
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDivergent total syntheses of pyrroloiminoquinone alkaloids enabled by the development of a Larock/Buchwald–Hartwig annulation/cyclization†
Samir P.
Rezgui
 ,
Jonathan
Farhi
,
Hao
Yu
,
Zachary P.
Sercel
,
Scott C.
Virgil
and
Brian M.
Stoltz
*
,
Jonathan
Farhi
,
Hao
Yu
,
Zachary P.
Sercel
,
Scott C.
Virgil
and
Brian M.
Stoltz
*
Warren and Katharine Schlinger Laboratory for Chemistry and Chemical Engineering, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA 91125, USA. E-mail: stoltz@caltech.edu
First published on 27th June 2024
Abstract
Pyrroloiminoquinone alkaloids are a large class of natural products that display a wide range of biological activities. Synthetic approaches to these natural products typically rely on a common late-stage C10-oxygenated pyrroloiminoquinone intermediate, but these strategies often lead to lengthy synthetic sequences that are not amenable to divergent syntheses. We devised an alternative approach aimed at the early introduction of the C10 nitrogen, which we hypothesized would enable late-stage diversification. This strategy hinged upon a Larock/Buchwald–Hartwig annulation/cyclization to quickly access the core of these alkaloids. We report the development of this cascade process, which was facilitated by a dual ligand system in addition to selective functionalization of the key intermediate, to provide efficient syntheses of makaluvamines A, C, and D and isobatzelline B, and the first total synthesis of makaluvamine N.
Pyrroloiminoquinone alkaloids are a large class of marine- and fungal-derived natural products that have received significant attention within the scientific community for over 30 years.1 This is in part due to their broad biological activities such as antitumor, antiviral, and antimicrobial activities, among many others.2 Structurally, pyrroloiminoquinone alkaloids are defined by a conserved pyrrolo[4,3,2-de]quinoline ring system and differ by the functionality appended to this central ring. These natural products typically contain other complex ring systems and a variety of heteroatoms, as exemplified by aleutianamine (1, Fig. 1A).3 Their complex and unique structures have inspired synthetic efforts for decades and several successful total syntheses of these alkaloids have been reported.4,5 Strategies typically utilize a common late-stage C10-oxygenated pyrroloiminoquinone intermediate 2, with amination serving to introduce the final nitrogen atom. While this approach has proved successful, there is limited divergency from this common iminoquinone intermediate, and other functionality appended to the central core (i.e., R1–R4) must be introduced earlier in the synthesis. This lack of divergency could explain the paucity of structure–activity relationship (SAR) studies of pyrroloiminoquinone alkaloids.6 Additionally, access to the common intermediate 2 typically necessitates lengthy synthetic sequences due to the linear nature of arene functionalization steps (see ESI† for a detailed discussion of synthetic strategies toward the common intermediate 2).
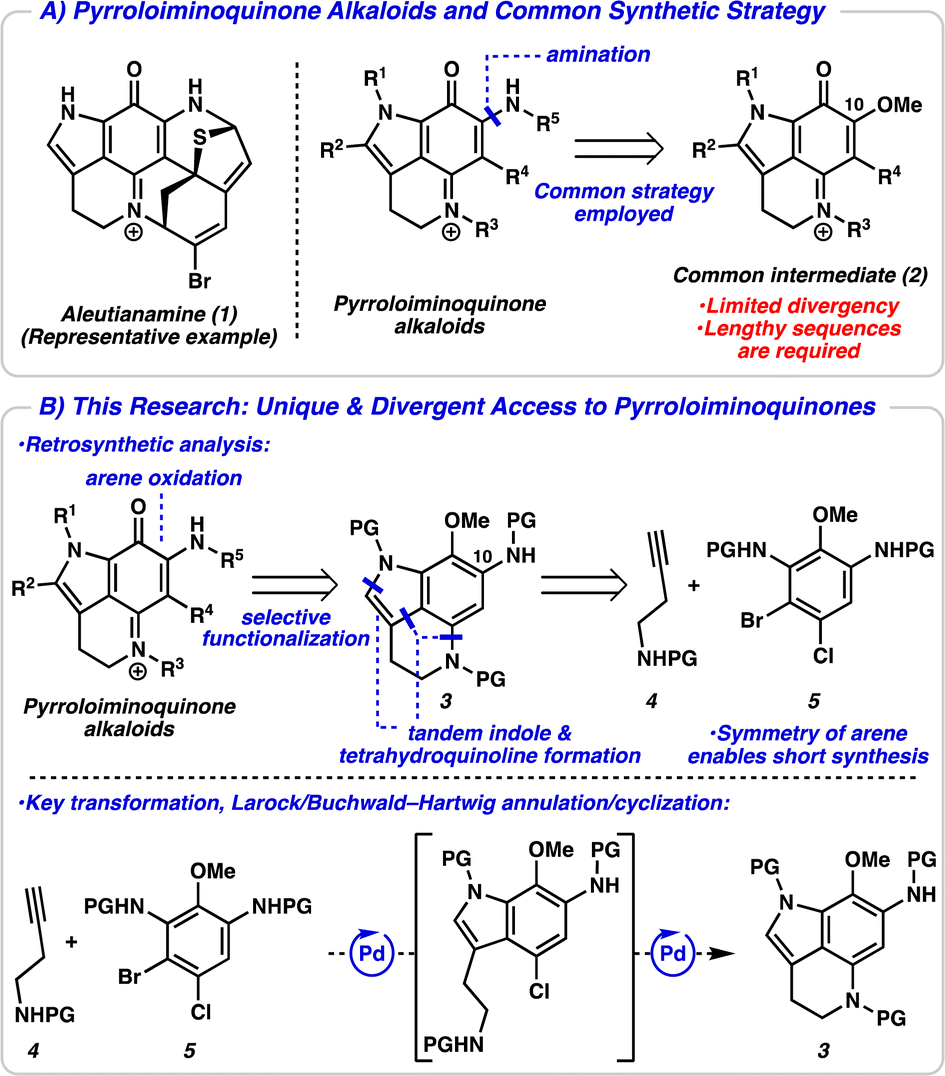 | ||
| Fig. 1 (A) Representative pyrroloiminoquinone and the common strategy employed toward these alkaloids. (B) Our strategy toward pyrroloiminoquinones and the key transformation to realize this strategy. | ||
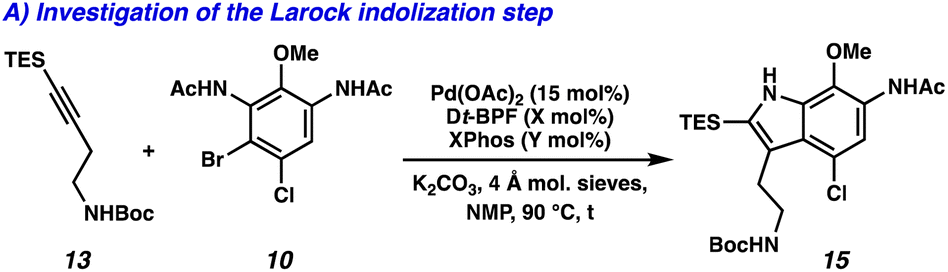
We devised an alternative approach in order to address the issues of divergence and step count, envisioning a direct arene oxidation state adjustment leading back to an aminoindole (3), an uncommon disconnection that avoids the use of the common intermediate 2 (Fig. 1B).7 We hypothesized that with judicious choice of nitrogen protecting groups, selective functionalization of all five possible positions of the pyrroloiminoquinone scaffold (R1–R5) could be achieved. We next disconnected both the indole and tetrahydroquinoline, revealing a pseudo symmetric arene (5). We imagined that the symmetry of this highly substituted arene would simplify its synthesis, precluding the need for a lengthy synthetic sequence. Critical to the success of this strategy is the indolization/tetrahydroquinoline formation step. We envisioned that the development of a tandem Larock/Buchwald–Hartwig annulation/cyclization could enable this approach. While both the Larock indole synthesis and Buchwald–Hartwig amination are well documented independently,8,9 to the best of our knowledge a tandem Larock/Buchwald–Hartwig annulation/cyclization has yet to be reported. To this end, we set out to access the Larock/Buchwald–Hartwig annulation/cyclization substrates.
Dinitration of 4-chloroanisole (6) proceeded smoothly to provide dinitro arene 7, which underwent reduction to the corresponding dianiline (8) (Scheme 1A). Phosphomolybdic acid (PMA)-mediated acetylation10 then yielded diacetanilide 9. Finally, monobromination served as the desymmetrizing step to provide the first Larock/Buchwald–Hartwig cyclization substrate 10 in just 4 steps. The synthesis of the alkyne coupling partner commenced from commercially available alkyne 11, which was silylated followed by substitution of the tosyl group with NaN3 to afford azide 12 (Scheme 1B). Azide 12 was then converted to the amine via Staudinger reduction, which was subsequently Boc protected to provide alkyne coupling partner 13 in 4 total steps.
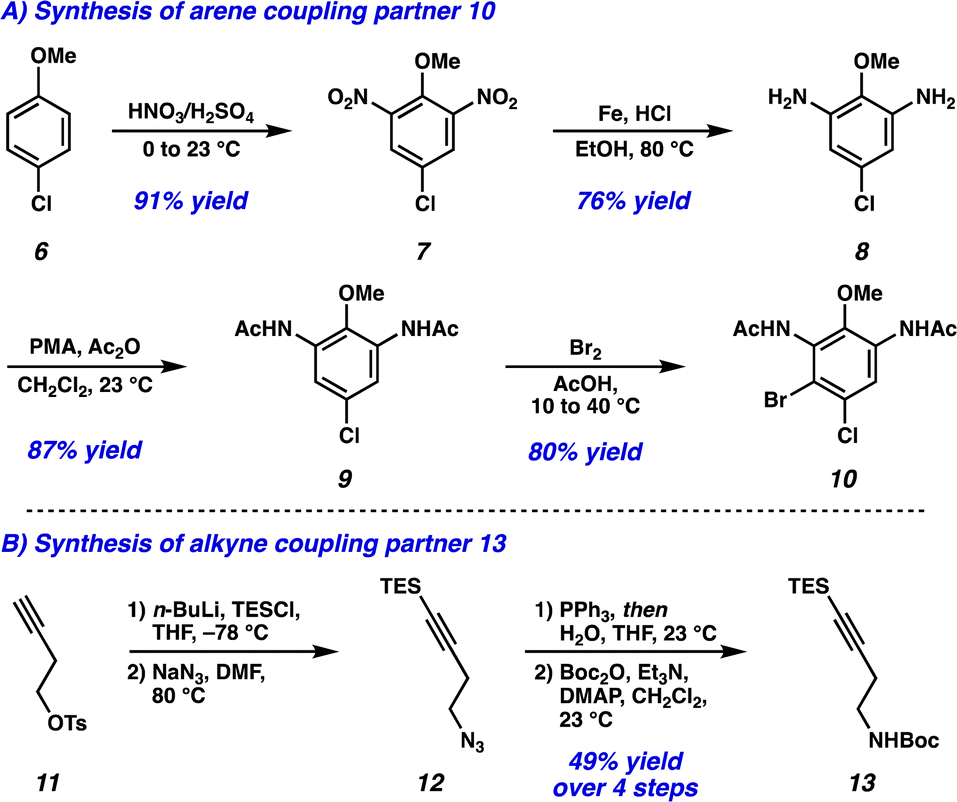 | ||
| Scheme 1 (A) Synthesis of arene coupling partner 10. (B) Synthesis of alkyne coupling partner 13. | ||
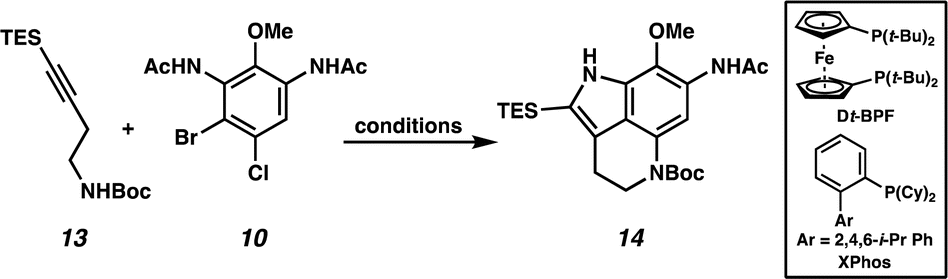
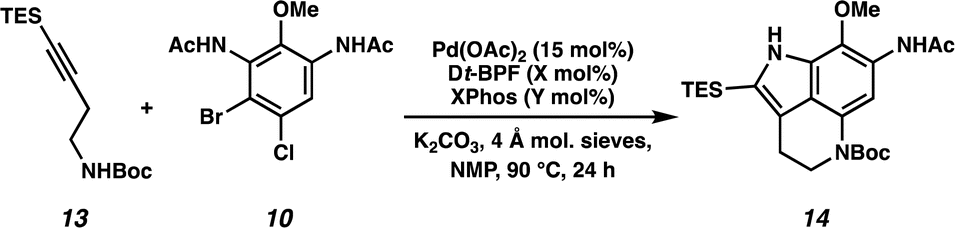
We next turned our attention to the development of the key Larock/Buchwald–Hartwig annulation/cyclization. Since Larock's original disclosure of the palladium-mediated indolization of alkynes and o-iodoanilines, the scope has been expanded to o-bromoanilines through catalyst and ligand design.9 Despite these efforts, examples of substrates bearing bis ortho substitution with respect to the halide are rare, so we anticipated that this would be a difficult transformation to realize. We first tested the conditions reported by Reisman and coworkers,11 which employ Pd[P(t-Bu)3]2 as the catalyst and ligand source along with a soluble amine base (Cy2NMe), but these led to no detectable product formation and primarily protodebromination of arene 10 (Table 1, entry 1). We next evaluated the conditions reported by Boger and coworkers12 which again utilize a Pd (0) precatalyst, now in combination with the electron rich 1,1′-bis(di-tert-butylphosphino)ferrocene (Dt-BPF) ligand and Et3N as the base, but protodebromination was similarly observed with no desired product formation (entry 2). Utilizing modified conditions reported by Senanayake and coworkers,13 which implement a Pd(II) precatalyst and Dt-BPF as the ligand, now using an inorganic base (K2CO3), we were encouraged to isolate 15% yield of desired tricycle 14 (entry 3). This represents a powerful transformation, as 3 bonds and 2 rings are formed in a single synthetic step. This yield could be improved to 32% by increasing the catalyst and ligand loading, using higher equivalents of alkyne 13, and freshly drying the K2CO3 (entry 4). After extensive investigation, we discovered that the addition of another ligand (XPhos) to the system improved the yield to 69% (entry 5, see ESI† for additional optimization details). Lastly, the addition of 4 Å molecular sieves addressed issues with reproducibility, likely caused by the presence of water. Overall, this transformation represents the first example of a tandem Larock/Buchwald–Hartwig annulation/cyclization and provides access to the core of the pyrroloiminoquinone alkaloids in a longest linear sequence (LLS) of just 5 steps, which to the best of our knowledge is the shortest synthetic sequence to such a tricyclic intermediate.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Ligand | Base | Solvent | Temp/time | Yield (%) |
| Reactions performed on a 0.1 mmol scale at 0.1 M. Yields refer to isolated yields.a 2.0 equiv. of alkyne 13, 2.5 equiv. of base.b 5.0 equiv. of alkyne 13, 5.0 equiv. of freshly dried powdered K2CO3.c 0.44 mmol scale, 5.0 equiv. of alkyne 13, 5.0 equiv. of freshly dried granular K2CO3, freshly activated 4 Å mol. sieves. | ||||||
| 1a | Pd[P(t-Bu)3]2 (10 mol%) | — | Cy2NMe | 1,4-Dioxane | 80 °C, 24 h | 0 |
| 2a | Pd2(dba)3 (10 mol%) | Dt-BPF (20 mol%) | Et3N | DMF | 130 °C, 30 h | 0 |
| 3a | Pd(OAc)2 (5 mol%) | Dt-BPF (10 mol%) | K2CO3 | NMP | 110 °C, 24 h | 15 |
| 4b | Pd(OAc)2 (15 mol%) | Dt-BPF (30 mol%) | K2CO3 | NMP | 110 °C, 24 h | 32 |
| 5c | Pd(OAc)2 (15 mol%) | Dt-BPF (15 mol%) + XPhos (15 mol%) | K2CO3 | NMP | 90 °C, 24 h | 69 |
We were next interested into gaining insight on how the dual ligand system was uniquely effective in this transformation. First, we investigated the effects of the ligand stoichiometry under the optimal reaction conditions. The ratio of Dt-BPF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :XPhos was varied, while keeping the total ligand percentage at 30 mol% (2 equivalents with respect to Pd). If an excess of Dt-BPF was used in respect to XPhos, the efficiency of the annulation/cyclization was diminished (Table 2, entries 1–3). In these cases, a large portion of the remaining mass balance was the intermediate indole product (vide infra). Efficient formation of tricyclic product 14 was restored under the optimal reaction conditions consisting of a 1:1 Dt-BPF:XPhos ratio (entry 4). Interestingly, a 1:2 Dt-BPF:XPhos mixture was also effective, providing 14 in a 64% yield (entry 5). However, once a larger excess of XPhos was employed the yield of 14 decreased (entries 6 and 7). In these cases, the reaction profile was more complicated with unidentifiable side/decomposition products.
:XPhos was varied, while keeping the total ligand percentage at 30 mol% (2 equivalents with respect to Pd). If an excess of Dt-BPF was used in respect to XPhos, the efficiency of the annulation/cyclization was diminished (Table 2, entries 1–3). In these cases, a large portion of the remaining mass balance was the intermediate indole product (vide infra). Efficient formation of tricyclic product 14 was restored under the optimal reaction conditions consisting of a 1:1 Dt-BPF:XPhos ratio (entry 4). Interestingly, a 1:2 Dt-BPF:XPhos mixture was also effective, providing 14 in a 64% yield (entry 5). However, once a larger excess of XPhos was employed the yield of 14 decreased (entries 6 and 7). In these cases, the reaction profile was more complicated with unidentifiable side/decomposition products.
To gain further insight, we studied each step of the tandem process individually while adjusting the ligand stoichiometry. We first studied the Larock indolization process by monitoring the consumption of bromoarene 10 by LC/MS. We found that either Dt-BPF or XPhos on their own could facilitate the Larock indolization with similar efficiencies, although a significant reaction rate difference was observed (Table 3A, entries 1 and 3). Interestingly, the yield of indole 15 was increased when a 1:1 Dt-BPF:XPhos ratio was employed, suggesting a synergistic effect of the dual ligand system on the Larock indolization process (entry 2). The lower yield of just the Larock indolization process (Table 3A, entry 2) compared to the tandem process (Table 1, entry 5) is likely due to quenching of intermediates on the catalytic cycle, as the reaction was stopped as soon as bromoarene 10 was consumed.‡ We next studied the Buchwald–Hartwig step by subjecting indole 15 to different ligand mixtures. Dt-BPF on its own only provided a 20% yield of tricycle 14 (Table 3B, entry 1). However, the addition of XPhos formed 14 in a 87% yield (entry 2). A similar result was obtained when XPhos was used on its own, however the rate of product formation was faster compared to the 1:1 Dt-BPF:XPhos mixture (entry 3). These results suggest that XPhos facilitates the Buchwald–Hartwig amination step of the tandem annulation/cyclization process.
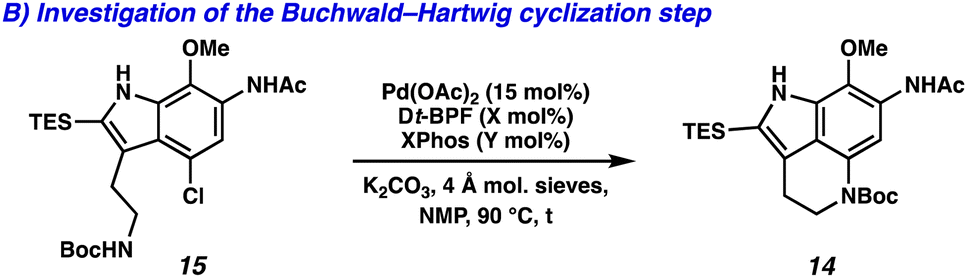
Taken together, we hypothesize that under the optimal reaction conditions a dynamic ligand exchange process between Dt-BPF and XPhos on the palladium center gives rise to a synergistic effect on the Larock indolization, a phenomenon that has been well documented for mixed ligand systems in palladium catalysis.14 Additionally, the ligand exchange allows for XPhos to facilitate the Buchwald–Hartwig amination step efficiently. This transformation highlights the utility of mixed ligand systems in palladium catalysis and suggests it should be explored more generally in reaction optimization.
With these results in hand, we were interested to see if this method could be applied more generally. Using commercially available 2-bromo-3-chloroaniline resulted in a 75% yield of tricycle 16 (Scheme 2). Additionally, the catalyst and ligand loading could be lowered to 5 mol% without a significant decrease in yield (i.e., 71% yield). N-Substitution on the aniline was tolerated, providing both N-Me (17) and N-Boc (18) cyclization products. Excitingly, employing 3-bromo-2-chloropyridine resulted in a 41% yield of azaindole derivative 19, demonstrating the compatibility of heterocyclic substrates in the transformation. Finally, an alkyne coupling partner with a longer carbon chain could be used to provide the 5,6,7-tricyclic product 20 in 57% yield.
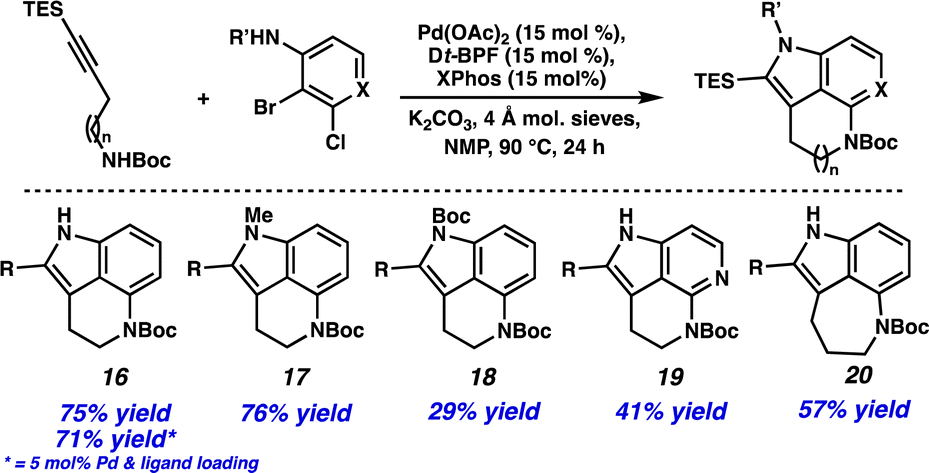 | ||
| Scheme 2 Selected substrate scope. R = TES. | ||
With the successful development of the Larock/Buchwald–Hartwig annulation/cyclization, we turned our attention toward selectively functionalizing all five positions on tricycle 14 to access an array of pyrroloiminoquinone natural products. To this end, the Larock/Buchwald–Hartwig annulation/cyclization could be followed by an acidic workup to provide desilylated tricycle 21 (Scheme 3A). Employing modified conditions from Ohshima and coworkers,15 the acetamide could be selectively removed in the presence of the carbamate, yielding aniline 22. Finally, lithium aluminum hydride (LAH) reduction of the carbamate to the N-Me moiety was accompanied by spontaneous aerobic oxidation to yield makaluvamine C (23) in just 7 steps (LLS).
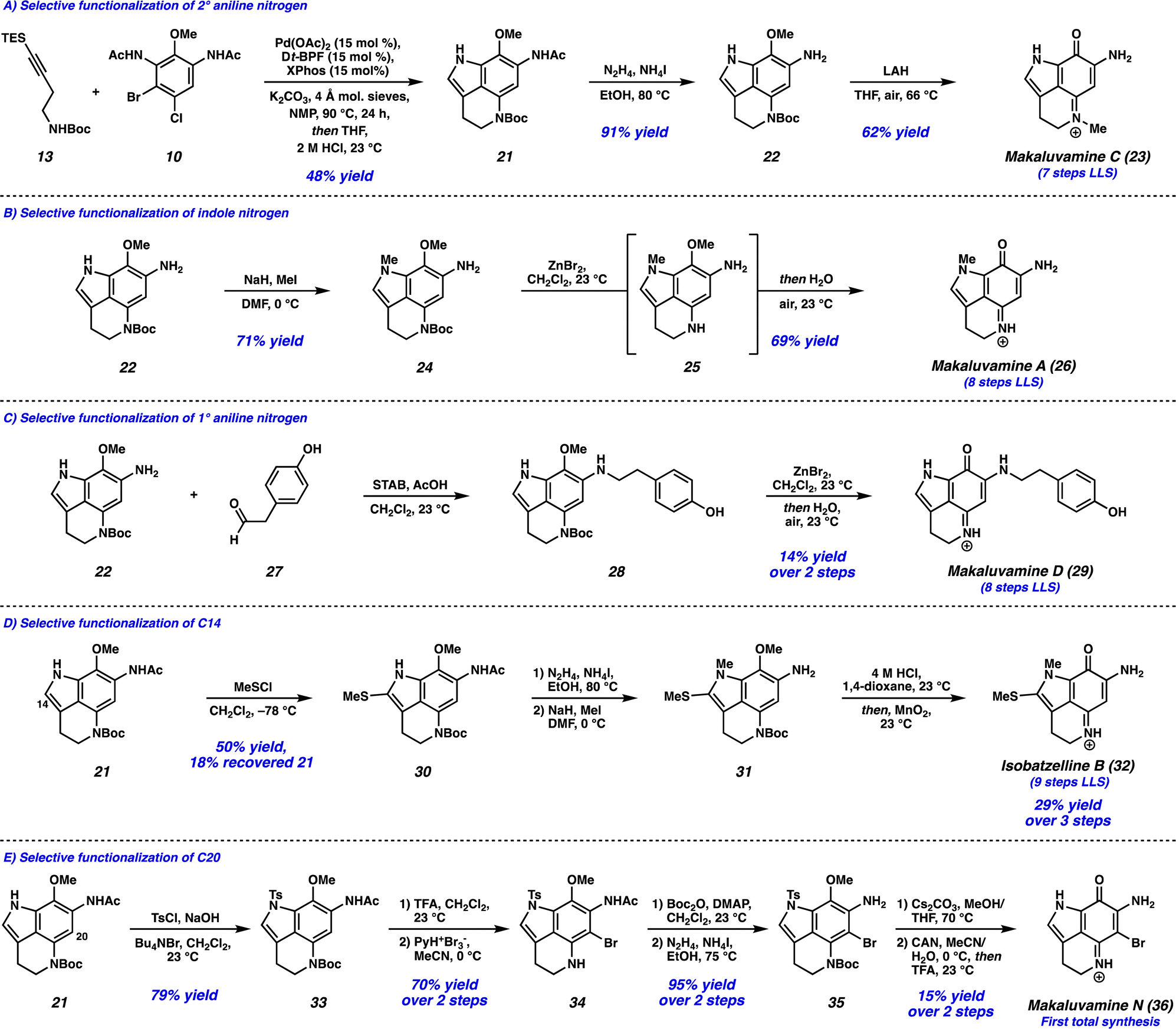 | ||
| Scheme 3 Divergent syntheses of 5 pyrroloiminoquinone natural products from key tricyclic intermediate 21. (A) Selective functionalization of the 2° aniline nitrogen. (B) Selective functionalization of the indole nitrogen. (C) Selective functionalization of the 1° aniline nitrogen. (D) Selective functionalization of C14. (E) Selective functionalization of C20. | ||
We next investigated the selective functionalization of the indole nitrogen (Scheme 3B). Starting with aniline 22, selective N-methylation of the indole nitrogen was achieved under basic conditions to give tricycle 24. We attempted canonical arene oxidation conditions to pyrroloiminoquinones such as CAN, Fremy's salt, or MnO2, but unfortunately non-specific decomposition of tricycle 24 was observed under these conditions. Excitingly, when the Boc group was first removed using ZnBr2,16 the resulting highly electron-rich aniline intermediate 25 underwent spontaneous aerobic oxidation, providing makaluvamine A (26) in 8 steps (LLS).
Next, we targeted the selective functionalization of the 1° aniline nitrogen (Scheme 3C). This was achieved using a reductive amination between aniline 22 and aldehyde 27, to yield N-alkylated aniline 28. Then, our mild arene oxidation protocol was employed to yield makaluvamine D (29) in 8 steps (LLS).
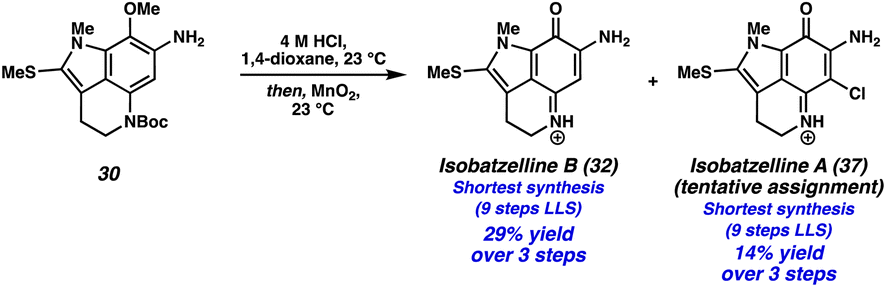
We then turned our attention to functionalizing the indole C14 position (Scheme 3D). Using modified conditions reported by Joule and coworkers,4i treatment of tricycle 21 with freshly prepared MeSCl (generated from Me2S2 and SO2Cl2) at −78 °C lead to selective thiomethylation at the C14 position, yielding sulfide 30. The use of an excess of Me2S2 to quantitively generate MeSCl, and performing the reaction at low temperatures was critical to suppressing arene chlorination or difunctionalization. From thiomethylated arene 30, acetamide cleavage followed by indole N-methylation provided tricycle 31. Unfortunately, this substrate performed poorly in the ZnBr2/O2 deprotection/oxidation step. Instead, we found that the Boc deprotection could be achieved using anhydrous HCl in dioxane, and the resulting intermediate (not shown) could undergo arene oxidation with MnO2 in the same flask to provide isobatzelline B (32) in 9 steps (LLS).§
Finally, we set out to selectively functionalize the C20 position of tricycle 21 (Scheme 3E). The indole nitrogen was first protected as a sulfonamide to yield tricycle 33, which then underwent Boc removal and selective C20 bromination to provide bromo-tricycle 34. Unfortunately, direct acetamide cleavage of this compound proved unsuccessful. Instead, the 2° aniline nitrogen of bromo-tricycle 34 was reprotected as the carbamate, which was followed by acetamide removal to yield bromoaniline 35. The tosyl group was then removed under basic conditions, and arene oxidation using CAN followed by Boc deprotection yielded makaluvamine N (36) in 12 steps (LLS), representing the first total synthesis of this natural product to date.
Conclusions
In summary, we devised a unique approach to five pyrroloiminoquinone alkaloids, which could likely be applied to additional natural products and analogs. Our strategy was enabled by the development of a novel Larock/Buchwald–Hartwig annulation/cyclization to rapidly access the core of these natural products. Critical to the success of this transformation was the employment of a dual ligand system. By judicious choice of nitrogen protecting groups, the key tricyclic intermediate was selectively functionalized in 5 different positions to provide efficient total syntheses of makaluvamines A (26), C (23), and D (29) and isobatzelline B (32), and the first total synthesis of makaluvamine N (36). We believe that this strategy will facilitate analog synthesis and the advancement of pyrroloiminoquinones as therapeutic candidates, which has historically been a challenge due to the lack of divergent synthetic routes. Additionally, we aim to use the key tricyclic intermediate 14 prepared by our tandem annulation/cyclization strategy to access more complex pyrroloiminoquinone natural products.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
S. P. R. conceived of and performed experiments. J. F., H. Y., Z. P. S., and S. C. V. also performed experiments. B. M. S. supported and supervised the research. S. P. R. wrote the original draft of the manuscript, which was edited by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge Caltech, the NSF (CHE-2247315), and the Heritage Medical Research Investigators Program for financial support. S. P. R. and Z. P. S. thank the NSF GRFP for predoctoral fellowships. Dr Mona Shahgholi (Caltech) is thanked for assistance with mass spectrometry. Dr David VanderVelde (Caltech) is acknowledged for NMR instrumentation. The authors also thank Prof. Sarah E. Reisman for helpful discussions.Notes and references
- For comprehensive reviews of pyrroloiminoquinone alkaloids see:
(a) J.-F. Hu, H. Fan, J. Xiong and S.-B. Wu, Chem. Rev., 2011, 111, 5465–5491 CrossRef CAS
; (b) E. M. Antunes, B. R. Copp, M. T. Davies-Coleman and T. Samaai, Nat. Prod. Rep., 2005, 22, 62–72 RSC
-
(a) J.-C. J. Kalinski, A. Polyzois, S. C. Waterworth, X. S. Noundou and R. A. Dorrington, Molecules, 2022, 27, 8724 CrossRef CAS
- Y. Zou, X. Wang, J. Sims, B. Wang, P. Pandey, C. L. Welsh, R. P. Stone, M. A. Avery, R. J. Doerksen, D. Ferreira, C. Anklin, F. A. Valeriote, M. Kelly and M. T. Hamann, J. Am. Chem. Soc., 2019, 141, 4338–4344 CrossRef CAS
- For representative syntheses of simpler pyrroloiminoquinones see:
(a) X. L. Tao, S. Nishiyama and S. Yamamura, Chem. Lett., 1991, 20, 1785–1786 CrossRef
- For representative syntheses of complex pyrroloiminoquinones see:
(a) S. Nishiyama, J.-F. Cheng, X. L. Tao and S. Yamamura, Tetrahedron Lett., 1991, 32, 4151–4154 CrossRef CAS
- Limited SAR studies have been reported focusing on diversification of R5. See:
(a) B. A. Shinkre, K. P. Raisch, L. Fan and S. E. Velu, Bioorg. Med. Chem. Lett., 2007, 17, 2890–2893 CrossRef CAS PubMed
- For a synthesis of pyrroloiminoquinones that does not use the common intermediate see: G. A. Kraus and N. Selvakumar, J. Org. Chem., 1998, 63, 9846–9849 CrossRef CAS
- For reviews of the Buchwald–Hartwig cyclization see:
(a) P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed
-
(a) R. C. Larock and E. K. Yum, J. Am. Chem. Soc., 1991, 113, 6689–6690 CrossRef CAS
- S. T. Kadam and S. S. Kim, Synthesis, 2008, 2, 267–271 Search PubMed
- K. V. Chuang, M. E. Kieffer and S. E. Reisman, Org. Lett., 2016, 18, 4750–4753 CrossRef CAS PubMed
- S. P. Breazzano, Y. B. Poudel and D. L. Boger, J. Am. Chem. Soc., 2013, 135, 1600–1606 CrossRef CAS
- M. Shen, G. Li, B. Z. Lu, A. Hossain, F. Roschangar, V. Farina and C. H. Senanayake, Org. Lett., 2004, 6, 4129–4132 CrossRef CAS
-
(a) W.-M. Cheng, R. Shang and Y. Fu, Nat. Commun., 2018, 9, 5215 CrossRef PubMed
- Y. Shimizu, M. Noshita, Y. Mukai, H. Morimoto and T. Ohshima, Chem. Commun., 2014, 50, 12623–12625 RSC
- S. C. Nigam, A. Mann, M. Taddei and C. G. Wermutha, Synth. Commun., 1989, 19, 3139–3142 CrossRef CAS
- F. Frank, L. M. Alice, P. Mauker, A. A. Alsimaree, P. G. Waddell, M. R. Probert, T. J. Penfold, J. G. Knight and M. J. Hall, Tetrahedron, 2020, 76, 131113 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02981j |
| ‡ This is supported by observation of large amounts of protodebromination (i.e.9) and other unidentifiable by-products in the LC/MS trace, which is not the case when the reaction is allowed to proceed for 24 h (i.e. optimized conditions). |
| § Interestingly, we also isolated isobatzelline A (37) from the reaction mixture, representing a 9 step synthesis (LLS) of this natural product. We report this as a tentative assignment as decomposition was observed during the 13C NMR experiment, although the 1H NMR spectra was in good agreement with those previously reported4n (see the ESI for additional details). We hypothesize that isobatzelline A (37) forms from isobatzelline B (32) under the reaction conditions through an oxidative nucleophilic substitution of hydrogen (ONSH) type transformation, wherein Cl− addition into isobatzelline B (32) is followed by oxidation. Alternatively, Cl2 could be generated from HCl and MnO2, which then chlorinates the pyrroloiminoquinone double bond in isobatzelline B (32), followed by elimination to provide isobatzelline A (37). For an example of ONSH with chloride nucleophiles see ref. 17. |
| This journal is © The Royal Society of Chemistry 2024 |