
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactive direct air capture of CO2 to C–C coupled products using multifunctional materials†
Shazia Sharmin
Satter
 ,
Johnny
Saavedra Lopez
,
Michael L.
Hubbard
,
Yuan
Jiang
,
Robert A.
Dagle
and
Jotheeswari
Kothandaraman
*
,
Johnny
Saavedra Lopez
,
Michael L.
Hubbard
,
Yuan
Jiang
,
Robert A.
Dagle
and
Jotheeswari
Kothandaraman
*
Pacific Northwest National Laboratory, Advanced Energy Systems, 902 Battelle Blvd, Richland, WA 99352, USA. E-mail: jotheeswari.kothandaraman@pnnl.gov
First published on 29th May 2024
Abstract
Current direct air capture (DAC) approaches require a significant amount of energy for heating CO2-sorbed materials for regeneration and for compressing CO2 for transportation purposes. Rationally designing materials offering both capture and conversion functionalities could enable more energy and cost-efficient DAC and conversion. We have developed a single sorbent-catalytic (non-noble metal) material for the Integrated Direct Air Capture and CATalytic (iDAC-CAT) conversion of captured CO2 into value-added products. Solid sorbents are integrated with catalytic components to first capture CO2 from air. Subsequently, captured CO2, with renewable H2 co-feed is converted into olefins and paraffins. To the best of our knowledge, this is the first proof-of-concept demonstration for production of C2 products such as olefins from captured CO2. Among the different sorbent-catalytic materials studied, Fe/K2CO3/Al2O3 showed the best performance for integrated CO2 capture and conversion to C2 products. CO2 capture capacity of 8.2 wt% was achieved under optimized capture conditions at 25 °C, and a conversion of >70% to paraffins and olefins was achieved at 320–400 °C. The hydrogenation of captured CO2 was facilitated by the in situ formation of Fe3O4 and Fe5C2 species. The proximity between K and Fe was identified to be critical for producing C2 products from the captured CO2. The preliminary technoeconomic and life-cycle assessments suggest that the cost of the DAC can be considerably decreased by adopting the suggested iDAC-CAT technology, while renewable olefins could potentially be produced with negative greenhouse gases emissions.
 Jotheeswari Kothandaraman | Dr Jothi Kothandaraman is a Scientist at the Pacific Northwest National Laboratory (PNNL), one of the US Department of Energy national laboratories. She obtained her Ph.D. in chemistry from the University of Southern California, focusing on catalytically driven reversible hydrogen storage materials. In 2017, she joined PNNL as a postdoctoral researcher, performing research on the catalytic conversion of CO2. In 2019, she accepted a full-time Scientist position at PNNL. She is interested in developing green and sustainable chemical processes for fuels and materials. Currently, she is leading projects on the upcycling of CO2 sourced from both air and flue gas emissions. Dr Kothandaraman holds two US patents and has authored invited book chapters and perspective articles on CO2 capture and conversion. Dr Kothandaraman's contributions have been recognized with the Phi Kappa Phi Award for Creative and Scholarly Achievements. In 2023, Dr Kothandaraman was elected Fellow of the Royal Society of Chemistry, and she also received the PNNL Laboratory Director's Award for Early Career Exceptional Achievement, also known as the Ronald L. Brodzinski Award. Currently, she serves as an advisory board member for the RSC Sustainability journal. |
Introduction
Given the increasing CO2 concentration in the atmosphere, rapid and massive deployment of negative emission technologies (NETs) will be needed to limit global temperature increase to 1.5–2 °C.1 NETs should be large enough to remove several gigaton quantities of CO2 from the atmosphere and in this context, DAC is expected to complement other NET options. DAC technologies remove CO2 from the atmosphere at any location to balance emissions that are unavoidable or technically difficult to avoid.2 A variety of sorbents are being investigated for CO2 capture, including physisorbents such as metal–organic frameworks,3 zeolites,4,5 and activated carbon,6 as well as chemisorbents such as amine-functionalized adsorbents that commonly contain polyamines.7,8 Particularly, chemisorbents are best suited for CO2 capture from ultra-dilute sources such as air due to strong chemical interactions between CO2 and sorbents. As a result, chemisorbents are the subject of extensive studies aimed at understanding and improving their CO2 adsorption and desorption processes. However, the economic feasibility of large-scale deployment of current DAC systems is uncertain due to high energy input needed for the desorption process (cost estimates are $200–1000 per tonne CO2 for DAC compared to $36–53 per tonne CO2 for coal-derived flue gas).9 Thus, innovative use opportunities, including synthetic fuels and chemicals are desired as a means to drive down costs and provide a market for DAC. However, there are no commercial technologies that can economically produce either value-added fuels and chemicals, or solid products for storage using CO2 captured from air.The capture of CO2 and conversion of CO2 have long been viewed as two independent processes. Recently, the benefits associated with integrating the capture and conversion processes have been realized by the scientific community.10–13 The direct conversion of captured CO2 into value-added products (coupled approach) has potential advantages over traditional decoupled CO2 capture and CO2 conversion because the coupled approach avoids the energy-intensive sorbent regeneration (CO2 desorption), compression and transportation steps. Importantly, new reactive pathways for the CO2 conversion can be realized in the capture media, leading to higher conversion, selectivity, and reduced cost.11 For example, typical gas-phase CO2 hydrogenation to methanol requires high temperatures due to slower kinetics. At high temperature, a competing reaction–the reverse water gas shift reaction–is also favored, which reduces the selectivity and consumes valuable H2. On the other hand, in the amine-based capture medium, CO2 hydrogenation to methanol followed a nontraditional route for conversion to methanol through a formamide intermediate.14–17 This nontraditional low-temperature methanol synthesis route was made possible by the presence of an amine-based capture solvent medium. However, amine-based aqueous/non-aqueous solvents are not suitable for DAC application due to high volatility, viscosity, and evaporative loss of water under realistic DAC conditions. For DAC, solid sorbents have several benefits (over well-studied liquid sorbents) such as increased adsorption capacities, lower regeneration energy penalties, relative ease of handling, and improved recyclability.18,19
Though the feasibility of integrating capture and conversion processes has been shown with liquid capture solvent systems,11,14,17,20–23 the material design principles are not transferable to solids because unlike liquid systems, the sorbent and catalyst need to be integrated into a single multifunctional material in solids. The solid-state iDAC-CAT approach is limited by the lack of design parameters for this multifunctional material with the cooperative sorbent and catalytic features to perform both capture and conversion. In traditional DAC approaches, solid or liquid sorbents with low reaction enthalpy, high capture capacity, and rapid kinetics are preferred. The strong binding of CO2via chemisorption is considered a limitation in traditional DAC approaches due to regeneration requirements. But in the iDAC-CAT approach, the strong binding will be considered an opportunity because the captured CO2 is undergoing chemical conversion. The strong CO2 binding will enhance the CO2 uptake kinetics, which is critical for DAC application.
Solid materials with dual functionalities have been reported for integrated CO2 capture and conversion to C1 products such as methane24–30 and methanol.31–34 Most of these materials are composed of sorbents (metal oxides and carbonates) and metal catalysts (such as Ru, Ni, and Rh).35,36 In a first step, the sorbent reacts with CO2 to form (bi)carbonate and in a second step, (bi)carbonate reacts with hydrogen at high temperature (>300 °C) to form methane. Most of these materials also require high temperature for capture, which is not an economical option.24,37–39Amine-functionalized silica and Pd catalyst combinations have been demonstrated to be active for the integrated capture and conversion to methanol.32,33 Recently, Cu/Zn catalyst and metal carbonate combinations were identified as effective for the reactive capture of CO2 to methanol.31,34 While these materials are effective for the formation of C1 products, the conversion of captured CO2 to C2+ products remains a challenge.
In this work, we report how combinations of catalytic components and sorbents can be integrated into a single material that can capture CO2 from air at ambient conditions, and then convert the captured CO2 into valuable C2 products such as olefins. Olefins are building blocks for producing a variety of products including plastics, paints, lubricants, and surfactants. Olefins can also be converted into hard-to-decarbonize jet and diesel fuels.40 In this work, Fe-based catalytic components were incorporated into the sorbent materials to facilitate the formation of C–C bonds. Upon studying different materials and conditions, we show a proof of concept using Fe/K2CO3/Al2O3 (Fe/KA) to produce C2–C4 olefins from CO2 derived from air. We also identified that these materials are effective at converting gas-phase CO2 to olefins, with olefin to paraffin ratio of 6.9 at 360 °C.
Results and discussion
CO2 capture studies using K2CO3/Al2O3
Inorganic chemisorbents are chosen for this study because they are more durable and low-cost materials compared to amine-based sorbents for DAC.41 The commonly used inorganic chemisorbents for DAC are CaO, MgO, and alkali metal carbonates.42 Among these sorbents, alkali metal carbonates can perform capture at ambient temperature.43,44 Alkali metal carbonates are usually dispersed on high-surface-area materials such as Al2O3, to increase the carbonation rate of alkali carbonates.45,46 Based on literature studies conducted on K2CO3 loading on various supports, including carbon, alumina, and ZrO2, an optimal K2CO3 loading between 25–35 wt% on supports was identified for CO2 capture. The adsorption capacity increases with higher K2CO3 loading; however, loading above 35 wt% results in decreased adsorption capacity due to reductions in surface area and pore volume.47–49 Thus, here 25 wt% of K2CO3/Al2O3 was synthesized,50 characterized, and evaluated at 25 °C at different capture conditions to identify suitable conditions for DAC (sections S1.2, 1.3 and 1.4†). As-synthesized K2CO3/Al2O3 was characterized by BET (Brunauer–Emmett–Teller) analysis. Type IV isotherms with a characteristic hysteresis loop for both Al2O3 and K2CO3/Al2O3 were realized in the BET analysis (shown in Fig. S1A†), indicating that alumina is mesoporous in nature. Impregnation of K2CO3 over alumina resulted in a decrease in both surface area and pore volume of the original support, but the average pore sizes were almost comparable as shown in Table S1.† This implies that smaller sizes of K2CO3 filled the pores of the mesoporous alumina, confirming the dispersion of K2CO3 over the alumina surface.44The effect of pretreatment conditions and water vapor content on the capture performance of the sorbent was studied. The K2CO3/Al2O3 sorbent was first pretreated at 200 °C for 1 h under N2 flow (100 mL min−1). The material was then cooled to room temperature and pre-saturated with both 0.5 and 1.0 mol% H2O vapor, followed by introduction of 400 ppm of CO2 (Fig. S2A and S2B†) with H2O vapor (0.5 or 1.0 mol%). The amount of CO2 per g of sorbent adsorbed during both the experiments was calculated from the molar flow concentration profile of CO2versus time. For 0.5 mol% of H2O, 850 μmol g−1 of CO2 was adsorbed, whereas in the case of 1.0 mol% of H2O, 770 μmol g−1 of CO2 was adsorbed. This indicates that the 0.5 mol% of H2O had a slightly higher adsorption capacity, possibly due to the K2CO3 phase transition in the presence of excess water.51
The effect of saturating the sorbent with water vapor during CO2 capture was investigated. Here, CO2 was co-fed with 0.5 mol% H2O vapor over the pretreated K2CO3/Al2O3 as shown in Fig. S3† and compared with the pre-saturated sample (0.5 mol% H2O vapor pretreatment). The water vapor co-fed sample shows the highest sorption capacity of 6.5 wt% compared to the water vapor pretreated samples, as shown in Fig. S3.† The amount of CO2 adsorbed by the 25 wt% K2CO3/Al2O3 sorbent is ∼6.5 wt%, surpassing the amounts reported in the literature, which are 3.6 wt% for K2CO3/Al2O3 and 4.1 wt% for K2CO3/Al2O3-750 (Al2O3 heated at 750 °C before K2CO3 impregnation) under similar capture conditions, as shown in Table S3.†52 The increased CO2 capture capacity could result from the fine dispersion of K2CO3 over Al2O3. The X-ray diffraction (XRD) analysis shown in Fig. S1B† illustrates the change in phase composition of the K2CO3/Al2O3 before and after CO2 capture at room temperature in the presence of water vapor. For fresh K2CO3/Al2O3, the main diffraction peaks were attributed to dawsonite, KAlCO3(OH)2, K2CO3, and γ-Al2O3. The formation of the dawsonite on the fresh samples takes place due to the exposure of as-synthesized K2CO3/Al2O3 to CO2 in air. This agrees with the Temperature Programmed Desorption (TPD) of the fresh material shown in Fig. S1C,† where the peak at 350 °C is due to the decomposition of the dawsonite.
Thermal decomposition of the CO2-captured K2CO3 using TPD shown in Fig. S1C† shows two characteristic peaks within 100–200 °C, which is likely due to the decomposition of the species containing bicarbonate, K2CO3·2KHCO3·1.5H2O, and KHCO3. This agrees with the XRD diffraction patterns of the air-captured sorbent. The higher-temperature peak is mainly due to the decomposition of the KAlCO3(OH)2, which was reported to take place between 260 and 320 °C.44
As activated carbon (AC) is recognized as a suitable support material for CO2 capture, K2CO3/AC was synthesized and tested to evaluate its CO2 capture capacity.49 Compared to K2CO3/Al2O3, the capture capacity of K2CO3/AC was 1.3 times lower, as shown in Fig. S4.† Due to the superior capture performance of K2CO3/Al2O3 under the optimized reaction conditions, K2CO3/Al2O3 was chosen as the sorbent material for the integrated capture and conversion studies.
Conversion of captured CO2 to C1 and C2 products
The direct conversion of captured CO2 from air or concentrated point sources to C1 products such as methane, methanol, and CO has been effectively demonstrated in earlier studies.24–26,29–31,34,53 However, due to the high energy barrier of C–C coupling reactions, conversion of captured CO2 to C2+ products is still a challenge. In the literature, combining the endothermic reverse water gas shift (RWGS) (CO2 + H2 → CO + H2O) reaction with the exothermic Fischer–Tropsch (FTS) (CO + H2 → CxHy) reaction has been identified as one of the strategies for converting concentrated streams of CO2 and H2 in the gas phase to C2+ products.54 Particularly, potassium (alkali metal) modified Fe-based catalysts are known to promote carbon-chain growth in the gas-phase CO2 hydrogenation reactions.55–58 We hypothesized that by combining the Fe-based catalysts and potassium-based sorbents the captured CO2 can be directly converted to C2+ products, bypassing the energy-intensive CO2 regeneration and compression steps. Additionally, alkali modification of metals can potentially develop optimal electronics that allow the selective formation of olefins by decreasing the reactivity of adsorbed H species.59,60 To test our hypothesis, we synthesized different combinations of iron and K2CO3/Al2O3 based sorbent-catalytic materials and evaluated the capture and conversion performance of these synthesized materials.CO2 capture was performed with 400 ppm of CO2 (1200 mL min−1) and 0.5 mol% of H2O at 25 °C. The capture performance was compared with K2CO3/Al2O3, which was activated under similar conditions. Under this condition, ∼100% of the K2CO3 was utilized during CO2 capture in the case of K2CO3/Al2O3, whereas in the case of Fe2O3–KA, only 81% of the K2CO3 was utilized in CO2 capture, as shown in Table S2.† High-temperature pretreatment enhanced the capture capacity through the dawsonite decomposition reaction.44 Then, hydrogenation of the captured CO2 was performed under hydrogen pressure of 1.0 MPa at 320 °C (hold for 2.5 h) and 360 °C (hold for 2 h) at a ramp rate of 5 °C min−1 under H2 flow (60 mL min−1). This resulted in desorption of CO2 with no detectable amount of hydrogenated CO2-derived products. Most of the CO2 was released at ∼320 °C, suggesting that dawsonite is the major species formed during CO2 capture.
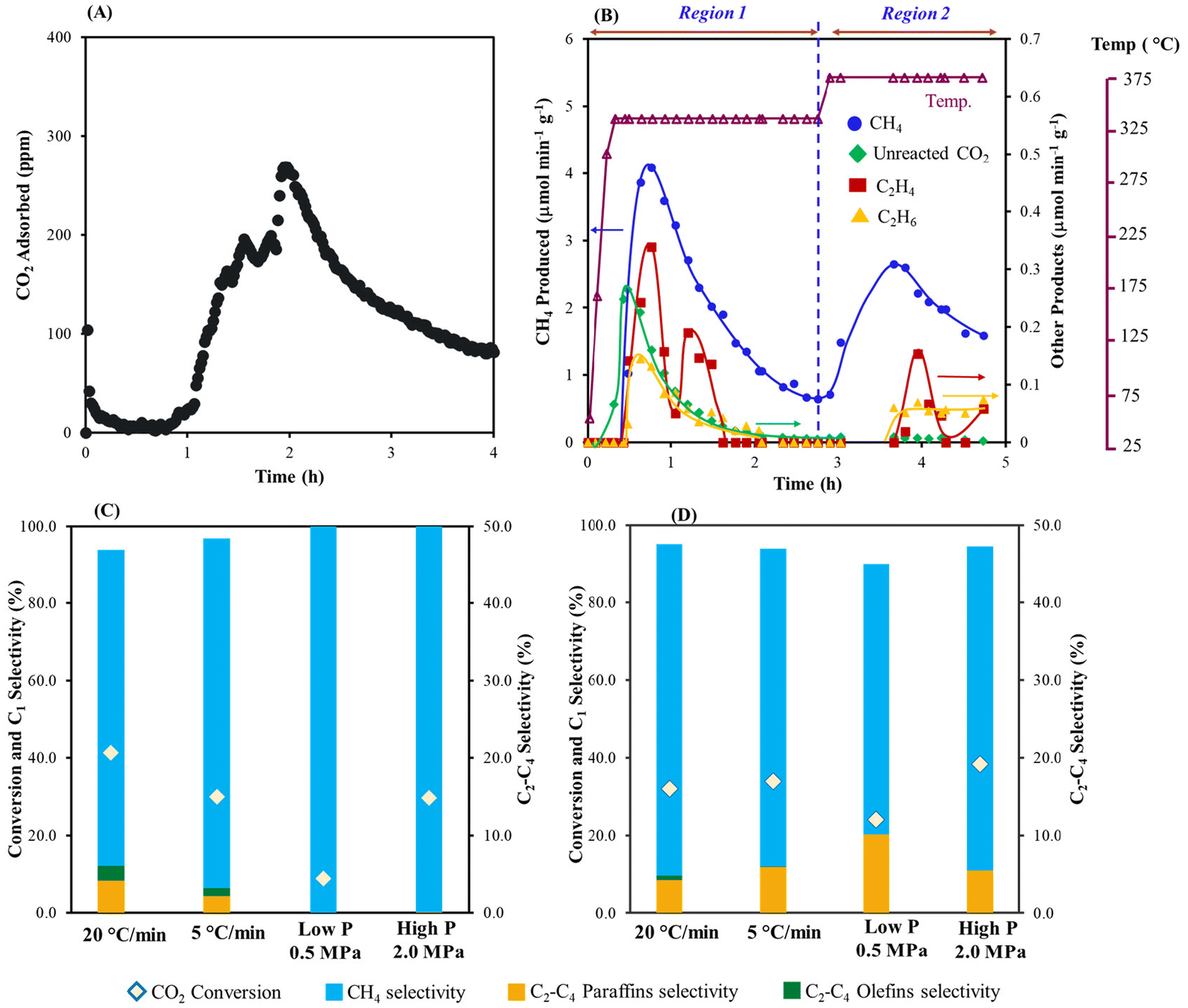 | ||
| Fig. 1 (A) CO2 capture profile over K2CO3/Fe/C at 25 °C, (B) hydrogenation profile of the captured CO2 (at heating rate of 20 °C min−1), (C) comparison of the CO2 conversion and selectivity of products formed in Region 1, at 320 °C, and (D) comparison of the CO2 conversion and selectivity of the products formed in Region 2, at 360 °C. Amount of material: 2 g; pretreatment conditions: H2 = 60 mL min−1, 400 °C, 10 h; CO2 capture conditions: CO2 = 400 ppm in N2 (flow rate = 1200 mL min−1), H2O vapor = 0.5 mol%, 25 °C, 4 h; hydrogenation: H2 = 60 mL min−1, 1.0 MPa, 320 °C for 2.5 h (5 °C min−1), followed by heating to 360 °C (5 °C min−1) for 2 h. The selectivity of CO is <5% during the hydrogenation. | ||
| Entry | Materials | Physical properties | CO2 capture | Catalytic activity | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SA (m2g−1) | PV (cm3g−1) | Average diameter (nm) | (μmol g−1) | (wt%) | CO2 conv. (%) | CH4 sel (%) | C2–C4 paraffins sel (%) | C2–C4 olefins sel (%) | C5+ sel (%) | ||
| Pretreatment conditions for materials: H2 = 60 mL min−1, 400 °C, 5 h (entry 3) and 10 h (for entries 4–6); CO2 capture conditions: CO2 = 400 ppm in N2 (flow rate = 1200 mL min−1), H2O vapor = 0.5 mol%, 25 °C, 4 h; hydrogenation: H2 = 60 mL min−1, 1.0 MPa, 320 °C for 2.5 h (5 °C min−1).a Heating rate at 5 °C min−1 during hydrogenation of captured CO2.b Heating rate of 20 °C min−1 during hydrogenation of captured CO2.c Average diameter is not given due to low surface area. | |||||||||||
| 1 | Fe/C | 33.16 | 0.4008 | —c | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| 2 | Al2O3 | 182.4 | 0.6001 | 11.4 | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| 3 | K2CO3/Al2O3 | 99.19 | 0.3262 | 10.09 | 1862 | 8.2 | N/A | N/A | N/A | N/A | N/A |
| 4 | K2CO3/Fe/Ca | — | — | — | 600–700 | 2.6–3.1 | 30.0 | 96.8 | 2.2 | 1.0 | 0.0 |
| 5 | K2CO3/Fe/Cb | — | — | — | 41.4 | 93.9 | 4.1 | 2.0 | 0.0 | ||
| 6 | K2CO3/Fe/C/Al2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
29.23 | 0.2476 | 8.95 | 1223 | 5.4 | 30.5 | 83.2 | 8.6 | 7.3 | 0.9 |
As shown in Fig. 1B, conversion of the captured CO2 was carried out with H2 feed at different temperatures. When the temperature was increased from room temperature (capture) to 320 °C (conversion) some unreacted CO2 began to desorb. Along with CO2, CH4 also formed and was the highest when the temperature reached 320 °C as shown in Fig. 1B. C2H4 and C2H6 were also produced at Region 1 (at 320 °C, 2.5 h). Further increasing the temperature to 360 °C resulted in additional CH4 production along with small amounts of ethylene and ethane (Fig. 1D). Overall, ∼74% of the total captured CO2 was converted to C1 and C2 products with ∼94.4% selectivity to methane, 4.2% selectivity to ethane, and 1.4% selectivity to ethene. To the best of our knowledge, this is the first demonstration for conversion of captured CO2 (air derived) to C1 and C2 based products in the presence of an Fe-based catalyst. Besides the formation of olefins, which are the target products, the production of renewable methane from the captured CO2 is also advantageous. This presents an alternative pathway for generating synthetic natural gas, and its utilization in existing infrastructure could lead to a lower carbon footprint. There were no detectable amounts of higher olefins or paraffins formed. Decreasing the heating rate from 20 to 5 °C min−1 decreased the overall conversion of CO2 along with a decrease in olefin selectivity at 320 °C (Fig. 1C). Increasing the hydrogen pressure further increased the selectivity to methane with a decrease in the conversion of the captured CO2 (see Fig. 1C and D).
A decrease in CO2 capture with K2CO3/Fe/C compared to K2CO3/Al2O3 is likely due to the smaller surface area of Fe/C (33.16 m2 g−1), which results in larger K2CO3 particles (Table 1). A lower CO2 loading could inhibit C–C bond formation because there are fewer carbons. To increase the surface area and eventually improve the capture performance, K2CO3/Fe/C/Al2O3 was synthesized via the wet impregnation method, as discussed in section S1.2,† and the adsorption capacity was compared with that of K2CO3/Al2O3 and K2CO3/Fe/C under similar capture conditions. The capture performance was significantly improved after the addition of Al2O3. The K2CO3/Fe/C/Al2O3 captured ∼1220 μmol g−1 of CO2 (vs. 600–700 μmol g−1 of CO2 for K2CO3/Fe/C) (Fig. 2A). This difference can be explained from the BET results of the support over which K2CO3 was impregnated. The BET isotherms of three materials are shown in Fig. 3A. Dispersion of K2CO3 on Al2O3 retained the mesoporosity of the support and showed a type IV isotherm despite a decrease in the surface area as shown in Table 1. The isotherm of Fe/C is a type II isotherm with no pronounced hysteresis loop, showing that the material is either non-porous or microporous. The surface area is very low compared to the Al2O3 support and has no pores, as shown in Table 1. Therefore, the impregnation of K2CO3 could have formed larger particles on Fe/C, leading to lower CO2 capture.49 Due to the presence of the Al2O3 pores, K2CO3 was well dispersed over a mixture of high-surface-area, mesoporous Al2O3 and non-porous Fe/C. This led to higher CO2 capture for K2CO3/Fe/C/Al2O3 compared to only K2CO3/Fe/C, as shown in Table 1.
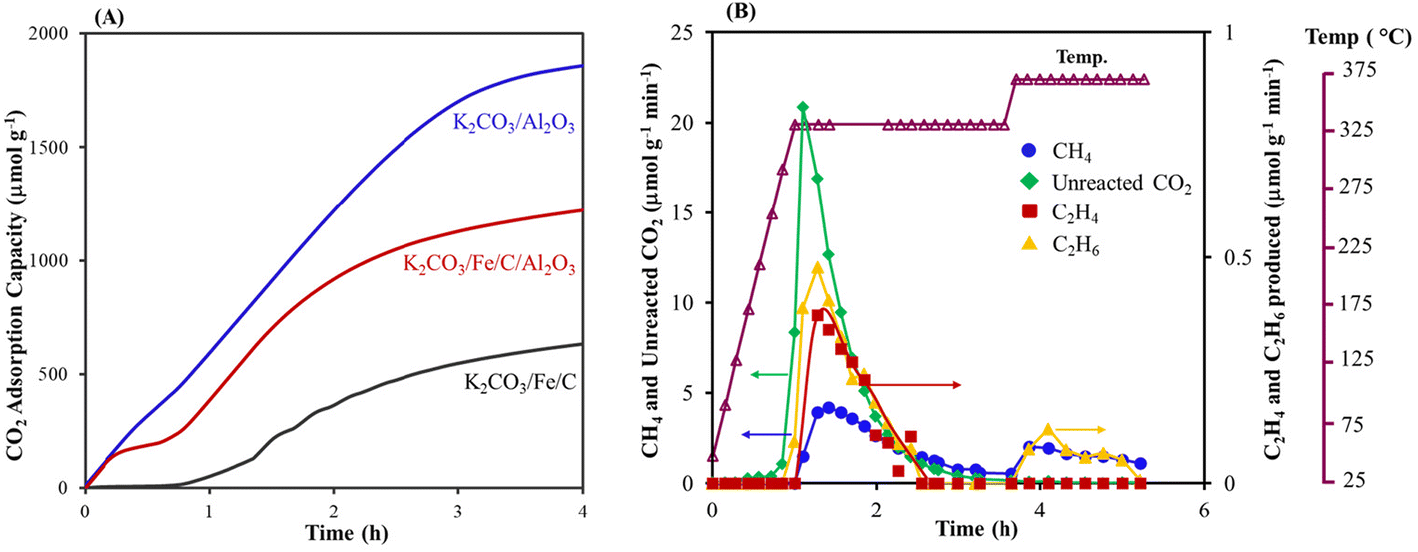 | ||
| Fig. 2 (A) Comparison of the CO2 adsorption capacity of K2CO3 on various materials pretreated at 400 °C under H2 flow, and (B) hydrogenation of the captured CO2 over K2CO3/Fe/C/Al2O3 at 320 and 360 °C. Amount of material: 2 g; pretreatment conditions: H2 = 60 mL min−1, 400 °C, 5 h (K2CO3/Al2O3) and 10 h (K2CO3/Fe/C/Al2O3 and K2CO3/Fe/C); CO2 capture conditions: CO2 = 400 ppm in N2 (flow rate = 1200 mL min−1), H2O vapor = 0.5 mol%, 25 °C, 4 h; hydrogenation: H2 = 60 mL min−1, 1.0 MPa, 320 °C for 2.5 h (5 °C min−1), followed by heating to 360 °C (5 °C min−1) for 2 h. The selectivity of CO is <5% during the hydrogenation. | ||
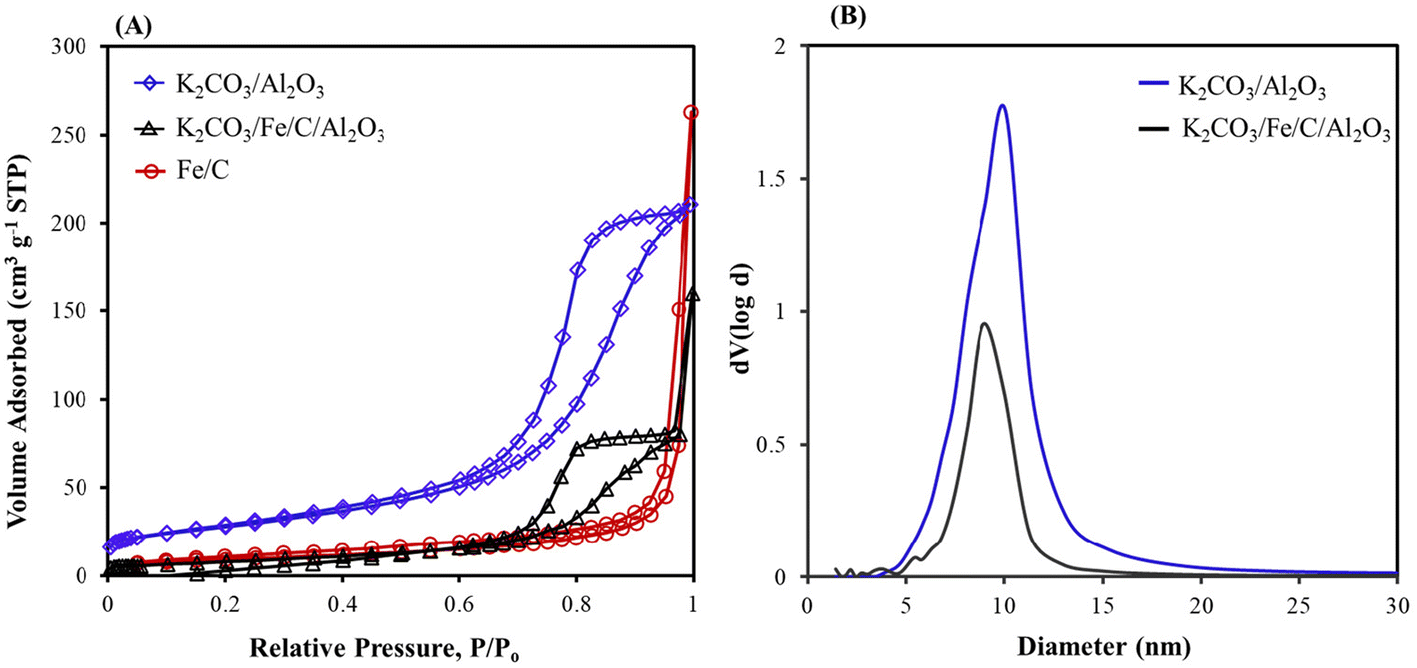 | ||
| Fig. 3 (A) Nitrogen adsorption–desorption isotherms of K2CO3/Al2O3, K2CO3/Fe/C/Al2O3, and Fe/C and (B) Barrett–Joyner–Halenda (BJH) curves for K2CO3/Al2O3, and K2CO3/Fe/C/Al2O3. | ||
With the improvement in capture performance, the CO2 captured in K2CO3/Fe/C/Al2O3 was converted in situ (Fig. 2B shows the conversion profile of captured CO2). A comparison of the conversion activities of K2CO3/Fe/C and K2CO3/Fe/C/Al2O3 is shown in Table 1. Interestingly, the C2–C4 olefin selectivity significantly improved to 7.3% in the case of K2CO3/Fe/C/Al2O3. The improved C–C coupled products formation could be because of the relatively high CO2 loading. In addition, a small amount of C5+ products (∼1%) was also detected. Increasing the hydrogenation temperature to 360 °C increased the conversion and selectivity further to methane. Increased methane formation at higher temperature could be due to a decrease in the chain growth probability of the Anderson–Schultz–Flory product distribution that governs the FTS reaction.61 Alternatively, it could be due to less carbon (i.e., captured CO2) content on the material, which could prevent C–C formation.
| Heating rate (°C min−1) | CO2 captured (μmol g−1) | CO2 conv. (%) | Selectivity to hydrocarbons | |||
|---|---|---|---|---|---|---|
| CH4 sel (%) | C2–C4 paraffins sel (%) | C2–C4 olefins sel (%) | ||||
| Amount of material: 2 g; pretreatment conditions: H2 = 60 mL min−1, 400 °C, 5 h; CO2 capture conditions: CO2 = 400 ppm in N2 (flow rate = 1200 mL min−1), H2O vapor = 0.5 mol%, 25 °C, 4 h; hydrogenation: H2 = 60 mL min−1, 1.0 MPa, 320 °C for 2.5 h (5 °C min−1). The selectivity of CO is <5% during the hydrogenation.a CO2 capture conditions: CO2 = 430 ppm CO2 (in 78% nitrogen, 21% oxygen and <1% other gases).b Pretreatment conditions: H2/CO (2:1) = 60 mL min−1, 400 °C, 3 h, followed by H2 = 60 mL min−1, 400 °C, 5 h. |
||||||
| Fe–Co/KA | 5 | 1970 | 21.3 | 88.3 | 5.1 | 6.7 |
| 20 | 1970 | 12.0 | 86.8 | 8.3 | 4.9 | |
| Fe/KA | 5 | 1645 | 22.4 | 79.7 | 8.9 | 11.4 |
| Fe/KAa | 5 | 1525 | 18.1 | 81.9 | 6.6 | 11.5 |
| Fe/KA (H2/CO pretreated)b | 5 | 1659 | 24.5 | 72.1 | 11.2 | 16.7 |
The Fe/KA captured ∼1600 μmol g−1 of CO2 at our standard capture conditions as shown in Fig. 4A. The hydrogenation results are shown in Fig. S5A† and Table 2. At 320 °C, C2–C4 olefins and paraffins started forming accompanied with the formation of CH4. The highest olefin selectivity of ∼11.4% was obtained with a CO2 conversion of 22.4% as shown in Fig. 4B. Next, to understand the effect of oxygen on the capture and conversion, CO2 capture was performed with real air (430 ppm CO2 containing 21% oxygen) using Fe/KA, which captured ∼1500 μmol g−1 of CO2. After the CO2 capture, the material was purged with N2 for 10 min to remove air. The subsequent hydrogenation produced C2–C4 olefins with a selectivity of 11.5% and a CO2 conversion of 18.1%, suggesting that the presence of oxygen during capture did not significantly affect the conversion and selectivity.
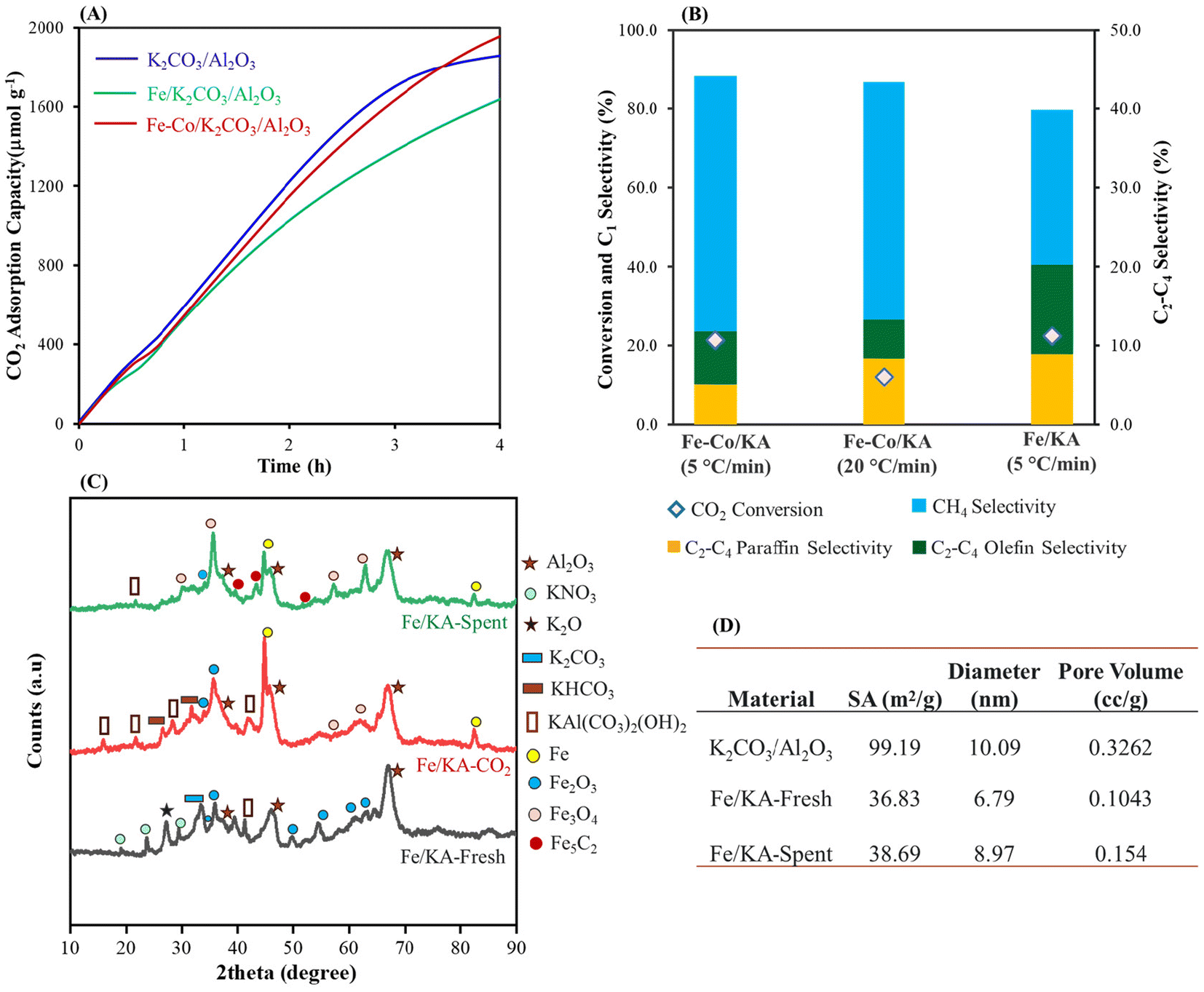 | ||
| Fig. 4 (A) Comparison of CO2 adsorption capacity of K2CO3/Al2O3, Fe–Co/KA and Fe/KA, (B) comparison of hydrogenation of Fe–Co/KA with different heating rates and Fe/KA (C) X-ray diffraction patterns of fresh, CO2 captured and spent Fe/KA, and (D) physicochemical properties of the K2CO3/Al2O3 and Fe/KA materials. Amount of material: 2 g; pretreatment conditions: H2 = 60 mL min−1, 400 °C, 5 h; CO2 capture conditions: CO2 = 400 ppm in N2 (flow rate = 1200 mL min−1), H2O vapor = 0.5 mol%, 25 °C, 4 h; hydrogenation: H2 = 60 mL min−1, 1.0 MPa, 320 °C for 2.5 h (5 °C min−1). The selectivity of CO is <5% during the hydrogenation. | ||
The BET isotherm shows that the mesoporosity of the K2CO3/Al2O3 is still maintained after impregnation of Fe particles (Fig. S5B†). Upon impregnating Fe, the surface area decreased from 99.19 (for K2CO3/Al2O3) to 36.83 m2 g−1 and the diameter of the mesopores decreased to 6.79 nm, confirming the formation of Fe particles inside the mesopores (Fig. 4D). The average pore size of the spent Fe/KA material (after hydrogenation) increased compared to the fresh material along with slight increase in the pore volume and surface area. This shows that after the hydrogenation, more dispersed particles were formed. This could be due to the formation of Fe5C2 and Fe3O4 particles during the high-temperature hydrogenation.
Fig. 4C shows wide-angle XRD of the fresh Fe/KA, CO2 captured Fe/KA and spent (after hydrogenation) Fe/KA. In the fresh sample, diffraction peaks corresponding to KNO3, dawsonite, and Fe2O3 particles were evident. XRD of the CO2-captured Fe/KA material shows peaks for KHCO3 and dawsonite along with some Fe2O3 and Fe particles. The Fe particles could form from Fe2O3 due to hydrogenation with H2 at high temperature.62 The spent (after hydrogenation) Fe/KA shows peaks for Fe5C2 along with Fe3O4, which were formed during hydrogenation of the captured CO2. The formation of these dispersed particles resulted in an increase of pore volume and of the average pore size of the material. The formation of the Fe5C2 phase shows the carburization of Fe3O4 particles.
The spent Fe/KA after the first cycle of capture and hydrogenation was reused to study the robustness of these materials (Fig. 5). The capture capacity was reduced in the second cycle to 1276 μmol g−1 (5.6 wt CO2%) compared to 1645 μmol g−1 (7.4 wt CO2%) in the first cycle. However, the capture performance was steady in the subsequent third (5.4 wt%), fourth (5.6 wt%), and fifth (∼5.03 wt%) cycles. The drop in the capture capacity could be because of the presence of K2O in the fresh Fe/KA, which consumed CO2 from air to form K2CO3. A similar drop in the capture capacity was observed between the first (6.5 wt% CO2) and second cycles (5.3 wt% CO2) for K2CO3/Al2O3 (Table S3†). However, in this case (K2CO3/Al2O3), the drop in performance could be because the low-temperature pretreatment conditions (at 200 °C for 1 h) prevented the conversion of dawsonite back to K2CO3. Prior to hydrogenation during the fifth cycle, the CO2 captured material was purged with N2 flow for 1 h to quantify physiosorbed CO2 content. Only trace amounts of CO2 were released during the N2 purge, and subsequent hydrogenation showed consistent conversion and selectivity to products, demonstrating that the material is stable for at least five cycles.
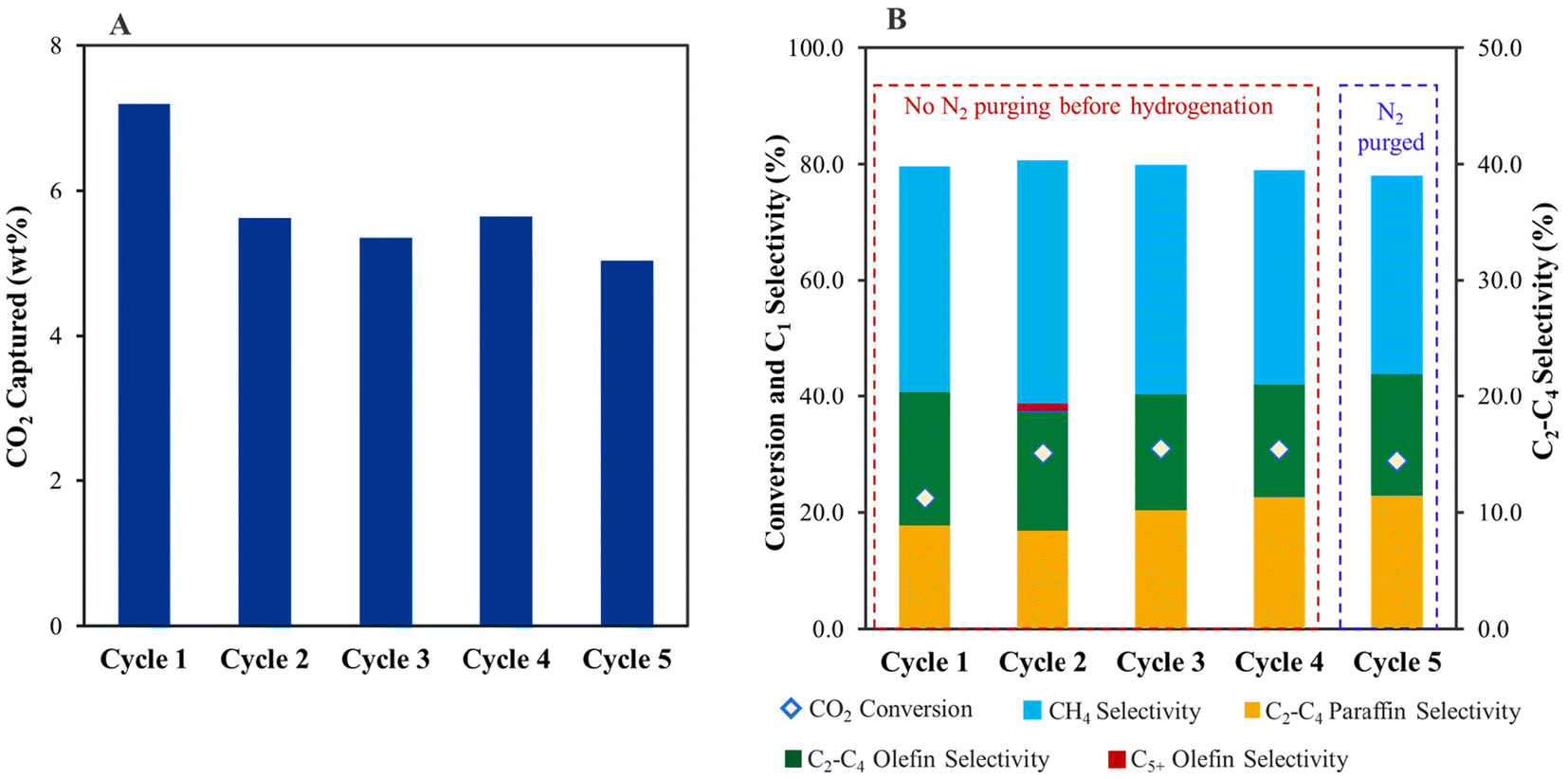 | ||
| Fig. 5 (A) CO2 capture and (B) hydrogenation of captured CO2 over five cycles using Fe/KA. Fe/KA: 2 g, pretreatment conditions: H2 = 60 mL min−1, 400 °C, 5 h; CO2 capture conditions: CO2 = 400 ppm in N2 (flow rate = 1200 mL min−1), H2O vapor = 0.5 mol%, 25 °C, 4 h; hydrogenation: H2 = 60 mL min−1, 1.0 MPa, 320 °C for 2.5 h (5 °C min−1) followed by heating to 400 °C (5 °C min−1) for 2.5 h to mimic the pretreatment conditions. For cycle 5, the spent catalyst was purged with N2 (30 mL min−1) for 1 h before hydrogenation. The selectivity of CO is <5% during the hydrogenation. | ||
To understand the effect of the CO2:H2 ratio and reaction temperature on the product distribution and conversion, the gas-phase hydrogenation studies were performed with Fe/KA using 1:3 and 1:10 ratios of CO2:H2. The conversion results for the Fe/KA at 320 °C and 360 °C are shown in Tables 3 and 4, along with Fig. 6. At 320 °C, in the case of the 1:10 ratio of CO2/H2, the selectivity to C2–C4 paraffins was higher compared to DAC and 1:3 ratio of CO2/H2 studies. The O/P (olefin/paraffin) ratio selectivity to C2–C4 olefins was not significantly altered by the CO2/H2 ratio at 320 °C. In addition to C2–4 products, C5+ products were detected by gas chromatography in the case of 1:3 ratio of CO2/H2. The reaction temperature played a significant role in O/P selectivity and CO2 conversion. The CO2 conversion was 66% and 15% for 1:10 and 1:3 ratios of CO2/H2, respectively, at 360 °C (Table 4). High olefin selectivity and O/P (olefin/paraffin) ratios were achieved for 1:3 ratio of CO2/H2 at 360 °C. In addition, the CO selectivity depends on the reaction temperature and the CO2:H2 ratio. As shown in Tables 3 and 4, a higher temperature (i.e., 360 °C) and lower CO2 concentrations significantly reduced the CO selectivity, suggesting that the reaction is proceeding via the CO intermediate.
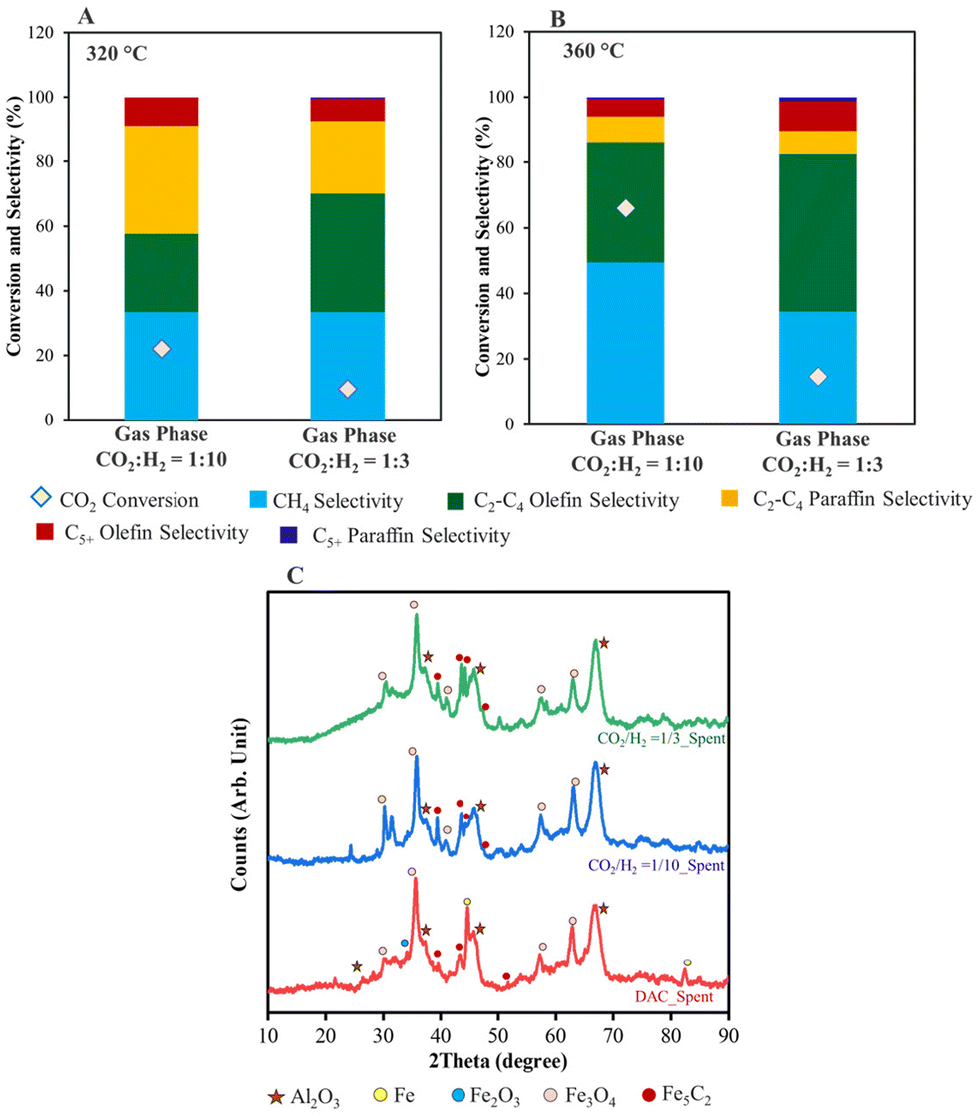 | ||
| Fig. 6 Comparison of gas-phase hydrogenation at (A) 320 °C and (B) 360 °C using Fe/KA. (C) Comparison of XRD of Fe/KA for DAC and gas-phase reactions carried out at 1:10 and 1:3 ratios of CO2:H2. Reaction Conditions: Fe/KA: 2 g, total flow rate = 60 mL min−1, CO2:H2 = 1:10 or 1:3 ratios; flow rate = 60 mL min−1, 1.0 MPa, 320 °C for 2.5 h (5 °C min−1), followed by heating to 360 °C for 2 h (5 °C min−1); GHSV = 1800 mL h−1 g−1. | ||
| CO2 conv. (%) | CO sel (%) | Selectivity of hydrocarbons (%) | O/P ratio | |||||
|---|---|---|---|---|---|---|---|---|
| CH4 sel (%) | C2–C4 paraffins sel (%) | C2–C4 olefins sel (%) | C5+ olefins (sel %) | C5+ (sel %) | ||||
| Fe/KA = 2 g, amount of material: 2 g; pretreatment conditions: H2 = 60 mL min−1, 400 °C, 5 h; CO2 capture conditions: CO2 = 400 ppm in N2 (flow rate = 1200 mL min−1), H2O vapor = 0.5 mol%, 25 °C, 4 h; hydrogenation: H2 = 60 mL min−1, 1.0 MPa, 320 °C for 2.5 h (5 °C min−1); CO2:H2 = 1:10 or 1:3 ratio; flow rate = 60 mL min−1, 1.0 MPa, 320 °C for 2.5 h (5 °C min−1); GHSV = 1800 mL h−1 g−1. |
||||||||
| DAC | 22.4 | <5 | 79.7 | 8.9 | 11.4 | 0.0 | 0.0 | 1.3 |
| CO2:H2 = 1:10 |
22 | 54.7 | 33.5 | 33.1 | 24.2 | 9.21 | 0.0 | 0.73 |
| CO2:H2 = 1:3 |
9.55 | 79.4 | 33.4 | 22.5 | 36.6 | 6.91 | 0.50 | 1.62 |
| CO2 conv. (%) | CO sel (%) | Selectivity of hydrocarbons (%) | O/P ratio | |||||
|---|---|---|---|---|---|---|---|---|
| CH4 sel (%) | C2–C4 paraffins sel (%) | C2–C4 olefins sel (%) | C5+ olefins (sel %) | C5+ (sel %) | ||||
| Fe/KA = 2 g, pretreatment conditions: H2 = 60 mL min−1, 400 °C, 5 h; CO2:H2 = 1:10 or 1:3 ratio; flow rate = 60 mL min−1, 1.0 MPa, 320 °C for 2.5 h (5 °C min−1) followed by heating to 360 °C (5 °C min−1) for 2 h; GHSV = 1800 mL h−1 g−1. |
||||||||
| CO2:H2 = 1:10 |
66.0 | 11.5 | 49.5 | 8.19 | 36.4 | 5.27 | 0.61 | 4.5 |
| CO2:H2 = 1:3 |
14.7 | 37.3 | 34.4 | 6.97 | 48.0 | 9.17 | 1.40 | 6.9 |
The XRD spectra of the spent DAC and gas-phase CO2 hydrogenation materials are shown in Fig. 6C. Fe3O4 was observed in all spent materials. The Fe5C2 diffraction patterns are more pronounced for the 1:3 CO2/H2 reaction compared to the 1:10 CO2/H2 reaction. This agrees with the decreased CH4 selectivity and increased O/P ratio of the 1:3 CO2/H2 reaction because both Fe3O4 (for the RWGS) and Fe5C2 are important for C–C formation (Table 4). Peaks for Fe were also observed in the spent DAC material, showing that not all of the Fe was carburized to Fe5C2. The formation of the carbide-phase reaction route is as follows: Fe2O3 → Fe3O4 → FeO → Fe, and then finally the Fe is carburized to Fe5C2.63
The Fourier transform infrared spectroscopy (FTIR) spectrum of the spent DAC and gas-phase CO2 hydrogenation materials were compared (Fig. S6†). The C–H vibrations were seen between 2960–2627 cm−1 corresponding to formate and other bound –CH species. The carbonyl vibration of formate was observed at the ∼1631 cm−1 region.64 The Fe–CO interactions were visible in the 1800–2100 cm−1 region, which corresponds to bound CO with different forms of Fe.65 In addition to formate and CO, there are additional bands visible for carbonates and bicarbonates in the IR spectrum.
To further enhance the formation of C–C coupled products, we pretreated the Fe/KA with an H2/CO gas mixture to improve iron carbide formation. The outlet gas stream during the pretreatment consisted of CO2, CH4 and C2–C4 hydrocarbons. After H2/CO pretreatment, Fe/KA was treated with H2 at 400 °C to remove CO2 and other hydrocarbons adsorbed on the Fe/KA prior to CO2 capture. The capture capacity of this H2/CO pretreated Fe/KA was 1659 μmol g−1 at our standard CO2 capture conditions, which is comparable to Fe/KA (Table 2). Subsequent hydrogenation of the captured CO2 resulted in an enhancement in C–C coupled products selectivity to 27.9% (16.7% selectivity to C2–C4 olefins and 11.2% selectivity to C2–C4 paraffins) with a slight improvement in the CO2 conversion to 24.5%. The selectivity of the hydrogenated products for the first hour at 320 °C is shown in Fig. S7.† It is evident that the selectivity to C–C coupled products was high, ∼50% (with >30% selectivity to C2–C4 olefins), initially, but decreased significantly as the concentration of the captured CO2 decreased.
Based on the selectivity of the products and the XRD and FTIR analyses of the spent samples, the conversion of captured CO2 to olefins occurs through the direct CO2 conversion pathway, where the CO2 is converted to CO via the RWGS in the presence of Fe3O4.66 Subsequently, the CO is converted to C–C products following the FTS mechanism in the presence of Fe5C2.66 A proposed pathway has been shown in Scheme 1. When CO2 (400 ppm) is captured in the presence of water vapor at room temperature, the K2CO3 of Fe/KA transforms into KHCO3 and KAlCO3(OH)2. This transformation leads to the formation of HCOOK and CO upon hydrogenation catalyzed by Fe3O4/KA. The Fe3O4/KA is derived from Fe2O3/KA in the presence of H2. Furthermore, the Fe3O4/KA facilitates the conversion of CO to *CH species, which undergo C–C coupling in the presence of Fe5C2 formed in situ during the reaction. The increased selectivity observed for the C–C coupled products (as depicted in Table 2 and Fig. S7†) following the pretreatment of Fe/KA with H2/CO gas mixture strongly suggests that the enhanced formation of Fe5C2 facilitates C–C coupling.
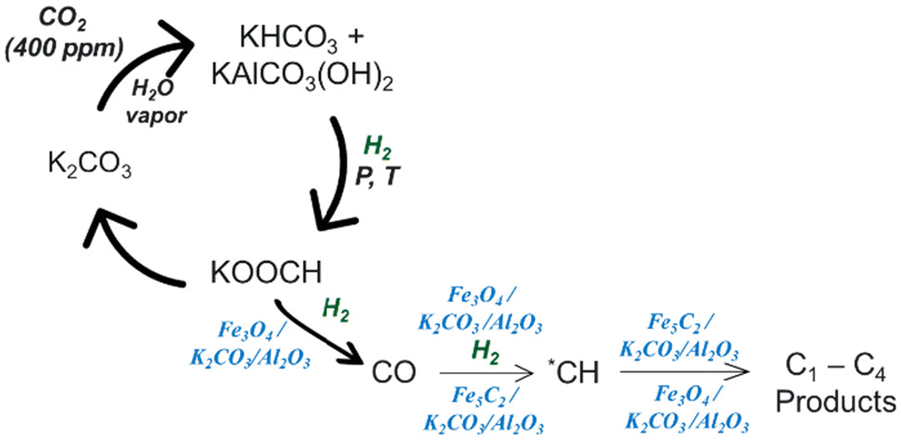 | ||
| Scheme 1 Proposed mechanism for the conversion of captured CO2 to C–C coupled products in the presence of Fe/KA. | ||
A preliminary techno-economic analysis (TEA) and life-cycle analysis (LCA) were conducted to evaluate the proposed iDAC-CAT technology for olefins production. In both TEA and LCA, it was assumed that renewable hydrogen, electricity, and fossil-based natural gas were used as main energy inputs. A process model was developed in Aspen Plus V14 to calculate the mass and energy balance and life-cycle inventory of the proposed technology based on the performance measures and assumptions listed in Table 5. The results were compared with NETL's case study for sorbent-based DAC67 and other CO2 to olefin technologies available in the literature.68
| Assumptions | Value | Assumptions | Value |
|---|---|---|---|
| Adsorption temperature (°C) | 25 | Conversion temperature (°C) | 320 |
| Adsorption pressure (bar) | 1 | Conversion pressure (bar) | 10 |
| Adsorption time (hr) | 2 | Conversion time with heating (hr) | 2 |
| CO2 capture (%) | 62 | CO2 conversion (%) | 80 |
| Sorbent loading (wt% CO2) | 5 | C2+ olefin selectivity (%) | 60 |
| Plant size (tonne CO2 per year) | 100000 |
CH4 selectivity (%) | 40 |
| H2 Price ($ per kg) | 5 | Excess hydrogen (ratio over stoic) | 4 |
Fig. 7 shows the process flow diagram of the technology, where air first enters the adsorption bed, and CO2 is adsorbed by the sorbent at ambient conditions. The bed is then heated to 320 °C and H2 is fed to the bed to produce CH4 and olefins from CO2. The product stream leaving the adsorption bed contains H2, CH4, and C2+ olefins. H2 is first separated in the pressure swing adsorption (PSA) unit. The remaining products are then sent to a de-methanization tower. This tower is of the design commonly used in the commercial ethylene plants, and operated at cryogenic conditions (−100 °C, 35 bar). The CH4 and C2+ olefin streams from the tower are depressurized and then used to pre-chill the inlet stream. In the TEA, the plant size was set the same as Case 0B in the NETL's case study for sorbent-based DAC.67 The capital cost of the iDAC-CAT unit was calculated by adjusting NETL Case 0B value67 based on flowrate and cycling time. The capital cost of the downstream product separation and purification section was calculated using Aspen Process Economic Analyzer V14. A simple annualized cost approach was used to calculate the minimum olefin selling price with 20-year depreciation and 10% per year return on investment. It was assumed that the renewable natural gas (RNG) produced as by-product can be sold at a price of $13 per MMBtu,69 roughly five times of the market price of fossil-based natural gas. For the Fe/KA material, CatCost™ tool70 was used to estimate its production cost as a pre-commercial material as well as the utility consumptions and emissions during the manufacturing step. For the LCA, a cradle-to-gate system boundary was used to evaluate the life-cycle greenhouse gases (GHG) emissions of olefin production using the iDAC-CAT technology, which was compared with conventional petrochemical process. The functional unit was set to per kg of olefins produced. Carbon Intensity data for each raw material and energy involved in the process were sourced from the GREET 202271 and Ecoinvent V3.8 databases.
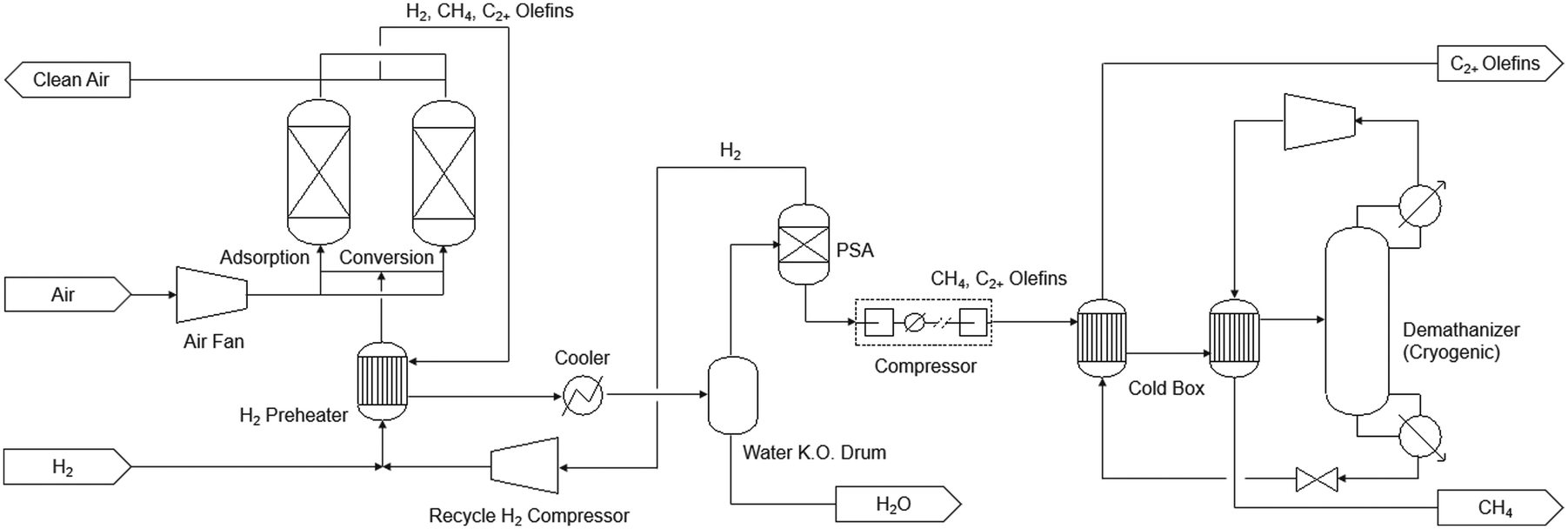 | ||
| Fig. 7 Process flow diagram of the proposed integrated DAC-CAT technology for olefins production. | ||
The preliminary TEA and LCA results were summarized in Fig. 8, while the mass and energy balance, life-cycle inventory and carbon intensity were provided in Table 6. Fig. 8(a) suggests the integrated iDAC-CAT technology can potentially produce renewable olefins at a cost 35% lower than that of a separated DAC and CO2 to olefins (S–DAC–C2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ) technology.67,68Fig. 8(b) indicates that the iDAC-CAT technology can significantly reduce the cost of DAC on a per tonne CO2 basis. The error bars in both Fig. 8(a) and (b) represent the uncertainties in TEA results from literature, as well as the market prices of RNG and olefins. Furthermore, Fig. 8(c) and Table 6 demonstrate that the CO2 adsorbed from the atmosphere and the GHG emissions avoided by producing RNG can completely offset the GHG emissions from upstream processes and the iDAC-CAT process when renewable H2 is used as a process input. A GHG emission reduction of 105% can be achieved compared to the petroleum baseline.
) technology.67,68Fig. 8(b) indicates that the iDAC-CAT technology can significantly reduce the cost of DAC on a per tonne CO2 basis. The error bars in both Fig. 8(a) and (b) represent the uncertainties in TEA results from literature, as well as the market prices of RNG and olefins. Furthermore, Fig. 8(c) and Table 6 demonstrate that the CO2 adsorbed from the atmosphere and the GHG emissions avoided by producing RNG can completely offset the GHG emissions from upstream processes and the iDAC-CAT process when renewable H2 is used as a process input. A GHG emission reduction of 105% can be achieved compared to the petroleum baseline.
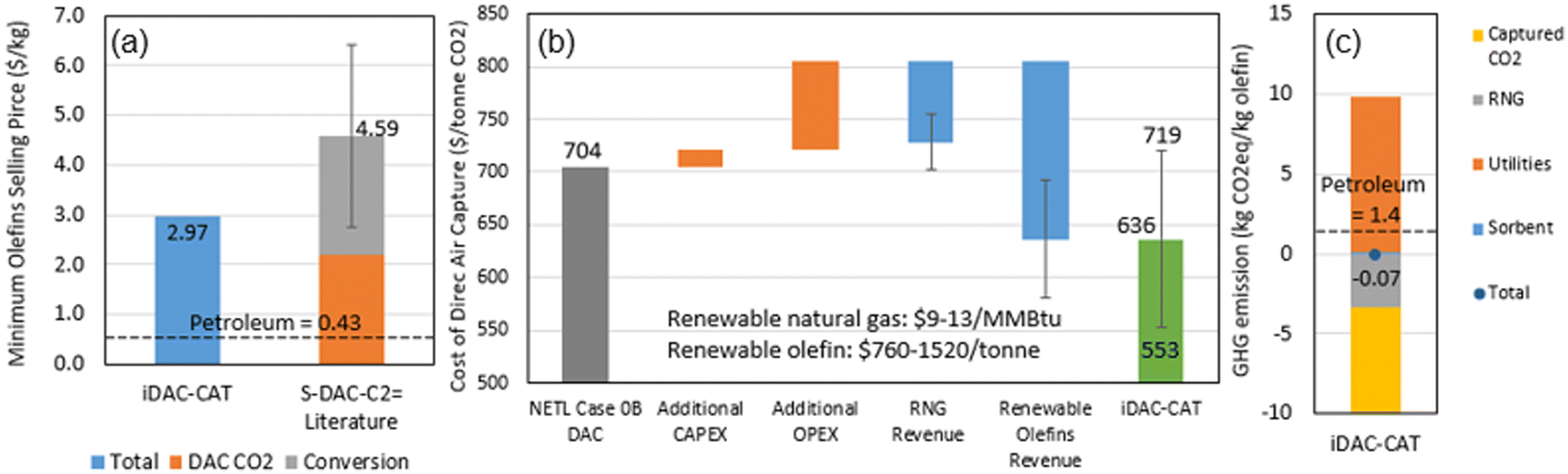 | ||
| Fig. 8 Economic and environmental performance: (a) minimum olefin selling price; (b) cost of direct air capture; (c) life-cycle GHG emissions. | ||
| Mass energy balance | Life cycle inventory | Carbon intensity | |
|---|---|---|---|
| Products | |||
| C2+ olefins | 2360 kg h−1 | ||
| RNG | 2015 kg h−1 | −0.045 MMBtu kg−1 C2+ olefins | 72.62 kg CO2 eq. MMBtu−1 NG |
| Feedstock | |||
| Renewable H2 | 2112 kg h−1 | 0.895 kg kg−1 C2+ olefins | 0 kg CO2 eq. kg−1 H2 |
| CO2 captured | 15553 kg h−1 |
6.590 kg kg−1 C2+ olefins | −1 kg CO2 eq. kg−1 CO2 |
| Sorbent (Fe/KA) | 116 kg h−1 | 0.049 kg kg−1 C2+ olefins | 2.20 kg CO2 eq. kg−1 sorbent |
| Utilities | |||
| Electricity | 32984 kW |
13.98 kW h kg−1 C2+ olefins | 0 kg CO2 eq. kW−1 h−1 electricity |
| NG | 315.3 MMBtu h−1 | 0.133 MMBtu kg−1 C2+ olefins | 72.62 kg CO2 eq. MMBtu−1 NG |
Conclusions
A series of materials have been evaluated for direct air capture and conversion to C–C coupled products for the first time. A novel multifunctional and multicomponent material for iDAC-CAT has been developed, employing a combination of non-noble metal and solid inorganic sorbent, Fe/K2CO3/Al2O3. Upon the impregnation of catalytic Fe particles to the sorbent (K2CO3/Al2O3), despite the decrease in surface area, pore size, and pore volume, high and consistent CO2 capture was realized at room temperature in presence of water vapor. This shows that the addition of Fe particles did not significantly change the CO2 capture property of K2CO3/Al2O3. On recycling, the material showed a consistent capture capacity of ∼5 wt% for up to five cycles, followed by consistent CO2 conversion into C–C products. In contrast, the physical mixture of Fe2O3 and K2CO3/Al2O3 desorbed the CO2 and showed no formation of C1–C4 products on hydrogenation. Based on this comparison and activity data of various combination of materials, along with XRD and BET results, it is evident that the proximity between the Fe and K on the Al2O3 is important for CO2 activation and subsequent conversion to C–C products.We have successfully developed an approach for integrated direct air capture and conversion to C–C coupled products using Fe/K2CO3/Al2O3. The utilization of this material for CO2 capture from the air and subsequent conversion to C2+ products represents an environmentally friendly approach. Despite the current breakthrough and success of the bench-scale experiment, scaling up poses multiple risks. Factors such as kinetics, material mechanical strength and stability, environmental conditions throughout the year, processing temperature range, and deployment site must be carefully considered during the scaling process. Our preliminary TEA analysis indicates that iDAC-CAT technology has the potential to substantially decrease the cost of DAC. The preliminary LCA suggests a 105% reduction in GHG emissions compared to the petroleum baseline, and indicates a negative cradle-to-gate GHG emission for renewable olefin production via iDAC-CAT when renewable H2 is used as the process input. Future efforts will focus on developing materials with enhanced reactivity for C–C coupling and stronger CO2 binding affinity to prevent desorption during conversion at the high temperatures required for C–C coupling reactions. Further exploration of Fe/K2CO3/Al2O3 under varying conditions, and experimentation with different material combinations, is needed to improve the conversion efficiency. This exploration should be accompanied by a full TEA and LCA to assess its feasibility for real-world applications.
Author contributions
Shazia Sharmin Satter: investigation, data curation, and writing – original draft; Johnny Saavedra Lopez: investigation, methodology, and writing – editing, Michael L. Hubbard: investigation and data curation, Yuan Jiang: TEA, LCA and writing – editing, Robert A. Dagle: conceptualization, and writing – editing; Jotheeswari Kothandaraman: supervision, funding acquisition, conceptualization, investigation, data curation, and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Laboratory Directed Research and Development Program (LDRD) at Pacific Northwest National Laboratory (PNNL). The authors would like to thank Dr Jaelynne A. King and Dr Austin D. Winkelman for performing the FTIR and XRD measurements, respectively. The Pacific Northwest National Laboratory is proudly operated by Battelle for the US Department of Energy.References
- J. Hilaire, J. C. Minx, M. W. Callaghan, J. Edmonds, G. Luderer, G. F. Nemet, J. Rogelj and M. del Mar Zamora, Clim. Change, 2019, 157, 189–219 CrossRef.
- W. J. Shaw, M. K. Kidder, S. R. Bare, M. Delferro, J. R. Morris, F. M. Toma, S. D. Senanayake, T. Autrey, E. J. Biddinger, S. Boettcher, M. E. Bowden, P. F. Britt, R. C. Brown, R. M. Bullock, J. G. Chen, C. Daniel, P. K. Dorhout, R. A. Efroymson, K. J. Gaffney, L. Gagliardi, A. S. Harper, D. J. Heldebrant, O. R. Luca, M. Lyubovsky, J. L. Male, D. J. Miller, T. Prozorov, R. Rallo, R. Rana, R. M. Rioux, A. D. Sadow, J. A. Schaidle, L. A. Schulte, W. A. Tarpeh, D. G. Vlachos, B. D. Vogt, R. S. Weber, J. Y. Yang, E. Arenholz, B. A. Helms, W. Huang, J. L. Jordahl, C. Karakaya, K. C. Kian, J. Kothandaraman, J. Lercher, P. Liu, D. Malhotra, K. T. Mueller, C. P. O’Brien, R. M. Palomino, L. Qi, J. A. Rodriguez, R. Rousseau, J. C. Russell, M. L. Sarazen, D. S. Sholl, E. A. Smith, M. B. Stevens, Y. Surendranath, C. J. Tassone, B. Tran, W. Tumas and K. S. Walton, Nat. Rev. Chem., 2024, 8, 376–400 CrossRef PubMed.
- P. M. Bhatt, Y. Belmabkhout, A. Cadiau, K. Adil, O. Shekhah, A. Shkurenko, L. J. Barbour and M. Eddaoudi, J. Am. Chem. Soc., 2016, 138, 9301–9307 CrossRef CAS PubMed.
- D. G. Boer, J. Langerak and P. P. Pescarmona, ACS Appl. Energy Mater., 2023, 6, 2634–2656 CrossRef CAS.
- P. Murge, S. Dinda and S. Roy, Langmuir, 2019, 35, 14751–14760 CrossRef CAS PubMed.
- R. V. Siriwardane, M.-S. Shen, E. P. Fisher and J. A. Poston, Energy Fuels, 2001, 15, 279–284 CrossRef CAS.
- S. A. Didas, S. Choi, W. Chaikittisilp and C. W. Jones, Acc. Chem. Res., 2015, 48, 2680–2687 CrossRef CAS PubMed.
- W. Chaikittisilp, H.-J. Kim and C. W. Jones, Energy Fuels, 2011, 25, 5528–5537 CrossRef CAS.
- N. G. McQueen, K. V. McCormick, C. Blumanthal, K. Pisciotta and M. Wilcox, J. Prog. Energy, 2021, 3, 032001 CrossRef.
- M. A. Sabri, S. Al Jitan, D. Bahamon, L. F. Vega and G. Palmisano, Sci. Total Environ., 2021, 790, 148081 CrossRef CAS PubMed.
- D. J. Heldebrant, J. Kothandaraman, N. M. Dowell and L. Brickett, Chem. Sci., 2022, 13, 6445–6456 RSC.
- R. E. Siegel, S. Pattanayak and L. A. Berben, ACS Catal., 2022, 13, 766–784 CrossRef.
- S. Kar, A. Goeppert and G. K. S. Prakash, Acc. Chem. Res., 2019, 52, 2892–2903 CrossRef CAS PubMed.
- J. Kothandaraman, A. Goeppert, M. Czaun, G. A. Olah and G. K. Prakash, J. Am. Chem. Soc., 2016, 138, 778–781 CrossRef CAS PubMed.
- N. M. Rezayee, C. A. Huff and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 1028–1031 CrossRef CAS PubMed.
- J. B. Jakobsen, M. H. Ronne, K. Daasbjerg and T. Skrydstrup, Angew. Chem., Int. Ed., 2021, 60, 9174–9179 CrossRef CAS PubMed.
- S. Kar, J. Kothandaraman, A. Goeppert and G. K. S. Prakash, J. CO2 Util., 2018, 23, 212–218 CrossRef CAS.
- X. Shi, H. Xiao, H. Azarabadi, J. Song, X. Wu, X. Chen and K. S. Lackner, Angew. Chem., Int. Ed., 2020, 59, 6984–7006 CrossRef CAS PubMed.
- R. Custelcean, Annu. Rev. Chem. Biomol. Eng., 2022, 13, 217–234 CrossRef CAS PubMed.
- J. Kothandaraman, J. S. Lopez, Y. Jiang, E. D. Walter, S. D. Burton, R. A. Dagle and D. J. Heldebrant, Adv. Energy Mater., 2022, 12(46), 2202369 CrossRef CAS.
- J. Kothandaraman, J. S. Lopez, Y. Jiang, E. D. Walter, S. D. Burton, R. A. Dagle and D. J. Heldebrant, ChemSusChem, 2021, 14, 4812–4819 CrossRef CAS PubMed.
- J. Kothandaraman, A. Goeppert, M. Czaun, G. A. Olah and G. K. S. Prakash, Green Chem., 2016, 18, 5831–5838 RSC.
- D. Wei, H. Junge and M. Beller, Chem. Sci., 2021, 12, 6020–6024 RSC.
- S. Jo, H. D. Son, T.-Y. Kim, J. H. Woo, D. Y. Ryu, J. C. Kim, S. C. Lee and K. L. Gilliard-AbdulAziz, Chem. Eng. J., 2023, 469, 143772 CrossRef CAS.
- T. Sasayama, F. Kosaka, Y. Liu, T. Yamaguchi, S.-Y. Chen, T. Mochizuki, A. Urakawa and K. Kuramoto, J. CO2 Util., 2022, 60, 102049 CrossRef CAS.
- M. S. Duyar, M. A. A. Treviño and R. J. Farrauto, Appl. Catal., B, 2015, 168–169, 370–376 CrossRef CAS.
- L. Hu and A. Urakawa, J. CO2 Util., 2018, 25, 323–329 CrossRef CAS.
- M. Abdallah, Y. C. Y. Lin and R. Farrauto, Appl. Catal., B, 2023, 339, 123105 CrossRef CAS.
- C. Jeong-Potter, M. Abdallah, C. Sanderson, M. Goldman, R. Gupta and R. Farrauto, Appl. Catal., B, 2022, 307, 120990 CrossRef CAS.
- C. Jeong-Potter and R. Farrauto, Appl. Catal., B, 2021, 282, 119416 CrossRef CAS.
- C. Jeong-Potter, M. A. Arellano-Treviño, W. W. McNeary, A. J. Hill, D. A. Ruddy and A. T. To, EES Catal., 2024, 2, 253–261 RSC.
- J. Pazdera, E. Berger, J. A. Lercher and A. Jentys, Catal. Commun., 2021, 159, 106347 CrossRef CAS.
- J. Pazdera, D. Issayeva, J. Titus, R. Gläser, O. Deutschmann and A. Jentys, ChemCatChem, 2022, 14, e202200620 CrossRef CAS.
- L. C. Wirner, F. Kosaka, T. Sasayama, Y. Liu, A. Urakawa and K. Kuramoto, Chem. Eng. J., 2023, 470, 144227 CrossRef CAS.
- A. Bermejo-López, B. Pereda-Ayo, J. A. González-Marcos and J. R. González-Velasco, J. CO2 Util., 2019, 34, 576–587 CrossRef.
- F. Kosaka, Y. Liu, S.-Y. Chen, T. Mochizuki, H. Takagi, A. Urakawa and K. Kuramoto, ACS Sustainable Chem. Eng., 2021, 9, 3452–3463 CrossRef CAS.
- S. C. Lee, H. J. Chae, S. J. Lee, B. Y. Choi, C. K. Yi, J. B. Lee, C. K. Ryu and J. C. Kim, Environ. Sci. Technol., 2008, 42, 2736–2741 CrossRef CAS PubMed.
- M. S. Cho, S. C. Lee, H. J. Chae, Y. M. Kwon, J. B. Lee and J. C. Kim, Process Saf. Environ. Prot., 2018, 117, 296–306 CrossRef CAS.
- S. B. Jo, S. C. Lee, H. J. Chae, M. S. Cho, J. B. Lee, J.-I. Baek and J. C. Kim, Korean J. Chem. Eng., 2016, 33, 3207–3215 CrossRef CAS.
- J. Holladay, Z. Abdullah and J. Heyne, Technical Report: Sustainable Aviation Fuel: Review of Technical Pathways, United States, 2020, DOI:10.2172/1660415.
- E. S. Sanz-Perez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef CAS PubMed.
- J. Wang, L. Huang, R. Yang, Z. Zhang, J. G. Wu, Y. Gao, Q. Wang, D. O’Hare and Z. Zhong, Energy Environ. Sci., 2014, 7, 3478–3518 RSC.
- J. V. Veselovskaya, V. S. Derevschikov, T. Y. Kardash and A. G. Okunev, Renew. Bioresour., 2015, 3, 1 CrossRef.
- J. V. Veselovskaya, V. S. Derevschikov, T. Y. Kardash, O. A. Stonkus, T. A. Trubitsina and A. G. Okunev, Int. J. Greenhouse Gas Control, 2013, 17, 332–340 CrossRef CAS.
- A. G. Okunev, V. E. Sharonov, Y. I. Aristov and V. N. Parmon, React. Kinet. Catal. Lett., 2000, 71, 355–362 CrossRef CAS.
- V. E. Sharonov, E. A. Tyshchishchin, E. M. Moroz, A. G. Okunev and Y. I. Aristov, Russ. J. Appl. Chem., 2001, 74, 409–413 CrossRef CAS.
- J. V. Veselovskaya, V. S. Derevschikov, A. S. Shalygin and D. A. Yatsenko, Microporous Mesoporous Mater., 2021, 310, 110624 CrossRef CAS.
- M. Yan, Y. Zhang, Q. Huan, Y. Song, X. Zhou, H. Wibowo and C. Yu, Biomass Convers. Biorefin., 2022, 14, 3667–3677 CrossRef.
- N. Masoud, G. Bordanaba-Florit, T. v. Haasterecht and J. H. Bitter, Ind. Eng. Chem. Res., 2021, 60, 13749–13755 CrossRef CAS.
- C. Zhao, X. Chen and C. Zhao, Energy Fuels, 2009, 23, 4683–4687 CrossRef CAS.
- S. C. Lee, H. J. Chae, B. Y. Choi, S. Y. Jung, C. Y. Ryu, J. J. Park, J.-I. Baek, C. K. Ryu and J. C. Kim, Korean J. Chem. Eng., 2011, 28, 480–486 CrossRef CAS.
- J. V. Veselovskaya, A. I. Lysikov, O. V. Netskina, D. V. Kuleshov and A. G. Okunev, Ind. Eng. Chem. Res., 2020, 59, 7130–7139 CrossRef CAS.
- J. Kothandaraman, J. S. Lopez, Y. Jiang, E. D. Walter, S. D. Burton, R. A. Dagle and D. J. Heldebrant, Adv. Energy Mater., 2022, 12, 2202369 CrossRef CAS.
- D. Wang, Z. H. Xie, M. D. Porosoff and J. G. G. Chen, Chem, 2021, 7, 2277–2311 CAS.
- T. Numpilai, N. Chanlek, Y. Poo-Arporn, C. K. Cheng, N. Siri-Nguan, T. Sornchamni, M. Chareonpanich, P. Kongkachuichay, N. Yigit, G. Rupprechter, J. W. Limtrakul and T. Witoon, ChemCatChem, 2020, 12, 3306–3320 CrossRef CAS.
- A. Ramirez, S. Ould-Chikh, L. Gevers, A. D. Chowdhury, E. Abou-Hamad, A. Aguilar-Tapia, J. L. Hazemann, N. Wehbe, A. J. Al Abdulghani, S. M. Kozlov, L. Cavallo and J. Gascon, ChemCatChem, 2019, 11, 2879–2886 CrossRef CAS.
- Y. Han, C. Fang, X. Ji, J. Wei, Q. Ge and J. Sun, ACS Catal., 2020, 10, 12098–12108 CrossRef CAS.
- C. G. Visconti, M. Martinelli, L. Falbo, A. Infantes-Molina, L. Lietti, P. Forzatti, G. Iaquaniello, E. Palo, B. Picutti and F. Brignoli, Appl. Catal., B, 2017, 200, 530–542 CrossRef CAS.
- H. Yu, C. Wang, T. Lin, Y. An, Y. Wang, Q. Chang, F. Yu, Y. Wei, F. Sun, Z. Jiang, S. Li, Y. Sun and L. Zhong, Nat. Commun., 2022, 13, 5987 CrossRef PubMed.
- J. Ren, N. Ai, D. Ou and Y. Yu, Mol. Catal., 2023, 538, 112990 CrossRef CAS.
- H. M. T. Galvis and K. P. de Jong, ACS Catal., 2013, 3, 2130–2149 CrossRef.
- J. Zieliński, I. Zglinicka, L. Znak and Z. Kaszkur, Appl. Catal., A, 2010, 381, 191–196 CrossRef.
- V. V. Ordomsky, B. Legras, K. Cheng, S. Paul and A. Y. Khodakov, Catal. Sci. Technol., 2015, 5, 1433–1437 RSC.
- J. Lee and J. Otomo, Ind. Eng. Chem. Res., 2023, 62, 12096–12108 CrossRef CAS.
- V. I. Bogdan, A. E. Koklin, A. L. Kustov, Y. A. Pokusaeva, T. V. Bogdan and L. M. Kustov, Molecules, 2021, 26, 2883 CrossRef CAS PubMed.
- B. Pawelec, R. Guil-Lopez, N. Mota, J. L. G. Fierro and R. M. Navarro Yerga, Materials, 2021, 14, 6952 CrossRef CAS PubMed.
- J. Valentine and A. Zoelle, Direct Air Capture Case Studies: Sorbent System, DOE/NETL-2021/2865, National Energy Technology Laboratory, 2022, DOI:10.2172/1879535.
- A. Somoza-Tornos, O. J. Guerra, A. M. Crow, W. A. Smith and B. M. Hodge, iScience, 2021, 24, 102813 CrossRef CAS PubMed.
- R. Gupta, Reactive CO2 Capture DAC-DFM, ARPA-E Reactive Carbon Capture Workshop, 2022 Search PubMed.
- https://catcost.chemcatbio.org/home .
- M. Wang, A. Elgowainy, U. Lee, K. H. Baek, A. Bafana, P. T. Benavides, A. Burnham, H. Cai, V. Cappello, P. Chen, Y. Gan, U. R. Gracida-Alvarez, T. R. Hawkins, R. K. Iyer, J. C. Kelly, T. Kim, S. Kumar, H. Kwon, K. Lee, X. Liu, Z. Lu, F. H. Masum, C. Ng, L. Ou, K. Reddi, N. Siddique, P. Sun, P. Vyawahare, H. Xu and G. G. Zaimes, Summary of Expansions and Updates in GREET® 2022, ANL/ESIA-22/1, Argonne National Laboratory, 2022 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc01244e |
| This journal is © The Royal Society of Chemistry 2024 |