
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Beyond serendipity: uncovering novel ratiometric urea·24DHBA cocrystals through mechanochemistry and MicroED
Ankur
Bonia
a,
Diptajyoti
Gogoi
a,
Nabadeep
Kalita
a,
Takanori
Nakane
 bc,
Akihiro
Kawamoto
bc,
Khaled
Althubeiti
d,
Toshiyuki
Sasaki
*e,
Genji
Kurisu
*bcf and
Ranjit
Thakuria
*ag
bc,
Akihiro
Kawamoto
bc,
Khaled
Althubeiti
d,
Toshiyuki
Sasaki
*e,
Genji
Kurisu
*bcf and
Ranjit
Thakuria
*ag
aDepartment of Chemistry, Gauhati University, Guwahati 781014, India. E-mail: ranjit.thakuria@gauhati.ac.in; ranjit.thakuria@gmail.com
bInstitute for Protein Research, Osaka University, 3-2 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: gkurisu@protein.osaka-u.ac.jp
cJEOL YOKOGUSHI Research Alliance Laboratories, Graduate School of Frontier Biosciences, Osaka University, 1-3 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: gkurisu@protein.osaka-u.ac.jp
dDepartment of Chemistry, College of Science, Taif University, Taif 21944, Saudi Arabia
eJapan Synchrotron Radiation Research Institute (JASRI), 1-1-1 Kouto, Sayo-cho, Sayo-gun, Hyogo 679-5198, Japan. E-mail: toshiyuki.sasaki@spring8.or.jp
fInstitute for Open and Transdisciplinary Research Initiatives, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan
gFaculty of Chemistry, University of Warsaw, 1 Pasteura Street, 02-093 Warsaw, Poland
First published on 3rd September 2025
Abstract
Mechanochemical synthesis of urea cocrystals with 2,4-dihydroxybenzoic acid (24DHBA) reveals two new stoichiometric forms: (urea)2·(24DHBA) and (urea)2·(24DHBA)2, not accessible via solution methods. The known (urea)0.5·(24DHBA) form remains the most stable. These findings provide critical insights into stoichiometric tuning of agro-cocrystals and pave the way for their practical application in sustainable agriculture.
Impurities pose a significant challenge not only during the synthesis but also throughout large-scale manufacturing.1,2 Their presence, whether as unwanted polymorphs, by-products, or unreacted starting materials, can adversely affect the physicochemical properties of the final product. This, in turn, may lead to batch rejection or, in severe cases, market withdrawal.3 The importance of impurity identification is thus paramount during process development and scale-up manufacturing.4 Well-known cases, such as the metastable polymorph of ritonavir,3 or rifaximin,5,6 highlight the critical impact that even trace impurities can have on drug performance and regulatory acceptance. To mitigate such risks, it is a standard practice to explore both the crystal structure landscape and the energy landscape of disappearing polymorphs,7–11 alongside the investigation of various solid-state formulations12 such as cocrystals, solvates, hydrates, and reaction intermediates.13 These strategies are routinely employed during scale-up manufacturing.3,14–19 While such approaches are well-established for single-component systems, the complexity increases considerably when dealing with multicomponent systems like cocrystals. This complexity arises from the wide range of possible solid forms generated during co-crystallization, including cocrystal hydrates, cocrystal solvates, salt–cocrystal hybrids, salt solvates,20 ratiometric cocrystals,21,22 and polymorphs of all possible multi-component solids,23 among others.24–28
The spontaneity of cocrystal nucleation is closely tied to the activation energy (Ea) of the system. Typically, solution crystallization favors the formation of thermodynamically stable cocrystals. In contrast, alternative techniques such as melt crystallization, sublimation, supercritical CO2 anti-solvent methods, freeze-drying, and mechanochemistry can often yield kinetically stable forms.29,30 This distinction has been well-articulated in a recent highlight by Wong et al.4 Recent investigations have increasingly focused on agro-based cocrystals due to their potential application as alternative fertilizers with sustained-release properties.31–33 As a result, the development of scalable synthesis methods for these materials is essential for enabling field trials. Among the various approaches, mechanochemical synthesis has emerged as one of the most efficient and environmentally benign techniques for large-scale preparation.8,11,19,26,28,30
In continuation to one of our recent studies34 on mechanochemical synthesis and sustained release behavior of urea–hydroxybenzoic acid cocrystals, we report here the discovery of two previously elusive urea·24DHBA cocrystals. Through high-throughput mechanochemical screening combined with microcrystal electron diffraction (MicroED), an efficient tool for small molecule characterization,35 we were able to resolve their crystal structures and determine their precise stoichiometries. Furthermore, we systematically explored the influence of various mechanochemical parameters such as milling time, milling frequency, nature and volume of liquid additives (η), and reactant stoichiometry on the product formation. These studies provide critical insights into the mechanistic aspects of cocrystal formation and open up new avenues for the rational design of agro-based cocrystal materials.
In our previous report, we characterized a urea cocrystal with 24DHBA, synthesized via mechanochemistry. The resulting cocrystal, with a stoichiometry of (urea)0.5·(24DHBA), demonstrates high stability under 75% relative humidity as well as during solubility analysis. Notably, it does not undergo any phase transformation or cocrystal dissociation under various environmental conditions. Since solution crystallization from even a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric mixture consistently yields a 0.5:1 cocrystal, we reinvestigated the potential for stoichiometric modulation using liquid-assisted grinding (LAG) (Scheme 1). Powder X-ray diffraction (PXRD) of ground samples prepared in a Retsch Mixer Mill (MM400) by mixing 1:1 stoichiometry of urea (20.2 mg, 0.33 mmol) and 24DHBA (51.37 mg, 0.33 mmol) in a 5 mL stainless-steel milling jar along with two 7 mm stainless-steel grinding balls in the presence of 150 μL (η = 2.1) of various added liquids for 30 min at a frequency of 20 Hz showed the appearance of a few new diffraction peaks (Fig. S1). After several batches of failed crystallization, MicroED was used as a characterization tool to determine the crystal structure of the elusive 1:1 cocrystal.
:1 stoichiometric mixture consistently yields a 0.5:1 cocrystal, we reinvestigated the potential for stoichiometric modulation using liquid-assisted grinding (LAG) (Scheme 1). Powder X-ray diffraction (PXRD) of ground samples prepared in a Retsch Mixer Mill (MM400) by mixing 1:1 stoichiometry of urea (20.2 mg, 0.33 mmol) and 24DHBA (51.37 mg, 0.33 mmol) in a 5 mL stainless-steel milling jar along with two 7 mm stainless-steel grinding balls in the presence of 150 μL (η = 2.1) of various added liquids for 30 min at a frequency of 20 Hz showed the appearance of a few new diffraction peaks (Fig. S1). After several batches of failed crystallization, MicroED was used as a characterization tool to determine the crystal structure of the elusive 1:1 cocrystal.
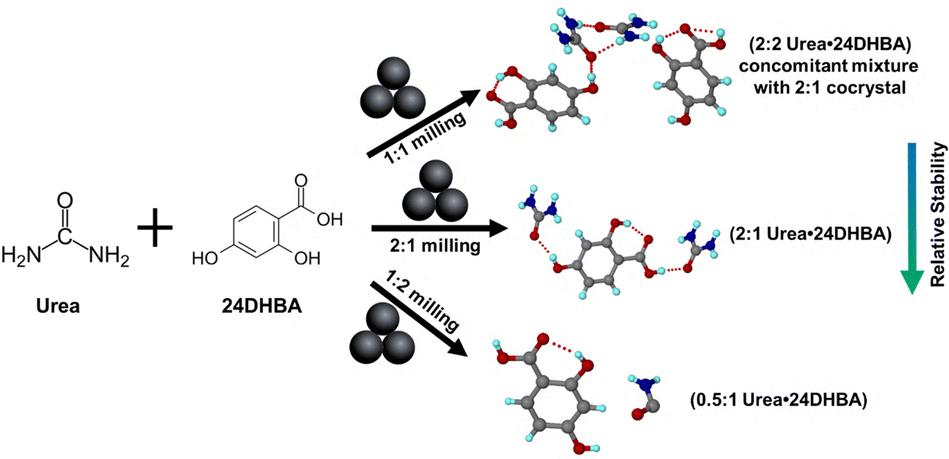 | ||
| Scheme 1 Formation of ratiometric cocrystals under different milling environments, summarizing the present study and our previous report.34 | ||
In the reported (urea)0.5·(24DHBA) cocrystal, the asymmetric unit comprises half a molecule of urea and one molecule of 24DHBA (Fig. 1(a)). The crystal structure was solved in the orthorhombic Pnma space group, with no direct interactions observed between urea molecules. Instead, the two amino groups and the carbonyl group of urea engage in hydrogen bonding with the para-hydroxyl group of 24DHBA, forming an infinite molecular tape parallel to the a-axis. The carboxylic acid groups of 24DHBA form centrosymmetric acid–acid homodimers, which link antiparallel urea–24DHBA tapes from different molecular planes, resulting in a three-dimensional (3D) network structure (Fig. 1(b)).
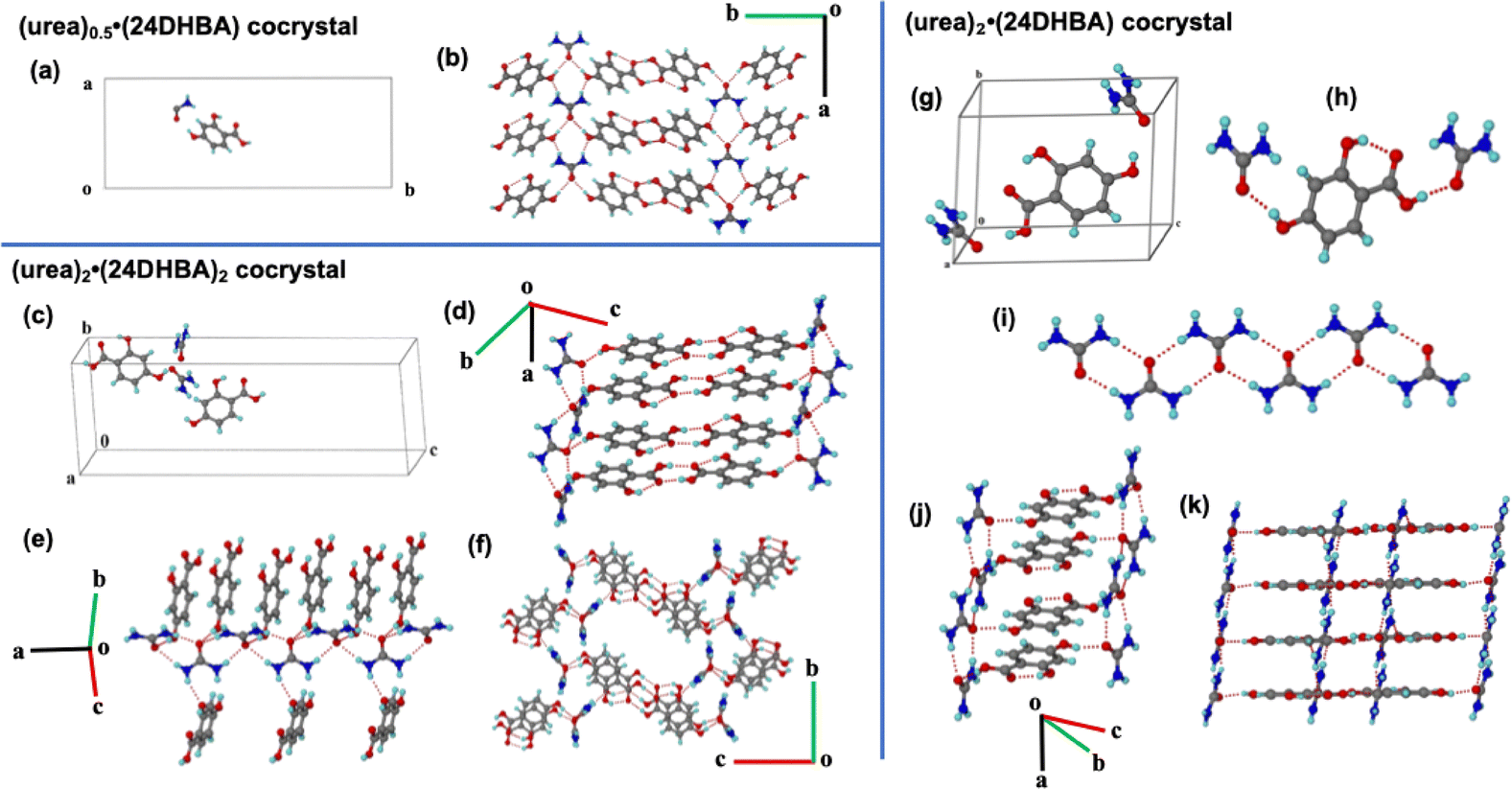 | ||
| Fig. 1 Crystal packing and hydrogen bond interactions present in the variable stoichiometric cocrystals of urea with 24DHBA, respectively (a) and (b) (urea)0.5·(24DHBA); (c)–(f) (urea)2·(24DHBA)2 and (g)–(k) (urea)2·(24DHBA). | ||
In contrast, MicroED structural analysis of the 1:1 stoichiometric powder mixture revealed the presence of two molecules each of urea and 24DHBA in the asymmetric unit (Fig. 1(c)). This new cocrystal, henceforth referred to as (urea)2·(24DHBA)2, crystallizes in the P21/n space group with unit cell parameters a = 6.9711 Å, b = 11.059 Å, c = 31.307 Å, α = 90°, β = 91.70°, γ = 90°, and V = 2412.6 Å3. Unlike the previously reported structure, this form features an infinite urea tape along the a-axis, in which urea molecules are connected via an amide–amide dimer synthon (Fig. 1(e)). The two symmetry-independent 24DHBA molecules link the urea tape through O–H⋯O and O⋯H–N hydrogen bonds via their hydroxyl groups. Parallel urea tapes are further bridged by these 24DHBA molecules through acid–acid homodimers, forming a layered structure. In 3D, the 24DHBA dimer units connect adjacent urea tapes, resulting in a 3D molecular grid architecture (Fig. 1(f)).
Interestingly, during MicroED structure elucidation of mechanically ground powder samples of the 1:1 urea–24DHBA mixture, some crystals had different unit cell parameters. These correspond to a 2:1 cocrystal, hereafter referred to as (urea)2·(24DHBA). The crystal structure, solved in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, contains two molecules of urea and one molecule of 24DHBA in the asymmetric unit (Fig. 1(g) and (h)). The unit cell parameters are a = 7.2558 Å, b = 8.8651 Å, c = 10.860 Å, α = 74.412°, β = 75.956°, γ = 71.464°, with a unit cell volume of V = 628.4 Å3. Similar to the (urea)2·(24DHBA)2 cocrystal, this (urea)2·(24DHBA) structure features infinite tapes of urea molecules aligned parallel to the a-axis, formed via amide–amide dimer synthons (Fig. 1(i)). However, unlike the other forms, this cocrystal does not exhibit the characteristic acid–acid dimer synthon. Instead, the carboxylic acid and para-hydroxyl groups of the 24DHBA coformer bridge two symmetry-independent urea molecules from opposite directions, forming a two-dimensional ladder-like motif (Fig. 1(j)). Additionally, an intramolecular hydrogen bond between the 2-hydroxyl group and the carbonyl oxygen of 24DHBA facilitates further linkage of neighboring urea tapes, resulting in a 2D square grid network (Fig. 1(k)). Crystallographic and hydrogen bond parameters of all the urea·24DHBA cocrystals are summarized in Tables S1 and S2, respectively.
space group, contains two molecules of urea and one molecule of 24DHBA in the asymmetric unit (Fig. 1(g) and (h)). The unit cell parameters are a = 7.2558 Å, b = 8.8651 Å, c = 10.860 Å, α = 74.412°, β = 75.956°, γ = 71.464°, with a unit cell volume of V = 628.4 Å3. Similar to the (urea)2·(24DHBA)2 cocrystal, this (urea)2·(24DHBA) structure features infinite tapes of urea molecules aligned parallel to the a-axis, formed via amide–amide dimer synthons (Fig. 1(i)). However, unlike the other forms, this cocrystal does not exhibit the characteristic acid–acid dimer synthon. Instead, the carboxylic acid and para-hydroxyl groups of the 24DHBA coformer bridge two symmetry-independent urea molecules from opposite directions, forming a two-dimensional ladder-like motif (Fig. 1(j)). Additionally, an intramolecular hydrogen bond between the 2-hydroxyl group and the carbonyl oxygen of 24DHBA facilitates further linkage of neighboring urea tapes, resulting in a 2D square grid network (Fig. 1(k)). Crystallographic and hydrogen bond parameters of all the urea·24DHBA cocrystals are summarized in Tables S1 and S2, respectively.
We investigated the formation of different stoichiometries by varying the amount of the organic liquid, THF, during LAG. Milling equimolar (1:1) amounts of urea and 24DHBA in the presence of varying amounts of THF yielded different product mixtures. Specifically, using THF volumes corresponding to η values between 0.54 and 2.1 resulted in a mixture of reported (urea)0.5·(24DHBA) and (urea)2·(24DHBA) cocrystals (Fig. S2a). In contrast, η values between 2.8 and 8.4 led to a mixture of (urea)2·(24DHBA)2 and (urea)2·(24DHBA) cocrystals (Fig. S2b). Interestingly, milling with a 2:1 stoichiometric ratio of urea to 24DHBA under various THF volumes consistently yielded a pure phase of the resulting material, i.e. a (urea)2·(24DHBA) cocrystal (Fig. S2c).
The hydration stability of the synthesized urea cocrystals was evaluated by storing the powder samples in a bell jar at 75% RH (achieved using a saturated aqueous sodium chloride (NaCl) solution) and ambient temperature. PXRD analysis was conducted periodically over a duration of 45 days. No significant changes were observed in the PXRD patterns of the phase pure (urea)0.5·(24DHBA) and (urea)2·(24DHBA) cocrystals upon prolonged storage, even after 45 days. Due to the challenges in obtaining a bulk pure sample of the (urea)2·(24DHBA)2 cocrystal, a powder mixture containing both (urea)2·(24DHBA)2 and (urea)2·(24DHBA) cocrystals was used. PXRD analysis revealed the emergence of peaks corresponding to the reported (urea)0.5·(24DHBA) cocrystal after 30 days, indicating a phase transformation and metastable nature of the (urea)2·(24DHBA)2 cocrystal (Fig. S3).
Hirshfeld surface analysis is a powerful tool to explore intermolecular interactions within a crystal structure, and visualize and quantify how molecules pack together and interact in the solid-state.36–39 The parameters di and de signify the distance from the Hirshfeld surface to the nearest internal and external atoms, respectively. And the normalized contact distance dnorm is defined as
| dnorm = d∣e∣ + d∣i∣ |
To investigate the relative stability of the ratiometric cocrystals, we performed Hirshfeld surface analysis on all the urea·24DHBA cocrystals and generated their corresponding 2D fingerprint plots (Fig. S4). By quantifying the contributions of various intermolecular interactions (Fig. 2), we observed that the O⋯H hydrogen bonding interactions (blue bar) exhibited the highest contribution of strong non-covalent interactions to the Hirshfeld surface for all cocrystals analyzed. Specifically, the (urea)0.5·(24DHBA) cocrystal showed the greatest contribution from O⋯H interactions at 39.3%, followed by (urea)2·(24DHBA) at 37.7%, and the lowest in the (urea)2·(24DHBA)2 cocrystal at 31.2%. These results highlight a trend in which the strength and prevalence of O⋯H interactions correlate with the relative thermodynamic stability of the cocrystals, i.e thermodynamic stability of the reported (urea)0.5·(24DHBA) cocrystal and metastable nature of the (urea)2·(24DHBA)2 cocrystal. Differential scanning calorimetry (DSC) analysis revealed a consistent trend, with the highest melting peak observed for the (urea)0.5·(24DHBA) cocrystal, which also contained trace amounts of the other two stoichiometric cocrystals as impurities. In contrast, the lowest melting point corresponded to the metastable (urea)2·(24DHBA)2 cocrystal (Fig. S5).
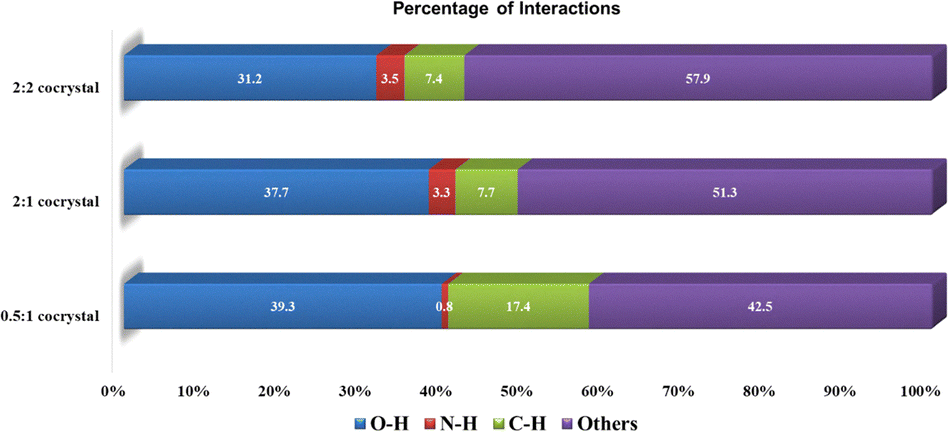 | ||
| Fig. 2 Percentage contributions of various intermolecular interactions present in the (urea)0.5·(24DHBA), (urea)2·(24DHBA) and (urea)2·(24DHBA)2 cocrystals calculated based on Hirshfeld surface analysis fingerprint plots. | ||
In summary, mechanochemical milling has enabled the discovery of two previously unreported ratiometric cocrystals alongside the well-established (urea)0.5·(24DHBA) form. While conventional solution-based crystallization consistently yields this thermodynamically stable 0.5:1 cocrystal, the integration of mechanochemistry with MicroED revealed (Fig. S6 and S7) two elusive forms and allowed their structural characterization. Hirshfeld surface analysis and stability assessments confirmed that (urea)0.5·(24DHBA) is the most stable form, with (urea)2·(24DHBA)2 identified as metastable. The markedly higher aqueous solubility of urea relative to 24DHBA likely drives the selective formation of the 0.5:1 cocrystal during solution crystallization, even when starting from 1:1 or 2:1 stoichiometric mixtures. Interestingly, the metastable (urea)2·(24DHBA)2 cocrystal was only observed during mechanochemical milling, consistently appearing as a minor, concomitant phase alongside the 1:2 and 2:1 cocrystals. However, extensive mechanochemical milling ultimately revealed the optimal conditions for the selective synthesis of the phase-pure (urea)2·(24DHBA) cocrystal. These findings highlight a critical consideration for the large-scale production of cocrystals and other multicomponent solids: the necessity of identifying and controlling potential phase impurities. Our work underscores the value of comprehensive high-throughput screening across various stoichiometries and polymorphs, supplemented by detailed mapping of the energy landscape. Such an approach is essential to ensure batch consistency and to mitigate risks associated with unexpected phase impurities, including ratiometric cocrystals and undesired polymorphic forms.
In this context, the combined mechanochemistry and MicroED proves to be a highly sensitive and effective characterization strategy, capable of detecting even nanomolar-scale molecular impurities. This approach is especially valuable for quality assurance in the industrial-scale production of pharmaceutical and agrochemical multicomponent solids.
R. T. thanks the Polish National Agency for Academic Exchange (Application no. BNI/ULM/2024/1/00042) for their support to work as an Ulam NAWA fellow at Faculty of Chemistry, University of Warsaw, Poland; and also thankfully acknowledges Crystallography in India Trust for donating Retsch Mixter Mill MM400. K. A. extends his appreciation to Taif University, Saudi Arabia, for supporting this work through project number (TU-DSPP-2024-59). T. S. thanks JSPS KAKENHI Grant Number JP22K05054. The electron microscope was partly supported by Research Support Project for Life Science and Drug Discovery (BINDS) from AMED under Grant Number JP24ama121001 and JP25ama121001. MicroED was performed under the Collaborative Research Program of Institute for Protein Research, Osaka University, MEDCR-24-02 and MEDCR-25-02. We thankfully acknowledge the DST-FIST program for supporting the Department of Chemistry, GU for the Rigaku powder X-ray diffractometer, basic instrumentation facility, and infrastructure.
Conflicts of interest
There are no conflicts to declare.Data availability
The detailed protocols and scripts for MicroED are in our GitHub repository (https://github.com/GKLabIPR/MicroED). Raw diffraction images are available at XRDa (ID: 390).Supplementary information: Methods, PXRD data, crystallographic parameters, 2D fingerprint plots, and MicroED data. See DOI: https://doi.org/10.1039/d5cc03628c.
CCDC 2465561 (24DHBA), 2465562 ((urea)2·(24DHBA)) and 2465472 ((urea)2·(24DHBA)2) contain the supplementary crystallographic data for this paper.40a–c Coordinates are also available from Crystallographic Open Database (accession: 3000604, 3000605, 3000606).
Notes and references
- W. Kras, A. Carletta, R. Montis, R. A. Sullivan and A. J. Cruz-Cabeza, Commun. Chem., 2021, 4, 38 CrossRef CAS PubMed.
- D. Zheltikova, E. Losev and E. Boldyreva, CrystEngComm, 2023, 25, 4879–4888 RSC.
- S. R. Chemburkar, J. Bauer, K. Deming, H. Spiwek, K. Patel, J. Morris, R. Henry, S. Spanton, W. Dziki, W. Porter, J. Quick, P. Bauer, J. Donaubauer, B. A. Narayanan, M. Soldani, D. Riley and K. McFarland, Org. Process Res. Dev., 2000, 4, 413–417 CrossRef CAS.
- S. N. Wong, M. Fu, S. Li, W. T. C. Kwok, S. Chow, K.-H. Low and S. F. Chow, CrystEngComm, 2024, 26, 1505–1526 RSC.
- G. C. Viscomi, M. Campana, M. Barbanti, F. Grepioni, M. Polito, D. Confortini, G. Rosini, P. Righi, V. Cannata and D. Braga, CrystEngComm, 2008, 10, 1074–1081 RSC.
- A. Xu, Y. Xue, Y. Zeng, J. Li, H. Zhou, Z. Wang, Y. Chen, H. Chen, J. Jin and T. Zhuang, Molecules, 2023, 28(5), 2415 CrossRef CAS.
- D.-K. Bučar, R. W. Lancaster and J. Bernstein, Angew. Chem., Int. Ed., 2015, 54, 6972–6993 CrossRef PubMed.
- L. Wang, G. Sun, K. Zhang, M. Yao, Y. Jin, P. Zhang, S. Wu and J. Gong, ACS Sustainable Chem. Eng., 2020, 8, 16781–16790 CrossRef CAS.
- Y. Park, S. X. M. Boerrigter, J. Yeon, S. H. Lee, S. K. Kang and E. H. Lee, Cryst. Growth Des., 2016, 16, 2552–2560 CrossRef CAS.
- P. Sacchi, S. E. Wright, P. Neoptolemou, G. I. Lampronti, A. K. Rajagopalan, W. Kras, C. L. Evans, P. Hodgkinson and A. J. Cruz-Cabeza, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2319127121 CrossRef CAS PubMed.
- D. Hasa, M. Marosa, D.-K. Bučar, M. K. Corpinot, D. Amin, B. Patel and W. Jones, Cryst. Growth Des., 2020, 20, 1119–1129 CrossRef CAS.
- S. Huang, V. K. R. Cheemarla, D. Tiana and S. E. Lawrence, Cryst. Growth Des., 2023, 23, 5446–5461 CrossRef CAS.
- D. Gogoi, T. Sasaki, T. Nakane, A. Kawamoto, H. Hojo, G. Kurisu and R. Thakuria, Cryst. Growth Des., 2023, 23, 5821–5826 CrossRef CAS.
- C. Heffernan, M. Ukrainczyk, J. Zeglinski, B. K. Hodnett and Å. C. Rasmuson, Cryst. Growth Des., 2018, 18, 4715–4723 CrossRef CAS.
- C. Darmali, S. Mansouri, N. Yazdanpanah and M. W. Woo, Ind. Eng. Chem. Res., 2019, 58, 1463–1479 CrossRef CAS.
- S. J. Urwin, G. Levilain, I. Marziano, J. M. Merritt, I. Houson and J. H. Ter Horst, Org. Process Res. Dev., 2020, 24, 1443–1456 CrossRef CAS PubMed.
- H. A. Moynihan and D. E. Horgan, Org. Process Res. Dev., 2017, 21, 689–704 CrossRef CAS.
- S. Mohamed and L. Li, CrystEngComm, 2018, 20, 6026–6039 RSC.
- S. N. Madanayake, A. Manipura, R. Thakuria and N. M. Adassooriya, Org. Process Res. Dev., 2023, 27, 409–422 CrossRef CAS.
- R. Thakuria and A. Nangia, Cryst. Growth Des., 2013, 13, 3672–3680 CrossRef CAS.
- N. Tumanova, N. Tumanov, F. Fischer, F. Morelle, V. Ban, K. Robeyns, Y. Filinchuk, J. Wouters, F. Emmerling and T. Leyssens, CrystEngComm, 2018, 20, 7308–7321 RSC.
- G. L. Perlovich, Cryst. Growth Des., 2020, 20, 5526–5537 CrossRef CAS.
- S. Aitipamula, P. S. Chow and R. B. H. Tan, CrystEngComm, 2014, 16, 3451–3465 RSC.
- M. R. Ahsan, L. Singh, B. Sar and A. Mukherjee, Cryst. Growth Des., 2024, 24, 1695–1704 CrossRef CAS.
- R. Kaur, S. Cherukuvada, P. B. Managutti and T. N. G. Row, CrystEngComm, 2016, 18, 3191–3203 RSC.
- D. Gogoi, K. J. Kalita, N. Biswakarma, M. Arhangelskis, R. C. Deka and R. Thakuria, RSC Mechanochem., 2024, 1, 452–464 RSC.
- B. Saikia, D. Pathak and B. Sarma, CrystEngComm, 2021, 23, 4583–4606 RSC.
- A. V. Trask, J. van de Streek, W. D. S. Motherwell and W. Jones, Cryst. Growth Des., 2005, 5, 2233–2241 CrossRef CAS.
- Z.-H. Li and W.-S. Kim, Cryst. Growth Des., 2024, 24, 5974–5989 CrossRef CAS.
- L. S. Germann, M. Arhangelskis, M. Etter, R. E. Dinnebier and T. Friščić, Chem. Sci., 2020, 11, 10092–10100 RSC.
- L. Casali, L. Mazzei, O. Shemchuk, L. Sharma, K. Honer, F. Grepioni, S. Ciurli, D. Braga and J. Baltrusaitis, ACS Sustainable Chem. Eng., 2019, 7, 2852–2859 CrossRef CAS.
- N. M. Adassooriya, S. P. Mahanta and R. Thakuria, CrystEngComm, 2022, 24, 1679–1689 RSC.
- N. H. Madanayake, D. Gogoi, G. K. M. G. Jayasuriya, A. T. D. Perera, R. Thakuria and N. M. Adassooriya, ACS Sustainable Chem. Eng., 2025, 13, 9481–9489 CrossRef CAS.
- T. Rajbongshi, S. Parakatawella, D. Gogoi, P. Deka, N. M. Adassooriya and R. Thakuria, RSC Sustain., 2023, 1, 1416–1422 RSC.
- C. G. Jones, M. W. Martynowycz, J. Hattne, T. J. Fulton, B. M. Stoltz, J. A. Rodriguez, H. M. Nelson and T. Gonen, ACS Cent. Sci., 2018, 4, 1587–1592 CrossRef CAS PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627–668 CrossRef PubMed.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814–3816 RSC.
- (a) A. Bonia, D. Gogoi, N. Kalita, T. Nakane, A. Kawamoto, K. Althubeiti, T. Sasaki, G. Kurisu and R. Thakuria, CCDC 2465561: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2nrm79; (b) A. Bonia, D. Gogoi, N. Kalita, T. Nakane, A. Kawamoto, K. Althubeiti, T. Sasaki, G. Kurisu and R. Thakuria, CCDC 2465562: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2nrm8b; (c) A. Bonia, D. Gogoi, N. Kalita, T. Nakane, A. Kawamoto, K. Althubeiti, T. Sasaki, G. Kurisu and R. Thakuria, CCDC 2465472: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2nrjcb.
| This journal is © The Royal Society of Chemistry 2025 |