
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Analysis of the impact of remote oxygen plasma treatment on the surface chemistry and electrochemical properties of graphite felt electrodes for redox flow batteries†
L. Mauricio
Murillo-Herrera
*a,
Carlos J.
Mingoes
a,
J.
Obrero-Pérez
b,
Juan R.
Sánchez-Valencia
 b,
Michael W.
Thielke
a,
Ángel
Barranco
b and
Ana B.
Jorge Sobrido
*a
b,
Michael W.
Thielke
a,
Ángel
Barranco
b and
Ana B.
Jorge Sobrido
*a
aSchool of Engineering and Materials Science, Queen Mary University of London, Mile End Rd, London E1 4NS, UK. E-mail: a.sobrido@qmul.ac.uk
bInstituto de Ciencia de Materiales de Sevilla, Consejo Superior de Investigaciones Cientificas, c/Américo Vespucio 49, 41092, Sevilla, Spain
First published on 19th August 2024
Abstract
The effects of a remote oxygen plasma (ROP) treatment on the surface of commercial graphite felts were investigated and compared against a conventional thermal treatment. In contrast to methodologies where the sample is directly exposed to the plasma, ROP allows for a high control of sample–plasma interaction, thereby avoiding extensive etching processes on the fibre surface. To assess the impact of ROP treatment time, the electrodes were subjected to three different periods (10, 60, and 600 seconds). X-ray photoelectron spectroscopy showed that the ROP treatment introduced nearly three times more surface oxygen functionalities than the thermal treatment. Raman spectroscopy measurements revealed a significant increase in amorphous carbon domains for the ROP samples. The thermal treatment favoured increases in graphitic defects and resulted in an order of magnitude larger ECSA compared to the ROP treated materials despite having lower content in oxygen functionalities. The electrochemical analysis showed enhanced charge-transfer overpotentials for GF400. The ROP samples exhibited a lower mass-transport overpotential than the thermally treated material and had similar permeabilities, which overall translated to the thermal treatment offering better performance at fast flow rates. However, at slow flow rates (∼10 mL min−1), the ROP treatment for the shortest period offered comparable performance to conventional thermal treatment.
Introduction
To reduce the dependence on fossil fuels and be able to rely on naturally intermittent renewable energies such as solar and wind, large-scale efficient energy storage technologies must further be developed at lower costs. This is essential to ensure a stable electrical grid supply to meet demand at any given time. Redox flow batteries (RFBs) are one of the most promising large-scale energy storage technologies to support intermittent power generation. One of the unique aspects of RFBs is their ability to decouple power and energy, which enables increasing energy capacity by increasing the volume and concentration of the electrolytes, while power output can be scaled by expanding the stack size.A key component of RFBs is the electrodes, whose role is to enable the electrochemical reaction between the half cells. RFB electrodes need to withstand the pressure arising from the flow of electrolytes, while providing a high electrochemically active surface area to promote performance, durability, and lifetime of the battery.
Graphite felts (GFs) are popular materials as electrodes for these batteries due to their commercial availability, high conductivity, and large surface area, while still being at a relatively low price.1–3 Nevertheless, pristine GFs exhibit poor wettability and electrochemical activity for most redox reactions in aqueous electrolytes, due to the lack of reactive sites and hydrophobic surfaces. This translates into large overpotentials, low efficiencies and reaction rates.4,5 Different approaches have been reported to overcome these drawbacks, primarily focused on the chemical modification of the surface. One strategy involves the introduction of heteroatom functionalities such as nitrogen6 or phosphorus,7,8 which promote the interaction with the electroactive species through built-in defects at the fibre surface. Other strategies involve the immobilisation of transition metal nanoparticles,9,10 carbon nanoparticles,11 and the deposition of biomass-derived carbon particles12 onto the surface area of the GF. The poor wettability of the GF electrodes has also been enhanced through thermal treatment,5,13–16 chemical modification17–19 or oxygen and air plasma treatments.20–22
Direct exposure to oxygen plasma has been reported to modify the surface of the GF, increasing roughness, leading to an increase of the electrochemical active surface area and the addition of oxygen-containing functional groups (phenolic, quinone, epoxy or carboxylic acids), increasing the number of electrochemically active sites.20–22 The use of reactive ROP treatments allows for a high degree of control of the interaction between the plasma and the sample, avoiding extensive etching processes, which are characteristic of protocols involving direct exposure to oxidative plasmas.23
In this report, we examined the effects of a ROP treatment on the surface of commercial GFs. The felts were exposed to the plasma for three different periods (10, 60, and 600 seconds per side) to determine the influence of the plasma treatment exposure on the surface properties. The ROP-treated materials were compared against a commonly employed thermal treatment method.5 Electrochemical and hydraulic experiments in a single-electrolyte flow cell were carried out to establish correlations between surface properties and flow cell performance. Our results showed that all plasma-treated samples developed larger surface oxygen populations than the thermally treated sample and different surface features. However, the electrochemical performance was not directly correlated with the amount of oxygen. Instead, an interplay between surface area, oxygen functionalities and graphite defects was responsible for the enhancements observed in the flow cell performance.
Experimental
Remote plasma treatments for the preparation of modified electrodes
GF samples were positioned within a remote electron cyclotron resonance microwave (ECR-MW) plasma reactor operating at 2.45 GHz with a power of 350 W. A complete description of the experimental set-up can be found elsewhere.23 The samples were placed at 6 cm from the glow discharge region. Before the plasma treatment, the system was evacuated to attain a base pressure of 10−6 mbar. Then, an oxygen flow was introduced using a calibrated mass flow controller set at 6 sccm. The gas pressure was controlled via an automated butterfly valve regulator, which controls the pumping flow of the system to maintain the pressure at 10−3 mbar. Three independent batches of graphite felt samples were subjected to O2 plasma conditions for three different periods: 10 seconds, 60 seconds, and 600 seconds. To ensure a uniform plasma treatment of the GF, both sides of the samples were exposed to the plasma for the specified duration on one side and then flipped and treated for the same duration on the other. The substrates remained at room temperature during the treatments, as determined by a thermocouple in the sample holder.Preparation of modified electrodes using the thermal treatment
A piece of GF of 3.18 mm thickness (Thermo Fisher Scientific) was placed inside a ceramic crucible and baked at 400 °C for 30 h using a Carbolite Gero box furnace and a heating ramp of 10 °C min−1.Electrolyte preparation
The 50% SoC iron electrolyte consisted of a 0.25 M solution of iron(II) chloride tetrahydrate 98% (Sigma Aldrich UK) and iron(III) chloride hexahydrate 97% (Sigma-Aldrich, UK) in 2 M hydrochloride acid (Fisher scientific, UK) for an overall 0.5 M iron concentration. The supporting electrolyte for the ECSA measurements consisted of 2.6 M sulfuric acid (Chem-Lab, Belgium).Surface characterization
Field emission scanning electron microscopy (SEM, FEI, Inspect F50) was used to analyse the morphology of the graphite felt surfaces. X-ray photoelectron spectroscopy (XPS, Thermo Scientific, Nexsa) was used to study the chemical composition of the fibre surface. Survey scan parameters: 50 eV pass energy, 10 scans, and 50 s dwell time. C1s, O1s, high resolution spectrum parameters: 50 eV pass energy, 30 scans, and 50 s dwell time. A set of asymmetric convoluted Gaussian and Lorentzian curves was used to model the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C contribution to the high resolution C1s spectrum. For the rest of the contributions, symmetric Gaussian and Lorentzian convoluted line shapes were used. BET analysis was performed in a Nova Quantachrome instrument, using N2 as an adsorbate. Raman spectroscopy was performed using a Renishaw in Via instrument, equipped with a 660 nm laser. The spectra were obtained by performing 10 acquisitions with 30 s of exposure between 850 and 3300 cm−1 using an ×20 objective lens. The Raman spectra were recorded using 5% of the laser power to avoid etching effects from biasing the ID/IG ratio of the fibres.
C contribution to the high resolution C1s spectrum. For the rest of the contributions, symmetric Gaussian and Lorentzian convoluted line shapes were used. BET analysis was performed in a Nova Quantachrome instrument, using N2 as an adsorbate. Raman spectroscopy was performed using a Renishaw in Via instrument, equipped with a 660 nm laser. The spectra were obtained by performing 10 acquisitions with 30 s of exposure between 850 and 3300 cm−1 using an ×20 objective lens. The Raman spectra were recorded using 5% of the laser power to avoid etching effects from biasing the ID/IG ratio of the fibres.
Single-electrolyte cell setup
The single-electrolyte cell active area size was 5.0625 cm2. The setup consisted of a couple of PAN-based graphite felt electrodes (Thermo Fisher Scientific) with a thickness of 3.18 mm separated by a Nafion 115 membrane (previously soaked in 2 M sulfuric acid for several days). The electrodes and membrane were compressed by PTFE gaskets of 2.132 mm thickness (33% compression). The flow fields consisted of two graphite plates, each with a single inlet and outlet, and six interdigitated channels with a 2.1 cm channel length and 0.1 cm channel width. The rest of the device consisted of copper current collectors, stainless-steel plates and PTFE tubes with an inner diameter of 3/16 inches. In a single-electrolyte setup, the cell is connected in such a way that the same electrolyte flows through both the cathode and the anode.24,25 A single-electrolyte cell schematic can be found in Fig. S22 in the ESI† document. A dual channel Watson Marlow 323 series peristaltic pump (Fisher Scientific, UK) with rubber tubes connected to the PTFE tubes using push-in fittings was used to distribute the electrolyte through the system. Before each experiment, a calibration curve for the pump was developed by recording five sets of volume and flow periods at different pump speeds.Single-electrolyte cell impedance
The potentiostatic impedance data were acquired using a Biologic SP300 potentiostat at the cell's open circuit potential with a sinusoidal amplitude of 10 mV within a frequency range of 1 MHz to 0.1 Hz and 8 points per decade. To fit the impedance spectra, a modified Randles equivalent circuit was used. It was composed of an inductor connected in series to a resistor representing the cell internal resistance (Rs), connected in series to a parallel circuit featuring a CPE element to describe the double layer capacitance of the electrode (CPEdl), and a resistor modelling charge-transfer contribution (Rct) in series to a Warburg short element (Wd) to describe mass-transfer impedance in porous electrodes.25 A schematic of the equivalent circuit is shown in Fig. 3. All experiments were performed in triplicate, for convenience, only the third Nyquist plot is represented.Single-electrolyte cell polarization
The polarisation plots were obtained using a Gamry E5000 series potentiostat. The experiment consisted of 24 chronoamperometric steps within a range of 25 mV to 600 mV. The timeframe of every chronoamperometric step was 30 s and the current data collection was done every second, from which the last 10 current values were averaged and used to build the polarisation plot. Before every set of 24 chronoamperometries, a high frequency impedance measurement was obtained to estimate the ohmic resistance of the cell to perform an ohmic-drop correction of the potential values. All the polarisation experiments were performed in triplicate.Single-electrolyte cell electrochemical active surface area determination
The electrochemical double layer capacitance of the materials was obtained through cyclic voltammetry experiments involving 2.6 M sulfuric acid at five different scan rates: experiments at 10, 20, 40, 80 and 160 mV s−1. A 400 mV potential window between −200 mV and 200 mV was used to avoid any faradaic processes and ensuring that all the current was due to the electrode's double layer capacitance. The absolute values of the currents corresponding to the middle region of the voltammograms were averaged and plotted against the scan rate to obtain the double layer capacitance according to eqn (1). To derive the qualitative ECSA, the relationship of eqn (2) was used, where Cs is 23 μF cm−2, with the specific capacitance value of glassy carbon in 4 M sulfuric acid.26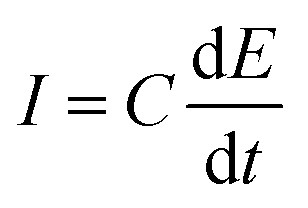 | (1) |
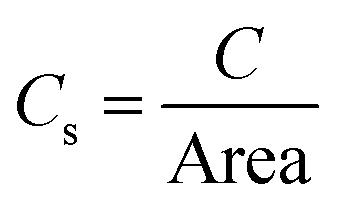 | (2) |
Pressure drop determination
The flow cell was rearranged to only distribute fluid through one of the sides. For this measurement, the membrane was replaced with a layer of plastic and aluminium foil and the acidic aqueous electrolyte was replaced with ultrapure water. A couple of Fluke 700RG29 digital pressure gauges (RS components, UK) were installed in the cell inlet and outlet to record the pressure difference. A schematic of a flow cell modified to perform pressure drop experiments can be found in Fig. S18 in the ESI† document. To obtain the permeability of the electrodes, the experimental pressure drop data were fitted using the Darcy–Forchheimer model displayed in eqn (3), where ΔP is the pressure drop in Pa, x is the characteristic path length, in this case the electrode length in m, μ is the fluid dynamic viscosity in Pa s, κ is the permeability in m2, ρ is the fluid density in kg m3, β is the Forchheimer coefficient in m−1 and v is the fluid linear velocity in m s−1.25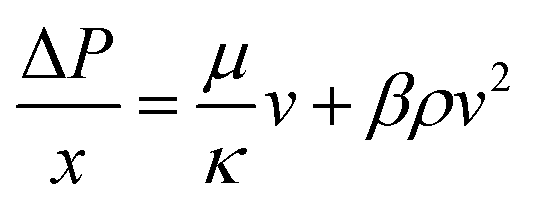 | (3) |
The fluid linear velocity was calculated considering an interdigitated flow field according to eqn (4), where Q is the flow rate in cm3 s, Ni is the dimensionless quantity corresponding to the number of interdigitated channels, hc is the thickness of the compressed electrode in cm, and lc is the length of the interdigitated channels.27
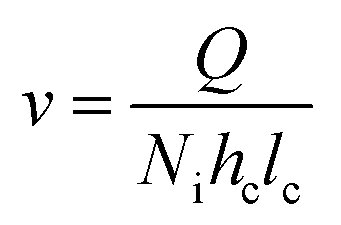 | (4) |
Results
Surface characterisation
The chemical composition of the surface of the various electrodes was characterised using XPS. The atomic percentages of oxygen in the pristine felt GF, thermally treated felt GF400, and plasma treated felts, P10s, P60s and P600s were found to be 2.15% ± 0.45%, 4.14% ± 0.54% 13.58% ± 0.24%, 10.42% ± 0.33% and 10.87% ± 0.78%, respectively. Both thermal and ROP treatments introduced oxygen on the surface of the carbon materials. All the plasma treatments resulted in higher amounts of oxygen functionalities compared to the thermal treatment, in agreement with previous research on the modification of graphite felt electrodes.5,13,20,21,28 This is relevant in the context of VRFBs, since previous research had identified oxygen functional groups such as C–O and O–CO as active or catalytic sites for outer-shell vanadium electrochemical reactions.5,8,15,20,28–30 However, there is still an open debate in relation to the role of the oxygen functionalities, whether these act as catalytic sites, improve the wettability of the electrodes, or both.14,15 The thermal and plasma treatments also introduced graphite defects and increase the surface area of the carbons, all of which can also contribute to the enhancement in performance.4,14,16,28
Among the plasma treated materials, the shortest period of exposure (10 seconds, P10s) resulted in the largest oxygen population. It must be noted that although the oxygen content of GF400 was found to be somewhat consistent across different measured samples, few super oxidised regions with oxygen contents between 11% and 21% and high population of carbonyl functionalities were observed (Table S1 and Fig. S3 in the ESI† document). The apparent inhomogeneity of the thermal treatment has been reported in previous thermal treatment studies and was caused by differences in graphitic edges or differences in the population of stable carbon oxides between samples of the same batch. Overall, these parameters impacted the wettability, capacitance and mass loss of the samples, however, the origin of these differences remained unclear.31,32
The distribution of oxygen-rich functional groups was analysed using the C1s and O1s high resolution spectra. The contributions arising from C(sp3), C(sp2), C–O associated with phenolic, ether or epoxy chemical shifts, CO associated with quinone chemical shifts and O–CO associated with carboxylate or lactone chemical shifts were used to deconvolute the C1s spectrum. The oxygen atomic percentage of the region associated with the high-resolution oxygen spectrum was used as a criterion to adjust the quantities of the chemical shifts related to oxygen-rich functional groups in the C1s spectrum. Once a satisfactory fitting of the C1s region was obtained, the O1s spectrum was deconvoluted into C–O and CO contributions considering the distribution of the different oxygen-rich functionalities found in the C1s spectrum. The feedback between the C1s and O1s was used as a way of confirming the validity of the deconvolution models. A breakdown of the C1s spectrum deconvolution for all materials is shown in Fig. 1a and Table S1 of the ESI† document.
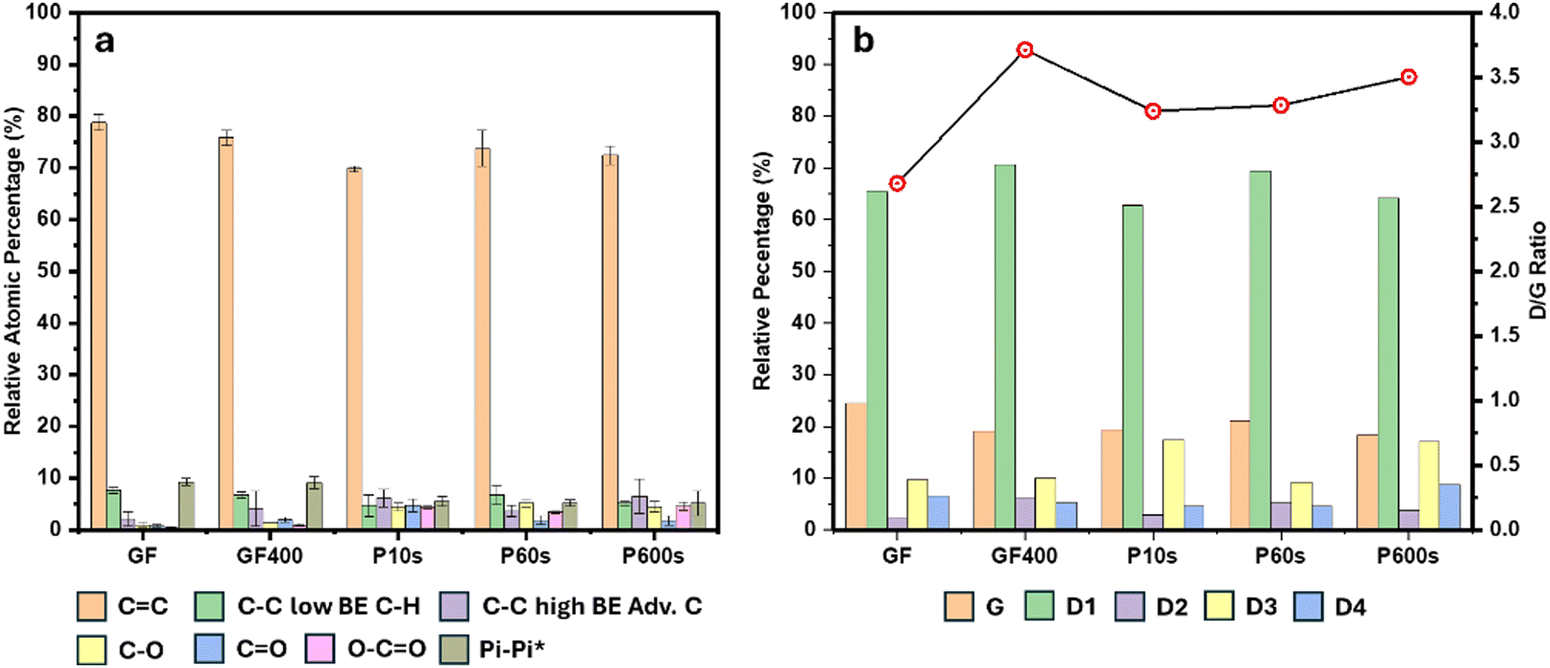 | ||
| Fig. 1 Bar charts showing, (a) relative percentages of the chemical functionalities deconvoluted from C1s XPS spectra of the treated and untreated felts. (b) Relative percentages of D and G bands deconvoluted from Raman spectra as well as the D1/G ratio for the treated and untreated felts. | ||
The surface of the pristine GF consisted of C–O, CO and O–CO functional groups (0.93% ± 0.48%, 0.76% ± 0.23% and 0.45% ± 0.12%, respectively). On the other hand, GF400 exhibited twice as much overall oxygen content on the surface. The populations of the C–O, CO and O–CO groups increased by 41%, 150% and 100%, respectively. On the other hand, the ROP treatment introduced two and three times more oxygen on the surface of the GF compared to the thermal treatment. A comparison of the distribution of oxygen functionalities between GF400 and the ROP-treated samples is displayed in Table S1 of the ESI† document. The distribution of oxygen functionalities for GF400 was CO > C–O > O–CO. On the other hand, P10s showed an even distribution of oxygen functionalities. For P60s, the distribution was C–O > O–CO > CO, whereas for P600s, C–O and O–CO dominated equally over CO.
P10s exhibited slightly lower population of C–O functional groups compared to the other two longer plasma treatments. Populations of 4.38% ± 0.72%, 5.18% ± 0.77% and 4.49% ± 0.93% were determined for P10s, P60s and P600s respectively. However, all three values resulted within the same margin of error, which could suggest that either after the first few seconds, no new significant amounts of C–O groups are formed or that there is a correlation between the formation of new C–O functionalities with subsequent reactions to higher oxidations states and eventual decomposition as CO2. This would render only few rich C–O regions in the fibres, with a larger presence of highly oxidized functional groups. CO functionalities resulted more prominent in P10s with a population of 4.75% ± 1.22% compared to 1.82% ± 0.84% and 1.79% ± 0.93% for P60s and P600s, respectively. These oxygen functional groups seemed to exhibit an initial increase to then experience a decrease and finally stabilize over a similar period. This trend suggests that the CO groups were readily oxidised to O–CO groups. Finally, for the O–CO functionalities, similar concentrations were observed for P10s and P600s, whereas P60s exhibited the lowest value. The drop in the O–CO atomic percentages, around the first minute of plasma exposure with further recovery, suggests a dynamic degradation of the carboxylate functionalities by CO2 evolution. In this scenario, the rate of degradation slows down over time, which gives opportunity to replenish the functionalities at extended periods of exposure.
Acidic oxygen groups such as phenolic and carboxylate have been reported to originate when carbon is thermally treated at temperatures close to its ignition point under an oxygen atmosphere. Alternatively, neutral groups like quinone, ether, epoxy and ester are formed by chemisorption at mild temperatures.32 Neutral and acidic oxygen functionalities can decompose as CO2 at lower temperatures.32,33
From these results, we can conclude that C–O functionalities develop up to a maximum concentration of around 4.7%. The concentration of CO functionalities decreased after a few seconds of plasma treatment, whereas the population of O–CO functionalities showed a fast development followed by a drop that then increases again with further exposure. Moreover, P10s exhibited the largest oxygen content (ca. 13.6%), while P60s and P600s showed slightly lower amounts (between 10.5% and 10.8%). These observations support the hypothesis that for the ROP treatment, there is a balance between CO2 decomposition and replenishment of the oxygen functionalities over long exposure periods.
The surface of the carbon fibres was further investigated via Raman spectroscopy to analyse the degree of graphitisation and the nature of defects produced during the electrode treatments. The Raman spectra of all samples showed two characteristic peaks associated to graphitic materials between 1330 cm−1 (D band) and 1590 cm−1 (G band). The G band is associated to the in-plain vibration of graphitic C(sp2) lattices. Conversely, the D can be decomposed into four contributions: D1 (∼1330 cm−1) associated to the total symmetric vibration of disordered graphitic lattice edges; D2 (∼1620 cm−1) associated to disordered graphitic superficial layer vibrations, D3 (1520 cm−1) associated to amorphous C(sp2) domains, and D4 (∼1200 cm−1) associated to polyene structures present at defect sites in the periphery of graphitic domains or ionic impurities.28,34 The analysis of the D1/G ratio is commonly used as a metric to determine the degree of defects on graphitic materials.
Several feasible sets of band contributions can be obtained by fitting the Raman spectrum employing different degrees of Gaussian and Lorentzian mixtures to define each Raman band. Allowing different ranges of full-width half maximum (FWHM) values will produce several potential suitable fittings. To avoid the bias associated with the choice of a model, the pristine material was used to define the parameters of the fitting model which were then applied to the rest of the samples. The fitting parameters are reported in Table S2 in the ESI† document.
A full breakdown of the relative percentages of the different bands as wells as the D1/G ratio for the respective materials is shown in Fig. 1b and Table S2 of the ESI† document. The GF sample exhibited a D1/G ratio of 2.68. Ordered graphitic materials tend to exhibit larger G bands, which bring the ID/IG values closer to zero. The analysis suggests that the origin of the D1/G ratio in the pristine material could be related to the fibre surface being composed mainly of disordered graphitic domains and amorphous C(sp2) carbon as opposed to ideal graphitic layers, in agreement with previous studies.31 The oxygen content of the pristine material obtained from XPS of around 2% would be more favourably bonded to carbon atoms located in the graphitic defective sites or the amorphous carbon domains, as oxygen functionalities are associated with these surface features (Fig. 2).28
 | ||
Fig. 2 Scanning electron microscopy images for the GF, GF400, P10s, P60s and P600s materials. p: ×2600 magnification. Bottom: ×20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 magnification. 000 magnification. | ||
GF400 exhibited the largest D1/G ratio of the studied materials (3.71). The most significant change in band contributions was the increase of the D1 and D2 bands and the decrease of the G band. The fitting contributions of the D3 and D4 bands were very similar to those from GF. This suggests that the thermal treatment leads to an increase in the number of disordered graphitic domains of the fibres, demonstrating that the surface modification mechanism of the thermal treatment predominantly induces the formation of defects and exposed edges with less significant impact on the amount of amorphous or polyene-like carbon.
P10s was characterised by a D1/G ratio of 3.24, while P60s and P600s exhibited D1/G ratios of 3.28 and 3.50, respectively. The increase in the D1/G ratio of P10s and P600s with respect to GF was due to a significant increase in the D3 band, thus reducing the size of the G band. These results indicate that a shorter exposure to plasma generates various sites of superficial amorphous carbon. These features were in correlation with the XPS results which showed a higher degree of carboxylate groups for these samples compared to P60s. On the other hand, the increase in the D1/G ratio of P60s was better explained by an increase of the D1 and D2 bands at the expense of the G band and a smaller contribution of the D3 band. Moreover, P600s showed the largest D4 band signal of the series, whereas P10s and P60s showed very similar and smaller signals compared to GF. This suggests that plasma and thermal treatments follow different surface modification mechanisms. Raman analysis showed the development of graphitic defective sites and edges for GF400 and to a lesser extend for P60s, whereas for P10s and P600s, the formation of amorphous carbon domains was predominant.
The SEM micrographs of the treated felts showed that the samples exhibiting an increase in amorphous carbon domains were characterised by rougher fibre surfaces. P10s exhibited a uniformly rough surface with large relief features of around 500 nm. P600s also exhibited similar features, but to a lesser extent, because the coverage in this case was not homogeneous and was limited to certain fibres. The inhomogeneity observed for P600s could be a consequence of the degradation of oxygen functionalities as CO2, as discussed in the XPS section. This could etch the fibres, thus exposing fresh carbon. Conversely, GF400 and P60s which were characterised by major increases in the graphitic edges and in plain defects exhibited smoother fibre surfaces. A potential explanation for the smoothing of P60s could be the same etching mechanism suggested for P600s. As discussed previously, a fast degradation rate over the first seconds of exposure could effectively remove the roughness. However, as the degradation rate slows down, it could give rise to a balance between the formation of new oxygen functionalities and their degradation. Therefore, at longer periods of exposure, the fibres could recover some of the surface features observed for P10s.
Single-electrolyte cell characterisation
The electrochemically active surface area (ECSA) of the electrodes was obtained via electrochemical double layer capacitance (EDLC) measurements in a single-electrolyte cell configuration using 2 M H2SO4. The EDLC is dependent on the quantity and mobility of the electrode charge carriers, which are associated to the defects on the graphitic surface.28,31 The plasma treated felts displayed larger ECSA values compared to GF, with P10s and P600s reporting the largest difference, being ca. 2 times that of GF. However, GF400 exhibited the largest ECSA of all samples, which is ca. 26 times that of GF and more than 12 times that of P10s. In principle, this result is consistent with the Raman analysis, which showed a greater D1/G ratio for the treated felts compared to GF, as it is expected that the graphitic defective sites and edges have larger EDLC values than graphitic basal planes. The differences in EDLC and ECSA values between GF400 and the ROP samples suggest that these may be related to the concomitant increase of graphitic defects observed via Raman spectroscopy.The EDLC experiments also confirmed that the mechanism of surface modification by ROP treatment is inherently different from the thermal treatment, as the introduction of oxygen functionalities was not accompanied by a proportional increase in ECSA as observed for the thermal treatment. This was supported by the BET analysis which produced similarly increased specific physical surface areas for the ROP and thermally treated electrodes over the untreated one (Table 1). However, a closer look at the pore size distribution of these materials (Fig. S17, ESI†) revealed that GF400 possesses a fraction of micropores (<1 nm) not present in the other materials. During electrochemical measurements, access to these sites could serve as explanation for the order of magnitude increase in ECSA for GF400.
| Sample | EDLC (μF mg−1) | ECSA (m2 g−1) | BET (m2 g−1) | Permeability × 10−11 (m2) |
|---|---|---|---|---|
| Pristine | 13.55 | 0.060 | 1.63 | — |
| GF400 | 376.88 | 1.639 | 6.81 | 2.55 |
| P10s | 29.94 | 0.131 | 5.87 | 2.28 |
| P60s | 18.69 | 0.082 | 4.51 | 2.33 |
| P600s | 29.72 | 0.129 | 2.31 | 2.33 |
Overall, the general increase in the specific surface area as shown in the BET data justifies the increase in ECSA observed for the treated materials. Notable however is how much lower the ECSAs are in comparison to these specific surface areas. This suggests that not all physical electrode surface areas are available/accessible for electrochemical species to react. In this regard, surface area measurements obtained under electrochemical reaction conditions are more representative of the electrode availability during real cell operation.35
Pressure drop experiments were carried out to assess the pumping energy losses through the electrochemical cell for all electrodes. From these datasets, permeability was extracted using the Darcy–Forchheimer equation.25 The permeabilities of the ROP electrodes all differed by ca. 2% difference from each other and were ca. 12% less than GF400 (Fig. S24–S26, ESI†). These results agree with the pore size distribution of the electrodes (Fig. S17, ESI†). All ROP electrodes have similar pore size distributions and thus electrolyte flow paths are expected to be similar as reflected by their permeabilities. The fact that GF400 displayed permeability similar to the ROP electrodes (only >12%) suggests that the primary flow paths are similar in both types of electrodes. In other words, the micropores may play little to no role in bulk electrolyte distribution though the electrode.
The performance of the thermally treated and ROP treated electrodes was analysed in a flow battery set up in single-electrolyte configuration. The electrolyte of choice was FeCl2/FeCl3 (0.25 M each for a total of 0.5 M iron) in 2M HCl, which is equivalent to a 50% state of charge (SoC). In single-electrolyte configuration, the electrolyte passes through both the cathode and the anode, carrying out the opposite electrochemical reaction on either side, thus ensuring that the battery operation proceeds always at a fixed SoC. This configuration ensures that the cell operates under steady-state conditions, which is an important requirement to obtain reliable impedance data. Moreover, this also ensures that there is no electrolyte crossover or self-discharge effects.24,25 Polarisation and impedance data were acquired at four different flow rates across the whole range of our peristaltic pumps (between 15 and 110 mL min−1).
The purpose of using iron for this test is to employ a redox couple exhibiting well defined one-electron redox processes to simplify the deconvolution of the electrochemical impedance data into clear ohmic, charge-transfer and mass-transport contributions. The Nyquist plots of the single-electrolyte cells were characterised by two arcs, corresponding to high and low frequency impedance associated with the charge transfer and mass transport impedance, respectively. The last few data points from the low frequency arc described a reduction in the magnitude of the real impedance. This was not the expected behaviour for the mass-transport impedance domain, and as such, the Warburg short circuit element employed to fit this segment was not able to model these data points. This anomalous behaviour is associated with the sluggish diffusion of ions through the fibrous carbon electrodes.
The ohmic resistance was obtained from the intercept of the impedance spectrum and the real impedance axis. The corresponding ohmic resistances were then subtracted from all Nyquist plots to normalise the onset of the impedance spectrum to simplify comparisons between samples. The ohmic resistance corresponds to the internal resistance of the cell, and since the same electrochemical setup was used to characterise all electrodes, the differences in this parameter were attributed to the conductivity differences of the electrodes. It was expected that the electrodes exhibiting higher degrees of oxygen coverage would be characterised by higher internal resistances. GF400 exhibited 8% less internal resistance than P10s, 6% less resistance than P60s, and 9% less resistance than P600s.
Limiting current densities were observed for the set of electrodes at low electrolyte velocity (Fig. 3c and d) and is indicative of a lack of readily available active reactant species.36 With the increasing electrolyte velocity, faster replenishment of active reactant species is enabled, diminishing this limitation and enhancing limiting current density.25 This was observed as the polarisation plots became more linear with increasing flow rates for all electrodes. Not surprisingly, the mass transfer resistance of the cells also decreased with the increase of the electrolyte flow rate, as the mass transfer resistance is associated with overpotential due to the limitations of supplying electroactive reactants to the active sites of the electrode surface, and the diffusion coefficient of the active species which is constant in this case.37,38 The charge transfer resistance contribution to the cell polarisation was also assessed. Charge-transfer resistance is associated with the kinetics of the iron redox process over the graphitic fibre surface, i.e. the kinetic overpotential. The charge transfer resistance contribution was observed to remain relatively consistent regardless of the flow rate for all electrodes.
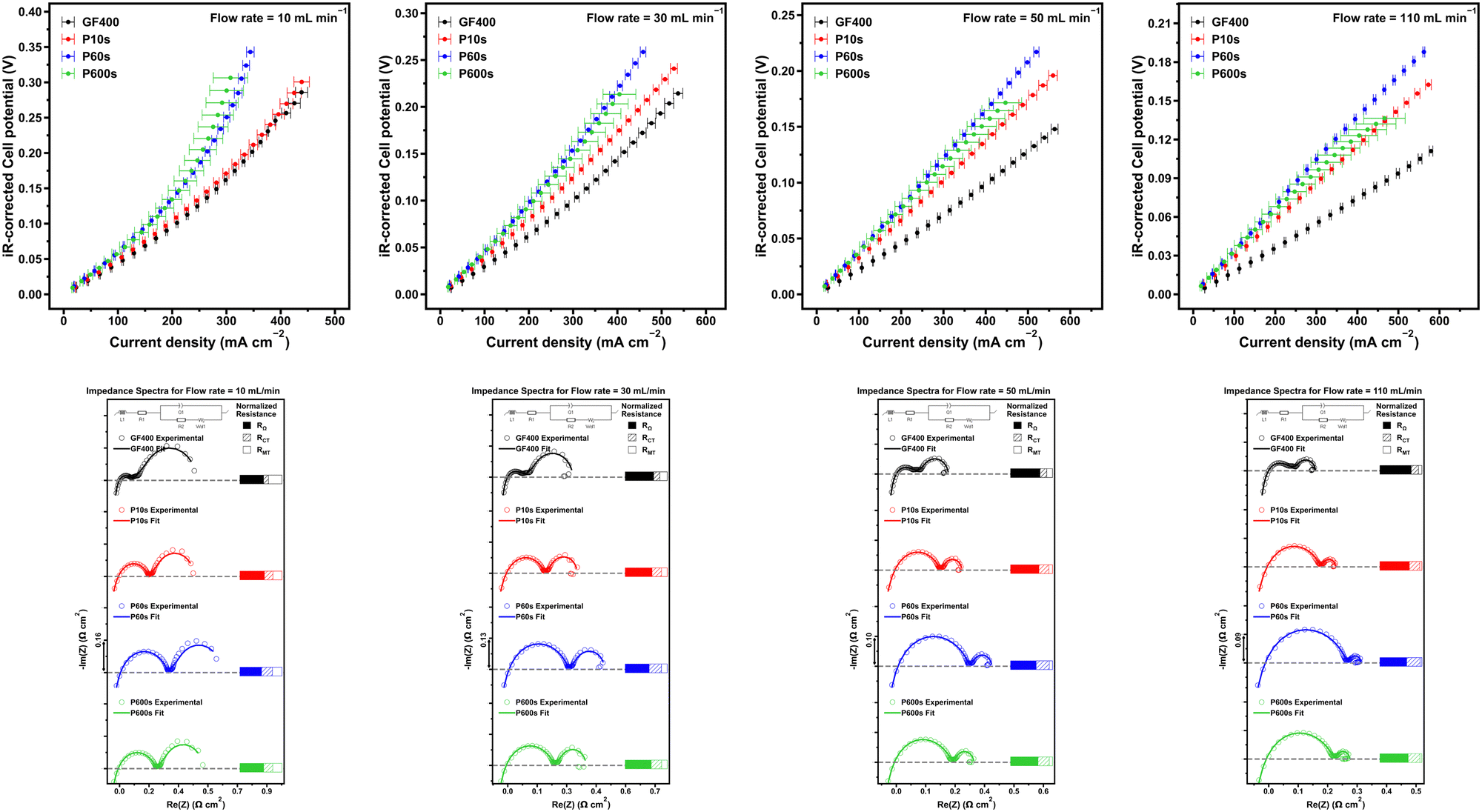 | ||
| Fig. 3 Single-electrolyte cell results. Top: Polarisation data. Bottom: Impedance data. | ||
When comparing polarisation plots of all electrodes, clear trends were observed. The increase in the flow rate resulted in more notable differences in performance among the studied carbon electrodes. The general trend in performances across electrodes follows the order: GF400 > P10s > P600s > P60s (between flow rates 30 mL min−1 and 110 mL min−1). The opposite trend is observed when the averaged charge-transfer resistances across flow rates are taken. A closer look at the mass transfer resistance reveals a more complex relationship, with the only clear trend being GF400 > P10s, P60s and P600s. In fact, all plasma treated electrodes exhibit ca. 35% less mass transfer resistance than GF400. Mass transfer is linked to diffusion coefficients, flow rates, and intrinsic reaction kinetic parameters. The higher O content in ROP samples may be responsible for higher diffusivity of ions and potential higher intrinsic reaction kinetics that can translate into lower mass transport resistance. If we consider that as the flow rate increases, we approach infinitely fast mass transfer, and then charge transfer resistance becomes the determining factor to limiting current density. As such, it makes sense for the polarisation performance to trend inversely with charge transfer resistance.
At lower flow rates, mass transfer resistance dominates, and therefore, due to its smaller mass-transport resistance, P10s is able to overcome the larger ECSA and superior charge transfer capabilities of GF400 (GF400 resulted in 43%, 59% and 50% smaller charge transfer resistance than P10s, P60s and P600s), thus leading to comparable performance. This also highlights another observation of GF400 that despite having a magnitude size increase in ECSA, this does not translate into a more superior activity at all flow rates. Previous studies in the literature have demonstrated this limitation, attributing it to a limited accessibility to the electrode micropores by the reactive species.4 This idea was explored earlier when discussing the permeabilities of the electrode, which points to all the electrodes having the same general electrolyte flow trajectory. This was an expected feature, as the treatments were intended to modify the graphitic felt fibres surface and not the mesostructure of the materials. This finding is relevant because the common operation flow rates for real life flow battery applications range between 10 and 30 mL min−1.
Interestingly, the magnitude of the charge transfer resistance followed the order P10s < P600s < P60s, which was the same trend observed for the ECSA of the electrodes. As mentioned before, although this trend also coincides with the amount of carboxylate functional groups and the prevalence of amorphous carbon domains, the origin of the differences in the charge transfer resistance between samples are most probably associated with the differences in ECSA. Otherwise, the ROP felts should have exhibited inferior charge transfer resistances than GF400 by virtue of their superior oxygen loading.
Conclusions
The surface of commercial GF electrodes was modified by a ROP using three different periods of exposure (10 s, 60s and 600 s) and their surface properties were compared to a common thermally treated felt. The XPS analysis of the samples indicated that all ROP-treated samples exhibited two and three times larger superficial oxygen populations than the thermally treated sample. The C–O oxygen functionalities were found to remain constant among the three treatments, whereas CO groups were found more prominent for P10s and in similar quantities for P60s and P600s. Conversely, a larger amount of O–CO functionalities were found for P10s and P600s compared to P60s. The analysis of the Raman spectra of the samples showed that the pristine material exhibited the smallest D1/G ratio, whereas GF400 exhibited the highest D1/G ratio. Among the ROP-treated materials, the D1/G ratio increased in correlation with the exposure period. The deconvolution of the D band showed that the thermal treatment favoured the increase of D1 and D2 bands associated with the vibrations of defective graphitic domains. For P10s, the major contribution to explain the D1/G ratio came from an increase of the D3 band, associated with amorphous carbon domains. P60s exhibited major increases in the D1 and D2 bands and minor increases in the D3 band. P600s also exhibited larger contributions from the D3 band, although an increase in the D4 band was also observed. The SEM micrographs of the samples showed rough features covering the majority of the P10s fibres and were present to a lesser extent in P600s. Conversely, GF400 and P60s were characterised by smoother fibres and absence of distinctive features.
EDLC and BET experiments showed that all treated materials were characterised by larger surface areas compared to the pristine material. However, for all samples, the ECSA was significantly lower than the BET area, illustrating the low accessibility of the electrode surface for electrochemical reactions. GF400 exhibited the largest physical surface area and ECSA among all the treated samples despite not being the sample with the largest number of oxygen functionalities. Moreover, GF400 showed a distinctive fraction of microporosity, which was not found in any of the ROP treatments. The permeability of all the treated materials showed GF400 with a 12% higher permeability than the ROP-treated materials which showed similar values regardless of the exposure period.
The single-electrolyte polarisation and impedance experiments revealed that GF400 exhibited 43–59% lower charge-transfer resistance than the ROP treated-samples. This indicated that the kinetic overpotential improvements were driven by the ECSA and not by the amount of superficial oxygen functionalities. The magnitude of the charge-transfer resistance among the ROP samples followed the same trend as the ECSA, further supporting that the kinetic overpotential improvements were due to increases in the ECSA. The mass-transport resistance of all the ROP-treated materials was ca. 35% lower than that of GF400. At fast flow rates, when the kinetic overpotential is the most important factor in electrode polarisation, GF400 showed the best performance. However, at slower flow rates, when the mass transport overpotential becomes the dominant factor for polarisation, the performance of GF400 and P10s became comparable. Overall, this report provides an insight into the effects of oxygen-enriching surface treatments on graphite felt electrodes in terms of surface chemistry, defects, ECSA, porosity and their correlation with flow cell performance.
Author contributions
L. M. M. H., C. M., and M. T.: characterisation of the materials, data analysis, and manuscript drafting. J. O. P. and J. R. S.: preparation of the materials. A. B. J. S.: conceptualisation of the project, writing – review & editing. A. B. J. S.: funding acquisition.Data availability
The data sets generated in this work are available upon request to the first and corresponding author.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
A. B. J. S. acknowledges the UK Research and Innovation for Future Leaders Fellowship no. MR/T041412/1.References
- K. E. Rodby, T. J. Carney, Y. A. Gandomi, J. L. Barton, R. M. Darling and F. R. Brushett, J. Power Sources, 2020, 460, 227958 CrossRef CAS.
- A. Fornier-Cuenca and F. R. Brushett, Curr. Opin. Electrochem., 2019, 18, 113–122 CrossRef.
- J. P. Tafoya, M. Thielke, G. Tian, R. Jervis and A. B. Sobrido, Curr. Opin. Chem. Eng., 2022, 38, 100876 CrossRef.
- K. V. Greco, J. K. Bonesteel, N. Chanut, C. T.-C. Wan, Y.-M. Chiang and F. R. Brushett, ACS Appl. Energy Mater., 2021, 4, 13516–13527 CrossRef CAS.
- B. Sun and M. Skyllas-Kazacos, Electrochim. Acta, 1992, 37, 1253–1260 CrossRef CAS.
- S. J. Yoon, S. Kim, H. Kim, D. K. Kim, S. So, Y. T. Hong and R. Hempelmann, Carbon, 2020, 166, 131–137 CrossRef CAS.
- R. Wang, Y. Li, Y. Wang and Z. Fang, Appl. Energy, 2020, 261, 114369 CrossRef CAS.
- K. J. Kim, H. S. Lee, J. Kim, M.-S. Park, J. H. Kim, Y.-J. Kim and M. Skyllas-Kazacos, ChemSusChem, 2016, 9, 1329–1338 CrossRef CAS PubMed.
- K. Amini, J. Gostick and M. D. Pritzker, Adv. Funct. Mater., 2020, 30, 1910564 CrossRef CAS.
- Y. Lv, C. Han, Y. Zhu, T. Zhang, S. Yao, Z. He, L. Dai and L. Wang, J. Mater. Sci. Technol., 2021, 75, 96–109 CrossRef.
- Y. Zhang, L. Liu, J. Zi, Z. Wu and X. Qiu, Appl. Energy, 2017, 204, 373–381 CrossRef CAS.
- M. W. Thielke, G. Tian and A. Jorge Sobrido, J. Phys.: Mater., 2022, 5, 024004 CAS.
- A. Kaur, K. I. Jeon, S. S. Kim and J. W. Lim, Compos. Struct., 2022, 290, 115546 CrossRef CAS.
- K. V. Greco, A. Forner-Cuenca, A. Mularczyk, J. Eller and F. R. Brushett, Appl. Mater. Interfaces, 2018, 10, 44430–44442 CrossRef CAS PubMed.
- A. M. Pezeshki, J. T. Clement, G. M. Veith, T. A. Zawodzinski and M. M. Mench, J. Power Sources, 2015, 294, 333–338 CrossRef CAS.
- P. C. Ghimire, R. Schweiss, G. G. Scherer, T. M. Lim, N. Wai, A. Bhattarai and Q. Yan, Carbon, 2019, 155, 176–185 CrossRef CAS.
- L. M. Murillo-Herrera, E. S. Aguilar, M. W. Thielke and A. Jorge Sobrido, Chem. – Asian J., 2023, 18 Search PubMed.
- L. Yue, W. Li, F. Sun, L. Zhao and L. Zing, Carbon, 2010, 48, 3079–3090 CrossRef CAS.
- B. Sun and M. Skyllas-Kazacos, Electrochim. Acta, 1992, 37, 2459–2465 CrossRef CAS.
- D. Dixon, D. J. Babu, J. Langner, M. Bruns, L. Pfaffmann, A. Bhaskar, J. J. Schneider, F. Scheiba and H. Ehrenberg, J. Power Sources, 2016, 332, 240–248 CrossRef CAS.
- C.-H. Lin, Y.-D. Zhuang, D.-G. Tsai, H.-J. Wei and T.-Y. Liu, Polymers, 2020, 12, 1372 CrossRef CAS.
- T. Jirabovornwisut, B. Singh, A. Chutimasakul, J.-H. Chang, J.-Z. Chen, A. Arpornwichanop and Y.-S. Chen, Materials, 2021, 14, 3487 CrossRef PubMed.
- M. Alcaire, F. J. Aparicio, J. Obrero, C. López-Santos, F. J. Garcia-Garcia, J. R. Sánchez-Valencia, F. Frutos, K. Ostrikov, A. Borrás and A. Barranco, Adv. Funct. Mater., 2019, 29, 1903535 CrossRef.
- R. M. Darling and M. L. Perry, ECS Trans., 2013, 53, 31–38 CrossRef.
- C. T.-C. Wan, R. R. Jacquemond, Y.-M. Chiang, A. Forner-Cuenca and F. R. Brushett, Energy Technol., 2023, 11, 2300137 CrossRef CAS.
- R. R. Jacquemond, C. T.-C. Wan, Y.-M. Chiang, Z. Borneman, F. R. Brushett, K. Nijmeijer and A. Forner-Cuenca, Cell Rep. Phys. Sci., 2022, 3, 100943 CrossRef CAS.
- J. D. Milshtein, K. M. Tenny, J. L. Barton, J. Drake, R. M. Darling and F. R. Brushett, J. Electrochem. Soc., 2017, 164, E3265–E3275 CrossRef CAS.
- J. Langner, M. Bruns, D. Dixon, A. Nefedov, C. Wöll, F. Scheiba, H. Ehrenberg, C. Roth and J. Melke, J. Power Sources, 2016, 321, 210–218 CrossRef CAS.
- J. Lee, J. T. Muya, H. Chung and J. Chang, ACS Appl. Mater. Interfaces, 2019, 11, 42066–42077 CrossRef CAS PubMed.
- D. Kil, H. J. Lee, S. Park, S. Kim and H. Kim, J. Electrochem. Soc., 2017, 164, A3011–A3017 CrossRef CAS.
- T. J. Rabbow, M. Trampert, P. Pokorny, P. Binder and A. H. Whitehead, Electrochim. Acta, 2015, 173, 17–23 CrossRef CAS.
- T. J. Rabbow, M. Trampert, P. Pokorny, P. Binder and A. J. P. White, Electrochim. Acta, 2015, 173, 24–30 CrossRef CAS.
- C. A. Frysz, X. Shui and D. D. L. Chung, Carbon, 1994, 32, 1499–1505 CrossRef CAS.
- M. W. Thielke, S. Lopez Guzman, J. P. Victoria Tafoya, E. García Tamayo, C. I. Castro Herazo, O. Hosseinaei and A. Jorge Sobrido, Front. Mater., 2022, 9, 1–10 Search PubMed.
- T. J. Rabbow and A. H. Whitehead, Carbon, 2016, 111, 782–788 CrossRef.
- D. Aaron, Z. Tang, A. B. Papandrew and T. A. Zawodzinski, J. Appl. Electrochem., 2011, 41, 1175–1182 CrossRef CAS.
- A. Forner-Cuenca, E. E. Penn, A. M. Oliveira and F. R. Brushett, J. Electrochem. Soc., 2019, 166, A2230–A2241 CrossRef CAS.
- M. Van Der Heijden, M. Kroese, Z. Borneman and A. Forner-Cuenca, Adv. Mater. Technol., 2023, 8, 2300611 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ya00383g |
| This journal is © The Royal Society of Chemistry 2024 |