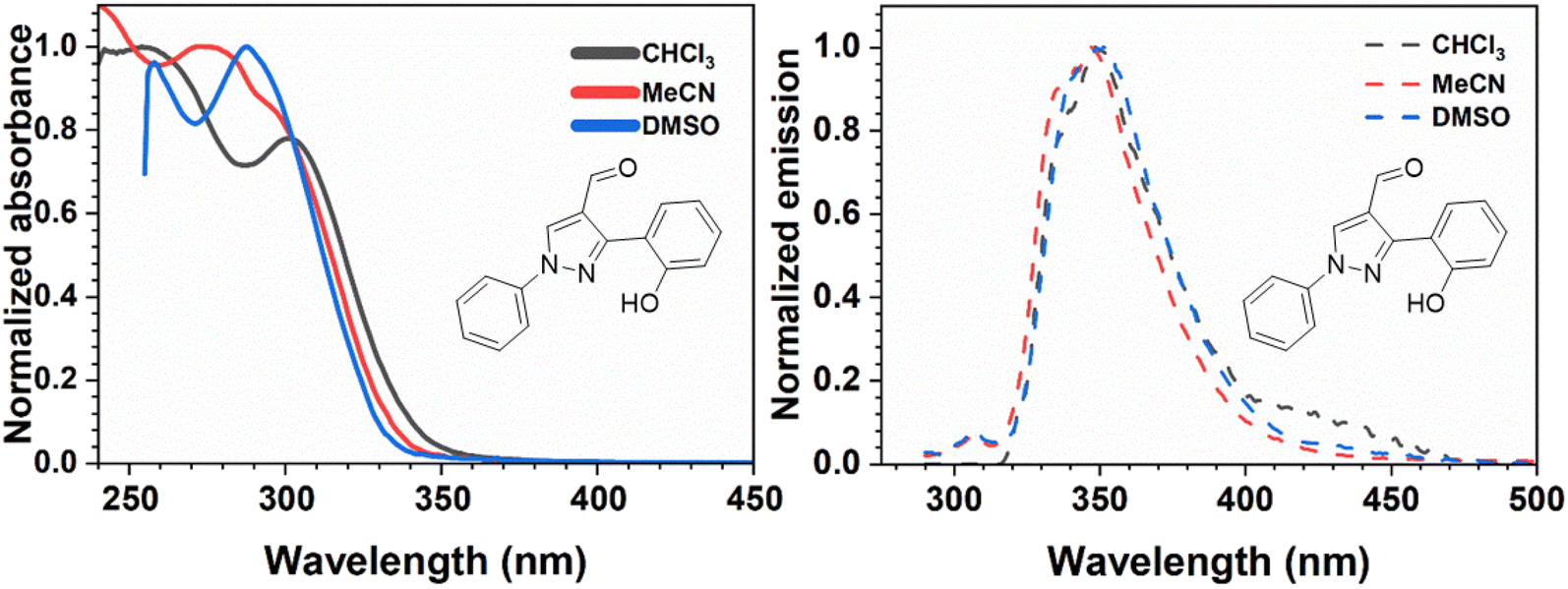 | ||
| Fig. 2 Absorption (left) and emission (right) spectra of the pyrazole aldehyde 1, measured at 254 nm, 275 nm, and 288 nm in CHCl3, MeCN, and DMSO, respectively. | ||
| Solvent | Absorption | Emission | Stokes shifts |
|---|---|---|---|
| λ max [nm] (ε) [M−1 cm−1] | λ max [nm] |
|
|
| CHCl3 | 254 (12568) |
350 | 10799 |
| MeCN | 275 (11637) |
348 | 7628 |
| DMSO | 288 (13342) |
354 | 6313 |

With these data, we proceeded to measure the normalized absorption and emission spectra of compounds 6a–f in CHCl3 at a concentration of 10−5 M (Fig. 3A and B) to compare their optical properties with those of the starting material 1. A prominent absorption peak at 278 nm, corresponding to the pyrrolo[3,4-b]pyridin-5-one core, was observed, along with a secondary, weaker absorption band between 297 and 303 nm, likely due to the pyrazole moiety. The polarity of the solvents had a minor effect on the absorption band positions. In MeCN and DMSO, the bands appeared at nearly identical wavelengths, while CHCl3 exhibited a slight bathochromic shift of 2 to 6 nm relative to MeCN and 4 to 5 nm compared to DMSO (Table 2). The emission spectra of compounds 6a–f in CHCl3 are shown in Fig. 3B.
 | ||
| Fig. 3 (A) Absorption, (B) emission espectra of pyrrolo[3,4-b]pyridin-5-ones 6a–f at 10−5 M in CHCl3, (C) solid-state emission of compounds 6a–fλex = 343 nm, (D) naked-eye-visible emission of compounds 6a–f under UV illumination λex = 365 nm, observed in the solid-state (top) and in solution (bottom). | ||
| Compound | Solvent | Absorbance | Emission | Stokes Shiftsa | Φ(%) |
|---|---|---|---|---|---|
| λ max [nm] (ε) [M−1 cm−1] | λ max [nm] |
|
|||
a The Stokes shifts were calculated as 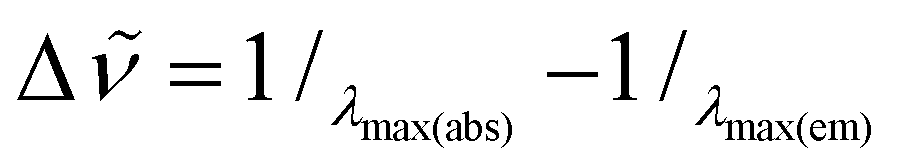 . .
|
|||||
| 6a | MeCN | 275 (24525) |
444 | 13841.1 |
— |
| CHCl3 | 279 (23827) |
419 | 11976.0 |
1.5 | |
| DMSO | 274 (23712) |
447 | 14125.0 |
— | |
| Solid | — | 420 | — | 21.5 | |
| 6b | MeCN | 275 (25943) |
441 | 13687.9 |
— |
| CHCl3 | 278 (24153) |
422 | 12274.5 |
2.4 | |
| DMSO | 274 (24034) |
445 | 14024.4 |
— | |
| Solid | — | 407 | — | 14.2 | |
| 6c | MeCN | 275 (23032) |
441 | 13687.9 |
— |
| CHCl3 | 278 (22497) |
422 | 12274.5 |
2.4 | |
| DMSO | 273 (24823) |
444 | 14107.5 |
— | |
| Solid | — | 407 | — | 13.2 | |
| 6d | MeCN | 278 (20892) |
442 | 13346.8 |
— |
| CHCl3 | 278 (22494) |
423 | 12330.6 |
1.5 | |
| DMSO | 278 (33064) |
448 | 13649.8 |
— | |
| Solid | — | 437, 532 | — | 7.1 | |
| 6e | MeCN | 272 (22453) |
441 | 14089.0 |
— |
| CHCl3 | 278 (23072) |
424 | 12386.3 |
1.4 | |
| DMSO | 278 (23537) |
447 | 13599.9 |
— | |
| Solid | — | 417 | — | 17.9 | |
| 6f | MeCN | 276 (21309) |
450 | 14009.7 |
— |
| CHCl3 | 278 (18859) |
435 | 12982.7 |
1.6 | |
| DMSO | 275 (17533) |
451 | 14190.7 |
— | |
| Solid | — | 435, 572 | — | 11.1 | |
As shown in Table 2, excitation of compounds 6a–f at 265 nm revealed that 6b and 6c exhibited the highest fluorescence intensity with emission maxima at 422 nm, while compound 6a displayed the lowest intensity at 419 nm. Notably, compound 6f demonstrated a bathochromic shift of approximately 11 nm. In general, these compounds exhibited bathochromic shifts with increasing solvent polarity. Compared to their emission in CHCl3, the respective emissions of compounds 6a–f in MeCN and DMSO occurred at significantly longer wavelengths, suggesting a higher dipole moment in the excited state.20 When a fluorophore is in the excited state, solvent molecules can relax around the excited dipole by reorienting themselves via electrostatic solute-solvent interactions. Higher solvent polarity and an increased excited state dipole moment of the fluorophore will enhance these interactions. This stabilizes and lowers the energy of the excited fluorophore, resulting in a red-shifted emission spectrum. Various excitation wavelengths were tested to acquire these spectra; however, the emission profiles consistently revealed a single emission band, indicating the predominance of one tautomer across the compounds. It is important to note that the Stokes shifts for compounds 6a–f exceeded 11900 cm−1 in all three solvents, particularly in MeCN and DMSO. This observation suggests the potential for an ESIPT, as compounds exhibiting ESIPT typically show large Stokes shifts due to structural differences between the emissive enol and keto forms.21 However, the presence of a single emission band for both the aldehyde and the products, combined with the large Stokes shifts observed in polar solvents, makes ESIPT unlikely in solution. Proton transfer is expected to be inhibited in such environments, particularly in DMSO. Instead, the observed Stokes shifts are more likely related to the integration of the fluorophore within the pyrrolo[3,4-b]pyridin-5-one framework, a phenomenon previously reported in similar systems.18
Additionally, the photoluminescence quantum yields (PLQYs) of compounds 6a–f in chloroform were measured as 1.5%, 2.4%, 2.4%, 1.5%, 1.4%, and 1.6%, respectively. It is noteworthy that all PLQYs were relatively low (<2.5%), indicating a strong preference for non-radiative relaxation pathways (Table 2).22 In the solid state, however, ESIPT can be modulated by molecular aggregation and restricted vibrational motion, potentially enhancing emission by minimizing non-radiative losses.23 This phenomenon is exemplified by aggregation-induced emission (AIE) materials, which are typically weakly emissive or non-emissive in solution but become strongly emissive in the solid state due to restricted molecular motion.5
For compounds 6b, 6c, and 6e, solid-state emissions were observed at λem = 407, 407, and 417 nm, respectively, showing blue shifts of 15 nm for 6b and 6c and 7 nm for 6e compared to their emissions in CHCl3 solution. Compound 6a, however, exhibited nearly identical fluorescence emission maxima in both solution and solid states. Notably, compounds 6d and 6f showed a prominent solid-state ESIPT effect, with dual emission bands at 437 and 532 nm for 6d, and at 435 and 572 nm for 6f, corresponding to the enol and keto tautomers, respectively, when excited at λex = 343 nm and 374 nm. Additionally, solid-state excitation at λex = 500 nm enhanced the longer-wavelength maxima, 532 nm for 6d and 572 nm for 6f (Fig. 4), supporting the presence of ESIPT and its role in solid-state emission.
 | ||
| Fig. 4 Solid-state emission spectra at λex = 343 nm, 374 nm and 500 nm of compound 6d (A) and compound 6f (B). | ||
In the solid state, compounds 6a–f exhibited PLQYs of 21.5%, 14.2%, 13.2%, 7.1%, 17.9%, and 11.1%, respectively, demonstrating a substantial enhancement of fluorescence compared to CHCl3 solutions, with increases ranging from 5-fold for 6d to 14-fold for 6a. The fluorescence enhancement observed in the solid state among 6a–f can be attributed to the restriction of molecular motions and the effective prevention of detrimental π–π stacking, which inhibits non-radiative decay pathways. In the crystal structure of compound 6e, the pyrazole moiety and the pyrrolo[3,4-b]pyridin-5-one core participate in π–π stacking interactions, with centroid-to-centroid distances of approximately 6.897 Å and 4.332 Å, respectively (see the ESI, Fig. S42–S44†). These distances are significantly larger than the typical detrimental π–π stacking distance (∼3.35–3.50 Å), supporting the conclusion that the solid-state emission is enhanced due to the absence of strong π–π interactions that would otherwise quench fluorescence.1,24 Also, similar results have been observed in squaraine-type ESIPT fluorophores reported by Xia and Wang, where solid-state conformations assisted for a ESIPT mechanism suppress π-stacking, leading to low emission in solution but strong fluorescence in the solid state.25
Electronic structure
The geometries of the ground-state structures of the molecules were optimized through DFT calculations using the Gaussian 09 program,26 employing the B3LYP functional27 and the 6-311G(d,p) basis set.28 The solvation model based on density (SMD) using CHCl3 as solvent was applied to allow comparison with the experimental results. A frequency analysis was performed to ensure that all structures were at an energy minimum and not in a transition state. The theoretical absorption spectra reproduced the experimental results with good accuracy, although all absorption maxima for the pyrrolo[3,4-b]pyridin-5-one showed a hypsochromic shift between 3 and 5 nm, while for the pyrazole, there was a bathochromic shift between 6 and 10 nm. The computed oscillator strength values are generally lower compared to those of previous series, indicating that the transitions are weak but allowed. In general, all absorptions are composed of electronic transitions between various orbitals, and notably, in no case is the HOMO → LUMO transition the most significant, unlike other reports of similar systems.29 The absorption of the pyrazole fragment is predominantly composed of a HOMO → LUMO+1 transition in all cases, while the absorption of the pyrrolo[3,4-b]pyridin-5-one is predominantly composed of a HOMO−3 → LUMO transition in compounds 6a, 6b, 6c, and 6e. For compound 6d, the predominant transition is HOMO−4 → LUMO, and for compound 6f, it is HOMO−2 → LUMO+1. These data are summarized in Table 3.| Compound | Experimental | Calculated | Oscillator strength f | Main contributions |
|---|---|---|---|---|
| 6a | 279 | 274 | 0.192 | HOMO−4 → LUMO (21%) |
| HOMO−3 → LUMO (31%) | ||||
| HOMO−2 → LUMO+1 (27%) | ||||
| 302 | 310 | 0.363 | HOMO → LUMO+1 (94%) | |
| 6b | 278 | 273 | 0.247 | HOMO−3 → LUMO (46%) |
| HOMO−2 → LUMO+1 (34%) | ||||
| HOMO → LUMO3 (10%) | ||||
| 301 | 308 | 0.344 | HOMO → LUMO+1 (89%) | |
| 6c | 278 | 275 | 0.260 | HOMO−8 → LUMO (16%) |
| HOMO−3 → LUMO (49%) | ||||
| HOMO−2 → LUMO+1 (13%) | ||||
| 303 | 309 | 0.361 | HOMO → LUMO+1 (92%) | |
| 6d | 278 | 274 | 0.193 | HOMO−8 → LUMO (8%) |
| HOMO−4 → LUMO (49%) | ||||
| HOMO−3 → LUMO+1 (20%) | ||||
| 303 | 309 | 0.323 | HOMO−1 → LUMO (10%) | |
| HOMO−1 → LUMO+1 (10%) | ||||
| HOMO → LUMO+1 (78%) | ||||
| 6e | 278 | 275 | 0.186 | HOMO−4 → LUMO (24%) |
| HOMO−3 → LUMO (52%) | ||||
| HOMO−2 → LUMO+1 (18%) | ||||
| 303 | 310 | 0.347 | HOMO → LUMO+1 (91%) | |
| 6f | 278 | 276 | 0.119 | HOMO−2 → LUMO+1 (53%) |
| HOMO → LUMO+2 (38%) | ||||
| 302 | 312 | 0.380 | HOMO → LUMO+1 (100%) |
In compounds 6a–c and 6e, the HOMO−3 orbital (HOMO−4 for compound 6d) is distributed over the pyrrolo[3,4-b]pyridin-5-one fragment with its substituents, including the benzyl group and the pyrrole substituent, while the LUMO is localized solely on the pyrrolo[3,4-b]pyridin-5-one structure with a node at the methine group. For compound 6f, the most intense absorption consists of two main contributions: one from the HOMO−2 → LUMO+1 orbital (53%), which is distributed over the pyrazole and its substituents, and the other contribution from the HOMO → LUMO+2 orbital (38%), where the HOMO is localized on the pyrazole and the two rings attached to it, while the LUMO+2 is located on the pyrrolo[3,4-b]pyridin-5-one, though in this case, the node is located on the nitrogen of the pyrrole. Fig. 5 shows the distribution of the HOMO and LUMO orbitals involved in the strongest absorption band in the synthesized compounds.
 | ||
| Fig. 5 Representation of the frontier molecular orbitals involved in the strongest absorption band for compounds 6a–f. For 6a–c and 6e HOMO−3 → LUMO is shown. For 6d HOMO−4 → LUMO is shown. For 6f HOMO−2 → LUMO+1 is shown. | ||
The HOMO distribution is primarily localized within the pyrazole ring, while the LUMO is situated in the pyrrolo[3,4-b]pyridin-5-one moiety, indicating that the pyrazole fragment is more electron-rich. Generally, the substituents have little effect on the HOMO energy; however, a greater impact is observed on the LUMO energy. For example, the LUMO of compounds 6b and 6d is destabilized by 0.038 eV and 0.036 eV, respectively, relative to compound 6a. Conversely, the LUMO of compound 6f is stabilized by 0.014 eV compared to 6a. These results suggest that no clear relationship can be established between the nature of the substituents and their influence on the LUMO, as no consistent trend is apparent (Fig. S51†).
Conclusions
In closing, in this study six new pyrazolyl-pyrrolo[3,4-b]pyridin-5-ones were successfully synthesized in a one-pot fashion coupling a multicomponent Ugi-Zhu reaction to a cascade sequence (aza Diels–Alder/N-acylation/decarboxylation/dehydration), achieving isolated yields between 42% and 79%, which are acceptable considering the structural complexity of synthesized polyheterocycles, and that only a couple of molecules of water and one carbon dioxide were released in all synthetic procedure. These compounds exhibited DSE properties. Comprehensive photophysical analysis revealed impressive fluorescence features, including large Stokes shifts in solution and intense solid-state emissions, attributed to restricted intramolecular motions. Notably, the PLQYs were significantly higher in the solid state than in solution, highlighting the beneficial role of molecular conformation in reducing π–π stacking interactions to enhance fluorescence emission. Additionally, some compounds demonstrated a prominent ESIPT, a valuable property for applications requiring stable dual-state emissions. Complementary TD-DFT calculations accurately predicted the absorption maxima, with minor deviations in certain transitions. Electronic structure analysis indicated that the HOMO is predominantly localized on the pyrazole fragment, while the LUMO is centered on the pyrrolo[3,4-b]pyridin-5-one scaffold. Substituent modifications had a minimal impact on HOMO energy but significantly influenced LUMO energy. These findings underscore the effectiveness of multicomponent reactions in constructing complex heterocyclic frameworks with tunable optical properties, emphasizing their potential in photonic and bioimaging applications. In summary, the synthetic efficiency, structural diversity, and unique photophysical properties of these pyrazole-derived pyrrolo[3,4-b]pyridin-5-ones position them as promising candidates for further investigation in optoelectronic applications, particularly in the development of luminescent materials.Data availability
The data supporting this article have been included in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors thank Mónica A. Rincón-Guevara (HRMS), and Atilano Gutierrez-Carrillo (NMR) for spectra. A. I.-J. acknowledges “Proyecto Apoyado por el Fondo Sectorial de Investigación para la Educación CONAHCyT-SEP CB-2017-2018 (A1-S-32582)” for financial support. E. G. Z acknowledges DCBI-UAM-I/PEAPDI 2024 for financial support. J. C. F.-R. thanks CONAHCYT for his PhD scholarship (886026). Single-crystal X-ray diffraction data were collected using a Rigaku XtaLAB Synergy-s diffractometer equipped with a HyPix-6000HE detector funded by the Australian Research Council (ARC LE210100163). We are grateful to Prof. A. Paulina Gómora-Figueroa for providing access to the FT-IR instrument.References
- J. L. Belmonte-Vázquez, Y. A. Amador-Sánchez, L. A. Rodríguez-Cortés and B. Rodríguez-Molina, Dual-State Emission (DSE) in Organic Fluorophores: Design and Applications, Chem. Mater., 2021, 33(18), 7160–7184 CrossRef.
- F. Yu, H. Zhao, Y. Li, G. Xia and H. Wang, D–A-Type fluorophores with efficient dual-state emission for imaging at ultralow concentration, Mater. Chem. Front., 2022, 6(2), 155–162 RSC.
- Z. Huang, A. Ding, J. Yang, C. Wang and F. Tang, Conjugating Coumarin with Tetraphenylethylene to Achieve Dual–State Emission for Reversible Mechanofluorochromism and Live Cell Imaging, Chem. – Eur. J., 2023, 29, e202203628 CrossRef CAS PubMed.
- Ein Konzentrationsumschlag der Fluoreszenz, Z. Phys. Chem., 1954, 1(56), 275–277 Search PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Aggregation-Induced Emission: Together We Shine, United We Soar!, Chem. Rev., 2015, 115(21), 11718–11940 CrossRef CAS PubMed.
- L. A. Rodríguez-Cortés, A. Navarro-Huerta and B. Rodríguez-Molina, One molecule to light it all: The era of dual-state emission, Matter, 2021, 4(8), 2622–2624 CrossRef.
- (a) G. Xia, Q. Shao, K. Liang, Y. Wang, L. Jiang and H. Wang, A phenyl-removal strategy for accessing an efficient dual-state emitter in the red/NIR region guided by TDDFT calculations, J. Mater. Chem. C, 2020, 8(39), 13621–13626 RSC; (b) M. Durko-Maciag, G. Ulrich, D. Jacquemin, J. Mysliwiec and J. Massue, Solid-state emitters presenting a modular excited-state proton transfer (ESIPT) process: recent advances in dual-state emission and lasing applications, Phys. Chem. Chem. Phys., 2023, 25(22), 15085–15098 RSC.
- V. Kumar, K. Kaur, G. K. Gupta and A. K. Sharma, Pyrazole containing natural products: Synthetic preview and biological significance, Eur. J. Med. Chem., 2013, 69, 735–753 CrossRef CAS PubMed.
- A. Burke, M. di Filippo, S. Spiccio, A. M. Schito, D. Caviglia, C. Brullo and M. Baumann, Antimicrobial Evaluation of New Pyrazoles, Indazoles and Pyrazolines Prepared in Continuous Flow Mode, Int. J. Mol. Sci., 2023, 24(6), 5319 CrossRef CAS PubMed.
- A. Tigreros and J. Portilla, Recent progress in chemosensors based on pyrazole derivatives, RSC Adv., 2020, 10(33), 19693–19712 RSC.
- (a) I. v. Taydakov, A. A. Akkuzina, R. I. Avetisov, A. v. Khomyakov, R. R. Saifutyarov and I. Ch, Avetissov, Effective electroluminescent materials for OLED applications based on lanthanide 1.3-diketonates bearing pyrazole moiety, J. Lumin., 2016, 177, 31–39 CrossRef CAS; (b) M. Jin, Y. Chen, W. Song, H. Lian, H. Guo, Q. Dong, J. Huang and J. Su, Synthesis, characterization, and electroluminescent properties of indazole, pyrazole, and triazole/triphenylamine-based compounds, Dyes Pigm., 2020, 173, 106912 CrossRef CAS.
- A. Dömling, W. Wang and K. Wang, Chemistry and Biology of Multicomponent Reactions, Chem. Rev., 2012, 112(6), 3083–3135 CrossRef PubMed.
- L. Levi and T. J. J. Müller, Multicomponent syntheses of functional chromophores, Chem. Soc. Rev., 2016, 45(10), 2825–2846 RSC.
- (a) I. A. Ibarra, A. Islas-Jácome and E. González-Zamora, Synthesis of polyheterocycles via multicomponent reactions, Org. Biomol. Chem., 2018, 16(9), 1402–1418 RSC; (b) J. C. Flores-Reyes, V. d. C. Cotlame-Salinas, I. A. Ibarra, E. González-Zamora and A. Islas-Jácome, Pseudo-multicomponent reactions, RSC Adv., 2023, 13(24), 16091–16125 RSC; (c) J. C. Flores-Reyes, A. Islas-Jácome and E. González-Zamora, The Ugi three-component reaction and its variants, Org. Chem. Front., 2021, 8(19), 5460–5515 RSC.
- D. Udhayakumari, P. Jerome, N. Vijay and T. H. Oh, ESIPT: An approach and future perspective for the detection of biologically important analytes, J. Lumin., 2024, 267, 120350 CrossRef CAS.
- X. Sun, P. Janvier, G. Zhao, H. Bienaymé and J. Zhu, A Novel Multicomponent Synthesis of Polysubstituted 5-Aminooxazole and Its New Scaffold-Generating Reaction to Pyrrolo[3,4-b]pyridine, Org. Lett., 2001, 3(6), 877–880 CrossRef CAS.
- (a) I. Morales-Salazar, F. P. Montes-Enríquez, C. E. Garduño-Albino, M. A. García-Sánchez, I. A. Ibarra, Y. Rojas-Aguirre, M. E. García-Hernández, R. E. Sarmiento-Silva, S. L. Alcaraz-Estrada, E. Díaz-Cervantes, E. González-Zamora and A. Islas-Jácome, Synthesis of bis-furyl-pyrrolo[3,4-b]pyridin-5-ones via Ugi–Zhu reaction and in vitro activity assays against human SARS-CoV-2 and in silico studies on its main proteins, RSC Med. Chem., 2023, 14(1), 154–165 RSC; (b) I. Morales-Salazar, C. E. Garduño-Albino, F. P. Montes-Enríquez, D. A. Nava-Tapia, N. Navarro-Tito, L. D. Herrera-Zúñiga, E. González-Zamora and A. Islas-Jácome, Synthesis of Pyrrolo[3,4-b]pyridin-5-ones via Ugi–Zhu Reaction and In Vitro–In Silico Studies against Breast Carcinoma, Pharmaceuticals, 2023, 16(11), 1562 CrossRef CAS PubMed; (c) I. Morales-Salazar, C. E. Garduño-Albino, F. P. Montes-Enríquez, A. Gutiérrez-Carrillo, Y. Rojas-Aguirre, N. V. Estrada-Toledo, J. Sandoval-Basilio, S. L. Alcaraz-Estrada, E. Díaz-Cervantes, E. González-Zamora and A. Islas-Jácome, In Vitro and In Silico Studies of Bis-furyl-pyrrolo[3,4-b]pyridin-5-ones on Dengue Virus, J. Mex. Chem. Soc., 2024, 68(1), 170–183 CrossRef CAS.
- (a) J. C. Flores-Reyes, S. M. Rojas-Montoya, L. Blancarte-Carrazco, E. Xochitiotzi-Flores, C. A. Guarin, N. Farfán, A. Galano, A. Islas-Jácome and E. González-Zamora, Multicomponent synthesis and photophysical properties of meso-thienyl BODIPY-pyrrolo[3,4-b]pyridin-5-ones. An experimental and theoretical study, J. Lumin., 2024, 273, 120698 CrossRef CAS; (b) J. C. Flores-Reyes, A. Galano, S. M. Rojas-Montoya, L. Blancarte-Carrazco, E. Xochitiotzi-Flores, H. García-Ortega, N. Farfán, A. Islas-Jácome and E. González-Zamora, Synthesis of BODIPY-pyrrolo[3,4-b]pyridin-5-ones via Ugi-Zhu/cascade reactions and studies of fluorescence response toward viscosity, Front. Chem., 2024, 12, 1488933 CrossRef CAS PubMed.
- I. Ali, A. Haque, K. Saleem and M. F. Hsieh, Curcumin-I Knoevenagel's condensates and their Schiff's bases as anticancer agents: Synthesis, pharmacological and simulation studies, Bioorg. Med. Chem., 2013, 21(13), 3808–3820 CrossRef CAS PubMed.
- A. Kawski, On the Estimation of Excited-State Dipole Moments from Solvatochromic Shifts of Absorption and Fluorescence Spectra, Z. Naturforsch., A: Phys. Sci., 2002, 57(5), 255–262 CrossRef CAS.
- A. C. Sedgwick, L. Wu, H.-H. Han, S. D. Bull, X.-P. He, T. D. James, J. L. Sessler, B. Z. Tang, H. Tian and J. Yoon, Excited-state intramolecular proton-transfer (ESIPT) based fluorescence sensors and imaging agents, Chem. Soc. Rev., 2018, 47(23), 8842–8880 RSC.
- K.-L. Wong, J.-C. G. Bünzli and P. A. Tanner, Quantum yield and brightness, J. Lumin., 2020, 224, 117256 CrossRef CAS.
- V. S. Padalkar and S. Seki, Excited-state intramolecular proton-transfer (ESIPT)-inspired solid state emitters, Chem. Soc. Rev., 2016, 45(1), 169–202 RSC.
- (a) J. Shi, L. E. Aguilar Suarez, S. J. Yoon, S. Varghese, C. Serpa, S. Y. Park, L. Lüer, D. Roca-Sanjuán, B. Milián-Medina and J. Gierschner, Solid State Luminescence Enhancement in π-Conjugated Materials: Unraveling the Mechanism beyond the Framework of AIE/AIEE, J. Phys. Chem. C, 2017, 121(41), 23166–23183 CrossRef CAS; (b) K. Shirai, M. Matsuoka and K. Fukunishi, Fluorescence quenching by intermolecular π–π interactions of 2,5-bis(N,N-dialkylamino)-3,6-dicyanopyrazines, Dyes Pigm., 1999, 42(1), 95–101 CrossRef CAS.
- Q. Shao, K. Liang, H. Ling, Y. Wang, Z. Yan, G. Xia and H. Wang, Tetraphenylethylene-incorporated squaraine dyes: structural and theoretical insights into the diverse emission behaviors in solution and solid state, J. Mater. Chem. C, 2020, 8(13), 4549–4556 RSC.
- M. J. Frisch, et al., Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- (a) A. D. Becke, Density–functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) C. Lee, W. Yang and R. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. People, Self–consistent molecular orbital methods. XX. A basis set for correlated wave functions, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- (a) H. Zhuang, W. Shi, G. Zhao and Y. Li, Regulating and controlling the stepwise ESDPT channel of BP(OH)2DCEt2 using the strategy of solvent polarity and external electric field, Phys. Chem. Chem. Phys., 2024, 26, 12016–12026 RSC; (b) X. Xin, W. Shi, G. Zhao and Y. Li, Effectively regulation of the ESIPT process via ring-annelation modification for schiff base derivatives: A theoretical study, Dyes Pigm., 2024, 224, 112020 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2402623. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qo02256d |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2025 |