
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pathway to a molecular calcium methyl†
Kyle G. Pearce *,
Samuel E. Neale,
Claire L. McMullin,
Mary F. Mahon and
Michael S. Hill
*,
Samuel E. Neale,
Claire L. McMullin,
Mary F. Mahon and
Michael S. Hill
Department of Chemistry, University of Bath, Claverton Down, Bath, UK. E-mail: kgp29@bath.ac.uk
First published on 2nd July 2024
Abstract
The dimeric β-diketiminato calcium hydride, [(BDI)CaH]2 (BDI = HC{(Me)CN-2,6-iPr2C6H3}2), reacts with ZnMe2 to afford the bimetallic calcium zincate complex, [(BDI)Ca(μ-CH3)2Zn(μ-H)]2, which subsequently undergoes an intramolecular reaction to effect the formation of [(BDI)CaMe]2, a notable omission from the homologous series of β-diketiminato alkylcalcium derivatives.
Since the discovery of Grignard reagents in 1900,1 organomagnesium derivatives have prevailed as some of the most widely employed compounds in chemical synthesis. In contrast, exploration of magnesium's heavier congener, calcium, lay largely dormant throughout the 20th Century, to the extent that it has even been deemed as the “sleeping beauty” of organometallic chemistry.2 Despite reports of σ–C–Ca-bonded calcium alkyl and aryl complexes from as early as 1905,3,4 the first crystallographically verified calcium σ-alkyl, [Ca{CH(SiMe3)2}2(Diox)2] (Diox = 1,4-dioxane), was only reported by Cloke and Lappert in 1991.5 Like this species, many subsequently characterised diorganocalcium compounds were dependent on the use of silylated and kinetically stabilising organic anions.6 More recently, however, a defined and characteristic chemistry has finally started to emerge for derivatives of wholly hydrocarbon ligands.2 Especially notable in this regard are routes to a variety of di- and monoaryl calcium reagents and dimethylcalcium devised, respectively, by the groups of Westerhausen and Anwander.7,8
While sufficiently remarkable to merit a mononymic account of its synthesis,8 the subsequent reactivity of [CaMe2]∞ in the absence of secondary donors is relatively limited as a consequence of its polymeric nature and consequent poor solubility in non-coordinating media.8 Further advancements have, thus, focused on heteroleptic derivatives,9 with the β-diketiminate ligand scaffold proving a particularly adept, hydrocarbon-soluble spectator anion.10 While σ-n-alkyl species had previously proved inaccessible via the analogous THF-adducted hydride, [(BDI)Ca(THF)H]2 (BDI = HC{(Me)CN-2,6-iPr2C6H3}2),11 its base-free variant, [(BDI)CaH]2 (1),12 was found to be reactive toward terminal alkenes to provide a wide range of dimeric σ-n-alkyl derivatives, [(BDI)Ca(R)]2 (Scheme 1).13 While a further impact of reduction in coordinative saturation at the calcium centre was manifest in the ability of such σ-alkyls to effect the nucleophilic alkylation of benzene (Scheme 1),12 a similar alkene insertion-based approach is clearly inapplicable to the synthesis of the simplest methylcalcium homologue.
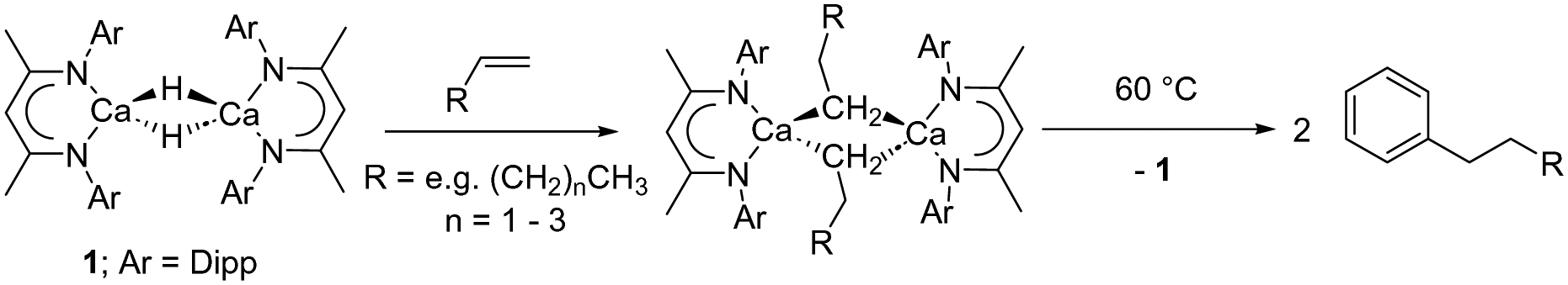 | ||
| Scheme 1 Preparation of calcium n-alkyl complexes from compound 1 and the nucleophilic alkylation of benzene. | ||
We have recently developed a transmetallation-based approach to prepare coordinatively unsaturated σ-aryl derivatives.14,15 Sequential reactions with arylmercuric reagents (Ar2Hg; Scheme 2) enabled the preparation of [(BDI)Ca(μ-H)(μ-Ar)Ca(BDI)] and [(BDI)Ca(μ-Ar)(μ-Ar′)Ca(BDI)] (Ar = C6H5, ortho-Me-C6H5, meta-Me-C6H5, para-Me-C6H5, 3,5-(tBu)2C6H3, Ar′ = C6H5, ortho-Me-C6H5, meta-Me-C6H5, para-Me-C6H5). These β-diketiminato arylcalcium derivatives detail a structural dependence between η1- and η6-aryl coordination and facilitate uncatalyzed access to biaryl molecules by direct SNAr displacement of halide from aryl bromides. While a similar mercury-based approach could be applicable to the preparation of a molecular calcium methyl complex, [(BDI)CaMe]2, and our previous use of arylmercurials was a necessary synthetic expedient, the severe toxicity of dimethylmercury renders this synthetic pathway unattractive.16 In this contribution, therefore, we describe the use of the less-toxic group 12 metal, zinc, to address this lacuna in the homologous chemistry of β-diketiminato alkylcalcium derivatives.
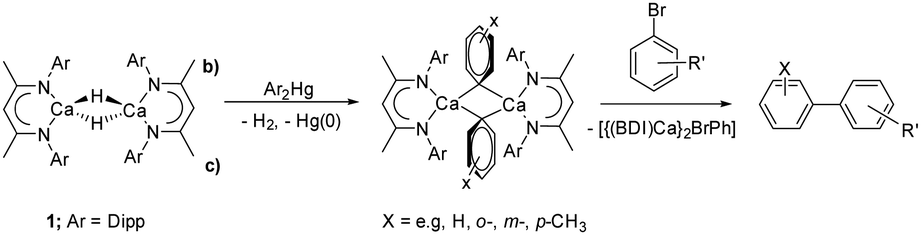 | ||
| Scheme 2 Use of compound 1 in the synthesis of arylcalcium compounds and the reaction of the phenyl derivative with aryl bromides to prepare biaryl molecules. | ||
Immediate analysis by 1H NMR spectroscopy of a reaction of compound 1 with one equivalent of ZnMe2 (1.0 M in heptane) in d8-toluene, identified three upfield shifted methyl resonances at δH −0.71, −0.81 and −1.39 ppm (Fig. S1, ESI†). The secondmost downfield of these signals was assigned to [(BDI)ZnMe] (2),17 which, accompanied by the appearance of a colourless precipitate, continued to emerge as the sole soluble component of the reaction over the subsequent 16 hours (Fig. S2, ESI†). Repetition of this reaction with two equivalents of ZnMe2 afforded 2 over a shorter time period (4 hours), albeit the methyl-containing species at δH −1.39 ppm was again observed to form but be subsequently consumed. In an attempt to trap the other methyl organometallic intermediates formed during this reaction, 1 was again reacted with two equivalents of ZnMe2 but immediately placed in the freezer (−35 °C). This procedure afforded a crop of colourless crystals of 3. Analysis of 3 by 1H NMR spectroscopy evidenced a single β-diketiminate environment characterized by a BDI γ-methine singlet at δH 4.77 ppm and an associated upfield signal at δH −0.66 ppm, which resonated with a respective 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 ratio of intensities by relative integration. A further (1H) singlet at 3.15 ppm was tentatively identified as a hydride signal, the incorporation of which alongside the apparent retention of both methyl groups of the zinc reagent was interpreted to imply a bimetallic structure. This inference was subsequently confirmed by single crystal X-ray diffraction analysis, which identified compound 3 as [(BDI)Ca(μ-CH3)2Zn(μ-H)]2 (Fig. 1).
:6 ratio of intensities by relative integration. A further (1H) singlet at 3.15 ppm was tentatively identified as a hydride signal, the incorporation of which alongside the apparent retention of both methyl groups of the zinc reagent was interpreted to imply a bimetallic structure. This inference was subsequently confirmed by single crystal X-ray diffraction analysis, which identified compound 3 as [(BDI)Ca(μ-CH3)2Zn(μ-H)]2 (Fig. 1).
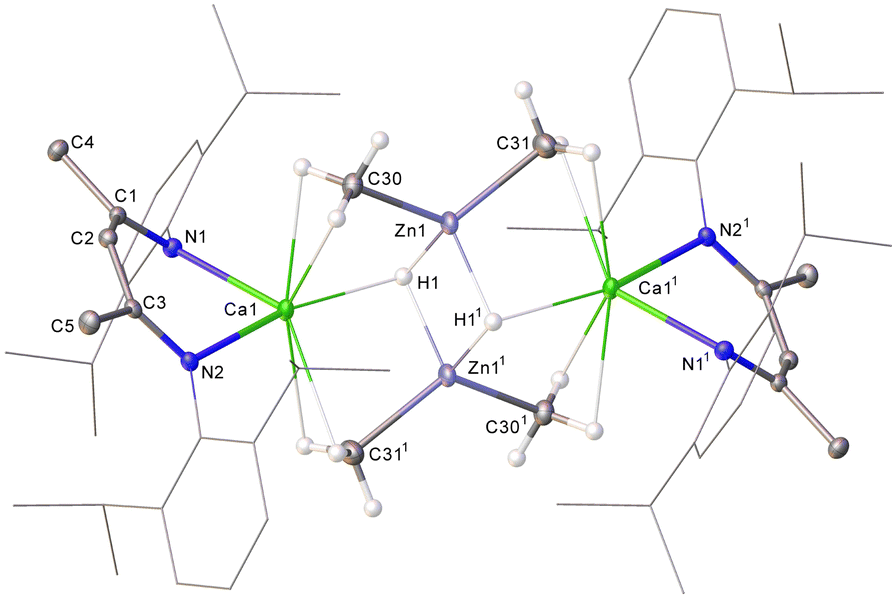 | ||
| Fig. 1 Molecular structure of the Ca1-containing component of 3, displacement ellipsoids at 30%. For clarity, hydrogen atoms, apart from the bridging hydrides and those attached to C30 and C301 are omitted, as are the minor components of disordered atoms and solvent. Dipp groups are displayed as wireframe, also for visual ease. Selected bond lengths (Å): Ca1–N1 2.3237(17), Ca1–N2 2.3253(16), Ca1–Zn1 3.0196(5), Ca1–Zn11 3.0188(5), Ca1–C30 2.608(2), Ca1–C311 2.629(2), Ca1–H1 2.37(3), Ca–H11 3.40(2), Zn1–C30 2.038(2), Zn1–C31 2.047(3), Zn1–H1 1.79(3), Zn1–H11 1.78(3), Zn1–Zn11 2.7222(5). Symmetry operations to generate primed atoms: 11 − x, 1 − y, 1 − z. | ||
The asymmetric unit of 3 contains two independent, but effectively identical, dimer halves and the following representative discussion refers only to the Ca1-containing molecule. Complex 3 provides Ca1 with two N–Ca contacts [2.3237(17) and 2.3253(16) Å], such that, in a manner reminiscent of the puckered geometry observed for [(BDI)Ca(o-C2B10H11)] and [(BDI)CaN(SiMe3)2],18–20 the calcium centre is situated ca. 1.088 Å above the least squares plane defined by the BDI-backbone. Ca1 is connected to Zn1 through a three-centre methyl-bridged bond, exhibiting Ca1–C30 and Zn1–C30 bond lengths of 2.608(2) and 2.038(2) Å, respectively. These distances are in line with the only other reported Ca–μ2–C–Zn species, [(THF)2Ca{(μ-CH2SiMe3)2Zn(CH2SiMe3)}2] (Ca–C 2.655(2), Zn–C 2.080(2) Å), reported by Westerhausen in 2002.21 The bridging hydrides display a significant asymmetry with respect to each calcium atom such that Ca1–H11 is elongated by ca. 1 Å compared to Ca1–H1. Nevertheless, both Zn–H distances are commensurate with precedented molecular zinc compounds comprising bridging hydrides (ca. 1.6–1.8 Å).22–26
Although atoms in molecules (AIM) analysis of 3 (PBE0/def2-TZVPP//TPSS/def2-SVP level, see the ESI† for full details) reveals an appreciable Zn1–H1 (ρ(r) = 0.0630, ∇2ρ(r) = +0.1620, H(r) = –0.0129) bond critical point (BCP), with Ca1–H1 (ρ(r) = 0.0214, ∇2ρ(r) = +0.0623, H(r) = 0.0003), this is comparatively weaker in nature with respect to those characterized in 1 (ρ(r) = 0.0326, ∇2ρ(r) = +0.0835, H(r) = –0.0017), indicating transfer of each hydride to Zn upon addition of ZnMe2. This deduction is further supported by mechanistic calculations (see the ESI† for more details). The Zn1–C30 BCP (ρ(r) = 0.0870, ∇2ρ(r) = +0.2180, H(r) = –0.0242), while slightly weaker with respect to ZnMe2 (ρ(r) = 0.1140, ∇2ρ(r) = +0.2100, H(r) = –0.0453), is still significantly stronger than Ca1–C30 (ρ(r) = 0.0242, ∇2ρ(r) = +0.0957, H(r) = +0.0199) which, along with Natural Bond Orbital analysis, indicates this structure is best ascribed as a dianionic {Zn2Me2H2}2− hydridomethylzincate(II) charge-compensated by flanking {(BDI)Ca}+ cations.
Although compound 3 is sufficiently stable in solution to allow the prompt acquisition of NMR spectra (Fig. S3–S6, ESI†), a further intramolecular transformation was apparent upon redissolution at ambient temperature in d8-toluene. After 2 hours the resultant 1H NMR spectrum (Fig. S7, ESI†) comprised an approximate 1:4 ratio of compounds 2 and 3 along with a further minor species (4), which was most clearly identified by the emergence of a broad upfield methyl resonance at −1.35 ppm. Reasoning that the breadth of this latter signal was most likely a consequence of its dynamic consumption to form 2 (Scheme 3), a further reaction was performed between 1 and a sub-stoichiometric amount (0.5 eq.) of ZnMe2. Under these conditions, compound 4 was observed as the major product after 2 hours (Fig. S8, ESI†). Removal of volatiles at this point, followed by extraction into hexane and immediate crystallization at −35 °C, thus, provided a pure sample of compound 4 in the form of colourless crystals.
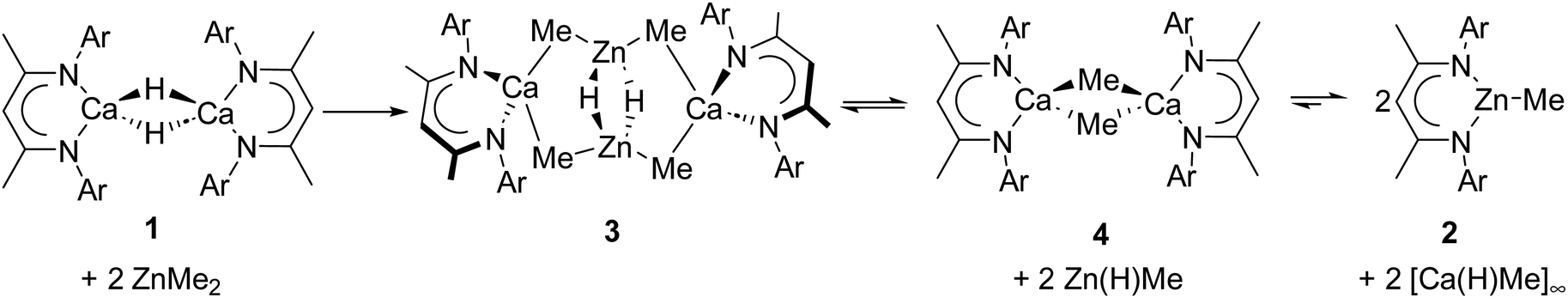 | ||
| Scheme 3 Proposed reaction pathway between ZnMe2 and [(BDI)CaH]2 (1), at ambient temperature. | ||
A solution of 4 in C6D6 displays single BDI γ-methine and Dipp isopropyl methine resonances in its 1H NMR spectrum at 4.74 and 3.07 ppm, respectively, indicative of a symmetrical species. Although somewhat downfield in comparison to the methyl resonance assigned to the THF-adduct of [CaMe2]∞, [(THF)10Ca7Me14] (−1.42 ppm),8 consistent with its exclusive binding to the more electropositive calcium, the now sharp (3H) resonance of 4 at δH −1.29 (δC 8.3 ppm) is significantly upfield with respect to the metallomethyl signal of 3. The constitution of compound 4 was ultimately confirmed by X-ray diffraction analysis, which established it as a centrosymmetric dimer in the solid state (Fig. 2).
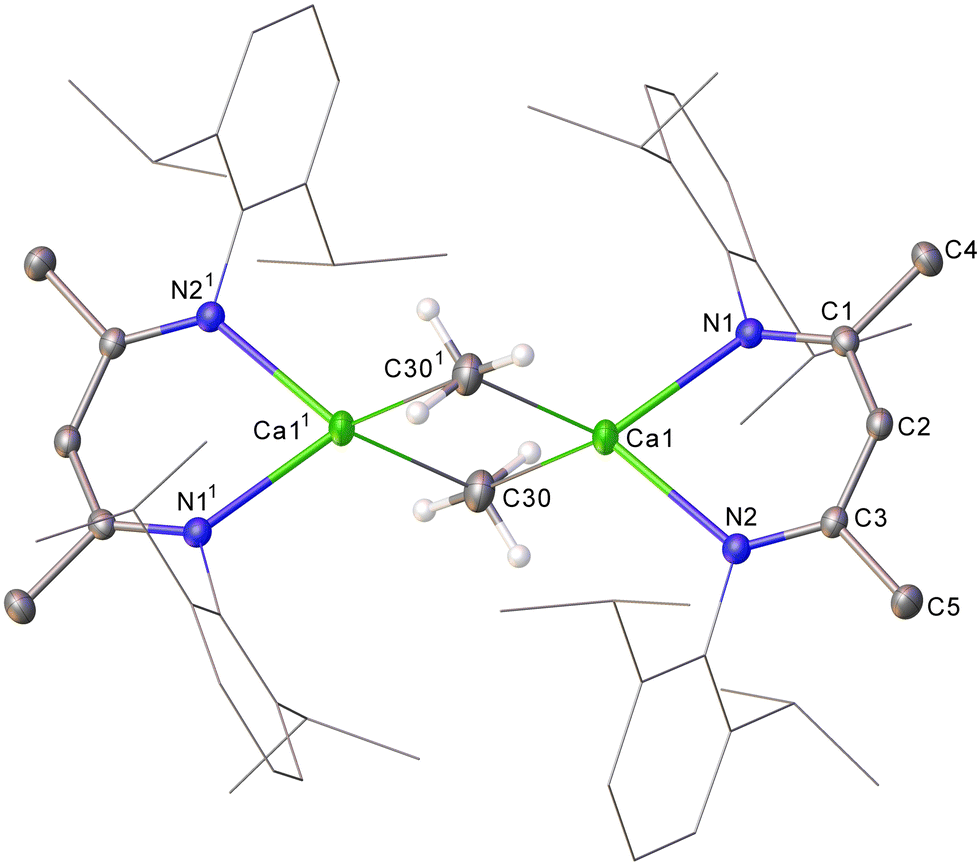 | ||
| Fig. 2 Molecular structure of 4, displacement ellipsoids at 30%. For clarity, an occluded molecule of hexane and the hydrogen atoms, apart from those attached to C30/C301, are omitted and the Dipp groups are displayed as wireframe. Selected bond lengths (Å) and angles (°): Ca1–N1 2.3285(15), Ca1–N2 2.3299(15), Ca1–C30 2.500(3), Ca1–C301 2.539(2), N1–Ca1–N2 81.53(5), N1–Ca1–C30 120.58(7), N1–Ca1–C301 120.70(7). Symmetry operations to generate primed atoms: 11 − x, 1 − y, 2 − z. | ||
The dimeric Ca–μ2–C–Ca structure of 4 is reminiscent of previously described longer-chain σ-alkyl calcium derivatives in which each calcium centre is similarly ligated by a bidentate BDI ligand.12,13 Both the Ca1–C30 [2.500(3) Å] and Ca1–C301 [2.539(2) Å] bond lengths in 4, however, are notably shorter than in any higher homologue (e.g. [(BDI)CaEt]2 2.5733(19) Å).6,12,13 Similarly, the contacts between the methyl carbon and the 4-coordinate calcium atom in 4 are shorter than the analogous three-centre Ca–μ2–C–Ca bonds in Anwander's structurally characterised derivative, [(THP)5Ca3(Me)5(I)] (THP = tetrahydropyran) [2.592(8)–2.673(9) Å],8 in which the calcium centres reside in a 6-coordinate pseudo-octahedral geometry, and the small number of heterometallic Ca–μ2–Me–Al-containing species that have been described. ([Ca(μ2-Me)2AlMe2)2(Phen)] (Phen = 1,10-phenanthroline) 2.571(7)–2.610(7) Å,27 [(Me5C5)2Ca(μ-Me3Al(THF)]2 2.948(7)–2.999(7) Å,28 and [(THF)4Ca(μ2-NR)(μ2-Me)AlMe2] R = Dipp 2.651(2) Å, SiPh3 2.668(2) Å).29 The hypothetical linear MeCaI molecule has been predicted to display a terminal Ca–C bond as short as 2.377 Å,2b while the analogous metric in gas phase dimethylcalcium itself had earlier been computed to be 2.487 or 2.416 Å, dependant on the adoption of a linear or bent geometry, respectively.30,31 While these estimates most likely represent unapproachable extremes, it is notable that the most closely comparable C–Ca bond lengths are provided by Anwander's 5-coordinate calcium complexes, (TptBu,Me)Ca(Me)(THF) (2.477(2), 2.482(2) Å) and [(TptBu,Me)Ca–(Me)(THP)], (2.485(2), 2.507(2) Å), which provide unique examples of terminal calcium-to-methyl interactions.8 Complementary AIM analysis of 4 was subsequently undertaken (PBE0/def2-TZVPP//TPSS/def2-SVP), revealing a suite of similar Ca–C BCPs (ρ(r) = 0.0339/0.0338, ∇2ρ(r) = +0.1100/+0.0990, H(r) = –0.0011/–0.0004) and computed atomic charges (qCa1 = +1.593, qC30 = –0.640), which are indicative of bridging methyl interactions dominated by electrostatic character.
In summary, we have shown that the reaction between ZnMe2 and 1 proceeds in a step-wise manner, affording the bimetallic hydridozincate complex, 3. Although, under appropriate conditions, the intermediates 3 and 4 can be isolated exclusively, compound 3 undergoes a subsequent intramolecular transformation to afford 4 and, ultimately, 2. We suggest that 2 is the thermodynamic product of an equilibration process that is rendered irreversible as a consequence of the insoluble polymeric nature of [Ca(H)Me]∞ (Scheme 3). Compound 4 corrects a notable omission from the homologous series of β-diketiminato alkylcalcium derivatives and we are currently exploring its reactivity and its potential as a potent methylating agent.
The authors acknowledge the EPSRC (EP/X01181X/1) and the University of Bath's Research Computing Group (doi.org/10.15125/b6cd-s854) for their support in this work.
Data availability
Experimental details, NMR spectra, X-ray crystallography, computational details, and atomic coordinates for the optimized geometries of the compounds can be found in the ESI.† Crystallographic data for all compounds have been deposited with the Cambridge Crystallographic Data Centre as supplementary publications CCDC 2360310 and 2360311 for 3 and 4, respectively. Copies of these data can be obtained free of charge on application to CCDC.Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) V. Grignard, C. R. Hebd. Seances Acad. Sci., 1900, 130, 1322 CAS; (b) D. Seyferth, Organometallics, 2009, 28, 1598–1605 CrossRef CAS.
- (a) S. Harder, Alkaline-Earth Metal Compounds: Oddities and Applications, Springer, 2013 CrossRef; (b) M. Westerhausen, A. Koch, H. Görls and S. Krieck, Chem. Eur. J., 2017, 23, 1456–1483 CrossRef CAS PubMed.
- E. Beckmann, Chem. Ber., 1905, 38, 904–906 CrossRef CAS.
- H. Gilman and F. Schulze, J. Am. Chem. Soc., 1926, 48, 2463–2467 CrossRef CAS.
- F. G. N. Cloke, P. B. Hitchcock, M. F. Lappert, G. A. Lawless and B. Royo, J. Chem. Soc., Chem. Commun., 1991, 724–726 RSC.
- See for example, (a) C. Eaborn, S. A. Hawkes, P. B. Hitchcock and J. D. Smith, Chem. Commun., 1997, 1961–1962 RSC; (b) F. Feil and S. Harder, Organometallics, 2000, 19, 5010–5015 CrossRef CAS; (c) S. Harder, F. Feil and A. Weeber, Organometallics, 2001, 20, 1044–1046 CrossRef CAS; (d) M. R. Crimmin, A. G. M. Barrett, M. S. Hill, D. J. MacDougall, M. F. Mahon and P. A. Procopiou, Chem. Eur. J., 2008, 14, 11292–11295 CrossRef CAS PubMed; (e) M. P. Coles, S. E. Sözerli, J. D. Smith, P. B. Hitchcock and I. J. Day, Organometallics, 2009, 28, 1579–1581 CrossRef CAS; (f) M. S. Hill, M. F. Mahon and T. P. Robinson, Chem. Commun., 2010, 46, 2498–2500 RSC; (g) A. Koch, M. Wirgenings, S. Krieck, H. Görls, G. Pohnert and M. Westerhausen, Organometallics, 2017, 36, 3981–3986 CrossRef CAS.
- (a) M. Westerhausen, M. Gärtner, R. Fischer and J. Langer, Angew. Chem., Int. Ed., 2007, 46, 1950–1956 CrossRef CAS PubMed; (b) R. Fischer, H. Görls and M. Westerhausen, Inorg. Chem. Commun., 2005, 8, 1159–1161 CrossRef CAS; (c) R. Fischer, M. Gartner, H. Görls and M. Westerhausen, Angew. Chem., Int. Ed., 2006, 45, 609–612 CrossRef CAS; (d) R. Fischer, M. Gartner, H. Görls and M. Westerhausen, Organometallics, 2006, 25, 3496–3500 CrossRef CAS; (e) M. Westerhausen, M. Gäertner, R. Fischer and J. Langer, Angew. Chem., Int. Ed., 2007, 46, 1950–1956 CrossRef CAS; (f) M. Westerhausen, M. Gäertner, R. Fischer, J. Langer, L. Yu and M. Reiher, Chem. Eur. J., 2007, 13, 6292–6306 CrossRef CAS PubMed; (g) J. Langer, H. Görls and M. Westerhausen, Inorg. Chem. Commun., 2007, 10, 853–855 CrossRef CAS; (h) J. Langer, S. Krieck, H. Görls and M. Westerhausen, Angew. Chem., Int. Ed., 2009, 48, 5741–5744 CrossRef CAS PubMed; (i) S. Krieck, H. Görls and M. Westerhausen, Chem. Asian J., 2010, 5, 272–277 CrossRef CAS PubMed; (j) R. Fischer, J. Langer, S. Krieck, H. Görls and M. Westerhausen, Organometallics, 2011, 30, 1359–1365 CrossRef CAS; (k) J. Langer, M. Kohler, R. Fischer, F. Dundar, H. Görls and M. Westerhausen, Organometallics, 2012, 31, 6172–6182 CrossRef CAS; (l) M. Kohler, J. Langer, H. Görls and M. Westerhausen, Organometallics, 2012, 31, 8647–8653 CrossRef; (m) J. Langer, M. Kohler, H. Görls and M. Westerhausen, Chem. Eur. J., 2014, 20, 3154–3161 CrossRef CAS PubMed; (n) M. Kohler, J. Langer, H. Görls and M. Westerhausen, Organometallics, 2014, 33, 6381–6388 CrossRef; (o) A. Koch, S. Krieck, H. Görls and M. Westerhausen, Organometallics, 2017, 36, 2811–2817 CrossRef CAS; (p) A. Koch, S. Krieck, H. Görls and M. Westerhausen, Dalton Trans., 2018, 47, 12534–12539 RSC; (q) J. Langer, M. Kohler, J. Hildebrand, R. Fischer, H. Görls and M. Westerhausen, Angew. Chem., Int. Ed., 2013, 52, 3507–3510 CrossRef CAS PubMed.
- B. M. Wolf, C. Stuhl, C. Maichle-Mössmer and R. Anwander, J. Am. Chem. Soc., 2018, 140, 2373–2383 CrossRef CAS PubMed.
- See, for example, (a) S. Harder, F. Feil and K. Knoll, Angew. Chem., Int. Ed., 2001, 40, 4261–4264 CrossRef CAS; (b) S. Harder and F. Feil, Organometallics, 2002, 21, 2268–2274 CrossRef CAS; (c) F. Feil, C. Müller and S. Harder, J. Organometal. Chem., 2003, 683, 56–63 CrossRef CAS; (d) D. F.-J. Piesik, K. Habe and S. Harder, Eur. J. Inorg. Chem., 2007, 5652–5661 CrossRef CAS; (e) A. Causero, H. Elsen, G. Ballmann, A. Escalona and S. Harder, Chem. Commun., 2017, 53, 10386–10389 RSC; (f) X. Shi, C. Hou, L. Zhao, P. Deng and J. Cheng, Chem. Commun., 2020, 56, 5162–5165 RSC; (g) L. Zhao, X. Shi and J. Cheng, ACS Catal., 2021, 11, 2041–2046 CrossRef CAS.
- For benzylic derivatives, see (a) M. R. Crimmin, A. G. M. Barrett, M. S. Hill, D. J. MacDougall, M. F. Mahon and P. A. Procopiou, Dalton Trans., 2009, 9715–9717 RSC; (b) X. Zheng, I. del Rosal, X. Xu, Y. Yao, L. Maron and X. Xu, Inorg. Chem., 2021, 60, 5114–5121 CrossRef CAS PubMed.
- S. Harder and J. Brettar, Angew. Chem., Int. Ed., 2006, 45, 3474–3478 CrossRef CAS PubMed.
- A. S. S. Wilson, M. S. Hill, M. F. Mahon, C. Dinoi and L. Maron, Science, 2017, 358, 1168–1171 CrossRef CAS PubMed.
- (a) A. S. S. Wilson, M. S. Hill and M. F. Mahon, Organometallics, 2019, 38, 351–360 CrossRef CAS; (b) A. S. S. Wilson, C. Dinoi, M. S. Hill, M. F. Mahon and L. Maron, Angew. Chem., Int. Ed., 2018, 57, 15500–15504 CrossRef CAS PubMed.
- K. G. Pearce, C. Dinoi, M. S. Hill, M. F. Mahon, L. Maron, R. J. Schwamm and A. S. S. Wilson, Angew. Chem., Int. Ed., 2022, 61, e202200305 CrossRef CAS PubMed.
- K. G. Pearce, C. Dinoi, R. J. Schwamm, L. Maron, M. F. Mahon and M. S. Hill, Adv. Sci., 2023, 10, 2304765 CrossRef CAS PubMed.
- R. W. Siegler, D. W. Nierenberg and W. F. Hickey, Hum. Pathol., 1999, 30, 720–723 CrossRef CAS PubMed.
- J. Prust, A. Stasch, W. Zheng, H. W. Roesky, E. Alexopoulos, I. Usón, D. Böhler and T. Schuchardt, Organometallics, 2001, 20, 3825–3828 CrossRef CAS.
- K. G. Pearce, L. J. Morris, T. P. Robinson, A. L. Johnson, M. F. Mahon and M. S. Hill, Dalton Trans., 2024, 53, 6653–6659 RSC.
- M. R. Crimmin, M. S. Hill, P. B. Hitchcock and M. F. Mahon, New J. Chem., 2010, 34, 1572–1578 RSC.
- A. Causero, G. Ballmann, J. Pahl, C. Färber, J. Intemann and S. Harder, Dalton Trans., 2017, 46, 1822–1831 RSC.
- M. Westerhausen, C. Gückel, H. Piotrowski and M. Wogt, Z. Anorg. Allg. Chem., 2002, 628, 735–740 CrossRef CAS.
- A. Hicken, A. J. P. White and M. R. Crimmin, Inorg. Chem., 2017, 56, 8669–8682 CrossRef CAS PubMed.
- H. Hao, C. Cui, H. W. Roesky, G. Bai, H.-G. Schmidt and M. Noltemeyer, Chem. Commun., 2001, 1118–1119 RSC.
- A. Rit, T. P. Spaniol, L. Maron and J. Okuda, Organometallics, 2014, 33, 2039–2047 CrossRef CAS.
- M. Chen, S. Jiang, L. Maron and X. Xu, Dalton Trans., 2019, 48, 1931–1935 RSC.
- A. Lennartson, M. Håkansson and S. Jagner, Angew. Chem., Int. Ed., 2007, 46, 6678–6680 CrossRef CAS PubMed.
- O. Michel, C. Meermann, K. W. Törnroos and R. Anwander, Organometallics, 2009, 28, 4783–4790 CrossRef CAS.
- P. S. Tanner, R. A. Williams and T. P. Hanusa, Inorg. Chem., 1993, 32, 2234–2235 CrossRef CAS.
- B. M. Wolf, C. Maichle-Mössmer and R. Anwander, Angew. Chem., Int. Ed., 2016, 55, 13893–13897 CrossRef CAS PubMed.
- M. Kaupp and P. V. R. Schleyer, J. Am. Chem. Soc., 1992, 114, 491–497 CrossRef CAS.
- I. Bytheway, R. J. Gillespie, T.-H. Tang and R. F. W. Bader, Inorg. Chem., 1995, 34, 2407–2414 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General synthetic experimental details, NMR spectra, X-ray analysis of compound 3 and 4. CCDC 2360310 and 2360311. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc02930e |
| This journal is © The Royal Society of Chemistry 2024 |