
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of pyrene orientation on the electronic properties and stability of graphene ribbons†
Tanner
Smith
a,
Karl
Thorley
a,
Kevin
Dimmitt
b,
Sean
Parkin
a,
Oksana
Ostroverkhova
b and
John
Anthony
 *a
*a
aCenter for Applied Energy Research, University of Kentucky, Lexington, USA. E-mail: anthony@uky.edu
bDepartment of Physics, Oregon State University, Corvalis, USA
First published on 18th September 2024
Abstract
We report the synthesis and study of trialkylsilylethyne-substituted “oblique” pyrene-fused acenes, carbon nanoribbons demonstrating near-IR absorption with reversible oxidation and reduction, and acene-like evolution of electronic properties upon extension of the aromatic core. Their electronic structures are investigated through DFT studies, which support the more delocalized nature of their frontier molecular orbitals compared to more common “vertical” pyrene systems. Despite a longer aromatic core, the more extended of the two examples demonstrates enhanced photostability compared to the shorter derivative, running counter to the trend in linear acenes. The unusual stability of the longer core is ultimately linked to its relatively low T1 energy inhibiting the generation of reactive O2 species. The byproduct generated upon photooxidation of the shorter nanoribbon appears to catalyze the generation of 1O2 due to its large T1 energy, leading to its relatively decreased stability.
Introduction
The enormous degree of structural diversity in polycyclic aromatic hydrocarbons (PAH) provides remarkable opportunities to systematically alter electronic properties, which are highly dependent on molecular structure and edge topology.1–3 The importance of molecular structure is prominently displayed in the acene series, in which the presence of only one Clar sextet, regardless of molecular length, results in rapid evolution of electrochemical properties with each linearly-fused ring.4 While the robust tunability of acenes makes them of significant interest for organic electronic and photonic applications, the diminished stability with each additional fused ring means that the longer acenes are especially prone to decomposition.5 The functionalization of acenes, particularly through substitution with trialkylsilylethyne (TAS) groups, is a common method to improve their stability as well as enhance solid-state ordering.6,7 With few exceptions,8 the poor stability of TAS-substituted acenes longer than pentacene has hindered their study under ambient conditions and largely prevents practical device applications.The development of structure–property relationships in larger, 2-D PAH systems, often referred to as graphene nanoribbons (GNRs) or nanographenes, is aided through the bottom-up synthesis of well-defined molecules.9,10 The incorporation of acene-like moieties onto other versatile molecular frameworks, such as pyrene, is an increasingly popular method used to efficiently tune the electronic structure of GNRs. The properties of pyrene-fused acenes are highly dependent on the orientation of the central pyrene moiety.11 The vast majority of previously reported pyrene-fused acenes may be considered “vertical pyrenes,” in which linear extension of the acene moieties occurs from the k-region of the pyrene moiety (Fig. 1). While numerous examples of vertical pyrenes have been reported, including systems that extend up to macromolecular size,12–14 in-depth studies have demonstrated that the interactions between the acene moieties are disrupted by the vertical orientation of the central pyrene.15 While some pyrene-fused acene systems hold unique photophysical properties,16 the disrupted conjugation may limit their use in charge transport applications.11
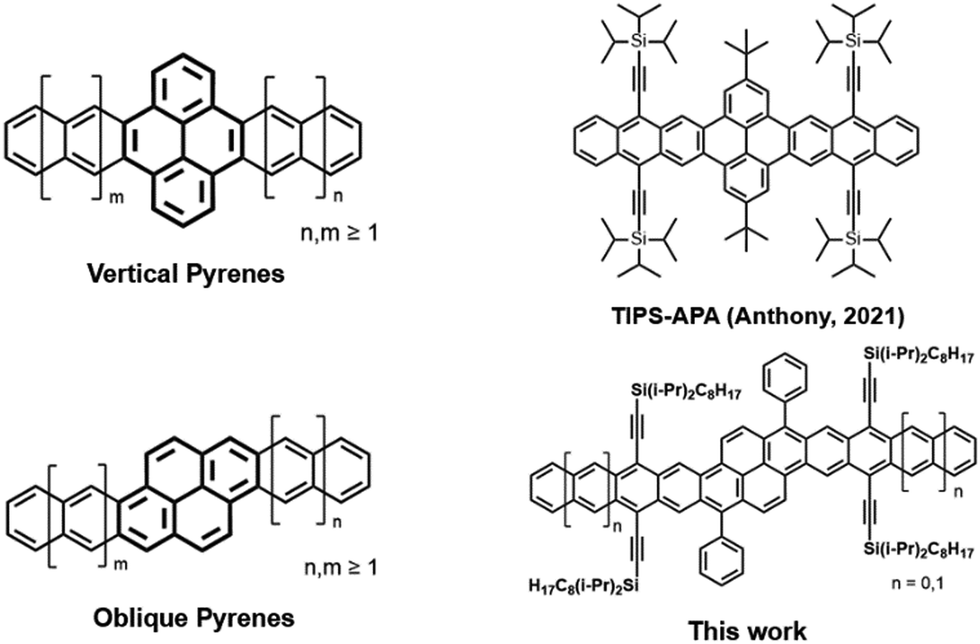 | ||
| Fig. 1 General structures of vertical and oblique pyrenes (left) and their functionalized derivatives (right). | ||
In contrast to vertical pyrenes, “oblique” pyrenes maintain full conjugation throughout the aromatic core, making them ideal candidates for applications that require fully-conjugated nanographenes.17–19 A notable example is a functionalized “bistetracene” originally reported by Briseno, which displayed remarkable stability and hole mobilities on the order of ∼4 cm2 V−1 s−1.20,21 As with other 2-D fused-acenes, the promising electronic performance of oblique pyrenes is aided by their large, planar aromatic cores, which can provide greater opportunity for significant orbital overlap in the solid-state.20,22,23 The band structures of oblique pyrenes are predicted to evolve with increased length of the acene moieties, meaning that high intrinsic charge mobilities are predicted for larger derivatives.11 However, stable oblique pyrene derivatives larger than 8 consecutively-fused rings have remained unexplored, primarily due to a lack of synthetic accessibility.
As an alternative to conventional pyrene-fused acene systems, we present the synthesis and electronic properties of extended oblique pyrene derivatives. The structures of our TAS-substituted bispentacene (BP) and bishexacene (BH) derivatives may be considered as a pyrene-fused anthracene and tetracene, respectively, allowing for straightforward comparison to functionalized vertical pyrenes such as a previously reported anthracene–pyrene–anthracene (APA) molecule featuring triisopropylsilylethyne (TIPS) substituents (TIPS-APA, Fig. 1).15 In contrast to vertical pyrenes, we demonstrate that the oblique orientation of the central pyrene provides for a fully conjugated ribbon. The synthetic route employed allows for relatively straightforward control over molecular length and functionalities. In contrast to vertical pyrenes, properties such as optical gap and ionization potential (IP) evolve with linear extension of the aromatic core in a manner similar to that seen in the linear acene series, allowing for excellent control over optical and electrochemical properties. The frontier molecular orbitals (FMOs) and aromaticity of our oblique pyrenes are investigated through DFT calculations, providing further insight into the role of ring annellation in larger pyrene-acene systems. We also demonstrate that functionalized oblique pyrenes as long as 12 consecutively fused rings exhibit prolonged stability under light exposure, and we examine the origins of such impressive photooxidative resistance through a combination of experimental and computational approaches.
Results and discussion
We employed a classical and versatile synthetic route to our oblique pyrenes (Scheme 1), utilizing a pyrene-dianhydride intermediate 1 which was originally reported by Clar.24 This starting material can be synthesized in large scale with inexpensive starting materials, and the aryl substituent can easily be varied to enable crystal packing tuning. The esterification of 1 provides the more soluble intermediate 2, which was reduced with LiBH4 to provide high yields of 3 with simple work-up. Bromination of 3 (PBr3) gave Cava precursor 4, and reaction between 4 and 1,4-naphthoquinone provided BP-Q, while a similar reaction with 1,4-anthraquinone gave BH-Q, showing the versatility of this approach to give polyaromatic cores of varying length.25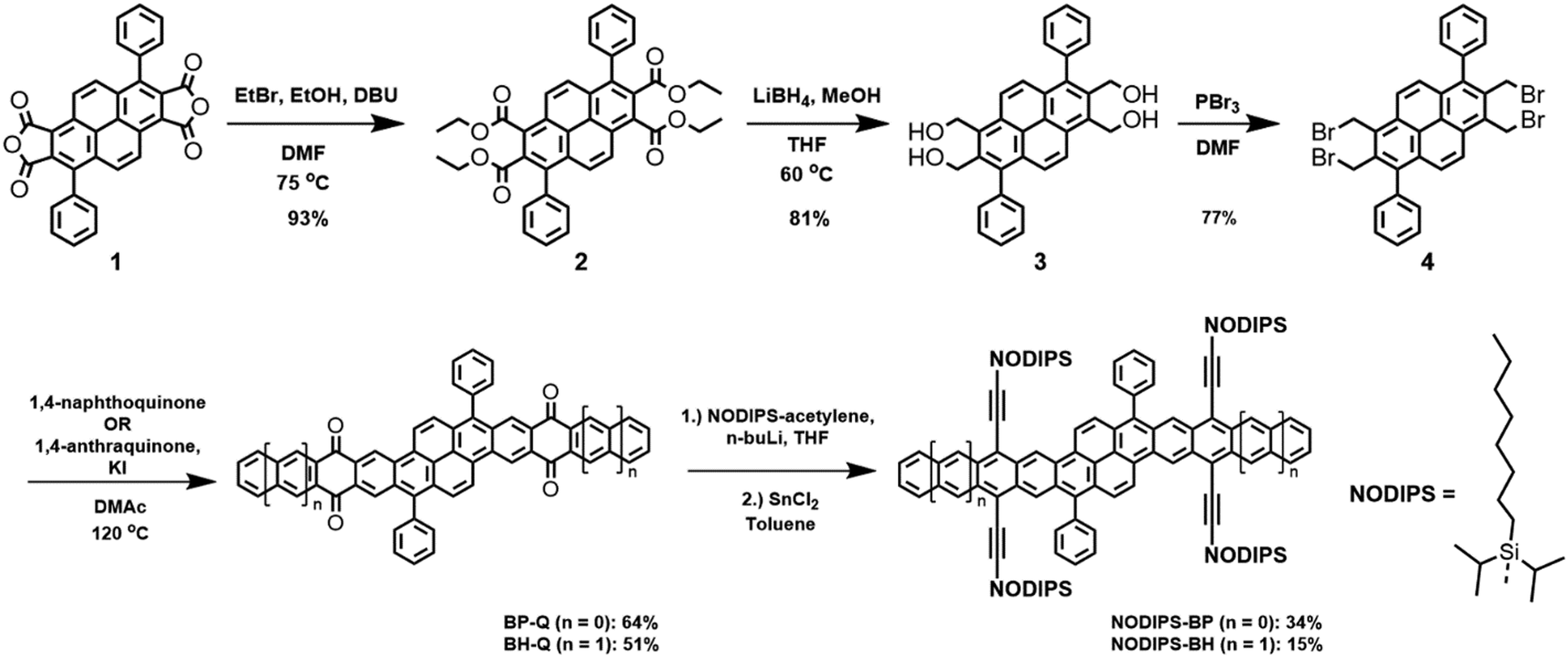 | ||
| Scheme 1 Generalized synthetic route to NODIPS-BP and NODIPS-BH. | ||
The formation of the fully aromatized oblique pyrenes was accomplished with our well-established silylethynylation chemistry, by addition of a lithiated silylacetylene to the quinone,26 followed by reductive deoxygenation with stannous chloride. In this case, we utilized the highly solubilizing n-octyldiisopropylsilyl (NODIPS) acetylene to provide NODIPS-BP and NODIPS-BH (Scheme 1). X-ray quality crystals of both NODIPS-BP and NODIPS-BH were obtained from ethyl acetate/toluene mixtures. The long octyl chains on the NODIPS groups provided ample solubility of NODIPS-BP and NODIPS-BH in most common organic solvents, allowing straightforward characterization. Surprisingly, NODIPS-BH appeared just as stable under ambient conditions as NODIP-BP despite its larger conjugated backbone. Thus, a similar synthetic route may allow for the future synthesis of even larger oblique pyrene derivatives. The structures of NODIPS-BP and NODIPS-BH were confirmed by X-ray crystallography (Fig. 2). While the long alkyl chains of the NODIPS substituents helped promote tight π-stacking in the previously reported bistetracene through the “zipper effect”,20 the crystal structures of both NODIPS-BP and NODIPS-BH are dominated by edge-to-face interactions. In both cases, the packing is driven primarily by C–H–π interactions between the NODIPS groups and the phenyl moieties, which direct the octyl chains parallel to the aromatic backbone. The crystal packing of our oblique pyrenes may be altered by tuning such intramolecular non-covalent interactions, with the goal of promoting long-range π-stacking. In the case of NODIPS-BH, the growth of X-ray quality crystals was also hindered by the incorporation of solvent into the crystal structure. Functionalization of the pendant phenyl groups may be a viable method to minimize solvent trapping and further tune the solid-state ordering in these compounds.
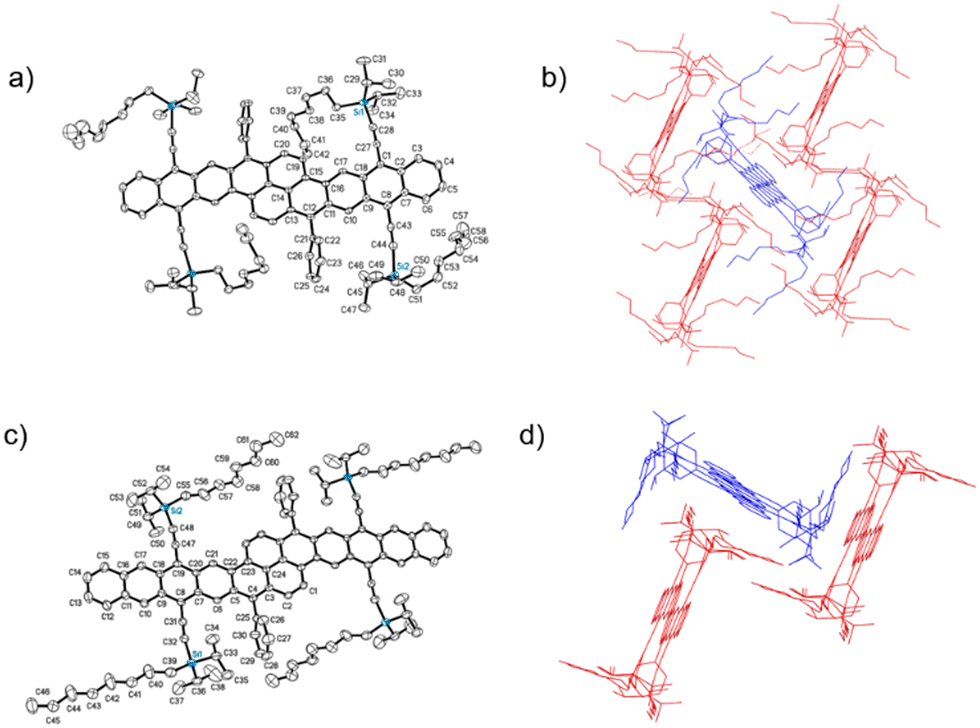 | ||
| Fig. 2 Crystal structure (a) and packing (b) of NODIPS-BP, and crystal structure (c) and packing (d) of NODIPS-BH. | ||
The UV-Vis-NIR absorbance spectra can be seen in Fig. 3. NODIPS-BP and NODIPS-BH feature onset absorbance peaks at 774 nm (1.60 eV) and 885 nm (1.40 eV), respectively. Both compounds feature a vibronic progression characteristic of acenes,27,28 which stretches from 800–600 nm in NODIPS-BP and from 900 nm to 700 nm in NODIPS-BH, indicating that extended oblique pyrenes display acene-like evolution of optical properties. The NIR absorbance profiles of both oblique-pyrenes are in stark contrast to vertical-pyrene derivatives of similar size. For example, TIPS-APA displays an onset absorbance peak at 505 nm, as the central pyrene disrupts electronic communication between the two anthracene moieties, resulting in an absorption profile between that of anthracene and tetracene.15 Thin-films of NODIPS-BP and NODIPS-BH both show absorption spectra similar to that observed in solution, consistent with a lack of aggregation effects in the solid-state. NODIPS-BP and NODIPS-BH display extremely weak emission peaks corresponding to S1 → S0 transitions in their respective photoluminescent spectra (Fig. S2, ESI†), with quantum yields < 1% for both compounds.
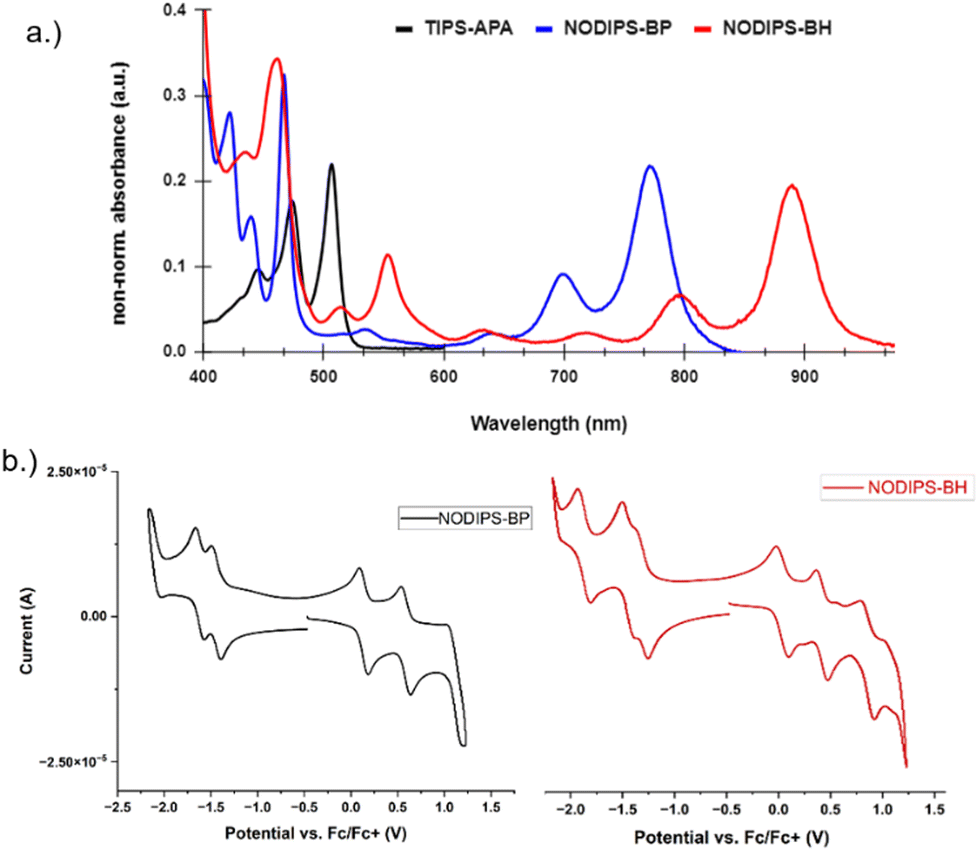 | ||
| Fig. 3 (a) UV-Vis-NIR absorbance spectrum of TIPS-APA, NODIPS-BP, and NODIPS-BH in CHCl3; (b) CV spectra of NODIPS-BP and NODIPS-BH in DCM. | ||
The electrochemical properties of both compounds were investigated using cyclic voltammetry (Fig. 3a). The onset oxidation/reduction potentials vs. Fc/Fc+ were 0.14/−1.44 eV for NODIPS-BP, and 0.037/−1.29 eV for NODIPS-BH. Linear fusion of additional benzene rings results in destabilization of the HOMO and stabilization of the LUMO, following the trend seen in the linear acene series. While NODIPS-BP displays two reversible oxidation and reduction peaks, extending the molecular length results in three reversible oxidation and reduction peaks for NODIPS-BH, consistent with greater delocalization of the HOMO and LUMO. The larger degree of separation between the first two oxidation peaks compared to the respective reduction peaks suggest that the radical anion is more localized than the radical cation, resulting in decreased charge repulsions in the dianion compared to the dication.29 The larger oxidation/reduction potentials observed for TIPS-APA (Fig. S1, ESI†) can be attributed to the localization of the cation/anion on the acene groups, with little-to-no interaction between the HOMOs and LUMOs of the anthracene moieties.15
To gain further insight into the electronic structure of our oblique pyrenes, density functional theory (DFT) calculations were carried out to examine the FMOs for TAS-substituted BP and BH (Fig. 4a). The HOMO and LUMO appear to fully delocalize across the aromatic core, which is consistent with the absorption and electrochemical properties observed in NODIPS-BP and NODIPS-BH. The HOMO−1 and LUMO+1 orbitals are localized primarily on the acene moieties (Fig. 4a and Fig. S7, ESI†), which offers a rationale for the acene-like absorption properties and rapid evolution of electronic character achieved through linear extension of the aromatic core. The FMOs of BP and BH consist of combinations of the FMOs of the equivalent acenes. The HOMO of BP can be considered as the bonding interaction between two pentacene HOMOs, while the HOMO−1 bears resemblance to two anthracene HOMOs, with conjugation across the pyrene unit disrupted by the mismatch in phase of the MO lobes. In analogy to bonding/antibonding interactions, relatively large electronic couplings between the HOMO and HOMO−1 for BP (0.64 eV) and BH (0.51 eV) results in a ground state electronic structure consistent with fully delocalized FMOs. While the HOMO and LUMO of APA (Fig. S8, ESI†) appear to fully delocalize across the pyrene moiety, in-depth computational studies have shown that a small energy splitting between the HOMO and the acene-like HOMO−1 result in limited electronic communication between the acene and pyrene segments.15
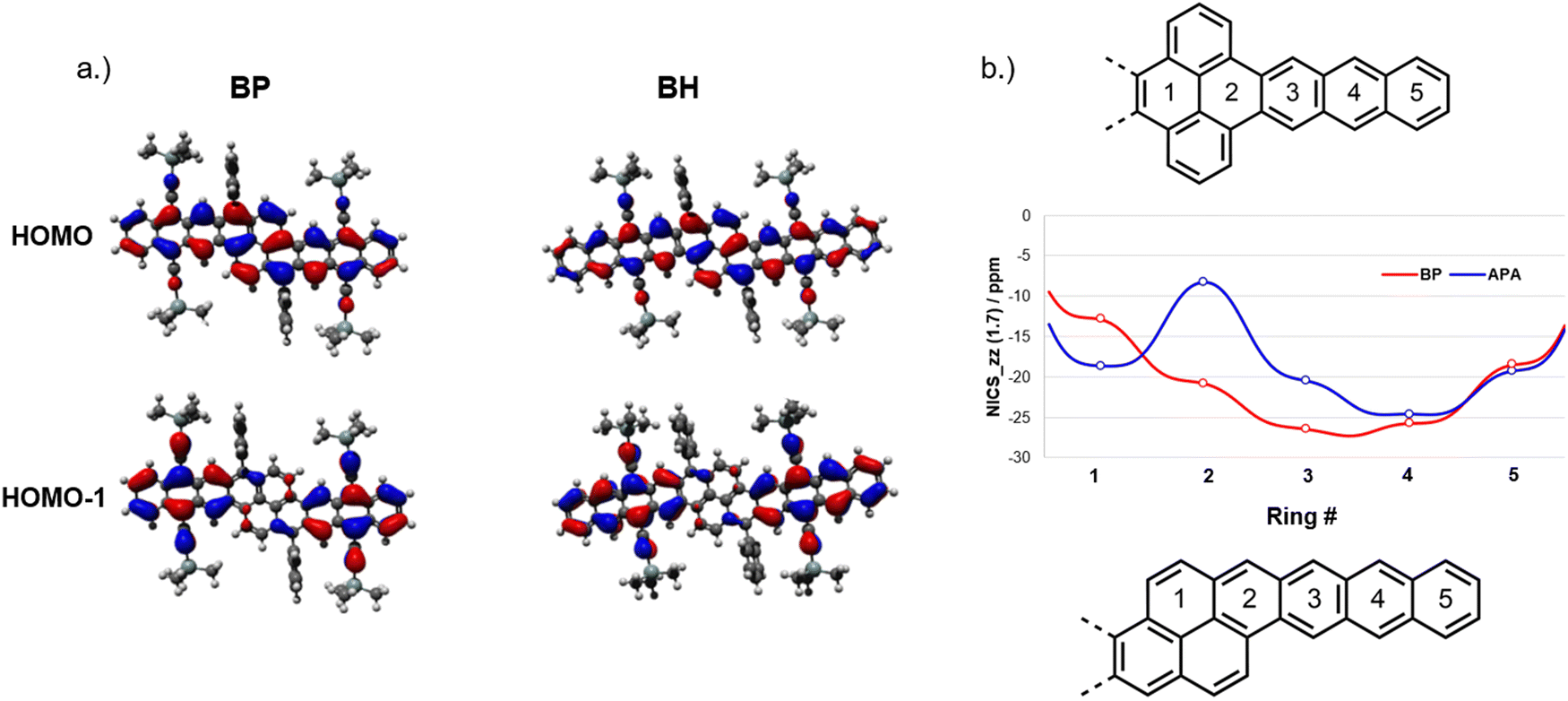 | ||
| Fig. 4 (a) HOMO and HOMO−1 of TAS-substituted BP and BH, and (b) NICS-xy scans of vertical and oblique pyrenes. | ||
The impact of pyrene-fusion on the aromaticity of oblique pyrene systems was investigated using nucleus-independent chemical shifts (NICS) calculations (Fig. 4b). For the oblique pyrenes, the ring directly adjacent to the acene segment (ring 2) sees only a modest reduction in aromatic character relative to those on the acene. Similarly, experimental bond lengths (Fig. S6, ESI†) obtained from XRD show decreased bond-length alternation along the zig-zag edges of rings 2–5 relative to ring 1, indicative of enhanced aromaticity on the acene segments. NICS scans of the analogous vertical pyrene (Fig. 4b) show diminished chemical shift in the ring adjacent to the acene segment, which results in limited electronic interactions between the acene and pyrene. The ACID plot (Fig. S9, ESI†) of BP shows a diatropic ring current largely delocalized onto the pyrene, while the analogous vertical pyrene maintains ring currents localized on the acene segments as well as the top and bottom rings on the pyrene moiety.
The biradical character (y0) was calculated for BP and BH at the UHF/6-31G level of theory.30,31 While BP displays a y0 of 0.42 and is predicted to maintain a closed-shell ground state, the large y0 of BH (0.69) predicts an open-shell ground state, which is supported by significant peak broadening observed in the 1H NMR spectrum of NODIPS-BH (Fig. S5, ESI†). The aromatic peaks gradually sharpen upon cooling from 30 °C to −15 °C, suggesting that NODIPS-BH has a triplet diradical form that is thermally populated near room temperature.32–34 The presence of a weak ESR signal at g = 2.0048 at room temperature (Fig. S3, ESI†) further supports diradical contribution to the ground state of NODIPS-BH. The BH aromatic core thus adds to the considerable and growing list of environmentally stable open-shell materials.22,35,36
Much like previously reported fused-acene derivatives,20,22,23NODIPS-BP and NODIPS-BH display enhanced stability in solution relative to TIPS-Pentacene, even upon prolonged exposure to an intense light source (Fig. 5a). Decomposition occurs much more slowly in toluene compared to CHCl3 (Fig. S4, ESI†), consistent with previous stability studies from the literature.37 Surprisingly, NODIPS-BH appears significantly more stable than NODIPS-BP in both solvents tested. This trend runs counter that seen in linear acenes, where extension of the aromatic core typically results in decreased stability.
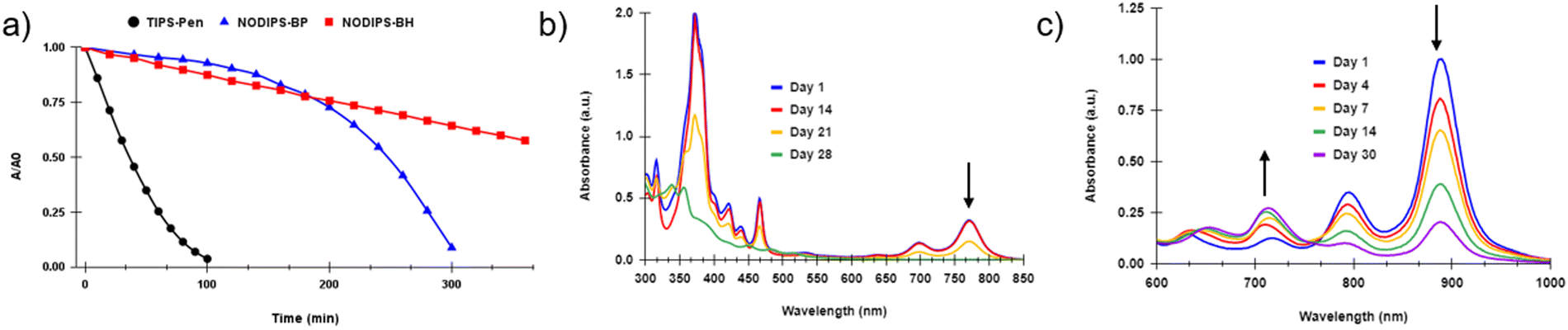 | ||
| Fig. 5 (a) Stability experiments in CHCl3, where onset absorbance normalized at t = 0 is monitored over time under exposure to 150-watt white light source. Absorbance spectra of (b) NODIPS-BP and (c) NODIPS-BH under ambient light exposure. | ||
Samples of NODIPS-BP and NODIPS-BH in CHCl3 were left under constant exposure to ambient laboratory light over the course of 1 month, and the decomposition of the samples was monitored by UV-Vis-NIR (Fig. 5b and c). While both compounds displayed half-lives between 1–2 weeks under such conditions, the manner in which they decomposed differed significantly. NODIPS-BP shows little apparent decay after two weeks, followed by rapid decay after 3 weeks and complete decay after 4 weeks. In contrast to the exponential rate of decay observed for NODIPS-BP, the decomposition of NODIPS-BH proceeds linearly with a half-life of approximately 14 days. Notably, the decay of the onset absorbance at 885 nm is accompanied by the steady rise of a feature at 710 nm, which can be attributed to the photodecomposition product generated upon continuous light exposure (vide infra).
Photooxidation of functionalized acenes commonly occurs due to reaction with singlet oxygen (1O2), which may be generated upon photoexcitation of the acene, provided that its T1 is larger than the singlet–triplet gap (ΔST) of O2.7 To better understand the stability of these compounds, DFT (M06-2X/cc-pVTZ, see ESI† for details) was employed to study the energetics of reactions of oblique pyrenes with 1O2. Fig. 6 (top) shows the activation energies for each ring of pentacene (PN), BP, and BH in the reaction with 1O2 relative to each ring. The most reactive ring in BP has a slightly higher activation energy than PN, while the activation energies for rings 3 and 4 in BH are significantly lower (4.6 and 7.1 kJ mol−1versus 16.0 kJ mol−1 for TIPS-PN). The reaction energies shown in Fig. 6 (bottom) underscore the importance of silylethyne substitution on stability: endoperoxide formation on ring 4, the ring featuring the silylethyne substituents (Fig. 6, filled circles), is thermodynamically less favorable compared to the same ring on the unsubstituted oblique pyrenes (Fig. 6, open circles). This effect has been well-documented in TIPS-pentacene,7 and is reflected in the reaction energies in Fig. 6 with a consistent stabilization of 55 kJ mol−1 for BP and BH.
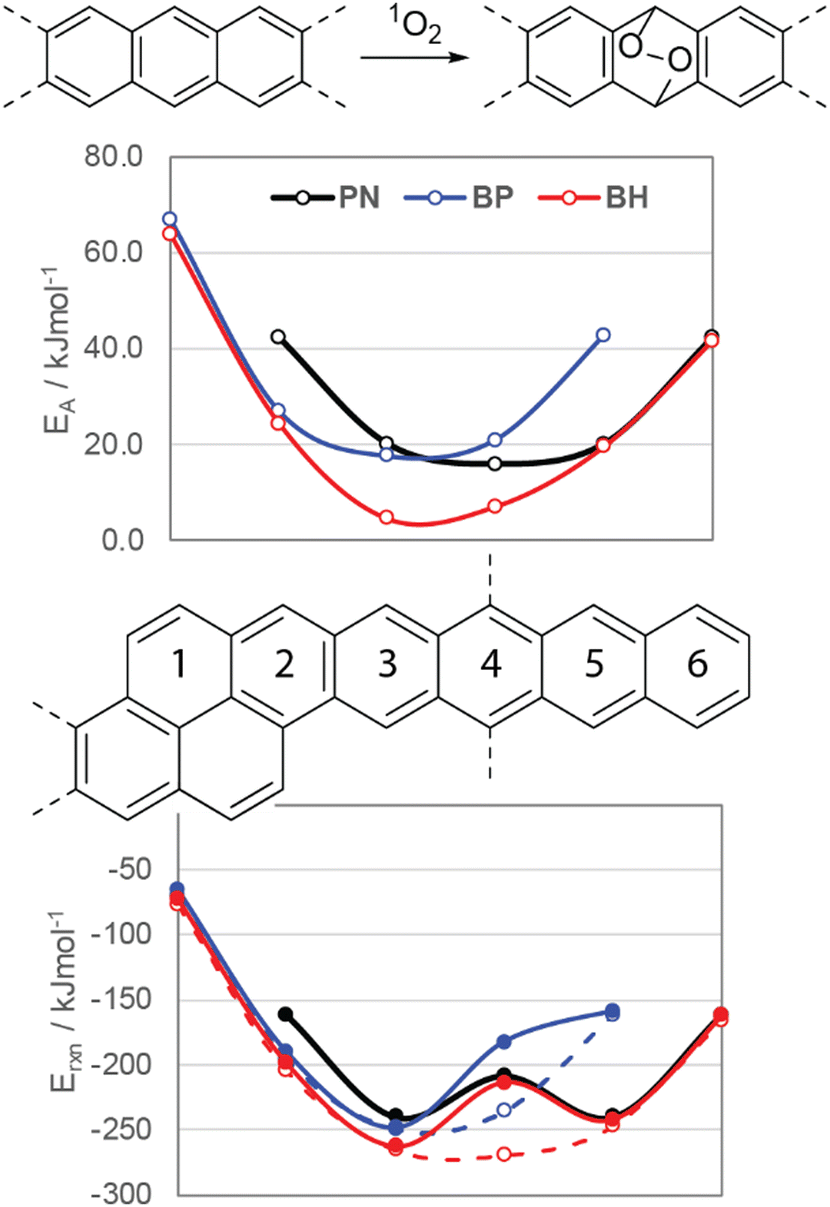 | ||
| Fig. 6 (top) Activation energies and (bottom) reaction energies of bisacenes with singlet oxygen at different ring positions. Open circles are without the silylethyne groups, and filled circles are with these groups included. All energies computed with M06-2X/cc-pVTZ. | ||
In the case of NODIPS-BP, the calculated energies in Fig. 6 are in good agreement with the experimentally observed stability in relation to TIPS-Pen. NODIPS-BH, however, displays even greater resistance to photooxidation compared to NODIPS-BP despite its substantially lower activation energy with 1O2. This can be rationalized by the low T1 of NODIPS-BH (Table 1), which is significantly lower than the ΔST of O2 (0.98 eV) and is sufficiently small to inhibit the generation of reactive 1O2 species, offering a likely explanation for improved photostability. The accelerating decomposition rate of NODIPS-BP over time suggests a different photooxidation mechanism may be in action. S1 and T1 were also calculated for decomposition products BP-EPO and BH-EPO, which feature endoperoxide formation on the most reactive ring (3) of BP and BH, respectively. The unique decomposition pattern seen in NODIPS-BP may be attributed to the initially slow formation of BP-EPO, which can more efficiently generate 1O2 due to its larger calculated T1 energy (1.20 eV) and thus accelerate the rate of decomposition of BPvia an energy-transfer mechanism. Notably, no absorbance feature near 597 nm corresponding to BP-EPO is observed following the decay of NODIPS-BP, suggesting that BP-EPO may promptly decompose to a second photooxidation product BP-EPO2 (Fig. S9, ESI†).
| Compound | S0–S1 (eV) | S0–S1 (nm) | S0–T1 (eV) |
|---|---|---|---|
| TIPS-Pen | 1.88 | 658 | 0.89 |
| BP | 1.64 | 755 | 0.84 |
| BH | 1.34 | 927 | 0.57 |
| BP-EPO | 2.08 | 597 | 1.20 |
| BH-EPO | 1.71 | 724 | 0.85 |
The reaction energy for the formation of BP-EPO2 (−230 kJ mol−1) is similar to that found for the formation of BP-EPO (−249 kJ mol−1), suggesting that the former process is slightly less thermodynamically favourable. Thus, the increased generation of reactive 1O2 by BP-EPO would be a plausible explanation for its prompt decomposition. Notably, an emission peak at 630 nm is observed in the photoluminescence spectrum of NODIPS-BP (Fig. S2, ESI†), potentially due to trace amounts of BP-EPO generated upon excitation. The absorbance feature near 710 nm observed upon decomposition of NODIPS-BH corresponds to the calculated absorbance of BH-EPO. The calculated T1 energy for BH-EPO (0.85 eV) is larger than that of NODIPS-BH but is still too small to efficiently generate 1O2, and thus should not play a significant role in the photooxidation of the starting material. The emission spectrum of NODIPS-BH features a prominent peak at 715 nm that intensifies upon further photobleaching (Fig. S2, ESI†), demonstrating that the decomposition products of both acenoacenes are highly emissive. These results highlight the importance of considering the properties of decomposition products when attempting to develop nanographenes with enhanced stability.
Conclusions
A series of oblique pyrenes/acene hybrids were prepared utilizing a synthetic route that allows for exquisite control over molecular length and functionalities. NODIPS-BP and NODIPS-BH display onset absorbance energies stretching into the near-IR, as well as decreased electrochemical gaps with increased molecular length, suggesting that linear extension of oblique pyrenes tunes their electronic structure in a manner similar to their linear acene counterparts. In contrast to the much more commonly reported vertical pyrenes, where the acene units on either side of the pyrene core are electronically isolated, the oblique pyrenes display experimental and computational properties consistent with fully conjugated aromatic ribbons. While acenes generally become less stable upon linear ring extension, BH appears more resistant to photodecomposition than BP, even upon prolonged light exposure. This counterintuitive trend is attributed to a low T1 level in BH that inhibits the generation of 1O2via an energy transfer mechanism, and thus substantially slows the rate of photodecomposition. The decomposition products play a significant role in the stability of BP and BH, as the endoperoxide formed upon photooxidation of NODIPS-BP appears to catalyse its further decay. Along with highlighting the critical importance of molecular orientation to the electronic properties of aromatic ribbons, this work demonstrates that oblique pyrene/acene hybrids with as many as 12-consecutively fused rings are remarkably resilient to photooxidation under ambient conditions, and opens the path to both larger oblique pyrene derivatives and potential graphitic ribbon polymers.Author contributions
T. S. and J. A. devised the project. K. T. carried out all theoretical investigations, while T. S. and J. A. contributed to molecular synthesis and characterization. S. P. solved the x-ray crystal structures. O. O. and K. D. carried out photophysical experiments. T. S., J. A., and K. T. wrote the article.Data availability
All data regarding the synthesis and characterization of the molecules presented in this work are available in the ESI.† Crystallographic data were deposited at the Cambridge Crystallographic Data Centre, with access numbers 2325313 for NODIPS-BP and 2325314 for NODIPS-BHConflicts of interest
The authors have no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation under Cooperative Agreement No. 1849213 (University of Kentucky) and grant CHE-1956431 (Oregon State University).Notes and references
- A. Narita, X. Y. Wang, X. Feng and K. Mullen, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC.
- O. Yazyev, Acc. Chem. Res., 2013, 46, 2319–2328 CrossRef CAS PubMed.
- K. B. Wiberg, J. Org. Chem., 1997, 62, 5720–5727 CrossRef CAS.
- K. J. Thorley and J. E. Anthony, Isr. J. Chem., 2014, 54, 642–649 CrossRef CAS.
- S. S. Zade and M. Bendikov, J. Phys. Org. Chem., 2012, 25, 452–461 CrossRef CAS.
- J. E. Anthony, J. S. Brooks, D. L. Eaton and S. R. Parkin, J. Am. Chem. Soc., 2001, 123, 9482–9483 CrossRef CAS.
- W. Fudickar and T. Linker, J. Am. Chem. Soc., 2012, 134, 15071–15082 CrossRef CAS.
- B. Purushothaman, S. R. Parkin, M. J. Kendrick, D. David, J. W. Ward, L. Yu, N. Stingelin, O. D. Jurchescu, O. Ostroverkhova and J. E. Anthony, Chem. Commun., 2012, 48, 8261–8263 RSC.
- Z. Liu, S. Fu, X. Liu, A. Narita, P. Samori, M. Bonn and H. I. Wang, Adv. Sci., 2022, 9, e2106055 CrossRef.
- Y. Gu, Z. Qiu and K. Mullen, J. Am. Chem. Soc., 2022, 144, 11499–11524 CrossRef CAS.
- Q. Ai, K. Jarolimek, S. Mazza, J. E. Anthony and C. Risko, Chem. Mater., 2018, 30, 947–957 CrossRef CAS.
- D. Cortizo-Lacalle, J. P. Mora-Fuentes, K. Strutynski, A. Saeki, M. Melle-Franco and A. Mateo-Alonso, Angew. Chem., Int. Ed., 2018, 57, 703–708 Search PubMed.
- F. Hernandez-Culebras, M. Melle-Franco and A. Mateo-Alonso, Angew. Chem., Int. Ed., 2022, 61, e202205018 Search PubMed.
- J. Li, S. Chen, Z. Wang and Q. Zhang, Chem. Rec., 2016, 16, 1518–1530 CrossRef CAS PubMed.
- Q. Ai, T. Smith, A. D. T. Liyanage, S. M. Mazza, S. R. Parkin, J. E. Anthony and C. Risko, J. Mater. Chem. C, 2021, 9, 16929–16934 RSC.
- L.-C. Lin, T. Smith, Q. Ai, B. K. Rugg, C. Risko, J. E. Anthony, N. H. Damrauer and J. C. Johnson, Chem. Sci., 2023, 14, 11554–11565 RSC.
- A. Celis, M. N. Nair, A. Taleb-Ibrahimi, E. H. Conrad, C. Berger, W. A. de Heer and A. Tejeda, J. Phys. D: Appl. Phys., 2016, 49, 143001 CrossRef.
- V. Saraswat, R. M. Jacobberger and M. S. Arnold, ACS Nano, 2021, 15, 3674–3708 CrossRef CAS PubMed.
- H. Wang, H. S. Wang, C. Ma, L. Chen, C. Jiang, C. Chen, X. Xie, A.-P. Li and X. Wang, Nat. Rev. Phys., 2021, 3, 791–802 CrossRef CAS.
- L. Zhang, A. Fonari, Y. Liu, A. L. Hoyt, H. Lee, D. Granger, S. Parkin, T. P. Russell, J. E. Anthony, J. L. Bredas, V. Coropceanu and A. L. Briseno, J. Am. Chem. Soc., 2014, 136, 9248–9251 CrossRef CAS.
- S. Thomas, J. Ly, L. Zhang, A. L. Briseno and J.-L. Bredas, Chem. Mater., 2016, 28, 8504–8512 CrossRef CAS.
- T. Jousselin-Oba, M. Mamada, J. Marrot, A. Maignan, C. Adachi, A. Yassar and M. Frigoli, J. Am. Chem. Soc., 2019, 141, 9373–9381 CrossRef CAS.
- T. Jousselin-Oba, M. Mamada, K. Wright, J. Marrot, C. Adachi, A. Yassar and M. Frigoli, Angew. Chem., Int. Ed., 2022, 61, e202112794 CrossRef CAS PubMed.
- E. Clar, M. M. Lovat and W. Simpson, Tetrahedron, 1974, 30, 3293–3298 CrossRef CAS.
- M. P. Cava and R. L. Shirley, J. Am. Chem. Soc., 1960, 82, 654–656 CrossRef CAS.
- J. E. Anthony, D. L. Eaton and S. R. Parkin, Org. Lett., 2002, 4, 15–18 CrossRef CAS PubMed.
- H. B. Klevens and J. R. Platt, J. Chem. Phys., 1949, 17, 470–481 CrossRef CAS.
- J. Tanaka, Bull. Chem. Soc. Jpn., 1964, 38, 86–102 CrossRef.
- C. A. M. Liberko, L. L. Katz and T. J. Liu, J. Am. Chem. Soc., 1993, 115, 2478–2482 CrossRef CAS.
- K. Yamaguchi, Chem. Phys. Lett., 1975, 33, 330–335 CrossRef CAS.
- M. K. Nakano, R. Nitta, T. Kubo, T. Nakasuji, K. Kamada, K. Ohta, K. Champagne, B. Botek and E. Yamaguchi, J. Phys. Chem., 2005, 109, 885–891 CrossRef CAS PubMed.
- J. Liu, P. Ravat, M. Wagner, M. Baumgarten, X. Feng and K. Mullen, Angew. Chem., Int. Ed., 2015, 54, 12442–12446 CrossRef CAS PubMed.
- W. Zeng, H. Phan, T. S. Herng, T. Y. Gopalakrishna, N. Aratani, Z. Zeng, H. Yamada, J. Ding and J. Wu, Chemistry, 2017, 2, 81–92 CrossRef CAS.
- J. J. Dressler, A. Cárdenas Valdivia, R. Kishi, G. E. Rudebusch, A. M. Ventura, B. E. Chastain, C. J. Gómez-García, L. N. Zakharov, M. Nakano, J. Casado and M. M. Haley, Chemistry, 2020, 6, 1353–1368 CrossRef CAS.
- J. Liu and X. Feng, Angew. Chem., Int. Ed., 2020, 59, 23386–23401 CrossRef CAS.
- W. Zeng and J. Wu, Chemistry, 2021, 7, 358–386 CrossRef CAS.
- L. Abu-Sen, J. J. Morrison, A. B. Horn and S. G. Yeates, Adv. Opt. Mater., 2014, 2, 636–640 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, characterization information, and supplemental spectroscopy. CCDC 2325313 and 2325314. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4tc03072a |
| This journal is © The Royal Society of Chemistry 2024 |