
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of a benzothiadiazole spacer on transport properties and N-doping of naphthalene-diimide-based copolymers†
Olivier
Bardagot
 *ab,
Yann
Kervella
b,
Asma Aicha
Medjahed
bc,
Stéphanie
Pouget
d,
Tamara Nunes
Domschke
e,
Alexandre
Carella
ef,
Cyril
Aumaître
b,
Patrick
Lévêque
g and
Renaud
Demadrille
*b
*ab,
Yann
Kervella
b,
Asma Aicha
Medjahed
bc,
Stéphanie
Pouget
d,
Tamara Nunes
Domschke
e,
Alexandre
Carella
ef,
Cyril
Aumaître
b,
Patrick
Lévêque
g and
Renaud
Demadrille
*b
aInstitute of Chemistry and Processes for Energy Environment and Health (ICPEES), CNRS University of Strasbourg, UMR 7515, 25 rue de Becquerel, Strasbourg Cedex 02, 67087, France. E-mail: olivier.bardagot@cnrs.fr
bUniversité Grenoble Alpes CEA CNRS IRIG-SyMMES, Grenoble 38000, France. E-mail: renaud.demadrille@cea.fr
cThe European Synchrotron, Avenue des Martyrs 71, Grenoble 38000, France
dUniversité Grenoble Alpes CEA, IRIG-MEM, Grenoble 38000, France
eUniversité Grenoble Alpes CEA-LITEN, Grenoble 38000, France
fCEA, DES, ISEC, DMRC, Univ. Montpellier, Marcoule, 30 207, France
gLaboratoire des Sciences de l’Ingénieur, de l’Informatique et de l’Imagerie (ICube Research Institute), Université de Strasbourg, CNRS, 23, Rue du Loess, Strasbourg Cedex 2 67037, France
First published on 4th October 2023
Abstract
We report the synthesis of two n-type semiconducting polymers containing a naphthalene diimide (NDI) monomer and thiophene spacers. The two polymers differ by the introduction of an additional 2,1,3-benzothiadiazole (BTD) moiety in the polymer backbone. Although there are examples of polymers combining these two moieties, the effects of introducing the BTD as a third co-monomer on the transport properties and, in particular, the doping mechanisms of these materials have not been elucidated. We describe the optoelectronic, structural organisation and transport properties of these two polymers and investigate the effect of BTD introduction on molecular doping with N-DMBI as a dopant using UV-Vis spectroscopy, DFT modelling, cyclic voltametry, and X-ray diffraction. We show that the incorporation of a BTD unit in the polymer backbone not only improves the charge transport in organic field effect transistors but also the doping efficiency with N-DMBI as well as the conductivity and stability in the conducting state. Our results highlight the need for post-deposition annealing to optimise the N-DMBI doping of the BTD-based polymer and ultimately its conductivity and thermoelectric properties.
Introduction
Organic semiconducting polymers have been extensively studied for more than three decades and are now emerging as the materials of choice for the development of the next generation of optoelectronic devices.1–3 Their unique ability to combine the mechanical properties of polymers with greater processability, chemically tuneable optoelectronic properties, potentially higher sustainability4 and lower production costs compared to inorganic materials has led to their adoption in many application areas.5,6 Like their inorganic counterparts, polymer-organic semiconductors can be divided into two categories: p-type materials, which primarily transport holes, and n-type materials, which primarily transport electrons.7Historically, the development of n-type polymers has lagged far behind their p-type counterparts in many organic electronic devices such as organic photovoltaics (OPVs)8 and organic field-effect transistors (OFETs)9 and more recently in organic electrochemical transistors (OECTs)10 and organic thermoelectric generators (OTEGs).11 This is mainly due to the dominant use of n-type molecular semiconductors such as fullerenes and its derivates and to inherent instability of doped n-type materials under ambient conditions.12 The synthesis of n-type polymers has gained momentum with the advent of the copolymer approach and the development of novel electron-rich (donor, D) and electron-deficient (acceptor, A) blocks, which allow the modulation of optoelectronic properties and energy levels to obtain more stable n-type materials.13
Among these classes of electron deficient moieties, naphthalene diimide (commonly abbreviated to NDI) is probably the most studied.14,15 Aromatic diimide species are known to undergo two reversible one-electron reductions.16 The resulting anions and dianions are indeed stabilised by a mesomeric effect. The electron density is mostly distributed over the oxygens of the two carbonyl groups.17 In addition to its remarkable electrochemical stability, this moiety has an advantageous packing ability due to its planar and rigid structure. The solubility and therefore the processability of the NDI derivatives can be tuned by alkylation of the nitrogen atoms. Most of the NDI-based polymers reported so far have β-branched octyldodecyl chains, allowing them to be processed with environmentally friendly solvents.18 Finally, NDI-based copolymers typically exhibit LUMO energy levels between −3.8 eV and −4.1 eV and some of them have shown remarkable air stability.19,20 For all these reasons, NDI copolymers have emerged as promising materials for future industrial developments in energy conversion devices.21,22 One example is their use in organic photovoltaic devices with power conversion efficiencies above 11%.18 In the field of thermoelectricity, NDI-based polymers have shown good conductivity of 1.8 ± 0.1 S cm−1 leading to a power factor of 4.6 ± 0.2 μW m−1 K−2.23 They also show the lowest thermal conductivity values reported for n-type doped conjugated polymers.24 There is a plethora of n-type materials based on NDI or BTD in the literature, but the combination of these two electron-deficient units within a single structure has rarely been explored.15,21,22 Although there are examples of polymers combining these two units, the effects of introducing the BTD unit as a third co-monomer on the transport properties and, in particular, the doping mechanisms of these materials have not been elucidated. In this study, we report on the synthesis of two NDI-based polymers, one of which contains a BTD unit, and we investigate in detail their optoelectronic and transport properties. We investigate the effect of BTD on molecular doping using 4-(2,3-dihydro-1,3-dimethyl-1H-benzimidazol-2-yl)-N,N-dimethylbenzenamine (also known as N-DMBI) as a dopant. We show that the introduction of a BTD unit does not significantly affect the optoelectronic properties, but helps to improve the charge transport and doping efficiency with N-DMBI and, hence, the conductivity and thermoelectric properties.
Results and discussion
Polymers synthesis
Fig. 1 shows the synthetic routes to the synthons and the two NDI-based copolymers. The preparation of the copolymers requires the synthesis of a di-brominated NDI precursor 6 starting from naphthalene di-anhydride (Fig. S1, ESI†). This step is limiting since the electrophilic aromatic substitution is hindered by the electron deficiency of the substrate. In addition, the bromination product is insoluble in common solvents, making it difficult to purify and analyse. Despite the widespread use of NDI-based compounds, the synthesis of 2,6-dibromonaphthalene dianhydride 6 is rarely reported. Our review of the literature shows a wide variation in reaction yields ranging from 12 to 42% (two steps, after N-alkylation) with the best yields described as poorly reproducible.25 The standard bromination methods are: (i) oleum (fuming sulphuric acid) with Br2, or (ii) concentrated sulphuric acid (H2SO4) with dibromo-isocyanuric acid as the brominating agent. Compared to Br2, dibromo isocyanuric acid is expensive. As for oleum, it is very toxic by inhalation and reacts via a dangerous exothermic reaction (explosively with water). In an attempt to reduce the use of oleum and to find a low cost route to produce gram scale dibrominated naphthalene dianhydride, we undertook a systematic study inspired by the work of Reczek et al.26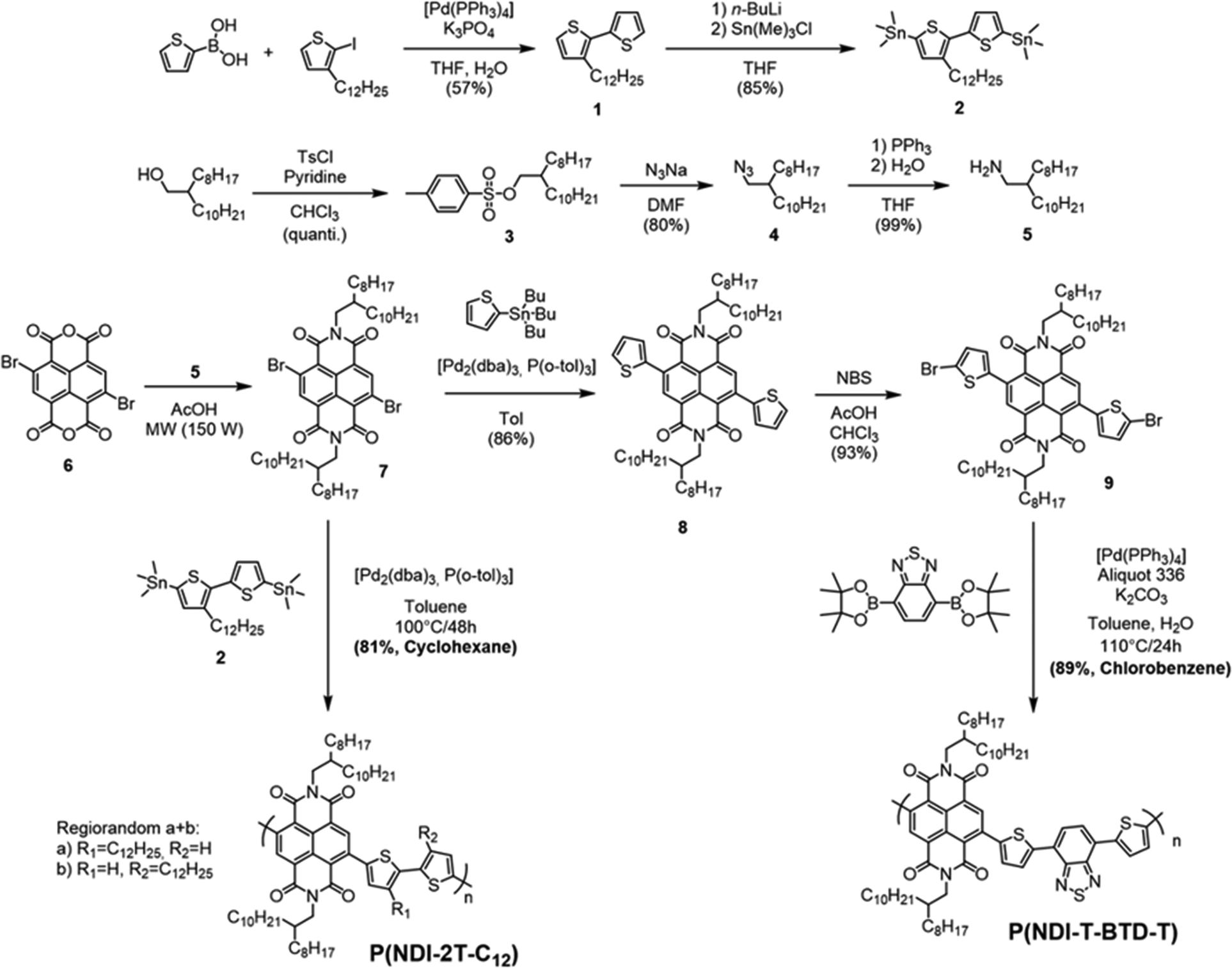 | ||
| Fig. 1 Synthetic route towards the precursors and the NDI-based copolymers. | ||
Firstly, we set up the conditions for preparing compound 7 by synthesising an analogue bearing an n-octyl chain. The synthesis routes and total yields obtained for the preparation of the compound 7a are summarised in Fig. S1 (ESI†). In a first step, three brominating agents were tested. The resulting crude products were then N-alkylated with commercial n-octylamine by microwave irradiation. The product 7a can be conveniently purified by filtration to quickly obtain a soluble product for routine NMR analysis. Note that a highly protic solvent (glacial AcOH) is required to avoid nucleophilic aromatic substitution of bromine by alkylamine.27 For reference, we obtained a total yield of 11% 7a using dibromo-isocyanuric acid. We first tried using NaBr as a brominating agent,28 which did not give any reaction in H2SO4, but gave 14% dibrominated NDI 7a in oleum (150 °C/16 h) with significant amounts of non-functionalised and mono-brominated compound. At a lower temperature (110 °C), a lower yield was obtained even after 69 hours of reaction. To reduce the hazard, we focused on optimising the conditions at 100 °C with Br2. Finally, we identified suitable Br2/oleum conditions (0.15 mol L−1, 100 °C/40 h) which gave pure 7a with a good yield of 37%. These conditions were then used to synthesise the desired monomer 7 with octyl-dodecyl side chains using amine 4. Prior to optimisation, residual brominated NDI was difficult to remove from the final product (Fig. S2, blue 1H NMR trace, 8.2–8.9 ppm, ESI†). Optimisation of the bromination step enhances the formation of the di-brominated product and thus facilitates the purification of the monomer. With the optimised route, we were able to synthesise compound 7 on a gram scale with a satisfactory yield (33%) and excellent purity (Fig. S2, red trace, ESI†).
Starting from compound 7, we synthesized the P(NDI-2T-C12) with a regiorandom dodecyl side chain on only one thiophene, being an analogue of the archetypal P(NDI-2T) without side chains on the thiophene, also known commercially as N2200. Interestingly, P(NDI-2T-C12) is highly soluble in common organic solvents and most of the formed copolymer was extracted from the cyclohexane fraction after Soxhlet purification. This fraction shows a moderate number average molar mass (Mn) of 8.6 kg mol−1 and a dispersity (Đ) of 1.9.
Starting from the compound 7, we can easily access the compound 8 where two thiophenes are introduced via Stille coupling.29 This compound can then be brominated and used as a precursor for the preparation of P(NDI-T-BTD-T). In this copolymer, the thiophenes (abbreviated T) act as spacers allowing higher number average molar mass to be achieved. Indeed, low Mn of only 5.2 kg mol−1 was reached for P(NDI-BTD)30 – without thiophene – while Fu and coll. managed to reach Mn up to 31.0 kg mol−1 by introducing a T-spacer to form P(NDI-T-BTD-T)31 (both synthesised by Suzuki coupling). We tentatively attribute the higher degree of polymerisation to a higher reactivity of the donor thiophene unit. We further optimised the polymerisation by reproducing the conditions used by Fu and coll.31 but with a shorter reaction time of 24 hours instead of 48 hours. We hence obtained P(NDI-T-BTD-T) with an excellent polymerisation yield of 89% (vs. 47%,31 chlorobenzene fraction) and a higher Mn estimated to be 64.1 kg mol−1 (vs. 31.0 kg mol−1). The dispersity (Đ) is 4.6 (see Fig. S11, ESI†). A higher Mn along with a higher polymerization yield, suggest that reducing the reaction time allows better control of the polymer chain length and prevents insoluble polymer aggregates. These differences may not only be due to the shorter reaction time, but also to the purity of the materials and the accuracy of the weighting according to Carothers's equation.32
Optical and electronic properties
The UV-Vis absorption spectra of the two polymers were recorded in solution and in thin films (Fig. 2). Spectral data are summarised in Table 1. In solution, P(NDI-2T-C12) and P(NDI-T-BTD-T) exhibit a defined absorption band at high energy (∼350 nm), which is attributed to the S0–S1 transition. For both D–A copolymers, a broad absorption band at lower energy (∼600–785 nm) is observed and attributed to the internal charge transfer (ICT) absorption band between the donor and acceptor units.33 When deposited as thin films, no obvious change in the S0–S1 transition bands is observed. As previously reported,34 no change in the ICT band is observed for P(NDI-T-BTD-T). However, significant bathochromic shifts of the ICT bands are observed for P(NDI-2T-C12) from solution to film. Such a red shift indicates a reduction in the energy gap associated with charge transfer. In solution, the ICT is associated to an intra-chain HOMO–LUMO transition, while in solid state it is attributed to an inter-chain charge transfer from the HOMO of one chain to the LUMO of another π-stacked chain, in accordance with the π-stacking observed in GIWAXS.35 This results in a strong red-shift of about 150 nm and 40 nm of the absorption onsets for P(NDI-2T-C12) and P(NDI-T-BTD-T), respectively.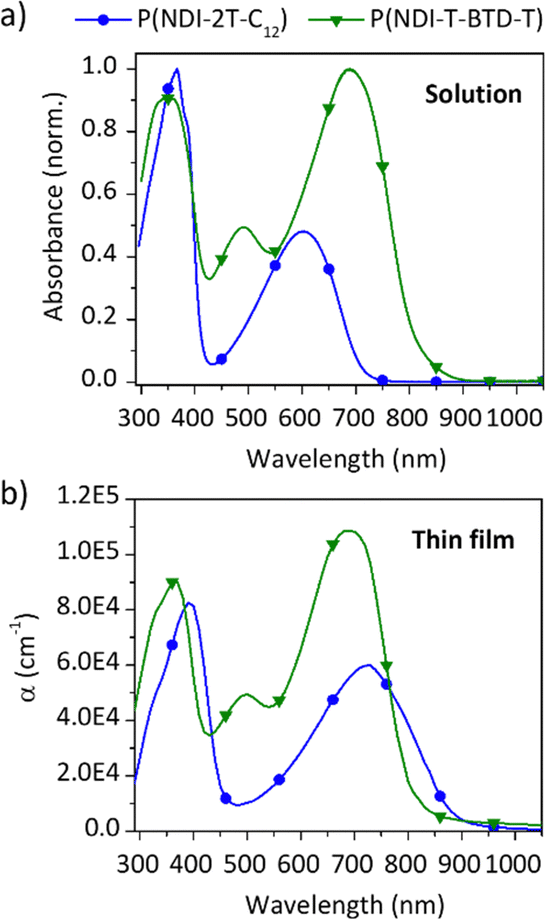 | ||
| Fig. 2 UV-Vis absorption spectra of both polymers (a) in chlorobenzene solution at 298 K normalized to the maximum absorption peak, and (b) as as-cast thin films in ambient conditions. | ||
| Copolymers | λ max_sol (nm) | λ max_film (nm) | λ onset (nm) |
E
g
opt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (eV)
(eV) |
E LUMO (eV) | E HOMO (eV) |
E
g
CV
(eV) |
E b (eV) |
μ
OFET
max
(cm2 V−1 s−1) |
σ (S cm−1) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Wavelength of peak maxima in chlorobenzene solution at 298 K. b Wavelength of peak maxima in as as-cast thin films. Brackets: absorption coefficients calculated by α = (2.303 abs)/thickness in cm−1.39 c Wavelength of absorbance onset in films. d Corresponding optical band gap calculated by hc/λonset. e Values extracted from CV on films (Fig. 3a). Calculated by ELUMO (eV) ≈ −[4.8 + Eredonset (V vs. Fc+/Fc)] f Values extracted from CV on films (Fig. 3a). Calculated by ELUMO (eV) ≈ −[4.8 + Eredonset (V vs. Fc+/Fc)]. g Values extracted from CV on films (Fig. 3a). Corresponding electrochemical band gap calculated by EgCV (eV) = ELUMO – EHOMO. Brackets: DFT results at the B3LYP/TZ2P level. h Exciton binding energy calculated by Eb = EgCV − Egopt.36 i Electron field-effect mobilities in saturation regime optimized by post-deposition thermal annealing in inert atmosphere. j Optimized electrical conductivity with N-DMBI doping. k Without thermal activation of the dopant. | ||||||||||
| P(NDI-2T-C12) | 365, 605 | 390 (8.3 × 104) | 890 | 1.4 | −3.8 (−3.62) | −5.8 (−5.36) | 2.0 (1.74) | 0.6 | (2.2 ± 0.4) × 10−3 | (1.3 ± 0.5) × 10−4k |
| 725 (5.9 × 104) | ||||||||||
| P(NDI-T-BTD-T) | 350, 490, 690 | 370 (9.0 × 104) | 810 | 1.5 | −3.8 (−3.63) | −5.7 (−5.39) | 1.9 (1.76) | 0.4 | (5.3 ± 0.3) × 10−2 | (1.2 ± 0.4) × 10−3 |
| 700 (1.1 × 105) | ||||||||||
| Values extracted from UV-Vis-NIR absorbance spectra (Fig. 2).abcd | ||||||||||
The difference with the transport bandgap of 2.0 eV (Eg measured by CV) indicates a rather high exciton binding energy (Eb) of about 0.6 eV.36
In thin films, P(NDI-2T-C12) exhibits two peaks in terms of absorption coefficients at 8.3 × 104 cm−1 at 390 nm and 5.9 × 104 cm−1 at 725 nm. In comparison, P(NDI-T-BTD-T) shows higher absorption coefficients for almost the entire visible range (450 to 750 nm) with two maxima at 9.0 × 104 cm−1 at 370 nm and 1.1 × 105 cm−1 at 700 nm (Table 1). This improvement is tentatively attributed to enhanced polaronic delocalisation due to the extended π conjugation afforded by the incorporation of the BTD spacer.
Cyclic voltammetry (CV) experiments were carried out to elucidate the optoelectronic properties of the two polymers (Fig. 3a and b). The voltammograms were recorded at a scan rate of 50 mV s−1 without stirring to limit analyte convection.37 The polymers were drop-cast films on the working electrode from chloroform solution for P(NDI-2T-C12) and chlorobenzene for P(NDI-T-BTD-T). The LUMO and HOMO energy levels were estimated from the reduction onset potential Ered and the oxidation onset potential Eox, respectively. For both polymers the oxidation waves show a lack of reversibility, which is not surprising for n-type materials. In agreement with the DFT-calculated values, the estimated HOMO energy levels are in the same range for both polymers, with P(NDI-2T-C12) at −5.8 eV and P(NDI-T-BTD-T) at −5.7 eV. When analysing the reduction process, we found that both polymers P(NDI-2T-C12) and P(NDI-T-BTD-T) exhibit stepwise one-electron transfers upon reduction.17
 | ||
| Fig. 3 (a) Cyclic voltammograms of both NDI-based copolymers. Experimental conditions: acetonitrile (MeCN), scan rate = 50 mV s−1, 0.1 M [NBu4][PF6], Pt electrode, 0.01 M Ag/AgNO3 calibrated Fc+/Fc. (b) Comparison of CV-measured (thick lines ±0.1 eV) and DFT-calculated (dashed lines) HOMO and LUMO energy levels. (c) Ground-state DFT calculations: (top) geometry-optimized structures and dihedral angles (VWN + PBE/TZ2P/Grimme 3/MeCN), (middle and bottom) LUMO and HOMO electronic density distributions, respectively (B3LYP/TZ2P/MeCN). The copolymers are approximated to dimers and the alkyl chains are replaced by ethyl chains. | ||
These remarkably stable and reversible reduction waves are attributed to the two successive reductions of the NDI moiety (Fig. S12, ESI†). Such stability of the anion and di-anion species is valuable for the long-term operation in thermoelectric devices. It should also be noted that for P(NDI-T-BTD-T), the introduction of a BTD unit has a minor influence on the band gap when compared to P(NDI-2T-C12). It should be noted that for P(NDI-T-BTD-T), the values reported by Michinobu and coll. are in accordance with our measurements (ELUMO = −3.8 vs. −3.52 eV,34EHOMO = −5.7 vs. −5.68 eV34).
These experimental observations are supported by modelling. density functional theory (DFT) calculations allowed us to estimate the overall molecular geometry, the electronic density distribution along the conjugated backbone and the energy levels of the frontier orbitals (Fig. 3b and c). Since the length of the alkyl side chains has a negligible effect on the position of the energy levels, phenyl-alkyl and alkyl chains have been substituted by phenyl-methyl and ethyl or propyl chains respectively.38 From the DFT calculations, it can be clearly seen that the HOMO energy level is spatially extended over the two thiophenes in P(NDI-2T-C12) and additionally delocalised on the benzene ring of the BTD spacer in P(NDI-T-BTD-T). The large torsional angles between the thiophenes and the NDI unit in the two polymers (comprised between 28° and 38°) prevent the delocalisation of the HOMO over the NDI units. The HOMO energy levels are found at −5.36 eV in P(NDI-2T-C12) and −5.39 eV in P(NDI-T-BTD-T). Regarding to the LUMO energy levels, they are strongly localised on the NDI units in both systems, and the energy level are found at around −3.6 eV.
Transport properties in pristine and doped states
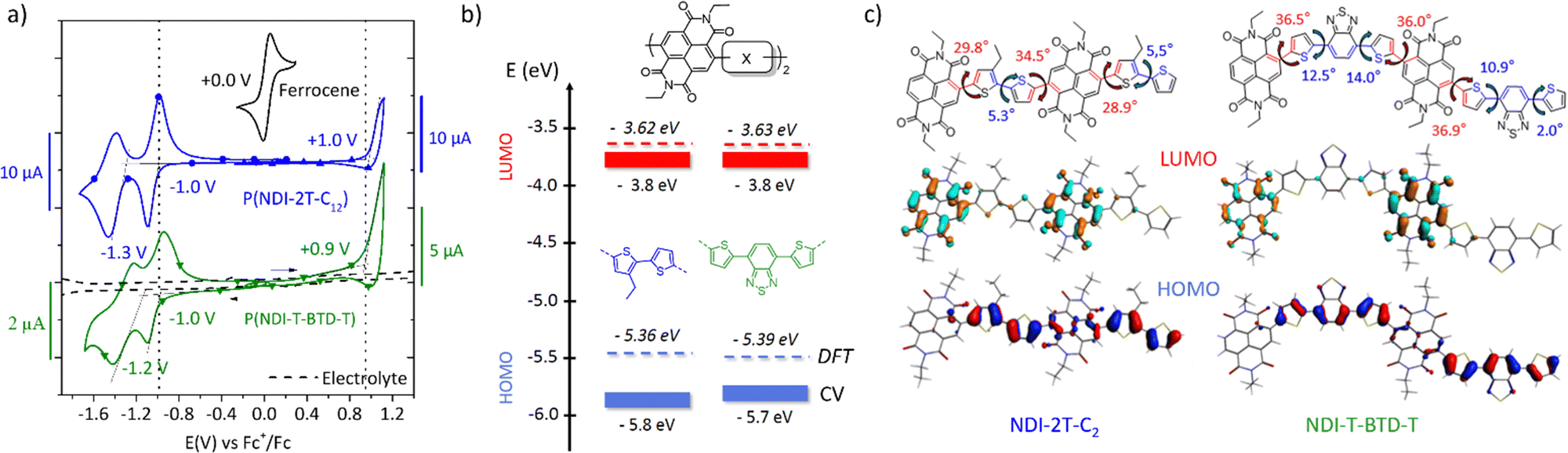
The evolution of the electron mobility versus annealing temperature in the saturation regime is shown in Fig. 4. The corresponding output and transfer characteristics are shown in Fig. S15c–f (ESI†). Interestingly, both materials show enhanced field-effect mobility after thermal annealing. P(NDI-2T-C12) shows a maximum mobility of (2.2 ± 0.4) × 10−3 cm2 V−1 s−1 after annealing at 120 °C. Above 120 °C, both the electron mobility and Ion/Ioff ratio decrease. This behaviour was confirmed on several devices. Considering the remarkable thermal stability up to 350 °C of P(NDI-2T-C12) measured by thermogravimetric analysis on powder (TGA, Fig. S13, ESI†), the decrease in mobility upon annealing therefore suggests a degradation of the OFET device rather than a degradation of the polymer itself. Potential causes of degradation include unfavourable morphological rearrangement of the polymer channel impeding long-range charge transport44 (e.g., charge trapping, grain boundaries, and lower molecular order) and degradation of the polymer/contact interface resulting in higher resistances to charge injection. Conversely, P(NDI-T-BTD-T) shows a steady improvement with increasing annealing temperature up to stability limit at 200 °C, reaching (5.3 ± 0.3) × 10−2 cm2 V−1 s−1, in excellent agreement with previously reported mobility.31 GIWAXS diffractograms show a decrease of the π-stacking signal after annealing at 200 °C, but a favourable enhancement of the edge-on lamellar orientation, which is likely responsible for the improved mobility (Fig. S15b and d, ESI†). Interestingly, annealing the OFETs also appears to be an effective post-processing method to increase the Ion/Ioff ratio and decrease hysteresis in-line with a charge-carrier trap reduction in the OFET channel after thermal annealing.45,46
 | ||
| Fig. 4 (a) and (b) Electron mobility in saturation regime (VDS = +100 V) as a function of the annealing temperature of P(NDI-2T-C12) and P(NDI-T-BTD-T) bottom gate bottom contacts OFETs. | ||
To reach sufficient electrical conductivity for thermoelectric applications, we have doped the polymers with the widely used n-dopant precursor N-DMBI.12,47 In our previous work, the HOMO energy level of N-DMBI was measured by CV at −4.4 eV.48 Using the same setup, the LUMO levels of the polymers were measured at −3.8 eV (Fig. 3a), which should hinder direct electron transfer from N-DMBI HOMO. Doping via SOMO-mediated electron and hydride transfers are two possible pathways.49–51
Electron paramagnetic resonance (EPR) measurements provide useful insights on the doping events occurring in solution prior to the deposition. EPR in solution allows to easily compare the reactivity of the two polymers in the presence of N-DMBI. The comparison is easier in solution than in thin films as the thickness and morphology of the films do not interfere. P(NDI-2T-C12) and P(NDI-T-BTD-T) were stirred with optimum amount of N-DMBI in toluene and chlorobenzene respectively. Both solutions have the same total concentration of polymer (5 mg mL−1). The EPR spectra of the solutions stirred at room temperature (RT) and after heating up at 120 °C/15 min are shown in Fig. 5. In both cases, no obvious radical signal is measured for neat undoped polymers. Conversely, doped films exhibit EPR signals with symmetric line shapes (mostly Lorentzian). P(NDI-T-BTD-T):N-DMBI solution stirred at RT (green line) clearly exhibits free radicals (signal at 3440 G, g ∼2.0). This is a clear evidence of its doping at room temperature using N-DMBI.52 By analogy with PCBM,47 all spontaneous doping observed at RT in dark conditions can be associated to the spontaneous hydride transfer from N-DMBI-H to P(NDI-T-BTD-T). This leads to the formation of the polymer anion (P–H−), followed by an electron transfer yielding to the neutral radical (P–H˙) and the radical anion (P˙−) stabilized by N-DMBI+. As expected, the quantity of spins is significantly increased (by approximately 650%) after heating the doped P(NDI-T-BTD-T) solution at 120 °C (Fig. 5d green line). Similarly, the thermal activation of the dopant allows efficient doping of P(NDI-2T-C12). In such conditions, the SOMO mediated doping pathway is enabled. According to the double integration of the signal, less spins are generated on doped-P(NDI-2T-C12), however a rate limited reaction (due to lower concentration of N-DMBI) and the presence of diamagnetic bipolaronic species (inactive in EPR) cannot be excluded. Interestingly, doped NDI-based polymers exhibit both air stability over 30 minutes of exposure (purple lines). The slight signal increase could originate from the kinetic of spin generation. These experiments show that: (i) both polymers can be n-doped with N-DMBI in solution, (ii) heating up the solution at 120 °C allows more efficient doping, (iii) the doping is stable in aerated solution over short period of time. The UV-Vis-NIR absorbance spectra of neat and doped films of P(NDI-2T-C12) and P(NDI-T-BTD-T) at various dopant concentrations of N-DMBI are presented in Fig. 6a–d. The new optical transitions can be used as a fingerprint to identify the formation of charge-transfer complexes (CTCs) or ion-pairs by UV-Vis-NIR spectroscopy.53 Neat films exhibit two neutral peaks with absorbance maxima at 400 nm/725 nm and 370 nm/700 nm for P(NDI-2T-C12)/P(NDI-T-BTD-T), assigned to S0–S1/intramolecular charge transfer (ICT) transitions respectively. The S0–S1 transition of the polymers overlap with the absorption feature of neat N-DMBI molecules. For doped P(NDI-T-BTD-T), the increase of dopant concentration induces a bleaching of the neutral absorption band concomitant with a growth of broad sub-gap absorption transitions in the near-infrared domain. Similar behaviour is observed for doped P(NDI-2T-C12) films, the bleaching of the neutral peak being accompanied by the emergence of a high-energy signal at circa 500 nm and a broadening of the ICT peak in the 900–1100 nm range – in accordance with previous studies on doped N2200 and NDI-based polymers.23,54 The sub-gap absorbing bands are characteristic of transitions to (bi)polaronic levels, indicating effective doping.55 The films were then annealed at 120 °C/1 h under inert atmosphere to thermally activate N-DMBI dopant.
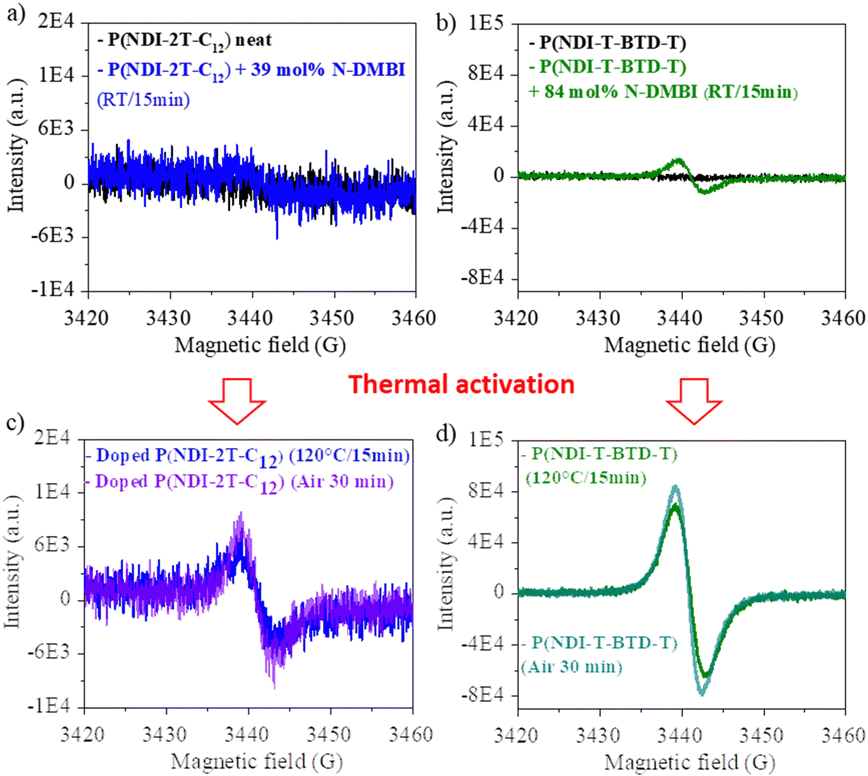 | ||
| Fig. 5 EPR spectra of (a) neat P(NDI-2T-C12) and P(NDI-2T-C12):N-DMBI mixed at RT in toluene (inert atmosphere, in dark). (b) Neat P(NDI-T-BTD-T) and P(NDI-T-BTD-T):N-DMBI mixed at RT in chlorobenzene (inert atmosphere, dark). (c) and (d) Same solutions heated up at 120 °C in inert atmosphere and exposed to air. Magnetic field power: 158.63 mW, modulation amplitude: 3.0 G. | ||
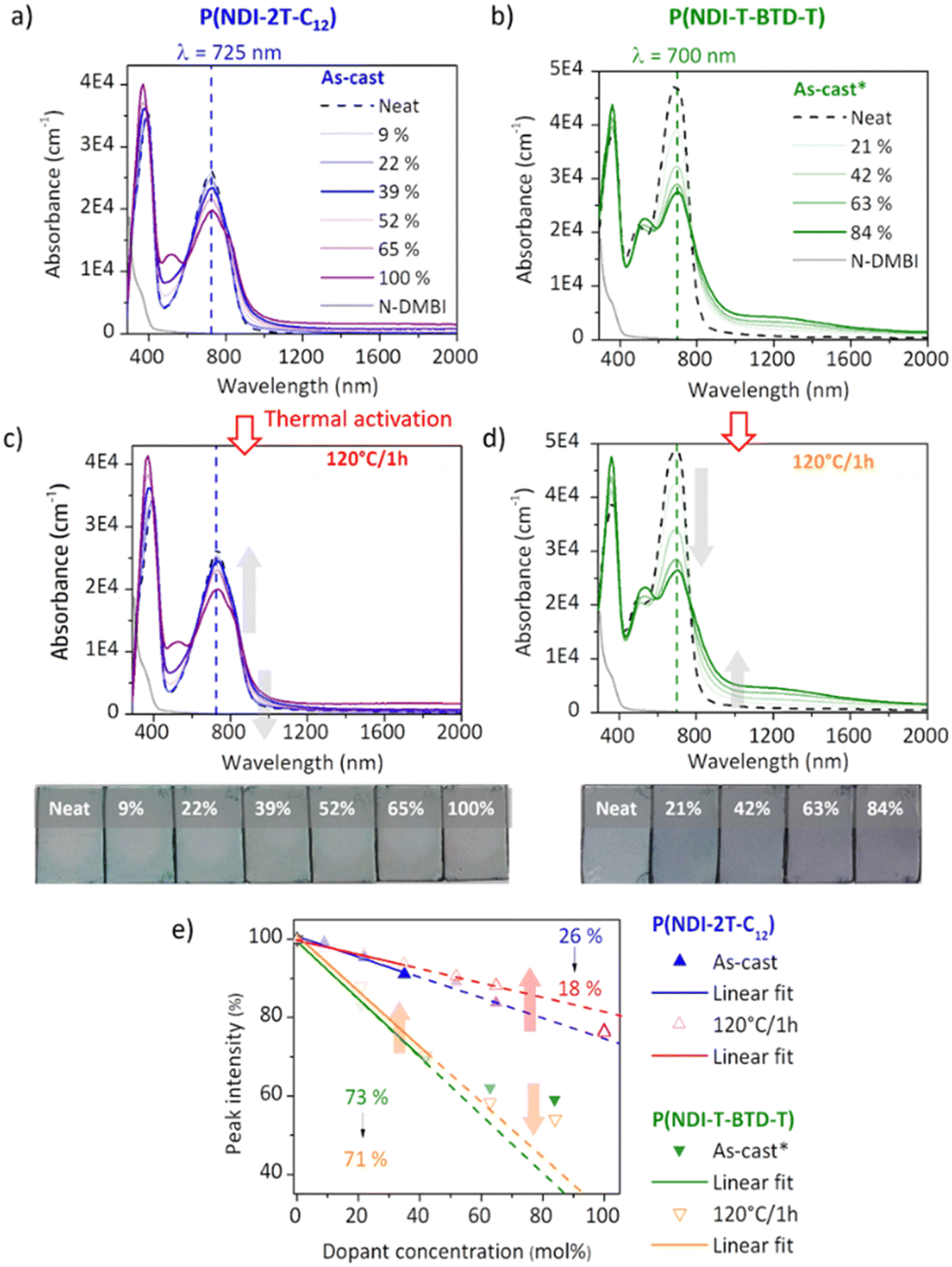 | ||
| Fig. 6 UV-Vis-NIR absorbance spectra of neutral and doped (+x mol% of N-DMBI) (a) and (c) P(NDI-2T-C12) (thickness = 30–40 nm) and (b) and (d) P(NDI-T-BTD-T) thin films (thickness = 40–45 nm). (a) and (b) As-cast (*from 110 °C solution). (c) and (d) After annealing at 120 °C/1h in glovebox. The arrows symbolize the evolution trend after annealing. Inset: Pictures of all the corresponding samples. (e) Absorption peak intensity at the neutral peak (725 nm for P(NDI-2T-C12) and 700 nm for P(NDI-T-BTD-T)) normalized to that of the neat film. Inset: Doping efficiency derived from the slope of the linear correlation between the peak intensity and the dopant concentration. The arrows highlight the evolution trends upon thermal activation. | ||
Surprisingly, it causes a slight dedoping of P(NDI-2T-C12) layers evidenced by a recovery of the neutral absorption band. In opposition, the doping level increases for annealed P(NDI-T-BTD-T) layers at high dopant concentration, as expected. To help analysing the data, we plotted the normalized neutral peak intensity evolution as a function of the dopant concentration for as-cast and annealed films. It provides useful insights on the doping efficiency.
At low dopant concentration, we observe a linear dependence of the peak intensity with the dopant concentration. It suggests a first order reduction reaction of neutral chromophore to polaronic species, as supported by the isosbestic point at ca. 800 nm.56 The doping efficiency is approximated to the slope of the linear fit.23 The sub-linear deviation at high dopant concentration (>50 mol%) observed for P(NDI-T-BTD-T) can be attributed to either the formation of higher reduction states that absorb at the sampled wavelengths (e.g. bipolaronic species),57 or the aggregation of N-DMBI within the polymer medium58 which would decrease the ionization efficiency.59 However, despite this, the doping efficiency of P(NDI-T-BTD-T) is significantly higher compared to P(NDI-2T-C12), with a doping yield of around 70% vs. 20–25% (see slope of linear fit of Fig. 6e). One should keep in mind that P(NDI-T-BTD-T) films are casted from hot solutions, hence improving the doping. Yet, the higher doping efficiency observed by absorbance spectroscopy is in good accordance with the higher spin density found in EPR. For P(NDI-2T-C12) the doping level remains low, indicating that most of the added dopants do not contribute to (bi)polaron formation.
Upon annealing, we could expect an increase of the doping efficiency similarly to what was observed in solution in EPR, but the doping yield is decreased from 27% to 18%. It hence seems that, in the solid state, N-DMBI activation is counterbalanced by another effect. We clarified this phenomenon by probing the topography of doped films (22 mol%) before and after annealing using atomic force microscopy (AFM) (Fig. S17, ESI†) and found aggregates on the top surface of the films regardless of the deposition solvent used. It appears that neutral N-DMBI-H and/or the cation N-DMBI+ are partly immiscible with P(NDI-2T-C12), similarly to N2200.60 Consequently, the low doping level evidenced by UV-Vis-NIR can be rationalized by the shielding of dopants, which remain electrically inactive inside the aggregates. Moreover, upon annealing the aggregates tend to cluster together. As a result, the dopant/polymer interface area decreases, leading to a reduction in the effective number of neat dopants and cations available to generate and stabilize doped states, respectively. This explains the unexpected loss of doping spectral signature despite the activation of N-DMBI upon annealing in the solid state. For P(NDI-T-BTD-T), a similar impact of thermal annealing can be predicted for low dopant concentration considering the slight decrease of efficiency from 73% to 71%. In opposition, for dopant concentration above 64 mol%, annealing increases the doping efficiency.
This result implies that the aggregates in as-cast layers – presumably identified by the sub-linear deviation – contain neutral N-DMBI-H molecules that are enabled to participate in doping by morphological rearrangement upon annealing.
These observations shed light on the many intertwined parameters that determine the effectiveness of doping with N-DMBI for this class of polymers. The processing conditions, including stirring time and temperature, should be explicitly reported and should not be overlooked. The conductivity associated with the processing conditions discussed above is presented in Fig. 7.
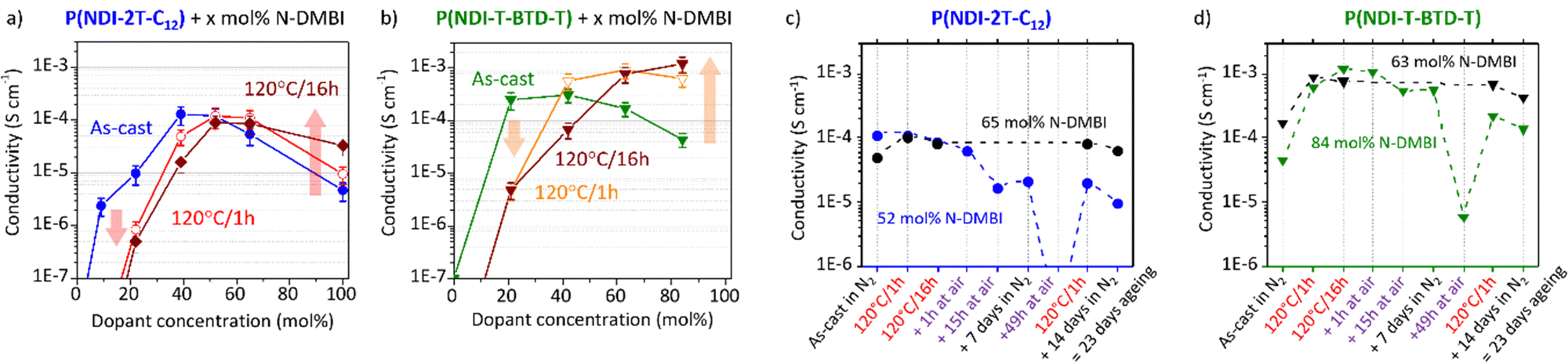 | ||
| Fig. 7 (a) and (b) Evolution of the conductivity of doped (a) P(NDI-2T-C12) and (b) P(NDI-T-BTD-T) films when exposed to ambient and inert atmospheres. Red lines correspond to the samples at optimal dopant concentration after annealing (thickness: 40 nm both). Black lines correspond to samples only aged under N2 atmosphere for reference (thickness: 45 nm and 40 nm respectively). (c) and (d) Evolution of the conductivity of doped (c) P(NDI-2T-C12) and (d) P(NDI-T-BTD-T) films. Red lines correspond to the samples at optimal dopant concentration after annealing (thickness: 40 nm both). Black lines correspond to samples only aged under N2 atmosphere for reference (thickness: 45 nm and 40 nm respectively). | ||
The mobility of undoped polymers is determined using OFET with gold contacts. For fair comparison, we used gold contacts for conductivity measurements to ensure similar contact resistances. Note that high injection barriers are expected considering the large energy offset between the LUMO levels of the polymers (−3.8 eV) and the work function of gold (−5.1 eV).61 The measured electrical properties may thus be underestimated.62 We tried to estimate the conductivity of the doped layers by four-point probe in ambient conditions.
Unfortunately, no reliable values were obtained despite brief air exposure less than 30 seconds and apparent air stability observed in solution using EPR. Consequently, instability caused by diffusion of oxidative ambient species within the solid polymer matrix can be speculated.63 Conductivity measurements in inert atmosphere are hence performed on both undoped and N-DMBI-doped films using the Transfer Length Method (TLM, Fig. S18, ESI†). All the neat undoped films exhibit intrinsic resistivity high enough compared to the input impedance of the setup to reach the measurement limit. This is consistent with the low carrier density expected for non-resonant structures. Considering the lowest conductivity measured on this setup, the conductivity of the neat polymers should be inferior to 10−8 S cm−1. The electrical conductivities of as-cast and annealed doped films as a function of the dopant concentration are shown in Fig. 7a and b. The samples discussed here were deposited from the same solutions than the UV-Vis-NIR samples on the same day, in the same conditions for direct comparison.
Considering as-cast doped films, we clearly see that in both cases (blue and green lines) the conductivity rises with respect to the dopant concentration before decreasing. This phenomenon is commonly observed for molecular-doped organic layers.23,60,64,65 The decrease of activation energy with the addition of dopant is usually counterbalanced by a synergetic effect of dopant segregation, in accordance to the AFM and UV-Vis-NIR experiments, and a loss of morphological order due to lattice disruption upon dopant uptake. Upon annealing at 120 °C/1 h (red and orange lines), two distinct behaviours are observed (highlighted by arrows). At low dopant concentration, the conductivity decreases by more than one order of magnitude, whereas at high concentration the conductivity increases. The evolution of the conductivity agrees with the formation of (bi)polaronic absorbance bands previously discussed. Further annealing the films up to 16 h exacerbates these behaviours. Hence, the annealing time should also be controlled with care.
Maximum conductivities of (1.3 ± 0.5) × 10−4 S cm−1 for as-cast P(NDI-2T-C12):N-DMBI (39 mol%) film and of (1.2 ± 0.4) × 10−3 S cm−1 for annealed (120 °C/16 h) P(NDI-T-BTD-T):N-DMBI (84 mol%) film are achieved. Note that the optimal dopant concentrations (39 and 84 mol%) are high compared to the high-performing NDI-based polymers reported,24,60,66 especially for P(NDI-T-BTD-T). Considering the high doping efficiency determined for P(NDI-T-BTD-T), this observation suggests that dopant/polymer miscibility is not the limiting factor but rather that the carriers generated at low doping level are poorly mobile at the macro-scale (μm-mm) probed by TLM. The origins can be diverse, for instance: low dissociation of charge transfer states leading to bound charges, high density of electron traps, grain boundaries hindering long-range hopping. The conductivities reached for P(NDI-T-BTD-T) are about one order of magnitude higher than the conductivities achieved with P(NDI-2T-C12). It corroborates with the higher mobility observed for undoped films in OFET. While the achieved conductivity values may not reach the level of the highest performing thermoelectric materials,11,15 we have successfully demonstrated an enhancement in conductivity using N-DMBI without the requirement of post-deposition thermal annealing. Avoiding post-deposition annealing is particularly interesting for polymer films deposited on flexible substrates being more sensitive to high temperature.
The values reported for N-DMBI-doped N2200 are in the range of 10−4 to 10−2 S cm−1 after thermal annealing.67,68 With P(NDI-2T-C12) we achieved a maximum conductivity that was only slightly inferior. One avenue for improvement is to optimize the synthesis route to produce higher molar masses. These are expected to improve the long-range interconnectivity of the polymer chains and provide higher charge transport properties. It should be noted that we deposited doped-P(NDI-2T-C12) films by a simple spin-coating method, from a non-halogenated solvent (toluene) and without heating at any step.
For as-cast films, both materials exhibit conductivities of about 10−4 S cm−1. After annealing, the conductivity of P(NDI-2T-C12) slightly decreases while the conductivity of P(NDI-T-BTD-T) is enhanced of one order of magnitude as explained. The samples were then exposed to air one hour in ambient dark conditions, demonstrating remarkable air-stability. We conclude that the characterization made in air are relevant for the doped state of these polymers with LUMO level at −3.8 eV. After 15 hours of exposure, a five-fold conductivity drop is observed for P(NDI-2T-C12) vs. a two-fold drop for P(NDI-T-BTD-T) under the same conditions. The electron affinity and layer thicknesses being similar (ca. 40 nm), the slower degradation kinetic is tentatively assigned to a higher degree of crystallinity of doped-P(NDI-T-BTD-T) films – as observed for neat polymer (see GIWAXS in Fig. S16, ESI†). However, the partial recovery of the conductivity when returned to inert atmosphere (9 days in N2) suggests a reversible process. Moisture and oxygen are known to act as electron traps, thereby reducing the overall conductivity.69 Consequently, the conductivity loss is mainly assigned to the reversible adsorption and diffusion of moisture and molecular oxygen inside the polymer matrix. To highlight this phenomenon, we exposed the layers to air more than two days (49 h). Most of the mobile charges are trapped in P(NDI-2T-C12) film – the conductivity being below the measurement limit – while the conductivity of P(NDI-T-BTD-T) dropped by two orders of magnitudes (Fig. 7c and d). Remarkably, the initial values of conductivity can be recovered after annealing at 120 °C in only one hour in the glovebox. In 2018, one article reported on a similar behavior for N-DMBI-doped polymer.66 They assigned the conductance recovery to the accelerated desorption of the electron-trapping species upon post annealing.
A similar claim is found for another n-type dopant.70 Yet, considering the low doping efficiency and the dopant propensity to aggregate, the hypothesis of activation of pristine N-DMBI-H molecules trapped within the aggregates due to morphological rearrangement cannot be ruled out. The black lines correspond to samples only aged in glovebox. After 23 days in the glove box, 78% and 56% of the conductivity was retained for doped-P(NDI-2T-C12) and doped-P(NDI-T-BTD-T) films respectively. It is noteworthy that the loss of spontaneous conductivity on ageing under N2 seems to be relatively similar for films exposed to air (red) or not (black) (see between 9 days and 23 days). Identical conclusions were drawn by monitoring the (bi)polaronic absorption band bleaching/growth upon air exposure/re-annealing using UV-Vis-NIR spectroscopy. Overall, good conductivities in the range of 10−3 to 10−4 S cm−1 have been achieved with NDI-based polymers. Improving the dopant:host interaction should be considered to increase the doping efficiency and achieve the carrier concentration required for high power factor.66
Conclusions
In this work, we have synthesized and characterized the optoelectronic properties of two NDI- and thiophene-based copolymers differing by the presence of a BTD spacer unit with the aim of elucidating its effect on the transport properties and doping mechanism with N-DMBI. We show that the introduction of the BTD unit does not change the LUMO energy level found at −3.8 eV. The electron mobility of the materials is studied in OFETs and reaches 5.3 ± 0.3 × 10−2 cm2 V−1 s−1 for the polymer embedding a BTD spacer between the thiophenes. Upon doping with N-DMBI, we found a higher doping efficiency for the BTD-containing copolymer, leading to a better conductivity of up to (1.2 ± 0.4) × 10−3 S cm−1 for annealed films (120 °C/16 h), which is an order of magnitude higher compared to the copolymer without BTD.Notably, we have observed spontaneous doping of the copolymers without thermal treatment when cast with N-DMBI at room temperature and we show that post-deposition annealing can be detrimental in certain cases, here for low dopant concentrations. These findings are in stark contrast to the typical approach to n-doping with N-DMBI and provide the field with nuances regarding the need for and effect of thermal activation of the dopant. Therefore, we argue that as-cast N-DMBI doped films should be characterised prior to any thermal treatment. Indeed, a systematic post-deposition annealing may not be optimal depending on the N-DMBI:host miscibility and the tendency of the semiconducting host to form C–H bonds.
The stability of the doped films over time and when exposed to ambient conditions was investigated. We show that the introduction of the BTD units provides the polymer with a remarkable stability in the conducting state, even after a short period of exposure to air. We also found that the degradation of conductivity after prolonged exposure to air is reversible and that the initial values can be recovered after annealing at 120 °C in only one hour in a glove box. A further improvement in stability could be achieved with adapted encapsulation layers. Finally, we have measured the thermoelectric parameters of the doped BTD-based films and found a power factor of 9 × 10−3 μW m−1 K−2 at 115 °C.
Author contributions
O. B.: conceptualization, investigation, formal analysis, data curation, methodology, visualization, writing original draft, writing review & editing. Y. K.: investigation. A. A. M.: investigation, methodology, visualization. S. P.: methodology. T. N. D.: investigation, methodology. A. C.: methodology, validation. C. A.: investigation, methodology, validation. P. L.: investigation, methodology. A. C conceptualization, project administration. R. D.: conceptualization, formal analysis, project administration, supervision, resources, writing review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the financial support of the ANR through the project Harvesters (ANR-16-CE05-0029-01). Dr S. Gambarelli is acknowledged for assistance during EPR measurements and helpful discussions. O. B. and R. D. acknowledge the LABEX Laboratoire d'Alliances Nanosciences-Energies du Futur (LANEF, ANR-10-LABX-51-44001) for funding and the Hybriden Facility at CEA-Grenoble.Notes and references
- F. Garnier, G. Horowitz and D. Fichou, Synth. Met., 1989, 28, 705–714 CrossRef.
- G. Horowitz, Adv. Mater., 1990, 2, 287–292 CrossRef CAS.
- Z. Qin, C. Gao, W. W. H. Wong, M. K. Riede, T. Wang, H. Dong, Y. Zhen and W. Hu, J. Mater. Chem. C, 2020, 8, 14996–15008 RSC.
- A. Marrocchi, A. Facchetti, D. Lanari, C. Petrucci and L. Vaccaro, Energy Environ. Sci., 2016, 9, 763–786 RSC.
- G. Horowitz, J. Mater. Res., 2004, 19, 1946–1962 CrossRef CAS.
- O. Ostroverkhova, Chem. Rev., 2016, 116, 13279–13412 CrossRef CAS PubMed.
- G. Horowitz, D. Fichou and F. Garnier, Solid State Commun., 1989, 70, 385–388 CrossRef CAS.
- C. Gu, X. Su, Y. Li, B. Liu, Y. Tian, W. Tan, J. Ma and X. Bao, Mol. Syst. Des. Eng., 2022, 7, 1364–1384 RSC.
- J. Quinn, J. Zhu, X. Li, J. Wang and Y. Li, J. Mater. Chem. C, 2017, 5, 8654–8681 RSC.
- S. Yu, C. J. Kousseff and C. B. Nielsen, Synth. Met., 2023, 293, 117295 CrossRef CAS.
- A. Tripathi, Y. Lee, S. Lee and H. Young Woo, J. Mater. Chem. C, 2022, 10, 6114–6140 RSC.
- D. Yuan, W. Liu and X. Zhu, Chem. Soc. Rev., 2023, 52, 3842–3872 RSC.
- Y. Zhang, Y. Wang, C. Gao, Z. Ni, X. Zhang, W. Hu and H. Dong, Chem. Soc. Rev., 2023, 52, 1331–1381 RSC.
- H. Jia and T. Lei, J. Mater. Chem. C, 2019, 7, 12809–12821 RSC.
- M. Li and Y. Shi, ChemPlusChem, 2023, 88, e202300215 CrossRef CAS PubMed.
- Y. Matsunaga, K. Goto, K. Kubono, K. Sako and T. Shinmyozu, Chem. – Eur. J., 2014, 20, 7309–7316 CrossRef CAS PubMed.
- S. K. Lee, Y. Zu, A. Herrmann, Y. Geerts, K. Müllen and A. J. Bard, J. Am. Chem. Soc., 1999, 121, 3513–3520 CrossRef CAS.
- K. Zhang, R. Xia, B. Fan, X. Liu, Z. Wang, S. Dong, H.-L. Yip, L. Ying, F. Huang and Y. Cao, Adv. Mater., 2018, 30, 1803166 CrossRef PubMed.
- Y. Xu, J. Yuan, S. Zhou, M. Seifrid, L. Ying, B. Li, F. Huang, G. C. Bazan and W. Ma, Adv. Funct. Mater., 2019, 29, 1806747 CrossRef.
- A. F. Paterson, A. Savva, S. Wustoni, L. Tsetseris, B. D. Paulsen, H. Faber, A. H. Emwas, X. Chen, G. Nikiforidis, T. C. Hidalgo, M. Moser, I. P. Maria, J. Rivnay, I. McCulloch, T. D. Anthopoulos and S. Inal, Nat. Commun., 2020, 11, 3004 CrossRef CAS PubMed.
- N. Zhou and A. Facchetti, Mater. Today, 2018, 21, 377–390 CrossRef CAS.
- Y. Song, J. Ding, X. Dai, C. Li, C. Di and D. Zhang, ACS Mater. Lett., 2022, 4, 521–527 CrossRef CAS.
- J. Liu, G. Ye, B. van der Zee, J. Dong, X. Qiu, Y. Liu, G. Portale, R. C. Chiechi and L. J. A. Koster, Adv. Mater., 2018, 30, 1804290 CrossRef PubMed.
- D. R. Villalva, S. Singh, L. A. Galuska, A. Sharma, J. Han, J. Liu, M. A. Haque, S. Jang, A. H. Emwas, L. J. A. Koster, X. Gu, B. C. Schroeder and D. Baran, Mater. Horiz., 2022, 9, 500–508 RSC.
- S. Sharma, N. B. Kolhe, V. Gupta, V. Bharti, A. Sharma, R. Datt, S. Chand and S. K. Asha, Macromolecules, 2016, 49, 8113–8125 CrossRef CAS.
- A. C. Thompson, H. M. Grimm, A. G. Bé, K. J. McKnight and J. J. Reczek, Synth. Commun., 2015, 45, 1127–1136 CrossRef CAS.
- C. Thalacker, C. Röger and F. Würthner, J. Org. Chem., 2006, 71, 8098–8105 CrossRef CAS PubMed.
- T. D. M. Bell, S. Yap, C. H. Jani, S. V. Bhosale, J. Hofkens, F. C. De Schryver, S. J. Langford and K. P. Ghiggino, Chem. – Asian J., 2009, 4, 1542–1550 CrossRef CAS PubMed.
- J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508–524 CrossRef.
- S. Vasimalla, S. P. Senanayak, M. Sharma, K. S. Narayan and P. K. Iyer, Chem. Mater., 2014, 26, 4030–4037 CrossRef CAS.
- C. Gu, W. Hu, J. Yao and H. Fu, Chem. Mater., 2013, 25, 2178–2183 CrossRef CAS.
- W. H. Carothers, Trans. Faraday Soc., 1936, 32, 39 RSC.
- T. E. Anderson, E. W. Culver, I. Badía-Domínguez, W. D. Wilcox, C. E. Buysse, M. C. R. Delgado and S. C. Rasmussen, Phys. Chem. Chem. Phys., 2021, 23, 26534–26546 RSC.
- Y. Wang, T. Hasegawa, H. Matsumoto, T. Mori and T. Michinobu, Adv. Mater., 2018, 30, 1707164 CrossRef PubMed.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191 RSC.
- B. P. Rand, D. P. Burk and S. R. Forrest, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 115327 CrossRef.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- G. Kim, A.-R. Han, H. Rang Lee, J. Lee, J. Hak Oh and C. Yang, Chem. Commun., 2014, 50, 2180–2183 RSC.
- V. S. Sangawar and N. A. Moharil, Chem. Sci. Trans., 2012, 1, 447–455 CrossRef.
- W. D. Gill, J. Appl. Phys., 1972, 43, 5033–5040 CrossRef.
- J. Veres, S. D. Ogier, S. W. Leeming, D. C. Cupertino and S. M. Khaffaf, Adv. Funct. Mater., 2003, 13, 199–204 CrossRef CAS.
- H. Sirringhaus, P. J. Brown, R. H. Friend, M. M. Nielsen, K. Bechgaard, B. M. W. Langeveld-Voss, A. J. H. Spiering, R. A. J. Janssen, E. W. Meijer, P. Herwig and D. M. de Leeuw, Nature, 1999, 401, 685 CrossRef CAS.
- J.-M. Verilhac, G. LeBlevennec, D. Djurado, F. Rieutord, M. Chouiki, J.-P. Travers and A. Pron, Synth. Met., 2006, 156, 815–823 CrossRef CAS.
- A. Zen, J. Pflaum, S. Hirschmann, W. Zhuang, F. Jaiser, U. Asawapirom, J. P. Rabe, U. Scherf and D. Neher, Adv. Funct. Mater., 2004, 14, 757–764 CrossRef CAS.
- W. Shi, Y. Zheng, A. D. Taylor, J. Yu and H. E. Katz, Appl. Phys. Lett., 2017, 111, 043301 CrossRef.
- Y.-H. Zhao, W. Li, T. Shen, Y. Zhao, Y. Liu and Y. Wang, Sci. China: Chem., 2023, 66, 548–561 CrossRef CAS.
- B. D. Naab, S. Guo, S. Olthof, E. G. B. Evans, P. Wei, G. L. Millhauser, A. Kahn, S. Barlow, S. R. Marder and Z. Bao, J. Am. Chem. Soc., 2013, 135, 15018–15025 CrossRef CAS PubMed.
- O. Bardagot, P. Kubik, T. Marszalek, P. Veyre, A. A. Medjahed, M. Sandroni, B. Grévin, S. Pouget, T. Nunes Domschke, A. Carella, S. Gambarelli, W. Pisula and R. Demadrille, Adv. Funct. Mater., 2020, 30, 2000449 CrossRef CAS.
- O. Bardagot, C. Aumaître, A. Monmagnon, J. Pécaut, P.-A. Bayle and R. Demadrille, Appl. Phys. Lett., 2021, 118, 203904 CrossRef CAS.
- S. Jhulki, H.-I. Un, Y.-F. Ding, C. Risko, S. K. Mohapatra, J. Pei, S. Barlow and S. R. Marder, Chem, 2021, 7, 1050–1065 CAS.
- F. Pallini, S. Mattiello, N. Manfredi, S. Mecca, A. Fedorov, M. Sassi, K. Al Kurdi, Y.-F. Ding, C.-K. Pan, J. Pei, S. Barlow, S. R. Marder, T.-Q. Nguyen and L. Beverina, J. Mater. Chem. A, 2023, 11, 8192–8201 RSC.
- J. Han, C. Ganley, Q. Hu, X. Zhao, P. Clancy, T. P. Russell and H. E. Katz, Adv. Funct. Mater., 2021, 31, 2010567 CrossRef CAS.
- I. Zozoulenko, A. Singh, S. K. Singh, V. Gueskine, X. Crispin and M. Berggren, ACS Appl. Poly. Mater., 2019, 1, 83–94 CrossRef CAS.
- Y. Wang, M. Nakano, T. Michinobu, Y. Kiyota, T. Mori and K. Takimiya, Macromolecules, 2017, 50, 857–864 CrossRef CAS.
- D. Huang, H. Yao, Y. Cui, Y. Zou, F. Zhang, C. Wang, H. Shen, W. Jin, J. Zhu, Y. Diao, W. Xu, C. Di and D. Zhu, J. Am. Chem. Soc., 2017, 139, 13013–13023 CrossRef CAS PubMed.
- P. Cavassin, I. Holzer, D. Tsokkou, O. Bardagot, J. Réhault and N. Banerji, Adv. Mater., 2023, 2300308 CrossRef CAS PubMed.
- G. Rebetez, O. Bardagot, J. Affolter, J. Réhault and N. Banerji, Adv. Funct. Mater., 2022, 32, 2105821 CrossRef CAS.
- J. Liu, L. Qiu, R. Alessandri, X. Qiu, G. Portale, J. Dong, W. Talsma, G. Ye, A. A. Sengrian, P. C. T. Souza, M. A. Loi, R. C. Chiechi, S. J. Marrink, J. C. Hummelen and L. J. A. Koster, Adv. Mater., 2018, 30, 1704630 CrossRef PubMed.
- J.-M. Kim, S.-J. Yoo, C.-K. Moon, B. Sim, J.-H. Lee, H. Lim, J. W. Kim and J.-J. Kim, J. Phys. Chem. C, 2016, 120, 9475–9481 CrossRef CAS.
- R. A. Schlitz, F. G. Brunetti, A. M. Glaudell, P. L. Miller, M. A. Brady, C. J. Takacs, C. J. Hawker and M. L. Chabinyc, Adv. Mater., 2014, 26, 2825–2830 CrossRef CAS PubMed.
- L.-L. Chua, J. Zaumseil, J.-F. Chang, E. C.-W. Ou, P. K.-H. Ho, H. Sirringhaus and R. H. Friend, Nature, 2005, 434, 194–199 CrossRef CAS PubMed.
- Y. Zhao, Y. Guo and Y. Liu, Adv. Mater., 2013, 25, 5372–5391 CrossRef CAS PubMed.
- T. Nunes Domschke, O. Bardagot, A. Benayad, R. Demadrille, A. Carella, R. Clerc and A. Pereira, Synth. Met., 2020, 260, 116251 CrossRef CAS.
- J. Liu, L. Qiu, G. Portale, S. Torabi, M. C. A. Stuart, X. Qiu, M. Koopmans, R. C. Chiechi, J. C. Hummelen and L. J. Anton Koster, Nano Energy, 2018, 52, 183–191 CrossRef CAS.
- K. Shi, F. Zhang, C.-A. Di, T.-W. Yan, Y. Zou, X. Zhou, D. Zhu, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2015, 137, 6979–6982 CrossRef CAS PubMed.
- D. Kiefer, A. Giovannitti, H. Sun, T. Biskup, A. Hofmann, M. Koopmans, C. Cendra, S. Weber, L. J. Anton Koster, E. Olsson, J. Rivnay, S. Fabiano, I. McCulloch and C. Müller, ACS Energy Lett., 2018, 3, 278–285 CrossRef CAS PubMed.
- Y. M. Gross, D. Trefz, C. Dingler, D. Bauer, V. Vijayakumar, V. Untilova, L. Biniek, M. Brinkmann and S. Ludwigs, Chem. Mater., 2019, 31, 3542–3555 CrossRef CAS.
- S. Wang, D. Fazzi, Y. Puttisong, M. J. Jafari, Z. Chen, T. Ederth, J. W. Andreasen, W. M. Chen, A. Facchetti and S. Fabiano, Chem. Mater., 2019, 31, 3395–3406 CrossRef CAS PubMed.
- R. T. Weitz, K. Amsharov, U. Zschieschang, E. B. Villas, D. K. Goswami, M. Burghard, H. Dosch, M. Jansen, K. Kern and H. Klauk, J. Am. Chem. Soc., 2008, 130, 4637–4645 CrossRef CAS PubMed.
- M. L. Tietze, B. D. Rose, M. Schwarze, A. Fischer, S. Runge, J. Blochwitz-Nimoth, B. Lüssem, K. Leo and J.-L. Brédas, Adv. Funct. Mater., 2016, 26, 3730–3737 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional results highlighted in the main text. See DOI: https://doi.org/10.1039/d3tc02816j |
| This journal is © The Royal Society of Chemistry 2023 |