
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, molecular docking analysis and in vitro evaluation of new heterocyclic hybrids of 4-aza-podophyllotoxin as potent cytotoxic agents†
Ha Thanh Nguyen *ab,
Ket Tran Vanbd,
Hai Pham-Thec,
Julien Brairee,
Phuong Hoang Thia,
Tuan Anh Nguyena,
Quynh Giang Nguyen Thia,
Tuyet Anh Dang Thiab,
Giang Le-Nhat-Thuyab,
Tu Anh Le Thia,
Doan Vu Ngocd and
Tuyen Nguyen Vanab
*ab,
Ket Tran Vanbd,
Hai Pham-Thec,
Julien Brairee,
Phuong Hoang Thia,
Tuan Anh Nguyena,
Quynh Giang Nguyen Thia,
Tuyet Anh Dang Thiab,
Giang Le-Nhat-Thuyab,
Tu Anh Le Thia,
Doan Vu Ngocd and
Tuyen Nguyen Vanab
aInstitute of Chemistry, Vietnam Academy of Science and Technology, 18 Hoang Quoc Viet, Cau Giay, Hanoi, Vietnam. E-mail: hathanhnguyen1512@gmail.com
bGraduate University of Science and Technology, Vietnam Academy of Science and Technology, 18 Hoang Quoc Viet, Cau Giay, Hanoi, Vietnam
cUniversity of Science and Technology of Hanoi, Vietnam Academy of Science and Technology, 18 Hoang Quoc Viet, Cau Giay, Hanoi, Vietnam
dMilitary Technical Academy, 236 Hoang Quoc Viet, Bac Tu Liem, Hanoi, Vietnam
eUniversité de Rennes 1, 2 Av. du Professeur Léon Bernard, 35042 Rennes, France
First published on 8th January 2024
Abstract
Two different synthetic approaches to novel heterocyclic hybrid compounds of 4-azapodophyllotoxin were investigated. The obtained products were characterized by infrared spectroscopy, nuclear magnetic resonance spectroscopy, and high-resolution mass spectrometry. MTT protocol was then performed to examine the cytotoxic activity of these products against KB, HepG2, A549, MCF7, and Hek-293 cell lines. The cytotoxic assessment indicated that all products displayed moderate to high cytotoxicity against all tested cancer cell lines. The most active compound 13k containing the 2-methoxypyridin-4-yl group exhibited selective cytotoxicity against KB, A549, and HepG2 cell lines with the IC50 values ranging from 0.23 to 0.27 μM, which were between 5- to 10-fold more potent than the positive control ellipticine. Compounds 13a (HetAr = thiophen-3-yl) and 13d (HetAr = 5-bromofuran-2-yl) displayed high cytotoxic selectivity for A549 and HepG2 cancer cell lines when compared to the other cancer cell lines and low toxicity to the normal Hek-293 cell line. Molecular docking study was conducted to evaluate the interaction of new synthesized compounds with the colchicine-binding-site of tubulin. Besides that, physicochemical and pharmacokinetic properties of the most active compounds 13h,k were predicted.
Introduction
Heterocycles, a class of cyclic organic compounds containing at least one ring hetero atom, have played an important role in pharmaceutical development due to their variety of biological activities such as anti-fungal,1 antimicrobial,2 anti-inflammatory,3,4 anti-diabetic,5 cytotoxic,6,7 anti-tumor, anti-cancer,8,9 anti-viral,10 acetylcholinesterase inhibitory,11 and SARS-CoV2 inhibitory activities.12 To date, heterocycles have presented themselves as the basic core of most marketed drugs. For examples, pyridine, a nitrogen-based heterocycle, has been known as the structural unit of several targeted cancer drugs such as sorafenib,13 regorafenib,14 and crizotinib.15 Ritonavir (1), a sulfur-based heterocycle drug, is an FDA-approved HIV protease inhibitor which can act as a potential cancer therapeutic agent (Fig. 1).16,17 Amiodarone (2), a benzofuran derivative, is an antiarrhythmic medication used in the treatment of heart rate problems, ventricular fibrillation and ventricular tachycardia (Fig. 1).18 The FDA has approved Jelmyto (mitomycin C, 3), a pyrrole-fused quinone molecule, for treatment of adult patients with low-grade upper tract urothelial cancer (Fig. 1).19 The quinone moiety of mitomycin C is enzymatically reduced into an active hydroquinone intermediate, which has an extraordinary ability to crosslink DNA with high efficiency.20 Owing to the significant role of heterocyclic ring systems in drug discovery and development, many studies have been undertaken to develop biologically active heterocyclic compounds.21–29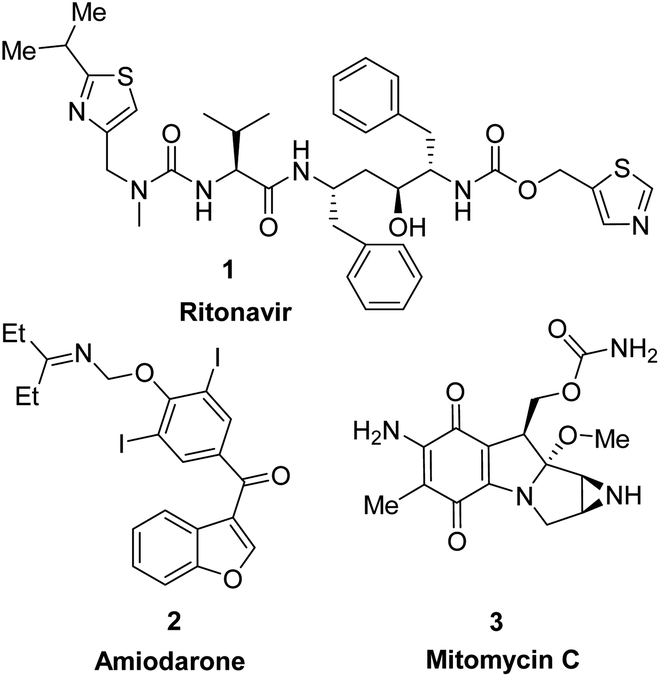 | ||
| Fig. 1 Chemical structure of FDA approved drugs containing heterocyclic ring system. | ||
Podophyllotoxin, an important plant-derived natural product, has several semisynthetic derivatives such as etoposide, teniposide and etophos, which have been employed in chemotherapy for various cancer types.30 Although podophyllotoxin and its derivatives have been known as tubulin polymerization or DNA topoisomerase II inhibitors, they are too toxic for therapeutic use and causes some side effects. To overcome such problems, great interest has been paid to the synthesis of 4-azapodophyllotoxin derivatives in order to obtain the new anticancer agents with little side effects (Fig. 2). 4-Aza-podophyllotoxins have exhibited a broad spectrum of biological activities including cancer cell growth inhibition,31 caspase-3 dependent apoptosis,32 cell cycle arrest in the G2/M phase,33 inhibition of tubulin polymerization and cellular microtubule disassembly34–36 and vascular disruption effect.37 Several hybrid compounds of 4-aza-podophyllotoxin with heterocycles have been developed and evaluated the anticancer activity.38–41 For instance, Kamal et al., has prepared a series of 4-aza-podophyllotoxin with heterocycles including benzothiazole-podophyllotoxin hybrids 6, pyrimidine-podophyllotoxin hybrids 7, and, indole-podophyllotoxin hybrids 8 which have inhibited tubulin polymerization, induced cell cycle arrest at G2/M phase and caspase-3 dependent apoptotic cell death in non-small lung A549 cell line.33 Pyrazole-podophyllotoxin hybrids which have been synthesized by Magedov et al., have showed apoptosis induction in cancerous Jurkat cells even after a short 24 h exposure. Azaanthraquinone-podophyllotoxin hybrids 11, which have been designed by replacing of γ-butyrolactone ring D of 4-aza-podophyllotoxin with naphthoquinone, have exhibited medium cytotoxic activity against hepatoma carcinoma HepG2 and Hela cell lines.42 Interestingly, azaanthraquinone-podophyllotoxin hybrids 12, reported in our previous study,43,44 have possessed excellent cytotoxic activity against hepatoma carcinoma HepG2 and non-small lung SK-Lu-1 cell lines with IC50 < 0.04 μM (Fig. 2). Hybrid compounds 12 were demonstrated to exhibit cytotoxicity by inducing cell cycle arrest at G2/M phase, activating caspase-3/7 activation, and promoting apoptosis in a concentration-dependent manner. In spite of the many researches focused on the synthesis of azapodophyllotoxin hybrids, the replacement of ring E with heterocycles has not attracted enough attention so far. Accordingly, in the view of the above mentioned facts and as a continuation of our efforts on the synthesis of biologically active compounds,45–49 herein, we have focused our research interest toward the synthesis of novel azaanthraquinone-podophyllotoxin hybrids with heterocyclic ring E 13 via microwave-assisted multicomponent reactions (Fig. 2), and evaluation of their cytotoxic activity against various cancer cell lines by using MTT colorimetric method. Besides, synthesized compounds 13 were docked into the binding sites of colchicine in tubulin and their binding energies as well as physicochemical and pharmacokinetic properties were determined.
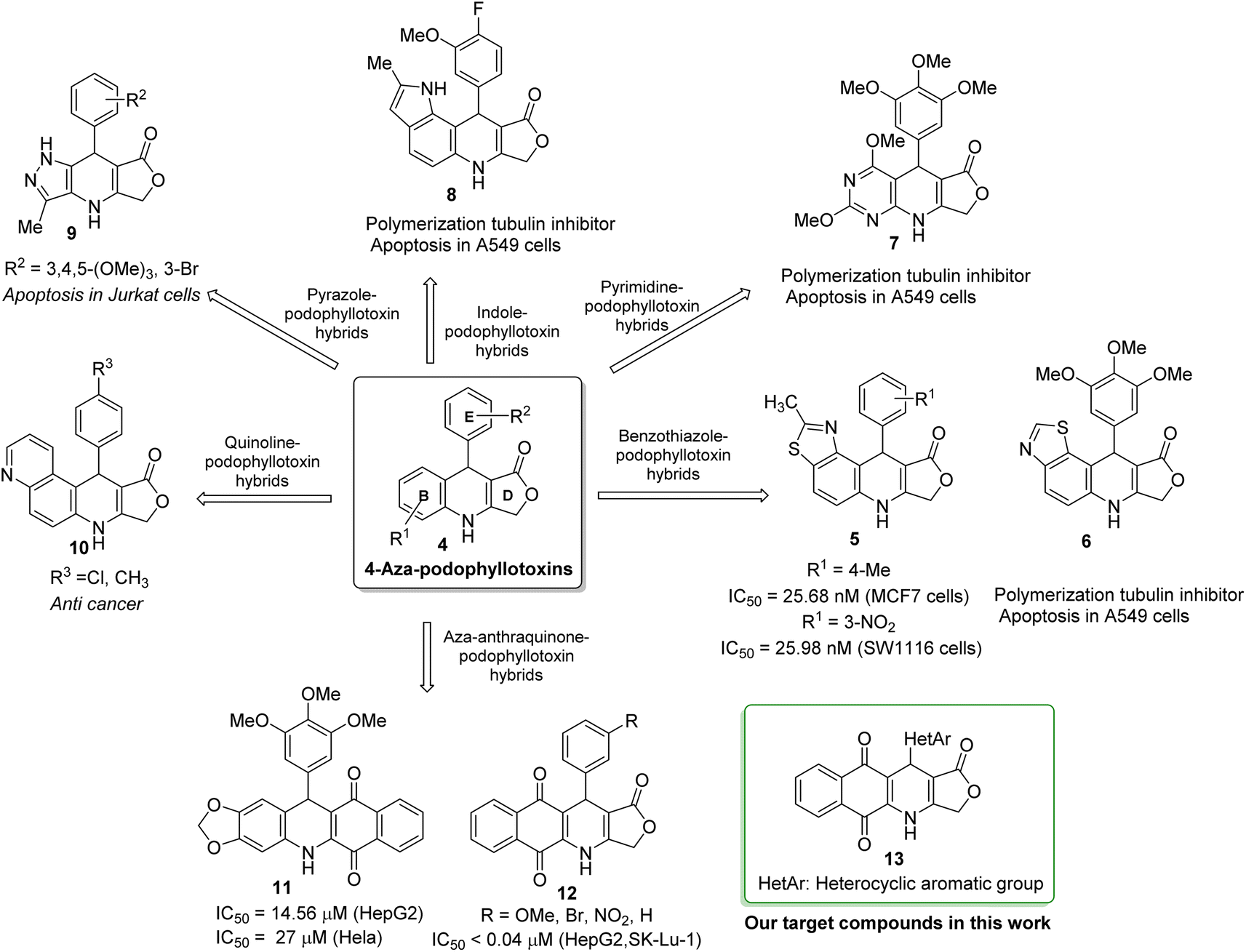 | ||
| Fig. 2 Hybrid compounds of 4-aza-podophyllotoxin with heterocycles. | ||
Results and discussion
Chemistry
Two convenient multicomponent synthetic approaches have been adopted to synthesize the target compounds 13 (Scheme 1). In the approach A, a range of structurally diverse heteroaromatic aldehydes 16 was subjected to the four-component reactions with 2-hydroxy-naphthoquinone (14), tetronic acid (15) and ammonium acetate (17) in glacial acetic acid (gl. AcOH) at 120 °C in 20 min under microwave irradiation, leading to products 13 in low yield ranging from 20 to 34% (Table 1). The formation of products 13 possibly started from Knoevenagel condensation of 2-hydroxy-1,4-naphthoquinone (14) with aldehydes, followed by Michael addition to 4-aminofuran-2(5H)-one 19, produced from the reaction of tetronic acid 15 with ammonia. The adduct 21 underwent tautomerization, intramolecular cyclization and dehydration to form products 13 (Scheme 2).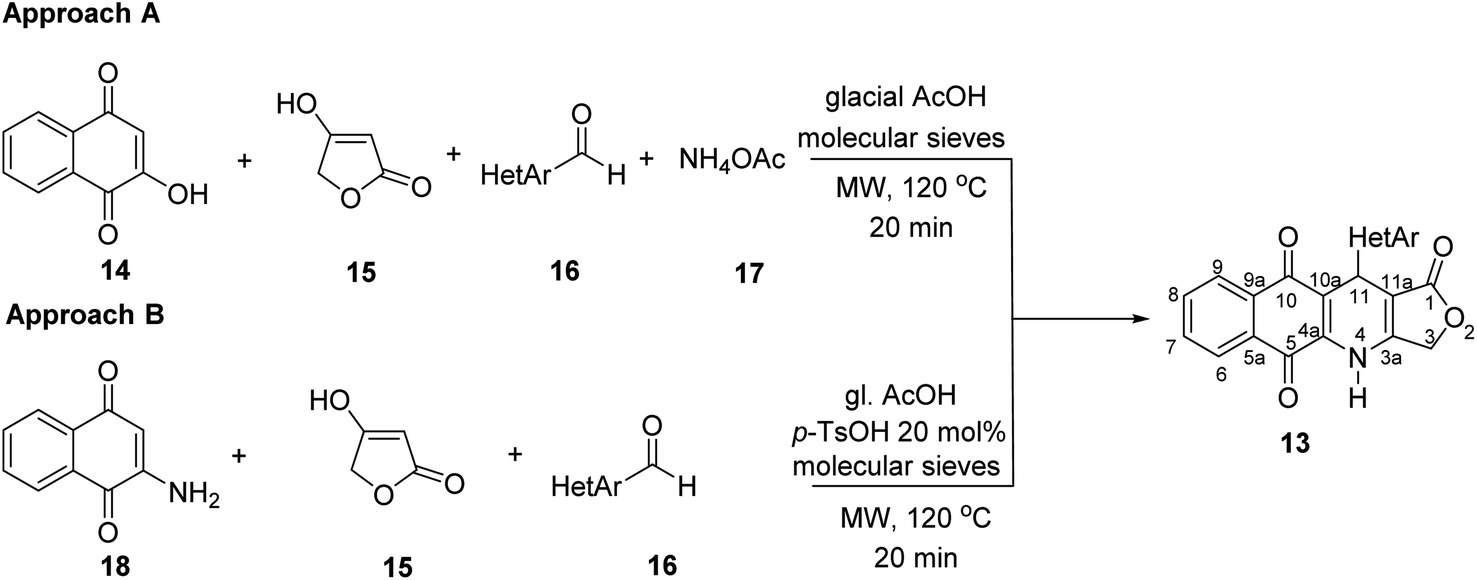 | ||
| Scheme 1 Synthesis of compounds 7 under microwave irradiation. | ||
|
|||
|---|---|---|---|
| Entry | Product | Yield (%) | |
| Approach A | Approach B | ||
| 1 | 13a | 29 | 64 |
| 2 | 13b | 25 | 62 |
| 3 | 13c | 20 | 60 |
| 4 | 13d | 20 | 62 |
| 5 | 13e | 33 | 70 |
| 6 | 13f | 32 | 68 |
| 7 | 13g | 33 | 69 |
| 8 | 13h | 28 | 64 |
| 9 | 13i | 27 | 63 |
| 10 | 13j | 20 | 61 |
| 11 | 13k | 34 | 74 |
| 12 | 13l | 32 | 72 |
| 13 | 13m | 33 | 72 |
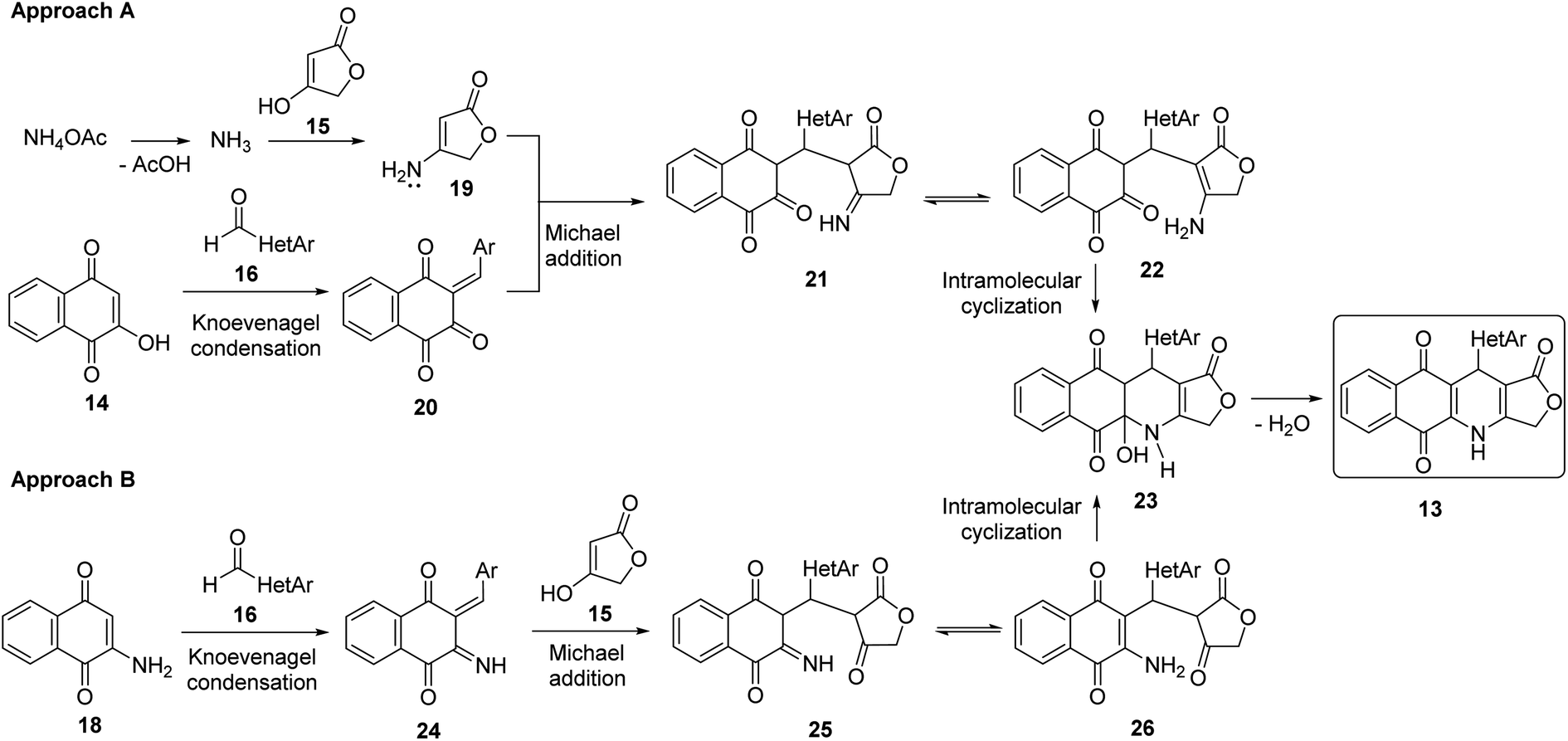 | ||
| Scheme 2 Plausible mechanism for the formation of obtained products 13. | ||
In the approach B, three-component synthesis of target products 13 from 2-amino-naphthoquinone, heteroaromatic aldehydes, and tetronic acid was investigated. In our preliminary studies, we indicated that the use of p-toluenesulfonic acid (p-TsOH) as an acid catalyst and gl. AcOH as a solvent promoted the three-component reactions to generate products in higher yields. Thus, p-TsOH (20 mol%) was supplemented to the reaction mixtures in gl. AcOH, and the reactions were then stirred at 120 °C, under microwave irradiation (150 W). The reactions proceeded rapidly to completion within 20 min. As shown in Table 1, under these optimized conditions, products 13 were afforded in good yields (60–74%). The synthetic approaches proceeded via a sequential steps including Knoevenagel condensation of 2-amino-1,4-naphthoquinone (14) with heteroaromatic aldehydes, Michael addition to tetronic acid, tautomerization, intramolecular cyclization and dehydration (Scheme 2).
It is worth mentioning that the approach B (synthesis by three-component reactions) is likely to be more efficient for the synthesis of target products 13 in comparison with the approach A (synthesis by four-component reactions). In approach B, the effect of heterocyclic substituents on aldehyde on the outcome of reactions could be observed more clearly. For instance, pyridinecarboxaldehyde derivatives 16k–m displayed the highest reactivity to give the desired products 13k–m in high yield (72–74%). The fusion of a benzene ring with five-membered aromatic heterocyclic ring of aldehydes improved the reaction yields compared to five-membered heterocyclic aldehydes. Benzo[b]thiophene-3-carbaldehyde (16e) furnished 70% yield of the product 13c, whereas, thiophene-3-carbaldehyde (16a) afforded 13a in 64% yield.
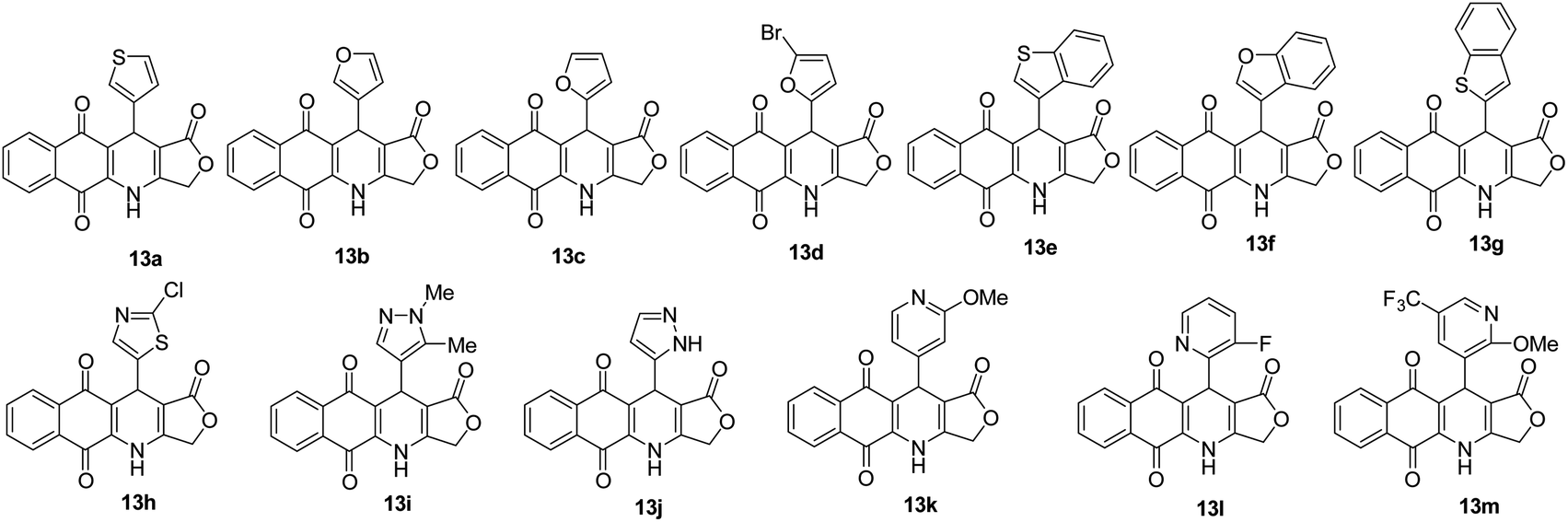
The structure of obtained compounds were examined by IR, 1H and 13C NMR, HSQC, HMBC, DEPT, and HRMS. Compound 13a was focused for illustrating the 1H and 13C assignments. In general, the 1H-NMR spectrum of 13a, presented a typical singlet at δH 10.60 ppm corresponding to an –NH group, two doublets of doublets at δH 8.05 (1H, dd, J = 1.2, 7.8 Hz, H6), and 7.93 (1H, dd, J = 0.6, 7.8 Hz, H9), two triples of doublets at δH 7.85 (1H, td, J = 1.2, 7.2 Hz, H8), 7.81 (1H, td, J = 1.2, 7.2 Hz, H7) corresponding to four aromatic protons in naphthoquinone ring. Three aromatic protons in thiophene ring appeared at δH 7.38 (1H, dd, J = 3.0, 4.8 Hz, H-5′), 7.24 (1H, dd, J = 0.6, 3.0 Hz, H-2′), 7.07 (1H, dd, J = 1.2, 4.8 Hz, H-4′). Moreover, the typical singlet at 5.11 ppm was referred to the H-11 proton, two doublets were observed at 4.94 ppm and 4.90 ppm with a coupling constant of 16.8 Hz assignable to the 3-CH2 protons in γ-butyrolactone ring. On the other hand, the 13C NMR spectrum of product 13a displayed 19 carbon resonances, which were fully assigned with DEPT, 1H–13C HSQC and 1H–13C HMBC support. The DEPT-135 shows 8 positive peaks and 1 negative peak. The negative peak at 66.1 ppm corresponded to methylene (CH2) carbon C-3. Proton signal (H-11) at 5.11 ppm showed proton-carbon couplings to C-10 at 182.1 ppm, C-1 at 171.2 ppm, C-3a at 156.1 ppm, C-4a at 139.2 ppm, C-3′ at 145.0 ppm, C-4′ at 127.7 ppm, C-2′ at 122.4 ppm, C-10a at 118.0 ppm, and C-11a at 101.6 ppm (Fig. 3). Moreover, under positive HR–ESI–MS conditions, [M + H]+ at m/z 350.0484, which confirms the molecular formula for compound 13a is C19H11NO4S.
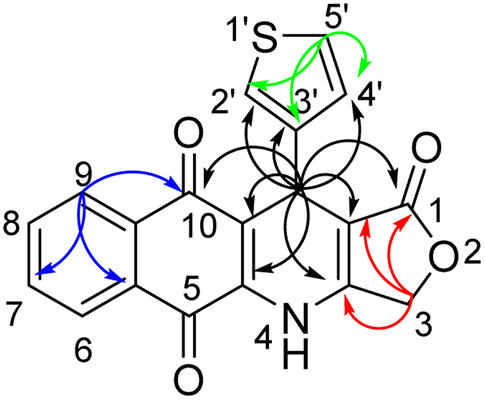 | ||
| Fig. 3 Key correlations of protons H-11 (black arrows), H-3 (red arrows), H-9 (blue arrows), and H-5′ (green arrows) in 1H–13C HMBC spectrum of compound 13a. | ||
Cytotoxicity of synthesized compounds
All of these products were further studied cytotoxicity evaluation in vitro against cultured A549 (human lung adenocarcinoma), MCF7 (human breast adenocarcinoma), KB (human mouth epidermal carcinoma), HepG2 (human hepatocellular carcinoma) and Hek-293 (human embryonic kidney 293) cell lines, in comparison to ellipticine, the positive control, by using MTT assay (Table 2). The selective index (SI) of products was calculated as the ratio of the IC50 value in Hek-293 (human embryonic kidney 293) cell line to the IC50 value in cancer cell line (Table 3). As illustrated in Table 2, all products showed high and moderate cytotoxic activity to all tested cancer cell lines with IC50 values ranging from 0.16 to 14.22 μM. Cytotoxic activity of compounds 13a,h,k was better than that of ellipticine against 4 tested cancer cell lines, a fact supporting their anti-cancer activity. Besides, products 13f (HetAr = benzofuran-3-yl) exerted almost 1.8-fold higher toxicity to KB and HepG2 cells when compared with ellipticine. Notably, product 13k (HetAr = 2-methoxypyridin-4-yl) exhibited the significantly potent and selective cytotoxicity against KB, A549, and HepG2 cell lines with the IC50 values ranging from 0.23 to 0.27 μM. The cytotoxic activity of product 13k toward KB, A549, and HepG2 cancer cells was approximately 10 times higher in relation to normal Hek-293 cells (Table 3). Product 13h (HetAr = 2-chlorothiazol-5-yl) showed the highest growth inhibitory activity against A549 and MCF7 cancer cell lines and normal Hek-293 cells with IC50 values of 0.16, 1.07, and 1.38 μM, respectively. Compound 13a (HetAr = thiophen-3-yl) displayed higher cytotoxic selectivity for A549 cancer cells when compared to the other cancer cell lines and low toxicity to normal Hek-293 cell line with IC50 = 22.90 μM, what can indicate their specificity to cancer cell lines. In particularly, compound 13d (HetAr = 5-bromofuran-2-yl) exhibited selective cytotoxicity against HepG2 cancer cells with IC50 value of 0.30 μM, which was 122-fold higher than its IC50 value for Hek-293 cells (Table 3).| Product | IC50, μM | ||||
|---|---|---|---|---|---|
| KB | A549 | HepG2 | MCF7 | Hek-293 | |
| 13a | 1.43 ± 0.14 | 0.48 ± 0.03 | 1.43 ± 0.15 | 2.06 ± 0.66 | 22.90 ± 2.18 |
| 13b | 10.05 ± 1.17 | 3.09 ± 0.39 | 12.36 ± 1.29 | 12.63 ± 1.14 | 13.80 ± 1.41 |
| 13c | 14.22 ± 1.23 | 3.06 ± 0.27 | 3.75 ± 0.69 | 12.15 ± 1.14 | 14.55 ± 1.35 |
| 13d | 2.81 ± 0.32 | 2.06 ± 0.22 | 0.30 ± 0.06 | 10.36 ± 1.29 | 37.12 ± 3.30 |
| 13e | 10.94 ± 1.40 | 9.94 ± 0.73 | 10.42 ± 1.18 | 13.07 ± 1.60 | 11.17 ± 0.85 |
| 13f | 1.10 ± 0.09 | 7.02 ± 0.91 | 1.30 ± 0.13 | 12.65 ± 1.20 | 19.69 ± 1.70 |
| 13g | 9.94 ± 0.65 | 3.13 ± 0.20 | 12.67 ± 1.40 | 11.27 ± 1.78 | 13.07 ± 1.70 |
| 13h | 1.12 ± 0.10 | 0.16 ± 0.02 | 0.81 ± 0.11 | 1.07 ± 0.10 | 1.38 ± 0.18 |
| 13i | 10.74 ± 1.27 | 2.27 ± 0.22 | 7.80 ± 0.53 | 10.41 ± 0.80 | 12.26 ± 1.13 |
| 13j | 6.00 ± 0.63 | 5.43 ± 0.39 | 4.35 ± 0.57 | 3.24 ± 0.36 | 3.75 ± 0.69 |
| 13k | 0.27 ± 0.24 | 0.27 ± 0.21 | 0.23 ± 0.02 | 1.34 ± 0.16 | 2.48 ± 0.32 |
| 13l | 4.69 ± 0.58 | 2.15 ± 0.25 | 5.52 ± 0.86 | 7.34 ± 0.94 | 10.21 ± 1.24 |
| 13m | 4.63 ± 1.04 | 2.83 ± 0.54 | 5.83 ± 0.86 | 7.26 ± 1.29 | 16.73 ± 1.94 |
| Ellipticine | 1.95 ± 0.12 | 1.42 ± 0.08 | 2.11 ± 0.16 | 2.09 ± 0.16 | 5.60 ± 0.37 |
| Entry | Compound | Selective index | |||
|---|---|---|---|---|---|
| KB | A549 | HepG2 | MCF7 | ||
| 1 | 13a | 16.00 | 47.90 | 16.00 | 11.12 |
| 3 | 13b | 1.37 | 4.47 | 1.12 | 1.09 |
| 4 | 13c | 1.02 | 4.75 | 3.88 | 1.20 |
| 5 | 13d | 13.19 | 18.00 | 122.40 | 3.58 |
| 6 | 13e | 1.02 | 1.12 | 1.07 | 0.85 |
| 7 | 13f | 17.98 | 2.81 | 15.10 | 1.56 |
| 8 | 13g | 1.31 | 4.18 | 1.03 | 1.16 |
| 9 | 13h | 1.23 | 8.55 | 1.71 | 1.29 |
| 10 | 13i | 1.14 | 5.40 | 1.57 | 1.18 |
| 11 | 13j | 0.63 | 0.69 | 0.86 | 1.16 |
| 12 | 13k | 9.30 | 9.30 | 10.81 | 1.86 |
| 13 | 13l | 2.18 | 4.74 | 1.85 | 1.39 |
| 14 | 13m | 3.61 | 5.92 | 2.87 | 2.31 |
| 15 | Ellipticine | 2.88 | 3.94 | 2.65 | 2.68 |
Molecular docking study
Based on experimental assessments, it can be initially identified that compounds 13a–13m are good cytotoxic agents. Due to crucial function in mitosis and cell division, microtubules, which are produced through the polymerization of heterodimers of α and β-tubulins, have been a desirable target in the development of novel anticancer medicines.50 Meanwhile, podophyllotoxin has long been regarded as a potent microtubule destabilizing agent that binds to the tubulin colchicine site, inhibiting tubulin polymerization and suppressing the production of microtubules.44,51 Therefore, in this study, we used molecular docking to evaluate the interaction of novel synthesized substances with this molecular target. The natural tubulin substrate colchicine will be used as a positive control.Firstly, colchicine were redocked into the active site in tubulin to validate the docking process. As the results, the redocked conformers tightly bound to the colchicine binding site of tubulin with a very high matching with the co-crystallized structure (RMSD = 0.3339 Å). Moreover, it preserves the key interactions such as hydrogen bonds with Val181, Ala180… of the α subunit through its methoxytropone ring, hydrophobic interactions with Leuβ242, Cysβ241, Alaβ354, and Alaβ316 (Fig. 4A),52 suggesting the validity of established docking protocol.
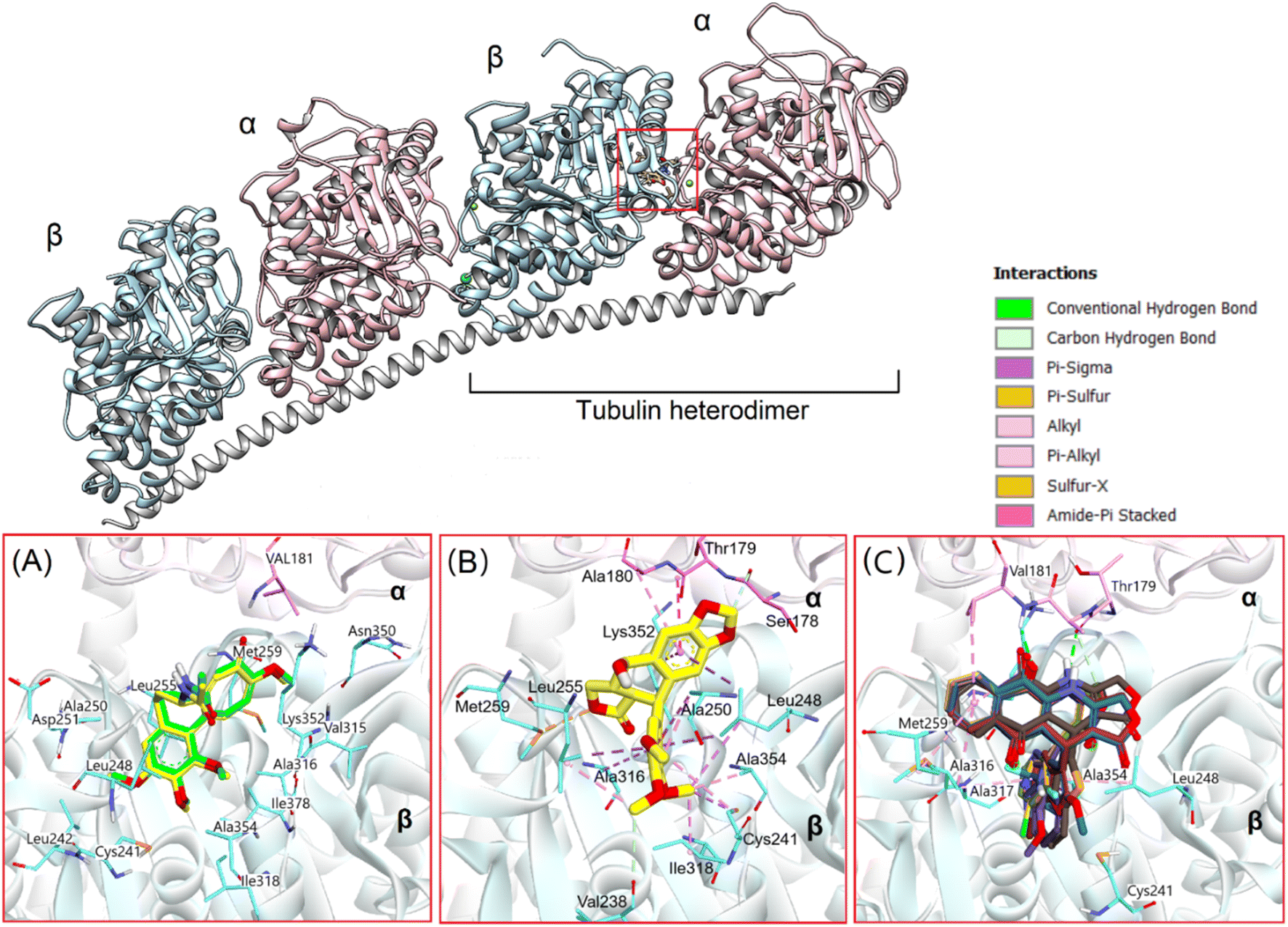 | ||
| Fig. 4 (A) Redocked poses (green) and original poses (yellow) of colchicine in tubulin; (B) docking pose of podophyllotoxin, and (C) docking poses of 13 synthesized compounds in colchicine binding site. | ||
In the next step, podophyllotoxin, which is considered reference compound was also docked into the binding site of tubulin. The results were similar to those previously determined.44 As can be seen in Fig. 4, this compound exhibited a large interaction network with residues in the binding site. Some of the key interactions should be mentioned here are the hydrogen bonds with Val178, Ala180… of the α subunit and numerous hydrophobic interactions with Cys241, Ala250, Leu248, Leu255, Ala354, and Ala316 of the β subunit (Fig. 4B). The binding energy was determined to be −18.68 kcal mol−1, and hydrogen bonding and hydrophobic stacking interactions the most contributed components (Fig. 5). The results obtained confirmed the similarity and accuracy of the docking protocol.
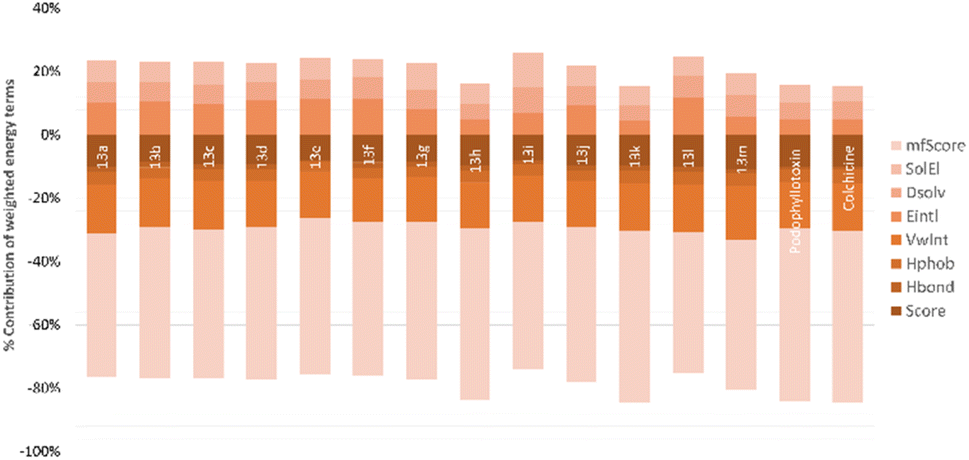 | ||
| Fig. 5 Contribution (%) of the different energy terms to the binding affinity (score) of tubulin-ligands: the van der Waals interaction energy (VwInt), Hydrogen Bond energy (Hbond), the hydrophobic energy (Hphob), internal conformation energy (Eintl), the desolvation of exposed H-bond donors and acceptors (Dsolv), the solvation electrostatics energy change upon binding (SolEl), the potential of mean force score (mfScore). | ||
After validating the docking protocol, all 13 synthesized compounds (13a–13m) were docked into the binding site of colchicine in tubulin, using the above validated protocol. The binding energies and the main interactions between the compounds and tubulin in comparison with podophyllotoxin are summarized in Table 4 and Fig. 4–6.
| Cpd | Binding energy (kcal mol−1) | Interaction with residues |
|---|---|---|
| 13a | −16.38 | H-bond: Val 181α, Thr 179α, Ala 180α |
| Hydrophobic: Ala 354β, Leu 248β, Ala 316β, Lys 352β, Met 259β | ||
| 13b | −13.79 | H-bond: Val 181α, Thr 179α, Ala 180α, Ala 354β, Ala 317β |
| Hydrophobic: Leu 248β, Ala 316β, Lys 352β, Met 259β | ||
| 13c | −14.55 | H-bond: Val 181α, Thr 179α, Ala 180α |
| Hydrophobic: Ala 354β, Leu 248β, Ala 316β, Lys 352β, Met 259β | ||
| 13d | −15.29 | H-bond: Val 181α, Thr 179α, Ala 180α |
| Hydrophobic: Ala 354β, Leu 248β, Ala 316β, Lys 352β, Met 259β, Ile 318β, Cys 241β | ||
| 13e | −14.09 | H-bond: Val 181α, Thr 179α, Ala 180α |
| Hydrophobic: Ala 354β, Leu 248β, Ala 316β, Lys 352β, Met 259β, Leu 255β, Cys 241β | ||
| 13f | −16.45 | H-bond: Val 181α, Thr 179α, Ala 180α, Asn 101α, Leu 255β, Ala 250β |
| Hydrophobic: Ala 354β, Leu 248β, Ala 316β, Lys 352β, Met 259β, Leu 255β, Asn 258β, Ala 250β | ||
| 13g | −15.08 | H-bond: Val 181α, Thr 179α, Ala 180α, Leu 255β, Lys 352β |
| Hydrophobic: Ala 354β, Leu 248β, Ala 316β, Lys 352β, Met 259β, Leu 255β, Asn 258β, Ala 250β, Cys 241β | ||
| 13h | −17.86 | H-bond: Val 181α, Thr 179α, Ala 180α |
| Hydrophobic: Ala 354β, Leu 248β, Ala 316β, Lys 352β, Met 259β, Ile 318β, Cys 241β | ||
| 13i | −13.83 | H-bond: Val 181α, Ala 180α, Asn 101α, Ala 317β |
| Hydrophobic: Ala 354β, Leu 248β, Leu 255β, Ala 316β, Lys 352β, Met 259β, Ile 318β | ||
| 13j | −15.64 | H-bond: Val 181α, Thr 179α, Ala 180α, Ala 317β, Lys 352β |
| Hydrophobic: Ala 354β, Leu 248β, Ala 316β, Lys 352β, Met 259β | ||
| 13k | −18.45 | H-bond: Val 181α, Thr 179α, Ala 180α, Ala 317β |
| Hydrophobic: Ala 354β, Leu 248β, Ala 316β, Lys 352β, Met 259β, Ile 318β, Cys 241β | ||
| 13l | −15.78 | H-bond: Val 181α, Thr 179α, Ala 180α, Ala 317β, Lys 352β |
| Hydrophobic: Ala 354β, Leu 248β, Ala 316β, Lys 352β, Met 259β | ||
| 13m | −16.8 | H-bond: Val 181α, Ala 180α, Ala 317β, Ala 316β, Asn 101α, Asp251β, Ala 250β |
| Hydrophobic: Ala 354β, Leu 248β, Ala 316β, Lys 352β, Met 259β, Leu255β, Thr 353β | ||
| Podophylotoxin | −18.68 | H-bond: Ser 178α, Ala 180α, Ala 317β, Ala 316β, Asn 101α, Asp251β, Ala 250β |
| Hydrophobic: Val 238β, Cis 241β, Leu 248β, Ala 316β, Ala 318β, Lys 352β, Met 259β, Leu255β, Thr 353β | ||
| Colchicin | −19.18 | H-bond: Val 181α, Ala 180α, Ala 250β, Asp251β, val 315β, asn 350β |
| Hydrophobic: Leu 255β, Leu 248β, Leu 242β, Cys 241β, Ile 318β, Ala 354β, Ile 378β, Ala 316β, Met 359β, Lys 352β |
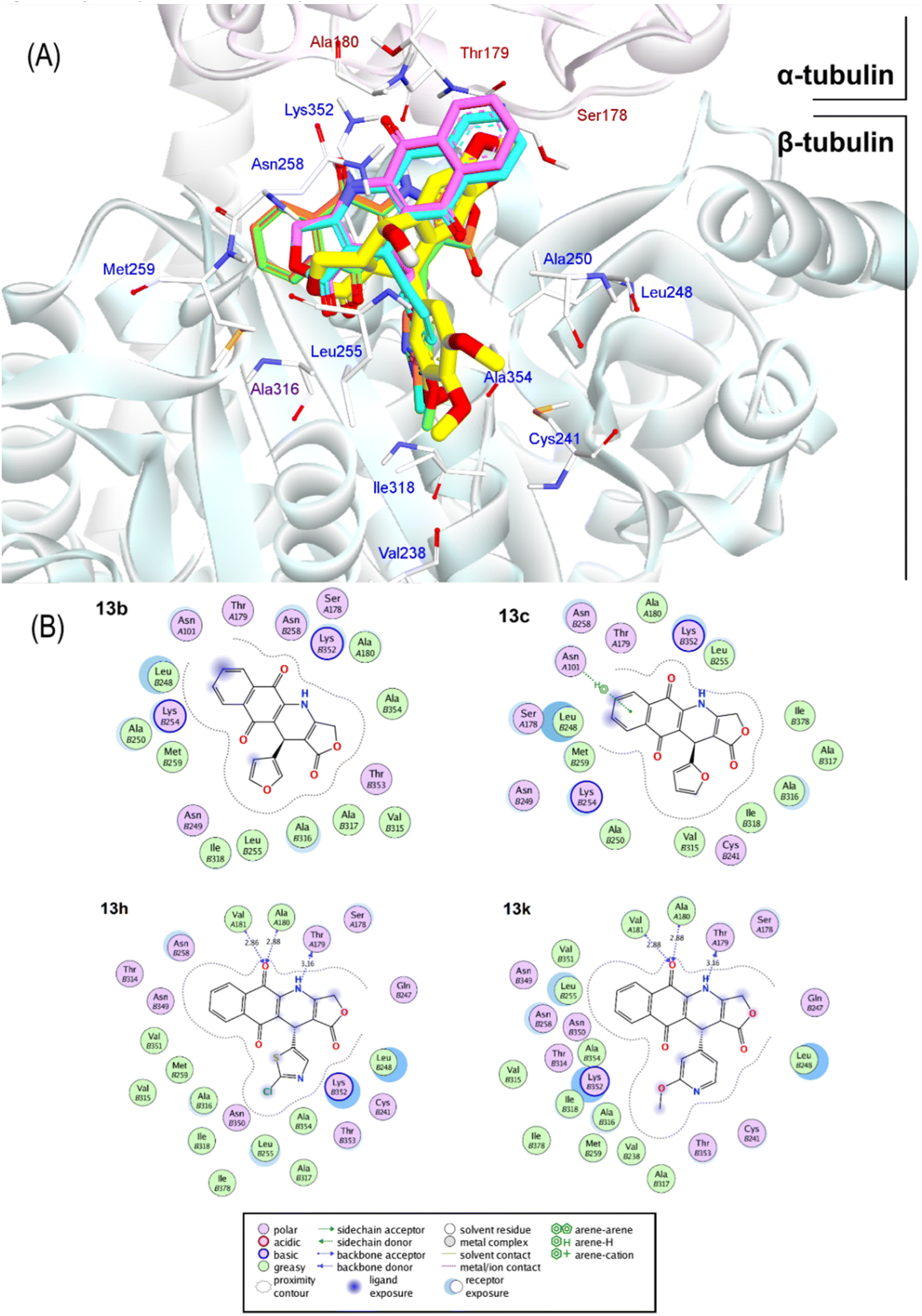 | ||
| Fig. 6 (A) Superimposition of docking poses of 13b (purple carbon), 13c (cyan carbon), 13h (orange carbon), and 13k (green carbon) against podophyllotoxin (yellow carbon). (B) Topological interactions of 13b, 13c, 13h, and 13k with the residues in the active sites of α and β-tubulin domains (labeled with A and B prefixes). | ||
Overall, the docking results of 13 substances 13a–13m were similar to colchicine and those previously published.44 They are all capable of binding to the active site of colchicine in tubulin. The colchicine binding site was divided into three zones including zones 1, 2, and 3 according to Massaroti et al.53 zone 1 is surrounded by the residues Valα181, Serα178, Metβ259, Asnα101, and is situated at the α subunit interface. Located in the β subunit, zone 2 is an accessory hydrophobic pocket made up of the residues Lysβ352, Valβ318, Alaβ317, Alaβ316, Leuβ255, Alaβ250, Leuβ248, Leuβ242, and Cysβ241 zone 3, which is buried deeper inside the β subunit, is formed by the residues Thrβ239, Valβ238, Tyrβ202, Gluβ200, Pheβ169, Asnβ167, Glnβ136, and Ileβ4… Therein, these studied conformations with rigid structures are capable of stretching into the deeper zone 2 of the β-subunit.54 Almost all the synthesized compounds conserved these key interactions such as the hydrogen bond between the carbonyl group of the quinone ring and Valα181, Alaα180; the hydrophobic interactions which are mainly pi-alkyl stacking with most residues of zone 2 like Lysβ352, Alaβ317, Alaβ316, Leuβ255, Alaβ250, and so on. Moreover, all compounds formed hydrogen bonds between N and Thrα179 that do not exist in colchicine,52 making it bind to the active site more strongly.
The docking poses and binding energies also showed good ability to discriminate the good ligands from the bad ones. 13b and 13c were determined to be two of the weakest ligands of tubulin. As can be seen in Fig. 6, these compounds were quite different from 13k and 13h in alignment with podophyllotoxin. In fact, fewer interactions were observed in 13b and 13c, being several stacking interactions with HetAr with residues in the β subunit, such as Asn101, Ala250, Leu248, and Ala316 the most important ones. In particular, the orientation of HetAr in 13b and 13c were different from 13k and 13h, suggesting the change in their target interactions.
The relative binding energies were determined based on a GBSA/MM-type function implemented into ICM pro.66 In addition, other energy terms extracted from the final complex, including Hbond, Vwin. Accordingly, compound 13k has the strongest binding energy at −18.05 kcal mol−1. Besides general interactions, it also formed hydrogen bonds between the lactone ring and Asn101α of zone 1, and between the carbonyl group of the quinone ring and Leuβ255 of zone 2. Additionally, it interacts better with more residues of zone 2 of colchicine binding site through its substituent. Compound 13h had good binding energy of −17.86 kcal mol−1 resulted by key hydrogen bonds with residues of zone 1 and hydrophobic interactions which are similar to compound 13k (Fig. 6). The results also highlighted the highest values of Hbond and Hphob energies of 13h and 13k compared to the other derivatives, which correlated well with the order ranking of all the compounds (Fig. 5). There is a concurrence between the binding energy and the ICM docking mfScore which refers to the strength of the inhibitor–tubulin interaction. The mfScore values of 13h and 13k were −102.91 and −103.1 kcal mol−1, which are similar to podophyllotoxin. Interestingly, Hbonds appear to have small impact on the binding ability of synthesized compounds. While hydrogen bonding energies of 13b and 13c are quite high, their overall energies computed by GBSA/MM-type and mfScore functions showed significantly lower than podophyllotoxin and colchicine (see Table S1†). For the generated docking conformations, the hydrophobic and van der Waals interaction appear to be the driving forces for binding ability of tested compounds. Over podophyllotoxin all, the docking results are in consistency with our previous findings, and in turn match well with experimental ranking using the cytotoxicity tests.67,69
Physicochemical and pharmacokinetic properties prediction
The above experimental test revealed that all 13 compounds exhibit good activity against a variety of cancer cell lines. In which, the two chemicals 13h and 13k have the most potential. So, we performed assessments related to physicochemical and pharmacokinetic feature as a criteria for drug-likeness evaluation.First, radar plot of the physicochemical properties in Fig. 7 showed that both compounds met most of the criteria, excepting logP and logS. This issue may affect to their membrane permeability. However, according to the prediction of SwissADME, the LogPo/w value of them are less than 5.0 that is suitable for absorption.55,56 In terms of solubility, based on ESOL topological model, they classified as moderately soluble chemicals (Table 4). In addition, 13h and 13k were predicted to have zero alert according to the Pan Assay Interference Compounds (PAINS) which is desired to avoid promiscuous behavior and nonselective reactivity with proteins.
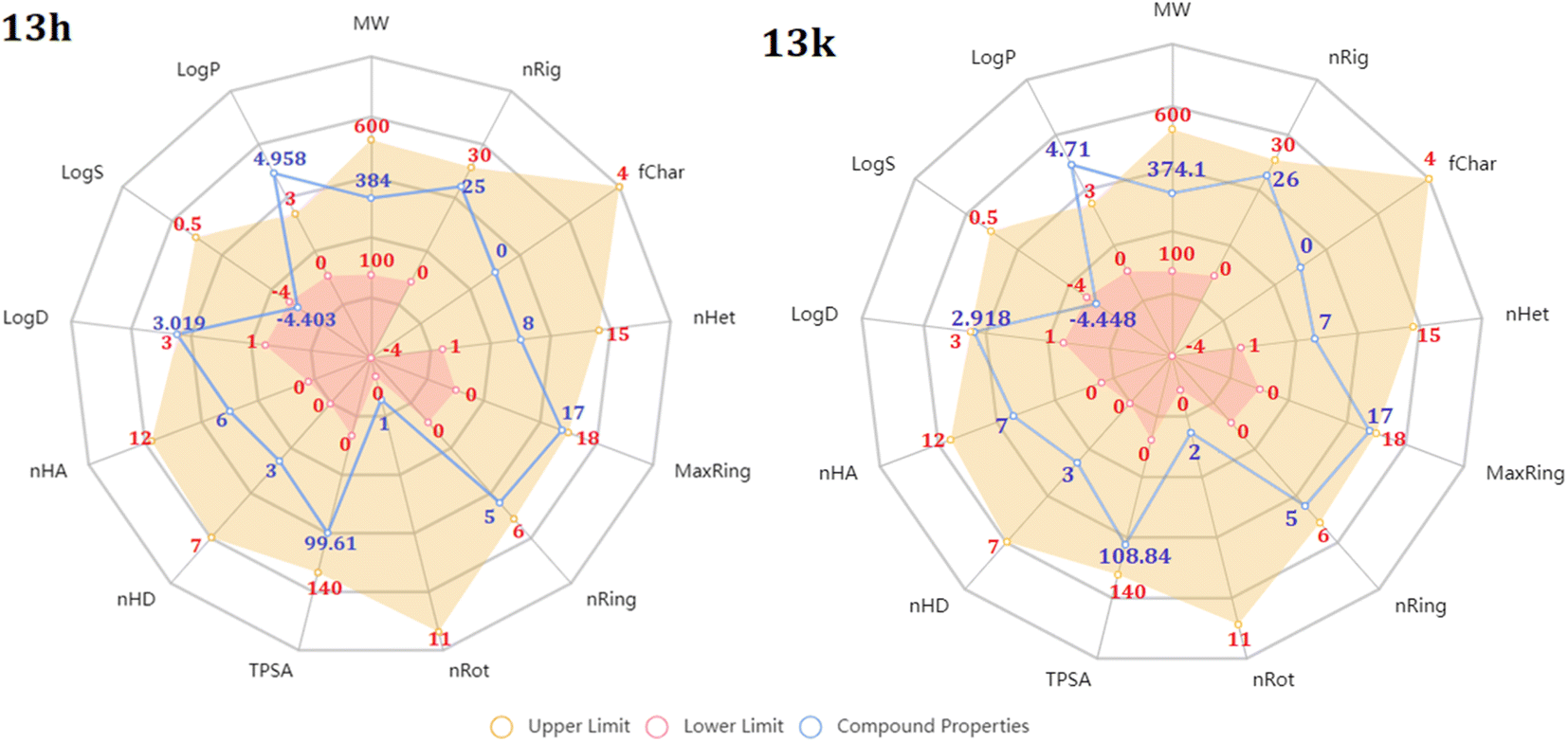 | ||
| Fig. 7 Radar diagram illustrating the physicochemical characteristics of 13h and 13k. MW: molecular weight, nRig: number of rigid bonds, fChar: formal charge, nHet: number of heteroatoms, MaxRing: number of atoms in the biggest ring, nRing: number of rings, nRot: number of rotatable bonds, TPSA: topological polar surface area (Å2), nHD: number of hydrogen bond donors, nHA: number of hydrogen bond acceptors, logD: log of octanol partition coefficient at physiological pH 7.4, logS: log of aqueous solubility (mol L−1), and logP: log of octanol partition coefficient. | ||
In addition, these two compounds were subjected to drug-likeness prediction. Table 5 demonstrates that 13h and 13k successfully accomplished all the drug-likeness rules with no violations but failed to pass lead-likeness rule due to their molecular weight.57
| Predicted parameters | 13h | 13k |
|---|---|---|
| Drug likeness | ||
| Lipinski | Accept | Accept |
| Goshe | Accept | Accept |
| Veber | Accept | Accept |
| Egan | Accept | Accept |
| Muegge | Accept | Accept |
| Bioavailability score | 0.55 | 0.55 |
| Lead-likeness | No, (MW > 350) | No, (MW > 350) |
| PAINS | 0 alert | 0 alert |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Absorption | ||
| Gastrointestinal absorption | High | High |
| Pgp-substrate | No | No |
| Log Kp (skin permeation) | −6.72 cm s−1 | −6.72 cm s−1 |
|
||
| Distribution | ||
| Plasma protein binding | 100.0% (not optimal) | 97.87% (not optimal) |
| Volume distribution | 0.402 | 0.413 |
| Blood–brain barrier (BBB) penetration | No | No |
|
||
| Metabolism | ||
| CYP interaction* | CYP1A2 inhibitor (0.94) | CYP1A2 inhibitor (0.92) |
| CYP2C9 inhibitor (0.66) | CYP2C9 inhibitor (0.75) | |
| CYP2D6 substrate (0.58) | ||
|
||
| Excretion | ||
| Clearance (CL) | 3.043 mL min−1 kg (low) | 3.317 mL min−1 kg (low) |
| Half-life (T1/2) | 0.066 h (short) | 0.194 (short) |
Second, the properties related to Absorption, Distribution, Metabolism and Excretion (ADME) were analyzed. According to the absorption, the predicted values showed that both compounds have high human intestinal absorption.58 According to the BOILED-Egg plot (Fig. 8), they were not glycoprotein-P substrate and shas high intestinal absorption percent.59,60 Prediction of skin permeability showed that they are less permeable across the skin. In addition, 13f and 13h could not across the blood–brain barrier, suggesting a little effect on the central nervous system (CNS). These compounds were estimated to have high plasma protein binding which may narrow their therapeutic index. Both compounds showed optimal volume of distribution. In accordance with the first-pass metabolism, hepatic cytochrome plays a pivotal role in catalytic reactions. Our predictions showed that 13h and 13k mainly interacted with CYP1A2 and CYP2C9, suggesting potential drug–drug interactions with some medications and food.61 These compounds were also estimated to have low clearance together with a short half-life in human body.
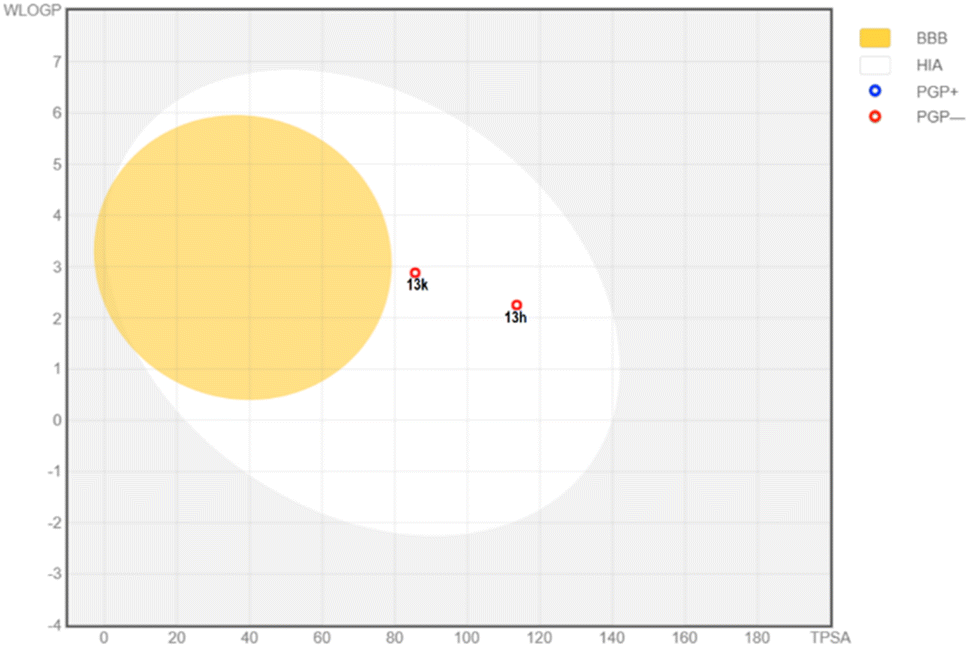 | ||
| Fig. 8 BOILED-Egg diagram of two potent compounds 13h and 13k. Abbreviation: BBB: blood–brain barrier; HIA: human intestinal absorption; PGP+: P-glycoprotein substrate; PGP-: P-glycoprotein non-substrate. | ||
ADMETlab 2.0 was subsequently used to anticipate toxicity of 13h and 13k (Table 6).62 Accordingly, both compounds are not hERG blockers, suggesting low cardiotoxicity effect.44 Besides, they did not show human hepatoxicity, mutagenic effect, and eye corrosion. However, 13k may cause reactions with the skin and respiratory tract. Meanwhile 13h showed no interaction with most organs excepting the respiratory system. In general, these derivatives provided suitable physicochemical and ADMET profiles to be considered as good anticancer compounds for further hit-to-lead optimization stages.
| Toxicitya | 13h | 13k |
|---|---|---|
| a The values in parentheses display the probability of being toxic. | ||
| hERG blockers | Inactive (0.002) | Inactive (0.009) |
| Human hepatotoxicity | Negative (0.425) | Negative (0.12) |
| AMES toxicity | No (0.324) | No (0.273) |
| Rat oral acute toxicity | No (0.412) | No (0.436) |
| FDA maximum (recommended) daily dose | Negative (0.11) | Negative (0.235) |
| Skin sensitization | No (0.497) | Yes (0.747) |
| Respiratory toxicity | Yes (0.687) | Yes (0.893) |
| Eye irritation | No (0.263) | No (0.135) |
Experimental
Materials and methods
All chemicals were used as received without any further purification and obtained from Aldrich or Merck. Microwave irradiation experiments were performed using an Anton Paar Microwave Synthetic Reactor Monowave 400. Merck silica gel 60 F254 plates and Merck silica gel 60 (240–400 mesh) were used for thin-layer chromatography and column flash chromatography, respectively. Melting points were measured in open capillaries on a Buchi melting point B-545 apparatus (Buchi Instrument, Switzerland) and the values reported are uncorrected. A SCIEX X500 QTOF mass spectrometer in ESI (+) or ESI (−) mode was used to calculate the HRMS spectra. IR spectra have been recorded as KBr pellets, with a PerkinElmer Spectrum Two FT-IR spectrometer. NMR experiments were acquired using a Bruker Avance III spectrometer (600 and 125/150 MHz).General procedure for synthesis of compounds 13a–m
Approach A: Under microwave irradiation (150 W), a mixture of 2-hydroxy-1,4-naphthoquinone (14, 1 mmol), tetronic acid (15, 1 mmol), heteroaromatic aldehyde (16a–m, 1 mmol), and NH4OAc (17, 3.0 mmol) in glacial acetic acid (3 mL) was stirred for 20 min at 120 °C. The reaction mixture was then poured into water (20 mL) and extracted with dichloromethane (3 × 20 mL), washed with brine (3 × 10 mL). The combined organic layer was dried over anhydrous Na2SO4, concentrated in vacuo, and purified by column chromatography using a dichloromethane-acetone eluent (20:1, 25:2, v/v) to furnish products 13a–m.
Approach B: Under microwave irradiation (150 W), a mixture of 2-amino-1,4-naphthoquinone (18, 1 mmol), tetronic acid (15, 1 mmol), heteroaromatic aldehyde (16a–m, 1 mmol), and p-TsOH (0.02 mmol) in glacial acetic acid (3 mL) was stirred for 20 min at 120 °C. The reaction mixture was then poured into water (20 mL), extracted with dichloromethane (3 × 20 mL), and washed with brine (3 × 10 mL). The combined organic layer was dried over anhydrous Na2SO4, concentrated in vacuo, and purified by column chromatography using a dichloromethane-acetone eluent (20:1, 25:2, v/v) to furnish products 13a–m.
MTT assay
The investigated cancer cell lines were purchased from the American Type Culture Collection (ATCC, USA). The cytotoxicity of the newly synthesized series of compounds was studied against non-small lung (A549, CCL-185™), epidermoid carcinoma (KB, CCL-17™), breast (MCF7, HTB-22™), hepatocellular carcinoma (HepG2, HB-8065™), cancer cells, and human embryonic kidney (Hek-293, CRL-1573™) cells, which were cultured in DMEM (Dulbeccos Modified Eagle Medium) medium, supplemented with 10% fetal bovine serum, 100 U ml−1 penicillin, and 100 μg mL−1 streptomycin, at 37 °C in humidified atmosphere (95% air and 5% CO2). Solutions of compounds 13a–m in DMSO at different concentratrions (1.00, 0.25, 0.0625, and 0.0156 μg mL−1) were added to human cancer cell lines (3 × 104 cells per ml). After 3 days-incubation, 10 μl solution of 5 mg mL−1 MTT in sodium phosphate buffer (PBS, 0.1 M, pH 7.4) was then supplemented, the cells were incubated at 37 °C for 4 h, and then removed medium. The obtained formazan crystals were dissolved by DMSO (150 μl). A Biotek Epoch 2 microplate reader was used to measure the absorbance of the solutions at 540 nm.63–65Molecular docking study
Molecular docking studies were performed for all the synthesized compounds against the tubulin target. To do so, the tubulin-colchicine complex (PDB ID: 4O2B) was taken from the Protein Data Bank.52 They were prepared for docking assays following the standard protocol implemented into ICM Pro (x64) software.44,66 The chains α and β tubulin were retained, followed by deleting all the water molecules, optimizing all hydrogens and optimizing HisProAsnGlnCys, setting MMFF forcefield, and predicting the binding site of Colchicine by using ICMpocketfinder.67 The 2D structures of synthesized compounds 13a–13m were generated using ChemDraw 20.1.1, then imported to ICM Pro software and convert to 3D conformations for docking. Before docking synthesized compounds, the co-crystallized ligand colchicine was removed, and redocked into the binding site of Tubulin. Conformational sampling is based on the biased probability Monte Carlo (BPMC) procedure. The ICM scoring function is a GBSA/MM-type,66 which is weighted according to the following parameters (i) internal force-field energy of the ligand, (ii) entropy loss of the ligand between bound and unbound states, (iii) ligand-receptor hydrogen bond interactions, (iv) polar and non-polar solvation energy differences between bound and unbound states, (v) electrostatic energy, (vi) hydrophobic energy, and (vii) hydrogen bond donor or acceptor desolvation. The binding energy (kcal mol−1) is calculated via equation: ΔG = ΔEIntFF + TΔSTor + α1ΔEHBond + α2ΔEHBDesol + α3ΔESolEl + α4ΔEHPhob + α5QSize.66 For each ligand, 50 conformations were generated, and the conformations with better binding scores and key interactions similar to colchicine were selected for further studies. The docking results were then visualized using BIOVIA Discovery Studio Visualizer 2021.Physicochemical and pharmacokinetic properties prediction
Calculations of the physicochemical parameters of synthesized compounds relevant to ADME were performed using SwissAMDE, a free online cheminformatics tool.57 Computed parameters are related to the evaluation of drug-likeness, lead-likeness characteristics such as Lipinski, Veber, Goshe, Egan, and Muegge rules.68 Additionally, their toxicity was also predicted by another web-based tool – ADMETlab 2.0.62,69 The parameters can be rapidly determined by easily inputting the SMILES codes of the compounds into the website.Conclusions
A series of new heterocyclic hybrid compounds of 4-azapodophyllotoxin were successfully synthesized via microwave-assisted multicomponent reactions. The efficient synthesis of novel hybrid compounds has been expected to facilitate the discovery of numerous classes of bioactive heterocyclic compounds. Obviously, the in vitro cytotoxic assessment and molecular docking study revealed that all products showed cytotoxic activity and they are all capable of binding to the active site of colchicine in tubulin. Compounds 13a (HetAr = thiophen-3-yl), and 13d (HetAr = 5-bromofuran-2-yl) displayed high cytotoxic selectivity for A549 and HepG2 cancer cell lines when compared to the other cancer cell lines and low toxicity to normal Hek-293 cell line with IC50 = 22.90 ± 2.18 and 37.12 ± 3.30 μM, respectively. The most active compound 13k containing 2-methoxypyridin-4-yl group exhibited significant cytotoxicity against human lung adenocarcinoma cells, human mouth epidermal carcinoma cells, and human hepatocellular carcinoma cells with IC50 ranging from 0.23 to 0.27 μM. Compound 13h (HetAr = 2-chlorothiazol-5-yl) displayed the highest toxicity against A549 cells with IC50 = 0.16 ± 0.02 μM. Notably, compounds 13h,k, which have strong binding energy with the active site of tubulin at −17.86, and −18.05 kcal mol−1, respectively, provided suitable physicochemical and ADMET profiles to be considered as good anticancer compounds. Taken together, these potent cytotoxic compounds have potential for investigation into the anticancer activity.Author contributions
Ha Thanh Nguyen: project administration: conceptual design; writing – original draft. Ket Tran Van, and Phuong Hoang Thi: investigation: synthesis of compounds. Julien Braire and Quynh Giang Nguyen Thi: investigation: characterisation of compounds. Tu Anh Le Thi and Giang Le-Nhat-Thuy: investigation: biological evaluation of compounds. Tuyet Anh Dang Thi and Doan Vu Ngoc: investigation: revised draft. Hai Pham-The and Tuan Anh Nguyen: investigation: molecular docking studies. Tuyen Van Nguyen: supervision: conceptual design; writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are indebted to National Foundation for Science and Technology Development (NAFOSTED) Grant number: 104.01-2021.50 for financial support. Ha Thanh Nguyen was also funded by the Postdoctoral Scholarship Programme of Vingroup Innovation Foundation (VINIF), code VINIF.2023.STS.29.Notes and references
- G. Le-Nhat-Thuy, T. A. Dang Thi, Q. G. Nguyen Thi, P. Hoang Thi, T. A. Nguyen, H. T. Nguyen, T. H. Nguyen Thi, H. S. Nguyen and T. V. Nguyen, Synthesis and biological evaluation of novel benzo[a]pyridazino[3,4-c]phenazine derivatives, Bioorg. Med. Chem. Lett., 2021, 43, 128054 CrossRef PubMed.
- V. Bhardwaj, M. N. Noolvi, S. Jalhan and H. M. Patel, Synthesis, and antimicrobial evaluation of new pyridine imidazo [2,1b]-1,3,4-thiadiazole derivatives, J. Saudi Chem. Soc., 2016, 20, S406–S410 CrossRef CAS.
- V. Kamat, R. Santosh, B. Poojary, S. P. Nayak, B. K. Kumar, M. Sankaranarayanan, Faheem, S. Khanapure, D. A. Barretto and S. K. Vootla, Pyridine- and thiazole-based hydrazides with promising anti-inflammatory and antimicrobial activities along with their in silico studies, ACS Omega, 2020, 5, 25228–25239 CrossRef CAS PubMed.
- H. T. Nguyen, T. H. Nguyen Thi, J. Braire, T. A. Dang Thi and T. Nguyen Van, Microwave-assisted multicomponent synthesis of new 6-arylated 5-hydroxy-benzo[ a ]phenazine derivatives and their potential anti-inflammatory activity, ChemistrySelect, 2023, 8, e202204376 CrossRef CAS.
- H. T. Nguyen, A. N. Tuan, T. A. D. Thi, K. T. Van, G. Le-Nhat-Thuy, P. H. Thi, Q. G. N. Thi, C. B. Thi, H. T. Quang and T. Van Nguyen, Synthesis, in vitro α-glucosidase, and acetylcholinesterase inhibitory activities of novel indol-fused pyrano[2,3-d]pyrimidine compounds, Bioorg. Med. Chem. Lett., 2023, 129566 Search PubMed.
- H. T. Nguyen, G. Le-Nhat-Thuy, P. H. Thi, Q. G. N. Thi, T. A. Nguyen, T. H. N. Thi, T. A. D. Thi and T. V. Nguyen, Microwave-Assisted Three-Component Synthesis of Novel N-Arylated-Dihydrobenzo[g]quinoline-5,10-Diones and Their Potential Cytotoxic Activity, Chem. Biodiversity, 2022, 19, e202200359 CrossRef CAS PubMed.
- N. T. Dung, L. N. T. Giang, P. H. Thu, N. H. Thuong, D. T. Tuyet Anh, L. Q. Tan, N. H. Thanh, L. T. T. Anh, N. T. Anh, B. L. Giang, N. V. Tuyen and P. V. Kiem, Synthesis and cytotoxic evaluation of carboxylic acid-functionalized indenoisoquinolines, Nat. Prod. Commun., 2019, 14, 1934578x1984978 CrossRef.
- A. A. Abbas and K. M. Dawood, Anticancer therapeutic potential of benzofuran scaffolds, RSC Adv., 2023, 13, 11096–11120 RSC.
- T. A. Dang Thi, T. H. Vu Thi, H. Thi Phuong, T. Ha Nguyen, C. Pham The, C. Vu Duc, Y. Depetter, T. Van Nguyen and M. D’hooghe, Synthesis and anticancer properties of new (dihydro)pyranonaphthoquinones and their epoxy analogs, Bioorg. Med. Chem. Lett., 2015, 25, 3355–3358 CrossRef CAS.
- M. Ashraf-Uz-Zaman, X. Li, Y. Yao, C. B. Mishra, B. K. Moku and Y. Song, Quinazolinone Compounds Have Potent Antiviral Activity against Zika and Dengue Virus, J. Med. Chem., 2023, 66, 10746–10760 CrossRef CAS PubMed.
- R. J. Obaid, N. Naeem, E. U. Mughal, M. M. Al-Rooqi, A. Sadiq, R. S. Jassas, Z. Moussa and S. A. Ahmed, Inhibitory potential of nitrogen, oxygen and sulfur containing heterocyclic scaffolds against acetylcholinesterase and butyrylcholinesterase, RSC Adv., 2022, 12, 19764–19855 RSC.
- X. Li and Y. Song, Structure and function of SARS-CoV and SARS-CoV-2 main proteases and their inhibition: A comprehensive review, Eur. J. Med. Chem., 2023, 260, 115772 CrossRef CAS PubMed.
- C. H. Takimoto and A. Awada, Safety and anti-tumor activity of sorafenib (Nexavar®) in combination with other anti-cancer agents: a review of clinical trials, Cancer Chemother. Pharmacol., 2008, 61, 535–548 CrossRef CAS PubMed.
- F. Fondevila, C. Méndez-Blanco, P. Fernández-Palanca, J. González-Gallego and J. L. Mauriz, Anti-tumoral activity of single and combined regorafenib treatments in preclinical models of liver and gastrointestinal cancers, Exp. Mol. Med., 2019, 51, 1–15 CrossRef CAS PubMed.
- K. V. M. Huber, E. Salah, B. Radic, M. Gridling, J. M. Elkins, A. Stukalov, A.-S. Jemth, C. Göktürk, K. Sanjiv, K. Strömberg, T. Pham, U. W. Berglund, J. Colinge, K. L. Bennett, J. I. Loizou, T. Helleday, S. Knapp and G. Superti-Furga, Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy, Nature, 2014, 508, 222–227 CrossRef CAS PubMed.
- A. Srirangam, M. Milani, R. Mitra, Z. Guo, M. Rodriguez, H. Kathuria, S. Fukuda, A. Rizzardi, S. Schmechel, D. G. Skalnik, L. M. Pelus and D. A. Potter, The human immunodeficiency virus protease inhibitor ritonavir inhibits lung cancer cells, in part, by inhibition of survivin, J. Thorac. Oncol., 2011, 6, 661–670 CrossRef PubMed.
- R. Batchu, O. Gruzdyn, C. Bryant, A. Qazi, S. Kumar, S. Chamala, S. Kung, R. Sanka, U. Puttagunta, D. Weaver and S. Gruber, Ritonavir-mediated induction of apoptosis in pancreatic cancer occurs via the rb/e2f-1 and akt pathways, Pharmaceuticals, 2014, 7, 46–57 CrossRef CAS PubMed.
- S. J. Connolly, Evidence-based analysis of amiodarone efficacy and safety, Circulation, 1999, 100, 2025–2034 CrossRef CAS PubMed.
- A. T. Lenis, P. M. Lec, K. Chamie and M. MSHS, Bladder Cancer: A Review, JAMA, 2020, 324, 1980–1991 CrossRef CAS PubMed.
- M. Belcourt, P. Penketh, W. Hodnick, D. Johnson, D. Sherman, S. Rockwell and A. Sartorelli, Mitomycin resistance in mammalian cells expressing the bacterial mitomycin C resistance protein MCRA, Proc. Natl. Acad. Sci. U.S.A., 1999, 96, 10489–10494 CrossRef CAS PubMed.
- T. Nguyen Van, G. Verniest, S. Claessens and N. De Kimpe, Total synthesis of four naturally occurring 2-azaanthraquinone antibiotics, 6-deoxy-8-methylbostrycoidin, 6-deoxybostrycoidin, 7-O-demethyl-6-deoxybostrycoidin and scorpinone, Tetrahedron, 2005, 61, 2295–2300 CrossRef.
- H. T. Nguyen, H. Nguyen Van, P. Hoang Thi, T. A. D. Thi, G. Le-Nhat-Thuy, Q. G. Nguyen Thi, A. N. Tuan, C. Ba Thi, H. Tran Quang and T. Van Nguyen, Synthesis and cytotoxic evaluation of new fluoro and trifluoromethyl substituents containing chromeno[2,3- d ]pyrimidines, ChemistrySelect, 2023, 8, e202300227 CrossRef CAS.
- N. H. Thanh, H. T. Phuong, L. N. T. Giang, N. T. Q. Giang, N. T. T. Ha, D. T. T. Anh, V. D. Cuong, N. Van Tuyen and P. Van Kiem, 4-(Dimethylamino)pyridine as an efficient catalyst for one-pot synthesis of 1,4-pyranonaphthoquinone derivatives via microwave-assisted sequential three component reaction in green solvent, Nat. Prod. Commun., 2021, 16, 1934578X2110539 CrossRef.
- N. H. Thanh, N. T. Q. Giang, N. V. Ha, H. T. Phuong, L. N. Thuy Giang, N. T. Anh, B. T. Cham, L. D. Huy, D. T. T. Anh, P. Van Kiem and N. V. Tuyen, Synthesis and evaluation of acetylcholinesterase inhibitory and cytotoxic activities of pyrano[2,3- d ]pyrimidines, Nat. Prod. Commun., 2023, 18, 1934578X231201037 CrossRef CAS.
- M. M. Abdelshaheed, H. I. El Subbagh, M. A. Tantawy, R. T. Attia, K. M. Youssef and I. M. Fawzy, Discovery of new pyridine heterocyclic hybrids; design, synthesis, dynamic simulations, and in vitro and in vivo breast cancer biological assays, RSC Adv., 2023, 13, 15689–15703 RSC.
- G. Le-Nhat-Thuy, T. V. Dinh, H. Pham-The, H. Nguyen Quang, N. Nguyen Thi, T. A. Dang Thi, P. Hoang Thi, T. A. Le Thi, H. T. Nguyen, P. Nguyen Thanh, T. Le Duc and T. V. Nguyen, Design, synthesis and evaluation of novel hybrids between 4-anilinoquinazolines and substituted triazoles as potent cytotoxic agents, Bioorg. Med. Chem. Lett., 2018, 28, 3741–3747 CrossRef CAS PubMed.
- T. A. Dang Thi, Y. Depetter, K. Mollet, H. Thi Phuong, D. Vu Ngoc, C. Pham The, H. Thanh Nguyen, T. H. Nguyen Thi, H. Huy Nguyen, M. D’hooghe and T. Van Nguyen, Expedient stereoselective synthesis of new dihydropyrano- and dihydrofuranonaphthoquinones, Tetrahedron Lett., 2015, 56, 2422–2425 CrossRef CAS.
- G. Le-Nhat-Thuy, T. A. Dang Thi, P. Hoang Thi, Q. G. Nguyen Thi, H.-T. Nguyen, D. Vu Ngoc, T.-A. Nguyen and T. Van Nguyen, Multicomponent synthesis of novel 3-benzoyl-4h-benzo[g]chromene-5,10-dione derivatives, Tetrahedron Lett., 2021, 75, 153215 CrossRef CAS.
- I. Dine, E. Mulugeta, Y. Melaku and M. Belete, Recent advances in the synthesis of pharmaceutically active 4-quinolone and its analogues: a review, RSC Adv., 2023, 13, 8657–8682 RSC.
- Y. You, Podophyllotoxin derivatives: current synthetic approaches for new anticancer agents, Curr. Pharm. Des., 2005, 11, 1695–1717 CrossRef CAS PubMed.
- N. Jeedimalla, M. Flint, L. Smith, A. Haces, D. Minond and S. P. Roche, Multicomponent assembly of 4-aza-podophyllotoxins: A fast entry to highly selective and potent anti-leukemic agents, Eur. J. Med. Chem., 2015, 106, 167–179 CrossRef CAS PubMed.
- I. V. Magedov, M. Manpadi, E. Rozhkova, N. M. Przheval’skii, S. Rogelj, S. T. Shors, W. F. A. Steelant, S. V. slambrouck and A. Kornienko, Structural simplification of bioactive natural products with multicomponent synthesis: Dihydropyridopyrazole analogues of podophyllotoxin, Bioorg. Med. Chem. Lett., 2007, 17, 1381–1385 CrossRef CAS PubMed.
- A. Kamal, P. Suresh, A. Mallareddy, B. A. Kumar, P. V. Reddy, P. Raju, J. R. Tamboli, T. B. Shaik, N. Jain and S. V. Kalivendi, Synthesis of a new 4-aza-2,3-didehydropodophyllotoxin analogues as potent cytotoxic and antimitotic agents, Bioorg. Med. Chem., 2011, 19, 2349–2358 CrossRef CAS PubMed.
- A. Kamal, J. R. Tamboli, V. L. Nayak, S. F. Adil, M. V. P. S. Vishnuvardhan and S. Ramakrishna, Synthesis of a terphenyl substituted 4-aza-2,3-didehydropodophyllotoxin analogues as inhibitors of tubulin polymerization and apoptosis inducers, Bioorg. Med. Chem., 2014, 22, 2714–2723 CrossRef CAS PubMed.
- M. N. Semenova, A. S. Kiselyov, D. V. Tsyganov, L. D. Konyushkin, S. I. Firgang, R. V. Semenov, O. R. Malyshev, M. M. Raihstat, F. Fuchs, A. Stielow, M. Lantow, A. A. Philchenkov, M. P. Zavelevich, N. S. Zefirov, S. A. Kuznetsov and V. V. Semenov, Polyalkoxybenzenes from plants. 5. parsley seed extract in synthesis of azapodophyllotoxins featuring strong tubulin destabilizing activity in the sea urchin embryo and cell culture assays, J. Med. Chem., 2011, 54, 7138–7149 CrossRef CAS PubMed.
- I. V. Magedov, M. Manpadi, S. Van slambrouck, W. F. A. Steelant, E. Rozhkova, N. M. Przheval’skii, S. Rogelj and A. Kornienko, Discovery and investigation of antiproliferative and apoptosis-inducing properties of new heterocyclic podophyllotoxin analogues accessible by a one-step multicomponent synthesis, J. Med. Chem., 2007, 50, 5183–5192 CrossRef CAS PubMed.
- R. Labruère, B. Gautier, M. Testud, J. Seguin, C. Lenoir, S. Desbène-Finck, P. Helissey, C. Garbay, G. G. Chabot, M. Vidal and S. Giorgi-Renault, Design, Synthesis, and biological evaluation of the first podophyllotoxin analogues as potential vascular-disrupting agents, ChemMedChem, 2010, 5, 2016–2025 CrossRef PubMed.
- N. H. Thanh, H. T. Phuong, L. T. Tu Anh, L. N. Thuy Giang, N. T. Quynh Giang, N. T. Anh, D. T. Tuyet Anh and P. Van Kiem, Synthesis and cytotoxic evaluation of fluoro and trifluoromethyl substituents containing novel naphthoquinone-fused podophyllotoxins, Nat. Prod. Commun., 2022, 17, 1934578X2211331 CrossRef.
- F. Shi, X.-N. Zeng, G. Zhang, N. Ma, B. Jiang and S. Tu, Facile synthesis of new 4-aza-podophyllotoxin analogs via microwave-assisted multi-component reactions and evaluation of their cytotoxic activity, Bioorg. Med. Chem. Lett., 2011, 21, 7119–7123 CrossRef CAS PubMed.
- C. Shi, J. Wang, H. Chen and D. Shi, Regioselective synthesis and in vitro anticancer activity of 4-aza-podophyllotoxin derivatives catalyzed by l -proline, J. Comb. Chem., 2010, 12, 430–434 CrossRef CAS PubMed.
- N. H. Thanh, L. Q. Bao, H. Pham-The, D. T. T. Anh and P. Van Kiem, Synthesis, molecular docking, and cytotoxic evaluation of fluorinated podophyllotoxin derivatives, Nat. Prod. Commun., 2023, 18, 1934578X2311537 CrossRef.
- X. Yang, C. Zhang and L. Wu, l -Proline catalyzed three-component synthesis of para-naphthoquinone–4-aza-podophyllotoxin hybrids as potent antitumor agents, RSC Adv., 2015, 5, 18945–18951 RSC.
- Q. G. Nguyen Thi, G. Le-Nhat-Thuy, T. A. Dang Thi, P. Hoang Thi, A. Nguyen Tuan, T. H. Nguyen Thi, T. T. Nguyen, T. Nguyen Ha, H. Hoang Mai and T. V. Nguyen, Synthesis of novel potent cytotoxicy podophyllotoxin-naphthoquinone compounds via microwave-assited multicomponent domino reactions, Bioorg. Med. Chem. Lett., 2021, 37, 127841 CrossRef CAS PubMed.
- H. T. Nguyen, Q. G. Nguyen Thi, T. H. Nguyen Thi, P. H. Thi, G. Le-Nhat-Thuy, T. A. Dang Thi, B. Le-Quang, H. Pham-The and T. Van Nguyen, Synthesis and biological activity, and molecular modelling studies of potent cytotoxic podophyllotoxin-naphthoquinone compounds, RSC Adv., 2022, 12, 22004–22019 RSC.
- H.-T. Nguyen, P. H. Thi, G. Le-Nhat-Thuy, Q. G. N. Thi, T. A. Nguyen, T. A. D. Thi and T. V. Nguyen, Synthesis of novel γ-butyrolactone-based phenazine compounds via microwave-assisted multicomponent domino reactions, Chem. Heterocycl. Compd., 2022, 58, 749–755 CrossRef CAS.
- H.-T. Nguyen, T. A. Dang Thi, P. Hoang Thi, G. Le-Nhat-Thuy, Q. G. Nguyen Thi, A. Nguyen Tuan, T. A. Le Thi and T. Van Nguyen, A new approach for the synthesis of novel naphthoquinone chalcone hybrid compounds, Tetrahedron Lett., 2021, 81, 153337 CrossRef CAS.
- H. T. Nguyen, T. H. Nguyen Thi, J. Braire, T. A. Dang Thi and T. Nguyen Van, Microwave-assisted multicomponent synthesis of new 6-arylated 5-hydroxy-benzo[ a ]phenazine derivatives and their potential anti-inflammatory activity, ChemistrySelect, 2023, 8, e202204376 CrossRef CAS.
- B. Kesteleyn, T. N. Van and N. De Kimpe, Synthesis of 3-alkyl- and 3-aryl-2-aza-anthraquinones, Tetrahedron, 1999, 55, 2091–2102 CrossRef CAS.
- N. Van Tuyen, B. Kesteleyn and N. De Kimpe, Synthesis of 2-alkoxymethyl-3-trifluoromethyl-1,4-naphthoquinones, Tetrahedron, 2002, 58, 121–127 CrossRef.
- R. A. Stanton, K. M. Gernert, J. H. Nettles and R. Aneja, Drugs that target dynamic microtubules: A new molecular perspective, Med. Res. Rev., 2011, 31, 443–481 CrossRef CAS PubMed.
- J. D. Loike and S. B. Horwitz, Effects of podophyllotoxin and VP-16-213 on microtubule assembly in vitro and nucleoside transport in HeLa cells, Biochemistry, 1976, 15, 5435–5443 CrossRef CAS PubMed.
- A. E. Prota, F. Danel, F. Bachmann, K. Bargsten, R. M. Buey, J. Pohlmann, S. Reinelt, H. Lane and M. O. Steinmetz, The novel microtubule-destabilizing drug bal27862 binds to the colchicine site of tubulin with distinct effects on microtubule organization, J. Mol. Biol., 2014, 426, 1848–1860 CrossRef CAS PubMed.
- A. Massarotti, A. Coluccia, R. Silvestri, G. Sorba and A. Brancale, The tubulin colchicine domain: a molecular modeling perspective, ChemMedChem, 2012, 7, 33–42 CrossRef CAS PubMed.
- W. Li, H. Sun, S. Xu, Z. Zhu and J. Xu, Tubulin inhibitors targeting the colchicine binding site: a perspective of privileged structures, Future Med. Chem., 2017, 9, 1765–1794 CrossRef CAS PubMed.
- H. Pham-The, M. Á. Cabrera-Pérez, N.-H. Nam, J. A. Castillo-Garit, B. Rasulev, H. Le-Thi-Thu and G. M. Casañola-Martin, In silico assessment of adme properties: advances in caco-2 cell monolayer permeability modeling, Curr. Top. Med. Chem., 2019, 18, 2209–2229 CrossRef.
- H. Pham-The, I. González-Álvarez, M. Bermejo, T. Garrigues, H. Le-Thi-Thu and M. Á. Cabrera-Pérez, The use of rule-based and qspr approaches in adme profiling: a case study on caco-2 permeability, Mol. Inf., 2013, 32, 459–479 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- H. Pham-The, T. Garrigues, M. Bermejo, I. González-Álvarez, M. C. Monteagudo and M. Á. Cabrera-Pérez, Provisional classification and in silico study of biopharmaceutical system based on caco-2 cell permeability and dose number, Mol. Pharmaceutics, 2013, 10, 2445–2461 CrossRef CAS.
- A. Daina and V. Zoete, A BOILED-Egg to predict gastrointestinal absorption and brain penetration of small molecules, ChemMedChem, 2016, 11, 1117–1121 CrossRef CAS.
- A. J. Castillo-Garit, M. G. Casanola-Martin, H. Le-Thi-Thu, H. Pham-The and J. S. Barigye, A simple method to predict blood–brain barrier permeability of drug- like compounds using classification trees, Med. Chem., 2017, 13, 664–669 Search PubMed.
- M. Á. Cabrera-Pérez and H. Pham-The, Computational modeling of human oral bioavailability: what will be next?, Expert Opin. Drug Discovery, 2018, 13, 509–521 CrossRef PubMed.
- G. Xiong, Z. Wu, J. Yi, L. Fu, Z. Yang, C. Hsieh, M. Yin, X. Zeng, C. Wu, A. Lu, X. Chen, T. Hou and D. Cao, ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties, Nucleic Acids Res., 2021, 49, W5–W14 CrossRef CAS PubMed.
- P. T. Tham, P. T. Chinh, N. V. Tuyen, D. N. Bang, D. T. Van, V. T. Kien, H. T. Thanh, D. H. Quynh, V. D. Cuong, N. H. Thanh and A. Pérez-Encabo, Synthesis and cytotoxic evaluation of novel simplified plinabulin-quinoline derivatives, Mendeleev Commun., 2021, 31, 213–215 CrossRef CAS.
- H. T. Pham, G. Le-Nhat-Thuy, T. A. D. Thi, P. H. Thi, T. A. Nguyen, T. H. Nguyen, T. D. Nguyen, H. T. Ngo, T. A. Le and T. Van Nguyen, Synthesis and evaluation of the cytotoxicity of indeno[1,2-c]isoquinoline derivatives bearing ester functional group and 1,2,3-triazole moiety, Chem. Heterocycl. Compd., 2020, 56, 1167–1172 CrossRef CAS.
- T. Pham Thi, T. G. Le Nhat, T. Ngo Hanh, T. Luc Quang, C. Pham The, T. A. Dang Thi, H. T. Nguyen, T. H. Nguyen, P. Hoang Thi and T. Van Nguyen, Synthesis and cytotoxic evaluation of novel indenoisoquinoline-substituted triazole hybrids, Bioorg. Med. Chem. Lett., 2016, 26, 3652–3657 CrossRef CAS PubMed.
- M. A. C. Neves, M. Totrov and R. Abagyan, Docking and scoring with ICM: the benchmarking results and strategies for improvement, J. Comput.-Aided Mol. Des., 2012, 26, 675–686 CrossRef CAS PubMed.
- L. C. Huan, H. Pham-The, H. Le-Thi-Thu, T. P. Thao, D. N. Que, N.-T. Trang, P. T. Phuong Dung, M. Pyo, S.-B. Han, N. T. Thuan and N.-H. Nam, Exploration of some thiazolidine-2,4-dione and 2-oxoindoline derivatives incorporating 3,4,5-trimethoxybenzyl moiety as novel anticancer agents, Lett. Drug Des. Discovery, 2018, 15, 375–387 CrossRef CAS.
- M. A. Cabrera-Perez, H. Pham-The, M. Bermejo, I. G. Alvarez, M. G. Alvarez and T. M. Garrigues, QSPR in oral bioavailability: specificity or integrality?, Mini-Rev. Med. Chem., 2012, 12, 534–550 CrossRef CAS PubMed.
- G. Xiong, Z. Wu, J. Yi, L. Fu, Z. Yang, C. Hsieh, M. Yin, X. Zeng, C. Wu, A. Lu, X. Chen, T. Hou and D. Cao, ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties, Nucleic Acids Res., 2021, 49, W5–W14 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07396c |
| This journal is © The Royal Society of Chemistry 2024 |