
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-catalyzed silylation of aryl and alkenyl triflates with silylboronic esters avoiding base-mediated borylation†
Shintaro
Kamio
abc,
Masaaki
Nakamoto
a,
Takehiro
Yamagishi
b,
Martin
Oestreich
 *c and
Hiroto
Yoshida
*a
*c and
Hiroto
Yoshida
*a
aGraduate School of Advanced Science and Engineering, Hiroshima University, Higashi-Hiroshima 739-8526, Japan. E-mail: yhiroto@hiroshima-u.ac.jp
bDepartment of Pharmacy, Faculty of Pharmaceutical Sciences, Hokkaido University of Science, Sapporo 006-8585, Japan
cInstitut für Chemie, Technische Universität Berlin, Strasse des 17. Juni 115, 10623 Berlin, Germany. E-mail: martin.oestreich@tu-berlin.de
First published on 2nd May 2024
Abstract
Silylation of aryl and alkenyl triflates is found to occur readily with silylboronic esters as a silicon source under copper catalysis. The silyl moieties are exclusively installed into the organic frameworks through the preferential generation of a silylcopper species, wherein base-mediated direct borylation is totally suppressed. The combined use of tri-n-butylphosphine and 4,4′-diphenyl-2,2′-bipyridine as a ligand combination turned out to be indispensable for achieving the high catalytic activity.
Silylboronic esters, versatile unsymmetrical bimetal reagents, play a pivotal role in modern synthetic organic chemistry, and have been demonstrated to be convertible into a variety of synthetically important organosilicon and -boron compounds.1 The Si–B σ-bond is readily activated by transition metal catalysts; for instance, copper alkoxides undergo σ-bond metathesis to produce silylcopper species (Cu–Si),2 where the selective interaction between the Lewis acidic boryl group and the alkoxy moiety determines the chemoselectivity. The Cu–Si species thus generated serves as a silicon nucleophile for addition reactions across C–C multiple bonds3 as well as C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X bonds4 and is a coupling partner in ionic and radical reactions with alkyl (pseudo)halides.5 While the Cu–Si species have been well established to be coupled with sp3 carbon electrophiles,6,7 their coupling reaction with sp2 counterparts, especially aryl/alkenyl halides, has continued to be a challenging subject. We inferred that the difficulties in the copper-catalyzed silylation of aryl/alkenyl halides with silylboronic esters should be mainly due to inevitable borylation that concomitantly takes place in the presence of a metal alkoxide, commonly used as a base in the above Cu-catalyzed silylations, giving aryl/alkenyl boron side-products as reported by the Ito group.8 Because the base-mediated borylation was reported to facilely proceed even at 30 °C under transition metal-free conditions through a halogen–metal exchange pathway, where a halogenate-type intermediate is generated as a key intermediate (Scheme 1),8 C(sp2)–Br/I that can easily be converted into the respective hypervalent forms are basically unsuitable electrophiles for the selective silylation. Under these circumstances, aryl carboxylic acid derivatives and aryl aldehydes were reported to serve as C(sp2) electrophiles by use of Ni/Cu cooperative catalysts, undergoing decarbonylative silylation with silylboronic esters to give arylsilanes selectively (Scheme 2).9 Herein, we disclose for the first time that silylboronic esters selectively act as silyl-installing reagents into C(sp2) frameworks under Cu-only catalysis by utilizing aryl and alkenyl triflates as electrophiles.
X bonds4 and is a coupling partner in ionic and radical reactions with alkyl (pseudo)halides.5 While the Cu–Si species have been well established to be coupled with sp3 carbon electrophiles,6,7 their coupling reaction with sp2 counterparts, especially aryl/alkenyl halides, has continued to be a challenging subject. We inferred that the difficulties in the copper-catalyzed silylation of aryl/alkenyl halides with silylboronic esters should be mainly due to inevitable borylation that concomitantly takes place in the presence of a metal alkoxide, commonly used as a base in the above Cu-catalyzed silylations, giving aryl/alkenyl boron side-products as reported by the Ito group.8 Because the base-mediated borylation was reported to facilely proceed even at 30 °C under transition metal-free conditions through a halogen–metal exchange pathway, where a halogenate-type intermediate is generated as a key intermediate (Scheme 1),8 C(sp2)–Br/I that can easily be converted into the respective hypervalent forms are basically unsuitable electrophiles for the selective silylation. Under these circumstances, aryl carboxylic acid derivatives and aryl aldehydes were reported to serve as C(sp2) electrophiles by use of Ni/Cu cooperative catalysts, undergoing decarbonylative silylation with silylboronic esters to give arylsilanes selectively (Scheme 2).9 Herein, we disclose for the first time that silylboronic esters selectively act as silyl-installing reagents into C(sp2) frameworks under Cu-only catalysis by utilizing aryl and alkenyl triflates as electrophiles.
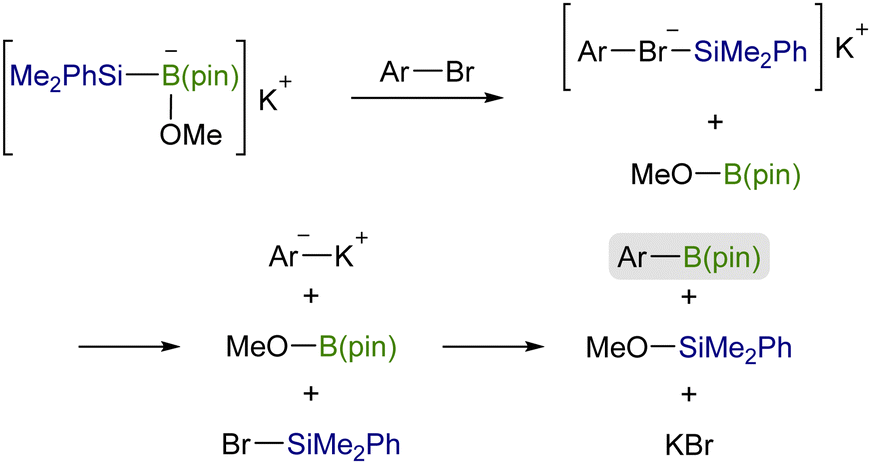 | ||
| Scheme 1 Base-mediated, metal catalyst-free direct borylation of aryl bromides. | ||
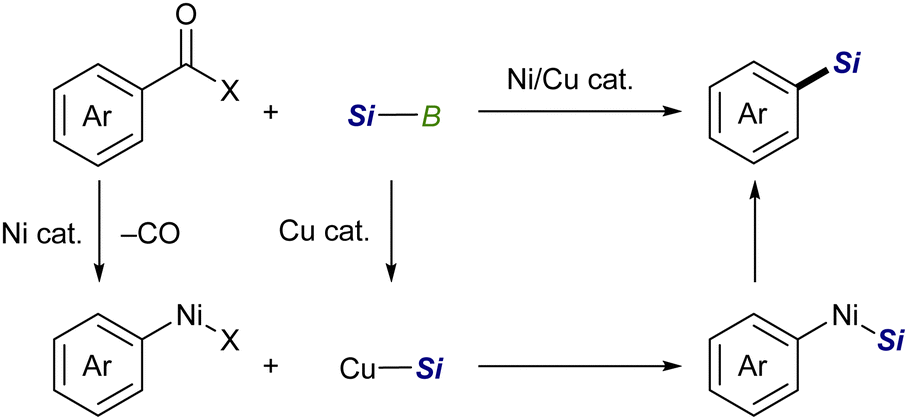 | ||
| Scheme 2 Cooperative catalytic silylation of aryl electrophiles. | ||
Trialkylsilylboronic esters employed in this study could readily be prepared according to our gram-scale synthesis, in which key trialkylsilyl lithium reagents were generated with the aid of less toxic tris(N,N-tetramethylene)phosphoric acid triamide (TPPA) (Scheme 3).10 With Et3Si–B(pin) thus prepared, our effort was initially focused on the investigation of a proper leaving group on the C(sp2) electrophiles that can avoid the base-mediated borylation while exhibiting enough reactivity toward a Cu–Si species. As was expected, the reactions of p-tolyl bromide/iodide with Et3Si–B(pin) in the presence of CuI–P(n-Bu)3 as a catalyst afforded p-tolyl–B(pin) (2a) as the major product, showing that chemoselective installation of a silicon functionality is indeed difficult with C(sp2)–Br/I bonds even under copper catalysis (Table 1, entries 1 and 2). On the other hand, the borylation was completely suppressed with N-hydroxyphthalimide ester (COONPth) or such pseudohalides as mesylate (OMs) and chloromethane sulfonate (OMc: OSO2CH2Cl), but the silylation did not take place either (entries 3–5). Finally, the desired silylation was found to proceed exclusively by employing tosylate (OTs) or triflate (OTf) as leaving groups to provide 1a in 18% and 34% yield, respectively (entries 6 and 7).
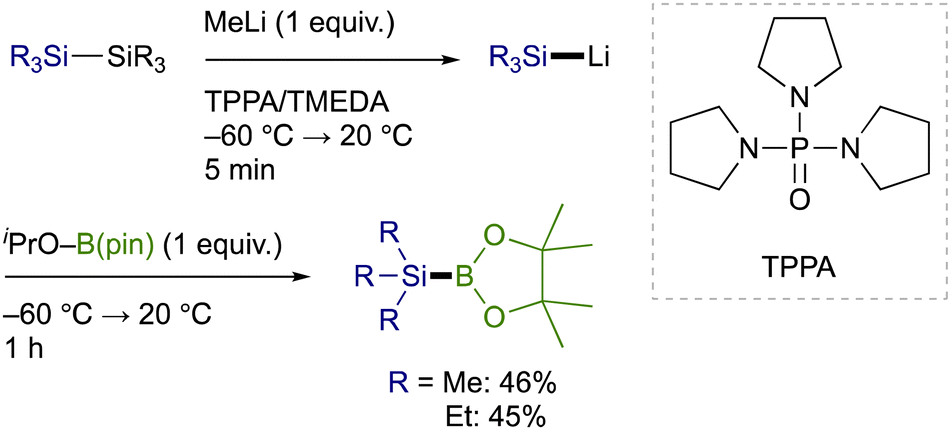 | ||
| Scheme 3 Synthesis of trialkylsilylboronic esters via TPPA-assisted generation of trialkylsilyl lithium reagents. | ||
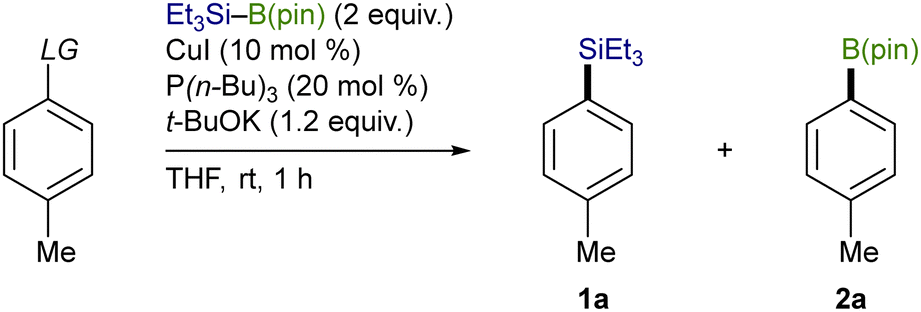
Using an OTf moiety as the better leaving group, we next carried out the silylation with various copper salts in the presence of t-BuOK (base) and P(n-Bu)3 (ligand) and found that CuI was optimal (Table 2, entries 1–7). The use of t-BuOK turned out to be indispensable for the silylation, and thus reactions with other alkoxides of lower basicity were totally unsuccessful (entries 8–11). Among the ligands surveyed, the combined use of P(n-Bu)3 (10 mol%) and 4,4′-diphenyl-2,2′-bipyridine (4,4′-Phbpy) (10 mol%) proved to be the most effective as was the case of our previously reported Cu-catalyzed silylation of C(sp3)–COONPth (entry 12).7b,11 No desired product was obtained in the absence of the ligands and/or CuI, verifying the necessity of the copper catalysis together with the ligand system for the smooth silylation (entries 13 and 14).
|
|
||||
|---|---|---|---|---|
| Entry | [Cu] | Ligand | Base | GC yield (%) |
| a Isolated yield. Conditions: p-tolyl–OTf (0.3 mmol), Et3Si–B(pin) (0.6 mmol), copper salt (0.03 mmol), ligand (0.06 mmol), base (0.36 mmol), THF (1 mL), rt, 1 h. | ||||
| 1 | CuI | P(n-Bu)3 | t-BuOK | 34a |
| 2 | CuCl | P(n-Bu)3 | t-BuOK | 29 |
| 3 | CuBr·SMe2 | P(n-Bu)3 | t-BuOK | 27 |
| 4 | CuTc | P(n-Bu)3 | t-BuOK | 9 |
| 5 | CuSCN | P(n-Bu)3 | t-BuOK | 10 |
| 6 | Cu(MeCN)4·PF6 | P(n-Bu)3 | t-BuOK | 7 |
| 7 | Cu(MeCN)4·BF4 | P(n-Bu)3 | t-BuOK | 26 |
| 8 | CuI | P(n-Bu)3 | MeONa | 0 |
| 9 | CuI | P(n-Bu)3 | MeOK | 0 |
| 10 | CuI | P(n-Bu)3 | t-BuOLi | 0 |
| 11 | CuI | P(n-Bu)3 | t-BuONa | Trace |
| 12 | CuI | P(n-Bu)3/4,4′-Phbpy | t-BuOK | 67a |
| 13 | CuI | None | t-BuOK | 0 |
| 14 | None | None | t-BuOK | 0 |
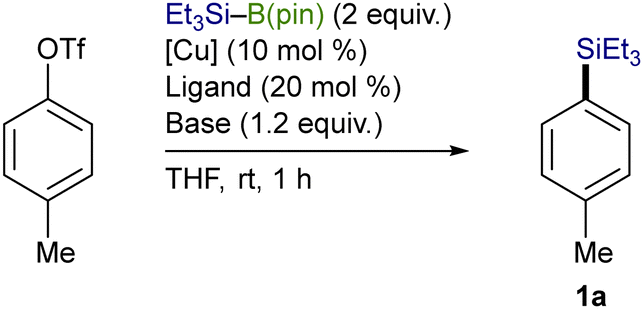
A variety of aryl triflates bearing an electron-donating group could facilely undergo the silylation under the optimum conditions to furnish the respective aryl silanes (1a–1h) with exclusive chemoselectivity, and borylation-based side-products were not generated at all (Scheme 4). In addition, the reaction of naphthyl and biphenyl triflates also took place to afford the corresponding arylsilanes (1i–1k) in moderate to good yields. Although the silylation of functionalized aryl triflates with Cl, B(dan) (dan: naphthalene-1,8-diaminato), and B(pin) (pin: pinacolato) also proceeded under the present reaction conditions, the yield became somewhat lower (1l–1n). The reaction was applicable to a bis-triflate, whose C–OTf bonds could both be transformed into C–SiEt3 bonds to give 1o. Furthermore, alkenyl triflates, readily prepared from the respective ketones, could also participate in the present reaction, providing good yields of various alkenylsilanes (1p–1r).
 | ||
Scheme 4 Substrate scope on triflates. Conditions: triflate (0.3 mmol), Et3Si–B(pin) (0.6 mmol), CuI (0.03 mmol), P(n-Bu)3 (0.03 mmol), 4,4′-Phbpy (0.03 mmol), t-BuOK (0.36 mmol), THF (1 mL), rt, 1 h. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction was carried out with a ditriflate (0.15 mmol). Reaction was carried out with a ditriflate (0.15 mmol). | ||
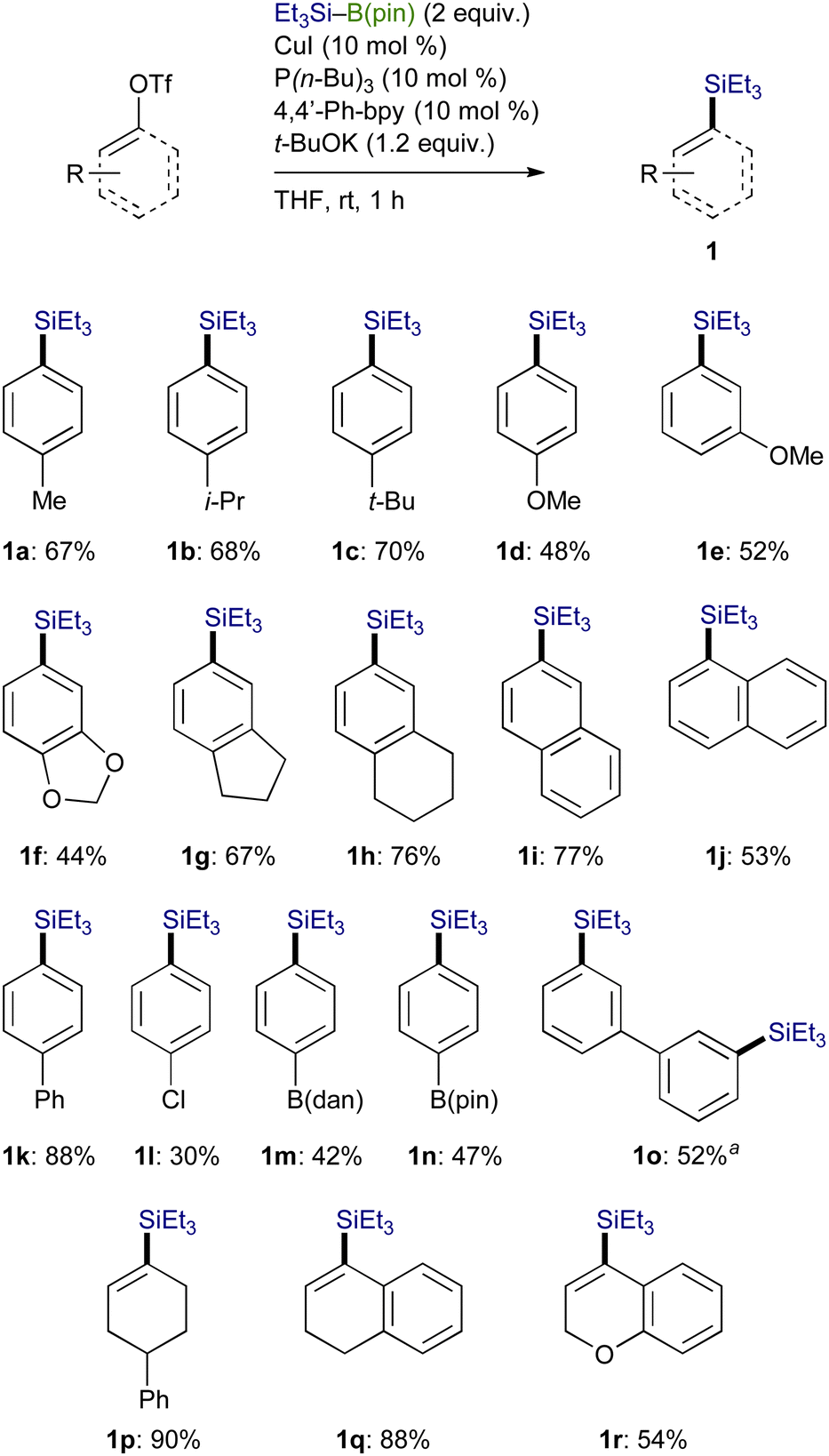
The catalytic silylation was also applicable to Me2PhSi–B(pin), giving an arylsilane (1s) in 70% yield (Scheme 5). It should be noted that Me3Si–B(pin), which can practically be prepared by our method (Scheme 3, vide supra), has proven to be convertible into the corresponding product (1t) in a straightforward manner, providing an alternative approach for the Me3Si-installing process.12
 | ||
| Scheme 5 Silylation with other silylboronic esters. Conditions: biphenyl–OTf (0.3 mmol), silylboronic ester (0.6 mmol), CuI (0.03 mmol), P(n-Bu)3 (0.03 mmol), 4,4′-Phbpy (0.03 mmol), t-BuOK (0.36 mmol), THF (1 mL), rt, 1 h. | ||
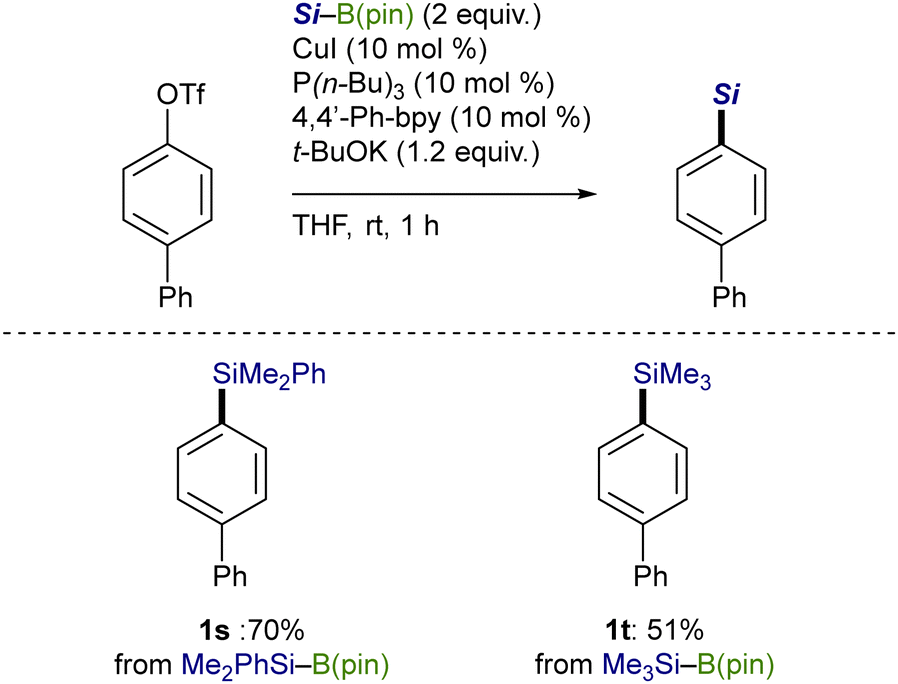
As depicted in Scheme 6, control experiments were conducted to gain mechanistic insights into the C(sp2)–Si bond-forming process. The stereochemical outcome with an acyclic alkenyl triflate (isomeric ratio = 58:42) indicates that a radical pathway would be operative in the present substitution: (E)-alkenylsilane (1u) became enriched through a stereoconvergent pathway, where an alkenyl radical intermediate rapidly isomerized before the silylation.13 Besides, the addition of 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO), a radical scavenger, to the reaction of 2-naphthyl triflate reduced the yields of 1i as the amount of TEMPO increased, which also supports the possibility of the radical pathway.14
 | ||
| Scheme 6 Control experiments. | ||
Based on the above results, the silylation would be triggered by the generation of a CuI–Si species via σ-bond metathesis between a copper alkoxide [CuI–Ot-Bu] and a silyboronic ester (Scheme 7).7a Then t-BuO− coordinates to CuI–Si to provide an electron-rich silylcuprate species [t-BuO–CuI–Si]− (3a), which may serve as a single-electron reductant for an aryl/alkenyl triflate. The resulting radical anion then releases TfO− of good leaving ability to generate an isomerizable free radical (cf.Scheme 6), which readily recombines with [t-BuO–CuII–Si] (3b), affording a CuIII complex (3c).15 Finally, 3c undergoes reductive elimination to provide a C(sp2)–Si product with the regeneration of the copper alkoxide.
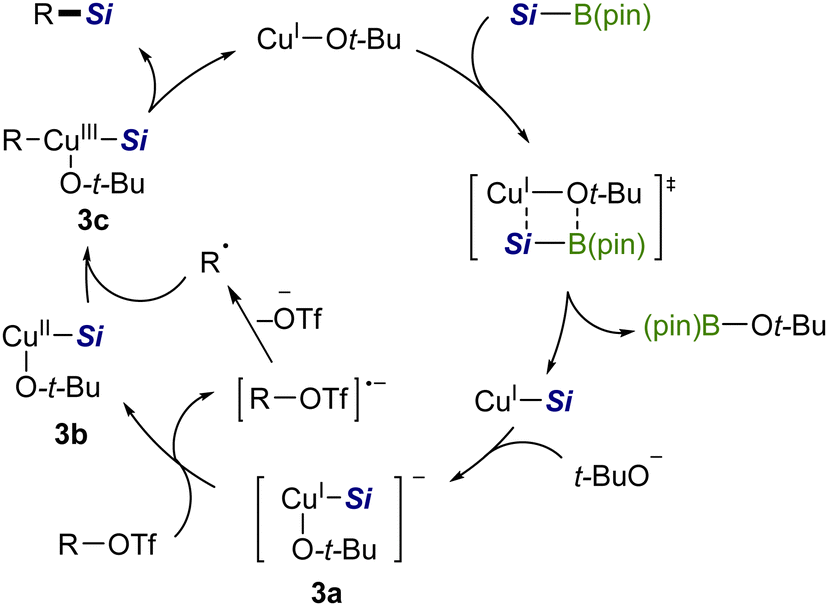 | ||
| Scheme 7 A proposed catalytic cycle. | ||
In conclusion, we have demonstrated for the first time that C(sp2) electrophiles undergo selective silylation with silylboronic esters under copper-only catalysis by using a triflate moiety as an optimal leaving group, which leads to the exclusive formation of various aryl/alkenyl–silanes with complete suppression of undesired base-mediated borylation. Moreover, the mechanistic studies suggested that the present silylation may involve a radical pathway. A similar silylation of aryl and alkenyl carbamates under iron catalysis was reported by the Feng group;12e however, the present silylation is a meaningful expansion of the reactivity as this proceeds under milder conditions and permits the use of easily accessible triflates as substrates. Further studies on the catalytic utilization of silylboronic esters as well as on details of the reaction mechanism are in progress.
S. K., M. O, and H. Y. designed the study. M. N. and T. Y. aided in interpreting the results. H. Y. supervised the project. S. K. collected all data and wrote the manuscript with support from M. O. and H. Y. All authors have approved the final version of the manuscript.
This work was supported by the Natural Science Center for Basic Research and Development, Hiroshima University (NBARD-00001). S. K. acknowledges the JSPS fellowship for young scientists, JSPS KAKENHI grant number JP20J14589 and JSPS Overseas Challenge Program for Young Researchers.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For reviews on the chemistry of silylboranes, see: (a) J. Feng, W. Mao., L. Zhang and M. Oestreich, Chem. Soc. Rev., 2021, 50, 2010 RSC; (b) M. Oestreich, E. Hartmann and M. Mewald, Chem. Rev., 2013, 113, 402 CrossRef CAS PubMed; (c) T. Ohmura and M. Suginome, Bull. Chem. Soc. Jpn., 2009, 82, 29 CrossRef CAS.
- For a review on the reactions of silylcopper species, see: W. Xue and M. Oestreich, ACS Cent. Sci., 2020, 6, 1070 CrossRef CAS PubMed.
- For the seminal works with alkynes, see: (a) T. Fujihara, Y. Tani, K. Semba, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2012, 51, 11487 CrossRef CAS; (b) F. Meng, H. Jang and A. H. Hoveyda, Chem. – Eur. J., 2013, 19, 3204 CrossRef CAS PubMed.
- For the seminal works with carbonyl compounds, see: (a) C. Kleeberg, E. Feldmann, E. Hartmann, D. J. Vyas and M. Oestreich, Chem. – Eur. J., 2011, 17, 13538 CrossRef CAS PubMed; (b) J. Plotzitzka and C. Kleeberg, Inorg. Chem., 2017, 56, 667 CrossRef PubMed.
- For the seminal works with allylic electrophiles, see: (a) D. J. Vyas and M. Oestreich, Angew. Chem., Int. Ed., 2010, 49, 8513 CrossRef CAS PubMed; (b) L. B. Delvos, D. J. Vyas and M. Oestreich, Angew. Chem., Int. Ed., 2013, 52, 465 CrossRef PubMed; (c) C. K. Hazra, E. Irran and M. Oestreich, Eur. J. Org. Chem., 2013, 4903 CrossRef CAS; (d) M. Takeda, R. Shintani and T. Hayashi, J. Org. Chem., 2013, 78, 5007 CrossRef CAS PubMed ; for the seminal works with propargylic electrophiles, see: ; (e) D. J. Vyas, C. K. Hazra and M. Oestreich, Org. Lett., 2011, 13, 4462 CrossRef CAS PubMed.
- For ionic reactions with alkyl electrophiles, see: (a) J. Scharfbier and M. Oestreich, Synlett, 2016, 127 Search PubMed; (b) J. Scharfbier, H. Hazrati and M. Oestreich, Org. Lett., 2017, 19, 656 CrossRef PubMed; (c) J. Scharfbier, B. M. Gross and M. Oestreich, Angew. Chem., Int. Ed., 2020, 59, 1577 CrossRef CAS PubMed.
- For radical reactions with alkyl electrophiles, see: (a) W. Xue, Z.-W. Qu, S. Grimme and M. Oestreich, J. Am. Chem. Soc., 2016, 138, 14222 CrossRef CAS PubMed; (b) W. Xue and M. Oestreich, Angew. Chem., Int. Ed., 2017, 56, 1164 CrossRef PubMed ; for a review, see: ; (c) S. Bähr, W. Xue and M. Oestreich, ACS Catal., 2019, 9, 16 CrossRef.
- (a) E. Yamamoto, K. Izumi, Y. Horita and H. Ito, J. Am. Chem. Soc., 2012, 134, 19997 CrossRef CAS PubMed; (b) E. Yamamoto, K. Izumi, Y. Horita, S. Ukigai and H. Ito, Top. Catal., 2014, 57, 940 CrossRef CAS; (c) E. Yamamoto, S. Ukigai and H. Ito, Chem. Sci., 2015, 6, 2943 RSC; (d) R. Uematsu, E. Yamamoto, S. Maeda, H. Ito and T. Taketsugu, J. Am. Chem. Soc., 2015, 137, 409 CrossRef PubMed; (e) E. Yamamoto, K. Izumi, R. Shishido, T. Seki, N. Tokodai and H. Ito, Chem. – Eur. J., 2016, 22, 17547 CrossRef CAS PubMed ; for a review, see: ; (f) E. Yamamoto, S. Maeda, T. Taketsugu and H. Ito, Synlett, 2017, 1258 CAS.
- (a) L. Guo, A. Chatupheeraphat and M. Rueping, Angew. Chem., Int. Ed., 2016, 55, 11810 CrossRef CAS PubMed; (b) X. Pu, J. Hu, Y. Zhao and Z. Shi, ACS Catal., 2016, 6, 6692 CrossRef CAS; (c) S.-C. Lee, L. Guo, H. Yue, H.-H. Li and M. Rueping, Synlett, 2017, 2594 CAS; (d) X. Wang, Z. Wang and Y. Nishihara, Chem. Commun., 2019, 55, 10507 RSC; (e) W. Srimontree, L. Guo and M. Rueping, Chem. – Eur. J., 2020, 26, 423 CrossRef CAS PubMed.
- S. Kamio, T. Imagawa, M. Nakamoto, M. Oestreich and H. Yoshida, Synthesis, 2021, 4678 CAS.
- For the effects of other ligands and full optimization of the reaction conditions, see ESI† for details.
- For the seminal works on catalytic Me3Si-installation reactions, see: (a) H. Matsumoto, S. Nagashima, K. Yoshihiro and Y. Nagai, J. Organomet. Chem., 1975, 85, C1 CrossRef CAS; (b) D. Azarian, S. S. Dua, C. Eaborn and D. R. M. Walton, J. Organomet. Chem., 1976, 117, C55 CrossRef CAS; (c) H. Matsumoto, K. Yoshihiro, S. Nagashima, H. Watanabe and Y. Nagai, J. Organomet. Chem., 1977, 128, 409 CrossRef CAS; (d) L. J. Gooßen and A. S. Ferwan, Synlett, 2000, 1801 Search PubMed ; for recent works on constructing C(sp2)–SiEt3 bonds with Et3Si–B(pin), see: ; (e) J. Zhang, Y. Zhang, S. Geng, S. Chen, Z. Liu, X. Zeng, Y. He and Z. Feng, Org. Lett., 2020, 22, 2669 CrossRef CAS PubMed; (f) J. Jia, X. Zeng, Z. Liu, L. Zhao, C.-Y. He, X.-F. Li and Z. Feng, Org. Lett., 2020, 22, 2816 CrossRef CAS PubMed.
- Less steric repulsion between a Ph group of the cis-alkenyl radical and an incoming 3b may determine the stereoselectivity.
- The naphthyl moiety was not trapped by TEMPO while a Et3Si-TEMPO adduct was generated as a side-product. In sharp contrast to the results obtained with TEMPO, the addition of 9,10-dihydroanthracene, a known radical scavenger, hardly influenced the course of the silylation, implying that an ionic mechanism involving direct σ-bond metathesis between Cu-SiEt3 and aryl/alkenyl triflates might still be conceivable.
- For our previous reports on similar copper-catalyzed bolylations of alkyl, alkenyl and aryl halides with diborons, see: (a) H. Yoshida, Y. Takemoto, S. Kamio, I. Osaka and K. Takaki, Org. Chem. Front., 2017, 4, 1215 RSC; (b) H. Yoshida, S. Kamio and I. Osaka, Chem. Lett., 2018, 47, 957 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data. See DOI: https://doi.org/10.1039/d4cc01005a |
| This journal is © The Royal Society of Chemistry 2024 |