
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Graphene-based materials are not skin sensitizers: adoption of the in chemico/in vitro OECD test guidelines†
Michela
Carlin
 a,
Marc
Morant-Giner
bc,
Marina
Garrido
bd,
Silvio
Sosa
a,
Alberto
Bianco
e,
Aurelia
Tubaro
a,
Maurizio
Prato
bfg and
Marco
Pelin
*a
a,
Marc
Morant-Giner
bc,
Marina
Garrido
bd,
Silvio
Sosa
a,
Alberto
Bianco
e,
Aurelia
Tubaro
a,
Maurizio
Prato
bfg and
Marco
Pelin
*a
aDepartment of Life Sciences, University of Trieste, Via Fleming 22, 34127, Trieste, Italy. E-mail: mpelin@units.it
bDepartment of Chemical and Pharmaceutical Sciences, University of Trieste, Via Giorgieri 1, 34127 Trieste, Italy
cInstituto de Ciencia Molecular (ICMol), Universitat de València, C/Catedrático José Beltrán 2, 46980, Paterna, Spain
dIMDEA Nanociencia, C/Faraday, 9, Ciudad Universitaria de Cantoblanco, 28049, Madrid, Spain
eCNRS, Immunology, Immunopathology and Therapeutic Chemistry, UPR3572, University of Strasbourg, ISIS, 67000 Strasbourg, France
fCenter for Cooperative Research in Biomaterials (CIC biomaGUNE), Basque Research and Technology Alliance (BRTA), Parque Científico y Tecnológico de Gipuzkoa, Paseo Miramón 194, 20014, Donostia/San Sebastián, Spain
gBasque Foundation for Science (IKERBASQUE), Plaza Euskadi 5, 48009, Bilbao, Spain
First published on 9th April 2025
Abstract
The boost in the market size of graphene-based materials (GBMs) requires a careful evaluation of their impact on human health, acquiring robust and reliable data, also suitable for regulatory purposes. Considering cutaneous contact as one of the most relevant GBM exposure routes, this study is focused on skin sensitization, aimed at assessing the possibility to adopt the three in chemico/in vitro test guidelines (TGs) defined by the Organization for Economic Cooperation and Development (442C, D and E) to predict the first three phases of the skin sensitization adverse outcome pathway. Being originally validated for chemicals, modifications allowing their adoption for GBMs were evaluated. TG 442C was found to be not suitable for testing GBMs due to their reactivity, leading to possible misclassifications. In contrast, TG 442D and E can generally be applied for GBMs. However, protocol adjustments were required to assess cell viability reducing interferences for TG 442D, whereas caution should be exercised regarding dose-finding selection and GBM dispersion stability for TG 442E. When applying these modifications, GBMs were found to be unable to activate keratinocytes and promote dendritic cell differentiation, so they can be considered non-sensitizers. Overall, these results significantly contribute to understanding the safety profiles of GBMs and to improve testing methodologies to obtain reliable toxicological data.
Introduction
Two-dimensional (2D) nanomaterials have attracted increasing interest and expectations in the scientific community during the last few decades. Notably, graphene-based materials (GBMs) represent the most renowned members of 2D nanomaterials1 to such an extent that a recent meta-market analysis anticipated a significant graphene market growth over the next few years, reaching 1.5 billion US$ in 2027.2 GBMs, including few layer graphene (FLG), graphene oxide (GO), reduced GO (rGO), graphene nanoplatelets (GNPs) and other functionalized and/or composite derivatives, show unique physicochemical properties.3 Considering their high surface-area-to-volume ratio, mechanical resistance, thermal and electrical conductivity, light weight, flexibility, chemical inertness, atomic thickness and optical transparency, GBMs are widely used as advanced materials for a large variety of applications spanning from nanoelectronics and energy technology to biosensing and biomedicine.4,5 An active field of GBM investigation is their use at the skin level, mainly as artificial and electronic skin, wound healing applications, wearable technologies, skin sensors and scaffolds for tissue engineering.6–10 In all these cases, skin exposure to GBMs, either as direct contact with GBM-enriched devices or released GBM particles, is highly feasible. Moreover, cutaneous exposure can be definitely assumed as one of the most important exposure routes to GBMs considering both occupational and consumer exposure.11,12 In this scenario, skin irritation, corrosion and sensitization may be considered the most feasible adverse outcomes after cutaneous exposure to GBMs. In this framework, an emerging area of concern is associated with human exposure to these manufactured nanomaterials, making the assessment of their safety profile a challenging research field. Our previous studies demonstrated that GBMs are non-corrosive and non-irritant for the skin;13,14 however, skin sensitization potential cannot be excluded, also considering the ability of GBMs to interact with proteins and, therefore, behaving as possible haptens. In fact, it has been reported that GBMs can be detected by immune system components through attachment of proteins, such as the complement factors and/or damage- and stress-signals produced by cells in contact with the materials.15,16 On top of that, literature data already reported the ability of different GBMs to be internalized into the outer layers of epidermis13,17 and in keratinocytes,18–20 ultimately leading to different events such as oxidative stress,21 metabolic alterations22,23 and other effects such as those on cell proliferation,24 probably due to alteration of the cell cycle, as assessed on other cell models.25,26 Nevertheless, it should be noted that skin sensitization does not necessarily require substance internalization into keratinocytes, since haptenization of the substance may also occur as a result of reaction with extracellular or cell membrane proteins.27,28In agreement with European Union regulations, manufacturing companies need to comply with the REACH (registration, evaluation, authorization and restriction of chemicals) regulation for all chemical substances produced and/or imported at a volume of 1 ton per year or more.29 To this aim, reliable toxicological data should be collected following robust, repeatable, predictive, and accurate toxicity studies. In this context, a significant tool is represented by test guidelines (TGs) given by the Organization for Economic Co-operation and Development (OECD), a series of international consolidated methods that should be adopted to assess the potential toxic effects of marketed substances. Considering skin exposure, several OECD TGs are currently available to evaluate the main adverse outcomes at the skin level, including irritation, corrosion, sensitization and phototoxicity, using approaches both in vitro and in vivo.30–33 Nevertheless, these TGs were originally set up and validated only for chemicals, but their suitability for GBMs, and in general nanomaterials, needs to be evaluated due to their unique physicochemical properties. The OECD started a preliminary revision about the applicability of OECD TGs to manufactured nanomaterials already in 2009, highlighting these drawbacks.34 Regarding skin sensitization, many efforts have been currently made to assess the possibility to adopt the relevant OECD TGs, introducing, if necessary, appropriate modification(s) to the original procedures when not adoptable for manufactured nanomaterials. For instance, the OECD Chemicals and Biotechnology Committee recently published a report on the applicability of TG 442D for in vitro skin sensitization testing of selected manufactured nanomaterials on the basis of the knowledge gained in several EU projects [OECD Series on Testing and Assessment No. 382; ENV/CBC/MONO(2023)18]. Hence, herein we assessed if the three OECD TGs able to evaluate in chemico/in vitro skin sensitization potential (OECD TGs 442C, D and E) can also be adopted for GBMs. These TGs allow the assessment of the first three key events of the adverse outcome pathways (AOP) of skin sensitization, including the molecular initiation event (reactivity towards skin peptides; OECD TG 442C) and cellular responses (keratinocyte and dendritic cell activation; OECD TG 442D and E, respectively).
Materials and methods
Materials
Three representative GBMs were evaluated for their skin sensitization potential: (i) GO, considered a reactive material, (ii) rGO, the reduced form of GO, and (iii) GNPs, materials with potentially lower oxidizing and reactivity properties. GO (Batch #GOP20010) and rGO (Batch #rGOP20003) powders were kindly provided by Graphenea S.A. (Spain) and their complete physicochemical characterization is reported in our previous study.35 GNPs (Grade 4) were purchased from CheapTube (Grafton, VT, USA) and their complete physicochemical characterization can be found in our previous study.14OECD TG 442C – direct peptide reactivity assay (DPRA)
Skin sensitization potential was initially assessed using an in chemico test, namely the direct peptide reactivity assay (DPRA), able to determine the activation of the first AOP key phase. Following OECD TG 442C,36 the DPRA consists of the quantitation of the reactivity of a test substance with specific synthetic peptides by high performance liquid chromatography (HPLC) after the exposure time stated by the guideline (i.e. 24 h). According to this TG, a 100 mM stock solution of each test substance should be prepared in a suitable solvent. However, being not soluble, powdered GBMs can form only suspensions in appropriate buffers. In order to prepare approximately 100 mM suspensions of GBMs, the molecular weight (MW) of each test material was calculated according to the following formula:where [element%] is the weight percentage extracted from elemental analysis of each tested GBM and m is the atomic mass of each element.

As reported in the TG, an appropriate solvent that completely dissolves (in this case, disperses) the test substance should be selected. Hence, according to the list of suitable solvents reported in the TG, water was selected to obtain homogeneous dispersions of GO and rGO. In contrast, acetonitrile (ACN) was used for preparing cinnamic aldehyde solution at a concentration of 100 mM (positive control) and was chosen as the solvent for GNP dispersion due to the low aqueous dispersibility of this GBM. Stock solution of a cysteine peptide (C32H50N10O9S1, Ac-RFAACAA-OH, MW = 750.87 g mol−1, purity = 98.06%; RS Synthesis; Louisville, KY, USA) was prepared at 0.667 mM in phosphate buffer (pH 7.5). Samples for HPLC analysis were prepared in glass vials at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 ratio (0.5 mM cysteine peptide and 5 mM test substance) and incubated for 24 h in the dark at 25 °C. To maintain stable suspensions of the GBM samples during 24 h of incubation, magnetic stirring (150 rpm) was used, as an additional tip not provided by the TG. After the incubation period, the GBM samples were filtered (PTFE syringe filter, 0.2 μm) prior to HPLC injection to avoid any damage related to the presence of solid particles to the system. In addition, reference controls that only contained the peptide solution in the appropriate solvent were included to examine protein stability and confirm that solvents did not negatively affect peptide depletion. Furthermore, a calibration curve was determined. Serial dilutions of the peptide stock solution, encompassing the range of 0.0167–0.534 mM, were prepared using a 20% solution of ACN in phosphate buffer. A system was considered suitable if the standard calibration curve was linear with r2 > 0.99 and the mean of peptide concentration of the reference control was 0.50 ± 0.05 mM.
:10 ratio (0.5 mM cysteine peptide and 5 mM test substance) and incubated for 24 h in the dark at 25 °C. To maintain stable suspensions of the GBM samples during 24 h of incubation, magnetic stirring (150 rpm) was used, as an additional tip not provided by the TG. After the incubation period, the GBM samples were filtered (PTFE syringe filter, 0.2 μm) prior to HPLC injection to avoid any damage related to the presence of solid particles to the system. In addition, reference controls that only contained the peptide solution in the appropriate solvent were included to examine protein stability and confirm that solvents did not negatively affect peptide depletion. Furthermore, a calibration curve was determined. Serial dilutions of the peptide stock solution, encompassing the range of 0.0167–0.534 mM, were prepared using a 20% solution of ACN in phosphate buffer. A system was considered suitable if the standard calibration curve was linear with r2 > 0.99 and the mean of peptide concentration of the reference control was 0.50 ± 0.05 mM.
After incubation, the samples (40 μL) were injected in an HPLC-UV Agilent 1260 Infinity II equipped with a C18 reverse-phase column (Poroshell 120 EC-C18; 4.6 × 100 mm, 4 μm) and coupled with a UV detector operating at 220 nm. A binary mobile phase composed of 0.1% (v/v) trifluoroacetic acid (TFA) in water (A) and 0.085% (v/v) TFA in ACN (B) was used. The HPLC analysis was conducted at a flow rate of 0.35 mL min−1 according to the gradient program reported in Table 1.
| Time | Flow | A (H2O + TFA) | B (ACN + TFA) |
|---|---|---|---|
| 0 | 0.35 mL min−1 | 90 | 10 |
| 10 min | 0.35 mL min−1 | 75 | 25 |
| 11 min | 0.35 mL min−1 | 10 | 90 |
| 13 min | 0.35 mL min−1 | 10 | 90 |
| 13.5 min | 0.35 mL min−1 | 90 | 10 |
| 18 min | 0.35 mL min−1 | 90 | 10 |
Quantitation of the free cysteine peptide was measured and compared to that of the reference control prepared in the appropriate solvent. The percentage of cysteine peptide depletion was calculated using the following equation:

Data are presented as the mean % of peptide depletion of three independent experiments conducted in triplicate. A substance can be considered positive to the DPRA if it determines a cysteine peptide depletion above 13.89%. Positive results could also be used for classifying a substance based on its reactivity with the cysteine peptide, as reported in the prediction model in Table 2.
| Cysteine (Cys) peptide % depletion | Reactivity class | DPRA prediction |
|---|---|---|
| 0% ≤ Cys peptide % depletion ≤ 13.89% | No or minimal reactivity | Negative |
| 13.89% ≤ Cys peptide % depletion ≤ 23.09% | Low reactivity | Positive |
| 23.09% ≤ Cys peptide % depletion ≤ 98.24% | Moderate reactivity | Positive |
| 98.24% ≤ Cys peptide % depletion ≤ 100% | High reactivity | Positive |
:10 ratio (5 mM cysteine and 50 mM test substance). The samples were incubated at 25 °C under magnetic stirring (150 rpm) for 24 h in the dark and filtered (PTFE syringe filter, 0.2 μm) prior to the 1H-NMR analysis. In addition, as a reference control, 5 mM L-cysteine solution in D2O was incubated in the absence of the tested materials under the same conditions. NMR spectra were recorded using a Varian 400 spectrometer (1H: 400 MHz). The chemical shift (δ) for 1H is given in ppm relative to residual signals of the solvents. The concentration of the samples was increased ten-fold to perform 1H-NMR analyses.
OECD TG 442D – ARE-Nrf2 Luciferase KeratinoSens™ test method
where Ca is the lowest concentration with >1.5-fold luciferase induction, Cb is the highest concentration with <1.5-fold luciferase induction, Ia is the fold induction measured at the lowest concentration with >1.5-fold luciferase induction, and Ib is the fold induction measured at the highest concentration with <1.5-fold luciferase induction. The results are the mean of 3 independent experiments.
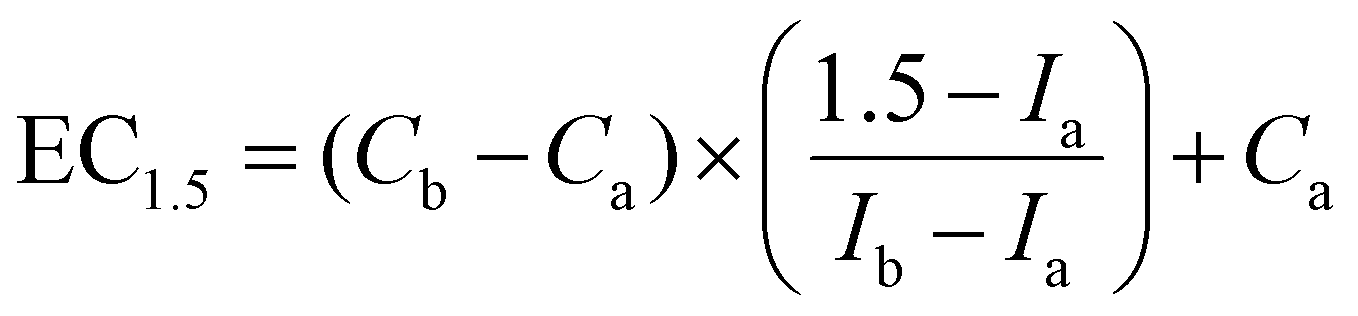
As done for cell viability assessment, as an additional step, not included in OECD TG 442D, two washing steps with 200 μL per well of PBS were performed after GBM treatment.
OECD TG 442E – human cell line activation test (h-CLAT)

For CD54/CD56 expression measurement, each test substance was tested in at least two independent runs to derive the prediction. A human cell line activation test (h-CLAT) prediction was considered positive if at least one of the following conditions was met in 2 out of 2 or in at least 2 out of 3 independent runs, otherwise the h-CLAT prediction was considered negative: (i) the RFI of CD86 is equal or greater than 150% in at least one of the tested concentrations (with cell viability ≥50%) and (ii) the RFI of CD54 is equal or greater than 200% in at least one of the tested concentrations (with cell viability ≥50%).
Results and discussion
In the last few years, alternative methods to the use of animals, named new approach methodologies (NAMs), have been pushed forward in hazard prevention strategies and regulatory frameworks. In compliance with the 3Rs principles (replacement, reduction, and refinement), NAMs provide information on chemical hazard and risk assessment while reducing or avoiding the use of animal models in laboratories.40 In detail, several NAMs for skin sensitization prediction were included in OECD TGs 442 (442C, 442D and 442E). However, none of them are considered sufficiently stand-alone replacements of animal test to conclude on skin sensitization potential or to provide information for potency sub-categorization according to the United Nations Globally Harmonized System of Classification and Labelling of Chemicals. OECD TG 497 presents the defined approaches (DAs) on skin sensitization that consist of a selection of information sources used in a specific combination and the resulting data are understood using a fixed data interpretation procedure to overcome some limitations of the stand-alone methods.41 DAs either provide the same level of information or are more informative than the murine local lymph node assay (LLNA); OECD TG 42942 for hazard identification (i.e., sensitizer versus non-sensitizer). In detail, the 2-out-of-3 (2o3) DA is intended for the discrimination between sensitizers and non-sensitizers, but it is not designed to provide information on the potency of a sensitizer. The combination of test methods included in the 2o3 DA covers at least two of the first three key events of the skin sensitization AOP as formally described: (i) the first key event (protein binding via the DPRA; OECD TG 442C), (ii) the second key event (keratinocyte activation via the KeratinoSens™ test method; OECD TG 442D), and (iii) the third key event (dendritic cell activation via the h-CLAT test method; OECD TG 442E). According to the 2o3 DA, two concordant results obtained from methods addressing at least two of the first three key events of the AOP determined the final classification of a substance.However, it should be considered that important limitations can be foreseen when adopting these TGs for substances other than chemicals. For instance, when testing nanomaterials, the test substance should be analyzed in dispersion form, not as powders. It is noteworthy that the dispersion of 2D nanomaterials, including GBMs, in different buffers may alter their characteristics, including agglomeration/aggregation, surface charge and others. Hence, it is fundamental to take into account that this limitation, together with others, could represent a bias when trying to adopt these TGs for 2D nanomaterials. Hence, according to the effort made by the Malta Initiative and the OECD Working Party on Manufactured Nanomaterials (WPMN), this study was aimed at: (i) assessing the possibility to adopt these OECD TGs to GBMs; (ii) in the case of limitations in this adoption, defining any modification to the procedures, if necessary; and (iii) assessing the skin sensitization potential of GBMs using robust and reliable animal-free approaches.
To this aim, we tested 3 representative GBMs that a priori may lead to different skin sensitization potential on the basis of their different physicochemical properties: (i) GO, an oxidized and reactive material due to the presence of oxygenated functional groups on its structure, (ii) rGO, the reduced form of GO with a significant lower content of oxygenated moieties, and (iii) GNPs, materials with lower oxidizing and reactivity properties.
OECD TG 442C – direct peptide reactivity assay (DPRA)
The DPRA is the main method reported in OECD TG 442C aiming at determining if a chemical substance is sufficiently electrophilically reactive towards peptides to initiate a skin sensitization process. According to the exposure time and the prediction threshold given by the TG, after 24 h of incubation of the test substance (5 mM) with a cysteine-enriched peptide (0.5 mM) containing reactive thiol units as surrogates of nucleophiles, a peptide depletion equal to 13.89% or above predicts activation of the first key event of the skin sensitization AOP.36,43In OECD TG 442C, several limitations for DPRA application were already reported.36 Among the known restrictions, some might be crucial when considering the adoption of this method for GBMs. First, the tested chemical should be soluble in an appropriate solvent at a final concentration of 100 mM and should not precipitate in the reaction solution. However, it should be noted that for GBMs, being insoluble materials, it is not possible to obtain solutions, but only homogeneous nanomaterial dispersions were obtained. Magnetic stirring was found to be essential to maintain a stable GBM dispersion during 24 h of incubation with the cysteine peptide, avoiding the formation of precipitates and ensuring continuous contact between the GBM particles and the cysteine peptide.
Second, OECD TG 442C reports that predicting the correct calculation of peptide depletion for test substances absorbing at 220 nm is not feasible. This characteristic is met in the case of GBMs,44per se potentially restricting the adoption of this TG for these materials. Hence, to overcome this issue, after 24 h of incubation with the peptide, the GBM samples were filtered before HPLC injection to prevent possible interferences of the nanomaterials with the absorbance observed at 220 nm: in this manner, in the solution injected in the HPLC system, only the non-interacting peptide fraction, but no interacting and/or free-standing GBMs, is present, allowing a potentially unbiased calculation of peptide depletion. In addition, filtration was found to be also necessary to avoid any damage (e.g., blocking the capillary tubes or the pumps) provoked by the presence of solid particles to the HPLC system. Once this modification was applied, HPLC analyses were carried out.
Before performing HPLC analysis, the system performance was assessed. Calibration standards of the cysteine peptide (0.0167–0.534 mM) were prepared and analyzed in triplicate; the calibration curve was constructed by plotting the peptide concentrations versus the areas of each corresponding peak, resulting in a calibration linearity of r2 = 0.9972, as shown in Fig. S1.† The reference control standards were also analyzed in each analytical run and their stability over analysis time resulted in a coefficient of variation (CV) of 4.9%, well below the acceptance threshold stated in the TG (<15%).
Once verified, the suitability of the system performance, HPLC-UV analyses of the GBM-treated samples were performed. After 24 h of incubation of GBMs (5 mM) with the cysteine peptide (0.5 mM) under magnetic stirring in the dark, the samples were filtered and loaded into the HPLC autosampler. Fig. S2† shows the chromatograms of cysteine peptide reference controls dissolved in ACN (a) or deionized water (b). The characteristic peak of the cysteine peptide has a retention time of 14.6 min in both solvents, proving that the dissolution solvent does not influence the HPLC retention time of the peptide. Fig. 1 shows representative chromatograms of the cysteine peptide incubated with GO (a), rGO (b), GNPs (c) or cinnamic aldehyde used as the positive control (d). Peptide depletion for each sample was calculated referring to the corresponding untreated control (cysteine peptide in water for GO and rGO or in ACN for GNPs and cinnamic aldehyde). Specifically, the mean percentage of peptide depletion of the three biological replicates is shown in Table 3. After 24 h of incubation, peptide depletions above the 13.89% threshold were recorded for both GO and rGO. Hence, these materials can be considered positive according to the OECD TG 442C DPRA prediction model. In particular, GO induced a peptide depletion of 14.52%, suggesting a low reactivity towards the cysteine peptide, whereas rGO induced a higher peptide depletion (56.65%), suggesting a moderate reactivity. In contrast, GNPs induced an extremely low peptide depletion (8.34%); being below the threshold given by the TG, GNPs can be considered negative to the DPRA. As expected, the positive control cinnamic aldehyde induced 73.53% cysteine peptide depletion within the acceptability range criteria defined by OECD TG 442C (between 60.8% and 100% peptide depletion).
 | ||
| Fig. 1 Representative chromatograms obtained by HPLC analysis showing the retention times and peak intensities of the free cysteine peptide (marked with *) incubated with GO ((a); retention time: 14.616 min), rGO ((b); retention time: 14.686 min), GNPs ((c); retention time: 14.590 min) or the positive control cinnamic aldehyde ((d); retention time: 14.712 min). | ||
| Sample | % Cysteine peptide depletion | Reactivity class | DPRA prediction |
|---|---|---|---|
| GO | 14.52 | Low reactivity | Positive |
| rGO | 56.65 | Moderate reactivity | Positive |
| GNPs | 8.34 | Minimal reactivity | Negative |
| Cinnamic aldehyde | 73.53 | Moderate reactivity | Positive |
These results demonstrate that both GO and rGO are reactive towards cysteine-enriched peptides, at levels showing positivity to the DPRA. In contrast, the reactivity of GNPs is lower with respect to the threshold given by the TG. However, no definitive conclusions should be drawn from these results, since this prediction could not be reliable considering the physicochemical properties of GBMs, such as their oxidative potential. In fact, as reported by the TG, test substances that spontaneously promote cysteine-enriched peptide oxidation (i.e., cysteine dimerization, sulfone or sulfoxide formation) could lead to false positive predictions and overestimation of peptide depletion, resulting in a possible assignment to a higher reactivity class.36 The literature data already reported that GO can oxidize cysteine-containing peptides: indeed, GO is reported to be able to oxidize thiol groups of glutathione to disulfide bonds to form glutathione disulfide, leading to the reduction of GO, in different biological systems.45,46 To confirm the capability of GBMs to oxidize cysteine residues into disulfides, a control experiment was performed using L-cysteine instead of the cysteine peptide due to its simpler structure. The results of 1H-NMR analyses are presented in Fig. 2 and their integrals are presented in Fig. S3.† In particular, panel a of Fig. 2 shows the spectra of the control experiment (red, L-cysteine incubated in the absence of the tested materials) and the freshly prepared L-cysteine solution (black). After its incubation, L-cysteine was spontaneously partially oxidized to cystine (its disulfide form) under ambient conditions,47,48 with a cysteine/cystine ratio of 9. The results presented in panel b demonstrate that L-cysteine incubated with GO or GNPs was also partially oxidized, with the cysteine/cystine ratio being equal to 2.5 or 4, respectively. Therefore, the presence of both GBMs accelerates the L-cysteine oxidation. In the case of the incubation performed in the presence of rGO, it can be observed that L-cysteine was fully oxidized to cystine, with the acceleration of the oxidation process being higher. These findings clearly demonstrate the capability GBMs to oxidize cysteine to cystine, with different degrees depending on the material. Hence, we cannot exclude that the positive results of GO and rGO in the DPRA might be ascribed to their ability to oxidize cysteine. Therefore, even though OECD TG 442C can be slightly modified to overcome some technical limitations hindering its application for GBMs, it anyway cannot be adopted, since results could lead to false positive prediction of skin sensitization properties given the ability of GBMs to spontaneously oxidize cysteine. In addition, it should be noted that GO and rGO can also strongly adsorb peptides and this property can be another important limiting factor.49
 | ||
| Fig. 2 (a) Representative 1H-NMR spectra of 5 mM L-cysteine solution in D2O freshly prepared and after 24 h (black and red, respectively). The peaks highlighted in grey refer to L-cysteine and the peaks highlighted in red refer to cystine. (b) Representative 1H-NMR spectra of 5 mM L-cysteine solution in D2O after 24 h of incubation with GO, rGO or GNPs (green, purple, and blue, respectively). The peaks highlighted in grey refer to L-cysteine and the peaks highlighted in red refer to cystine. | ||
However, it should be noted that some studies already predicted the skin sensitization potential of other related carbon-based nanomaterials through the DPRA. For instance, Bezerra et al. reported moderate skin sensitization properties of single walled carbon nanotubes and C60.50 Nonetheless, the methodological procedures were not reported and, considering the several criticisms and issues related to the suitability of this method, at least to carbon-based nanomaterials, these data should be considered with caution.50 Other studies assessed the reactivity of other types of metal-based nanomaterials, including TiO2, CeO2, Co3O4, NiO, Fe2O3, and gold nanoparticles, towards the cysteine peptide.51,52 However, considering the different physicochemical features and behaviors of metal-based nanoparticles with respect to GBMs, it is not possible to make the same assumptions when applying the DPRA protocol.
OECD TG 442D – ARE-Nrf2 Luciferase KeratinoSens™ test method
The skin sensitization potential of GBMs was subsequently assessed using the ARE-Nrf2 Luciferase KeratinoSens™ assay, addressing the second key event of the skin sensitization AOP, namely keratinocyte activation. The analysis was carried out after 48 h of exposure to the test substances, as suggested by the method described in OECD TG 442D.Although some limitations for testing water-insoluble substances were highlighted using this method, the TG reports the possibility to refer to weight per volume (w/v) concentrations (μg mL−1) instead of molarity, also allowing the testing of insoluble materials, such as GBMs. However, proper and homogeneous GBM dispersions in culture media are fundamental to correctly predict their effects. Initially, two parameters were considered to assess the suitability of OECD TG 442D: (i) the final readout to assess cytotoxicity, since GBMs may interfere with the OD measurement of formazan produced by MTT reduction53 and (ii) the possible unspecific interferences of GBMs, both with the cytotoxicity assay and with the luciferase measurement, since GBMs tend to precipitate above the cells, interacting with them and altering the signal.
First, as shown in Fig. S4(a, d and g),† the effects of GO, rGO and GNPs on the viability of KeratinoSens™ cells was evaluated after 48 h of exposure by the MTT assay, as stated in the TG. The results demonstrate that, strictly following the TG procedure, the effects of GBMs on cell viability were clearly concentration-independent, probably due to interferences with formazan measurement. As already reported in the literature,18 two washing steps with PBS (200 μL per well) could be sufficient to remove unbound GBMs from the cells, therefore reducing this interference and obtaining a correct prediction of the cytotoxic effect.
Second, the choice of the cytotoxicity test to be used was considered, since the MTT assay is reported to give artifacts with GBMs.53 Hence, the results obtained by the MTT test were compared to those obtained with two other related assays, WST-8 (Fig. S4b, e and h†) and MTS (Fig. S4c, f and i†), which were potentially affected to a lower extent by interferences with carbon-based nanomaterials. In all cases, when washing steps were not performed, unclear effects of GBMs on cell viability were recorded, since alterations of cell viability were not concentration-dependent and interferences between GBMs and cytotoxicity readouts were found. In contrast, after washing steps, clear concentration-dependent effects of GBMs were observed. However, the MTT assay underestimated cell viability reduction induced by 48 h of exposure to GBMs with respect to WST-8 and MTS assays, in line with the literature data.53 Hence, being already regulated by an ISO guideline for measuring the cytotoxic effect of nanoparticles,38 the MTS assay was chosen for the replacement of the MTT assay, to be performed after two washing steps with PBS, when OECD TG 442D is applied to GBMs.
As a second step, the activity of luciferase was measured in parallel to the MTS assay under the same experimental conditions. Considering the significant tendency of GBMs to precipitate the above cells, two washing steps with PBS (200 μL per well) were added before luciferase measurement to reduce at maximum any interference, and the results were compared to those obtained without performing the washing steps. A slight underestimation of luciferase induction was found under the second conditions, suggesting possible luminescence quenching properties of these materials,54,55 unlike luciferase measurements of cinnamic aldehyde-treated cells that were not affected by washing steps (Fig. S5†). Therefore, when measuring luciferase activity, two washing steps with PBS are also necessary to avoid any false negative results.
Hence, Fig. 3 shows the results obtained through the adoption of OECD TG 442D with the procedural modifications reported above (i.e., the MTS assay for cell viability assessment and two washing steps before cell viability and luciferase measurement). Only the positive control cinnamic aldehyde induced a significant increase in luciferase activity, higher than the threshold reported by OECD TG 442D (1.5-fold induction), confirming its ability to activate keratinocytes, with an Imax (maximal induction factor of luciferase activity compared to the negative control measured at any test chemical concentration) of 3.39-fold and an EC1.5 value (interpolated concentration for a 1.5-fold luciferase induction) of 10.74 μM. Acceptance criteria for the TG were met, since the luciferase activity induction recorded with the positive control should be above the threshold of 1.5-fold in at least one of the tested concentrations (4–64 μM).
 | ||
| Fig. 3 Effects on the viability of KeratinoSens™ cells and luciferase induction after 48 h of treatment with GO (a), rGO (b), GNPs (c) (0.2–400.0 μg mL−1) and the positive control ((d); cinnamic aldehyde: 4–64 μM) measured after performing two washing steps with PBS, as an additional step to the procedure described in OECD TG 442D. The dashed red line represents the 1.5-fold luciferase induction threshold defined by OECD TG 442D to predict keratinocyte-activating substances. The cell viability was evaluated using the MTS assay and the data are reported as % of cell viability with respect to negative controls. Luminescence activity is reported as fold induction. Data are the mean ± SE of three independent experiments performed in triplicate. | ||
GO, rGO and GNPs did not significantly increase luciferase activity, with Imax values being 1.01-, 1.06-, and 1.17-fold, respectively, all below the 1.5-fold threshold set by the TG (Fig. 3). In addition, the calculated EC1.5 values were >400 μg mL−1 for all GBMs. Moreover, the IC50 values (concentration inhibiting cell viability by 50%) were equal to 69.63 μg mL−1 and 81.80 μg mL−1 for GO and rGO, respectively. For GNPs, the IC50 value cannot be calculated (>400 μg mL−1) since no cytotoxic effect was found. A summary of GBM-relevant parameters to be considered in the KeratinoSens™ test method is reported in Table 4.
| KeratinoSens™ test method results | ||||
|---|---|---|---|---|
| Material | I max | EC1.5 | IC50 | Classification |
| GO | 1.01 | >400 μg mL−1 | 69.63 μg mL−1 | Negative |
| rGO | 1.06 | >400 μg mL−1 | 81.80 μg mL−1 | Negative |
| GNPs | 1.17 | >400 μg mL−1 | >400 μg mL−1 | Negative |
| Cinnamic aldehyde | 3.39 | 10.74 μM | >64 μM | Positive |
On the whole, these results demonstrate the necessity of introducing few minor modifications to the protocol, before adopting OECD TG 442D for testing GBMs, including: (i) the substitution of the MTT assay with the MTS assay for cell viability assessment to avoid interferences between the assay and the materials and (ii) the introduction of two washing steps with PBS to further minimize these interferences. When adopting these modifications, the application of OECD TG 442D showed that GO, rGO and GNPs did not significantly induce luciferase, demonstrating their inability to activate keratinocytes. These results are confirmed by previous studies, proving the absence of sensitizing properties of GNPs and carbon nanotubes using the same procedure.56,57 However, it should be noted again that the authors did not check any interference of materials with the readouts at any of the concentrations tested (up to 400 μg mL−1), an aspect that should be carefully evaluated to demonstrate the reliability of the data.
OECD TG 442E – human cell line activation test (h-CLAT)
The h-CLAT method was carried out following OECD TG 442E, addressing the third key event of the skin sensitization AOP, namely the differentiation of monocytes to dendritic cells (DCs). The assay quantifies the changes in the expression of specific cell surface markers (CD54 and CD86) associated with the activation of DCs derived from the human monocytic leukemia cell line THP-1 after the exposure time to a test substance suggested by the TG (i.e., 24 h). Cytotoxicity measurement was also performed concurrently to assess whether up-regulation of surface marker expression occurs at sub-cytotoxic concentrations.39Initially, a dose-finding step should be performed to identify the test chemical concentration, resulting in 75% cell viability compared to the solvent/vehicle control (CV75). OECD TG 442E indicates the use of the PI probe for the dose-finding step based on flow cytometry analysis starting from the highest concentration of 1000 μg mL−1 (dilution factor of 2 in 0.2% DMSO). However, the TG reported the possibility of using other cytotoxicity markers, such as trypan blue, if PI uptake cannot be performed. Hence, in the case of GBMs, considering their fluorescence quenching properties54,55 and the possibility of inducing instrumental damage related to the presence of particle aggregates in the fluidic system due to the high GBM concentrations suggested by the TG, the trypan blue exclusion assay was selected. THP-1 cells were exposed to GO, rGO or GNPs for 24 h in the concentration range suggested by the TG (7.8–1000 μg mL−1) and cell viability was evaluated by means of the trypan blue exclusion assay. Effects of GO, rGO and GNPs (7.8–1000 μg mL−1) on the viability of THP-1 cells were investigated and are presented in Fig. 4. Regarding GNPs, the highest concentration (1000 μg mL−1) was not tested due to the impossibility to form a homogeneous dispersion in complete culture medium. However, for all GBMs, it was not possible to determine the CV75 due to their low cytotoxicity. Even at the highest concentration (1000 μg mL−1), GO and rGO reduced cell viability at levels of 76.0% and 78.7%, respectively. GNPs reduced cell viability at a level equal to 89.8% at the highest concentration tested (500 μg mL−1).
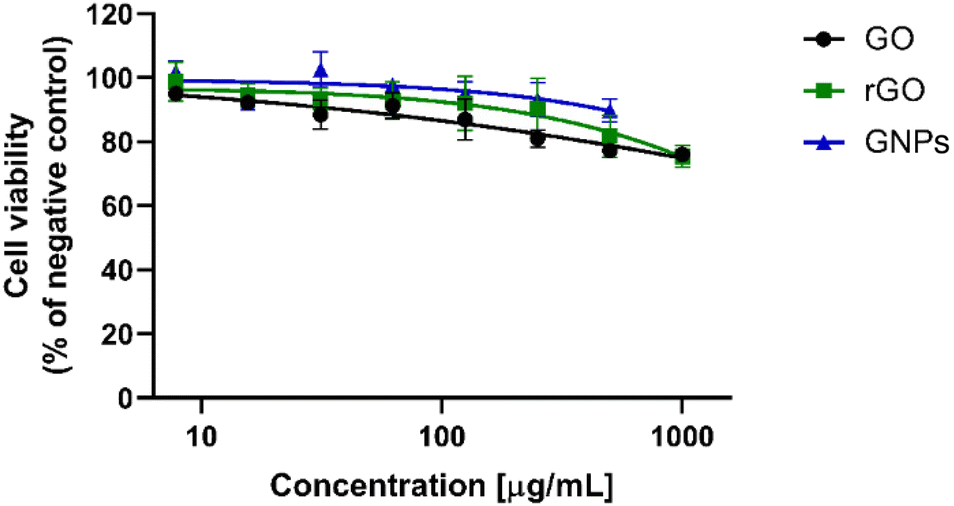 | ||
| Fig. 4 Effects of GO, rGO and GNPs (7.8–1000 μg mL−1) on the viability of THP-1 cells measured after 24 h using the trypan blue exclusion assay. The data are the mean ± SE of three independent experiments performed in triplicate. | ||
As stated by OECD TG 442E, since the CV75 could not be determined, the highest stably dispersed concentration of the test substance was chosen as the starting concentration to set the concentration range for the subsequent CD54/CD86 expression measurement. To this aim, the suspension stability of each material concentration (7.8–1000 μg mL−1) was assessed by UV-Vis analysis, measuring the absorbance at 660 nm up to 24 h. As shown in Fig. S6,† except for the concentration of 7.8 μg mL−1, the dispersion stability at all the other concentrations decreased starting from 2 to 4 h. Therefore, the concentration of 7.8 μg mL−1 is the most stable one for all the tested materials. Thus, this concentration was used to define the concentration range diluted by a 1:1.2 factor (2.6–9.4 μg mL−1) to be tested for the quantitation of CD54/CD86 markers.
Hence, THP-1 cells were exposed to GO, rGO or GNPs (2.6–9.4 μg mL−1) for 24 h. DNCB (4 μg mL−1) and NiSO4 (100 μg mL−1) were included as positive controls, while lactic acid (1000 μg mL−1) was included as the negative control. An additional negative control was represented by the vehicle alone (complete cell culture medium containing 0.2% DMSO). Table 5 shows the results obtained by flow cytometry analysis from three independent runs. All the experiments were concordant and the data were analyzed considering the changes in the surface marker (FL1) expression analyzed by flow cytometry. For all samples, the cell viability was ≥50% as assessed by PI uptake (FL3). In this last case, interferences given by GBMs with PI fluorescence measurement were excluded by preliminary experiments (data not shown) given the lower concentration range with respect to that used for the dose-finding assay. Anyway, cell viabilities ≥50% were confirmed also by the trypan blue exclusion assay. In particular, at the highest concentration tested (9.4 μg mL−1), RFI% values of CD54 with respect to the vehicle control were 84.3% for GO, 81.4% for rGO and 94.7% for GNPs. At the highest concentration tested, RFI% values of CD86 with respect to the vehicle control were 120.5%, 91.4% and 92.7% for GO, rGO and GNPs, respectively. These values are lower than the relevant RFI% thresholds given by the TG (RFI% ≥ 200 and ≥150 for CD54 and CD86, respectively, with the corresponding cell viabilities ≥50%). Thus, in accordance with OECD TG 442E, the tested GBMs were not able to induce monocyte differentiation to dendritic cells, the third key event of the skin sensitization AOP. In contrast, as expected, RFI% values of CD54 and CD86 calculated for the positive controls DNCB (205.3% and 324.7%, respectively) and NiSO4 (484.1% and 170.1%, respectively) and the negative control LA (87.3% and 71.2%, respectively) were correspondingly higher and lower than the respective thresholds. Similar results were obtained by testing all the other selected concentrations of all the tested GBMs (see Table S1†).
| Material | Concentration (μg mL−1) | RFI% CD86 | RFI% CD54 | Viability (PI) | Viability (trypan blue) | Classification |
|---|---|---|---|---|---|---|
| LA | 1000 | 71.2% | 87.3% | 98.4% | 99.3% | Negative |
| DNCB | 4 | 324.7%*** | 205.3%*** | 63.5% | 65.9% | Positive |
| NiSO4 | 100 | 170.1%*** | 484.1%*** | 79.3% | 76.1% | Positive |
| GO | 9.4 | 120.5% | 84.3% | 97.5% | 98.6% | Negative |
| rGO | 9.4 | 91.4% | 81.4% | 98.2% | 99.4% | Negative |
| GNPs | 9.4 | 92.7% | 94.7% | 97.7% | 98.1% | Negative |
Overall, these data suggest that OECD TG 442E can be adopted for GBMs with the following cautions: (i) the dose-finding step should be carried out using the trypan blue exclusion assay instead of PI uptake measurement, due to the fluorescence quenching properties of GBMs at high concentrations and possible instrumental damage related to the presence of particle aggregates in the fluidic system due to the high GBM concentrations suggested by OECD TG 442E for this step and (ii) in the case of a low cytotoxic potential preventing CV75 calculation, a careful check of the dispersion stability should be required for the determination of the optimal concentration range for CD54/CD86 expression measurements. Once these precautions are applied, considering the thresholds given by OECD TG 442E, the present results demonstrate that GO, rGO and GNPs seem unable to activate DCs, as the third key event of the skin sensitization AOP. Indeed, the relative expressions of CD54 and CD86 were far lower than the 200% and 150% thresholds, but were almost equal to the vehicle controls, suggesting the absence of sensitization properties in terms of DC activation. This observation is in line with previous in vitro findings demonstrating, albeit using other methods, that GO has a minimal impact on DC activation58 and that it is able to regulate differentiation of dendritic cells by suppressing their maturation.59–61 In contrast, other carbon-based nanomaterials seem to induce DC activation in vitro, such as functionalized carbon nanotubes in THP-1 cells,62 carbon black nanoparticles in mouse bone marrow-derived DCs63 and fullerene derivatives in myeloid DCs.64 These data demonstrate that, in comparison with other carbon-based nanomaterials, GBMs may be considered safer at least as they concern the third phase of skin sensitization.
Conclusions
Mechanistically-based in chemico and in vitro methods, addressing the first three key events of the skin sensitization AOP, can be used to assess skin sensitization potential with a battery of predictive tests completely substituting the use of animals. According to the 2o3 DA defined by OECD TG 497, two concordant results obtained from methods addressing at least two of the first three key events of the skin sensitization AOP (OECD TG 442C, D and E) determine the final classification of a substance. Therefore, on the basis of these considerations, our results allow a clear conclusion that the tested GBMs (i.e., GO, rGO and GNPs) are not skin sensitizers since at least 2 out of the 3 OECD TGs for skin sensitization prediction were reliably negative. This conclusion was achieved testing doses specifically defined by the OECD for each TG, being able to correctly predict the activation of the first key events of the skin sensitization AOP. These results are also confirmed and supported by previous in vivo studies assessing the fourth and last AOP key event, namely lymphocyte proliferation in the local lymph nodes draining the site of test substance application. Indeed, FLG and GO17 as well as GNPs56 are non-sensitizers, according to OECD TG 442B. These results, together with previous studies reporting the inability of a wide range of GBMs (including the same materials tested in the present manuscript) to induce skin irritation and skin corrosion through the adoption of the relevant OECD TGs,13,14 highlight a high biocompatibility of these materials with the skin and represent a significant advancement in the characterization of their safety profile at the cutaneous level.Beside this important aspect, a central outcome of this study is represented by the careful evaluation of OECD TG suitability to predict skin sensitization of GBMs. In general, being originally validated for chemicals, several technical limitations can be expected when health-related OECD TGs are adopted for nanomaterials because of their peculiar and diverse physicochemical properties, as clearly reported by the OECD WPMN, already established in 2006.65 In this scenario, the data reported in our study represent a significant gain of knowledge, considering GBMs as representative 2D nanomaterials. In particular, we clearly demonstrated that several cautions should be taken when adopting the in chemico/in vitro OECD TGs for skin sensitization prediction of GBMs. In particular: (i) even though OECD TG 442C can be slightly modified to overcome some technical limitations associated with GBM testing, it anyway cannot be adopted, since the results could lead to false positive prediction of skin sensitization properties given the ability of GBMs to spontaneously oxidize cysteine; (ii) regarding OECD TG 442D, few minor modifications to the protocol should be introduced before its adoption for GBM testing, including assay substitution for cell viability assessment and introducing washing steps to avoid interferences between the assays and the materials; and (iii) OECD TG 442E can be adopted for GBMs with few minor modifications to the procedure, such as particular cautions related to the dose-finding step associated with the cell viability assay and the careful check of GBM dispersion stability.
On the whole, these results represent significant progress in the hazard characterization of GBMs at the skin level, being particularly useful not only for assessing the safety profile of skin-related GBM-based nanotechnologies, but also for improving the knowledge on GBM testing to obtain reliable, reproducible and consistent toxicological data.
Author contributions
M. Carlin: investigation, formal analysis, data curation, and writing – original draft; M. Morant-Giner: investigation and writing – original draft; M. Garrido: investigation and writing – original draft; S. Sosa: formal analysis and writing – review & editing; A. Bianco: formal analysis and writing – review & editing; A. Tubaro: writing – review & editing; M. Prato: funding acquisition, project administration, and writing – review & editing; M. Pelin: conceptualization, supervision, data curation, and writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the European Commission Graphene Flagship Core 3 (grant agreement no. 881603). M. M.-G. thanks the Margarita Salas grant (MS21-041) from the Universitat de València, funded by the Spanish Ministry of Science and European Union (NextGenerationEU). The authors are grateful to the Flow Cytometry Service of the Department of Life Sciences of the University of Trieste (Prof. Sabrina Pacor). The graphics for the Graphical Abstract were created using BioRender.com.References
- S. S. Ashok Kumar, S. Bashir, K. Ramesh and S. Ramesh, J. Mater. Sci., 2022, 57, 12236–12278 CrossRef CAS.
- T. Schmaltz, L. Wormer, U. Schmoch and H. Döscher, 2D Mater., 2024, 11, 022002 CrossRef.
- P. Wick, A. E. Louw-Gaume, M. Kucki, H. F. Krug, K. Kostarelos, B. Fadeel, K. A. Dawson, A. Salvati, E. Vázquez, L. Ballerini, M. Tretiach, F. Benfenati, E. Flahaut, L. Gauthier, M. Prato and A. Bianco, Angew. Chem., Int. Ed., 2014, 53, 7714–7718 CrossRef CAS PubMed.
- H. Huang, H. Shi, P. Das, J. Qin, Y. Li, X. Wang, F. Su, P. Wen, S. Li, P. Lu, F. Liu, Y. Li, Y. Zhang, Y. Wang, Z.-S. Wu and H.-M. Cheng, Adv. Funct. Mater., 2020, 30, 1909035 CrossRef CAS.
- S. Song, H. Shen, Y. Wang, X. Chu, J. Xie, N. Zhou and J. Shen, Colloids Surf., B, 2020, 185, 110596 CrossRef CAS PubMed.
- A. Shariati, S. M. Hosseini, Z. Chegini, A. Seifalian and M. R. Arabestani, Biomed. Pharmacother., 2023, 158, 114184 CrossRef CAS PubMed.
- T. Das, B. K. Sharma, A. K. Katiyar and J.-H. Ahn, J. Semicond., 2018, 39, 011007 CrossRef.
- N. Karim, S. Afroj, D. Leech and A. M. Abdelkader, in Oxide Electronics, John Wiley & Sons, Ltd, 2021, pp. 21–49 Search PubMed.
- S. Chen, K. Jiang, Z. Lou, D. Chen and G. Shen, Adv. Mater. Technol., 2018, 3, 1700248 CrossRef.
- Y. Qiao, X. Li, J. Jian, Q. Wu, Y. Wei, H. Shuai, T. Hirtz, Y. Zhi, G. Deng, Y. Wang, G. Gou, J. Xu, T. Cui, H. Tian, Y. Yang and T.-L. Ren, ACS Appl. Mater. Interfaces, 2020, 12, 49945–49956 CrossRef CAS PubMed.
- M. Pelin, S. Sosa, M. Prato and A. Tubaro, Nanoscale, 2018, 10, 15894–15903 RSC.
- H. Lin, T. Buerki-Thurnherr, J. Kaur, P. Wick, M. Pelin, A. Tubaro, F. C. Carniel, M. Tretiach, E. Flahaut, D. Iglesias, E. Vázquez, G. Cellot, L. Ballerini, V. Castagnola, F. Benfenati, A. Armirotti, A. Sallustrau, F. Taran, M. Keck, C. Bussy, S. Vranic, K. Kostarelos, M. Connolly, J. M. Navas, F. Mouchet, L. Gauthier, J. Baker, B. Suarez-Merino, T. Kanerva, M. Prato, B. Fadeel and A. Bianco, ACS Nano, 2024, 18, 6038–6094 CrossRef CAS PubMed.
- L. Fusco, M. Garrido, C. Martín, S. Sosa, C. Ponti, A. Centeno, B. Alonso, A. Zurutuza, E. Vázquez, A. Tubaro, M. Prato and M. Pelin, Nanoscale, 2020, 12, 610–622 RSC.
- M. Carlin, M. Garrido, S. Sosa, A. Tubaro, M. Prato and M. Pelin, Nanoscale, 2023, 15, 14423–14438 RSC.
- S. P. Mukherjee, M. Bottini and B. Fadeel, Front. Immunol., 2017, 8, 673 CrossRef PubMed.
- S. Achawi, J. Pourchez, B. Feneon and V. Forest, Chem. Res. Toxicol., 2021, 34, 2003–2018 Search PubMed.
- S. Sosa, A. Tubaro, M. Carlin, C. Ponti, E. Vázquez, M. Prato and M. Pelin, NanoImpact, 2023, 29, 100448 Search PubMed.
- M. Pelin, L. Fusco, V. León, C. Martín, A. Criado, S. Sosa, E. Vázquez, A. Tubaro and M. Prato, Sci. Rep., 2017, 7, 40572 Search PubMed.
- M. Pelin, H. Lin, A. Gazzi, S. Sosa, C. Ponti, A. Ortega, A. Zurutuza, E. Vázquez, M. Prato, A. Tubaro and A. Bianco, Nanomaterials, 2020, 10, 1602 Search PubMed.
- L. Fusco, M. Pelin, S. Mukherjee, S. Keshavan, S. Sosa, C. Martín, V. González, E. Vázquez, M. Prato, B. Fadeel and A. Tubaro, Carbon, 2020, 159, 598–610 CrossRef CAS.
- M. Pelin, L. Fusco, C. Martín, S. Sosa, J. Frontiñán-Rubio, J. M. González-Domínguez, M. Durán-Prado, E. Vázquez, M. Prato and A. Tubaro, Nanoscale, 2018, 10, 11820–11830 RSC.
- J. Frontiñan-Rubio, M. V. Gomez, V. J. González, M. Durán-Prado and E. Vázquez, Sci. Rep., 2020, 10, 18407 CrossRef PubMed.
- J. Frontiñan-Rubio, E. Llanos-González, V. J. González, E. Vázquez and M. Durán-Prado, J. Proteome Res., 2022, 21, 1675–1685 CrossRef PubMed.
- B. Salesa and Á. Serrano-Aroca, Coatings, 2021, 11, 414 CrossRef CAS.
- E. Hashemi, O. Akhavan, M. Shamsara, S. A. Majd, M. H. Sanati, M. D. Joupari and A. Farmany, Int. J. Nanomed., 2020, 15, 6201–6209 CrossRef CAS PubMed.
- Z. Zheng, A. Halifu, J. Ma, L. Liu, Q. Fu, B. Yi, E. Du, D. Tian, Y. Xu, Z. Zhang and J. Zhu, Environ. Pollut., 2023, 330, 121817 CrossRef CAS PubMed.
- M. Divkovic, C. K. Pease, G. F. Gerberick and D. A. Basketter, Contact Dermatitis, 2005, 53, 189–200 CrossRef CAS PubMed.
- N. S. Sharma, R. Jindal, B. Mitra, S. Lee, L. Li, T. J. Maguire, R. Schloss and M. L. Yarmush, Cell. Mol. Bioeng., 2012, 5, 52–72 CrossRef CAS PubMed.
- REACH Legislation – ECHA, https://echa.europa.eu/regulations/reach/legislation, (accessed March 10, 2023).
- OECD, Test No. 439: In Vitro Skin Irritation: Reconstructed Human Epidermis Test Method, Organisation for Economic Co-operation and Development, Paris, 2021 Search PubMed.
- OECD, Test No. 431: In vitro skin corrosion: reconstructed human epidermis (RHE) test method, Organisation for Economic Co-operation and Development, Paris, 2019 Search PubMed.
- OECD, Test No. 406: Skin Sensitisation, Organisation for Economic Co-operation and Development, Paris, 2022 Search PubMed.
- OECD, Test No. 498: In vitro Phototoxicity – Reconstructed Human Epidermis Phototoxicity test method, Organisation for Economic Co-operation and Development, Paris, 2023 Search PubMed.
- OECD, Preliminary Review of OECD Test Guidelines for Their Applicability to Manufactured Nanomaterials, Organisation for Economic Co-operation and Development, Paris, 2009 Search PubMed.
- M. Pelin, C. Passerino, A. Rodríguez-Garraus, M. Carlin, S. Sosa, S. Suhonen, G. Vales, B. Alonso, A. Zurutuza, J. Catalán and A. Tubaro, Nanomaterials, 2023, 13, 2189 CrossRef CAS PubMed.
- OECD, Test No. 442C: In Chemico Skin Sensitisation: Assays addressing the Adverse Outcome Pathway key event on covalent binding to proteins, Organisation for Economic Co-operation and Development, Paris, 2024 Search PubMed.
- OECD, Test No. 442D: In Vitro Skin Sensitisation: Assays addressing the Adverse Outcome Pathway Key Event on Keratinocyte activation, Organisation for Economic Co-operation and Development, Paris, 2024 Search PubMed.
- ISO 19007, https://www.iso.org/standard/63698.html, (accessed December 20, 2022).
- OECD, Test No. 442E: In Vitro Skin Sensitisation: In Vitro Skin Sensitisation assays addressing the Key Event on activation of dendritic cells on the Adverse Outcome Pathway for Skin Sensitisation, Organisation for Economic Co-operation and Development, Paris, 2023 Search PubMed.
- L. Díaz, E. Zambrano, M. E. Flores, M. Contreras, J. C. Crispín, G. Alemán, C. Bravo, A. Armenta, V. J. Valdés, A. Tovar, G. Gamba, J. Barrios-Payán and N. A. Bobadilla, Rev. Invest. Clin., 2020, 73, 199–209 Search PubMed.
- OECD, Guideline No. 497: Defined Approaches on Skin Sensitisation, Organisation for Economic Co-operation and Development, Paris, 2023 Search PubMed.
- OECD, Test No. 429: Skin Sensitisation: Local Lymph Node Assay, Organisation for Economic Co-operation and Development, Paris, 2010 Search PubMed.
- D. W. Roberts, Crit. Rev. Toxicol., 2022, 52, 420–430 CrossRef CAS PubMed.
- M. Hu, Z. Yao, X. Wang, M. Hu, Z. Yao and X. Wang, AIMS Mater. Sci., 2017, 4, 755–788 Search PubMed.
- B. Ma, S. Guo, Y. Nishina and A. Bianco, ACS Appl. Mater. Interfaces, 2021, 13, 3528–3535 CrossRef CAS PubMed.
- S. Liu, M. Hu, T. H. Zeng, R. Wu, R. Jiang, J. Wei, L. Wang, J. Kong and Y. Chen, Langmuir, 2012, 28, 12364–12372 CrossRef CAS PubMed.
- M.-H. Morel, J. Bonicel, V. Micard and S. Guilbert, J. Agric. Food Chem., 2000, 48, 186–192 CrossRef CAS PubMed.
- X. Zhao, A. Kong, X. Zhang, C. Shan, H. Ding and Y. Shan, Catal. Lett., 2010, 135, 291–294 CrossRef CAS.
- Y. Zhang, C. Wu, S. Guo and J. Zhang, Nanotechnol. Rev., 2013, 2, 27–45 CAS.
- S. F. Bezerra, B. dos Santos Rodrigues, A. C. G. da Silva, R. I. de Ávila, H. R. G. Brito, E. R. Cintra, D. F. M. C. Veloso, E. M. Lima and M. C. Valadares, Contact Dermatitis, 2021, 84, 67–74 CrossRef CAS PubMed.
- E.-N. Kim, J.-A. Seo, B.-H. Kim and G.-S. Jeong, Toxicol. Res., 2023, 39, 485–495 CrossRef CAS PubMed.
- T. U. Noh and A. Abdul-Aziz, J. Res. Nanosci. Nanotechnol., 2021, 4, 1–12 Search PubMed.
- K.-H. Liao, Y.-S. Lin, C. W. Macosko and C. L. Haynes, ACS Appl. Mater. Interfaces, 2011, 3, 2607–2615 CrossRef CAS PubMed.
- J. Yang, Z. Zhang, W. Pang, H. Chen and G. Yan, Sens. Actuators, B, 2019, 301, 127014 CrossRef CAS.
- M. Garrido, E. Martínez-Periñán, J. Calbo, L. Rodríguez-Pérez, J. Aragó, E. Lorenzo, E. Ortí, N. Martín and M. Á. Herranz, J. Mater. Chem. C, 2021, 9, 10944–10951 RSC.
- S.-H. Kim, S.-H. Hong, J. H. Lee, D. H. Lee, K. Jung, J.-Y. Yang, H.-S. Shin, J. Lee, J. Jeong and J.-H. Oh, Toxics, 2021, 9, 62 CrossRef CAS PubMed.
- S.-H. Kim, D. H. Lee, J. H. Lee, J.-Y. Yang, H.-S. Shin, J. Lee, K. Jung, J. Jeong, J.-H. Oh and J. K. Lee, Toxics, 2020, 8, 122 CrossRef CAS PubMed.
- H. Parker, A. Maria Gravagnuolo, S. Vranic, L. Elena Crica, L. Newman, O. Carnell, C. Bussy, R. S. Dookie, E. Prestat, S. J. Haigh, N. Lozano, K. Kostarelos and A. S. MacDonald, Nanoscale, 2022, 14, 17297–17314 RSC.
- S. V. Uzhviyuk, M. S. Bochkova, V. P. Timganova, P. V. Khramtsov, K. Yu. Shardina, M. D. Kropaneva, A. I. Nechaev, M. B. Raev and S. A. Zamorina, Bull. Exp. Biol. Med., 2022, 172, 664–670 CrossRef CAS PubMed.
- A. V. Tkach, N. Yanamala, S. Stanley, M. R. Shurin, G. V. Shurin, E. R. Kisin, A. R. Murray, S. Pareso, T. Khaliullin, G. P. Kotchey, V. Castranova, S. Mathur, B. Fadeel, A. Star, V. E. Kagan and A. A. Shvedova, Small, 2013, 9, 1686–1690 CrossRef CAS PubMed.
- Z. Yang, Y. Pan, T. Chen, L. Li, W. Zou, D. Liu, D. Xue, X. Wang and G. Lin, Front. Pharmacol., 2020, 11, 1206 CrossRef CAS PubMed.
- M. Pescatori, D. Bedognetti, E. Venturelli, C. Ménard-Moyon, C. Bernardini, E. Muresu, A. Piana, G. Maida, R. Manetti, F. Sgarrella, A. Bianco and L. G. Delogu, Biomaterials, 2013, 34, 4395–4403 CrossRef CAS PubMed.
- E. Koike, H. Takano, K. Inoue, R. Yanagisawa and T. Kobayashi, Chemosphere, 2008, 73, 371–376 CrossRef CAS PubMed.
- D. Yang, Y. Zhao, H. Guo, Y. Li, P. Tewary, G. Xing, W. Hou, J. J. Oppenheim and N. Zhang, ACS Nano, 2010, 4, 1178–1186 CrossRef CAS PubMed.
- K. Rasmussen, H. Rauscher, P. Kearns, M. González and J. Riego Sintes, Regul. Toxicol. Pharmacol., 2019, 104, 74–83 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nr00307e |
| This journal is © The Royal Society of Chemistry 2025 |